
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomic/molecular layer deposition of cerium(III) hybrid thin films using rigid organic precursors†
Parmish
Kaur
 a,
Arbresha
Muriqi
b,
Jan-Lucas
Wree
a,
Ramin
Ghiyasi
c,
Muhammad
Safdar
c,
Michael
Nolan
b,
Maarit
Karppinen
c and
Anjana
Devi
*a
a,
Arbresha
Muriqi
b,
Jan-Lucas
Wree
a,
Ramin
Ghiyasi
c,
Muhammad
Safdar
c,
Michael
Nolan
b,
Maarit
Karppinen
c and
Anjana
Devi
*a
aInorganic Materials Chemistry, Ruhr University Bochum, Universitätsstraße 150, 44801 Bochum, Germany. E-mail: anjana.devi@rub.de
bTyndall National Institute, University College Cork, Lee Maltings, Cork T12 R5CP, Ireland
cDepartment of Chemistry and Materials Science, Aalto University, Kemistintie 1, 00076 Aalto, Espoo, Finland
First published on 15th March 2022
Abstract
An atomic/molecular layer deposition (ALD/MLD) process for the fabrication of cerium-based metal–organic hybrid films is demonstrated for the first time. The highly reactive cerium(III) guanidinate precursor [Ce(dpdmg)3] was employed in combination with organic precursors composed of rigid backbones, terephthalic acid (TPA) and hydroquinone (HQ) for the growth of the respective hybrid films. Growth rates of the films as high as 5.4 Å per cycle for Ce-TPA and 4.8 Å per cycle for Ce-HQ at a deposition temperature of 200 °C were obtained. Density functional theory (DFT) investigations confirm the favorable interaction between the cerium precursor and the organic co-reactants and predict that Ce maintains its +3 oxidation state in the films. This was also confirmed experimentally by X-ray photoelectron spectroscopy (XPS). Additionally, the films are highly UV absorbing. Hence, we envision that these films could find future application as promising redox active materials and/or UV absorbing materials.
Introduction
Rare-earth based metal–organic hybrid materials are gaining considerable interest nowadays due to their promising applications including information storage,1 gas storage/separation,2 colorimetric luminescent thermometers,3 luminescent probes for sensing of chemical species,4 and heterogeneous catalysts,5 among others.6 They offer synthetic tunability of their physical and chemical properties with potential for numerous metal and organic linker combinations. The major advantage offered by such hybrid materials is the variety of intrinsic properties of the individual building blocks i.e., the metal and organic components, which can be further tuned according to the requirements of the application by the choice of the components.Cerium is the most accessible rare-earth metal, with an abundance of 66 ppm in the Earth's crust (copper at 68 ppm).7 It has a unique electronic configuration and redox behavior among the rare-earths. Ce4+ has the [Xe] noble gas configuration and Ce3+ is the only stable rare-earth ion with a single 4f1 electron. The presence of one electron in the low energy 4f orbital for Ce3+ and unique redox active nature of the Ce(III/IV) couple, make it promising for catalysis and luminescent materials.8 The rather interchangeable oxidation state of cerium finds importance in applications where redox activity is crucial such as automotive three-way catalysts, immediate temperature solid oxide fuel cells, etc. On the other hand, organics provide a natural and environmentally benign material resource with the possibility to tune the properties by employing redox-active organic molecules or through the addition of various functional groups using standard synthesis approaches.
Considering potential applications such as sensors, information storage, catalysis, etc., it would be advantageous to produce cerium-organic hybrid materials as thin films. In addition, the organic component can enhance the flexibility of the inorganic films which can help to build flexible devices for advanced applications. However, despite these appealing properties and applications, these materials are to date not common as thin films.
The atomic/molecular layer deposition (ALD/MLD) technique is uniquely suited for the gas-phase deposition of high-quality thin films of hybrid materials.9 This method is derived from ALD where metalorganic precursors are chemisorbed on the surface in well-separated pulses for reaction with co-reactants to form inorganic films with atomic-level control over film thickness, homogeneity, uniformity, and composition, even on structurally challenging substrates. In MLD, these precursors are replaced by organic precursors which results in the formation of purely organic films. ALD and MLD together imply the involvement of a metalorganic ALD precursor together with an organic MLD precursor for the deposition of hybrid organic–inorganic films.9 An additional benefit of employing this technique is the exclusion of unwanted solvent molecules from these hybrid films which cannot be avoided by using standard synthesis routes like solvothermal synthesis, hydrothermal synthesis, etc.8 Hence, for ALD/MLD processes, the choice of precursors plays an important role in determining the film properties and functionalities in the films. Furthermore, the building blocks (organic and inorganic) should have similar thermal properties so that they do not undergo decomposition or condensation and are volatile enough to be able to saturate the surface at given deposition temperatures. Additionally, they must be reactive towards each other.
Earlier studies on ALD/MLD deposition of hybrid materials have used alkyl diols, e.g., ethylene glycol or glycerol with reactive terminal hydroxyl groups. A common problem with such organic precursors is their tendency to undergo the so-called ‘double reactions’ in which both hydroxyl ends interact with the inorganic component and thereby terminating the surface. For diols this eliminates the active hydroxyls needed for growth, while for glycerol this is yielding a lower growth per cycle.10 On the other hand, recent work using rigid aromatic backbones alleviates this problem, since, as demonstrated in recent DFT studies,11 aromatic species prefer to interact in an upright mode rather than undergoing ‘double reactions’ and this results in higher growth rates.
For tetravalent cerium in ALD, the precursors recently employed include [Ce(thd)4]12 and [Ce(mmp)4],13 both of which have poor volatility and the former one has additionally poor reactivity as it requires strong oxidizing agent such as ozone for the reaction to take place. For trivalent cerium, the known precursors are [Ce(iPrCp)3],14 [Ce(dpamd)3],15 and [Ce(iPrCp)2dpamd],16 among which [Ce(iPrCp)3] and [Ce(dpamd)3] have better volatility than the cerium +4 precursors, but they require strong oxidizing agents such as O2 plasma for [Ce(iPrCp)3] and ozone for [Ce(dpamd)3] due to their low reactivity. The heteroleptic precursor [Ce(iPrCp)2dpamd] was shown to have improved thermal properties than the above mentioned ones and was shown to be reactive with water but it is cost expensive due to the poor reaction yields.16 Recently, a highly reactive and volatile cerium tris-guanidinate precursor [Ce(dpdmg)3] with Ce +3 oxidation state was reported for water-assisted thermal ALD yielding cerium oxide films.17 The high reactivity of all nitrogen coordinating cerium guanidinate complex [Ce(dpdmg)3] was additionally confirmed by density functional theory (DFT). Therefore, ALD/MLD with all nitrogen coordinated guanidinate ligands for cerium precursor and oxygen-based rigid organic precursors possessing similar thermal properties seems promising as rare-earths are highly oxyphilic. The guanidinate precursor of erbium was previously demonstrated for the growth of ALD/MLD hybrid films with 3,5-pyridine dicarboxylic acid as the organic precursor.18 The rigidity of the organic backbone is advantageous to avoid blocking the surface sites by double reactions.
Herein, new ALD/MLD processes for cerium-based hybrid thin films using [Ce(dpdmg)3] as a metal precursor and benzene-1,4-dicarboxylic acid (1,4-BDC) also known as terephthalic acid (TPA) and benzene-1,4-diol (BDO) also known as hydroquinone (HQ) as organic precursors, respectively, are demonstrated. Fig. 1 shows the chemical structures of the precursors employed. These organic precursors contain the rigid aromatic phenyl ring which will promote growth of thicker films when compared to alkyl diols. Computational DFT calculations showed the feasibility of the reaction between the precursors and predicts the oxidation state of cerium in the films which are further confirmed by X-ray photoelectron spectroscopy (XPS). Additionally, the resulting thin films are analyzed for their composition and optical properties.
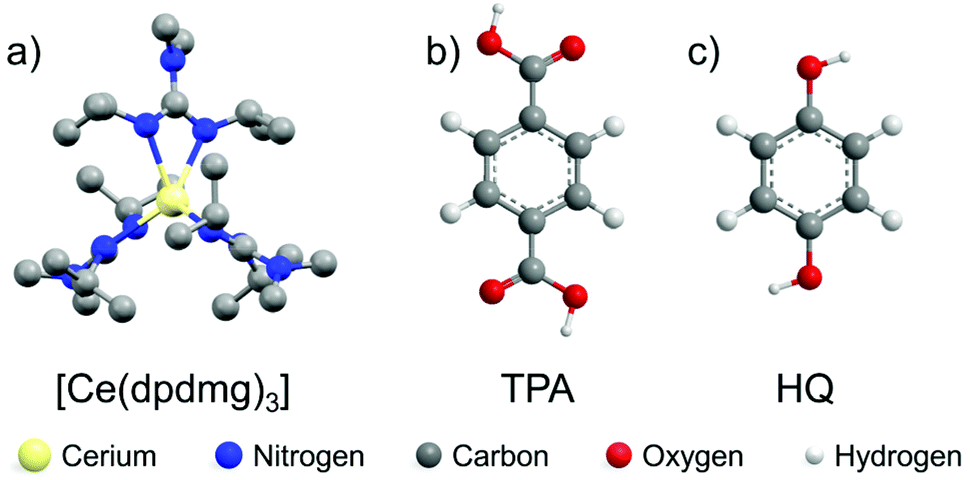 | ||
| Fig. 1 Molecular structures of the precursors (a) [Ce(dpdmg)3] (hydrogens are omitted for clarity) (CCDC 2023020†) (b) terephthalic acid (TPA) (c) hydroquinone (HQ). | ||
Results and discussion
In order to develop an efficient ALD/MLD process, it is important to address the compatibility of the organic and inorganic counterparts. In this context, physicochemical properties such as volatility, reactivity and thermal stability of the precursors [Ce(dpdmg)3], TPA and HQ were thoroughly investigated.Thermal investigations of the precursors
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed to study the volatility and thermal stability of the precursors.The TGA results in Fig. 2 (solid line) show the onset of volatilization (1% weight loss) for [Ce(dpdmg)3] at 90 °C; at 264 °C for TPA and at 139 °C for HQ under atmospheric pressure. The endothermic peaks in the DSC correspond to the melting points of the precursors which are found to be 104 °C for [Ce(dpdmg)3] and 172 °C for HQ. TPA sublimes without melting and the nearly 100% weight loss step in TGA for TPA and HQ can be attributed to sublimation of the respective precursors without any decomposition. However, a high residual weight of 25% for [Ce(dpdmg)3] precursor is obtained which corresponds to precursor decomposition at temperatures above 270 °C. From the thermal analysis studies it can be inferred that both the inorganic and organic precursors are volatile and thermally stable at given deposition temperatures and possess a compatible window for sublimation that can be beneficial ALD/MLD processing.
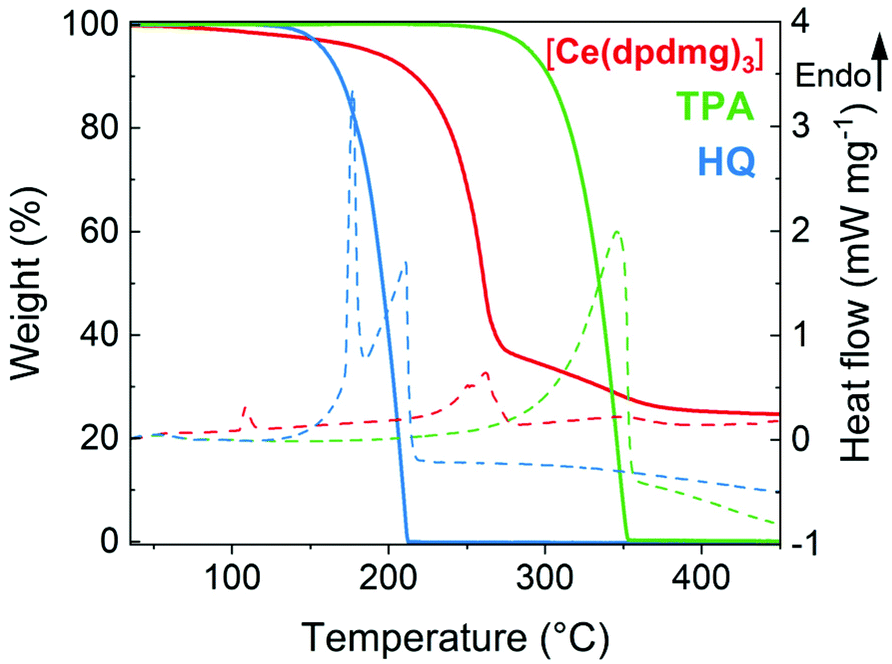 | ||
| Fig. 2 Thermogravimetric analysis (solid line) and differential scanning calorimetry (dashed line) of [Ce(dpdmg)3] in red, TPA in green and HQ in blue. | ||
ALD/MLD process development
Based on the promising thermal properties and being reactive, they were utilized to develop new ALD/MLD processes for Ce containing hybrid films. The films were deposited on Si(100) substrates at a deposition temperature of 200 °C and the self-limiting behaviour of the film growth was investigated, first for the terephthalic acid-based films. The [Ce(dpdmg)3] and TPA precursors were kept at vaporisation temperatures of 140 °C and 185 °C respectively. The TPA pulse length was varied from 2 to 10 s while all other parameters were kept constant: the [Ce(dpdmg)3] pulse length at 8 s, the N2 purge length at 30 s after both precursor pulses, and the number of deposition cycles at 75. It can be seen from Fig. 3a, that the 6 s pulse length for TPA was long enough to achieve surface saturation. The corresponding film growth rate expressed as growth-per-cycle (GPC) was 5.94 Å. After this, the [Ce(dpdmg)3] pulse length was varied from 4 to 12 s.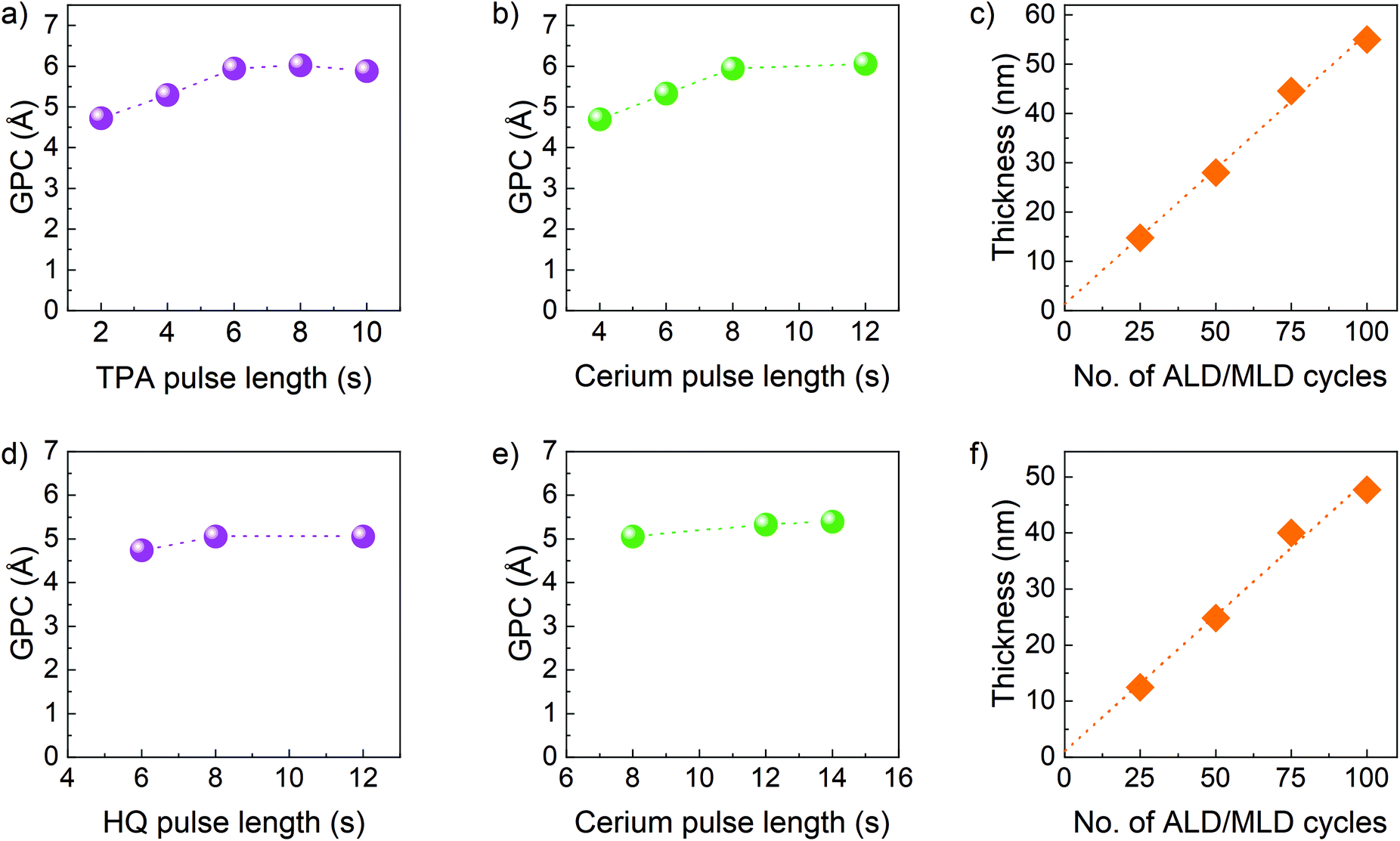 | ||
| Fig. 3 Precursor saturation studies by varing: (a) TPA pulse length and (b) [Ce(dpdmg)3] pulse length for Ce-TPA film growth, (d) HQ pulse length and (e) [Ce(dpdmg)3] pulse length for Ce-HQ film growth. Thickness of the film vs. number of applied ALD/MLD cycles for: (c) Ce-TPA film and (f) Ce-HQ film. The estimated error bars are smaller than the data point symbols. | ||
As shown in Fig. 3b the [Ce(dpdmg)3] saturation is achieved when the pulse length is 8 s or longer. The linear dependency of the film thickness on the number of ALD/MLD cycles was confirmed using the following precursor pulsing cycle: 8 s [Ce(dpdmg)3]/30 s N2 purge/6 s TPA/30 s N2 purge (Fig. 3c). From XRR, the estimated film density was found to be 2.037 g cm−3. The temperature dependency of the GPC was also investigated for the Ce-TPA films with the GPC increasing from 4.7 Å at 190 °C deposition temperature to 13.8 Å at 240 °C deposition temperature, Fig. S2.†
Similarly, the growth of Ce-HQ films was investigated keeping the deposition temperature fixed at 200 °C and the vaporisation temperatures of [Ce(dpdmg)3] and HQ at 140 °C and 120 °C, respectively. The HQ pulse length was varied from 6 to 12 s, maintaining all other parameters constant, [Ce(dpdmg)3] pulse 8 s, N2 purge lengths 30 s, 75 cycles. A saturation behaviour was observed as shown in Fig. 3d, at 8 s of HQ pulse having GPC of 5.06 Å. The cerium precursor pulse was varied from 8 s to 12 s, keeping all other parameters constant, HQ pulse 8 s and purge lengths 30 s. The saturation was observed at 12 s of [Ce(dpdmg)3] (Fig. 3e). Finally, the number of ALD cycles was varied using the optimized precursor pulsing cycle, 12 s [Ce(dpdmg)3]/30 s N2 purge/8 s HQ/30 s N2 purge, to demonstrate the linearity of the film growth (Fig. 3f). The density of the Ce-HQ films as determined by XRR was 2.302 g cm−3.
Fourier-transform infrared (FTIR) spectroscopy
The bonding in the films and the presence of organic groups were investigated by FTIR measurements (Fig. 4a) for the Ce-TPA films grown at 200 °C and 240 °C on Si(100). TPA was used as a reference for comparision. The absence of the –OH peak and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak at around 3000 cm−1 and 1671 cm−1, respectively, along with the appearance of the characteristic asymmetric and symmetric COO− bands at 1547 cm−1 and 1390 cm−1, respectively,19 confirms that the TPA molecules have reacted with [Ce(dpdmg)3] through the acid groups, as anticipated, and also discussed in light of DFT calculations in the next section. Fig. 4b shows the magnified spectra in the range 2000 cm−1–670 cm−1. The difference between the asymmetric and symmetric COO− bands (at 1547 cm−1 and 1390 cm−1) is Δ = 157 cm−1 which corresponds to bridging type bonding (130 < Δ < 200 cm−1) for metal–carboxylate bond and is in line with other reports on rare-earth-TPA films.20 The band at 751 cm−1 is due to in-plane deformation of the carboxylate group.19 The other prominent features seen in the spectra can be assigned as follows: features around 832 cm−1, 812 cm−1 due to C–H bond out of plane deformation, at 1154 cm−1 and 1017 cm−1 due to C–H bond in-plane deformation and at 1308 cm−1 due to benzene ring stretching.19,21
O peak at around 3000 cm−1 and 1671 cm−1, respectively, along with the appearance of the characteristic asymmetric and symmetric COO− bands at 1547 cm−1 and 1390 cm−1, respectively,19 confirms that the TPA molecules have reacted with [Ce(dpdmg)3] through the acid groups, as anticipated, and also discussed in light of DFT calculations in the next section. Fig. 4b shows the magnified spectra in the range 2000 cm−1–670 cm−1. The difference between the asymmetric and symmetric COO− bands (at 1547 cm−1 and 1390 cm−1) is Δ = 157 cm−1 which corresponds to bridging type bonding (130 < Δ < 200 cm−1) for metal–carboxylate bond and is in line with other reports on rare-earth-TPA films.20 The band at 751 cm−1 is due to in-plane deformation of the carboxylate group.19 The other prominent features seen in the spectra can be assigned as follows: features around 832 cm−1, 812 cm−1 due to C–H bond out of plane deformation, at 1154 cm−1 and 1017 cm−1 due to C–H bond in-plane deformation and at 1308 cm−1 due to benzene ring stretching.19,21
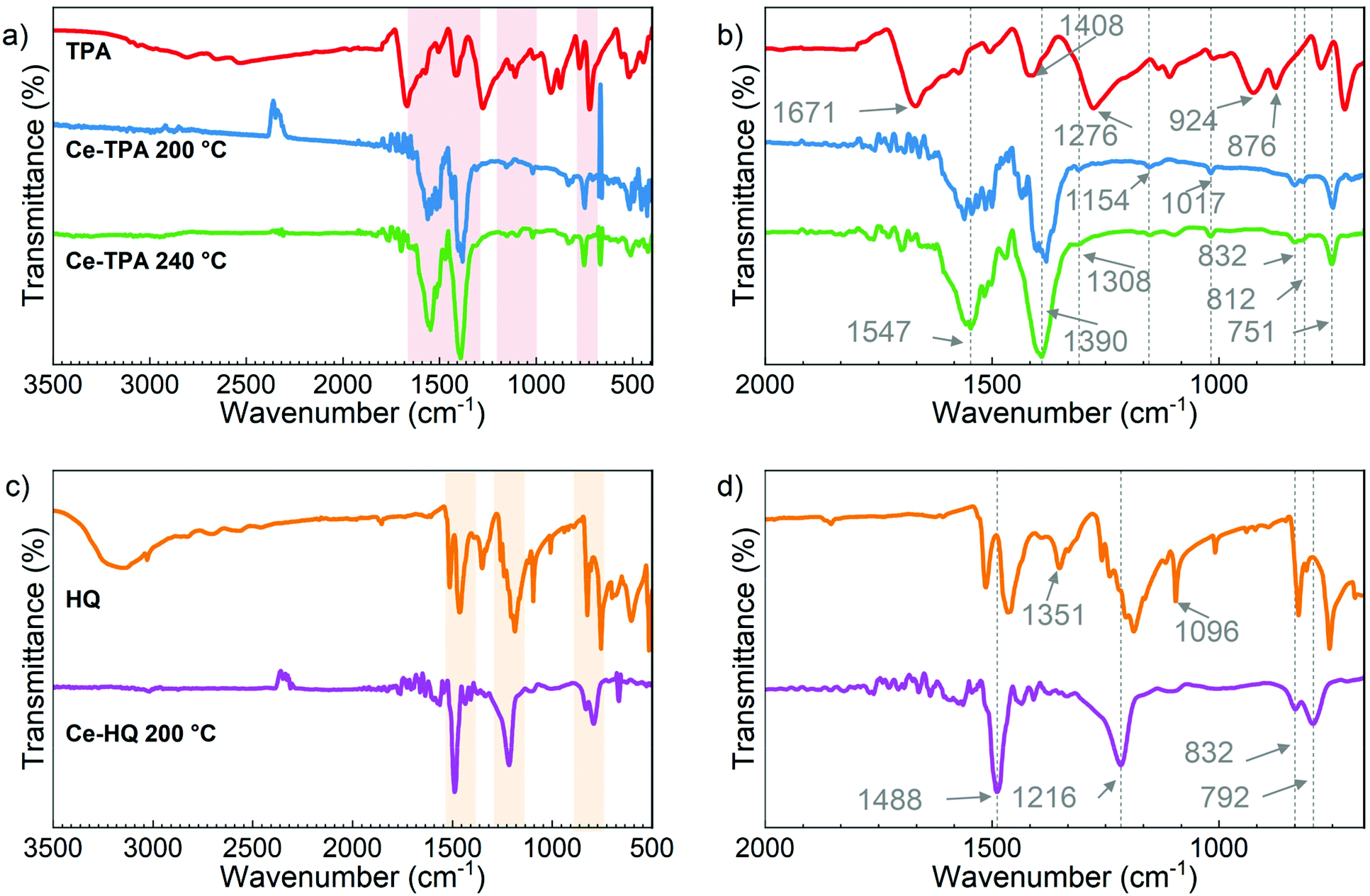 | ||
| Fig. 4 (a) and (b) FTIR spectra for Ce-TPA hybrid films deposited on Si(100) at 200 °C and 240 °C and reference TPA; (c) and (d) FTIR spectra for Ce-HQ hybrid films deposited on Si(100) at 200 °C and reference HQ. | ||
Likewise, the FTIR spectra for the Ce-HQ films and HQ as a reference are shown in Fig. 4c and d. In this case too, the absence of –OH stretching around 3200 cm−1 in the Ce-HQ hybrid films which confirms the deprotonation of HQ and is also shown favourable by DFT as discussed in the next section. The well-defined bands at 1488 cm−1, 1218 cm−1 correspond to CC aromatic stretch, C–O stretch, respectively and at 832 cm−1, 792 cm−1 are due to C–H bending vibrations22 (Fig. 4d) which verifies the presence of deprotonated HQ in the Ce-HQ hybrid films deposited at 200 °C.
Theoretical studies
DFT methods were employed to gain deeper insights into the atomistic mechanism of the growth of cerium-organic hybrid films using TPA and HQ as organic precursors and [Ce(dpdmg)3] as the metal source. In the simplified model, a hydroxylated CeO2(111) surface, with a coverage of 0.38 OH nm−2 formed by water dissociation at the surface is used. Firstly, the [Ce(dpdmg)3] precursor was chemisorbed on the hydroxylated CeO2 surface giving a large exothermic interaction energy change of −2.84 eV. Then the first and second ligand loss reactions of the [Ce(dpdmg)3] precursor when interacting with the hydroxylated CeO2 surface is examined. This takes place by a proton transfer from surface OH groups to precursor ligand. These reactions leave the surface covered with Ce bonded to one dpdmg ligand that can exchange with one organic molecule during the organic precursor pulse.Bader charge calculations were performed to determine any change in the oxidation state of Ce. The computed Bader charge of 9.8 electrons (a net charge of +2.2 electrons) for the Ce atom on adsorbed [Ce(dpdmg)3] corresponds to the Ce3+ oxidation state. Fig. S1† shows the atomic structure of the hydroxylated CeO2 surface after interaction with [Ce(dpdmg)3] precursor during the inorganic precursor pulse.
Next, the MLD reactions using HQ and TPA as organic reactants were analysed. Fig. 5 shows the proposed reaction mechanism between the Ce-dpdmg terminated CeO2 surface with TPA and HQ. During the reaction with TPA, a proton from the terminal COOH group of TPA transfers to dpdmg to release a new H-dpdmg molecule while the remaining O binds to the Ce atom of the precursor, with a Ce–O distance 2.45 Å, slightly longer than typical Ce–O distance of 2.37 Å in CeO2(111). The calculated interaction energy −1.96 eV relative to the Ce-dpdmg terminated CeO2 surface confirms that this reaction is exothermic and formation of the Ce–O bond linking cerium and TPA is favourable. After interaction with the TPA, a new Ce–O bond is formed between Ce atom of the precursor with the surface oxygen. The two existing Ce–O bonds are lengthened from 1.93 Å to 1.97 Å and 1.95 to 2.00 Å while the third Ce–O bond has a distance of 2.41 Å. A Bader charge of 9.8 electrons is calculated for the Ce atom bound to TPA, again indicating that Ce remains in the Ce3+ oxidation state.
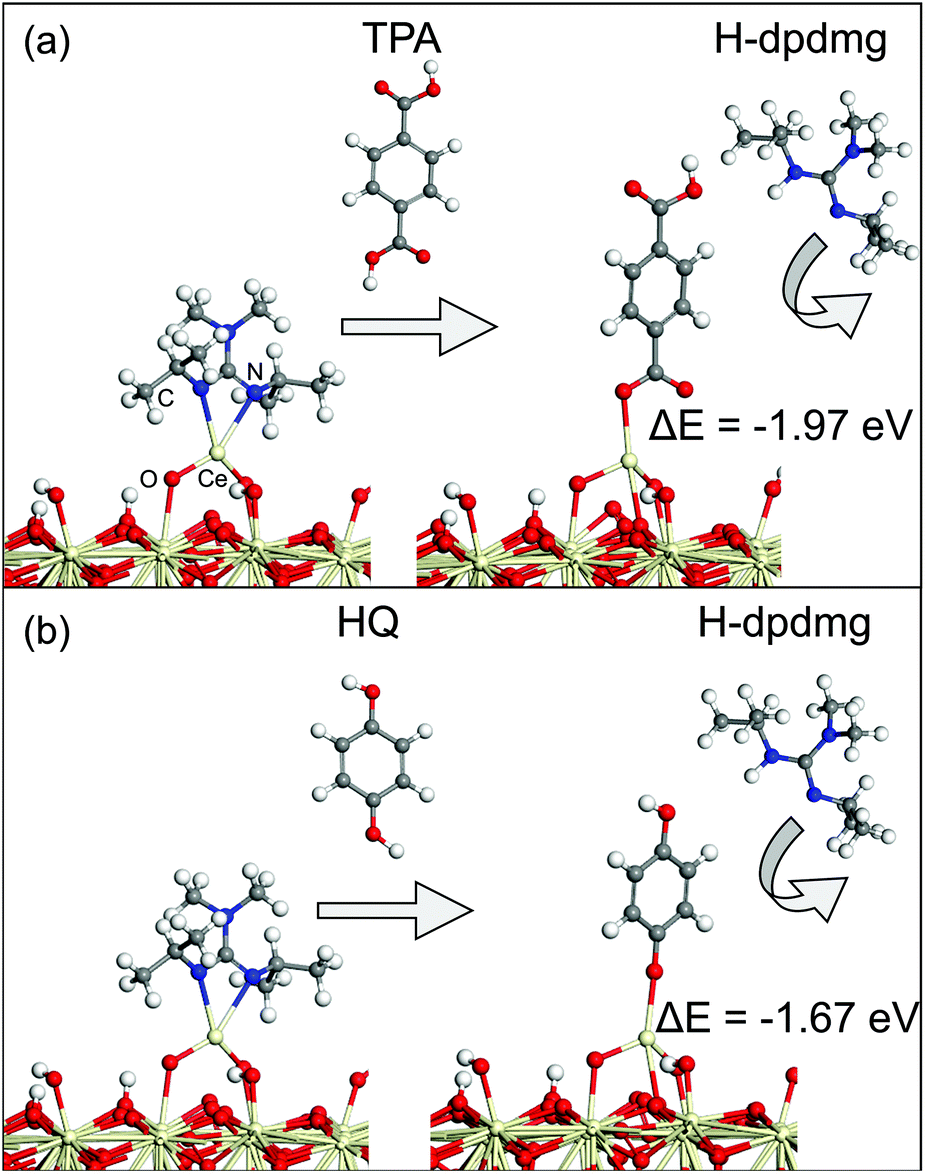 | ||
| Fig. 5 (a) Atomic structure of the reaction between Ce(dpdmg)-terminated CeO2 surface with TPA. (b) Atomic structure of the reaction between Ce(dpdmg)-terminated CeO2 surface with HQ. In this figure Ce is represented by light grey spheres, oxygen by red spheres, nitrogen by blue spheres, carbon by dark grey spheres and hydrogen by white spheres. | ||
The reaction mechanism between the Ce-dpdmg terminated CeO2 surface with HQ is similar to that with TPA. During this reaction a proton from the terminal OH group of HQ transfers to the dpdmg ligand to form a new H-dpdmg molecule while the remaining O binds to Ce site of Ce-dpdmg with a Ce–O distance 2.39 Å, shorter than that found for Ce-TPA. The calculated interaction energy −1.67 eV showed that HQ also reacts favourably with the Ce-dpdmg terminated CeO2 surface. Again, a new bond between the Ce atom of the precursor with the surface oxygen is formed. In addition to the Ce–O bond formed with HQ, the Ce atom is bonded to three surface oxygen sites. The two existing Ce–O bonds were lengthened from 1.93 Å and 1.95 Å to 1.98 Å while the new Ce–O bond had a distance of 2.50 Å. The computed Bader charge of the Ce atom that is bonded to the HQ molecule is 9.8 electrons, which indicates that the Ce3+ oxidation state persists.
The favourable adsorption and decomposition of Ce(dpdmg)3 precursor on the CeO2 surface and the favourable reactions with TPA and HQ aromatic molecules indicate that the formation of oxide/metal–organic films of CeO2/Ce-TPA and CeO2/Ce-HQ is feasible.
In summary, DFT calculations show that HQ and TPA are probably very reactive towards [Ce(dpdmg)3]. Combined with the fact that aromatic molecules, because of their stiff backbone prevent the double reactions on the surface that hinder the film growth, the use of HQ and TPA as organic precursors is a good solution to produce thicker and more flexible films.9,11,23
Composition analysis
XPS analysis was conducted for Ce-TPA and Ce-HQ on Si(100) as-deposited at 200 °C. From the survey spectra as shown in Fig. S3 and S4,† typical signals for Ce, O and C were found. For the compositional analysis, high-resolution spectra were measured for the C 1s, O 1s and Ce 3d core levels. The Ce 3d spectra was analysed by the method developed by Romeo et al.24 and was used to fit all spin-orbital and splitting components of the Ce 3d signal, resulting in two multiplets for the Ce hybrid films (Fig. 6a and d). Interestingly, the spectra clearly indicate the formation of Ce3+ without any signs of Ce4+ for both materials. The typical signal around 916–917 eV, the fingerprint region for Ce4+ species,25 was not observed (Fig. 6a and d) and moreover, only four peaks corresponding to the pairs of spin–orbit doublets, could be fitted into the Ce 3d spectrum of both Ce hybrid films. The corresponding binding energies of the Ce-TPA films (Fig. 6a) and the Ce-HQ films (Fig. 6d) are in good agreement with typical values for Ce3+ species in literature.24,25 The deviation of 0.2 eV between the Ce core levels of Ce-TPA and Ce-HQ is within the experimental uncertainty.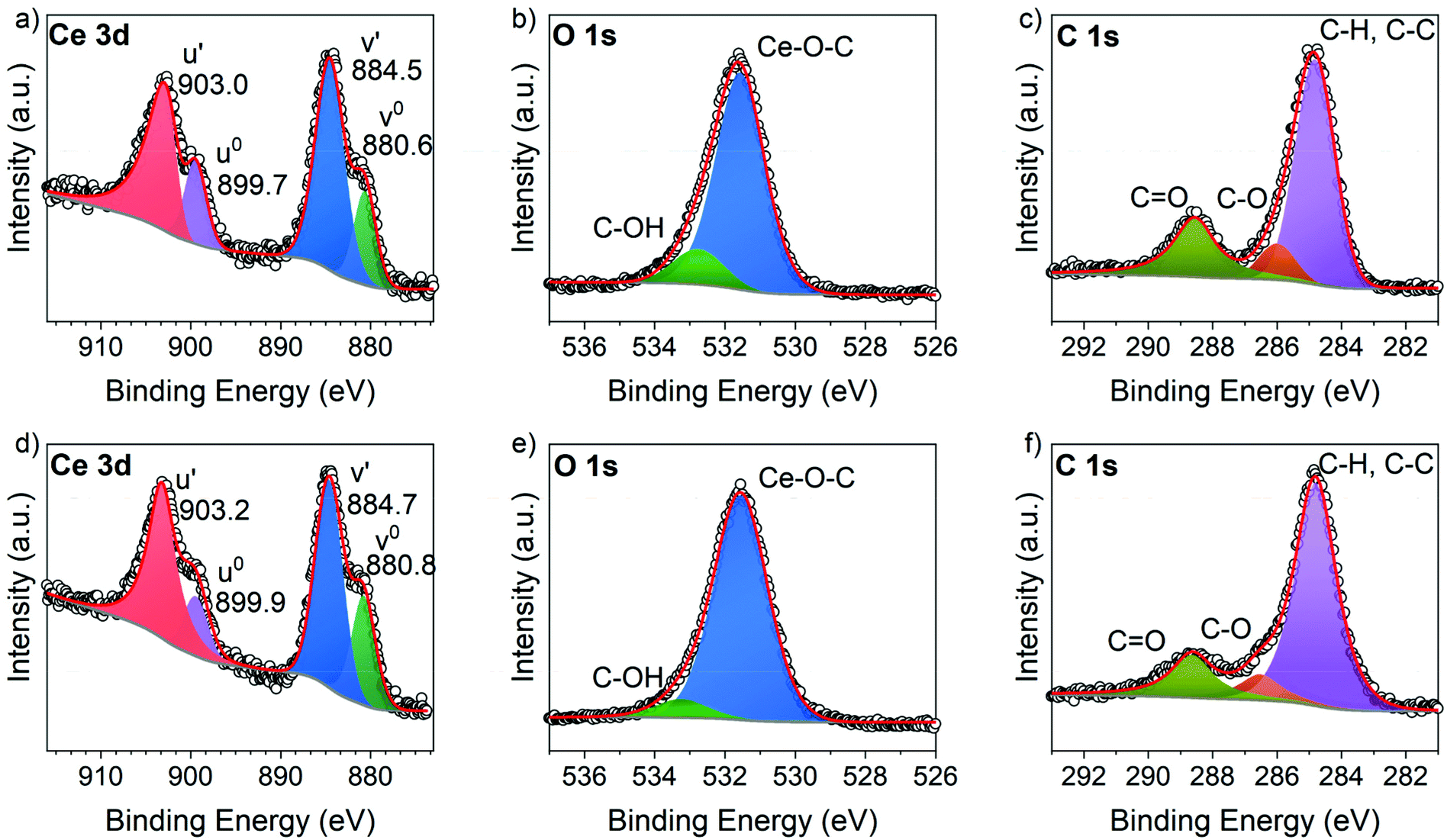 | ||
| Fig. 6 High resolution XPS spectra for Ce-TPA thin films deposited on Si(100) at 200 °C (a) Ce 3d core level spectrum (b) O 1s spectrum (c) C 1s spectrum, and Ce-HQ thin films deposited on Si(100) (d) Ce 3d core spectrum (e) O 1s spectrum (f) C 1s spectrum; the hollow circles represent the data points and the red curve shows the total fit. | ||
From the C 1s core level spectrum (Fig. 6f) three different carbon species could be identified for both material systems, namely C–H/C–C bonds with a binding energy of 284.8 eV, C–O bonds with a binding energy of 286.6 eV and carboxylic acid functions with a binding energy of 288.6 eV.26 For the Ce-TPA films roughly 74% of the carbon atoms have a contribution from C–C/C–H functionalities and other C–O functionalities, while 26% of the carbon atoms could be assigned to carboxylic carbon as estimated from the fits. For Ce-HQ films 71% of the carbon atoms could be assigned to C–C and C–H functions, while 29% was estimated to the combined amount of all M–O–C bonds. The slight deviations from the ideal case, (assuming optimal stoichiometry) where 75%/67% of C–C/C–H and 25%/33% of C–O functions would be found for Ce-TPA/Ce-HQ films respectively, arise from the contribution of adventitious carbon.
Another indication for organic Ce–O bonds is given by the O 1s spectra. While typical O 1s binding energies for lattice oxygen in Ce(IV) oxides (CeO2) are around 529.8 eV,17,25 binding energies for lattice oxygen in Ce3+ species are often shifted towards higher energies (531.5 eV).17,27 The O 1s spectra of the films show both the prominent signal at 531.5 eV indicating Ce–O bonds with Ce in the oxidation state +3. Additionally, a small contribution (14% for Ce-TPA and 8% for Ce-HQ) of C–OH functions at binding energies between 532.8 eV and 533.2 eV was found in the O 1s spectra respectively. Since the data was collected from the as-deposited layers and no surface sputtering was performed, it cannot be excluded that those contributions could partly originate from adventitious species on the surface. This is also important for the overall compositions which are shown in Table S1† with the corresponding values calculated for optimal stoichiometries. While the composition for the Ce-TPA films is in close proximity to the theoretical values, assuming only Ce3+ cations, the Ce-HQ seem to be slightly oxygen deficient.
To sum up, the successful formation of Ce–O–C bonds within Ce-HQ and Ce-TPA can be confirmed by XPS analysis. Although the contribution from adventitious species cannot be excluded completely, the calculation of the composition yielded accurate amounts of Ce, O and C for Ce-TPA and slightly oxygen deficient Ce-HQ films. Moreover, it could be confirmed that the as deposited films consists mainly of Ce3+ species while Ar+ sputtering led to a degradation of the layers, yielding CeO2 species. For further insight into the films, mild sputtering with C60 sources should be considered to apply in order to confirm the composition in bulk.28
UV-Visible spectroscopy
The optical properties of the Ce-TPA and Ce-HQ hybrid thin films deposited on quartz substrates were analysed by UV-Vis spectroscopy, and the spectra are displayed in Fig. 7. The Ce-TPA film shows a prominent strong absorption maximum at 252.5 nm which corresponds to π–π* transition of the aromatic ring. The π–π* transition for the Ce-HQ film at 244.5 nm is hypsochromic shifted (blue shift) which could be due to the presence of a different functional group. Another prominent peak for Ce-HQ is at 298.5 nm which can be attributed to ligand to metal charge transfer (LMCT) from O 2p to Ce 4f orbitals which is less prominent for Ce-TPA and shown by a shoulder around 298 nm. The Ce-TPA film is transparent i.e., >90% transmittance in the range 350 nm–800 nm while the Ce-HQ film is >85% transmitting above 400 nm and >90% in the region 665 nm–800 nm.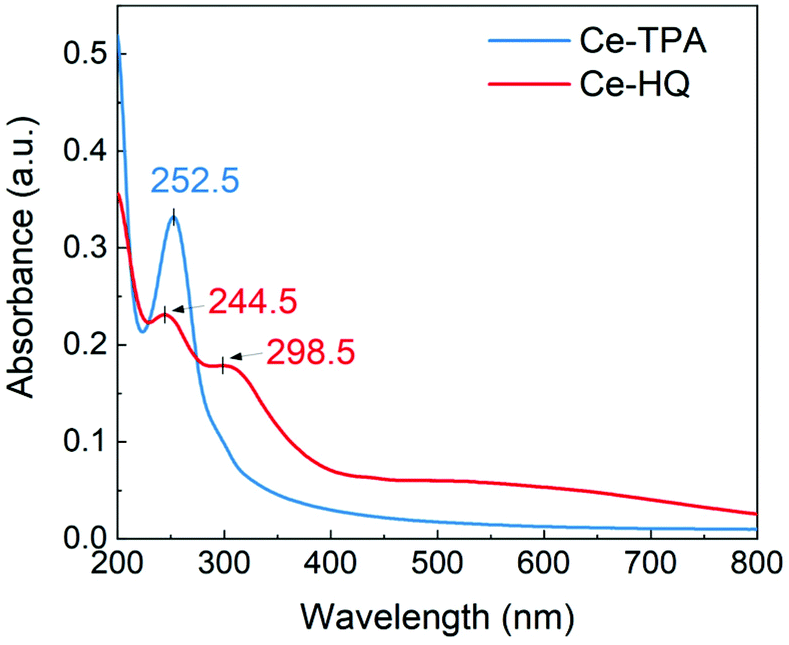 | ||
| Fig. 7 UV-Vis spectra of the Ce-TPA and Ce-HQ hybrid films grown on quartz substrate @200 °C. | ||
From the above observations, the growth of Ce-TPA and Ce-HQ hybrid thin films by ALD/MLD opens new pathways for potential applications. The presence of Ce3+ in the films, with its 4f1 electronic configuration, makes it interesting to study these materials further for photo-emitting applications. The aromatic backbones act as good antennas due to their strong binding tendency towards the cerium ions and can influence the luminescent properties of the Ce3+ ions.29
Conclusion
A new pathway for the development of cerium-organic hybrid materials is successfully demonstrated by ALD/MLD processing. The compatibility of the inorganic precursor [Ce(dpdmg)3] and the organic precursors terephthalic acid (TPA) and hydroquinone (HQ) was thoroughly analysed in terms of their physicochemical properties. Thermal studies showed that the precursors are sufficiently volatile and thermally stable at the given deposition temperatures. Moreover, the DFT calculations predicted the reactivity of HQ and TPA precursors towards the chemisorbed [Ce(dpdmg)3] inorganic precursor to be favourable. Hence, the thin films of the Ce-based hybrid materials were successfully grown using [Ce(dpdmg)3] precursor as a source of cerium along with TPA and HQ as the organic linkers. Furthermore, DFT analysis revealed the charge of the cerium in the films to be +3, which was confirmed by XPS. The bonding in the films was studied by FTIR analysis, to confirm the successful deprotonation of the organic precursors and the formation of metal–organic bonds which could be seen by the absence of –OH functionalities near 3000 cm−1 and the presence of the expected features related to the Ce–O bonding motifs, and the benzene backbone in the films. Furthermore, XPS analysis confirmed the formation of Ce–O bonds. UV-Vis spectroscopy showed the π–π* transition and LMCT for both films.Our future work will focus on testing the layers for potential applications in photoemission and sensing.
Experimental
Precursors
The synthesis of [Ce(dpdmg)3] was carried out following the procedure reported earlier17 and was scaled up to 15 g. TPA (99%) and HQ (>99.5%) were obtained from commercial sources (Sigma Aldrich).TGA was done using Netzsch STA409 PC/PG at ambient pressure under nitrogen atmosphere using 10 mg of the precursors, with a heating ramp of 5 °C min−1 and under nitrogen atmosphere. Melting points were determined by simultaneous differential scanning calorimetry (DSC).
Thin film deposition and analysis
The films were deposited in F-120 ASM Microchemistry flow-type ALD reactor on 2 cm × 2 cm Si(100) and quartz substrates. Nitrogen (99.999% purity) gas was implemented as a carrier and purging gas at 300 sccm. The sublimation temperature for [Ce(dpdmg)3] was set at 140 °C, TPA at 185 °C and HQ at 120 °C.The film thickness was measured by X-ray reflectivity (XRR). Fourier-transform infrared (FTIR) spectra were collected on Bruker Alpha 2 in transmittance mode on Si(100) substrates. The UV/Vis absorption spectroscopy was measured by Shimadzu UV/2600 spectrometer in the range of 200–800 nm on the quartz substrate.
XPS measurements were done on a PHI5000 instrument. The X-ray source was operated at 10 kV and 24.6 W using Al Kα (1486.6 eV) radiation with a 45° electron take-off angle. The kinetic energy of the electrons was analysed by a spherical Leybold EA-10/100 analyser using a pass energy of 18 eV. Step widths of 0.5 eV and 0.05 eV were adjusted for the survey and core level scans, respectively. The pressure of the analysis chamber was maintained at <10−7 mbar. The binding energies of the Ce 3d and O 1s core levels were charge referenced to the C 1s core level of adventitious carbon at 284.8 eV. A deconvolution analysis of the core level signals was completed with a Shirley background processing and Gaussian functions using UniFit 2017 software.
Computational methods
All reported DFT calculations were carried out using the Vienna ab initio simulation package (VASP) version 5.4.30 The exchange–correlation functional is approximated by the Perdew–Burke–Ernzerhof (PBE) approximation31 while the valence electrons are described using the projector augmented wave (PAW) potentials.32 For Ce, we use 12 valence electrons, for O 6 valence electrons, for C 4 valence electrons and for H 1 valence electron. We use the well-known GGA + U approach to consistently describe the Ce 4f electronic states,33 with U = 5 eV, typical of values used in the literature. The energy convergence criteria is set to 1 × 10−4 eV and the geometry convergence criterion for the forces is set to 1.5 × 10−2 eV Å−1. The ceria hybrids were modelled using a hydroxylated CeO2(111) surface, at a coverage of 0.38 OH nm−2 which is modified with [Ce(dpdmg)3] as inorganic precursor and HQ and TPA as organic precursors. The geometry was optimized by relaxing the ionic positions, using a Monkhorst–Pack k-point sampling grid of (1 × 1 × 1). The cut-off for the kinetic energy is 396 eV. The lattice parameters for this surface model are a = 30.95 Å, b = 20.47 Å, c = 40.27 Å, and it is four O–Ce–O layers thick.Adsorption energies for the example of [Ce(dpdmg)3] adsorption on the hydroxylated CeO2 were calculated using:
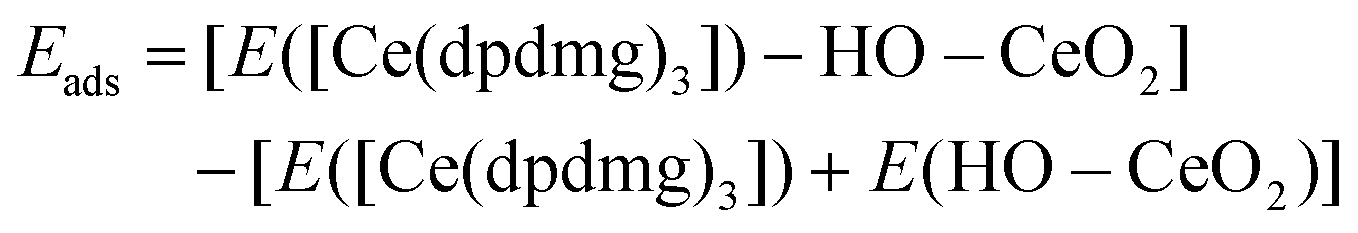
Interaction energies of HQ/TPA with Cedpdmg terminated CeO2 were calculated using:
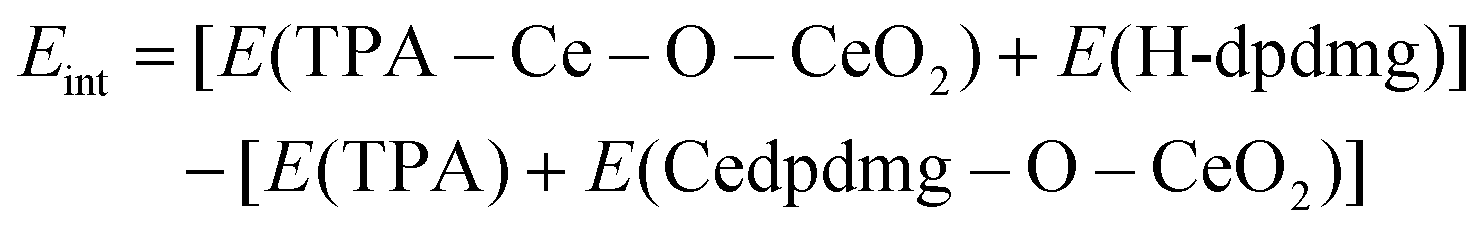
Given the magnitude of the computed adsorption energies, we do not include van der Waals interactions in the calculations.
Author contributions
The author contribution are as follows: Conceptualization: P. K. A. D.; visualization, validation, ALD experiments, thin film characterization, project administration, MS first draft preparation: P. K.; support for ALD experiments: R. G., M. S.; XRR fittings: R. G.; DFT studies and data interpretation: A. M., M. N.; XPS analysis and data interpretation: J.-L. W.; final MS writing: P. K., A. M., J.-L. W., M. N., M. K., A. D.; supervision: M. N., M. K., A. D.; all the authors have reviewed the results and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement (No. 765378). P. K., A. M. and R. G. thank HYCOAT for the early-stage researcher (ESR) fellowships. AD acknowledges the additional funding support from the DFG-SFB-TR-87. P. K., M. S., R. G. and M. K. acknowledge the use of the RawMatTERS Finland Infrastructure (RAMI) at Aalto University. MN acknowledges support from Science Foundation Ireland, through the SFI-NSFC Partnership Program, project NITRALD 17/NSFC/5279. A. M. and M. N. acknowledge support from Science Foundation Ireland for computational resources at Tyndall National Institute.References
- (a) L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092–3104 RSC; (b) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed; (c) K. Liu, X. Zhang, X. Meng, W. Shi, P. Cheng and A. K. Powell, Chem. Soc. Rev., 2016, 45, 2423–2439 RSC.
- S. Roy, A. Chakraborty and T. K. Maji, Coord. Chem. Rev., 2014, 273–274, 139–164 CrossRef CAS.
- X. Rao, T. Song, J. Gao, Y. Cui, Y. Yang, C. Wu, B. Chen and G. Qian, J. Am. Chem. Soc., 2013, 135, 15559–15564 CrossRef CAS PubMed.
- (a) Y. Cui, B. Chen and G. Qian, Coord. Chem. Rev., 2014, 273–274, 76–86 CrossRef CAS; (b) K. Müller-Buschbaum, F. Beuerle and C. Feldmann, Microporous Mesoporous Mater., 2015, 216, 171–199 CrossRef.
- X. Sun, K. Yuan and Y. Zhang, J. Rare Earths, 2020, 38, 801–818 CrossRef CAS.
- M. Safdar, A. Ghazy, M. Lastusaari and M. Karppinen, J. Mater. Chem. C, 2020, 8, 6946–6965 RSC.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Elsevier Science, 2012 Search PubMed.
- J. Jacobsen, A. Ienco, R. D'Amato, F. Costantino and N. Stock, Dalton Trans., 2020, 49, 16551–16586 RSC.
- P. Sundberg and M. Karppinen, Beilstein J. Nanotechnol., 2014, 5, 1104–1136 CrossRef PubMed.
- (a) K. van de Kerckhove, F. Mattelaer, D. Deduytsche, P. M. Vereecken, J. Dendooven and C. Detavernier, Dalton Trans., 2016, 45, 1176–1184 RSC; (b) K. van de Kerckhove, F. Mattelaer, J. Dendooven and C. Detavernier, Dalton Trans., 2017, 46, 4542–4553 RSC; (c) K. van de Kerckhove, M. K. S. Barr, L. Santinacci, P. M. Vereecken, J. Dendooven and C. Detavernier, Dalton Trans., 2018, 47, 5860–5870 RSC; (d) J. Kint, F. Mattelaer, S. S. T. Vandenbroucke, A. Muriqi, M. M. Minjauw, M. Nisula, P. M. Vereecken, M. Nolan, J. Dendooven and C. Detavernier, Chem. Mater., 2020, 32, 4451–4466 CrossRef CAS; (e) A. Muriqi and M. Nolan, Dalton Trans., 2020, 49, 8710–8721 RSC.
- A. Muriqi, M. Karppinen and M. Nolan, Dalton Trans., 2021, 50, 17583–17593 RSC.
- J. Päiväsaari, M. Putkonen and L. Niinistö, J. Mater. Chem., 2002, 12, 1828–1832 RSC.
- J. S. Wrench, K. Black, H. C. Aspinall, A. C. Jones, J. Bacsa, P. R. Chalker, P. J. King, M. Werner, H. O. Davies and P. N. Heys, Chem. Vap. Deposition, 2009,(15), 259–261 CAS.
- W.-H. Kim, M.-K. Kim, W. J. Maeng, J. Gatineau, V. Pallem, C. Dussarrat, A. Noori, D. Thompson, S. Chu and H. Kim, J. Electrochem. Soc., 2011, 158, G169 CrossRef CAS.
- L. Du, K. Wang, Y. Zhong, B. Liu, X. Liu and Y. Ding, J. Mater. Sci., 2020, 55, 5378–5389 CrossRef CAS.
- M. Golalikhani, T. James, P. van Buskirk, W. Noh, J. Lee, Z. Wang and J. F. Roeder, J. Vac. Sci. Technol., A, 2018, 36, 51502 CrossRef.
- P. Kaur, L. Mai, A. Muriqi, D. Zanders, R. Ghiyasi, M. Safdar, N. Boysen, M. Winter, M. Nolan, M. Karppinen and A. Devi, Chem. – Eur. J., 2021, 27, 4913–4926 CrossRef CAS PubMed.
- L. Mai, Z. Giedraityte, M. Schmidt, D. Rogalla, S. Scholz, A. D. Wieck, A. Devi and M. Karppinen, J. Mater. Sci., 2017, 52, 6216–6224 CrossRef CAS.
- C. A. Téllez, S. E. Hollauer, M. Mondragon and V. M. Castaño, Spectrochim. Acta, Part A, 2001, 57, 993–1007 CrossRef.
- (a) A. Ghazy, M. Safdar, M. Lastusaari and M. Karppinen, Chem. Commun., 2020, 56, 241–244 RSC; (b) J. Penttinen, M. Nisula and M. Karppinen, Chem. – Eur. J., 2017, 23, 18225–18231 CrossRef CAS PubMed; (c) M. Safdar, A. Ghazy, M. Tuomisto, M. Lastusaari and M. Karppinen, J. Mater. Sci., 2021, 56, 12634–12642 CrossRef CAS.
- H. T. Varghese, C. Y. Panicker, D. Philip, K. Sreevalsan and V. Anithakumary, Spectrochim. Acta, Part A, 2007, 68, 817–822 CrossRef PubMed.
- J. C. Evans, Spectrochim. Acta, 1960, 16, 1382–1392 CrossRef CAS.
- A. Tanskanen, P. Sundberg, M. Nolan and M. Karppinen, Thin Solid Films, 2021, 736, 138896 CrossRef CAS.
- M. Romeo, K. Bak, J. El Fallah, F. Le Normand and L. Hilaire, Surf. Interface Anal., 1993, 20, 508–512 CrossRef CAS.
- E. Bêche, P. Charvin, D. Perarnau, S. Abanades and G. Flamant, Surf. Interface Anal., 2008, 40, 264–267 CrossRef.
- T. L. Barr and S. Seal, J. Vac. Sci. Technol., A, 1995, 13, 1239–1246 CrossRef CAS.
- (a) E. J. Preisler, O. J. Marsh, R. A. Beach and T. C. McGill, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct. – Process., Meas., Phenom., 2001, 19, 1611 CrossRef CAS; (b) D. Mullins, S. Overbury and D. Huntley, Surf. Sci., 1998, 409, 307–319 CrossRef CAS.
- G. L. Fisher, M. Dickinson, S. R. Bryan and J. Moulder, Appl. Surf. Sci., 2008, 255, 819–823 CrossRef CAS.
- A. F. Kateshali, S. G. Dogaheh, J. Soleimannejad and A. J. Blake, Coord. Chem. Rev., 2020, 419, 213392 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953–17979 CrossRef PubMed.
- (a) V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter, 1991, 44, 943–954 CrossRef CAS PubMed; (b) M. Nolan, S. Grigoleit, D. C. Sayle, S. C. Parker and G. W. Watson, Surf. Sci., 2005, 576, 217–229 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2dt00353h |
| This journal is © The Royal Society of Chemistry 2022 |