
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and computational aspects of Al(II)–Al(II) and Ga(II)–Ga(II) dihalides based on an amidinate scaffold†
Arun
Kumar
a,
Samya
Banerjee
 *b,
Nishant
Sharma
c,
Mohd
Nazish
a,
Nico
Graw
a,
Regine
Herbst-Irmer
a,
Dietmar
Stalke
*a,
Upakarasamy
Lourderaj
*c and
Herbert W.
Roesky
*a
*b,
Nishant
Sharma
c,
Mohd
Nazish
a,
Nico
Graw
a,
Regine
Herbst-Irmer
a,
Dietmar
Stalke
*a,
Upakarasamy
Lourderaj
*c and
Herbert W.
Roesky
*a
aInstitut für Anorganische Chemie, Georg-August-Universität Göttingen, 37077 Göttingen, Germany. E-mail: hroesky@gwdg.de; dstalke@chemie.uni-goettingen.de
bDepartment of Chemistry, Indian Institute of Technology (BHU), Varanasi, Uttar Pradesh 221005, India. E-mail: samya.chy@itbhu.ac.in
cSchool of Chemical Sciences, National Institute of Science Education and Research (NISER) Bhubaneswar, HBNI, P. O. Jatni, Khurda, Odisha, India. E-mail: u.lourderaj@niser.ac.in
First published on 28th February 2022
Abstract
Amidinate compounds with stabilized aluminium(II) and gallium(II) elements of composition L2M2X2 (3 and 4) have been prepared from their LMX2 (1 and 2) precursor, where M = Al (1 and 4) and Ga (2 and 3); L = PhC(NiPr2C6H3)2 (1 and 4) and PhC(NtBu)2 (2 and 3); and X is I (1 and 4) and Cl (2 and 3) and insights into their bonding are gained. The M–M bond lengths are reported along with the single-crystal X-ray structures of 1–4.
Introduction
Compounds of low valent aluminium and gallium in the formal oxidation state of (II) date back to the pioneering discovery of gallium(II) dihalides by Worrall and co-workers in 1979.1 This was achieved by the recrystallization of Ga2Cl4 from dioxane at 0 °C to obtain Ga2Cl4(dioxane)2. In a similar manner, Ga2Br4(dioxane)2 was also isolated and characterized.2 In the regime of compounds with low valent aluminium, Uhl was the first to isolate and demonstrate the structure of a tetrakis[bis(trimethylsilyl)methyl]dialane in 1988 by a reaction of AlCl3 with LiCH(SiMe3)2.3Since then a considerable number of Al(II) and Ga(II) atoms have been reported with a wide array of ligand environments (Chart 1). The bond distances vary from 2.5–2.75 Å among Al(II) atoms whereas they are 2.3–2.6 Å among gallium atoms. For example, the substitution of 2,4,6-(iPr)3C6H2-(Trip) powder resulted in the isolation of the dialane Trip2 Al-AITrip2via the reduction of Trip2AlBr with potassium.4 In addition, the reduction of [C6H3-2,6-(C6H3-2,6-iPr)2]AlI2 with KC8 generates the aryl-based 1,2-diiodoalane.5 Klimek et al. described the preparation of compounds with Al–Al and Ga–Ga bonds as dihalide dimers containing the 1-azaallyl ligand [(Me3Si)2C(Ph)C(Me3Si)N].6 Arnold and coworkers synthesized and characterized dialane including hypercoordinated Al atoms with the C5Me5 (Cp*) ring, [Al2I2(η5-Cp*)2].7 This was followed by the synthesis of [Al2NBr2(η5-Cp*)2] by Braunschweig and co-workers.8
 | ||
| Chart 1 Selected examples of Al(II)–Al(II) and Ga(II)–Ga(II) compounds. | ||
Recently, Stalke and co-workers reported an Al(II) complex [(4-MeBox2CH)HAl–AlH(DippNacNac)] based on the bis(4-methyl-benzoxazole-2-yl)methanide and β-diketiminate (NacNac) ligand.9 To the best of our knowledge, the shortest distance among Al(II) atoms is reported by Andrada and co-workers; they have reduced the dicarba-bridged dicyclopentadienylaluminium chloride dimer with 1,3-β-diketiminate magnesium(I) to produce bis(dicarba[2]aluminocenophane) bearing an Al(II)–Al(II) distance of 2.5018(5) Å.10 In contrast, the longest distance among Al(II) centers is calculated as 2.751(2).11 This was achieved by the reaction of aluminium trihalide with supersilylsodium to give [(R)2Al–Al(R)2], where R is the supersilyl, Si(t-Bu)3. Braun and coworkers utilized the sterically demanding β-diketiminate ligand Ldmp = [HC{(CMe)N(dmp)}2], where dmp = C6H3-2,6-Me2 and synthesized a gallium complex of composition [Ga2I2(Ldmp)2] in the oxidation state +II.12 Fedushkin and coworkers reported a digallane of formula [Ga2(Ldmp)2] consisting of a redox active dpp-Bian ligand, where dpp-Bian is (1,2-bis[(2,6-diisopropyl phenyl)imino]acenaphthene).13 So far, only three compounds with an Al![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Al double bond have been reported. The first is a silyl-substituted dialumene by Inoue et al.14 followed by an aryl analogue15 and an amidophosphine-stabilized AlAl double bond.16
Al double bond have been reported. The first is a silyl-substituted dialumene by Inoue et al.14 followed by an aryl analogue15 and an amidophosphine-stabilized AlAl double bond.16
In spite of the several reports of compounds with Al–Al and Ga–Ga bonds, PhC(NtBu)2 (amidinate) stabilized double bonds are not known in the literature, although amidinates are widely used to stabilize compounds of low valent elements. It has been reported that an amidinate ligand stabilized the Si(I)–Si(I) bond.17 In contrast, the 1,3-β-diketiminate aluminium AlAl has not been reported; therefore, we assumed that the amidinate might function as a better electron donor for the preparation of an AlAl species. This unexplored synthetic route prompted the background of this work. Herein, we have synthesized amidinate-stabilized aluminium(II) and gallium(II) compounds of formula L2M2X2 (3 and 4) from their LMX2 (1 and 2) precursors, where M = Al (1 and 4) and Ga (2 and 3); L = PhC(NiPr2C6H3)2 (1 and 4) and PhC(NtBu)2 (2 and 3); X is I (1 and 4) and Cl (2 and 3). However, we were not successful in isolating a crystalline [PhC(NiPr2C6H3)2]2 (AlAl) compound.18
Results and discussion
Complex 1 was synthesized by treating one equivalent of AlI3 with one equivalent of [PhC(NiPr2C6H3)2]Li in Et2O under a nitrogen atmosphere.19 In a similar manner, complex 2 was prepared from GaCl3 and [PhC(NtBu)2]Li (Scheme 1(a)). Colourless crystals of 1 and 2 were isolated from the concentrated solution in Et2O at −4 °C. Compounds 3 and 4 were synthesized via the reduction of complexes 1 and 2 with 2 equivalents of KC8 in toluene at room temperature for 24 hours (Scheme 1(b)). Colourless crystals of 3 and 4 were isolated from the concentrated solution in toluene at −30 °C. Compounds 3 and 4 remain stable for months under an inert atmosphere in toluene at room temperature and also in the solid state at room temperature. | ||
| Scheme 1 Synthetic route for the synthesis of compounds 1–4. | ||
As a side product from the synthesis of 2, crystals of [PhC(HNtBu)2][GaCl4] (2a) were isolated and were characterized by single-crystal X-ray diffraction (see the ESI†).
Compound 1 crystallizes in the trigonal space group P3121. The asymmetric unit contains half of the molecule (Fig. 1). Crystals of 1 showed remarkable colour changes under polarized light from violet to yellow depending on the orientation of the crystals.
 | ||
| Fig. 1 Molecular unit of 1. The anisotropic displacement parameters are depicted at the 50% probability level. The hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: C1–N1 1.341(3), Al1–N1 1.890(2), Al1–I1 2.4700(7), N1–Al1–N1A 70.86(12), N1–Al1–I1 117.49(6), and I1–Al1–I1A 113.96(4). | ||
1 is so far only the second crystallographically characterized benzamidinate aluminium diiodide complex apart from [(I2Al)2(μ-{Phamd2})] (Phamd2 = 1,6-({2,6-iPrC6H3N}2C)C6H4 reported by Jones et al.20 The aluminium atom is coordinated by the amidinate chelate ligand and two iodine atoms in a distorted tetrahedral fashion. The Al–N distances (1: 1.890(2) Å) are slightly shorter than those in [(I2Al)2(μ-{Phamd2})] (1.901(5)–1.913(5) Å), whereas the mean Al–I distances are equal (1: 2.470(7) Å), [(I2Al)2(μ-{Phamd2})]: 2.471(5) Å).
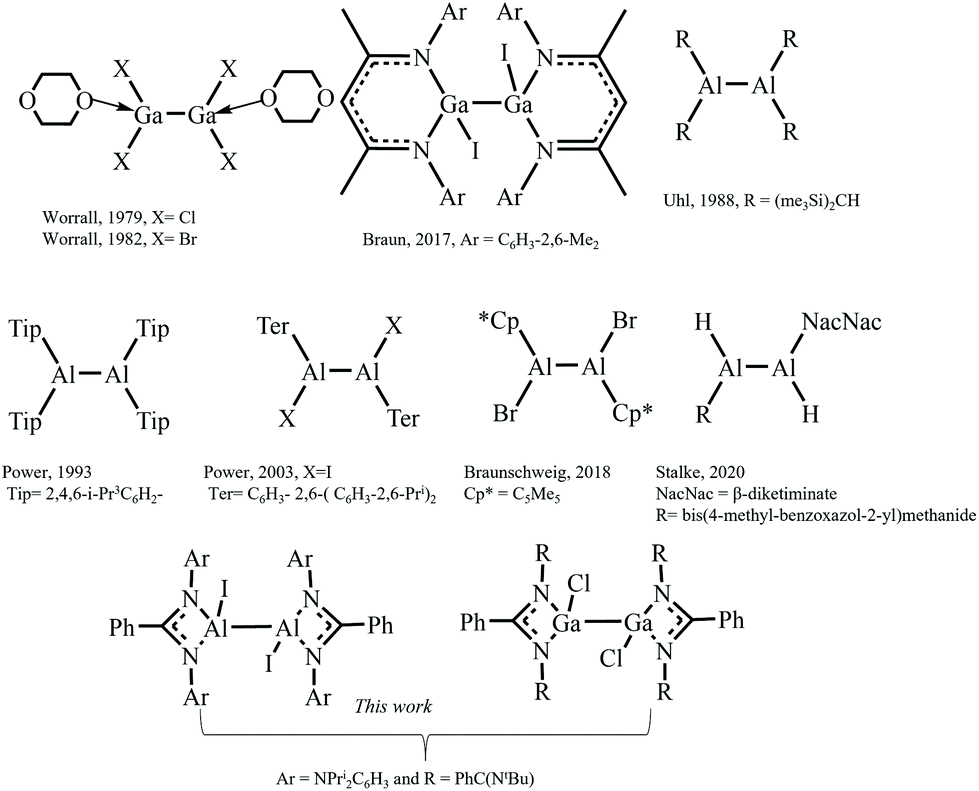
Compound 2 crystallizes in the monoclinic space group C2/c with half a molecule in the asymmetric unit. The central gallium atom has a distorted tetrahedral coordination environment. The Ga–N (2: 1.9415(12) Å) and Ga–Cl bonds (2: 2.1424(5) Å) are slightly shorter than those in the related PhC(NiPr)2GaCl2 (1.950(3) Å and 2.1520(11) Å) (Fig. 2).21
 | ||
| Fig. 2 Molecular unit of 2. The anisotropic displacement parameters are depicted at the 50% probability level. The hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: C1–N1 1.3288(16), Ga1–N1 1.9415(12), Ga1–Cl1 2.1424(5), N1–Ga1–N1A 68.10(7), N1–Ga1–Cl1 118.55(5), and Cl1–Ga1–Cl1A 109.10(3). | ||
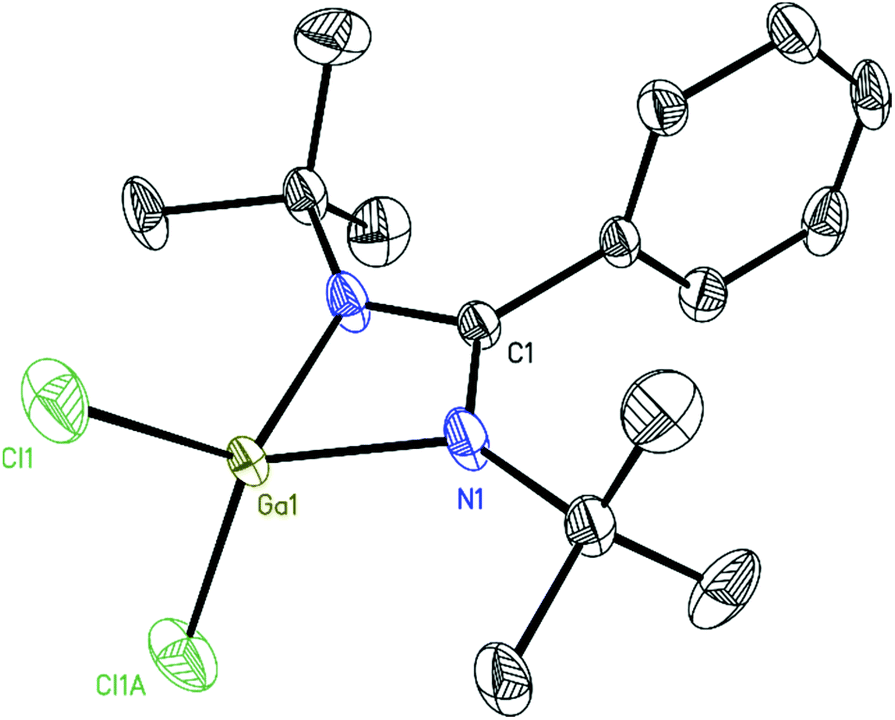
Compound 3 crystallizes in the monoclinic space group C2/c. The asymmetric units contain half of a molecule. The overall coordination environment of the gallium center remains similar to the starting material 2. However, the Ga–N distances (3: 1.9795(19) Å to 1.9807(19) Å and 2: 1.9415(12) Å) and the Ga–Cl distance (3: 2.2126(8) Å and 2: 2.1424(5) Å) are slightly elongated. The Ga–Ga distance (2.4053(6) Å) lies well within the range of the reported values for compounds with Ga–Ga single bonds (Fig. 3 and Fig. S11†).22
 | ||
| Fig. 3 Molecular unit of 3. The anisotropic displacement parameters are depicted at the 50% probability level. The hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: C1–N1 1.344(3), Ga1–N1 1.9807(19), Ga1–N2 1.9795(19), Ga1–Cl1 2.2126(8), Ga1–Ga1A 2.4053(6), N1–Ga1–N2 67.26(8), N1–Ga1–Cl1 111.23(6), N2–Ga1–Cl1 109.22(6), and Cl1–Ga1–Ga1A 116.01(3). | ||
Compound 4 crystallizes in the monoclinic space group P21/c. The asymmetric unit contains half of a molecule. Compared to Al(III) precursor 1, the Al–N distances in 4 are slightly elongated (1: 1.890(2) Å and 4: 1.9145(14) Å to 1.9466(14) Å). The same is true for the Al–I distance (1: 2.4700(7) Å and 4: 2.5469(6) Å). The Al–Al distance (2.5803(10) Å) fits well to the already reported compounds with Al–Al single bonds (Fig. 4 and Fig. S10†).23
 | ||
| Fig. 4 Molecular unit of 4. The anisotropic displacement parameters are depicted at the 50% probability level. The hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: C1–N1 1.341(2), Al1–N1 1.9466(14), Al1–N2 1.9145(14), Al1–I1 2.5469(6), Al1–Al1A 2.5803(10), N1–Al1–N2 69.39(6), N1–Al1–I1 109.92(4), N2–Al1–I1 113.53(4), and I1–Al1–Al1A 118.11(3). | ||
To understand the structure and bonding in the Al(II) and Ga(II) complexes, density functional theory (DFT) calculations were carried out for the systems 1–4 at the B3LYP/ECP(I),6-311++G**24–27 level of theory, where the effective core potential (ECP) with double-ζ LANL2DZ was used for iodine and the 6-311++G** basis set was used for the other atoms. The optimized geometries of these molecules are shown in Fig. 5. The geometrical parameters (Table S8†) obtained from the calculations are in good agreement with that of the crystal structures. It can be seen that the metal–metal (M–M) bonds are formed in the dimers with bond lengths of 2.46 and 2.63 Å for Ga–Ga and Al–Al bonds, respectively. It should be noted that the formation of the M–M bonds is accompanied by the elongation (∼0.1 Å) of the metal–halide (M–X) bonds.
 | ||
| Fig. 5 The optimized geometries of 1, 2, 3, and 4 were obtained at the B3LYP/ECP(I),6-311++G** level of theory and the σ-type NBOs (isosurface = 0.06 a.u.) of the Ga–Cl, Al–I, Ga–Ga, and Al–Al bonds in these compounds. The hydrogen atoms are not shown for clarity. The important bond distances are indicated in Å units. | ||
The nature of bonding in these molecules was investigated by performing natural bond orbital (NBO) calculations28 at the B3LYP/ECP(I),6-311++G** level of theory for the optimized geometries obtained at the same level of theory. The resulting σ-type NBOs of the Ga–Cl, Al–I, Ga–Ga, and Al–Al bonds in 1, 2, 3, and 4 are shown in Fig. 5. The atomic orbital contribution and the NBO occupancies are given in Table S9.† It was found that the Al–I bonds in 1 and 4 and the Ga–Cl bonds in 2 and 3 are highly polarized towards the halogen atoms as seen from the orbital contribution of the bonds (I: 74.6% (1), Cl: 80.3% (2), Cl: 83.0% (3), and I: 83.0% (4)). The orbital occupancies of the M–X bonds are in the range 1.936–1.965 e. The polarized nature of these bonds can also be seen from the natural population analysis (NPA) charges on the Al and Ga atoms (Al: +1.199/+1.084 e in 1/4 and Ga: +1.394/+1.005 e in 2/3, and Table S10†). The calculations also indicate that the M–X bonds (Al–X and Ga–X) in the monomers 1 and 2 are single bonds quantified by the Wiberg bond index (WBI) values (Al–I: 0.967 (1) and Ga–Cl: 0.780 (2)). The M–M bonds in 3 (Ga–Ga) and 4 (Al–Al) were also found to be single bonds with the WBI values of 0.851 and 0.819, respectively. The Ga–Ga bond in 3 was found to have a major contribution from the s (41.89%) and p (57.81%) orbitals of each Ga atom, while in 4, each Al atom contributed s (40.59%) and p (59.01%) orbitals to the Al–Al bond (Table S1†).
To further characterize the nature of the important bonds in 1, 2, 3, and 4, QTAIM calculations29 were carried out at the B3LYP/ECP(I),6-311++G** level of theory for the optimized geometries. The presence of (3, −1) bond critical points (BCPs) between the metal centers and the metal–halogen centers reiterate the existence of the M–M and M–X bonds (Table S11 and Fig. S13†). The electron densities at the BCPs for the Al–I bonds in 1 and 4 and the Ga–Cl bonds in 2 and 3 were in the range of 0.047–0.089 e Å−3. In addition, the Laplacian ([∇2ρ(r)]) values at the BCPs for these bonds were found to be positive indicating that these bonds are highly polarized as seen in the NBOs.
Conclusions
In summary, we present a successful use of an amidinate scaffold for the synthesis of novel amidinate-based aluminium(II) and gallium(II) dihalide dimers. X-ray crystal structures of compounds 3 and 4 fall into the examples of gallium(II) dichloride and aluminium(II) diiodide dimers. This indicates the ability of the amidinate ligand for stabilizing Al(II)–Al(II) and Ga(II)–Ga(II) bonds.Experimental section
X-ray crystallography
Single crystals of compounds 1–4, suitable for X-ray analysis, were mounted in inert oil. The diffraction data were collected at 100(2) K on a Bruker D8 three-circle diffractometer equipped with a SMART APEX II CCD detector and an INCOATEC Mo microsource with INCOATEC Quazar mirror optics (λ = 0.71073).30 The data were integrated with SAINT31 and an empirical absorption correction with SADABS32 was applied. For 2, TWINABS33 was used. The structures were solved using SHELXT32 and refined on F2 using SHELXL34 in the graphical user interface ShelXle.35 All non-hydrogen atoms were refined with anisotropic-displacement parameters.Crystal data for 1 at 100(2) K: C31H39AlI2N2, Mr = 720.42 g mol−1, 0.251 × 0.213 × 0.190 mm, trigonal, P3121, a = 15.276(3) Å, c = 12.183(2) Å, V = 2462.1(10) Å3, Z = 3, μ(Mo Kα) = 1.963 mm−1, θmax = 26.425°, 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 077 reflections measured, 3387 independent (Rint = 0.0273), R1 = 0.0157 [I > 2σ(I)], wR2 = 0.0369 (all data), res. density peaks: 0.389 to −0.220 e Å−3, CCDC: 2144443.†
077 reflections measured, 3387 independent (Rint = 0.0273), R1 = 0.0157 [I > 2σ(I)], wR2 = 0.0369 (all data), res. density peaks: 0.389 to −0.220 e Å−3, CCDC: 2144443.†
Crystal data for 2 at 100(2) K: C15H23Cl2GaN2, Mr = 371.97 g mol−1, 0.462 × 0.306 × 0.060 mm, monoclinic, C2/c, a = 14.553(3) Å, b = 11.340(2) Å, c = 12.589(2) Å, β = 119.16(2), V = 1814.3(6) Å3, Z = 4, μ(Mo Kα) = 1.804 mm−1, θmax = 26.408°, 15526 reflections measured, 1871 independent (Rint = 0.0267), R1 = 0.0208 [I > 2σ(I)], wR2 = 0.0538 (all data), res. density peaks: 0.390 to −0.345 e Å−3, CCDC: 2144444.†
Crystal data for 2a at 100(2) K: C15H25Cl4GaN2, Mr = 444.89 g mol−1, 0.387 × 0.245 × 0.238 mm, orthorhombic, P212121, a = 8.412(2) Å, b = 15.856(3) Å, c = 15.903(3) Å, V = 2121.2(8) Å3, Z = 4, μ(Mo Kα) = 1.799 mm−1, θmax = 26.355°, 17672 reflections measured, 4334 independent (Rint = 0.0223), R1 = 0.0175 [I > 2σ(I)], wR2 = 0.0430 (all data), res. density peaks: 0.482 to −0.322 e Å−3, CCDC: 2144445.†
Crystal data for 3 at 100(2) K: C30H46Ga2N4, Mr = 673.05 g mol−1, 0.376 × 0.171 × 0.163 mm, monoclinic, C2/c, a = 23.069(3) Å, b = 8.896(2) Å, c = 17.634(2) Å, β = 114.36(2), V = 3296.7(10) Å3, Z = 4, μ(Mo Kα) = 1.821 mm−1, θmax = 26.475°, 146235 reflections measured, 3406 independent (Rint = 0.0776), R1 = 0.0337 [I > 2σ(I)], wR2 = 0.0797 (all data), res. density peaks: 0.505 to −0.350 e Å−3, CCDC: 2144446.†
Crystal data for 4 at 100(2) K: C76H94Al2I2N4, Mr = 1371.31 g mol−1, 0.721 × 0.714 × 0.216 mm, monoclinic, P21/c, a = 14.825(2) Å, b = 10.706(2) Å, c = 22.175(3) Å, β = 93.54(2), V = 3512.8(9) Å3, Z = 2, μ(Mo Kα) = 0.964 mm−1, θmax = 26.374°, 46646 reflections measured, 7180 independent (Rint = 0.0248), R1 = 0.0225 [I > 2σ(I)], wR2 = 0.0598 (all data), res. density peaks: 0.756 to −0.383 e Å−3, CCDC: 2144447.†
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
HWR thanks the DFG for financial support (RO 224/71-1). NS and UL acknowledge NISER Bhubaneswar for the computational facility. UL also acknowledges the support from SERB, Department of Science and Technology (EMR/2017/004843).Notes and references
- J. C. Beamish, R. W. H. Small and I. J. Worrall, Inorg. Chem., 1979, 18, 220–223 CrossRef CAS.
- R. W. H. Small and I. J. Worrall, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, B38, 250–251 CrossRef CAS.
- W. Uhl, Z. Naturforsch., 1988, 43b, 1113–1118 CrossRef.
- R. J. Wehmschulte, K. R. Senge, M. M. Olmstead, H. Hope, B. E. Sturgeon and P. P. Power, Inorg. Chem., 1993, 32, 2983–2984 CrossRef CAS.
- R. J. Wright, A. D. Phillips and P. P. Power, J. Am. Chem. Soc., 2003, 125, 10784–10785 CrossRef CAS PubMed.
- K. S. Klimek, C. Cui, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Organometallics, 2000, 19, 3085–3090 CrossRef CAS.
- S. G. Minasian and J. Arnold, Chem. Commun., 2008, 4043–4045 RSC.
- A. Hofmann, A. Lamprecht, O. F. González-Belman, R. D. Dewhurst, J. O. C. Jiménez-Halla, S. Kachel and H. Braunschweig, Chem. Commun., 2018, 54, 1639–1642 RSC.
- J. Kretsch, A. Kreyenschmidt, T. Schillmöller, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2020, 59, 13690–13699 CrossRef CAS PubMed.
- W. Haider, D. M. Andrada, I.-A. Bischoff, V. Huch and A. Schäfer, Dalton Trans., 2019, 48, 14953–14957 RSC.
- N. Wiberg, K. Amelunxen, T. Blank, H. Nöth and J. Knizek, Organometallics, 1998, 17, 5431–5433 CrossRef CAS.
- R. Laubenstein, M. Ahrens and T. Braun, Z. Anorg. Allg. Chem., 2017, 643, 1723–1729 CrossRef CAS.
- I. L. Fedushkin, A. S. Nikipelov and K. A. Lyssenko, J. Am. Chem. Soc., 2010, 132, 7874–7875 CrossRef CAS PubMed.
- P. Bag, A. Porzelt, P. J. Altmann and S. Inoue, J. Am. Chem. Soc., 2017, 139, 14384–14387 CrossRef CAS PubMed.
- C. Weetman, A. Porzelt, P. Bag, F. Hanusch and S. Inoue, Chem. Sci., 2020, 11, 4817–4827 RSC.
- R. L. Falconer, K. M. Byrne, G. S. Nichol, T. Krämer and M. J. Cowley, Angew. Chem., Int. Ed., 2021, 60, 24702–24708 ( Angew. Chem. , 2021 , 133 , 24907–24913 ) CrossRef CAS PubMed.
- S. S. Sen, A. Jana, H. W. Roesky and C. Schulzke, Angew. Chem., Int. Ed., 2009, 48, 8536–8538 ( Angew. Chem. , 2009 , 121 , 8688–8690 ) CrossRef CAS PubMed.
- H. W. Roesky, et al., unpublished results.
- D. Stalke, M. Wedler and F. T. Edelmann, J. Organomet. Chem., 1992, 431, C1–C5 CrossRef CAS.
- P. Garg, D. Dange and C. Jones, Eur. J. Inorg. Chem., 2020, 4037–4044 CrossRef CAS.
- A. V. Protchenko, J. Urbano, J. A. B. Abdalla, J. Campos, D. Vidovic, A. D. Schwarz, M. P. Blake, P. Mountford, C. Jones and S. Aldridge, Angew. Chem., Int. Ed., 2017, 56, 15098–15102 ( Angew. Chem. , 2017 , 129 , 15294–15298 ) CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, B72, 171–179 CrossRef PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Phys. Chem., 1993, 98, 5648–5652 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. v. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2018 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Schulz, K. Meindl, D. Leusser, D. Stern, M. Ruf, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2009, 42, 885–891 CrossRef CAS.
- Bruker Apex CCD, SAINT v8.38C, Bruker AXS Inst. Inc., WI, USA, Madison, 2017 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- M. Sevvana, M. Ruf, I. Usón, G. M. Sheldrick and R. Herbst-Irmer, Acta Crystallogr., Sect. D: Struct. Biol., 2019, D75, 1040–1050 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, A71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3–8 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, tables, crystallography details and additional figures. CCDC 2144443–2144447. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2dt00317a |
| This journal is © The Royal Society of Chemistry 2022 |