
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of the anthraquinone drug rhein as an axial ligand in bifunctional Pt(IV) complexes to obtain antiproliferative agents against human glioblastoma cells†
Elisabetta
Gabano‡
 a,
Marzia Bruna
Gariboldi‡
b,
Giulia
Caron
c,
Giuseppe
Ermondi
c,
Emanuela
Marras
b,
Maura
Vallaro
c and
Mauro
Ravera
*a
a,
Marzia Bruna
Gariboldi‡
b,
Giulia
Caron
c,
Giuseppe
Ermondi
c,
Emanuela
Marras
b,
Maura
Vallaro
c and
Mauro
Ravera
*a
aDipartimento di Scienze e Innovazione Tecnologica, Università del Piemonte Orientale, Viale Michel 11, 15121 Alessandria, Italy. E-mail: mauro.ravera@uniupo.it
bDipartimento di Biotecnologie e Scienze della Vita (DBSV), Università dell'Insubria, via Dunant 3, Varese, Italy
cCASSMedChem, Dipartimento di Biotecnologie Molecolari e Scienze per la Salute, Università di Torino, Via Quarello 15, 10135 Torino, Italy
First published on 18th March 2022
Abstract
Octahedral Pt(IV) prodrugs are an effective way to combine cisplatin-like moieties and a second drug to obtain selective and stimuli responsive bifunctional antiproliferative compounds. Recently, two bifunctional Pt(IV) complexes have shown interesting in vitro and in vivo effects in glioblastoma, the most aggressive primary brain tumor. An interesting observation indicates that 4,5-dihydroxy-9,10-dioxo-9,10-dihydroanthracene-2-carboxylic acid (rhein) can inhibit in vivo glioma tumor progression. Furthermore, a prodrug in which cisplatin was combined with two molecules of rhein showed a potency higher than that of cisplatin toward cisplatin-resistant lung carcinoma cells. However, the high lipophilicity of this type of complex affects their solubility and bioavailability. To overcome these limits, in the present work, three Pt(IV) derivatives were obtained by differently linking one molecule of rhein and one acetato ligand at the axial position to a cisplatin core. The complexes proved to be similar to or more potent than the parent cisplatin and rhein, and the reference drug temozolomide on two human glioblastoma cell lines (U87-MG and T98G). They retained their activity under hypoxia and caused a significant reduction in the motility of both cell lines, which can be related to their ability to inhibit MMP2 and MMP9 matrix metalloproteinases. Finally, physicochemical and computational studies indicated that these Pt(IV) derivatives are more prone than rhein to cross the blood–brain barrier.
Introduction
Malignant gliomas are the most common tumors of the central nervous system (about 6 cases per 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people are diagnosed worldwide every year),1 with glioblastoma multiforme (GBM) accounting for about 60% of all gliomas (i.e., 3.5 cases per 100000 people in developed countries).2 The adjective multiforme (multifaceted) indicates the inter- and intra-tumor heterogeneity that is present in this kind of glioma, as well as hypermutation that appears as a consequence of chemotherapy. This mutability explains why these tumors are sadly associated with a dismal prognosis and poor quality of life, and remain difficult to treat, although therapeutic options have been improved in the last few years.3 The disease course is rapid and overall estimates of survival among patients with GBM are 25% patients alive 2 years after diagnosis and only 5–10% of patients alive at 5 years.4
000 people are diagnosed worldwide every year),1 with glioblastoma multiforme (GBM) accounting for about 60% of all gliomas (i.e., 3.5 cases per 100000 people in developed countries).2 The adjective multiforme (multifaceted) indicates the inter- and intra-tumor heterogeneity that is present in this kind of glioma, as well as hypermutation that appears as a consequence of chemotherapy. This mutability explains why these tumors are sadly associated with a dismal prognosis and poor quality of life, and remain difficult to treat, although therapeutic options have been improved in the last few years.3 The disease course is rapid and overall estimates of survival among patients with GBM are 25% patients alive 2 years after diagnosis and only 5–10% of patients alive at 5 years.4
The current standard treatment of GBM includes surgery, followed by radio- and concurrent chemo-therapy with the oral alkylating agent temozolomide (TMZ). With this therapeutic protocol, a little progress in median overall survival from approx. 7 months to 17 months was observed.4 Understandably, the extensive surgical resection of GBM is difficult because of its location. Moreover, infiltrating tumor cells remain invariably within the surrounding tissue, leading to inadequate recovery and recurrence of the disease.5 Since the cures for GBM are not always effective, new strategies are needed to overcome the resistance to standard therapeutic treatments.6
Naturally occurring rhein (4,5-dihydroxy-9,10-dioxo-9,10-dihydroanthracene-2-carboxylic acid or cassic acid, Fig. 1) has a number of pharmacological effects (e.g., anti-inflammatory, anti-fibrosis, and anti-oxidant activity),7 and it exerts anticancer effects by modulating cellular proliferation, apoptosis, migration, and invasion.8–10 Interestingly, nasal administration of the potassium salt of rhein appreciably inhibited tumor progression in mice intracranially injected with GL261 glioma cells.11 This result was associated with the inhibitory effect of rhein on the ectoenzyme CD38, which has several functions as both a nicotinamide adenine dinucleotide glycohydrolase and a cell surface receptor, and its deficiency was correlated with increased cell death in the tumor mass.12,13
 | ||
| Fig. 1 Synthesis of the complexes under investigation: (i) rhein, HATU and DIPEA; (ii) Fmoc-β-alanine or 7-(Fmoc-amino)heptanoic acid, HATU and DIPEA; (iii) piperidine. Fmoc = 9-fluorenylmethoxycarbonyl group; HATU = 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate; DIPEA = N,N-diisopropylethylamine. | ||
It is now well known that cancer is a multifactorial disease and combination therapy, using two or more therapeutic agents, may target several key pathways in an additive or a synergistic way, enhancing the treatment's efficacy. Thus, two drugs or two pharmacophores with complementary action can be combined in a single molecule, obtaining a “bifunctional drug” able to elicit two different primary pharmacological actions. For example, rhein was successfully combined with the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) in a rhein–SAHA hybrid that was more effective than rhein and SAHA alone in the inhibition, proliferation, invasion and migration of glioma cells.14
Nevertheless, the bifunctional drug can be designed for the concurrent delivery and release of the two moieties. One of these moieties can be represented by cisplatin ([PtCl2(NH3)2], (SP-4-2)-diamminedichloridoplatinum(II), CDDP), a well-known antitumor agent that is clinically used against a wide variety of solid tumors. An effective way to combine CDDP-like moieties and a second drug may be represented by the octahedral Pt(IV) complexes. They comprise a class of compounds that are particularly well suited to obtain selective and stimuli responsive bifunctional compounds, due to their ability to act as prodrugs. In fact, they should be designed to be specifically reduced in the hypoxic tumor tissue, to give the cytotoxic Pt(II) metabolite with the concomitant loss of the two axial ligands (activation by reduction).15–21 In this regard, two bifunctional Pt(IV) complexes, containing 2-(2-propynyl)octanoate and a methyltetrahydropyridin derivative, respectively, were reported to be effective against GBM in both in vitro22–24 and in vivo25 models. In addition, a recent review discussed the anticancer effects and advantages of platinum-based drugs in the field of brain tumors.26
In this context, CDDP has been combined with two molecules of rhein to obtain a prodrug that showed a potency higher than that of cisplatin toward lung carcinoma cells (A549 and cisplatin resistant A549/DDP subline) and higher antitumor efficacy in the A549/DDP xenograft model.27 However, Pt(IV) complexes with two very lipophilic axial ligands, such as rhein, usually suffer from low solubility and limited bioavailability. Therefore, we synthesized three different Pt(IV) compounds in which one molecule of rhein was linked in different ways at the axial position to a CDDP core. The coordination geometry was completed by a hydrophilic acetato ligand. The resulting complexes 1–3 (Fig. 1) were then tested on two human GBM cell lines to evaluate their effects on cell viability and on migration. Moreover, the estimated ability of 1–3 to cross the blood–brain barrier was also studied.
Results and discussion
Synthesis and solution properties of the Pt(IV) complexes 1–3
The investigated complexes were designed to link one molecule of rhein in two different ways, either directly or through a spacer of two lengths (Fig. 1). The common starting molecule was (OC-6-44)-acetatodiamminedichloridohydroxidoplatinum(IV), A, obtained by oxidizing CDDP with hydrogen peroxide in acetic acid.28 In the case of complex 1, rhein was directly reacted with A, upon its activation with the coupling agent HATU (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate) in the presence of DIPEA (N,N-diisopropylethylamine). In the case of complexes 2 and 3, intermediate A was firstly converted into the corresponding intermediates B and C upon reaction with Fmoc-β-alanine and 7-(Fmoc-amino)heptanoic acid, respectively, and following deprotection with piperidine29 (Fmoc = 9-fluorenylmethoxycarbonyl group). Finally, rhein, activated with HATU and DIPEA, was reacted with complexes B and C, to obtain 2 and 3, respectively.Complexes 1–3 were challenged with cell culture medium to investigate their behavior in solution by following the RP-HPLC peak of the compounds over 72 h, the time used in the cytotoxicity tests (see the ESI, Fig. S20†). All the complexes proved to be not completely stable, as the original peaks disappeared within 72 h, with the formation of hydrolyzed species. This behavior was observed for several other Pt(IV) compounds, undermining the paradigm that Pt(IV) is kinetically stable.30–32 However, the conversion of the complexes in cell culture medium is limited in the first hours and does not affect the overall accumulation and final effect of the prodrugs, which is ruled by the fast reduction of the complexes within the cells.33,34
The latter hypothesis was checked by studying the reduction of complexes 1–3 by means of [1H, 15N] HSQC (Heteronuclear Single Quantum Coherence) NMR spectroscopy. The 15N-labeled 1–3 were prepared by using [PtCl2(15NH3)2] as the starting material,35 and challenged with the cytosolic extract of A2780 ovarian cancer cells as a model. After 2 h of reaction, the spectra showed only the signal of 15N-CDDP (15N δ = −66.9 ppm; 1H δ = 3.99 ppm) confirming the expected fast activation by reduction mechanism mediated by the cytosolic content (see the ESI, Fig. S24–S29†).36–39
Effects on cell viability
The effect of the complexes 1–3 on cell viability was determined on the two human GBM cell lines T98G and U87-MG. Tumor cells were incubated with the compounds for 72 h, and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reduction assay was used to detect viable cells. The results were expressed as the half inhibitory concentration, IC50, and compared to those obtained after treatment of the cells with the reference compounds temozolomide (TMZ), rhein and CDDP (Fig. 2 and Table 1).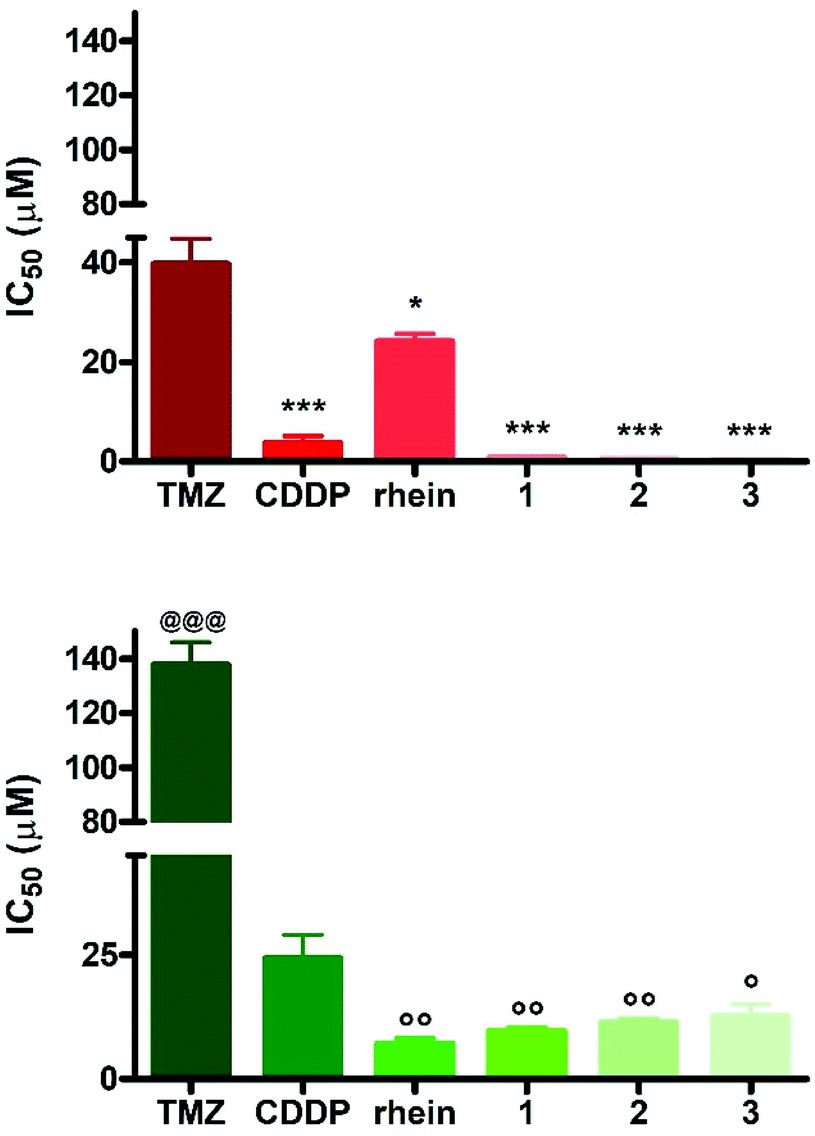 | ||
| Fig. 2 IC50 values obtained by MTT assay in (top) U87-MG and (bottom) T98G cells following 72 h treatment with rhein and its Pt(IV) derivatives 1–3. TMZ and CDDP were used as reference compounds. Data are mean ± standard deviation (sd) of six independent experiments; *p < 0.05 vs. TMZ; ***p < 0.001 vs. TMZ and rhein; *p < 0.05 vs. TMZ; @@@p < 0.001 vs. all the others; °°p < 0.01 vs.CDDP; °p < 0.05 vs.CDDP. | ||
| Compound | Normoxia | Hypoxia | ||
|---|---|---|---|---|
| U87-MG | T98G | U87-MG | T98G | |
| TMZ | 39.72 ± 5.17 | 137.85 ± 20.14 | 17.10 ± 1.50# | 148.92 ± 30.73# |
| CDDP | 3.70 ± 1.17*** | 24.30 ± 4.37*** | 2.73 ± 0.09*** | 19.85 ± 1.88*** |
| rhein | 24.19 ± 3.74* | 7.08 ± 2.34*** | 24.93 ± 0.64* | 61.73 ± 2.33***,# |
| 1 | 0.87 ± 0.19*** | 9.62 ± 1.13*** | 1.20 ± 0.21*** | 12.40 ± 0.17***,§ |
| 2 | 0.49 ± 0.14*** | 11.40 ± 1.37*** | 0.54 ± 0.07*** | 13.01 ± 0.86***,§ |
| 3 | 0.38 ± 0.08*** | 12.65 ± 1.17*** | 0.43 ± 0.03*** | 15.81 ± 1.28***,§ |
Fig. 2 shows at a glance that both cell lines were significantly more sensitive to all Pt complexes and rhein than TMZ, with IC50 ratios [IC50(TMZ)/IC50(Pt(IV) or rhein)] ranging from 1.6 (rhein) to 102 (3) on U87-MG, and from 5.6 (CDDP) to 19.5 (rhein) on T98G. Furthermore, T98G cells were less sensitive than U87-MG to Pt(IV) derivatives and to TMZ. This latter result agrees with the data reported by other authors.40 In general, the p53 status has been correlated with drug resistance in many tumors.41 Furthermore, a correlation between the p53 gene status and cisplatin response was reported, with the greatest response observed in p53 wild-type tumors and a relatively lower response rate observed in p53 mutated tumors.42 Thus, the different response of the two cell lines to reference compounds and Pt(IV) derivatives can be, at least in part, explained considering that U87-MG cells carry a wild-type form, while T98G is a mutated form, of p53.43,44
Interestingly, in the T98G cell line, rhein showed similar efficacy to the Pt derivatives, whereas in U87-MG cells, 1, 2 and 3 were significantly more potent than the organic compound.
Effect on cell migration
As already mentioned in the Introduction, aggressive invasiveness is a common feature of GBM.45–47 Although cell migration is required for physiological functions, cancer cell motility and invasion are among the crucial, though not sufficient, hallmarks of metastasis development.48With that in mind, the effect of CDDP, rhein and 1–3 derivatives on cellular migration was assessed using the scratch wound healing assay. This is a simple and low-cost method to evaluate cellular migration in 2D models and to study in vitro the anti- or pro-migration effects of a variety of experimental conditions and drugs.49–51 Briefly, in this assay, a cell-free area (wound gap) is physically created in a confluent monolayer of cells by scratching. The healing of the gap by migration and growth of the cells towards the center of the gap is monitored, quantified, and expressed as the percentage of closure of the “wounded” area with time. Cell motility or growth alterations or both can lead to a modified rate of closure of the gap.52
U87-MG and T98G cells have been shown to possess intrinsic migratory capacity;53–55 thus, the effect of a subtoxic concentration of the compounds, corresponding to the respective IC20 values, on the migration of the cells was evaluated on both cell lines. Images of the wounded area were acquired immediately after the production of the scratch and 24 h later through a camera connected to an inverted microscope (Fig. 3).
 | ||
| Fig. 3 Migratory activity of (left column) U87-MG and (right column) T98G cells following treatment with subtoxic concentrations (IC20) of rhein, CDDP and 1–3 derivatives. Pictures of the scratch wound were taken immediately after the production of the scratch (0) and 24 h later. | ||
To better represent scratch wound healing results, percentages of the open scratch wound detected after 24 h were normalized vs. the same percentage at 0 h and are shown in Fig. 4. In general, in the absence of treatment, U87-MG cells migrated faster than T98G cells, as indicated by the smaller uncovered surface at 24 h, Fig. 3. As shown in the same figure, in both cell lines, CDDP treatment unveiled similar results to the controls. In the literature, an antimigratory effect of CDDP has been reported in ovarian cell lines; however, CDDP concentrations used in that study were higher than those used in the present one to achieve subtoxic effects.56
 | ||
| Fig. 4 Percentage of open scratch wound, normalized vs. 0 h, in (top) U87-MG and (bottom) T98G cells following 24 h incubation with subtoxic concentrations (IC20) of rhein, CDDP and 1–3 derivatives. Data are mean ± SD of three independent experiments; *p < 0.05 vs. control; (a) p < 0.05 vs. control, CDDP, and rhein; (b) p < 0.05 vs. control, CDDP, and rhein; (c) p < 0.001 vs. control, CDDP, and rhein and p < 0.01 vs.1 and 2; (d) p < 0.01 vs. control, CDDP, and rhein and p < 0.05 vs.2 and 3. | ||
Different authors reported that rhein exerted inhibitory effects on migration in several cancer cell lines, including T98G cells.14,57,58 In agreement, a significant inhibition of T98G cell migration, compared to controls, was observed in the present work following exposure at a concentration that affects cell viability only minimally. In U87-MG cells, under the same conditions, only a slight increase in the % of open wound was observed.
Concerning the Pt(IV) derivatives, a significant reduction in cell motility was observed following treatment with subtoxic concentrations of 1–3 in both cell lines. In particular, complexes 3 (in U87-MG cells) and 1 (in T98G cells) were significantly more potent than all the other compounds in reducing cell migration (Fig. 4).
Extracellular matrix (ECM, the macromolecular network that constitutes the scaffold of tissues and organs) and basal membrane degradation is the first step for cancer cell migration and invasion. For maintaining normal tissue architecture and functions, the balance between ECM destruction and deposition is important. This is modulated by matrix metalloproteinases (MMPs, a family of enzymes whose activity is directed against the components of the ECM) and their tissue inhibitors. The high expression of different MMPs, including MMP-1, -2, -3, -7, -9, -13, -14, facilitates tumor cell invasion and metastasis.59 In particular, MMP2 (gelatinase-A) and MMP9 (gelatinase-B) are deeply associated with the presence of metastatic tumors.60,61 In human gliomas, a strong correlation has been reported between high MMP levels (MMP2 and MMP9, in particular) and invasiveness.62
It has been previously reported that rhein significantly inhibited the migration of ovarian cancer cells by down regulating MMP1, MMP2 and MMP9 expression.58,63 In agreement, the results in Fig. 5 and in Fig. 6 show that also in U87-MG and T98G GBM cell lines rhein was able to significantly downregulate MMP2 and MMP9 protein levels. Furthermore, also equitoxic concentrations of 1, 2 and 3 inhibited MMP2 and MMP9 to a similar extent, while no alterations in the levels of these proteins were observed following treatment with CDDP in both cell lines. Thus, the observed inhibitory effect on cellular migration exerted by 1, 2 and 3 could be, at least in part, attributed to their inhibitory activity against MMP2 and MMP9.
 | ||
| Fig. 5 (A) MMP9 and MMP2 protein levels and (B) densitometric analysis in U87-MG cells treated 72 h with CDDP, rhein and 1–3 derivatives at concentrations corresponding to the respective IC20. (A) Images of a representative experiment out of three independent experiments with similar results; (B) Results of all the experiments performed, that were normalized vs. actin protein levels ((a) p < 0.01 vs. control and CDDP; (b) p < 0.001 vs. control and CDDP and p < 0.05 vs.1 and 2). | ||
 | ||
| Fig. 6 (A) MMP-9 and MMP-2 protein levels and (B) densitometric analysis in T98G cells treated 72 h with CDDP, rhein and 1–3 derivatives at concentrations corresponding to the respective IC20. (A) Images of a representative experiment out of three independent experiments with similar results; (B) Results of all the experiments performed, that were normalized vs. actin protein levels (*p < 0.05 vs. control; (a) p < 0.01 vs. control and CDDP; (b) p < 0.05 vs. control and CDDP). | ||
Cell response in hypoxia
Hypoxia, defined as the decreased availability of oxygen, is one of the major features characterizing solid tumors. Hypoxic regions may serve as a niche for the maintenance of cancer stem cells,64 and may induce large adaptive changes at the cellular level favoring tumor progression.65–68 In particular, a hypoxic microenvironment is protective for glioblastoma, thus inducing cells to undergo malignant progression.69 Furthermore, cancer hypoxia has been associated with resistance to radio- and chemo-therapy, leading to an insufficient response to classic drug treatment.70For this reason, the relative effect of hypoxia on the response of U87-MG and T98G cells to the compounds under investigation was evaluated by the MTT assay also under hypoxic conditions (O2 1%, see the Experimental section). A comparison of the IC50 values obtained under normoxic and hypoxic conditions is reported in Table 1.
A number of experimental pieces of evidence suggest that the antiproliferative activity of CDDP is reduced under hypoxic conditions.71–73 However, no significant differences in U87-MG and T98G response to CDDP under hypoxia, compared to normoxia, was observed. Regarding the other compounds, a similar ratio of activity, compared to references, was maintained in both cell lines either under hypoxia or normoxia. The only deviation was the result obtained with rhein in T98G cells; as a matter of fact, rhein was significantly more potent under normoxia than under hypoxia, thus changing the ratio.
A particularly intriguing result concerns compounds 1–3, which under hypoxia retained their effects on cell viability, as observed for other Pt(IV) derivatives on different cancer cell lines.39,74–79
Physicochemical properties and estimation of blood–brain barrier crossing
The blood–brain barrier (BBB) is a specialized system of capillary endothelial cells that regulates ion trafficking, as well as the nutrient and oxygen supply.80It prevents approximately 98% of small molecules (smaller than 500 Da) and nearly 100% of large molecules from entering the brain, thus strictly limiting therapeutic intervention by impeding the systemic delivery of chemotherapeutics and other molecular-targeting assemblies. Although the tumors are known to compromise the integrity of the BBB and to increase its permeability, it still contributes in hampering drug accumulation in brain tumors.81
Lipophilicity and ionization have been related to the ability of compounds to cross the BBB, although there is a general agreement in the literature that the main molecular property affecting drug BBB passage is the H-bonding potential (i.e., the skill of a given molecule to form H bonds, HBs, with BBB components).82 Therefore, the ionization, lipophilicity and H-bonding potential of the investigated compounds were determined in the work at this stage.
The ionization behavior of rhein was firstly assessed; MarvinSketch v. 20.19.0 predicted three acidic centers with the following pKa values: 3.40 (COOH); 7.89 (OH close to COOH); 8.54 (OH distant from COOH). Although potentiometric titrations in water and in water/methanol mixtures were biased by solubility issues, one pKa between 4 and 5, a second pKa at 8.7 and a third pKa above 12 were experimentally observed for rhein. Therefore, at pH = 7.0 rhein is largely in its anionic form. On the contrary, pKa prediction could not be performed for 1–3, since Pt(IV) is not parameterized in any software developed for pKa calculations. However, potentiometric titrations showed an acidic pKa = 8.1 for 1. Derivatives 2 and 3 were largely insoluble in the whole pH range but structural considerations suggest that they share a similar ionization profile to 1, thus they are expected to be neutral at pH = 7.0.
Rhein and Pt(IV) derivatives were then characterized for their lipophilicity in three systems at pH = 7.0 (Table 2). First, the lipophilicity in the reference system n-octanol/water (logDoct) was measured. The different ionization pattern supports the lower logDoct of rhein (anion) when compared with the Pt(IV) derivatives (neutral). Similar results were obtained in the toluene/water system (logDtol). Notably, 3 is the most lipophilic Pt(IV) derivative in both biphasic systems whereas 1 is more lipophilic than 2 in n-octanol/water but not in toluene/water.
| Cmpd | logDoct |
logDtol |
logkIAMw |
ΔlogDoct-tol |
ΔlogkIAMw |
|---|---|---|---|---|---|
| rhein | −0.27 | <−2.5 | 1.62 | >2.5 | 2.36 |
| 1 | 1.85 | −0.85 | 2.50 | 2.7 | 1.51 |
| 2 | 1.02 | −0.52 | 2.13 | 1.54 | 1.54 |
| 3 | 2.27 | 0.07 | 3.06 | 2.20 | 1.92 |
Finally, a chromatographic lipophilicity index in a biomimetic environment was measured (logkIAMw, IAM standing for Immobilized Artificial Membrane. In IAM chromatographic columns, the covalent binding of a monolayer of phospholipids to silica particles permits to obtain a stationary phase able to mimic the lipid environment of a cell membrane).83 IAM chromatography is sensible to the presence of charges, and this could explain the higher logkIAMw value of rhein compared to those of 1, 2 (very close one with the other) and 3 (Table 2).
Two main experimental descriptors of the molecular HB potential have been reported in the literature and related to BBB permeability: ΔlogPoct-alk and ΔlogkIAMw. ΔlogPoct-alk is the difference between the partition coefficients in the octanol/water and hydrocarbon/water systems. Young et al. in 1988 showed a highly significant correlation between ΔlogPoct-alk and the logarithms of the brain/blood ratios (logBB) for 20 structurally different compounds involving twenty H2 antagonists.84 In another study, the H-donor capability was also related to the CNS (central nervous system) entering potential of seven phenylalanine oligomers esterified with carboxylic moieties.85,86
ΔlogkIAMw can be obtained when experimental logDoct and logkIAMw values are available; the higher the ΔlogkIAMw, the more polar the compound.87 Highly significant inverse linear relationships between ΔlogkIAMw and logBB values were observed in two works studying 14 structurally unrelated basic drugs and eight acidic compounds, respectively.88,89
In Table 2 ΔlogDoct-tol and ΔlogkIAMw are reported. For 1–3, but not for rhein, the partition coefficient P is equal to D, thus in this study we prefer to refer to ΔlogD rather than ΔlogP. Moreover, to limit solubility issues in alkanes, toluene replaced alkanes and thus ΔlogDoct-tol was measured instead of ΔlogDoct-alk, as suggested in a previous report.90
For interpretative purposes, it should be recalled that the higher the ΔlogDoct-alk/ΔlogkIAMw, the more likely the compound forms HBs with BB components, the less prone it is to cross the BBB. Furthermore, the presence of intramolecular HBs (IMHBs) causes a decrease in ΔlogDoct-alk/ΔlogkIAMw values. ΔlogDoct-tol and ΔlogkIAMw values in Table 2 indicate that rhein has a higher capacity to form HBs with BBB components than the Pt(IV) derivatives. This result is compatible with the formation of IMHBs that masks the polarity of the Pt(IV) amino groups. To explore the capacity of IMHB formation, Pt(IV) complexes and rhein were investigated with molecular modeling strategies. In particular, the compounds were subjected to a conformational sampling procedure and the resulting conformers were minimized using an implicit solvent treatment to mimic (a) the water (polar) environment and (b) the interior of membranes (nonpolar, see the Experimental section for details). The results are shown in Fig. 7 which shows that the amino ligands present in the three Pt(IV) complexes are involved in the formation of IMHBs with the carboxylic group in both the nonpolar and polar environments. Focusing only on minimum-energy conformers could leave doubts about the presence of IMHBs in the real biological environment, where the temperature and interaction with the solvent and membrane-forming molecules can provide the energy needed to break IMHBs. The persistence of IMHBs in conformers with a higher energy than the lowest energy conformer shown in Fig. 7 strengthens the hypothesis that the three complexes can form IMHBs even in a real environment. In particular, the energy-weighted average number of IMHBs of the three complexes ranges from 3.2 to 4.1 in the nonpolar environment and from 3.4 to 4.1 in the polar environment. These results are consistent with the presence of the above-said network of IMHBs.
 | ||
| Fig. 7 For each Pt(IV) derivative reported, from the left to the right side: the minimum energy conformer (MECs) in (i) a nonpolar environment (yellow rectangle) and (ii) in a polar environment (blue rectangle); (iii) the number of IMHBs vs. the difference of energy of the conformer respect to the MEC (E − EMEC) graph. In each plot yellow and cyan bullets refer to conformers present in nonpolar and polar environments, respectively. Pink bullet refers to rhein. | ||
Overall, the physicochemical analysis combined with the computational study supports that Pt(IV) derivatives are more prone to cross the BBB than rhein.
Experimental section
General procedures
4,5-Dihydroxy-9,10-dioxo-9,10-dihydroanthracene-2-carboxylic acid (rhein, Tokyo Chemical Industry) and all the other chemicals (Alfa Aesar-Thermo Fisher Scientific or Sigma Aldrich-Merck, except where otherwise specified) were used as received. Complexes (SP-4-2)-diamminedichloridoplatinum(II) (cisplatin),91 (OC-6-44)-acetatodiamminedichloridohydroxidoplatinum(IV), A,28,92 (OC-6-44)-acetato(β-alaninato)diamminedichloridoplatinum(IV), B,29 and (OC-6-44)-acetato[7-(Fmoc-amino) heptanoato] diammine dichlorido platinum(IV), C,93 were prepared according to established procedures. Complexes 1–3 were also obtained in their 15N-labeled form by using the same synthetic procedure except for the use of 15NH3-labeled cisplatin as the starting material (see the ESI†).28,35,36,38The purity of all the compounds was routinely verified by analytical RP-HPLC (>95%, see below for the instrumental details) and elemental analysis. Elemental analyses (C, H, and N) were carried out with an EA3000 CHN Elemental Analyzer (EuroVector, Milano, Italy) and were within ±0.4% of the theoretical values.
Chromatographic analyses were performed on a Waters HPLC-MS instrument (Waters Alliance 2695 separations module) by using a C18 Phenomenex Phenosphere-NEXT column (5 μm, 250 × 4.6 mm ID). The UV-visible detector (Waters 2487 dual lambda absorbance) was set at 210 nm. The ESI-MS spectra (Waters 3100 mass detector) were recorded using the source and desolvation temperatures set to 150 and 250 °C, respectively; N2 was used both as a drying and as a nebulizing gas. The cone and the capillary voltages were usually +30 V (positive ion mode) and 2.70 kV, respectively. The m/z values and the simulated isotope distribution patterns were used to assign the quasi-molecular ion peaks [M + H]+.
The NMR spectra were measured on a Bruker Advance III NMR spectrometer operating at 500 MHz (1H), 125.7 MHz (13C) and 107.2 MHz (195Pt with a spectral window of 2000 ppm). The chemical shifts in the 1H and 13C spectra were reported in parts per million (ppm) referenced to residual solvent resonances. In the 195Pt NMR spectra, a solution of K2[PtCl4] in saturated aqueous KCl was used as the external reference (δ = −1628 ppm from Na2PtCl6). The 15N NMR spectra were referenced to a solution of 15NH4Cl in 1 M HCl. [1H, 15N] Heteronuclear Single Quantum Correlation (HSQC) spectra were recorded with the standard Bruker sequence hsqcetgpsiz (with 0.2 s acquisition time, 8 scans, 1.3 s relaxation delay, and 128 F1 points) at 300 K.
Synthesis of the complexes
15NH3-1. 15N NMR (50.70 MHz, DMF/D2O/H2O): δ −38.8 ppm (1H δ = 6.59 ppm) with satellite peaks at −41.7 (1H δ = 6.63 ppm) and −36.3 (1H δ = 6.53 ppm) ppm (1JPt–N = 137 Hz and 2JPt–H = 25 Hz). ESI-MS: m/z calcd for C17H17Cl215N2O8Pt [M + H]+ 645; found, 645.
15NH3-2. 15N NMR (50.70 MHz, DMF/D2O/H2O): δ −40.0 ppm (1H δ = 6.44 ppm) with satellite peaks at −42.3 (1H δ = 6.48 ppm) and −37.5 (1H δ = 6.39 ppm) ppm (1JPt–N = 122 Hz and 2JPt–H = 22 Hz). ESI-MS: m/z calcd for C20H22Cl2N15N2O9Pt [M + H]+ 716; found, 716.
15NH3-3. 15N NMR (50.70 MHz, DMF/D2O/H2O): δ −39.7 ppm (1H δ = 6.39 ppm) with satellite peaks at −42.5 (1H δ = 6.44 ppm) and −37.5 (1H δ = 6.34 ppm) ppm (1JPt–N = 127 Hz and 2JPt–H = 25 Hz). ESI-MS: m/z calcd for C24H30Cl2N15N2O9Pt [M + H]+ 772; found 772.
Stability in solution and reduction with cytosol
Complexes 1–3 were dissolved in methanol and these solutions were diluted with complete RPMI 1640 cell culture medium (10% v/v CH3OH; final [Pt] = 0.1 mM). The behavior in solution was monitored at 37 °C for 72 h by measuring the area of the RP-HPLC chromatographic peaks of the Pt complexes. The analyses were performed using the Waters HPLC-MS instrument described in the General procedures section (Phenomenex Phenosphere-NEXT, 250 × 4.6 mm, 5 μm C18 column; 70% v/v of methanol and 30% v/v of 15 mM formic acid as an eluent; flow rate was 0.5 mL min−1; UV-vis detector set at 210 nm).To simulate the intracellular reduction, the 15N-labeled complexes 15N-1, 15N-2, and 15N-3, were challenged with cytosol obtained from A2780 cells by using the FractionPrep kit (BioVision, Milpitas, CA, USA). [1H, 15N] HSQC spectra were recorded on samples prepared as follows: the 15N-labeled complexes were dissolved in 20 μL of DMF and these solutions were diluted with 450 μL of cytosol and 30 μL of D2O (10 mM final concentration).
Cell cultures
U87-MG and T98G glioblastoma cell lines were obtained from ATCC (American Type Culture Collection, Manassas, VA, USA) and maintained in DMEM medium (Euroclone, Milan, Italy), supplemented with 10% fetal calf serum (Euroclone, Milan, Italy), 1% glutamine and 1% antibiotics mixture, 1% sodium pyruvate and 1% non-essential amino acids (both from Sigma-Aldrich, Milan, Italy) at 37 °C in a humidified 5% CO2 atmosphere. Cells were routinely checked for Mycoplasma (Molecular Biology Reagent Set Mycoplasma species, Euroclone, UK) and all experiments were performed within 15 passages from thawing. For all the experiments, cells were exposed to TMZ, CDDP, rhein and 1–3 derivatives for 72 h. Platinum derivatives were dissolved in DMSO and immediately diluted with buffer or cell culture media prior to the treatment. The final concentration of the organic solvent never exceeded 0.1% v/v. This concentration was found to be non-toxic to the cells tested (control). For hypoxia experiments, treated cells were incubated for 24 h under normoxia and then placed into a modular incubator chamber (Billups Rothenberg Inc., Del Mar, CA, USA) flushed with a mixture of 1% O2, 5% CO2 and 94% N2 at 37 °C during the last 48 h.Growth inhibition assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed on U87-MG and T98G according to previously published procedures.94 Briefly, 3 × 103 cells were plated in each well of a 96-well plate in 0.1 mL of complete culture medium and allowed to attach for 24 h. Cells were then exposed at 37 °C for 72 h to the compounds studied at concentrations ranging between 0.05 and 300 μM. At the end of the period of incubation, MTT (0.05 mL of a 2 mg mL−1 stock solution in PBS) was added to each well and cells were incubated for 3 h at 37 °C. Cell supernatants were then carefully removed, and the blue formazan crystals formed through MTT reduction by metabolically active cells were dissolved in 0.120 mL of DMSO and the corresponding optical densities were measured at 570 nm, using an iMARK Microplate Reader (Bio-RAD). IC50 values were calculated based on nonlinear regression analysis of dose–response data performed with the Calcusyn software (Biosoft, Cambridge, UK). Differences between IC50 values were evaluated statistically by analysis of variance with Bonferroni post-test for multiple comparisons.Scratch wound healing assay
U87-MG and T98G cells were seeded onto 6-well plates and allowed to grow approximately to confluence before a scratch was produced in cell monolayers using a 100 mL pipette tip. The scratched monolayers were then washed with fresh PBS and exposed to subtoxic concentrations of the compounds, corresponding to the respective IC20 concentrations, at 37 °C for 24 h. Images were acquired directly after producing the scratch and 24 h thereafter, using a camera connected to an Olympus IX81 inverted microscope.Western blot analysis
The expression of MMP2 and MMP9 in whole cell lysates following 72 h treatment with the compounds under investigation, at concentrations corresponding to the respective IC20, was detected by western blot analysis. For whole cell lysates cells were resuspended in lysis buffer (NaCl 120 mM, NaF 25 mM, EDTA 5 mM, EGTA 6 mM, sodium pyrophosphate 25 mM in tris-buffered saline TBS 20 mM pH 7.4, phenylmethanesulfonyl fluoride PMSF 2 mM, Na3VO4 1 mM, phenylarsine oxide 1 mM, 1% NP-40 and 10% protease inhibitor cocktail) and incubated for 10 min on ice after adding Nonidet P-40 (final concentration 0.1%) and lysates were collected by centrifugation (12800 rpm for 20 min). The protein concentration was determined by BCA assay (Pierce, Italy) and 50 μg of protein per sample were loaded onto 8% polyacrylamide gels and separated under denaturing conditions. Protein bands were then transferred onto Hybond-P membranes (Amersham Biosciences, Italy) and western blot analysis was performed by standard techniques with mouse monoclonal antibody directed against MMP2 and MMP9 (Santa Cruz Biotechnology, Inc). Equal loading of the samples was verified by re-probing the blots with a mouse monoclonal anti-actin antibody (Santa Cruz Biotechnology, Inc.). Protein bands were visualized by G-box (Syngene, Chemi-XT4) using peroxidase-conjugated anti-mouse secondary antibodies (Sigma-Aldrich) and the Westar Supernova Substrate (Cyanagen). Densitometric analysis were performed using Image-J software and differences between obtained values were evaluated statistically by analysis of variance with Bonferroni post-test for multiple comparisons.
Ionization
The potentiometric technique was used for the determination of the pKa values using the Pion SiriusT3 apparatus (Pion-Inc., Billerica, MA, USA). The instrument is equipped with an Ag/AgCl double junction reference pH electrode, a stirrer, a microcapillary dispenser (water, 0.5 M KOH and 0.5 M HCl), a temperature probe and a turbidity sensor. All the titration experiments were conducted at ionic strength adjusted at 0.15 M KCl either in water, cosolvent/water mixtures or in the presence of partition solvents, under a N2 atmosphere at a controlled temperature (25 ± 0.1 °C).In this pH-metric method, pKa is measured by titrating a solution (obtained either by the addition of water or water/cosolvent mixtures) of the sample with an acid and a base, and the results are obtained by a complex computational process.95 About 1 mg of the sample was weighed and put into the vial. Analyte dilution, mixing, acid/base titration, and measurement of pH were automated by the Sirius T3 measurement protocol. The pH range of the titration was from pH 12.2 to 1.8 via the addition of acid (0.5 M HCl) and base (0.5 M KOH), targeting 0.2 pH steps between pH measurements. Three sequential pH titrations on the same sample solution were performed.
Lipophilicity and polarity descriptors
LogDoct and logDtol were determined using a shake-flask procedure. A stock solution of the compound was prepared in n-octanol saturated with water 0.15 M KCl or toluene. 1 mL of solution was put in a separate vial to which 1 mL of buffer pH = 7.0 (ammonium acetate 10 mM) was added. All the buffers were ionic strength adjusted at 0.15 M KCl. The vial was vortexed for 10 min and the two phases were separated and analyzed by HPLC. Measurements were performed in triplicate.
The method applied for the determination of logkIAMw is already described elsewhere.96 Briefly the analyses were performed at 30 °C with 20 mM ammonium acetate at pH 7.0 in a mixture with acetonitrile at various percentages. The stationary phase was IAM.PC.DD.2 (10 cm × 4.6 cm, 10 μm packing, 300 Å pore size) (Regis Technology, Morton Grove, IL; USA). The flow rate was 1.0 mL min−1, and the injection volume was 10 mL. Chromatographic retention data at a given amount of cosolvent, expressed as logkIAM (the logarithm of the retention factor), were calculated using the expression:
| logkIAM = log[(tr − t0)/t0] |
kIAM values are the average of at least three measurements. The indexes logkIAMw were calculated by an extrapolation method. LogkIAMw values were determined at least three different acetonitrile percentages (ϕ) in the mobile phases (from 10 to 40% v/v) and the intercept values of the linear relationships (R2 ≥ 0.98) between logk and ϕ values were assumed as logkIAMw values.
A HPLC Varian ProStar instrument equipped with a 410 autosampler with thermostable column compartment, a PDA 335 LC Detector, Galaxie Chromatography Data System Version 1.9.302.952 and CompassCDS Data System Version 4.1.0.296 was used.
ΔlogkIAMw is the difference between the logarithm of the experimental chromatographic retention factor (logkIAMw) and the value here named logkIAMw calculated using the following equation:96
| clogkIAMw = 0.92 × BRlogD − 1.03 |
Molecular modeling
The 3D structures of the investigated Pt complexes and rhein were obtained using editing tools in Spartan'20 molecular modeling software (version 1.1.0, Wavefunction, Inc., Irvine, CA, USA, https://www.wavefun.com/). Conformational sampling was performed using Spartan'20 using Molecular Mechanics (MMFF) with default torsional angles selected by the tool. Finally, all the molecules were minimized with MOPAC2016 (version 21.237 W, Stewart Computational Chemistry, https://openmopac.net/). To mimic a water environment (dielectric constant ε = 80) and the interior of the membrane (dielectric constant ε = 4.81) minimization was performed applying the Conductor-like Screening Model (COSMO)97 continuum approach implemented in MOPAC2016. The number of IMHBs was determined importing conformations in Chimera (version 1.15, UCSF, San Francisco, CA, USA, https://www.cgl.ucsf.edu/chimera/) and visual analysis and figures were obtained using VIDA (version 5.0.0.1, OpenEye, Santa Fe, NM, USA, https://www.eyesopen.com/). Finally, the IMHB plots were obtained with DataWarrior (version 5.5.0, https://openmolecules.org/).98Conclusions
The three Pt(IV) bifunctional prodrugs based on rhein and CDDP, here designed to be tested against GBM cell lines, showed significantly better performances on U87-MG and T98G cell viability than CDDP. Furthermore, the effects of Pt(IV) complexes were comparable to or better than those of rhein on T98G and U87-MG, respectively. This last result was not unexpected, since lipophilic Pt(IV) bifunctional complexes often demonstrate a higher activity, when compared to their free metabolites (in this case, CDDP and rhein). An important result is that Pt(IV) complexes were more potent than TMZ, which represents one of the standard treatments for GBM, and retained their activity under hypoxic conditions. Hypoxia is a common feature of a solid tumor microenvironment, and it has been associated with tumor progression and resistance to chemotherapy. Therefore, it is extremely important to identify new compounds whose effects are unaffected by adaptive mechanisms activated under hypoxic conditions.Perhaps the most interesting results in the present study concern the anti-migratory effect and the evaluation of BBB crossing of 1–3. The migratory behavior in tumor cells of epithelial origin is a sign of invasion and metastasis and strongly indicates that the cells have undergone the transition to a mesenchymal phenotype (epithelial-to-mesenchymal transition, EMT). In this context, the presence of rhein (and its intracellular release after activation by reduction) in 1–3 provides Pt(IV) complexes with the ability to inhibit cancer cell migration, which is absent in CDDP at least at the subtoxic concentrations tested here.
CDDP is unable to cross the intact BBB, thus the limited efficacy of CDDP-based drugs on the CNS tumors is related to the disruption of the barrier caused by the tumor itself. As an example, in a recent paper the therapeutic activity of carmustine, cyclophosphamide and CDDP on intracranial Ehrlich tumor in mice was shown to be associated with the BBB permeability for these drugs (90, 20 and 8%, respectively).99 On the other hand, in mice intracranially injected with glioma cells, rhein appreciably inhibited tumor progression, demonstrating its ability to cross the BBB. As far as it concerns the estimation of blood–brain barrier crossing performed here, Pt(IV) derivatives can be more prone to cross the BBB than rhein.
In conclusion, the conjugation between CDDP and rhein in a bifunctional Pt(IV) complex produced compounds in which the characteristics of the two moieties and their activity may be combined and enhanced in a sort of “intramolecular combination therapy”. The results reported indicate that complexes 1–3 are worthy of further studies and that these compounds could represent an interesting improvement for GBM treatment.
Conflicts of interest
There are no conflicts to declare.References
- M. Weller, M. van den Bent, M. Preusser, E. Le Rhun, J. C. Tonn, G. Minniti, M. Bendszus, C. Balana, O. Chinot, L. Dirven, P. French, M. E. Hegi, A. S. Jakola, M. Platten, P. Roth, R. Ruda, S. Short, M. Smits, M. J. B. Taphoorn, A. von Deimling, M. Westphal, R. Soffietti, G. Reifenberger and W. Wick, Nat. Rev. Clin. Oncol., 2021, 18, 170–186 CrossRef PubMed.
- T. F. Cloughesy, W. K. Cavenee and P. S. Mischel, in Annual Review of Pathology: Mechanisms of Disease, Vol 9, ed. A. K. Abbas, S. J. Galli and P. M. Howley, Annual Reviews, Palo Alto, 2014, vol. 9, pp. 1–25 Search PubMed.
- B. M. Alexander and T. F. Cloughesy, J. Clin. Oncol., 2017, 35, 2402–2409 CrossRef CAS PubMed.
- R. Stupp, S. Taillibert, A. Kanner, W. Read, D. M. Steinberg, B. Lhermitte, S. Toms, A. Idbaih, M. S. Ahluwalia, K. Fink, F. Di Meco, F. Lieberman, J. J. Zhu, G. Stragliotto, D. D. Tran, S. Brem, A. F. Hottinger, E. D. Kirson, G. Lavy-Shahaf, U. Weinberg, C. Y. Kim, S. H. Paek, G. Nicholas, J. Burna, H. Hirte, M. Weller, Y. Palti, M. Hegi and Z. Ram, JAMA, J. Am. Med. Assoc., 2017, 318, 2306–2316 CrossRef CAS PubMed.
- R. Batash, N. Asna, P. Schaffer, N. Francis and M. Schaffer, Curr. Med. Chem., 2017, 24, 3002–3009 CrossRef CAS PubMed.
- A. Shergalis, A. Bankhead, U. Luesakul, N. Muangsin and N. Neamati, Pharmacol. Rev., 2018, 70, 412–445 CrossRef CAS PubMed.
- H. Sun, G. W. Luo, D. H. Chen and Z. Xiang, Front. Pharmacol., 2016, 7, 16 Search PubMed.
- C. Wu, H. Y. Cao, H. Zhou, L. Sun, J. G. Xue, J. Y. Li, Y. Q. Bian, R. F. Sun, S. Dong, P. Liu and M. Y. Sun, Anti-Cancer Agents Med. Chem., 2017, 17, 1624–1632 CrossRef CAS PubMed.
- A. S. Tikhomirov, A. A. Shtil and A. E. Shchekotikhin, Recent Pat. Anti-Cancer Drug Discovery, 2018, 13, 159–183 CrossRef CAS PubMed.
- S. Henamayee, K. Banik, B. L. Sailo, B. Shabnam, C. Harsha, S. Srilakshmi, V. G. M. Naidu, S. H. Baek, K. S. Ahn and A. B. Kunnumakkara, Molecules, 2020, 25, 26 CrossRef PubMed.
- E. Blacher, B. Ben Baruch, A. Levy, N. Geva, K. D. Green, S. Garneau-Tsodikova, M. Fridman and R. Stein, Int. J. Cancer, 2015, 136, 1422–1433 CrossRef CAS PubMed.
- A. Levy, E. Blacher, H. Vaknine, F. E. Lund, R. Stein and L. Mayo, Neuro-Oncology, 2012, 14, 1037–1049 CrossRef CAS PubMed.
- Y. Wang, X. G. Fan, T. Tang, R. Fan, C. H. Zhang, Z. B. Huang, W. J. Peng, P. P. Gan, X. G. Xiong, W. Huang and X. Huang, Sci. Rep., 2016, 6, 37098 CrossRef CAS PubMed.
- J. Chen, B. Luo, S. Wen and R. Pi, Invest. New Drugs, 2020, 38, 755–764 CrossRef CAS PubMed.
- E. Gabano, M. Ravera and D. Osella, Dalton Trans., 2014, 43, 9813–9820 RSC.
- D. Gibson, Dalton Trans., 2016, 45, 12983–12991 RSC.
- R. G. Kenny, S. W. Chuah, A. Crawford and C. J. Marmion, Eur. J. Inorg. Chem., 2017, 1596–1612 CrossRef CAS.
- D. Gibson, J. Inorg. Biochem., 2019, 191, 77–84 CrossRef CAS PubMed.
- M. Ravera, E. Gabano, M. J. McGlinchey and D. Osella, Inorg. Chim. Acta, 2019, 492, 32–47 CrossRef CAS.
- D. Gibson, J. Inorg. Biochem., 2021, 217, 111353 CrossRef CAS PubMed.
- D. Gibson, ChemMedChem, 2021, 16, 2188–2191 CrossRef CAS PubMed.
- B. Rangone, B. Ferrari, V. Astesana, I. Masiello, P. Veneroni, I. Zanellato, D. Osella and M. G. Bottone, Life Sci., 2018, 210, 166–176 CrossRef CAS PubMed.
- B. Ferrari, F. Urselli, M. Gilodi, S. Camuso, E. C. Priori, B. Rangone, M. Ravera, P. Veneroni, I. Zanellato, E. Roda, D. Osella and M. G. Bottone, Neurotoxic. Res., 2020, 37, 183–197 CrossRef CAS PubMed.
- B. Ferrari, E. Roda, E. C. Priori, F. De Luca, A. Facoetti, M. Ravera, F. Brandalise, C. A. Locatelli, P. Rossi and M. G. Bottone, Front. Neurosci., 2021, 15, 589906 CrossRef PubMed.
- S. Raghavan, D. S. Baskin and M. A. Sharpe, Mol. Cancer Ther., 2020, 19, 2445–2453 CrossRef CAS PubMed.
- J. Jeon, S. Lee, H. Kim, H. Kang, H. Youn, S. Jo, B. Youn and H. Y. Kim, Int. J. Mol. Sci., 2021, 22, 5111 CrossRef CAS PubMed.
- M.-X. Tan, Z.-F. Wang, Q.-P. Qin, B.-Q. Zou and H. Liang, Dalton Trans., 2020, 49, 1613–1619 RSC.
- M. Ravera, E. Gabano, I. Zanellato, F. Fregonese, G. Pelosi, J. A. Platts and D. Osella, Dalton Trans., 2016, 45, 5300–5309 RSC.
- M. Ravera, E. Gabano, S. Tinello, I. Zanellato and D. Osella, J. Inorg. Biochem., 2017, 167, 27–35 CrossRef CAS PubMed.
- A. Kastner, I. Poetsch, J. Mayr, J. V. Burda, A. Roller, P. Heffeter, B. K. Keppler and C. R. Kowol, Angew. Chem., Int. Ed., 2019, 58, 7464–7469 CrossRef CAS PubMed.
- E. Wexselblatt, R. Raveendran, S. Salameh, A. Friedman-Ezra, E. Yavin and D. Gibson, Chem. – Eur. J., 2015, 21, 3108–3114 CrossRef CAS PubMed.
- E. Wexselblatt, E. Yavin and D. Gibson, Angew. Chem., Int. Ed., 2013, 52, 6059–6062 CrossRef CAS PubMed.
- E. Gabano, B. Rangone, E. Perin, G. Caron, G. Ermondi, M. Vallaro, V. Gandin, C. Marzano, A. Barbanente, N. Margiotta and M. Ravera, Dalton Trans., 2021, 50, 4663–4672 RSC.
- M. Ravera, E. Gabano, I. Zanellato, B. Rangone, E. Perin, B. Ferrari, M. G. Bottone and D. Osella, Dalton Trans., 2021, 50, 3161–3177 RSC.
- M. S. Davies, M. D. Hall, S. J. Berners-Price and T. W. Hambley, Inorg. Chem., 2008, 47, 7673–7680 CrossRef CAS PubMed.
- E. Wexselblatt and D. Gibson, J. Inorg. Biochem., 2012, 117, 220–229 CrossRef CAS PubMed.
- D. Gibson, Dalton Trans., 2009, 10681–10689 RSC.
- A. Nemirovski, Y. Kasherman, Y. Tzaraf and D. Gibson, J. Med. Chem., 2007, 50, 5554–5556 CrossRef CAS PubMed.
- E. Gabano, M. Ravera, F. Trivero, S. Tinello, A. Gallina, I. Zanellato, M. B. Gariboldi, E. Monti and D. Osella, Dalton Trans., 2018, 47, 8268–8282 RSC.
- S. Kohsaka, L. Wang, K. Yachi, R. Mahabir, T. Narita, T. Itoh, M. Tanino, T. Kimura, H. Nishihara and S. Tanaka, Mol. Cancer Ther., 2012, 11, 1289–1299 CrossRef CAS PubMed.
- R. R. Wallace-Brodeur and S. W. Lowe, Cell. Mol. Life Sci., 1999, 55, 64–75 CrossRef CAS PubMed.
- Z. H. Siddik, Oncogene, 2003, 22, 7265–7279 CrossRef CAS PubMed.
- K. Bajbouj, C. Mawrin, R. Hartig, J. Schulze-Luehrmann, A. Wilisch-Neumann, A. Roessner and R. Schneider-Stock, J. Neuro-Oncol., 2012, 107, 503–516 CrossRef CAS PubMed.
- C. M. Park, M. J. Park, H. J. Kwak, S. I. Moon, D. H. Yoo, H. C. Lee, I. C. Park, C. H. Rhee and S. I. Hong, Int. J. Oncol., 2006, 28, 119–125 CAS.
- G. P. Dunn, M. L. Rinne, J. Wykosky, G. Genovese, S. N. Quayle, I. F. Dunn, P. K. Agarwalla, M. G. Chheda, B. Campos, A. Wang, C. Brennan, K. L. Ligon, F. Furnari, W. K. Cavenee, R. A. Depinho, L. Chin and W. C. Hahn, Genes Dev., 2012, 26, 756–784 CrossRef CAS PubMed.
- V. A. Cuddapah, S. Robel, S. Watkins and H. Sontheimer, Nat. Rev. Neurosci., 2014, 15, 455–465 CrossRef CAS PubMed.
- T. T. Lah, M. Novak and B. Breznik, Semin. Cancer Biol., 2020, 60, 262–273 CrossRef CAS PubMed.
- D. R. Welch and D. R. Hurst, Cancer Res., 2019, 79, 3011–3027 CrossRef CAS PubMed.
- C. C. Liang, A. Y. Park and J. L. Guan, Nat. Protoc., 2007, 2, 329–333 CrossRef CAS PubMed.
- E. Monti, E. Marras, P. Prini and M. B. Gariboldi, Eur. J. Pharmacol., 2020, 881, 173210 CrossRef CAS PubMed.
- J. Luo, X. Luo, X. Liu, Z. Q. Fang, J. Xu and L. K. Li, OncoTargets Ther., 2020, 13, 1321–1330 CrossRef CAS PubMed.
- Y. Chen, Bio-Protoc., 2012, 2, e100 Search PubMed.
- B. X. Yu, L. Zou, S. Li and Y. L. Du, Eur. Rev. Med. Pharmacol. Sci., 2019, 23, 8456–8467 Search PubMed.
- J. Zhang and J. Y. Fan, Exp. Ther. Med., 2020, 20, 1145–1152 CrossRef CAS PubMed.
- I. Emanuelsson, K. Wikvall, T. Friman and M. Norlin, Basic Clin. Pharmacol. Toxicol., 2018, 123, 130–136 CrossRef CAS PubMed.
- P. Yin, G. Z. Song and Z. H. Jiang, Cancer Chemother. Pharmacol., 2018, 81, 863–872 CrossRef CAS PubMed.
- M. L. Lin, J. G. Chung, Y. C. Lu, C. Y. Yang and S. S. Chen, Oral Oncol., 2009, 45, 531–537 CrossRef CAS PubMed.
- B. Y. Ren, W. J. Guo, Y. W. Tang, J. Zhang, N. Xiao, L. Zhang and W. L. Li, Biol. Pharm. Bull., 2019, 42, 568–572 CrossRef CAS PubMed.
- J. A. Eble and S. Niland, Clin. Exp. Metastasis, 2019, 36, 171–198 CrossRef CAS PubMed.
- H. C. Zheng, H. Takahashi, Y. Murai, Z. G. Cui, K. Nomoto, H. Niwa, K. Tsuneyama and Y. Takano, Anticancer Res., 2006, 26, 3579–3583 CAS.
- F. Q. Wang, J. So, S. Reierstad and D. A. Fishman, Int. J. Cancer, 2005, 114, 19–31 CrossRef CAS PubMed.
- M. Nakada, Y. Okada and J. Yamashita, Front. Biosci., 2003, 8, E261–E269 CrossRef CAS PubMed.
- G. M. Zhou, F. H. Peng, Y. P. Zhong, Y. H. Chen, M. Tang and D. R. Li, Int. J. Oncol., 2017, 50, 933–941 CrossRef CAS PubMed.
- T. M. Yeung, S. C. Gandhi and W. F. Bodmer, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4382–4387 CrossRef CAS PubMed.
- J. T. de Oliveira, C. Ribeiro, R. Barros, C. Gomes, A. J. de Matos, C. A. Reis, G. R. Rutteman and F. Gartner, PLoS One, 2015, 10, e0134458 CrossRef PubMed.
- L. Schito and G. L. Semenza, Trends Cancer, 2016, 2, 758–770 CrossRef PubMed.
- G. L. Semenza, Oncogene, 2010, 29, 625–634 CrossRef CAS PubMed.
- G. L. Semenza, Cell, 2012, 148, 399–408 CrossRef CAS PubMed.
- D. A. Cavazos and A. J. Brenner, Neurobiol. Dis., 2016, 85, 227–233 CrossRef PubMed.
- C. A. Waker and R. M. Lober, in Myelin: Basic and Clinical Advances, ed. K. Sango, J. Yamauchi, T. Ogata and K. Susuki, Springer, Singapore, 2019, vol. 1190, pp. 281–297 Search PubMed.
- A. M. Shannon, D. J. Bouchier-Hayes, C. M. Condron and D. Toomey, Cancer Treat. Rev., 2003, 29, 297–307 CrossRef CAS PubMed.
- P. Vaupel and A. Mayer, Cancer Metastasis Rev., 2007, 26, 225–239 CrossRef CAS PubMed.
- N. Devarajan, R. Manjunathan and S. K. Ganesan, Crit. Rev. Oncol. Hematol., 2021, 162, 103327 CrossRef PubMed.
- S. Karmakar, S. Chatterjee, K. Purkait and A. Mukherjee, Dalton Trans., 2016, 45, 11710–11722 RSC.
- Z. C. Xu, J. Zhao, S. H. Gou and G. Xu, Chem. Commun., 2017, 53, 3749–3752 RSC.
- Z. Wang, Z. Deng and G. Zhu, Dalton Trans., 2019, 48, 2536–2544 RSC.
- H. R. Mellor, S. Snelling, M. D. Hall, S. Modok, M. Jaffar, T. W. Hambley and R. Callaghan, Biochem. Pharmacol., 2005, 70, 1137–1146 CrossRef CAS PubMed.
- E. Schreiber-Brynzak, V. Pichler, P. Heffeter, B. Hanson, S. Theiner, I. Lichtscheidl-Schultz, C. Kornauth, L. Bamonti, V. Dhery, D. Groza, D. Berry, W. Berger, M. Galanski, M. A. Jakupec and B. K. Keppler, Metallomics, 2016, 8, 422–433 CrossRef CAS PubMed.
- S. Goschl, E. Schreiber-Brynzak, V. Pichler, K. Cseh, P. Heffeter, U. Jungwirth, M. A. Jakupec, W. Berger and B. K. Keppler, Metallomics, 2017, 9, 309–322 CrossRef PubMed.
- O. van Tellingen, B. Yetkin-Arik, M. C. de Gooijer, P. Wesseling, T. Wurdinger and H. E. de Vries, Drug Resist. Updates, 2015, 19, 1–12 CrossRef CAS PubMed.
- C. D. Arvanitis, G. B. Ferraro and R. K. Jain, Nat. Rev. Cancer, 2020, 20, 26–41 CrossRef CAS PubMed.
- H. van de Waterbeemd, G. Camenisch, G. Folkers, J. R. Chretien and O. A. Raevsky, J. Drug Targeting, 1998, 6, 151–165 CrossRef CAS PubMed.
- C. Pidgeon, S. W. Ong, H. L. Liu, X. X. Qiu, M. Pidgeon, A. H. Dantzig, J. Munroe, W. J. Hornback, J. S. Kasher, L. Glunz and T. Szczerba, J. Med. Chem., 1995, 38, 590–594 CrossRef CAS PubMed.
- R. C. Young, R. C. Mitchell, T. H. Brown, C. R. Ganellin, R. Griffiths, M. Jones, K. K. Rana, D. Saunders and I. R. Smith, J. Med. Chem., 1988, 31, 656–671 CrossRef CAS PubMed.
- B. Testa, P.-A. Carrupt, P. Gaillard and R.-S. Tsai, in Lipophilicity in Drug Action and Toxicology, ed. V. Pliška, B. Testa and H. van de Waterbeemd, VCH, Weinheim, Germany, 1996, ch. 4, pp. 49–71 Search PubMed.
- R. A. Conradi, P. S. Burton and R. T. Borchardt, in Lipophilicity in Drug Action and Toxicology, ed. V. Pliška, B. Testa and H. van de Waterbeemd, VCH, Weinheim, Germany, 1996, ch. 14, pp. 233–252 Search PubMed.
- G. Ermondi, M. Vallaro and G. Caron, Drug Discovery Today, 2020, 25, 1585–1591 CrossRef CAS PubMed.
- L. Grumetto, C. Carpentiero and F. Barbato, Eur. J. Pharm. Sci., 2012, 45, 685–692 CrossRef CAS PubMed.
- L. Grumetto, C. Carpentiero, P. Di Vaio, F. Frecentese and F. Barbato, J. Pharm. Biomed. Anal., 2013, 75, 165–172 CrossRef CAS PubMed.
- M. Shalaeva, G. Caron, Y. A. Abramov, T. N. O'Connell, M. S. Plummer, G. Yalamanchi, K. A. Farley, G. H. Goetz, L. Philippe and M. J. Shapiro, J. Med. Chem., 2013, 56, 4870–4879 CrossRef CAS PubMed.
- S. C. Dhara, Indian J. Chem., 1970, 8, 193–194 Search PubMed.
- G. Pelosi, M. Ravera, E. Gabano, F. Fregonese and D. Osella, Chem. Commun., 2015, 51, 8051–8053 RSC.
- E. Gabano, G. Pinton, C. Balzano, S. Boumya, D. Osella, L. Moro and M. Ravera, Molecules, 2021, 26, 4740 CrossRef CAS PubMed.
- M. B. Gariboldi, E. Taiana, M. C. Bonzi, I. Craparotta, S. Giovannardi, M. Mancini and E. Monti, Cancer Lett., 2015, 364, 156–164 CrossRef CAS PubMed.
- A. Avdeef, K. J. Box, J. E. A. Comer, M. Gilges, M. Hadley, C. Hibbert, W. Patterson and K. Y. Tam, J. Pharm. Biomed. Anal., 1999, 20, 631–641 CrossRef CAS PubMed.
- G. Ermondi, M. Vallaro and G. Caron, Eur. J. Pharm. Sci., 2018, 114, 385–390 CrossRef CAS PubMed.
- A. Klamt and G. Schuurmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- T. Sander, J. Freyss, M. von Korff and C. Rufener, J. Chem. Inf. Model., 2015, 55, 460–473 CrossRef CAS PubMed.
- A. N. Stukov, V. G. Bespalov, V. A. Alexandrov, A. L. Semenov, G. S. Kireeva, T. Y. Semiglazova, L. V. Filatova and D. A. Baranenko, Drug Res., 2020, 70, 86–90 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: ESI-MS, 1H, 13C, 195Pt, [1H, 1H] COSY and [1H, 13C] HSQC NMR spectra of complexes 1–3; solution behavior of 1–3; ESI-MS spectra of 15N-labeled 1–3; [1H, 15N] HSQC NMR spectra of 15N-1–3 before and after reduction with cytosol. See DOI: https://doi.org/10.1039/d2dt00235c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |