
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
HgBrI: a possible tecton for NLO molecular materials?†
Alessia
Giordana
 a,
Emanuele
Priola‡
a,
Stefano
Pantaleone
ab,
Luca
Andreo
a,
Leonardo
Mortati
c,
Paola
Benzi‡
a,
Lorenza
Operti
a and
Eliano
Diana‡
*a
a,
Emanuele
Priola‡
a,
Stefano
Pantaleone
ab,
Luca
Andreo
a,
Leonardo
Mortati
c,
Paola
Benzi‡
a,
Lorenza
Operti
a and
Eliano
Diana‡
*a
aDepartment of Chemistry, Università degli Studi di Torino, Via Pietro Giuria 7, 10125 Torino, Italy. E-mail: eliano.diana@unito.it
bDipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia, Via Elce di Sotto 8, I-06123 Perugia, Italy
cINRIM, Istituto Nazionale di Ricerca Metrologica, Strada delle Cacce 91, 10135 Torino, Italy
First published on 7th March 2022
Abstract
Mixed mercury(II) halogenides have been known for a long time as good NLO (non-linear optic) materials. The NLO properties are due to the halogen disposition in the solid state and the electron distribution among the bonds formed by soft elements. We investigated the possibility of using HgBrI as a asymmetric tecton in the preparation of noncentrosymmetric crystalline compounds, by exploiting the coordinating power of Hg(II) toward N-donor ligands, and seven coordination complexes have been obtained. To unravel the nature of these complex systems we combined the data from different techniques: Raman spectroscopy, SC-XRD and Second Harmonic Generation, supported by a periodic DFT computational approach. In HgBrI crystalline products with low symmetry, the presence of substitutional disorder leads to a lack of the inversion center conferring NLO activity, which is absent in analogous complexes of Hg(II) halogenides. These results indicate HgBrI as an interesting tecton to obtain metallorganic NLO materials.
Introduction
Mercury halogenides have been known for a very long time. The antiseptic properties of corrosive sublimate (HgCl2) have been widely exploited in surgery interventions and the study of solid state mixtures of different mercury halogenides attracted great interest at the beginning of the XX century.1 This pioneering research allowed the description of reliable phase diagrams of the HgBr2–HgI2 mixed system which show a eutectic point at 59% of HgBr2 and 216.1 °C. Mercury(II) bromide iodide (HgBrI) was first reported in 1869 by Oppenheimer, and was obtained by a reaction between HgBr2 and ethyl iodide.2 Later studies observed the formation of HgBrI as a component of the equilibrium HgBr2 + HgI2 ⇄ 2HgBrI either in the gas3 or molten4 phase or in solution,5 and the formation constants in different solvents were studied.6–8 The first diffraction data were reported only in 1969, when a crystalline powder was obtained from a solid state reaction between Hg2Br2 and I2, suggesting the existence of pure HgBrI.9 In 1974 Ammlung and Brill10 carefully examined the literature about mixed mercury(II) halogenides and collected the Raman spectra of the products obtained by different synthetic routes and dissolved in different solvents. On the basis of these data, the authors indicate that a ternary solid solution (HgBr2–HgBrI–HgI2) is always formed, also as a crystalline product. A contemporary paper of Nakashima et al.11 reveals in detail the Raman scattering of (HgBr2)1−x(HgI2)x mixed crystals: the bands observed in the experimental spectrum were assigned to the ternary solid solution and a correlation was observed between the band intensity and the composition of the mixture.In the 1990s attention was again focused on mixed HgBrxI2−x crystals, proposed as candidate materials for the fabrication of X- and γ-ray detectors. Crystals of a stable phase (HgBr1.16I0.86, the same composition of eutectic) have been synthesised by Bridgman growth, with interesting photodetection properties.12 At the beginning of the new century, mercury halogenides and chalcogenides received renewed attention as possible infrared nonlinear optic (NLO) materials suitable for use in the MIR spectral region because: (i) they crystallise in noncentrosymmetric space groups; (ii) show a wide IR transparency; and (iii) possess a large band gap, which can improve the laser-induced damage thresholds.13 Three different compounds have been reported (HgBrI, Hg2BrI3 and Hg2Br3I)14 and the structural parameters of the obtained phases are quite similar. HgBrI crystallises in the noncentrosymmetric Cmc21 space group, and the experimental intensity of Second Harmonic Generation (SHG) of the powder samples was 1.4 times stronger than that of potassium titanyl phosphate (KTiOPO4), a common standard material in NLO devices.14a The NLO properties of HgBrI have been examined by means of a plane-waves periodic computational approach that allowed the characterisation of the second-order susceptibility tensor (χ2).15 The large NLO response of the HgX2 and HgBrI systems drove us to investigate the possibility of employing HgBrI as a tecton to prepare asymmetric molecules, with the goal to obtain noncentrosymmetric crystals.16 The four criteria for organometallic NLO compounds are: (i) highly polarisable π systems; (ii) easily polarisable metal atoms; (iii) asymmetric metal coordination giving rise to an asymmetric electronic distribution at the metal centre; (iv) intense low-energy MLCT (metal-to-ligand charge-transfer) bands.17 Mercury(II) is a d10 easily polarisable metal ion and its mixed halogenides exhibit asymmetric coordination, matching with three of the previous criteria. With the aim of understanding the effects of HgBrI as a building block, we decided to employ as π systems seven unsubstituted polypyridyl ligands: 2′-bipyridine (bpy); 4,4′-dimethyl-2,2′-bipyridine (dmbpy); 1,10-phenanthroline (phen); 2,9-dimethyl-1,10-phenathroline (dmphen); 2,2′;6′,2′′-terpyridine (terpy); 2,2′-bipyrimidine (bpym); 2-(2′-pyridyl)-1,8-naphthyridine (pyNP) (shown in Fig. 1). Mononuclear [Hg(L)X2] complexes with these ligands have been reported18–27 and complexes [Hg(phen)Br2] (9) and [Hg(terpy)Br2] (10) have been synthesised and characterized in this work. The use of the bpym ligand resulted in the formation of coordination polymers, as previously described.28 All the compounds of Hg(II) halogenides with the employed ligands crystallise in centrosymmetric space groups (see Table 1).
 | ||
| Fig. 1 Pyridyl based ligands L and their corresponding Hg(II) complexes [Hg(L)BrI]. | ||
| a General formula [Hg2(bpym)X4]n. b From experimental results (SHG). | ||||
|---|---|---|---|---|
| L | [Hg(L)Cl2] | [Hg(L)Br2] | [Hg(L)I2] | [Hg(L)BrI] |
| bpy | C2/c18 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 19 19 |
P20 |
P1b |
| dmbpy | — |
P21 |
Pbca 22 | Pbca |
| phen | — |
P |
P20 |
P1b |
| dmphen | P21/c23 | P21/c24 | C2/c20 | C2/c |
| terpy | P21/c25 | I2/a | C2/c26 | I2/a |
| pyNP | P21/c27 |
P27 |
P27 |
P1b |
| bpym | P21/n28 | P21/n28 | Pnma 28 | P21/c |
In this paper, we propose an alternative preparation of HgBrI (1) by means of a mechano-chemical synthesis approach followed by a recrystallization from acetone, and employed pyridyl based ligands to obtain seven coordination complexes: [Hg(bpy)BrI] (2); [Hg(dmbpy)BrI] (3); [Hg(phen)BrI] (4); [Hg(dmphen)BrI] (5); [Hg(terpy)BrI] (6); [Hg(bpym)BrI] (7) and [Hg(pyNP)BrI] (8). All [Hg(L)BrI] complexes appear to be a solid solution with [Hg(L)Br2] and [Hg(L)I2], as observed in HgBrI. To reach a proper description of these systems it was necessary to combine structural SC-XRD characterization with Raman spectroscopy and SHG measurements, supported by a periodic DFT computational study. We tried to evaluate the relationship between the structure and the second order properties thanks to a NLO microscopy procedure.29
Experimental
Materials and methods
All chemicals are commercial products of reagent-grade purity and were used without further purification. HgBr2 was prepared and purified according to the literature procedure.30 The ligand pyNP was synthesized according to the published method.31 All products have been characterised by SC-XRD, Raman and ATR-FTIR spectroscopy and elemental analyses. SHG emission was measured using two different methods: a NLO multimodal microscope to get the SHG signal from a single crystal at different excitation angles with a tested procedure,29 and a specific excitation geometry over the powder samples. Periodic simulations on the experimental supercells were carried out with the CRYSTAL17 code.32 Details of the employed procedures are described in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, were ground in an agate mortar for 10 minutes, and then some drops of acetone were added. Kneading proceeded for about 60 minutes to allow for completion of the reaction. A yellow crystalline powder was finally obtained. The product was purified by sublimation, but no difference with respect to the ground products was observed. Powder diffraction data were recorded to check the absence of starting reagents and the purity of the ground and sublimed products by comparing the experimental and calculated powder patterns (shown in Fig. S1†). Crystals suitable for SC-XRD were obtained from slow evaporation of the acetone solution.
:1, were mixed and allowed to slowly evaporate. Magenta crystals suitable for SC-XRD were obtained after a few days. Elemental analysis (%): calc. for HgBrIC13N3H9 (8): C, 25.4; H, 1.5; N, 6.8; found: C, 26.1; H, 2.1; N, 6.4. ATR-FTIR (cm−1) 1602 s, 1591 s, 1574 m, 1551 s, 1534 sh, 1467 m, 1453 s, 1421 m, 1384 w, 1171 m, 1143 m, 1130 m, 1114 w, 1007 m, 999 sh, 846 s, 640 m.
:1, were ground in an agate mortar for 10 minutes, and then some drops of acetone were added. Kneading proceeded for about 60 minutes to allow for completion of the reaction. A yellow crystalline powder was finally obtained. The product was purified by sublimation, but no difference with respect to the ground products was observed. Powder diffraction data were recorded to check the absence of starting reagents and the purity of the ground and sublimed products by comparing the experimental and calculated powder patterns (shown in Fig. S1†). Crystals suitable for SC-XRD were obtained from slow evaporation of the acetone solution.
:1, were mixed and allowed to slowly evaporate. Magenta crystals suitable for SC-XRD were obtained after a few days. Elemental analysis (%): calc. for HgBrIC13N3H9 (8): C, 25.4; H, 1.5; N, 6.8; found: C, 26.1; H, 2.1; N, 6.4. ATR-FTIR (cm−1) 1602 s, 1591 s, 1574 m, 1551 s, 1534 sh, 1467 m, 1453 s, 1421 m, 1384 w, 1171 m, 1143 m, 1130 m, 1114 w, 1007 m, 999 sh, 846 s, 640 m.
Results and discussion
Identification of the complexes by Raman spectroscopy
For the synthesis of HgBrI (1) we propose an exchange reaction in the solid state between HgBr2 and HgI2, promoted by grinding: a homogeneous product has been obtained in about 60 minutes. This mechano-chemical procedure is able to produce 100% yield, to reduce reaction times, to limit solvent use and to avoid waste. The progress of the reaction can be observed by colour change: the starting orange mixture, made by white HgBr2 and red HgI2 (α phase), which turned gradually to a yellowish homogeneous powder. Raman spectroscopy was used to verify the progress of the reaction, by recording their spectra after 10–20 minutes of grinding. The Raman spectrum of crystalline HgBrI has been investigated in detail.10,11 Five bands are detected in the Raman spectra at 42, 140, 156, 181 and 226 cm−1 that have been interpreted by attributing the signals to a ternary solid solution HgBr2–HgBrI–HgI2: signals at 156 and 226 cm−1 are ascribed to symmetric and asymmetric Br–Hg–I stretching modes, the bands at 181 and 140 cm−1 respectively to HgBr2 and HgI2 (β phase, yellow polymorph) symmetric stretching and the band at 42 cm−1 to librational modes. The intensity of the bands in the range from 140 to 190 cm−1 has been correlated with the composition of the HgBr2–HgBrI–HgI2 mixture.11 As shown in Fig. 2, in the spectrum of the starting mixture characteristic signals of reagents are detectable: at 114 cm−1 for HgI2 (α phase, red polymorph)35 and at 187 cm−1 for HgBr2.35 Gradually, the strong iodide peak decreases in intensity and disappears, while the solid solution signals increase in intensity. It is interesting to notice that, in agreement with the formation of HgBrI, the polymorphic transformation of HgI2 is observed, suggesting a correlation between these two reactions. Grinding was continued until no more change was observed in the consecutive Raman spectra. The spectra of the ground product are compatible with x = 0.50 composition, suggesting that HgBrI is the most abundant compound in the obtained solid solution. The spectra of the sublimed product (obtained to check purity and stability) show a variation of band intensities, especially of the 156 cm−1 band, and a little shift of the band position, that can be explained with a compositional variation of the mixture as a consequence of the sublimation (see Fig. S2†). The attribution of the 156 and 226 cm−1 signals to the Br–Hg–I component is confirmed also by a periodic DFT computation of the vibrational spectra of a pure HgBrI phase. As shown in Fig. S2,† two main Raman features are computed: a strong band centred at 142 cm−1 (A1 asymmetric Br–Hg–I stretching mode) and a weak band at 218 cm−1 (A1 symmetric Br–Hg–I stretching). | ||
| Fig. 2 Raman spectra recorded at different times during the mechano-chemical synthesis of HgBrI (1): in blue the starting mixture and in red the final product. | ||
Using HgBrI as the starting material, a series of N donor-based complexes have been synthesised. After mixing the solutions of HgBrI and L (see Fig. 1), a crystalline product precipitates immediately, while for [Hg2(bpym)Br2I2] (7) and [Hg(pyNP)BrI] (8) single crystals were obtained after a few days. Reactions have been carried out at room temperature to favour the formation of HgBrI complexes, with the equilibrium HgBr2 + HgI2 ⇄ 2HgBrI being exothermic.8 Identical products have been obtained using ground, sublimed or crystalline HgBrI as the reagent.
Raman spectroscopy was employed not only to confirm the product formation, but also to understand the nature of the synthesised complexes on the basis of the analysis of Hg–X modes (Fig. S4–S10†). Compared to the spectra of the corresponding ligands,27,36 ring stretching modes (1600–1450 cm−1) are usually slightly shifted to higher wavenumbers by the rigidity imposed by metal coordination.37 The breathing modes of aromatic fragments (around 1000 cm−1 for py fragment) show a shift of 20 cm−1 in 2–10 with respect to the corresponding ligand (see Table S1†), thus suggesting the formation of a chelating ring. Below 300 cm−1 it is possible to observe the stretching of Hg–X and Hg–N bonds. As shown in Table 2, the ligand coordination does not significantly affect the ν(Hg–X) modes of HgBrI. The complexes with bpy (2), phen (4), and pyNP (8) show a strong νasym(HgBrI) signal and both the ν(HgBr2) and ν(HgI2) modes, suggesting the preservation of the HgBr2–HgBrI–HgI2 mixture also in the crystalline structure of these compounds. Similar signals are observed for [Hg(dmbpy)BrI] (3), [Hg(dmphen)BrI] (5) and [Hg(bpym)BrI] (7). A more sensible shift of Hg–X modes is detected in the [Hg(terpy)BrI] complex (6), the only one with tri-coordinated Hg(II).
| ν(HgI2) | ν asym (HgBrI) | ν(HgBr2) | ν(Hg–N) | |
|---|---|---|---|---|
| HgBrI (1) | 141 vs | 156 vs (142 calc) | 181 m | |
| HgBr2 | 188 vs | |||
| HgI2 (β)11 | 141 vs | |||
| Hg(bpy)BrI (2) | 144 s, sh | 155 vs | 188 m | 218 w |
| Hg(bpy)Br2 | 187 vs | 220 w | ||
| Hg(bpy)I2 | 143 vs | 220 w | ||
| Hg(dmbpy)BrI (3) | 139 s | 152 s | 178 m | |
| Hg(phen)BrI (4) | 139 vs | 153 vs | 179 m | 214 vw |
| Hg(phen)Br2 (9) | 177 vs | 215 vw | ||
| Hg(phen)I2 | 138 vs | 216 vw | ||
| Hg(dmphen)BrI (5) | 137 s | 151 s | 178 m | 218 vw |
| Hg(terpy)BrI (6) | 145 vs | 170 s | 228 w | |
| Hg(terpy)Br2 (10) | 169 vs | 232 w | ||
| Hg(terpy)I2 | 138 vs | 224 w | ||
| Hg(pyNP)BrI (8) | 141 vs | 153 vs | 181 s | 198 w |
| Hg(pyNP)Br227 | 180 vs | 200 vw | ||
| Hg(pyNP)I227 | 139 vs | 196 vw | ||
| Hg2(bpym)Br2I2 (7) | 142 vs | 156 vs | 183 m | 215 w |
| 230 w | ||||
| [Hg2(bpym)Br4]n28 | 183 vs | 209 m | ||
| 229 m | ||||
| [Hg2(bpym)I4]n28 | 137 vs | 216 m | ||
| 145 m | 239 m |
Analysis of the substitutional disorder
All crystal structures of the obtained complexes of HgBrI show Br/I positional disorder. SC-XRD data of the solid solution are nominally indistinguishable from those of pure molecular [Hg(L)BrI] with a 50%–50% disordered composition of the two sites, but the Raman spectra of the crystals support the formation of a ternary solid solution [Hg(L)Br2]–[Hg(L)BrI]–[Hg(L)I2].We obtained the crystals of 1 from an acetone solution to evaluate the similarities and differences in the positional disorder between the complexes and HgBrI phase. HgBrI (1) crystallises in the noncentrosymmetric Cmc21 space group. Merohedral twinning for the inversion centre has been detected and considered in resolution. The structure is similar to those of the corresponding isomorphic HgBr238 and HgI2 (β).39 There are four Hg–X bonds, two of which are shorter, determining a characteristic coordination number of 2 with a linear geometry (I2/Br1–Hg1–I1/Br2 = 179.27(17)°) and a bisfenoidic effective coordination number of 4 ([2 + 2] coordination number), according to Grdenić40 (see Fig. 3a). However, differently from crystal structures recently reported,14 there is no site preference for iodide and bromide: different crystallographic clues support that axial mercury coordination sites show a 50%–50% Br/I composition. First, the electron densities detected in the two positions are very similar, and do not fit with the presence of iodide or bromide alone, but are intermediates between them. In fact, by replacing only iodide or bromide in the two sites, the R parameters are much higher and the thermal ellipsoids are too big or too small compared to Hg1. In addition, the presence of a substitutional disorder in these sites is supported by the distance between Hg1 and the two sites: the modeled long range value obtained by SC-XRD is a weighted average of the Hg–I and Hg–Br distances. As shown in Fig. 4, there is a linear dependence of the Hg–X distance versus the fraction of iodide. On the basis of the Raman spectra and of the Hg–X distances, we suggest that also the previously reported phases (HgBrI, Hg2Br3I, and Hg2BrI3)14 should be better described as ternary solid solutions, with different proportions of the three components. Similarly, in the crystals of the complexes the Hg–X intermediate distances and the intermediate Br/I electron density demonstrate the presence of a substitutional disorder. XRD data indicate that for all compounds there is a 50% occupancy for each halogenide, and no difference is observed between inorganic and metallo-organic compounds. The Raman spectra indicate that HgBrI is the more abundant compound in our product and to simplify the description of the crystalline structure, the molecular unit will be indicated as HgBrI. In the crystal lattice of 1, we can observe that the HgBrI molecules interact through long Hg⋯X contacts (d(Hg1⋯I2/Br1) = 3.387(3) Å, in Fig. 3a) forming an hexagonal net in the (001) plane (see Fig. 3b), while the apical I1/Br2 site outside of the plane interacts weakly with opposite Hg atoms (d(Hg1⋯I1/Br2) = 3.411(3) Å). The planes are stacked along the [001] direction (see Fig. 3b) and are weakly connected by dispersion forces. The crystal of 1 was analysed at different temperatures: passing from 299 K to 200 K all distances are shortened, especially the weaker equatorial contacts that became lower than the sum of VdW radii, so the coordination sphere of HgBrI at 200 K became [2 + 4] (see Fig. 3c). A compression effect is evident between the layers: the distance between them, perpendicular to the [001] axis, changes from 6.686 Å at 299 K to 6.324 Å at 200 K. This indicates that the c axis is more sensitive to temperature effects with respect to the other two (dc/dT = 7.3 × 10−3vs. da/dT = 8 × 10−4 and db/dT = 2.5 × 10−3). This is due to the absence of strong directional interactions between the layers, similarly to the case of layers of mica minerals.41
 | ||
| Fig. 3 HgBrI (1): chemical environment around the disordered HgBrI molecular unit in the asymmetric unit at room temperature (a) and at 200 K (c) and packing along the [100] direction (b) (violet: iodine; grey: mercury; brown: bromide – ORTEP plot 50%). | ||
 | ||
| Fig. 4 Dependence of the Hg–X distance versus iodine fraction in the crystal structures of HgBrI (1) and reported Hg(II) mixed halogenides. | ||
The impossibility to resolve the disorder by SC-XRD data drove us to perform DFT periodic simulations to evaluate the effect of substitutional disorder in the Raman spectrum of solid HgBrI (1) and [Hg(bpy)BrI] (2), simulated as pure phases. Raman scattering is a versatile and efficient tool for probing long- and short-wavelength lattice vibrations. It is known that there is a composition dependence of optical modes in the presence of substitutional disorder, that randomly affects the periodic arrangement, and a relationship has been observed between disorder and the line shape of the Raman bands (as width and asymmetry).42 Different models have been proposed to explain this behaviour,43 and a description of these systems as a weighted average of ordered configurations has provided promising results.44 All the possible permutations on bromide and iodide positions were taken into account when calculating the Raman spectra of the disordered solution. The spectra were merged together, rescaling the intensities by the proper Boltzmann factor, calculated from the corresponding enthalpies of each configuration. In the final spectrum of HgBrI the νasym(BrHgI) mode shows an asymmetric broadening, with an evident shoulder. Nakashima et al.11 found that mixed crystals (HgBr2)1−x(HgI2)2 show a two-mode behaviour, in which the zone-centre optical phonons of each end member are present in intermediate compositions. To explain this behaviour, a model based on the nearly free approximation was proposed, with very weak intermolecular forces, which seems to be valid for the A1 mode. This model is supported by the SC-XRD data of 1 recorded at different temperatures, which indicate the presence of weak intermolecular forces and weak interlayer interactions. It is known that substitutional disorder results in broad spectra and the effect is more evident for composition x = 0.5 (in ABxC1−x system) that shows the maximum disorder.43a A study of positional disorder of GaAsxP1−x (x = 0.5), using a linear chain model, was performed using five different models (random chain, disordered chain, ordered chain, virtual ion, and isodisplacement) to calculate the reflectance IR spectrum. The results indicate that the numbers of bands increase with the increase of disorder in the model.43b Our periodic calculation also presents a major number of signals when the disorder is simulated in the systems, giving rise to the broad asymmetric band in the average spectrum (see Fig. S3†). No evident asymmetric broadening is observed in the experimental spectra recorded at room temperature. A behaviour more similar to the calculated one could be observed at lower temperature.
For [Hg(bpy)BrI] (2) the disorder of halogenide atoms in the crystallographic cell gives rise to four different permutations and their Raman patterns at low wavenumbers are quite different. Focusing on vibrational modes involving Hg atoms, all the signals related to the four permutations (at 130, 145, 155 and 176 cm−1) are present in the experimental spectrum (the black line in Fig. 5), suggesting that the halogenide atoms have a random disposition in the crystalline lattice. The preservation of a solid solution in the coordination product is suggested by signals attributable to the bromide and iodide complexes (at 143 and 188 cm−1, indicated with a star in Fig. 5) that are not present in the calculated spectra: in particular, ν(HgBr2) is not superimposed to any HgBrI mode. So the Raman intensities and XRD data suggest that [Hg(L)BrI] is the dominant component of the ternary solid solutions of metallo-organic derivatives.
 | ||
| Fig. 5 (a) Calculated Raman spectra of modes involving Hg in different permutations of the dimeric [Hg(bpy)BrI] unit cell (centrosymmetric in red and noncentrosymmetric in blue) and the experimental spectrum of [Hg(bpy)BrI] (2) in black; (b) centrosymmetric (p1 and p4, in the red boxes) and noncentrosymmetric (p2 and p3, in the blue boxes) permutation views along the ac plane; dashed and dotted lines are related to lines in the Raman spectrum. (Violet: iodine; yellow: mercury; green: bromide; grey: carbon; blue: nitrogen; white: hydrogen.) | ||
Symmetry of the crystals
As shown in Table 1, most of the obtained complexes crystallised in centrosymmetric space groups. In the compounds with lower symmetry ([Hg(bpy)BrI] (2), [Hg(phen)BrI] (4), and [Hg(pyNP)BrI] (8)) the choice of the correct space group has been very ambiguous. P shows better results in least squares refinement with respect to P1. In P1 space group resolution, the thermal ellipsoids and the distances show very high errors, and in the case of 4 and 8 no stable solution can be found with both a very high redundance and completeness of the data. Mercury(II) halogenides with similar ligands usually form dimers,20,45 and the dimeric interactions result always in the presence of the inversion centre, as shown in Fig. 6a. In the centrosymmetric model, the presence of the Br/I disorder in 2, 4 and 8 makes some of the cells noncentrosymmetric and so the crystal can be described as “not fully centrosymmetric”. Considering the possible disposition of disordered halogenides in the dimers, only 50% of the cells of the crystal (see Fig. 6b and c) still exhibit an inversion centre. However, this situation is made even more complicated by the solid solution formed with respective [Hg(L)I2] and [Hg(L)Br2], that can make dimers with all the complexes present in the crystals, exhibiting or not an inversion centre.
 | ||
| Fig. 6 (a) Example of (pseudo)-centrosymmetric dimeric interaction in [Hg(bpy)BrI] (2); models of noncentrosymmetric (b) and centrosymmetric (c) local disposition. (Violet: iodine; grey: mercury; orange: bromide; grey: carbon; blue: nitrogen; white: hydrogen – ORTEP plot 50%.) | ||
Being sometimes very complex to determine the correct symmetry between P and P1 from diffraction results alone, we decided to measure a property that inherently resolves this problem: SHG.46 Since only crystals with a lack of the inversion centre can show NLO properties, the experimental measurement of SHG demonstrates unambiguously the noncentrosymmetry of the crystal structures.47 SHG can occur with good efficiency only if a standard phase matching condition is satisfied (to compensate the dispersion due to the frequency dependence of the refractive indices), and this can often be done by using a birefringent material. To find out the transmission range the UV–Vis optical diffuse reflectance spectra were recorded and are shown in Fig. S11.† HgBrI (1) shows a relatively wide transparency in the visible region down to about 460 nm. Complexes 2, 4 and 8 are transparent above about 390 nm, but 8 shows a low absorption also in the visible region due to the intense magenta colour of its crystals. Qualitative measures have shown that the crystals of 2, 4 and 8 are birefringent and 8 in particular is pleochroic. SHG emission was detected on the powder crystalline samples of 1, 2, 4 and 8 (see Fig. S12†). This experimental evidence suggests that the average disposition in crystallographic cells is noncentrosymmetric. The great difficulty in obtaining a noncentrosymmetric model, notwithstanding its exactness, can be explained using the crystallographic orbit theory: the heaviest atoms (Hg and the I/Br positions) and most of the lightest atoms of the dimers are disposed in the unit cell in a way that simulates almost perfectly the presence of an inversion centre (belonging in pairs to a common crystallographic orbit in the P supergroup).48 As a consequence, XRD intensities are not affected enough by the deviation from centrosymmetry to ensure a clear noncentrosymmetric structure resolution, as found and confirmed in similar cases by the in-depth studies of Marsh.46a For these reasons, we decided to maintain the higher P symmetry in 4 and 8, for which no enough good refinement results have been obtained in P1, following the literature.46a,49 In this supergroup description, the resulting geometrical data are averaged between the non-equivalent molecules in the correct subgroup model.
With the aim of obtaining a more stable and more accurate noncentrosymmetric model of 2, two measurements at 200 K and at 299 K (both with resolution of 0.5 Å) have been performed on the same crystal. The effects due to the quasi-singularity in the least squares refinement have not been resolved, neither at low temperature nor at a high resolution limit, and the centrosymmetric model continued to be the best one on the basis of SC-XRD data. At low temperature there is a better resolution of the Br/I disorder in two stable distinct sites (d(Hg–Br) = 2.528(9) Å, d(Hg–I) = 2.659(5) Å).
Also for [Hg(dmbpy)BrI] (3) and [Hg(dmphen)BrI] (5) the dimeric disposition of the molecules in the crystal structure introduces a symmetry inversion centre, but previous considerations on the actual symmetry suggest the possibility of a lower space group symmetry associated with the disappearance of some symmetry elements. A centrosymmetric description has been preferred because the refinement in noncentrosymmetric space groups, considering all the possibilities derived by deleting the inversion centre, has been unsuccessful, and no anomalous behaviour of systematic absences has been observed (under the experimental conditions of measurements).
In the obtained compounds, we can observe that when the bromide and the iodide complexes are not isomorphous, solid solutions crystallise in one of the space groups of the analogous complexes, usually the iodide one, as observed for 3, 5 and [Hg(terpy)BrI] (6), but it can also show a different crystal packing with respect to the other halides, as in [Hg2(bpym)Br2I2] (7). When [Hg(L)Br2] and [Hg(L)I2] are isomorphous, the substitutional disorder leads to a lack of the inversion centre, thus conferring NLO activity, as observed for 2, 4 and 8. A similar behaviour is observed also in 1 that crystallises in the same space group of HgBr2, stabilising the isomorphous high temperature polymorph of HgI2.
Crystalline structures
The molecular structures of ([Hg(bpy)BrI] (2), [Hg(phen)BrI] (4), and [Hg(pyNP)BrI] (8) (in Fig. 7) are very similar to those of HgX2 (X = Cl, Br, and I) derivatives of the same ligands,18–20,27 with the exception of the Hg–X distance in the range of 2.550–2.590 Å that reflects the Br/I disorder. The crystal packing of these molecules can be usually considered an average of the characteristics of the iodide and bromide derivatives. It has been demonstrated that upon moving from the chloride to the iodide, the bridging behaviour is reduced and the bridging interactions are weaker.28 In the case of bpy complexes, the chloride forms a true 1D-coordination polymer,18 while the bromide and iodide form dimers that interact through spodium bonding,50 giving dimeric molecular entities19 and very weakly interacting dimers,20 respectively. The packing of 2 is an intermediate between that of bromide and iodide: very long spodium bonding interactions connect the molecules of the dimers, at the edge of the VdW sum (d(Br/I⋯Hg) = 3.404(8) Å and 3.377(8) Å, respectively). This distance is slightly shortened at low temperature, changing from 3.376(7) Å at 299 K to 3.364(6) Å at 200 K (in the centrosymmetric model). Dimers interact with each other through parallel π⋯π stacking between the bpy ligands (see Fig. 7d, an interplanar distance of 3.577(10) Å, a displacement of ca. 2.6 Å, and an intercentroid distance of 3.997(10) Å).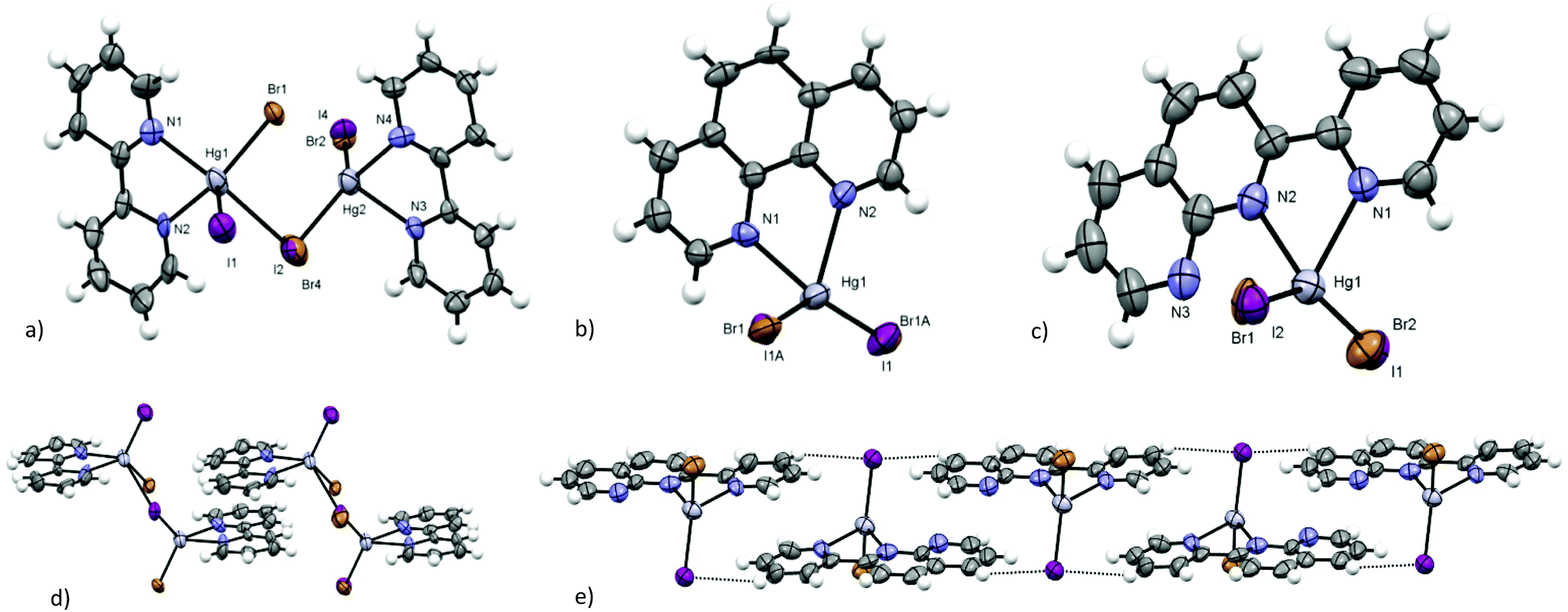 | ||
| Fig. 7 (a) Asymmetric unit and (d) crystal packing between dimers in the crystal structure of [Hg(bpy)BrI] (2); (b) asymmetric unit of [Hg(phen)BrI] (4); (c) asymmetric unit and (e) details of the structure of [Hg(pyNP)BrI] (8). (Violet: iodine; grey: mercury; brown: bromide; grey: carbon; blue: nitrogen; white: hydrogen – ORTEP plot 50%.) | ||
We have obtained the crystal structure of [Hg(phen)Br2] (9), isomorphous to [Hg(phen)I2],20 with the same space group, but with shorter intermolecular spodium interaction, as can be expected (d(Br⋯Hg) = 4.055(6) Å vs. d(I⋯Hg) = 4.400(6) Å). Also in the case of 4, the crystal packing is very similar to that of iodide analogues,20 although the dimeric spodium interaction is much shorter ( d(Br/I⋯Hg) = 4.247(7) Å). The differences in the Hg–Br and Hg–I bonds are in agreement with the weighted average in the case of Hg–(Br/I). All phen derivatives form π⋯π stacked dimers displaced of more than 3.6 Å, and place one of the lateral rings of an upper molecule on the top of the central one of a lower complex, with all ligands being disposed on parallel planes. Compound 4 shows one of the smaller values of the interplanar distance in the series of the synthesised complexes (an interplanar distance of 3.369(10) Å and an intercentroid distance of 3.671(10) Å).
Looking at the pyNP derivatives, the crystal packing of [Hg(pyNP)Cl2] is different from those of bromide, iodide and BrI analogues.27 [Hg(pyNP)Cl2] has a monoclinic structure, with the columns of inverted chloride complexes that stack together by placing the naphthyridine (NP) ring of the upper complex on the top of NP ring of the lower complex, and the same for the py substituent (an interplanar distance of 3.608(10) Å, a displacement of 1.38 Å, and an intercentroid distance of 3.602(10) Å). No intermolecular interactions can be observed, with the Cl⋯Hg contacts being very long (d(Cl⋯Hg) = 4.755(7) Å). In 8, isomorphous to the triclinic bromide and iodide derivatives, the ribbons of inverted molecules are π⋯π stacked and overlap along the ribbons of the NP ring of a molecule with one of the nearest molecules, and the same for py (see Fig. 7e, an interplanar distance of 3.489(9) Å; a displacement of 2.35 Å for the overlapping NP rings and of 1.39 Å for the overlapping py). The ribbons are stabilized by weak C–H⋯Br/I interactions (d(C–H⋯Br/I) = 3.164(9) Å). These interaction distances are intermediates between the values of the bromide and iodide derivatives.
[Hg(dmbpy)BrI] (3) and [Hg(dmphen)BrI] (5) show a higher reticular symmetry, and orthorhombic and monoclinic space groups respectively, due to the more symmetric disposition of the complexes in the crystal environment. In the crystal structure of 3, the π⋯π and the C–H⋯π aromatic interactions between parallel or perpendicular molecules determine a perpendicular herringbone packing motif isomorphic to that found in [Hg(dmbpy)I2],22 with similar interaction distances (see Fig. 8c, an interplanar distance of 3.715(9) Å, an intercentroid distance of 3.715(8) Å, and a displacement of 0.7 Å, d(CH3⋯π) = 3.601(8) Å). In [Hg(dmbpy)Br2], different from iodide and 3, a dimeric disposition is observed21 and the crystal packing is very similar to that of [Hg(bpy)X2] (X = Br, I). The crystal structure of 5 is isomorphic to that of [Hg(dmphen)I2]20 (C2/c space group), with diagonal columns of inverted dimers displaced on π⋯π stacked molecules (see Fig. 8d, an interplanar distance of 3.360(9) Å, an intercentroid distance of 3.605(9) Å and a displacement of 3.682(8) Å). The chloride and bromide complexes show a more opened crystal packing and a herringbone pattern,23,24 while in the iodide derivative and in 5, the ligands are disposed on parallel planes.
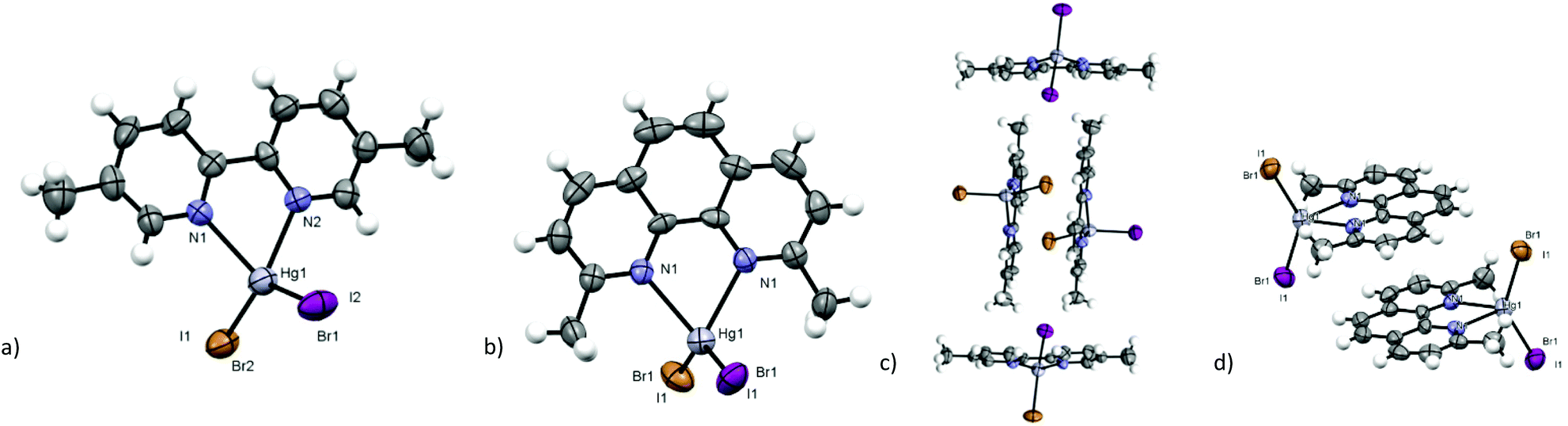 | ||
| Fig. 8 (a) Asymmetric unit and (c) packing interactions between molecules in the crystal structure of [Hg(dmbpy)BrI] (3); (b) asymmetric unit and (d) packing interactions between dimers of [Hg(dmphen)BrI] (5). (Violet: iodine; grey: mercury; brown: bromide; grey: carbon; blue: nitrogen; white: hydrogen – ORTEP plot 50%.) | ||
[Hg(terpy)BrI] (6) (see Fig. 9a) and [Hg(terpy)Br2] (10) are isomorphous to iodide analogues26 and crystallise in the I2/a space group. In 6 the Hg–X distance is longer than those found for the previous synthesised compounds (d(Hg–Br/I) = 2.657(5) Å), but is fairly shorter than the Hg–I bond reported for [Hg(terpy)I2]26 (d(Hg–I) = 2.697(5) Å) and longer than Hg–Br in 10 (d(Hg–Br) = 2.556(3) Å). In 6 the disorder is constrained to 50%–50% Br/I for symmetry reasons, excluding any preconcentration of one of the two halides or a pseudosymmetry nature of the twofold axis passing through Hg, but the Raman spectra and Hg–X distance support the presence of both halogenides. The crystal packing of 6 is similar to that of the iodide and bromide complexes but differs from the chloride one.25 [Hg(terpy)Cl2] shows a lower symmetry (P21/c space group) and a herringbone disposition similar to those of [Hg(dmphen)X2] (X = Cl, Br), while the heavier halogenides show diagonal columns of the overlapped but slightly shifted terpy ligands on parallel planes (see Fig. 9b for 6, interplanar distances of 3.540(9) Å, an intercentroid distance of 3.736(9) Å and a displacement of 4.203(8) Å, similar to those of iodide and bromide complexes).
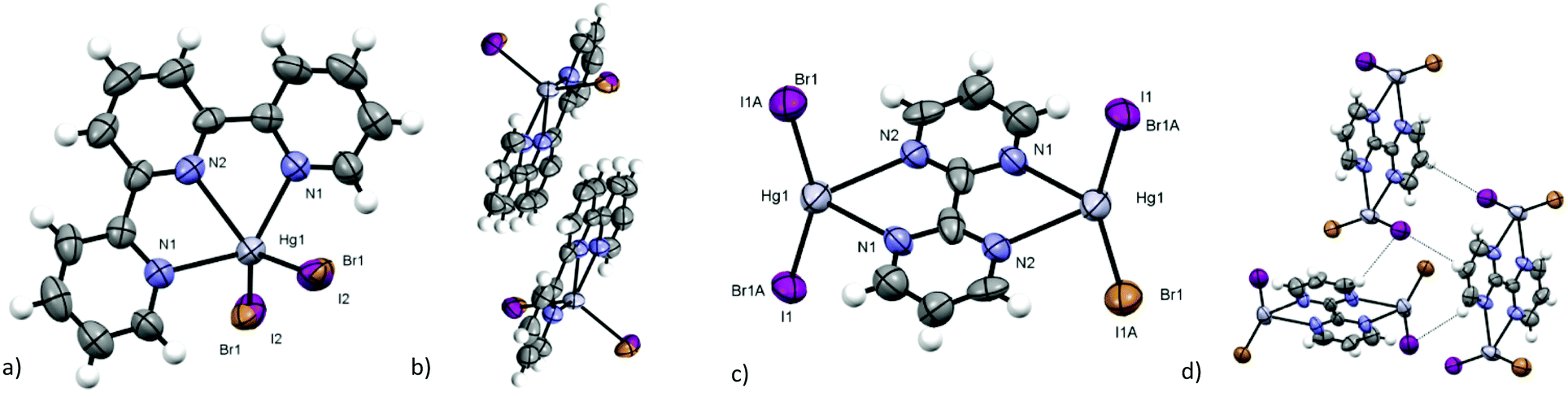 | ||
| Fig. 9 (a) Asymmetric unit and (b) dimeric interaction of [Hg(terpy)BrI] (6); (c) asymmetric unit and (d) crystal packing interactions of [(Hg2(bpym)Br2I2] (7). (Violet: iodine; grey: mercury; brown: bromide; grey: carbon; blue: nitrogen; white: hydrogen – ORTEP plot 50%.) | ||
The dinuclear [Hg2(bpym)Br2I2] (7) (see Fig. 9c) differs strongly from bromide and iodide analogues,28 adding further demonstration of the greater percentage of the HgBrI derivatives in the ternary mixture, and of its influence in crystal packing. Homo-halogen compounds are coordination polymers (staggered in the case of bromide and chloride and ribbon-like for iodide), while 7 crystallises in the molecular form without any bridging Hg⋯X or π⋯π interactions, forming a herringbone packing motif (see Fig. 9d) stabilised only by C–H⋯Br/I weak contacts (d(C–H⋯Br/I) = 3.042(9) Å and 3.081(10) Å).
SHG on single crystals
The measurement on the powder samples shows that the efficiency of SHG emission of the [Hg(L)BrI] complexes decreases with respect to 1 (see Fig. S12†). This may be attributed to different factors. First, it is known that the SHG response depends on band gaps, and lower band gaps generate higher hyperpolarizability.51 The calculated band gap for (1) (3.14 eV, similar to reported experimental value12e) is lower than that in (2) (3.73 eV). Second, as discussed previously, the noncentrosymmetric local disposition related to the Br/I disorder can be correlated to SHG emission. A recent study revealed that centrosymmetric crystals can generate the Second Harmonic, in the presence of twinning by inversion or local asymmetry.52 The components of hyperpolarizability and the second order susceptibility matrix have been calculated for HgBrI (1) and [Hg(bpy)BrI] (2), according to Kleinman's symmetry. The assumptions for Kleinman's symmetry are always valid for SHG.51 The β third rank tensor has 27 matrix components. Kleinman's symmetry demonstrates that the same indices of the matrix can be permuted and so the matrix is reduced to 10 components. The periodical simulation performed on 2 indicates that only for noncentrosymmetric permutations the components of hyperpolarizability and the second order susceptibility matrix are not zero (see Table S2†). This suggests that the noncentrosymmetry in these systems is related to the substitutional disorder that generates noncentrosymmetric local disposition.Further analysis on SHG emission has been performed along seven angles using a NLO microscope procedure29 on a crystal of 2, with the aim to correlate the crystalline structure and second order NLO properties. The single crystal of 2 gives an intense signal at the arbitrary angle 0 and two less intense emissions by rotating the crystal of 45 degrees in both directions, meaning that signals can be observed only in one specific direction (see Fig. S13†). The elaboration of the SHG signal through the microscopy technique allowed us to acquire an image of the emission and to highlight the crystal face responsible for that emission. The crystal faces of the measured sample have been determined with the metrical tensor derived from an XRD experiment, using the CrysAlisPro software.53 Therefore, it is possible to verify that the crystal emitted on the crystal face (010), as shown in Image S1.† Trying to understand the correlation between the structure and hyperpolarizability tensor, the crystal morphology was obtained with the BFDH approach, by using the Mercury software.54 By combining the morphological and microscopy data, it is possible to observe that the signal is detected in the direction parallel to π⋯π stacking, as shown in Fig. S14a.† Only the experimental data on the powder sample of 1 are available, but the periodic simulation suggests that only components along the z axis are not zero (xxz, yyz, zzz) and these components are parallel to the HgBrI molecular fragment (see Fig. S14b and Table S3†).
These results suggest that the direction of propagation of the Second Harmonic is different between pure inorganic HgBrI and its complex. HgBrI gives a signal in the direction of the molecular fragment, while in [Hg(bpy)BrI] (2) the signal is propagated in the direction parallel to the π⋯π stacking. According to Levine's bond charge model and to anionic group theory, the bulk second-order susceptibility of a noncentrosymmetric crystal is additive, and is the vector sum of the microscopic second-order susceptibilities of the constituent basic structural units.55 According to these models, the anharmonicity results from electronegativity and size differences between bonded atoms, so in complex 2 we can presume that hyperpolarizability originates from the HgBrI unit. Thanks to the employed NLO microscope procedure we can observe that the main direction of propagation of SHG emission in a single crystal of 2 is along π⋯π stacking interactions. It is known that the aromatic molecules conjugated by means of the π-orbital may exhibit microscopic SHG effects larger than the σ-type molecules of the same size. Thus it is not surprising that the strongest signal has been detected in the π⋯π more polarisable direction. Considering that [Hg(bpy)X2] (X = Br, I) do not show NLO activity, we can affirm that the HgBrI unit is responsible for this phenomenon.
Conclusions
The non-conformity of the nature of mixed mercury(II) halogenides in the literature is related to the challenging characterisation of these systems. In this work, we proved that HgBrI (1) obtained from mechano-chemical synthesis has the same characteristics of the previously reported phases, and we provided further evidence of the formation of a ternary solid solution HgBr2–HgBrI–HgI2. The interesting physico-chemical properties of HgBrI, especially its large NLO response, drove us to explore its coordination chemistry toward N-donor ligands. We prepared the first coordination compounds of HgBrI, whose characterisation was as challenging as that of the starting material. The Raman spectra confirmed that ternary solid solutions [Hg(L)Br2]–[Hg(L)BrI]–[Hg(L)I2] are formed as single crystal and powder crystalline products. SC-XRD data indicate that substitutional disorder is present in the crystal structures, resulting in Hg–X distances intermediate between those of the two halogenides. Due to this disorder, the assignment of the correct space group was very ambiguous, especially for compounds with the less symmetric space group. The crystalline powder of these compounds generates the Second Harmonic, confirming that the average crystalline structure is better described as noncentrosymmetric. The NLO activity could be related to the presence of substitutional disorder that generates local asymmetry, as supported by DFT periodic calculations.Although the products are solid solutions, they are reproducible and with a constant composition. The inorganic ternary solid solution, due to the presence of HgBrI, shows properties that are different from those of its components: for example it is the stabilised one of its component (HgI2) in its high temperature polymorph, isomorphic to HgBr2. In the coordination complexes, solid solutions are obtained as well, and they show properties different from those of the isolate component: in particular, [Hg(bpy)BrI] (2), [Hg(phen)BrI] (4) and [Hg(pyNP)BrI] (8) emit SHG.
These results suggest that HgBrI could represent an interesting tecton for NLO materials. In fact, the intrinsic disorder in the crystalline structures can generate local asymmetry and enhance the NLO activity, if derivatives have suitable ligands.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. P. acknowledges the Italian Space Agency for co-funding the Life in Space Project (ASI N. 2019-3-U.O).References
- (a) W. Reinders, Über die Bildung und Umwandlung der Mischkrystalle von Quecksilberbromid und quecksilbeijodid, Z. Phys. Chem., 1900, 32, 494–536 CrossRef CAS; (b) L. Losana, Sull'allotropia dello joduro mercurico, Gazz. Chim. Ital., 1900, 56, 301–331 Search PubMed.
- A. Oppenheim, Ueber Jod-Brom-quecksilber, Berquem, 1869, 2, 1571–1573 Search PubMed.
- A. Malt'sev, G. Selivanov, V. Yampolsky and N. I. Zavalishin, Far Infrared Absorption Spectra of Mercury Dihalide Vapours, Nat. Phys., 1971, 231, 157–158 CrossRef.
- J. H. R. Clarke and C. Solomons, Raman Spectra of Mercuric Iodide, Iodochloride, and Iodobromide in the Molten State, J. Chem. Phys., 1968, 48, 528–529 CrossRef CAS.
- M. L. Delwaulle, Quantitative study of chemical equilibrium using the Raman effect, in Proceedings of the Colloquium Spectroscopicum Internationale VI, ed. W. van Tongeren, et al., Pergamon Press, London, 1957, pp. 565–567 Search PubMed.
- Y. Marcus, Mercury(II) halide mixed complexes in solution, Acta Chem. Scand., 1957, 11, 329–339 CrossRef CAS.
- T. G. Spiro and D. N. Hume, The Uncharged Mixed Halides of Mercury(II). Equilibrium Constants and Ultraviolet Spectra, J. Am. Chem. Soc., 1961, 83, 4305–4310 CrossRef CAS.
- T. R. Griffiths and R. A. Anderson, Computer-Based Study of the Electronic Absorption Spectra of Neutral Mercury(II) Mixed Halides in Methanol: Equilibrium Constants and Band Assignments, Inorg. Chem., 1979, 18(9), 2506–2511 CrossRef CAS.
- (a) R. P. Rastogi and B. L. Dubey, Solid-State Reaction between Iodine and Mercurous Halides, J. Am. Chem. Soc., 1967, 89(2), 200–209 CrossRef CAS; (b) R. P. Rastogi and B. L. Dubey, Crystal structure of HgBrI, J. Inorg. Nucl. Chem., 1969, 31, 1530–1531 CrossRef CAS.
- R. L. Ammlung and T. B. Brill, The Nature of the Mixed Halides of Mercury(II), Inorg. Chim. Acta, 1974, 11, 201–205 CrossRef CAS.
- S. Nakashima, H. Mishima and H. Tai, Raman scattering from mixed crystals (HgBr2)1−x (HgI2)x, J. Phys. Chem. Solids, 1974, 35, 531–536 CrossRef CAS.
- (a) K. S. Shah, L. P. Moy, J. Zhang, F. Olscher, J. C. Lund and M. R. Squillante, HgBrxI2−x photodetectors for use in scintillation spectroscopy, Nucl. Instrum. Methods Phys. Res., Sect. A, 1992, 322, 509–513 CrossRef; (b) M. Veleva, M. M. Gospodinov, M. Daviti, A. N. Anagnostopoulos, K. Paraskevopoulos, E. K. Polychroniadis and I. Yanchev, Dielectric Behavior Of Mixed HgBrxI2−x Single Crystals, J. Mater. Sci. Lett., 2000, 19, 1019–1020 CrossRef CAS; (c) V. Marinova, S. Shurulinkov, M. Daviti, K. Paraskevopoulos and A. Anagnostopoulos, Refractive index measurements of mixed HgBrxI2−x single crystals, Opt. Mater., 2000, 14, 95–99 CrossRef CAS; (d) V. Marinova, I. Yanchev, M. Daviti, K. Kyritsi and A. N. Anagnostopoulos, Electron- and hole-mobility of Hg(BrxI1−x)2 crystals (x = 0.25, 0.50, 0.75), Mater. Res. Bull., 2002, 37(12), 1991–1995 CrossRef CAS; (e) M. Daviti and K. M. Paraskevopoulos, Temperature dependence of the fundamental absorption edge of HgBr2 and HgBrI single crystals, Mater. Res. Bull., 1999, 34, 381–388 CrossRef CAS.
- (a) Y. Li, W. Wang, H. Wang, H. Lin and L. Wu, Mixed-Anion Inorganic Compounds: A Favorable Candidate for Infrared Nonlinear Optical Materials, Cryst. Growth Des., 2019, 19, 4172–4192 CrossRef CAS; (b) H. Lin, W. Wei, H. Chen, X. Wu and Q. Zhu, Rational design of infrared nonlinear optical chalcogenides by chemical substitution, Coord. Chem. Rev., 2020, 406, 213150 CrossRef CAS; (c) H. Chen, W. Wei, H. Lin and X. Wu, Transition-metal-based chalcogenides: A rich source of infrared nonlinear optical materials, Coord. Chem. Rev., 2021, 448, 214154 CrossRef CAS; (d) M. Li, Z. Ma, B. Li, X. Wu, H. Lin and Q. Zhu, HgCuPS4: An Exceptional Infrared Nonlinear Optical Material with Defect Diamond-like Structure, Chem. Mater., 2020, 32, 4331–4339 CrossRef CAS.
- (a) Q. Wu, Y. Li, H. Chen, K. Jiang, H. Li, C. Zhong, X. Chen and J. Qin, HgBrI: A promising nonlinear optical material in IR region, Inorg. Chem. Commun., 2013, 34, 1–3 CrossRef; (b) Y. Huang, X. Meng, L. Kang, Y. Li, C. Zhong, Z. Lin, X. Chen and J. Qin, Hg2Br3I: a new mixed halide nonlinear optical material in the infrared region, CrystEngComm, 2013, 15, 4196–5000 RSC; (c) Y. Li, M. Wang, T. Zhu, X. Meng, C. Zhong, X. Chen and J. Qin, Synthesis, crystal structure and properties of a new candidate for nonlinear optical material in the IR region: Hg2BrI3, Dalton Trans., 2012, 41, 763–766 RSC.
- X. Shi, Z. Ma, C. He and K. Wu, Strong SHG responses predicted in binary metal halide crystal HgI2, Chem. Phys. Lett., 2014, 608, 219–223 CrossRef CAS.
- K. Y. Suponitsky and A. E. Masunov, Supramolecular step in design of nonlinear optical materials: Effect of π⋯π stacking aggregation on hyperpolarizability, J. Chem. Phys., 2013, 139, 094310 CrossRef PubMed.
- O. R. Evans and W. Lin, Crystal engineering of NLO materials based on metal-organic coordination networks, Acc. Chem. Res., 2002, 35, 511–522 CrossRef CAS PubMed.
- H. L. Chen, W. T. Chen, Q. Y. Luo and Y. P. Xu, A One-Dimensional Compound [HgCl2(2,2′-bipy)]n (bipy = bipyridine): Synthesis and Crystal Structure, Asian J. Chem., 2015, 27, 2005–2007 CrossRef CAS.
- D. C. Craig, Y. Farhangi, D. P. Graddon and N. C. Stephenson, Cryst. Struct. Commun., 1974, 155, 3–5 Search PubMed.
- E. Freire, S. Baggio, R. Baggio and L. Suescun, Mercury(II) halide complexes with N-donor organic ligands: crystal and molecular structure of HgI2R, R = 1,10-phenanthroline, 2,9-dimethyl-1,10-phenanthroline, bipyridine, J. Chem. Crystallogr., 1999, 29(7), 825–830 CrossRef CAS.
- R. Alizadeh, V. Amani, A. A. Farshady and H. R. Khavasi, Synthesis, characterization, and crystal structure determination of two mercury(II) dimer complexes with 4,7-diphenyl-1,10-phenanthroline, 5,5′-dimethyl-2,2′-bipyridine and bromide, J. Coord. Chem., 2010, 63, 2122–2131 CrossRef CAS.
- N. Tadayon Pour, A. Ebadi, A. Abedi, V. Amani and H. R. Khavasi, (5,5′-Dimethyl-2,2′-bipyridine-к2N,N′)-diiodidomercury(II), Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m1305 CrossRef CAS PubMed.
- I. Warad, M. Al-Noaimi, S. F. Haddad and R. Othman, Dichlorido(2,9-dimethyl-1,10-phenanthroline-к2N,N′) mercury(II), Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, m109 CrossRef CAS PubMed.
- R. Alizadeh, A. Heidari, R. Ahmadi and V. Amani, Dibromido(2,9-dimethyl-1,10-phenanthroline-к2N,N′)mercury(II), Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m483–m484 CrossRef CAS PubMed.
- D. Matkovic-Calogovic, N. Davidovic, Z. Popovic and Z. Zugaj, Dichloro(2,2′: 6′,2′′-terpyridyl-N,N′,N′′)mercury(II), Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 1766–1768 CrossRef.
- C. Yan, Q. Chen, L. Chen, R. Feng, X. Shan, F. Jiang and M. Hong, Crystal Structures and Luminescence Behaviour of d10 Metal-Organic Complexes with Multipyridine Ligands, Aust. J. Chem., 2011, 64, 104–118 CrossRef CAS.
- A. Giordana, E. Priola, E. Bonometti, P. Benzi, L. Operti and E. Diana, Structural and spectroscopic study of the asymmetric 2-(2′-pyridyl)-1,8-naphthyridine ligand with closed-shell metals, Polyhedron, 2017, 138, 239–248 CrossRef CAS.
- E. Priola, E. Bonometti, V. Brunella, L. Operti and E. Diana, Luminescent coordination polymers of 2,2′-bipyrimidine and mercury(II) salts: a structural and computational study, Polyhedron, 2016, 104, 25–36 CrossRef CAS.
- D. Marabello, P. Antoniotti, P. Benzi, C. Canepa, E. Diana, L. Operti, L. Mortati and M. P. Sassi, Non-linear optical properties of β-D-fructopyranose calcium chloride MOFs: an experimental and theoretical approach, J. Mater. Sci., 2015, 50, 4330–4341 CrossRef CAS.
- G. Brauer, Handbook of preparative inorganic chemistry, Academic Press, New York London, 2nd edn, 1963, vol. 2 Search PubMed.
- (a) S. K. Patra, N. Sadhukhan and J. K. Bera, Effects of Axial Coordination on the Ru−Ru Single Bond in Diruthenium Paddlewheel Complexes, Inorg. Chem., 2006, 45, 4007–4015 CrossRef CAS PubMed; (b) C. S. Campos-Fernández, L. M. Thomson, J. R. Galán-Mascarós, X. Ouyang and K. R. Dunbar, Homologous Series of Redox-Active, Dinuclear Cations [M2(O2CCH3)2(pynp)2]2+ (M = Mo, Ru, Rh) with the Bridging Ligand 2-(2-Pyridyl)-1,8-naphthyridine (pynp), Inorg. Chem., 2002, 41, 1523–1533 CrossRef PubMed.
- (a) R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich-Wilson, B. Civalleri, L. Maschio, M. Rerat, S. Casassa, J. Baima and B. Salustro, et al., CRYSTAL17 User Manual, Int. J. Quantum Chem., 2014, 114(19), 1287–1317 CrossRef CAS; (b) R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich-Wilson, B. Civalleri, L. Maschio, M. Rérat, S. Casassa, J. Baima and S. Salustro, et al., Quantum-Mechanical Condensed Matter Simulations with CRYSTAL, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8(4), 1–36 CAS.
- G. J. Sutton, Some Studies in Inorganic Complexes. V. Mercury(II), Aust. J. Chem., 1959, 12, 637–642 CrossRef CAS.
- J. E. Douglas and C. J. Wilkins, Terpyridyl complexes of zinc, cadmium, and mercury, Inorg. Chim. Acta, 1969, 3(4), 635–638 CrossRef CAS.
- J. R. Ferraro, Low frequency vibration of Inorganic and coordination compounds, Plenum Press, New York, 1971 Search PubMed.
- (a) L. Ould-Moussa, M. Castellà-Ventura, E. Kassab, O. Poizat, D. P. Strommen and J. R. Kincaid, Ab initio and density functional study of the geometrical, electronic and vibrational properties of 2,2′-bipyridine, J. Raman Spectrosc., 2000, 31, 337–390 CrossRef; (b) M. Reiher, G. Brehm and S. Schneider, Assignment of Vibrational Spectra of 1,10-Phenanthroline by Comparison with Frequencies and Raman Intensities from Density Functional Calculations, J. Phys. Chem. A, 2004, 108, 734–742 CrossRef CAS; (c) N. Neto, G. Sbrana and M. Muniz-Miranda, Vibrational analysis and molecular structure of 2,2′-bipyrimidine, Spectrochim. Acta, Part A, 1990, 46, 705–715 CrossRef.
- K. Burger, F. E. Wagner, A. Vértes, É. Bencze, J. Mink, I. Labàdi and Z. Nemes-Vetéssy, Structural study of terpyridine and o-phenanthroline complexes of iridium(III), J. Phys. Chem. Solids, 2001, 62, 2059–2068 CrossRef CAS.
- V. I. Pakhomov, A. V. Goryunov, A. A. Boguslavskii, R. S. Lotfullin and I. N. Ivanova-Korfini, Refinement of HgBr2 structure, Zh. Neorg. Khim., 1990, 35, 2476–2478 CAS.
- M. Hostettler, H. Birkedal and D. Schwarzenbach, The yellow polymorphs of mercuric iodide HgI2, Helv. Chim. Acta, 2003, 86, 1410–1422 CrossRef CAS.
- D. Grdenić, The structural chemistry of mercury, Q. Rev., Chem. Soc., 1965, 19, 303–328 RSC.
- P. Z. Zanazzi and A. Pavese, Behavior of Micas at High Pressure and High Temperature, Rev. Mineral. Geochem., 2002, 46, 99–116 CrossRef CAS.
- L. Y. Lin, C. W. Chang, W. H. Chen, Y. F. Chen, S. P. Guo and M. C. Tamargo, Raman investigation of anharmonicity and disorder-induced effects in Zn1−xBexSe epifilms, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 075204 CrossRef.
- (a) S. Sahoo, S. Dhara, V. Sivasubramanian, S. Kalavathi and A. K. Arora, Phonon confinement and substitutional disorder in Cd1−xZnxS nanocrystals, J. Raman Spectrosc., 2009, 40, 1050–1054 CrossRef CAS; (b) A. S. BarkerJr and A. J. Sievers, Optical studies of the vibrational properties of disordered Solids, Rev. Mod. Phys., 1975, 47(2), 1–179 CrossRef; (c) K. Hayashi, N. Sawaki and I. Akasaki, Raman Scattering in ZnSxSe1−x alloys, Jpn. J. Appl. Phys., 1991, 30(3), 501–505 CrossRef CAS; (d) P. Dean, Vibrational Properties of Disordered Systems: Numerical Studies, Rev. Mod. Phys., 1972, 44(2), 127–168 CrossRef CAS.
- P. D'Arco, S. Mustapha, M. Ferrabone, Y. Noël, M. De La Pierre and R. Dovesi, Symmetry and Random Sampling of Symmetry Independent Configurations for the Simulation of Disordered Solids, J. Phys.: Condens. Matter, 2013, 25, 355401 CrossRef PubMed.
- (a) W. T. Chen, M. S. Wang, X. Liu, G. C. Guo and J. S. Huang, Investigations of Group 12(IIB) Metal Halide/Pseudohalide-Bipy Systems: Syntheses, Structures, Properties, and TDDFT Calculations (Bipy = 2,2′-bipyridine or 4,4′-bipyridine), Cryst. Growth Des., 2006, 6(10), 2289–2300 CrossRef CAS; (b) A. J. Canty, C. L. Raston, B. W. Skelton and A. H. White, Structural studies of mercury(II) halide pyridine complexes [HgX2(py)2], X=Cl, Br, or I, J. Chem. Soc., Dalton Trans., 1982, 15–18 RSC.
- (a) R. E. Marsh, Some thoughts on choosing the correct space group, Acta Crystallogr., Sect. B: Struct. Sci., 1995, 51, 897–907 CrossRef; (b) F. H. Herbstein and R. E. Marsh, More Space-Group Corrections: From Triclinic to Centred Monoclinic and to Rhombohedral; Also From P1 to P-1 and From Cc to C2/c, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 677–686 CrossRef; (c) R. E. Marsh, P1 or P-1? Or something else?, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 931–936 CrossRef PubMed.
- B. Boulanger and J. Zyss, International Tables for Crystallography, 2013, vol. D, pp. 181–222 Search PubMed.
- L. Suescun and M. Nespolo, From patterns to space groups and the eigensymmetry of crystallographic orbits: a re-interpretation of some symmetry diagrams in IUCr Teaching Pamphlet No. 14, J. Appl. Crystallogr., 2012, 45, 834–837 CrossRef CAS.
- D. Watkins, The control of difficult refinements, Acta Crystallogr., Sect. A: Found. Crystallogr., 1994, A50, 411–437 CrossRef.
- (a) A. Bauzá, I. Alkorta, J. Elguero, T. J. Mooibroek and A. Frontera, Spodium Bonds: Noncovalent Interactions Involving Group 12 Elements, Angew. Chem., Int. Ed., 2020, 59, 17482–17487 CrossRef PubMed; (b) G. Ciancaleoni and L. Rocchigiani, Assessing the Orbital Contribution in the “Spodium Bond” by Natural Orbital for Chemical Valence–Charge Displacement Analysis, Inorg. Chem., 2021, 60(7), 4683–4692 CrossRef CAS PubMed; (c) T. Xia, D. Li and L. Cheng, Theoretical analysis of the spodium bonds in HgCl2⋯L (L = ClR, SR2, and PR3) dimers, Chem. Phys., 2020, 539, 1109782 Search PubMed; (d) G. Mahmoudi, E. Zangrando, B. Miroslaw, A. V. Gurbanov, M. G. Babashkin, A. Frontera and D. A. Safin, Spodium bonding and other non-covalent interactions assisted supramolecular aggregation in a new mercury(II) complex of a nicotinohydrazide derivative, Inorg. Chim. Acta, 2021, 519, 120279 CrossRef CAS.
- R. W. Boyd, Nonlinear Optics, 3th edn, 2008 Search PubMed.
- T. Y. Tou and S. W. Ng, SHG in a centrosymmetric crystal – can it be possible?, Acta Crystallogr., Sect. C: Struct. Chem., 2021, 77, 586–590 CrossRef CAS PubMed.
- CrysAlis PRO 1.171.38.46, Rigaku OD, 2015 Search PubMed.
- (a) C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS; (b) C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS; (c) I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed; (d) R. Taylor and C. F. Macrae, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 815–827 CrossRef CAS PubMed.
- (a) C. Chen, Y. Wu and R. Li, The anionic group theory of the non-linear optical effect and its applications in the development of new high-quality NLO crystals in the borate series, Int. Rev. Phys. Chem., 1989, 8(1), 65–91 Search PubMed; (b) D. Xue and S. Zhang, Chemical bond analysis of the correlation between crystal structure and nonlinear optical properties of complex crystals, Phys. B, 1999, 262, 78–83 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2086278–2086292. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2dt00201a |
| ‡ CriSDi, Interdepartmental Center for Crystallography, Via Pietro Giuria 7, 10125, Torino, Italy. |
| This journal is © The Royal Society of Chemistry 2022 |