
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeteroleptic mer-[Cr(N∩N∩N)(CN)3] complexes: synthetic challenge, structural characterization and photophysical properties†
Julien
Chong
a,
Céline
Besnard
 b,
Carlos M.
Cruz
c,
Claude
Piguet
*a and
Juan-Ramón
Jiménez
*ad
b,
Carlos M.
Cruz
c,
Claude
Piguet
*a and
Juan-Ramón
Jiménez
*ad
aDepartment of Inorganic and Analytical Chemistry, University of Geneva, 30 quai E. Ansermet, CH-1211 Geneva 4, Switzerland. E-mail: Claude.Piguet@unige.ch
bLaboratory of Crystallography, University of Geneva, 24 quai E. Ansermet, CH-1211 Geneva 4, Switzerland
cDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland
dDepartment of Inorganic Chemistry, University of Granada and “Unidad de Excelencia en Química” (UEQ), Avda. Fuentenueva, E-18071 Granada, España. E-mail: jrjimenez@ugr.es
First published on 17th February 2022
Abstract
The substitution of three water molecules around trivalent chromium in CrBr3·6H2O with the tridentate 2,2′:6′,2′′-terpyridine (tpy), N,N′-dimethyl-N,N′-di(pyridine-2-yl)pyridine-2,6-diamine (ddpd) or 2,6-di(quinolin-8-yl)pyridine (dqp) ligands gives the heteroleptic mer-[Cr(L)Br3] complexes. Stepwise treatments with Ag(CF3SO3) and KCN under microwave irradiations provide mer-[Cr(L)(CN)3] in moderate yields. According to their X-ray crystal structures, the associated six-coordinate meridional [CrN3C3] chromophores increasingly deviate from a pseudo-octahedral arrangement according to L = ddpd ≈ dpq ≪ tpy; a trend in line with the replacement of six-membered with five-membered chelate rings around CrIII. Room-temperature ligand-centered UV-excitation at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 170 cm−1 (λexc = 350 nm), followed by energy transfer and intersystem crossing eventually yield microsecond metal-centered Cr(2E → 4A2) phosphorescence in the red to near infrared domain 13150–12650 cm−1 (760 ≤ λem ≤ 790 nm). Decreasing the temperature to liquid nitrogen (77 K) extends the emission lifetimes to reach the millisecond regime with a record of 4.02 ms for mer-[Cr(dqp)(CN)3] in frozen acetonitrile.
170 cm−1 (λexc = 350 nm), followed by energy transfer and intersystem crossing eventually yield microsecond metal-centered Cr(2E → 4A2) phosphorescence in the red to near infrared domain 13150–12650 cm−1 (760 ≤ λem ≤ 790 nm). Decreasing the temperature to liquid nitrogen (77 K) extends the emission lifetimes to reach the millisecond regime with a record of 4.02 ms for mer-[Cr(dqp)(CN)3] in frozen acetonitrile.
Introduction
The [Ar]3d3 electronic configuration found in pseudo-octahedral trivalent chromium complexes is famous for its orbitally non-degenerate paramagnetic 4A2 ground state, which is ideally suited (i) for maximizing kinetic inertness with respect to ligand exchange reactions,1 a prerequisite for the design of heteroleptic complexes,2 and (ii) for programming magnetic exchange interactions with adjacent open-shell partners3 as found, for instance, in ferromagnetic Prussian blue-like CsINiII[CrIII(CN)6]·2H2O4 or NaIMnII[CrIII(CN)6]·2H2O5 phases. Moreover, the energy of the metal-centered excited states Cr(2E), Cr(2T1) and Cr(4T2), which control the optical properties, can be easily tuned by the primary coordination sphere around the chromium center via the ligand field (Δ) and Racah (B, C) parameters (Scheme 1 and Table S1†).6–13 Whereas the design of inert heteroleptic trivalent chromium complexes has been systematically exploited within the frame of the ‘complex-as-ligand and complex-as-metal’ strategy for producing heteropolymetallic paramagnetic assemblies and clusters working as ferromagnets or single-molecule magnets (SMMs),3,14–19 the alternative building of optical devices using the same strategy remained scarce.20–23 Under weak ligand field conditions, as found in [CrX6]3− (X = F, Cl) and in [CrO6] chromophores possessing Δ/B < 30 (Scheme 1a, Table S1†), the excited Cr(4T2) and Cr(2E,2T1) levels are close in energy and involved in back-intersystem crossing (BISC) processes.24–29 These circumstances produce: (i) Cr(4T2 → 4A2) fluorescence (green arrow in Scheme 1b) and (ii) Cr(2T1,2E → 4A2) phosphorescence (red arrows in Scheme 1b) with short excited-state lifetimes.2,11 The situation changes for strong field [CrN6] and [CrC6] chromophores with Δ/B > 30 (Scheme 1a) because (i) the large energy gap ΔE = E(4T2) − E(2T1,2E) prevents BISC and (ii) the intersystem crossing Cr(4T2 → 2T1,2E) process is fast.30,31 This ensures an efficient feeding of the long-lived doublet Cr(2T1,2E) excited states (Scheme 1b) and phosphorescence dominates the emission process. Competition between phosphorescence and intermetallic energy transfers thus becomes accessible in d–f heterometallic assemblies,20,21 which can be exploited for linear light-downshifting32–34 or for light-upconversion.35–39 Furthermore, energy (or electron) transfer can be also applied in bimolecular reactions, e.g. singlet oxygen formation or photoredox catalysis.40–42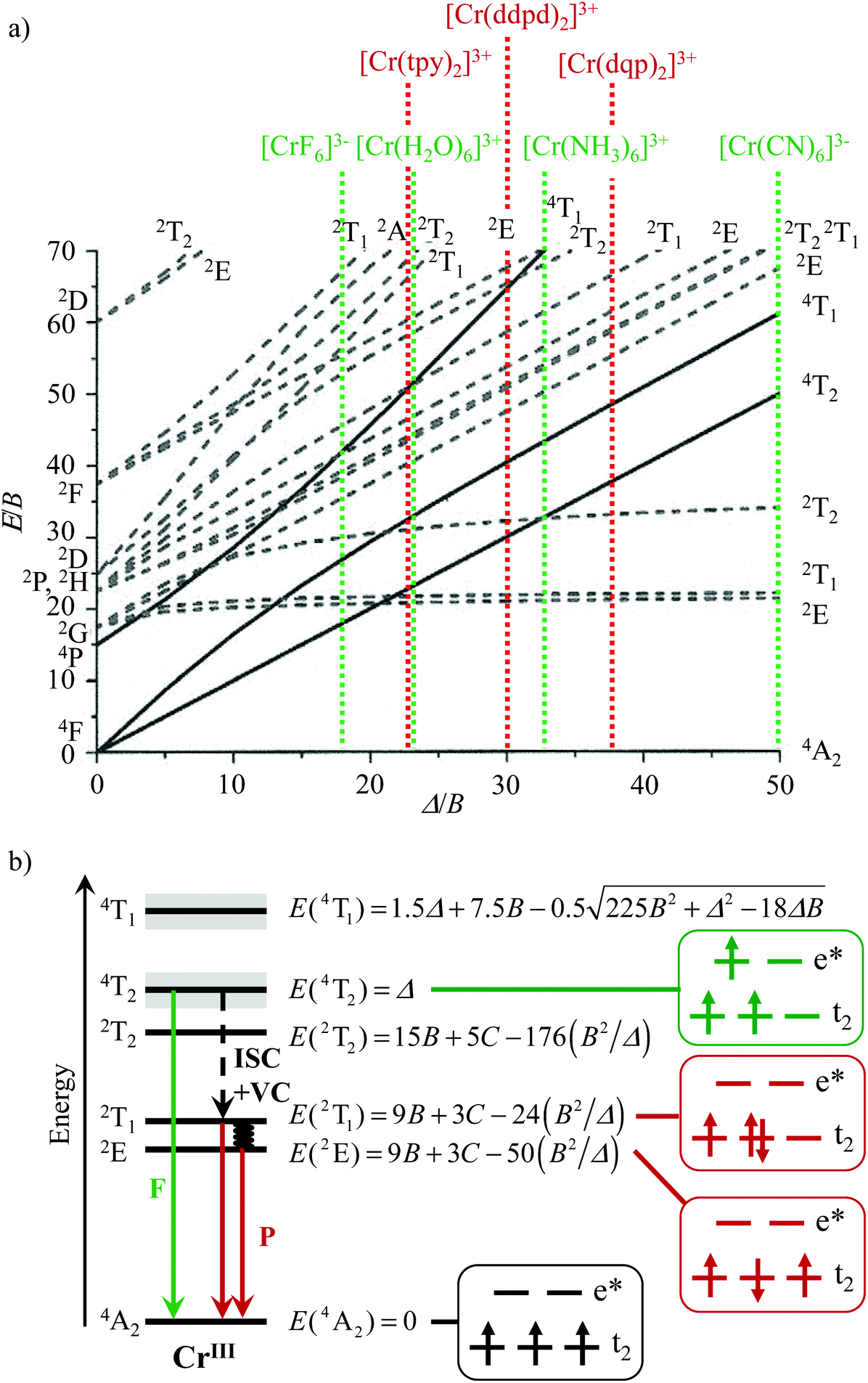 | ||
| Scheme 1 a) Tanabe–Sugano diagram (computed for C/B = 4.5)13 for the d3 configuration in an octahedral ligand field highlighting some selected trivalent chromium complexes (Δ/B taken from Table S1,† tpy, ddpd and dqp are shown in Fig. 2) and (b) Perrin–Jablonski diagram showing (i) the energy levels of [CrIIIX6] chromophores with strong ligand-field, (ii) the microstates and (iii) the energies associated with the low lying metal-centred levels computed in terms of ligand field Δ and Racah parameters B and C.10 F = fluorescence, P = phosphorescence, ISC = intersystem crossing. | ||
For using these strong-field pseudo-octahedral CrIII coordination complexes as sensitizers or activators in polymetallic (supra)molecular assemblies, the parameters of the intermetallic energy transfer processes (EnT), which rely on through-space (Förster) and/or through-bond (Dexter) mechanisms, should be optimized.43 Förster EnT is based on coulombic interactions operating between the multipole changes involved during the simultaneous relaxation process of the donor and the excitation of the acceptor.43,44 Its efficiency decreases as d−n, where d is the distance between the donor and the acceptor and n = 6, 8, 10 reflects the order of the multipole involved in the interaction. Its efficiency depends on the spectral overlap between the emission spectrum of the donor and the absorption spectrum of the acceptor. A spin rule is therefore implicit in this: for benefiting from an intense absorption spectrum, the associated transitions at each site must be spin-allowed. In contrast, Dexter EnT corresponds to a double electron-exchange which requires a strong electronic coupling, often obtained via chemical bonding between the donor and the acceptor. It involves the exchange integrals that account for the indistinguishability of the electrons in many-electron wavefunctions,45 and it is therefore facilitated by good orbital overlap and short distance (decreases as e−d). It does not collapse for local spin selection rules according that the spin of the whole assembly must be preserved.43,46–48 Consequently, Dexter EnT is sometimes called ‘triplet–triplet’ EnT.49 With this in mind, the building of short and directional molecular connectors between strong-field CrIII and its optical partners is highly wished. Simple two-atoms cyanide anions, previously used for the programming of magnetic exchange coupling in related systems,3,4,14 appears to be a logical choice since the considerable covalence of the Cr–CN bonds is known to give strong-field and kinetically inert [CrC6] chromophores in [Cr(CN)6]3−, while the peripheral nitrogen atoms are able to interact with solvated trivalent lanthanide centers to give very efficient CrIII → NdIII and CrIII → YbIII energy transfers (kEnT ≥ 108 s−1, Fig. 1).33
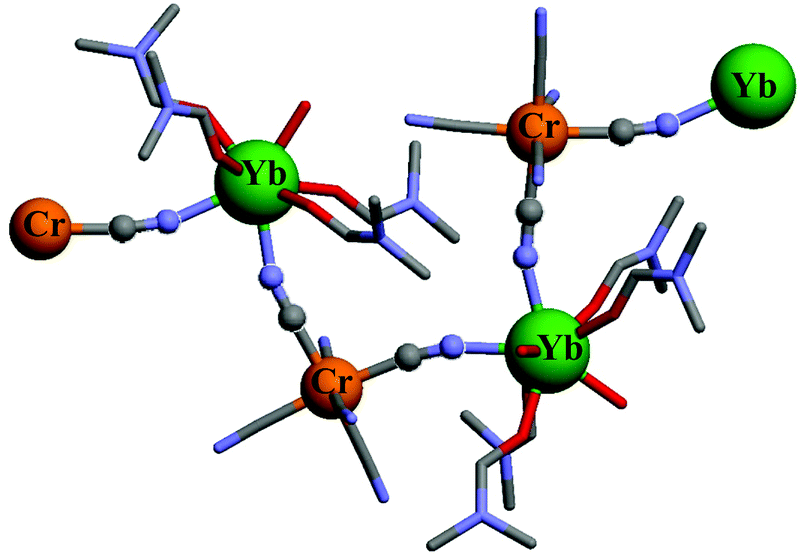 | ||
| Fig. 1 Structure of part of the one-dimensional chain of {[Cr(CN)6][Yb(dmf)4(H2O)2]·H2O}∞ highlighting the Cr–CN–Yb bridges.33 Color code: C = grey, N = blue, O = red, Cr = orange, Yb = green. | ||
Whereas [CrLx(CN)(6–2x)](3–2x)− (L = neutral didentate ligand, for example, 1,10-phenanthorline (phen) or 2,2′ bipyridine (bipy)) or [CrL(CN)3] (L = neutral facial tridentate ligand, for example, 1,4,7-triazacyclononane (tacn)) building blocks have been developed extensively for the design of magnetic clusters and assemblies,2–4,14,19 their use as potential optical partners remained confidential. We are aware of one contribution exploring the systematic low-energy shift of the Cr(2T1,2E → 4A2) phosphorescence emission spectra upon replacement of water with cyanide ligands around CrIII in [Cr(H2O)n(CN)(6−n)](n−3)+ (n = 1–6)50 and a second report dealing with the emission spectra of cis-[Cr(phen)(CN)4]−, cis-[Cr(bpy)2(CN)2]+ and fac-[Cr(tren)(CN)3].51 On the other side, the recent need for replacing the costly and rarest 4d (Ru) and 5d (Re, Os, Ir) metals with earth abundant 3d (Cr),27,28,35,52,54 Mn(I)52,54 or 4d (Mo)55 analogues boosted the use of the rigid tridentate polyaromatic ligands 2,2′:6′,2′′-terpyridine (tpy), N,N′-dimethyl-N,N′-di(pyridine-2-yl)pyridine-2,6-diamine (ddpd) or 2,6-di(quinolin-8-yl)pyridine (dqp) ligands for tuning ligand-field and Racah parameters via chelate ring sizes, donor atoms and sterical constraints around CrIII in pseudo-octahedral homoleptic [CrL2]3+ (L = tpy, ddpd or dqp; Scheme 1a)56–58 and heteroleptic [Cr(L)(L′)]3+ complexes.59–63 Some preliminary efforts have been described for exploiting these strong-field [CrN6] chromophores as complex-as-ligands for connecting d and/or f-block partners,64,65 but, to the best of our knowledge, the design of heteroleptic [Cr(L)(CN)3] building blocks is currently unknown despite the attractive perspectives of using short cyanide bridges for intermetallic connections while tuning the electronic properties of CrIII with the tridentate ligands. In this contribution we report on the explorative synthesis, characterization and photophysical properties of mer-[Cr(L)(CN)3] (L = tpy, ddpd and dqp, Fig. 2–4).
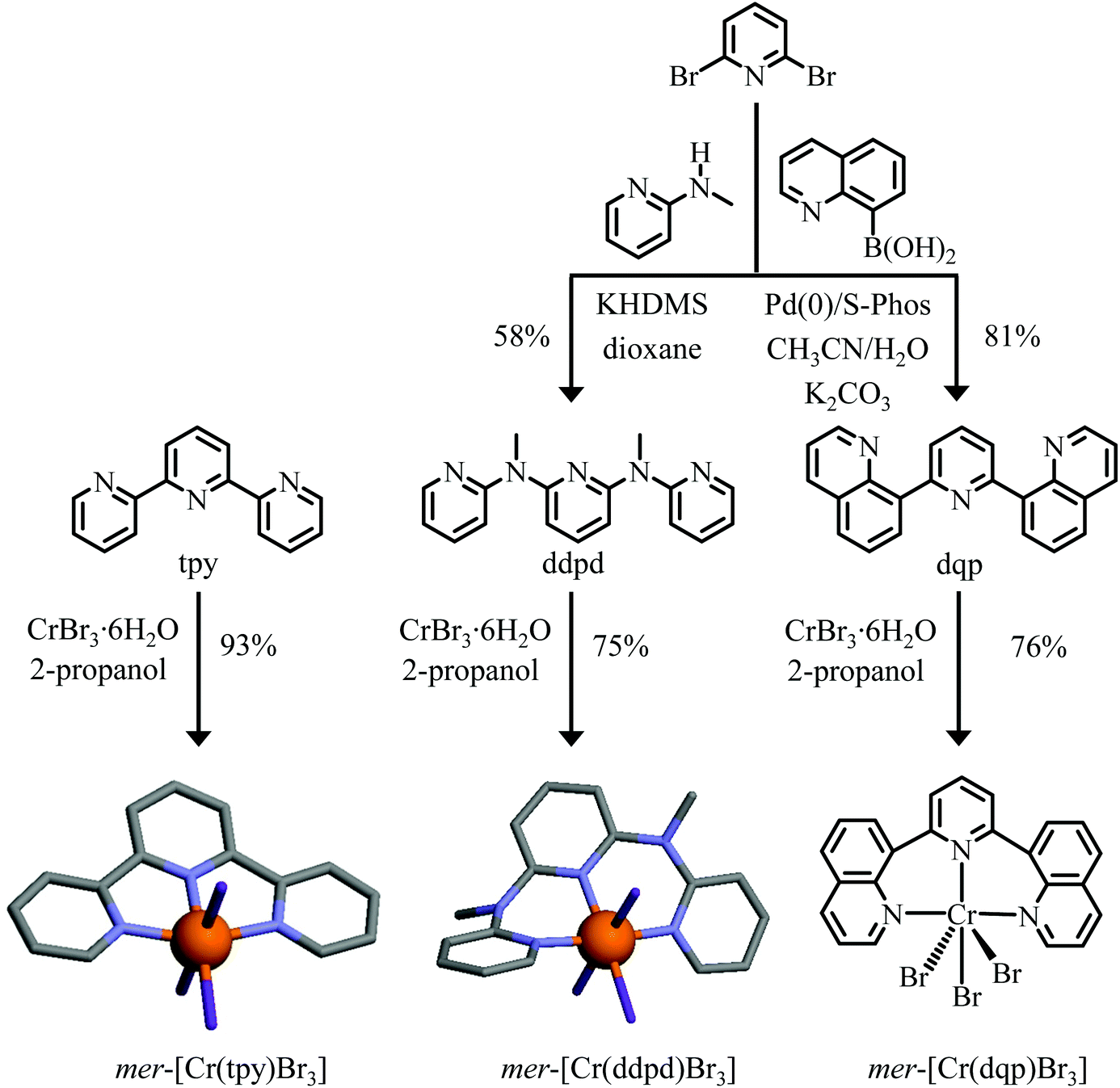 | ||
| Fig. 2 Syntheses and structures of mer-[Cr(tpy)Br3], mer-[Cr(ddpd)Br3]62 and mer-[Cr(dqp)Br3]. Color code: C = grey, N = blue, Br = purple, Cr = orange. H atoms have been omitted for clarity. | ||
 | ||
| Fig. 3 Syntheses and structures of mer-[Cr(L)(CN)3] (L = tpy, ddpd, dqp). Color code: C = grey, N = blue, Cr = orange. H atoms have been omitted for clarity. | ||
 | ||
| Fig. 4 IR spectra of mer-[Cr(L)(CN)3] (L = tpy, ddpd, dqp) recorded in the solid state at 293 K. | ||
Results and discussion
Synthesis and characterization of the heteroleptic mer-[Cr(L)(CN)3] (L = tpy, ddpd and dqp) complexes
Modern strategies used for the preparation of heteroleptic CrIII complexes are indebted to Kane-Maguire and co-workers, who took advantage of the unusual lability of the CrIII–O(triflate) bond in Cr(CF3SO3)n adducts for the controlled synthesis of tri-didentate complexes.66,67 Its extension for connecting two different tridentate binding units around six-coordinate CrIII was first reported by Constable and co-workers59–61 and required the preparation of the mer-[Cr(L)X3] (X = Cl, Br) intermediates prior to react them with HCF3SO3 to form labile mer-[Cr(L)(CF3SO3)3] and HX. Although this method was proved to be effective with five-membered tpy-type chelating ligands,59–61 its application with more basic six-membered ddpd and dqp ligands resulted in partial decomplexation and isolation of the protonated [Hddpd]+ and [Hdqp]+ ligands.62,63 An alternative strategy relies on the weak solubility of the silver salts AgX in acetonitrile (Ksp(AgCl) = 10−12.4, Ksp(AgBr) = 10−13.2)68 that can be easily separated from the soluble triflate [Cr(L)(CF3SO3)3] complex when [Cr(ddpd)X3] or [Cr(dqp)X3] are reacted with AgCF3SO3 in acetonitrile. Original attempts with [Cr(ddpd)Cl3] led to incomplete anion exchange, but starting from [Cr(ddpd)Br3] provided quantitative amount of [Cr(ddpd)(CF3SO3)3] and AgBr.62 With this in mind, the preparation of the heteroleptic mer-[Cr(L)Br3] (L = tpy, ddpd and dqp) precursors was conducted with CrBr3·6H2O as the metallic source and tpy (commercially available), ddpd69 and dqp70 as the tridentate ligands (Fig. 2). The characterization of the poorly soluble mer-[Cr(L)Br3] complexes (only slightly soluble in DMF or in acetonitrile) is challenging because (i) high resolution NMR spectroscopy is hampered by the long electronic relaxation time of the Cr(4A2) paramagnetic ground state and (ii) the lack of permanent charge limits mass spectrometry to the detection of dissociated [Cr(L)Brn](3−n)+ (n = 1, 2) derivatives.The IR spectra (Fig. 4 and Fig. S1†) combined with satisfying elemental analyses (Table S2†) confirmed the presence of both ligand L and CrBr3 in the isolated microcrystalline powders. Slow diffusion of diethyl ether into DMF solutions provided single crystal suitable for X-ray diffraction studies for [Cr(tpy)Br3] (Fig. S2† and Tables S3–S5†) and [Cr(ddpd)Br3],62 but we were unable to crystallize [Cr(dqp)Br3] (Fig. 2). In line with the molecular structures reported previously for [Cr(L)Cl3] (L = tpy,71 ddpd64 and dqp,63Table 1 entries 1–3), the tridentate ligands are all meridionally bound to CrIII (Fig. 2), but only the fused five-membered chelate rings in [Cr(tpy)Br3] are compatible with a rough planar arrangement of the aromatic units (3.6° ≤ interplanar angles ≤ 7.2°, Fig. 2 and Table S5†), whereas the six-membered chelate rings in [Cr(ddpd)Br3] produce an helical twist of the polyaromatic ligand (46.8° ≤ interplanar angles ≤ 46.9°, Fig. 2).62
| Complex | Cr–Ncentral /Å | Cr–Ndistal/Å | Cr–X/Å | Bite angle /° trans-N–Cr–N | Bite angle /° trans-N–Cr–X | Bite angle /° trans-X–Cr–X | Σ/° | Ref. |
|---|---|---|---|---|---|---|---|---|
a
Æ© is calculated with the formula 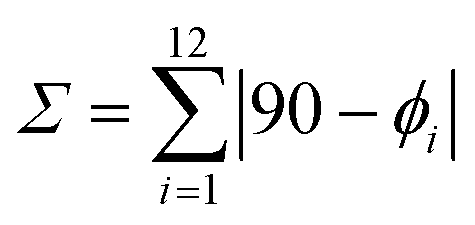 where ϕi are the 12 cisoid bond angles around the CrIII metal.
b The values given between parentheses correspond to the standard deviation when several distances or bond angles are considered.
c dpc− is the tridentate 3,6-di-tert-butyl-1,8-di(pyridine-2-yl)-carbazolate ligand. where ϕi are the 12 cisoid bond angles around the CrIII metal.
b The values given between parentheses correspond to the standard deviation when several distances or bond angles are considered.
c dpc− is the tridentate 3,6-di-tert-butyl-1,8-di(pyridine-2-yl)-carbazolate ligand.
|
||||||||
| [Cr(tpy)Cl3] | 1.992 | 2.075(5) | 2.32(2) | 157.02 | 178.48 | 174.57 | 59.9 | 71 |
| [Cr(ddpd)Cl3] | 2.071 | 2.068(5) | 2.33(1) | 172.82 | 178.17 | 179.11 | 21.6 | 64 |
| [Cr(dqp)Cl3] | 2.059 | 2.077(0) | 2.34(2) | 173.81 | 180 | 178.21 | 30.0 | 63 |
| [Cr(tpy)Br3] | 1.989 | 2.074(2) | 2.47(2) | 156.71 | 176.02 | 176.30 | 46.7 | This work |
| [Cr(ddpd)Br3] | 2.074 | 2.07(1) | 2.49(1) | 173.06 | 177.17 | 178.62 | 22.3 | 62 |
| [Cr(tpy)(CN)3] | 1.993 | 2.07(4) | 2.07(1) | 156.84 | 175.17 | 176.93 | 65.4 | This work |
| [Cr(ddpd)(CN)3] | 2.041 | 2.087(0) | 2.085(4) | 174.59 | 180 | 178.90 | 19.8 | This work |
| [Cr(dqp)(CN)3] | 2.044 | 2.072(1) | 2.076(8) | 176.71 | 175.94 | 173.59 | 25.6 | This work |
| [Cr(tpy)2](PF6)3 | 1.986(1) | 2.054(9) | — | 157.12(8) | — | — | 102.0 | 59 |
| [Cr(ddpd)2](BF4)3 | 2.042(4) | 2.046(5) | — | 170.87(2) | — | — | 36.0 | 56 |
| [Cr(dqp)2](CF3SO3)3 | 2.049(4) | 2.063(3) | — | 174.8(3) | — | — | 28.9 | 58 |
| [Cr(dqc)2]PF6c |
1.962(4) | 2.109(3) | — | 173.69(8) | — | — | 39.0 | 72 |
| K3[Cr(CN)6] | — | — | 2.08(2) | — | — | 179.0(6) | 6.6 | 73 |
Since the three halogen atoms occupy the three missing vertices of the pseudo-octahedron around CrIII with mainly no constraint, [Cr(tpy)X3] (X = Cl, Br) are close to fit the criteria of the non-chiral C2v point group whereas [Cr(ddpd)X3] and [Cr(dqp)Cl3] belong to the chiral C2 point group and crystallize as racemic mixtures. The amount of deviation from a perfect octahedral arrangement of the six donor atoms around the central metal can be obtained by computing  , where ϕi are the 12 cisoid bond angles around the Cr3+ cation (Table 1, column 8). The latter parameter is twice larger for [Cr(tpy)X3] (46.7 ≤ Σ ≤ 59.9°) than for [Cr(ddpd)X3] (21.6 ≤ Σ ≤ 22.3°) and [Cr(dqp)Cl3] (Σ = 30°), a distortion highlighted by the trans-N–Cr–N bite angles which reach 156–157° for [Cr(tpy)X3], but are close to be linear for [Cr(ddpd)X3] and [Cr(dqp)Cl3] (172–173°). As expected, the replacement of X = Cl with X = Br in [Cr(L)X3] (L = tpy and ddpd) produces only very minor changes except for a rough 6% increase of the Cr–X bond lengths (Table 1, column 4 and Fig. S3†).
, where ϕi are the 12 cisoid bond angles around the Cr3+ cation (Table 1, column 8). The latter parameter is twice larger for [Cr(tpy)X3] (46.7 ≤ Σ ≤ 59.9°) than for [Cr(ddpd)X3] (21.6 ≤ Σ ≤ 22.3°) and [Cr(dqp)Cl3] (Σ = 30°), a distortion highlighted by the trans-N–Cr–N bite angles which reach 156–157° for [Cr(tpy)X3], but are close to be linear for [Cr(ddpd)X3] and [Cr(dqp)Cl3] (172–173°). As expected, the replacement of X = Cl with X = Br in [Cr(L)X3] (L = tpy and ddpd) produces only very minor changes except for a rough 6% increase of the Cr–X bond lengths (Table 1, column 4 and Fig. S3†).
Treatment of mer-[Cr(L)Br3] with AgCF3SO3 was carried out in acetonitrile under microwave irradiation. The resulting precipitates of AgBr were removed by filtration, while peaks observed at m/z = 583 ([Cr(tpy)(CF3SO3)2]+), m/z = 641 ([Cr(ddpd)(CF3SO3)2]+) and m/z = 683 ([Cr(dqp)(CF3SO3)2]+) in the ESI-MS spectra confirmed the formation of the soluble [Cr(L)(CF3SO3)3] complexes in the filtrates. The latter complexes were not isolated, and subsequent reactions with KCN under microwave heating provided the final complexes [Cr(L)(CN)3] (L = tpy, ddpd, dqp, Table S2†) in low to moderate yields (Fig. 3). These complexes are reasonably soluble in polar solvents (DMF, water, acetonitrile, methanol), but insoluble in less polar organic solvents. Single crystals suitable for X ray diffraction (XRD) were obtained after recrystallization in hot water for [Cr(tpy)(CN)3] (Fig. S4, and Tables S6–S8†) and after slow evaporation of solutions containing complexes in acetone/water (1:1) or methanol/water (5:1) for [Cr(ddpd)(CN)3]·2H2O (Fig. S5, and Tables S9–S11†) and [Cr(dqp)(CN)3]·3.28H2O (Fig. S6, and Tables S12–S14†), respectively. The molecular structures of mer-[Cr(L)(CN)3] (L = tpy, ddpd, dqp, Fig. 3) are very similar to those previously discussed for related mer-[Cr(L)X3] complexes (X = Cl, Br, Fig. S7†), except for the observation of shorter Cr–C bonds (2.07 to 2.09 Å) compared with Cr–X bonds which cover the 2.32–2.49 Å range, but similar to 2.08(2) Å reported for Cr–C bonds in K3[Cr(CN)6] (Table 1). A thorough analysis of the packing of the crystal structures of [Cr(tpy)(CN)3], [Cr(ddpd)(CN)3]·2H2O and [Cr(dqp)(CN)3]·3.28H2O show only weak CN⋯HO interactions when water molecules are present in the crystal structure together with some weak interligand π-stacking involving layers of complexes (Fig. S8–S11†). No trace of strong intermolecular bridges involving cyanide anions could be detected so that mer-[Cr(L)(CN)3] exist as well-defined molecular entities in the solid state. The IR spectra display very weak CN stretching bands in the 2100–2170 cm−1 domain where three IR-active bands are expected for [Cr(tpy)(CN)3] (C2v symmetry: 2A1 + B1, Fig. 4a) and for [Cr(ddpd)(CN)3] or [Cr(dqp)(CN)3] (C2 symmetry: 2A + B, Fig. 4b and c).74 These stretching vibrations are located at higher energy than those found for non-complexed cyanide anion in KCN (2080 cm−1)75 because the poor π-accepting CN− ligands maximize their σ-donation upon complexation to a d-block cation. This tends to raise the v(CN) stretching frequencies since electrons are removed from the slightly anti-bonding cyanide-based σ orbital, a process which reinforces the CN− force constant.75 Following the thorough analysis of Jones,76 one expects that the changes in dipole moment accompanying the characteristic oscillations of cyanides bound to electron-poor CrIII are weak, but the faint absorptions recorded for mer-[Cr(L)(CN)3] merit to be stressed here (Fig. 4) and compared with the much stronger peaks observed for K3[Cr(CN)6].76
Photophysical properties of the heteroleptic mer-[Cr(L)(CN)3] (L = tpy, ddpd and dqp) complexes
Based on their molecular structures observed in the solid state, mer-[Cr(tpy)(CN)3] can be assigned to the C2v point group and mer-[Cr(ddpd)(CN)3] and mer-[Cr(dqp)(CN)3] to C2 point group, both possessing only mono-dimensional irreducible representations. Consequently, the three standard d–d transitions, i.e. the two spin-forbidden Cr(2E ↔ 4A2), Cr(2T1 ↔ 4A2) and the spin-allowed Cr(4T2 ↔ 4A2) transitions, which are detected as unsplit bands in the absorption and/or emission spectra of strong-field octahedral CrIII complexes (Oh symmetry, Scheme 1a), are expected to be split in mer-[Cr(L)(CN)3] because the excited E and T levels transform into A1, A2, B1 and B2 (C2v) or A and B (C2) representations. However, as observed for the [CrN6] chromophores found in the homoleptic D2(d) symmetrical [Cr(L)2]3+ (L = tpy, ddpd and dqp) analogues,56–58,64 the deviation from local Oh symmetry is small for the [CrC3N3] chromophores found in mer-[Cr(L)(CN)3] (Fig. 3 and Table 1). The detection of only minor additional splitting in the absorption and emission spectra (Fig. 5–7 and S21–S23†) justifies the use of (i) a simplified mathematical treatment of the optical spectra pertinent to Oh symmetry (Scheme 1b) and (ii) original labels reminiscent of Oh symmetry for these complexes.6,8–10,64,77 With this in mind, the ligand field and Racah parameters are obtained from the experimental energies of the excited levels by solving eqn (1)–(3) (Table 2, entries 8–10).58,64| E(4T2) = Δ | (1) |
| E(2T1) − E(2E) = 26(B2/Δ) | (2) |
| E(2T1) = 9B + 3C − 24(B2/Δ) | (3) |
 | ||
Fig. 5 Absorption (black full traces) and normalized excitation (red dotted traces, analysis fixed on the Cr(2E → 4A2) spin–flip emission band) spectra recorded for (a) [Cr(tpy)(CN)3] (c = 1.40 × 10−5 M in DMF at 293 K, for excitation  ), (b) [Cr(ddpd)(CN)3] (c = 1.75 × 10−5 M in CH3CN at 293 K, for excitation ), (b) [Cr(ddpd)(CN)3] (c = 1.75 × 10−5 M in CH3CN at 293 K, for excitation  ) and (c) [Cr(dqp)(CN)3] (c = 1.75 × 10−5 M in CH3CN at 293 K, for excitation ) and (c) [Cr(dqp)(CN)3] (c = 1.75 × 10−5 M in CH3CN at 293 K, for excitation  ). The NIR inset spectra correspond to transmittance absorption spectra recorded for solid state single crystals at 293 K. * represents the hypothetical OH overtone. ). The NIR inset spectra correspond to transmittance absorption spectra recorded for solid state single crystals at 293 K. * represents the hypothetical OH overtone. | ||
 | ||
Fig. 6 (a)–(c) Emission spectra ( ) recorded for [Cr(L)X3] (L = tpy, ddpd, dqp; X = Cl, Br, CN) in frozen solution at 77 K (c = 2 × 10−5 M; [Cr(L)Cl3], [Cr(L)Br3] and [Cr(tpy)(CN)3] in DMF; [Cr(ddpd)(CN)3] and [Cr(dqp)(CN)3] in CH3CN:C2H5CN 3:1). ) recorded for [Cr(L)X3] (L = tpy, ddpd, dqp; X = Cl, Br, CN) in frozen solution at 77 K (c = 2 × 10−5 M; [Cr(L)Cl3], [Cr(L)Br3] and [Cr(tpy)(CN)3] in DMF; [Cr(ddpd)(CN)3] and [Cr(dqp)(CN)3] in CH3CN:C2H5CN 3:1). | ||
 | ||
Fig. 7 Emission spectra ( ) recorded for [Cr(L)(CN)3] (L = tpy, ddpd, dqp) solution at 293 K (c = 2 × 10−5 M; [Cr(tpy)(CN)3] in DMF, [Cr(ddpd)(CN)3] and [Cr(dqp)(CN)3] in CH3CN). ) recorded for [Cr(L)(CN)3] (L = tpy, ddpd, dqp) solution at 293 K (c = 2 × 10−5 M; [Cr(tpy)(CN)3] in DMF, [Cr(ddpd)(CN)3] and [Cr(dqp)(CN)3] in CH3CN). | ||
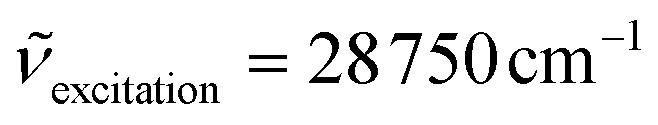 ) of the emissive CrIII-centered levels and estimated ligand-field Δ and Racah parameters B and C for the heteroleptic mer-[Cr(L)X3] (X = Cl, Br, CN) and their homoleptic parent [Cr(L)2]3+ (L = tpy, ddpd and dqp) complexes
) of the emissive CrIII-centered levels and estimated ligand-field Δ and Racah parameters B and C for the heteroleptic mer-[Cr(L)X3] (X = Cl, Br, CN) and their homoleptic parent [Cr(L)2]3+ (L = tpy, ddpd and dqp) complexes
| Complex | Solvent | E(2T1) /cm−1 | E(2E) /cm−1 | τ(2E) /μs 77 Kb | τ(2E)/μs 293 Ka | τ(2E)/μs 293 Kb | Δ/cm−1 | B/cm−1 | C/cm−1 | Δ/B | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Deaerated solution. b Non-deaerated solution. c Recorded at 10 K. d dpc− is the tridentate 3,6-di-tert-butyl-1,8-di(pyridine-2-yl)-carbazolate ligand. | |||||||||||
| [Cr(tpy)Cl3] | DMF | 13602 |
13175 |
690(10)c | — | — | 16694 |
697 | 2786 | 24.0 | 64 |
| [Cr(ddpd)Cl3] | DMF | 14040 |
13755 |
1580(15) | — | — | — | — | — | — | This work |
| [Cr(dqp)Cl3] | DMF | 14040 |
13670 |
1560(15) | — | — | — | — | — | — | This work |
| [Cr(tpy)Br3] | DMF | 13460 |
13210 |
110(5) | — | — | — | — | — | — | This work |
| [Cr(ddpd)Br3] | DMF | — | 13736 |
1560(15) | — | — | — | — | — | — | This work |
| [Cr(dqp)Br3] | DMF | — | 13698 |
1540(15) | — | — | — | — | — | — | This work |
| [Cr(tpy)(CN)3] | DMF | 13262 |
12610 |
1020(20) | 3.12(1) × 10−3 | 2.94(1) × 10−3 | 22370 |
670 | 2565 | 33.4 | This work |
| [Cr(ddpd)(CN)3] | CH3CN | 13605 |
13089 |
3590(20) | 79(3) | 10.1(2) | 24540 |
520 | 3030 | 47.2 | This work |
| [Cr(dqp)(CN)3] | CH3CN | 13513 |
13192 |
4020(20) | 13.0(6) | 6.8(2) | 24100 |
480 | 3140 | 50.2 | This work |
| [Cr(tpy)2](PF6)3 | CH3CN | 12953 |
13584 |
670(10) | 0.14 | - | 18750 |
790 | 2512 | 23.7 | 62 and 64 |
| [Cr(ddpd)2](BF4)3 | CH3CN | 13387 |
12788 |
1300(15) | 975(10) | 51(5) | 22990 |
659 | 2638 | 34.9 | 56 and 64 |
| [Cr(dqp)2](CF3SO3)3 | H2O | 13949 |
12903 |
3070(20) | 1270(10) | 83(5) | 24937 |
656 | 2791 | 38.0 | 58 and 62 |
| [Cr(dqc)2]PF6d |
CH3CN | — | 9370 | 2.0 | — | — | 19200 |
470 | 1880 | 40.9 | 53 and 72 |
| K3[Cr(CN)6] | H2O | — | 12400 |
4200 | — | — | 26600 |
480 | 2800 | 55.4 | 50 and 79 |
The situation is more tricky for the heteroleptic mer-[Cr(L)X3] (X = Cl, Br) complexes because the [CrN3X3] coordination spheres deviate significantly from octahedral symmetry and we prefer to not derive oversimplified ligand parameters for these low-symmetrical complexes (Table 2).
The absorption spectra are dominated by intense ligand-centered π* ← π transitions together with LMCT and MC transitions covering the UV-visible domain (Fig. 5); an assignment confirmed by theoretical TD-DFT calculations (Tables S15–S20 and Fig. S12–S20†). At lower energies, the TD-DFT calculations predict three sets of spin-allowed metal-centered d–d transitions located at 22170–22620 cm−1, 25445–28190 cm−1 and 31350–32570 cm−1 for [Cr(tpy)(CN)3] (Fig. S13†), which are reminiscent of the three Cr(4T1,2 ← 4A2) transitions expected in octahedral symmetry (Scheme 1a). Comparison with the experimental absorption spectrum (Fig. S13b†) allows to assign the weak band at 22370 cm−1 to Cr(4T2 ← 4A2), which gives a direct estimation of Δ (eqn (1), Table 2 column 8). The same procedure can be applied for [Cr(ddpd)(CN)3] (Fig. S16 and S17†) and [Cr(dqp)(CN)3] (Fig. S19 and S20†) to give slightly larger ligand-field parameters Δ = 24000–24500 cm−1 (Table 2, column 8); a result in line with the replacement of five-membered chelate rings in tpy with six-membered rings in ddpd and dqp.56–58 The forbidden Cr(2T1,2E ← 4A2) spin–flip transitions cannot be detected in solution at the largest accessible concentrations (10–50 mM due to limited solubility), which implies ε < 0.1 M−1 cm−1.
Excitations at λexc = 350 nm 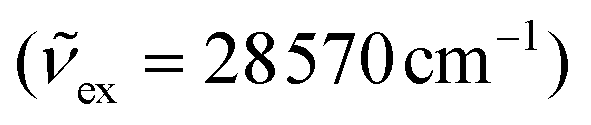 of all heteroleptic [Cr(L)X3] (L = tpy, ddpd, dqp; X = Cl, Br, CN) complexes in frozen solution at 77 K induce red to near-infrared emissions, which can be assigned to the spin–flip Cr(2T1,2E → 4A2) transitions, the energies of which follow the trends [Cr(L)Cl3] ≈ [Cr(L)Br3] > [Cr(L)(CN)3] for a given tridentate ligand L and [Cr(dqp)X3] ≥ [Cr(ddpd)X3] ≫ [Cr(tpy)X3] for a given anion X− (Fig. 6).
of all heteroleptic [Cr(L)X3] (L = tpy, ddpd, dqp; X = Cl, Br, CN) complexes in frozen solution at 77 K induce red to near-infrared emissions, which can be assigned to the spin–flip Cr(2T1,2E → 4A2) transitions, the energies of which follow the trends [Cr(L)Cl3] ≈ [Cr(L)Br3] > [Cr(L)(CN)3] for a given tridentate ligand L and [Cr(dqp)X3] ≥ [Cr(ddpd)X3] ≫ [Cr(tpy)X3] for a given anion X− (Fig. 6).
While the phosphorescence of the halogenido complexes [Cr(L)Cl3] and [Cr(L)Br3] is too weak to be detected at room temperature with our setup, detectable emissions can be observed in these conditions for [Cr(L)(CN)3] (Fig. 7). The larger intensities observed for the high-energy Cr(2T1 → 4A2) bands at room temperature results from the thermally-induced population of the Cr(2T1) excited levels by the close Cr(2E) states.56,57,78 The overall quantum yields recorded in de-aerated solutions at 293 K upon 22990 cm−1 excitation (λexc = 435 nm) are 0.2% for [Cr(ddpd)(CN)3], 0.08% for [Cr(dqp)(CN)3] and 0.0001% for [Cr(tpy)(CN)3].
The associated room-temperature excitation spectra recorded upon monitoring the Cr(2E → 4A2) phosphorescence in [Cr(L)(CN)3] match the absorption spectra (Fig. 5) and demonstrate that the Cr-centered emission can be sensitized by both LMCT, metal-centered and ligand-centered π* ← π transitions. In order to better characterize the spin–flip Cr(2T1,2E ↔ 4A2) transitions, the transmittance spectra of solid state samples of pure [Cr(L)X3] (L = tpy, ddpd, dqp; X = Cl, Br, CN) were recorded in the NIR region (shown as insets in Fig. 5) and compared with the corresponding emission spectra in Fig. S21–S23.† Firstly, it is worth stressing here that the intensities of the spin-forbidden Cr(2T1,2E ← 4A2) transitions are so weak that they correspond to only minor distortions from the baselines of the solid-state transmittance spectra and require thorough mathematical corrections to separate true spectroscopic signals from the background. This being said, we nevertheless observe that the intensities of the Cr(2T1,2E ← 4A2) absorption bands in solid state samples depend on the nature of the anions bound to CrIII and decrease with their electronegativities X = Cl− > Br− > CN−. As expected for these low-symmetry C2 or C2v complexes, the splitting of the 2E and 2T1 excited levels produce more than the two spin–flip transitions reminiscent of Oh point group (see for instance mer-[Cr(dqp)Cl3] in Fig. S23†), but the comparison of emission/absorption spectra allows to state that (i) the minor Stokes shifts rarely exceed 50 cm−1, (ii) room temperature emission spectra magnify the emission of the Cr(2T1) level located at higher energy (thermal equilibrium) and (iii) the pseudo-octahedral description of the spin–flip transitions as arising from Cr(2T1,2E ↔ 4A2) is satisfying for the [CrC3N3] chromophores found in [Cr(L)(CN)3]. Having the estimations of the energies of the Cr(2E) and Cr(2T1) levels in hand (Table 2, column 3 and 4), eqn (2) and (3) provide the nephelauxetic parameters collected in Table 2 (columns 9 and 10). The stepwise replacement of the two tridentate chelate N3 units found in [CrL2]3+ with monodentate CN− ligands to give mer-[CrL(CN)3] and [Cr(CN)6]3− is accompanied by an average 150 cm−1 decrease of the Racah B parameter (i.e. an increase of the nephelauxetic effect and charge delocalization) for the coordination of the three first cyanide anions and a further, but smaller, reduction of B for the substitution of the remaining tridentate ligand to eventually reach B = 480 cm−1 for [Cr(CN)6]3− (ref. 79) This trend is obviously more pronounced for the successive replacement of the constrained five-membered chelate rings found in [Cr(tpy)2]3+ than of the looser six-membered rings in [Cr(ddpd)2]3+ and [Cr(dqp)2]3+.
Surprisingly, the solid-state transmittance spectra recorded for single crystals of [Cr(ddpd)(CN)3] (Fig. 5b) and of [Cr(dqp)(CN)3] (Fig. 5c) display an additional weak absorption band centered at 12600 cm−1 (794 nm, marked with an asterisk), the intensity of which is still one order of magnitude larger than that measured for the spin-forbidden Cr(2T1,2E ← 4A2) transitions. These absorption bands are fully reproducible for the latter two complexes, but they are lacking for all the other studied heteroleptic [Cr(L)X3] complexes (L = tpy, X = Cl, Br, CN and L = ddpd, dqp, X = Cl, Br; Fig. S21–S23†) and for the free ligands. We must confess that we are not able to propose a fully convincing explanation, but one notices that crystals of [Cr(ddpd)(CN)3] (Table S9†) and [Cr(dqp)(CN)3] (Table S12†) contain co-crystallized water molecules, the fourth harmonic of which is expected to give a weak broad absorption band centered at 13300 cm−1 in the pure liquid (750 nm, ε ≈ 2 × 10−4 M−1 cm−1).80,81 On the contrary, crystals of [Cr(tpy)(CN)3] do not contain residual water molecules in the unit cell (Table S6†).
Finally, the Cr(2E) excited lifetimes of 1.0–4.0 ms observed for [Cr(L)(CN)3] in frozen solution at 77 K (Table 2, column 5) are intermediate between 4.2 ms reported for [Cr(CN)6]3− (an octahedral [CrC6] chromophore, Table 2, entry 13)50 and 0.67–3.0 ms characterizing [Cr(L)2]3+ ([CrN6] chromophores Table 2, entries 10–12).56,58,62,64 As usual, the longest lifetimes are found for L = dqp, which forms six-membered chelate rings in [Cr(dqp)(CN)3] and [Cr(dqp)2]3+, displays minor distortions from octahedral symmetry for the six bound nitrogen donor atoms and possesses only two rotational degrees of freedom.58 At room temperature in solution, the non-radiative deactivation pathways become particularly efficient in the heteroleptic [Cr(L)(CN)3] complexes and the Cr(2E) excited lifetimes drop to a few microseconds for L = ddpd, dqp and a few nanoseconds for L = tpy (Table 2, columns 6 and 7).
Conclusion
Although some rare cases of facial isomers have been reported for homoleptic [M(ddpd)2]z+ (M = CoII or VIII),82 none has been described with M = CrIII with any of the tridentate polyaromatic ligands tpy, ddpd and dqp used in this work. In agreement with these observations the tridentate ligands in the studied heteroleptic [Cr(L)X3] complexes (L = tpy, ddpd, dqp, X = Cl, Br, CN) are systematically meridionally tri-coordinated around CrIII, thus leaving three meridional positions for the anionic co-ligands. As found for the parent homoleptic [Cr(L)2]3+ complexes, the deviation from octahedral symmetry of the six donor atoms in the first coordination sphere depends on the size of the chelate rings. It severely decreases in going from five-membered chelates produced by bound L = tpy, to six-membered chelates found with L = ddpd, dqp. The photophysical properties reflect these distortions with a smaller ligand-field strength for mer-[Cr(tpy)(CN)3] compared with mer-[Cr(L)(CN)3] (L = ddpd, dqp). Additionally, the binding of cyanide anions to CrIII reinforces the nephelauxetic effect and shift the Δ/B values of mer-[Cr(L)(CN)3] toward the right of the Tanabe–Sugano diagram (Scheme 2). Consequently, the spin–flip phosphorescence appears at low-energy (in the red to near-infrared domain) and the associated emission lifetimes reach a few milliseconds at low-temperature where vibrational quenching mechanisms are negligible. Whereas maximum ligand-field strengths have been early recognized as a crucial tool for inducing detectable phosphorescence in CrIII complexes,24–26 the rational exploitation of the nephelauxetic effect for tuning the energy range of this delayed emission is more recent,28,58,62–65,72 but it appears to be particularly promising as illustrated in Scheme 2. Replacing X = CN−, with X = Cl−, Br− significantly reduces the energy gap ΔE = E(4T2) − E(2T1) from ca. 10000 cm−1 to 3000 cm−1 (Table 2). This accelerates the quenching of the spin–flip emissive levels through back intersystem crossing (BISC) and the photoluminescence cannot be detected at room temperature for the heteroleptic halogenido complexes.
 | ||
| Scheme 2 Tanabe–Sugano diagram for the d3 configuration in an octahedral ligand field (computed for C/B = 4.5) highlighting the pseudo-octahedral [CrN6] (red), [CrC3N3] (blue) and [CrC6] (green) chromophores. Δ/B taken from Table 2. | ||
Although the heteroleptic [Cr(L)(CN)3] complexes match most of the structural and photophysical requirements for being exploited as building block in (supra)molecular assemblies containing compact LnIII–X–CrIII light-converting units, their difficult synthesis and isolation combined with their low solubility and their sensitivity to thermally-activated emission quenching limit the perspectives for their use as sensitizers for molecular light upconversion. On the other side, the specific divergent meridional distribution of three CN– groups in the chromium complex might be cleverly exploited for designing d–f heterometallic magnetic materials according that some solid-state synthetic approaches could be developed.
Experimental section
General
Reagent grade acetonitrile (ACN) was distilled from CaH2. N,N-Dimethylformamide was dried through an alumina cartridge. All other chemicals such as CrBr3·6H2O, AgCF3SO3, 2,2′:6′,2′′-terpyridine (tpy) and organic solvents were purchased from commercial suppliers and used without further purification. The ligand N,N′-dimethyl-N,N′-dipyridin-2-ylpyridine-2,6-diamine (ddpd),69 2,6-di(quinolin-8-yl)pyridine (dqp)70 and the complexes [Cr(tpy)Cl3],71 [Cr(ddpd)Cl3],64 [Cr(dqp)Cl3]63 were prepared according to published methods. Silica-gel plates (Merck, 60 F254) were used for thin-layer chromatography, SiliaFlash® silica gel P60 (0.04–0.063 mm), and Acros silica gel 60 (0.035–0.07 mm) were used for preparative column chromatography.
Caution. Cyanide waste solutions are harmful and toxic and must be carefully destroyed (see https://sensorex.com/blog/2016/04/26/cyanide-wastewater-treatment/). We systematically decontaminated all solutions or waste containing cyanide ions by treatment with 1:1 mixtures of sodium hypochlorite (35% in water) and sodium hydroxide (2 M in water).
Preparation of [Cr(tpy)Br3]62
2,2′:6′,2′′-Terpyridine (tpy, 415 mg, 1.78 mmol) was dissolved in 50 mL of isopropanol. A solution of CrBr3·6H2O (708 mg, 1.77 mmol) in isopropanol (10 mL) was added dropwise and the resulting mixture was refluxed for 5 hours. The resulting green precipitate was filtered, washed with isopropanol (15 mL), hot ethanol (30 mL), dichloromethane (15 ml), diethylether (15 mL) and dried. Elemental analyses (Table S2†) were compatible with the formation of [Cr(tpy)Br3] (865 mg, 1.75 mmol, 93%) as a green powder. ESI-MS (CH3CN) m/z: [Cr(tpy)Br2]+ calc.: 444.87, found: 444.7; [Cr(tpy)Br]+ calc.: 363.95, found: 363.7. Small crystals suitable for XRD were obtained after a slow diffusion of Et2O into a solution of the complex in DMF.Preparation of [Cr(ddpd)Br3]62
N,N′-Dimethyl-N,N′-dipyridin-2-ylpyridine-2,6-diamine (ddpd, 372 mg, 1.28 mmol) was dissolved in 50 mL of isopropanol. A solution of CrBr3·6H2O (489 mg, 1.22 mmol) in isopropanol (10 mL) was added dropwise and the resulting mixture was refluxed for 5 hours. The resulting green precipitate was filtered, washed with isopropanol (15 mL), hot ethanol (30 mL), dichloromethane (15 ml), diethylether (15 mL) and dried. Elemental analyses (Table S2†) were compatible with the formation of [Cr(ddpd)Br3] (534 mg, 0.92 mmol, 75%) as a green powder. ESI-MS (CH3CN) m/z: [Cr(ddpd)Br2]+ calc.: 502.92, found: 502.5. Slow diffusion of diethyl ether into a concentrated solution of the complex in dimethylformamide led to the formation of green crystals suitable for X-ray diffraction.Preparation of [Cr(dqp)Br3]
2,6-Di(quinolin-8-yl)pyridine (dqp, 167 mg, 0.50 mmol) and the CrBr3·6H2O (200 mg, 0.50 mmol) were charged in a vial with isopropanol (15 mL). The vial was sealed and heated in a microwave oven at 160° for 6 h. The solid was filtered and washed with isopropanol (15 mL), hot ethanol (30 mL), dichloromethane (15 mL), diethyl ether (15 mL) and dried. Elemental analyses (Table S2†) were compatible with the formation of [Cr(dqp)Br3] (236 mg, 0.38 mmol, 76% yield) as an olive green powder. ESI-MS (CH3CN) m/z: [Cr(dqp)Br2]+ calc.: 544.90, found: 544.7.Preparation of [Cr(tpy)(CN)3]
AgCF3SO3 (160 mg, 0.62 mmol) and [Cr(tpy)Br3] (103 mg, 0.20 mmol) were charged in a vial with dry acetonitrile (2 mL). The vial was sealed and the solution was heated in a microwave oven at 140 °C for 30 min. The white precipitate (AgBr) was separated by filtration and the clear orange red solution was used directly for the next step. ESI-MS (CH3CN) m/z: [Cr(tpy)(CF3SO3)2]+ calc.: 582.94, found: 582.6, [Cr(tpy)(CF3SO3)3 + NH4]+ calc.: 749.93, found: 750.0.The solution of [Cr(tpy)(CF3SO3)3] was charged into a second vial containing a solution of KCN (43 mg, 0.66 mmol) partially dissolved in ethanol (5 mL). The sealed vial was heated in the microwave oven at 90 °C for 4 h. The resulting orange powder was filtered, and recrystallized from hot water to give [Cr(tpy)(CN)3]·1.7H2O (31 mg, 7.88 × 10−5 mol, 39% yield). Small crystals suitable for XRD were obtained after recrystallization in hot water.
Preparation of [Cr(ddpd)(CN)3]
AgCF3SO3 (353 mg, 1.37 mmol) and [Cr(ddpd)Br3] (250 mg, 0.43 mmol) were charged in a vial with dry acetonitrile (5 mL). The vial was sealed and the solution was heated in a microwave oven at 140 °C for 30 min. The white precipitate (AgBr) was separated by filtration and the clear orange red solution was used directly for the next step. ESI-MS (CH3CN) m/z: [Cr(ddpd)(CF3SO3)2]+ calc.: 640.99, found: 640.7.The solution of [Cr(ddpd)(CF3SO3)3] was charged into a second vial containing a solution of KCN (95 mg, 1.47 mmol) partially dissolved in acetonitrile (5 mL). The sealed vial was heated in the microwave oven at 120 °C for 30 min. The resulting orange powder was filtered, then suspended in water to remove the KCF3SO3 and the excess of KCN. The solid was again filtered, washed with dichloromethane to remove the possible traces of free ddpd and dried to give [Cr(ddpd)(CN)3]·0.85H2O·1.3CH2Cl2 (135 mg, 0.25 mmol, 58% yield). Small crystals suitable for XRD were obtained after a slow evaporation of a solution of complex in a 1:1 acetone/water mixture.
Preparation of [Cr(dqp)(CN)3]
AgCF3SO3 (267 mg, 1.04 mmol) and [Cr(dqp)Br3] (201 mg, 0.32 mmol) were charged in a vial with dry acetonitrile (5 mL). The vial was sealed and the solution was heated in a microwave oven at 140 °C for 30 min. The white precipitate (AgBr) was separated by filtration and the clear orange red solution was used directly for the next step. ESI-MS (CH3CN) m/z: [Cr(dqp)(CF3SO3)2]+ calc.: 682.97, found: 683.1.The solution of [Cr(dqp)(CF3SO3)3] was charged into a second vial containing a solution of KCN (71 mg, 1.09 mmol) partially dissolved in acetonitrile (5 mL). The sealed vial was heated in the microwave oven at 90 °C for 1 h. The red/brown mixture was filtered to remove a brown solid. The red solution was evaporated under reduced pressure and a light red solid was obtained. The red solid was suspended in H2O to remove KCF3SO3 and the excess of KCN. The suspension was filtered and the red solid suspended in dichloromethane in order to remove the possible traces of free ligand. Final filtration and drying led to [Cr(dqp)(CN)3]·1.5H2O·1.7CH2Cl2 (51 mg, 8.03 × 10−5 mol, 25% yield) as a red solid. Small crystals suitable for XRD were obtained by slow evaporation of a solution of complex in a 5:1 methanol/water mixture.
Spectroscopic and analytical measurements
1H and 13C NMR spectra were recorded at 298 K on a Bruker Avance 400 MHz spectrometer. Chemical shifts are given in ppm with respect to TMS. Pneumatically-assisted electrospray (ESI-MS) mass spectra were recorded from ∼1 × 10−4 M (ligands) and ∼1 × 10−3 M (complexes) solutions on an Applied Biosystems API 150EX LC/MS System equipped with a Turbo Ionspray source. Elemental analyses were performed by K. L. Paglia using a Microchemical Laboratory of the University of Geneva (Switzerland). FT-IR spectra were recorded with a Bruker Tensor 27 spectrometer equipped with a platinum ATR. Electronic spectra in the UV-Vis region were recorded at 293 K from CH3CN solutions with a PerkinElmer Lambda 1050 using quartz cells of 0.1 or 1.0 mm path length. Solid-state diffuse reflectance spectra were recorded using UV/Vis/NIR PerkinElmer Lambda 900 in reflectance (R) mode fitted with an integrating sphere. The pure samples were placed between two 1.0 mm path length quartz cells whose background cell was filled with pure MgO. The signal of pure MgO was used as the baseline. Emission spectra (excitation at 355 nm) and excitation spectra were recorded from frozen solution samples at 77 K (c ≈ 2 × 10−5 mol L−1) or from acetonitrile solution (c ≈ 2 × 10−5 mol L−1) at 293 K, with a Fluorolog (Horiba Jobin–Yvon), equipped with iHR320, a Xenon lamp 450 Watt Illuminator (FL-1039A/40A) and a water-cooled photo multiplier tube (PMT Hamamatsu R2658 or R928), and corrected for the spectral response of the system. For time-resolved experiments, the decay curves were recorded at 77 K and 293 K, using a photomultiplier (Hamamatsu R2658 or R928) and a digital oscilloscope (Tektronix MDO4104C). Pulsed excitation at 355 nm was obtained with the third harmonic of a pulsed Nd:YAG laser (Quantel Qsmart850) or by a pulsed diode light source NanoLED 375L (<200 ps pulse, HORIBA Scientific). Low temperature (77 K) was achieved using liquid quartz transparent Dewar filled with liquid N2 in the center of which samples were placed. Acetonitrile/propionitrile or DMF solutions (c ≈ 2 × 10−5 mol L−1) were introduced in quartz tube (4 mm interior diameter) introduced in sample holder of the Dewar. The oxygen free decay curve measurements were done at 293 K on acetonitrile or DMF solutions of the complex. The complexes in quartz tube were dissolved in acetonitrile or DMF then degassed by freeze pump thaw, and filled with argon. Aerated solutions were prepared by using no-degassed acetonitrile or DMF as solvent. Quantum yields were calculated using [Cr(ddpd)2]3+ as reference (λexc = 435 nm; Q.Y. = 12.1% in CH3CN).58X-ray crystallography
Summary of crystal data, intensity measurements and structure refinements for complexes [Cr(tpy)Br3], [Cr(tpy)(CN)3], [Cr(ddpd)(CN)3]·2H2O and [Cr(dqp)(CN)3]·3.28H2O were collected in Tables S3, S6, S9 and S12.† Pertinent bond lengths, bond angles and interplanar angles were collected in Tables S4, S5, S7, S8, S10, S11, S13 and S14† together with ORTEP views, pertinent numbering schemes and intermolecular interactions gathered in Fig. S2–S11.† The crystals were mounted on Hampton cryoloops with protection oil. X-ray data collections were performed with an XtaLAB Synergy-S diffractometer equipped with an hybrid pixel hypix arc 150 detector. The structures were solved by using dual space methods.83 Full-matrix least-square refinements on F2 were performed with SHELXL.84 within the OLEX2 refinement package.85 CCDC 2133575–2133578† contain the supplementary crystallographic data.Theoretical calculations
The Gaussian 16 software package was used to perform structural optimizations of the investigated complexes. Starting from the X-ray diffraction structures, the ground state, 4A2, structures of the different complexes were obtained from DFT optimizations using the unrestricted version of the Becke three-parameters exchange function in combination with the Lee–Yang–Parr correlation functional (UB3LYP). The basis set used for these optimizations was the 6-31G(d,p) for N, C and H and the LanL2DZ for the metal center. In addition, LANL2 pseudopotential was specified for the core electrons of the Cr atom. Solvent effects were included via the Polarizable Contiunuum Model (PCM) as implemented in Gaussian 16 with the dielectric constant of N,N-dimethylformamide (for [Cr(tpy)(CN)3]) and acetonitrile (for [Cr(ddpd)(CN)3] and [Cr(dqp)(CN)3]). Optimized geometries were confirmed to be stationary points by analysis of their vibrational frequencies. Orbital and spin density information was extracted from the optimized geometries. The 50 lowest energetic transitions were calculated by TD-DFT, using the unrestricted version of the Coulomb-attenuated B3LYP functional (UCAM-B3LYP), selecting the 6-31++G(d,p) basis set for N, C and H, and the LanL2DZ for the metal center. Same pseudopotential for the Cr atom and solvent effects were selected as for the ground state calculations. Electron Density Difference Maps (EDDM) were computed using GaussSum 3.086 and are represented with an isoval = 0.002 using GaussView 6.1.1.87Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Swiss National Science Foundation is gratefully acknowledged (grant numbers: 200020_178758 and 206021_183324). Carlos M. Cruz thankfully acknowledge Prof. Dr Michal Juríček for his support and access to the UZH Service and Support for Science IT (S3IT) computing facilities.References
- J. Ribas Gispert, Coordination Chemistry, Wiley-VCH Verlag, Weinheim, 2008, pp. 228–233 Search PubMed.
- J.-R. Jiménez, B. Doistau, M. Poncet and C. Piguet, Coord. Chem. Rev., 2021, 434, 213750 CrossRef.
- R. Lescouëzec, L. M. Toma, J. Vaissermann, M. Verdaguer, F. S. Delgado, C. Ruiz-Perez, F. Lloret and M. Julve, Coord. Chem. Rev., 2005, 249, 2691–2729 CrossRef.
- M. Verdaguer, A. Bleuzen, V. Marvaud, J. Vaissermann, M. Seuleiman, C. Desplanches, A. Scuiller, C. Train, R. Garde, G. Gelly, C. Lomenech, I. Rosenman, P. Veillet, C. Cartier and F. Villain, Coord. Chem. Rev., 1999, 190, 1023–1047 CrossRef.
- W. Dong, L. N. Zhu, H. B. Song, D. Z. Liao, Z. H. Jiang, S. P. Yan, P. Cheng and S. Gao, Inorg. Chem., 2004, 43, 2465–2467 CrossRef CAS PubMed.
- C. K. Jorgensen, Adv. Chem. Phys., 1963, 5, 33–146 CAS.
- Multiplets of Transition-Metal Ions in Crystals, Pure and Applied Physics, ed. S. Sugano, Y. Tanabe and H. Kamimura, 1970, 33, pp. 1–333 Search PubMed.
- J. Ferguson, Prog. Inorg. Chem., 1970, 12, 159–293 CAS.
- H. Witzke, Theor. Chim. Acta, 1971, 20, 171–185 CrossRef CAS.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam-Oxford-New York-Tokyo, 1984, pp. 417–429 Search PubMed.
- A. Hauser and C. Reber, Struct. Bonding, 2017, 172, 291–312 CrossRef.
- S. Adachi, J. Lumin., 2021, 232, 117844 CrossRef CAS.
- M. G. Brik and C.-G. Ma, Theoretical Spectroscopy of Transition Metal and Rare Earth Ions, Jenny Stanford Publishing, Singapore, 2020, pp 298–304 Search PubMed.
- J. M. Herrera, A. Bachschmidt, F. Villain, A. Bleuzen, V. Marvaud, W. Wernsdorfer and M. Verdaguer, Philos. Trans. R. Soc., A, 2008, 366, 127–138 CrossRef CAS PubMed.
- J. Dreiser, K. S. Pedersen, C. Piamonteze, S. Rusponi, Z. Salman, M. Ali, E. M. Schau-Magnussen, C. A. Thuese, S. Piligkos, H. Weihe, H. Mutka, O. Waldmann, P. Oppeneer, J. Bendix, F. Nolting and H. Brune, Chem. Sci., 2012, 3, 1024–1032 RSC.
- E. J. L. McInnes, G. A. Timco, G. F. S. Whitehead and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2015, 54, 14244–14269 CrossRef CAS PubMed.
- S. K. Langley, D. P. Wielechowski, B. Moubaraki and K. S. Murray, Chem. Commun., 2016, 52, 10976–10979 RSC.
- M. Darawsheh, L. A. Barrios, O. Roubeau, S. J. Teat and G. Aromi, Angew. Chem., Int. Ed., 2018, 57, 13509–13513 CrossRef CAS PubMed.
- C. Pichon, N. Suaud, C. Duhayon, N. Guihery and J. P. Sutter, J. Am. Chem. Soc., 2018, 140, 7698–7704 CrossRef CAS PubMed.
- F.-F. Chen, Z.-Q. Chen, Z.-Q. Bian and C.-H. Huang, Coord. Chem. Rev., 2010, 254, 991–1010 CrossRef CAS.
- L. Aboshyan-Sorgho, M. Cantuel, S. Petoud, A. Hauser and C. Piguet, Coord. Chem. Rev., 2012, 256, 1644–1663 CrossRef CAS.
- P. S. Wagenknecht, N. A. P. Kane-Maguire, D. G. Speece and N. Helwic, Inorg. Chem., 2002, 41(5), 1229–1235 CrossRef CAS PubMed.
- A. Cadranel, P. S. Oviedo, P. Alborés, L. M. Baraldo, D. M. Guldi and J. H. Hodak, Inorg. Chem., 2018, 57(6), 3042–3053 CrossRef CAS PubMed.
- L. S. Forster, Chem. Rev., 1990, 90, 331–353 CrossRef CAS.
- A. D. Kirk, Chem. Rev., 1999, 99, 1607–1640 CrossRef CAS PubMed.
- P. S. Wagenknecht and P. C. Ford, Coord. Chem. Rev., 2011, 255, 591–616 CrossRef CAS.
- O. S. Wenger, J. Am. Chem. Soc., 2018, 140, 13522–13533 CrossRef CAS PubMed.
- C. Forster and K. Heinze, Chem. Soc. Rev., 2020, 49, 1057–1070 RSC.
- P. A. Scattergood, Organomet. Chem., 2021, 43, 1–34 CAS.
- N. A. P. Kane-Maguire, Top. Curr. Chem., 2007, 280, 37–67 CrossRef CAS.
- L. S. Forster, Coord. Chem. Rev., 2006, 250, 2023–2033 CrossRef CAS.
- P. A. Brayshaw, J.-C. G. Bünzli, P. Froidevaux, J. M. Harrowfield, Y. Kim and A. N. Sobolev, Inorg. Chem., 1995, 34, 2068–2076 CrossRef CAS.
- T. Lazarides, G. M. Davies, H. Adams, C. Sabatini, F. Barigelletti, A. Barbieri, S. J. A. Pope, S. Faulkner and M. D. Ward, Photochem. Photobiol. Sci., 2007, 6, 1152–1157 CrossRef CAS PubMed.
- L. Aboshyan-Sorgho, H. Nozary, A. Aebischer, J.-C. G. Bünzli, P.-Y. Morgantini, K. R. Kittilstved, A. Hauser, S. V. Eliseeva, S. Petoud and C. Piguet, J. Am. Chem. Soc., 2012, 134, 12675–12684 CrossRef CAS PubMed.
- L. A. Büldt and O. S. Wenger, Chem. Sci., 2017, 8, 7359–7367 RSC.
- L. Aboshyan-Sorgho, C. Besnard, P. Pattison, K. R. Kittilstved, A. Aebischer, J.-C. G. Bünzli, A. Hauser and C. Piguet, Angew. Chem., Int. Ed., 2011, 50, 4108–4112 CrossRef CAS PubMed.
- Y. Suffren, D. Zare, S. V. Eliseeva, L. Guénée, H. Nozary, T. Lathion, L. Aboshyan-Sorgho, S. Petoud, A. Hauser and C. Piguet, J. Phys. Chem. C, 2013, 117, 26957–26963 CrossRef CAS.
- B. Golesorkhi, H. Nozary, A. Fürstenberg and C. Piguet, Mater. Horiz., 2020, 7, 1279–1296 RSC.
- J. Kalmbach, C. Wang, Y. You, C. Forster, H. Schubert, K. Heinze, U. Resch-Genger and M. Seitz, Angew. Chem., Int. Ed., 2020, 39, 18804–18808 CrossRef PubMed.
- S. M. Stevenson, M. P. Shores and E. M. Ferreira, Angew. Chem., Int. Ed., 2015, 54, 6506–6510 CrossRef CAS PubMed.
- R. F. Higgins, S. M. Fatur, S. G. Shepard, S. M. Stevenson, D. J. Boston, E. M. Ferreira, N. H. Damrauer, A. K. Rappe and M. P. Shores, J. Am. Chem. Soc., 2016, 138, 5451–5464 CrossRef CAS PubMed.
- S. Otto, A. M. Nauth, E. Emilov, N. Scholz, A. Friedrich, U. Resch-Genger, S. Lochbrunner, T. Opatz and K. Heinze, ChemPhotoChem, 2017, 1, 344–349 CrossRef CAS.
- M. D. Ward, Coord. Chem. Rev., 2010, 254, 2634–2642 CrossRef CAS.
- W. R. Browne, N. m. O'Boyle, J. J. McGarvey and J. G. Vos, Chem. Soc. Rev., 2005, 34, 641–663 RSC.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836 CrossRef CAS.
- P. A. Tanner, Coord. Chem. Rev., 2010, 254, 3026–3029 CrossRef CAS.
- X.-Y. Wang, R. R. Valiev, T. Y. Ohulchanskyy, H. Agren, C. Yang and G. Chen, Chem. Soc. Rev., 2017, 46, 4150–4167 RSC.
- P. A. Tanner, L. Zhou, C. Duan and K.-L. Wong, Chem. Soc. Rev., 2018, 47, 5234–5265 RSC.
- Z. Q. You, C. P. Hsu and G. R. Fleming, J. Chem. Phys., 2006, 124, 044506 CrossRef PubMed.
- A. Ghaith, L. S. Forster and J. V. Rund, Inorg. Chim. Acta, 1986, 116, 11–13 CrossRef CAS.
- C. K. Ryu, R. B. Lessard, D. Lynch and J. F. Endicott, J. Phys. Chem., 1989, 93, 1752–1759 CrossRef CAS.
- C. Wegeberg and O. S. Wenger, Dalton Trans., 2022, 51, 1297–1302 RSC.
- C. Wegeberg and O. S. Wenger, JACS Au, 2021, 1, 1860–1876 CrossRef CAS PubMed.
- P. Herr, C. Kerzig, C. B. Larsen, D. Häussinger and O. S. Wenger, Nat. Chem., 2021, 13, 956–962 CrossRef CAS PubMed.
- J. B. Bilger, C. Kerzig, C. B. Larsen and O. S. Wenger, J. Am. Chem. Soc., 2021, 143, 1651–1663 CrossRef CAS PubMed.
- S. Otto, M. Grabolle, C. Förtser, C. Kreitner, U. Resh-Genger and K. Heinze, Angew. Chem., Int. Ed., 2015, 54, 11572–11576 CrossRef CAS PubMed.
- S. Otto, M. Dorn, C. Förster, M. Bauer, M. Seitz and K. Heinze, Coord. Chem. Rev., 2018, 359, 102–111 CrossRef CAS.
- J. R. Jimenez, B. Doistau, C. M. Cruz, C. Besnard, J. M. Cuerva, A. G. Campana and C. Piguet, J. Am. Chem. Soc., 2019, 141, 13244–13252 CrossRef CAS PubMed.
- E. C. Constable, C. E. Housecroft, M. Neuburger, J. Schönle and J. A. Zampese, Dalton Trans., 2014, 43, 7227–7235 RSC.
- J. Schönle, E. C. Constable, C. E. Housecroft, M. Neuburger and J. A. Zampese, Inorg. Chem. Commun., 2015, 51, 75–77 CrossRef.
- J. Schönle, E. C. Constable, C. E. Housecroft, A. Prescimone and J. A. Zampese, Polyhedron, 2015, 89, 182–188 CrossRef.
- J.-R. Jiménez, B. Doistau, C. Besnard and C. Piguet, Chem. Commun., 2018, 54, 13228–13231 RSC.
- J.-R. Jiménez, M. Poncet, B. Doistau, C. Besnard and C. Piguet, Dalton Trans., 2020, 49, 13528–13532 RSC.
- D. Zare, B. Doistau, H. Nozary, C. Besnard, L. Guénée, Y. Suffren, A.-L. Pelé, A. Hauser and C. Piguet, Dalton Trans., 2017, 46, 8992–9009 RSC.
- B. Doistau, J.-R. Jiménez, S. Guerra, C. Besnard and C. Piguet, Inorg. Chem., 2020, 59, 1424–1435 CrossRef CAS PubMed.
- K. D. Barker, K. A. Barnett, S. M. Connel, J. W. Glaeser, A. J. Wallace, J. Wildsmith, B. J. Herbert, J. F. Wheeler and N. A. P. Kane-Maguire, Inorg. Chim. Acta, 2001, 316, 41–49 CrossRef CAS.
- E. G. Donnay, J. P. Schaeper, R. D. Brooksbank, J. L. Fox, R. G. Potts, R. M. Davidsdon, J. F. Wheeler and N. A. P. Kane-Maguire, Inorg. Chim. Acta, 2007, 360, 3272–3280 CrossRef CAS.
- D. C. Luehrs, R. T. Iwamoto and J. Kleinberg, Inorg. Chem., 1966, 5, 201–204 CrossRef CAS.
- A. Breivogel, C. Forster and K. Heinze, Inorg. Chem., 2010, 49, 7052–7056 CrossRef CAS PubMed.
- M. Jaeger, L. Eriksson, J. Bergquist and O. Johansson, J. Org. Chem., 2007, 72, 10227–10230 CrossRef CAS PubMed.
- N. Cloete, H. G. Visser and A. Roodt, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, M45–M47 CrossRef CAS.
- N. Sinha, J.-R. Jiménez, B. Pfund, A. Prescimone, C. Piguet and O. S. Wenger, Angew. Chem., Int. Ed., 2021, 60, 23722–23728 CrossRef CAS PubMed.
- S. Jagner, E. Ljungstrom and N. G. Vannerberg, Acta Chem. Scand., 1974, A28, 623–630 CrossRef.
- C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pearson Education Limited, England, 4th edn, 2021, p. 82 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds. Part B: Applicaton in Coordination, in Organometallic and Bioinorganic Chemistry, John Wiley & Sons, Inc., New York-Chichester-Weinheim-Brisbane-Singapore-Toronto, 1997, pp. 105–113 Search PubMed.
- L. H. Jones, Inorg. Chem., 1963, 2, 777–780 CrossRef CAS.
- D. L. Wood, J. Ferguson, K. Knox and J. F. Dillon, J. Chem. Phys., 1963, 39, 890–898 CrossRef CAS.
- S. Otto, N. Scholz, T. Behnke, U. Resh-Genger and K. Heinze, Chem. – Eur. J., 2017, 23, 12131–12135 CrossRef CAS PubMed.
- J. J. Alexander and H. B. Gray, J. Am. Chem. Soc., 1968, 90, 4260–4271 CrossRef CAS.
- J. Maillard, K. Klehs, C. Rumble, E. Vauthey, M. Heilemann and A. Fürstenberg, Chem. Sci., 2021, 12, 1352–1362 RSC.
- J. Maillard, C. A. Rumble and A. Fürstenberg, J. Phys. Chem. B, 2021, 125, 9727–9737 CrossRef CAS PubMed.
- C. Forster, M. Dorn, T. Reuter, S. Otto, G. Davarci, T. Reich, L. Carrella, E. Rentschler and K. Heinze, Inorganics, 2018, 6, 86 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6.1.1, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2133575–2133578. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2dt00126h |
| This journal is © The Royal Society of Chemistry 2022 |