
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactivity of a fluorine-containing dirhodium tetracarboxylate compound with proteins†
Domenico
Loreto
 a,
Anna
Esposito
b,
Nicola
Demitri
c,
Annalisa
Guaragna
b and
Antonello
Merlino
*a
a,
Anna
Esposito
b,
Nicola
Demitri
c,
Annalisa
Guaragna
b and
Antonello
Merlino
*a
aDepartment of Chemical Sciences, University of Naples Federico II, Complesso Universitario di Monte Sant'Angelo, via Cinthia 21, 80126 Naples, Italy. E-mail: antonello.merlino@unina.it
bDepartment of Chemical, Materials and Production Engineering, University of Naples Federico II, P.le V. Tecchio 80, 80125 Naples, Italy
cElettra–Sincrotrone Trieste, S.S. 14 km 163.5 in Area Science Park, 34149 Trieste, Italy
First published on 8th February 2022
Abstract
Dirhodium complexes of general formula [Rh2(O2CR)4]L2 are a well-known class of bimetallic compounds that are used as efficient catalysts for a variety of reactions and have been shown to be potent antibacterial and anticancer agents. The catalytic and biological properties of these complexes largely depend on the nature of the bridging carboxylate ligands. Trifluoroacetate (tfa)-containing dirhodium compounds have been used to build artificial metalloenzymes upon reaction with peptides and have been shown to be more cytotoxic than dirhodium tetraacetate. However, there is no structural information on the interaction between these compounds and proteins. Here, cis-Rh2(μ-O2CCH3)2(μ-O2CCF3)2 ([cis-Rh2(OAc)2(tfa)2]) has been synthesized and its reaction with bovine pancreatic ribonuclease (RNase A) and hen egg white lysozyme (HEWL) was analyzed using a combination of different techniques, including Fluorine-19 nuclear magnetic resonance spectroscopy and macromolecular X-ray crystallography, with the aim to unveil the differences in the reactivity of tfa-containing dihrodium complexes with proteins when compared to [Rh2(OAc)4]. [cis-Rh2(OAc)2(tfa)2] and [Rh2(OAc)4] bind the N atoms of His side chains of RNase A at the axial position; however the fluorine-containing compound rapidly loses its tfa ligands, while [Rh2(OAc)4] can retain the acetate ligands upon protein binding. The reactivity of [cis-Rh2(OAc)2(tfa)2] with HEWL is slightly distinct when compared to that of [Rh2(OAc)4] under the same experimental conditions; however, both [cis-Rh2(OAc)2(tfa)2] and [Rh2(OAc)4] degrade when soaked within HEWL crystals. These results provide a structural-based guide for the design of new heterogenous chiral dirhodium/peptide and dirhodium/protein adducts with application in the fields of organic synthesis and asymmetric catalysis.
Introduction
Dirhodium(II) paddlewheel complexes of general formula [Rh2(O2CR)4]L2 (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH3–, CH3CH2–, etc.) contain a Rh(II)–Rh(II) backbone, four bridging equatorial O2CR ligands, which surround the Rh atoms, and two axial ligands (L) coordinated through a dative bond to the dirhodium core.1 These molecules have been employed as efficient organometallic catalysts in organic chemistry and asymmetric catalysis.1 They can be utilized, for example, to produce C–C (or C–heteroatom) bonds via formation of rhodium carbenoid intermediates,2 as catalysts for cyclopropanation through decomposition of diazocompounds,3 hydrosilylation of alkynes,4 C−H amination and olefin aziridination,5 ylide transformation,6 photochemical dihydrogen production7 and conversion of CO2 to HCOOH.8 Interestingly, these molecules have been shown to be potent antibacterial agents9 and exhibit cytostatic activity against Ehrlich–Lettre ascites carcinoma,10 L1210 tumors,11 P388 leukemia and sarcoma 180.12 These properties could be related to the ability of dirhodium tetracarboxylates to inhibit 50% of the transcription by T7-RNA polymerase, by binding to the enzyme,13 or to their ability to bind nucleic acid bases in vitro14,15 and induce damage of nuclear DNA in cells.16
CH3–, CH3CH2–, etc.) contain a Rh(II)–Rh(II) backbone, four bridging equatorial O2CR ligands, which surround the Rh atoms, and two axial ligands (L) coordinated through a dative bond to the dirhodium core.1 These molecules have been employed as efficient organometallic catalysts in organic chemistry and asymmetric catalysis.1 They can be utilized, for example, to produce C–C (or C–heteroatom) bonds via formation of rhodium carbenoid intermediates,2 as catalysts for cyclopropanation through decomposition of diazocompounds,3 hydrosilylation of alkynes,4 C−H amination and olefin aziridination,5 ylide transformation,6 photochemical dihydrogen production7 and conversion of CO2 to HCOOH.8 Interestingly, these molecules have been shown to be potent antibacterial agents9 and exhibit cytostatic activity against Ehrlich–Lettre ascites carcinoma,10 L1210 tumors,11 P388 leukemia and sarcoma 180.12 These properties could be related to the ability of dirhodium tetracarboxylates to inhibit 50% of the transcription by T7-RNA polymerase, by binding to the enzyme,13 or to their ability to bind nucleic acid bases in vitro14,15 and induce damage of nuclear DNA in cells.16
The catalytic and biological properties of these types of dirhodium complexes largely depend on the nature of the bridging O2CR ligands. Interestingly, the products of the reaction of dirhodium complexes with peptides can retain the catalytic activity of the metal compounds. In this frame, the interaction of [Rh2(μ-O2CCH3)4] ([Rh2(OAc)4]) and its analogue cis-Rh2(μ-O2CCH3)2(μ-O2CCF3)2 ([cis-Rh2(OAc)2(tfa)2]) (Fig. 1) with peptides has been studied with the aim to obtain useful information for the design of new artificial metalloenzymes.17 Tfa-containing dirhodium compounds have been also shown to possess altered DNA-binding ability when compared to their parent compounds18,19 and increased cytotoxic activity.20 Despite several studies have been published on the cytotoxicity of dirhodium complexes as well as the reactivity of this class of molecules with DNA,14–16 little is known on their interaction with proteins and even less on interaction of tfa-containing dirhodium compounds with these biological macromolecules.
 | ||
| Fig. 1 Structure of the paddle-wheel compound [cis-Rh2(OAc)2(tfa)2]. Axial donor ligands (L) are shown. | ||
Upon reaction of [Rh2(OAc)4] with the model protein bovine pancreatic ribonuclease (RNase A), a variety of adducts with dirhodium moieties with two-four OAc ligands is formed and the intact compound can bind the protein with the dirhodium core coordinated to the side chain of His residues at the axial site.21
Reaction of [Rh2(OAc)4] with human serum albumin leads to metal compound decomposition and oxidation of Rh(II) to Rh(III).22 Rh centres then can bind to His, Lys and Cys side chains.23 Reaction of [Rh2(OAc)4] with hen egg white lysozyme (HEWL) also leads to complex decomposition and binding to the protein of monometallic, dimetallic Rh(II)–Rh(II) and dimetallic Rh(III)–Rh(III) fragments.24 This is in contrast to what has been observed in the structure of the adduct formed upon reaction of diruthenium tetraacetate compound [Ru2(OAc)4] with HEWL, where Asp side chains replace the acetate ligands.25
It has been suggested that the reaction of [cis-Rh2(OAc)2(tfa)2] with unstructured peptides containing Asp or Glu residues in positions i and i + 4 induces the formation of α-helical structures,26 where carboxylates of the Asp or Glu side chains replace the tfa ligands. Rh2(tfa)4 has been used to heterogenize dirhodium compounds anchoring the metal complex on the surface of the SBA-15 material bearing either amine or amine and carboxyl groups27 or on biocompatible cellulose nanocrystals (CNC) via ligand exchange by carboxyl groups of the CNC surface.28 These new materials overcome the disadvantages of the homogeneous catalysts.27–29
To evaluate if the substitution of OAc with tfa ligands could affect the reactivity of dirhodium tetracarboxylate compounds with proteins, the interaction of [cis-Rh2(OAc)2(tfa)2] with RNase A and HEWL has been herein investigated using a combination of physico-chemical techniques, which include UV-vis absorption spectroscopy, circular dichroism, X-ray crystallography and 19F nuclear magnetic resonance (NMR). Results are compared with those obtained when the same proteins react with [Rh2(OAc)4].21,24
Methods
Materials
[Rh2(OAc)4], CF3COOH (TFA), hen egg white lysozyme (HEWL) and bovine pancreatic ribonuclease (RNase A) were purchased from Sigma Chemical Co and used without further purification. All the organic solvents were obtained from VWR (Milan, Italy) or Merck Life Science S.r.l (Milan, Italy) at their highest degree of purity available and were used without further purification.Chemistry
[cis-Rh2(OAc)2(tfa)2] was synthetized using the procedure previously reported (see ESI† for further details).29 The reaction was monitored by TLC (precoated silica gel plate F254, Merck, Darmstadt, Germany) and the products were detected by ultraviolet radiation exposure and iodine vapor. Column chromatography was performed using Merck Kieselgel 60 (70–230 mesh). NMR spectra were acquired on NMR spectrometers operating at 300 MHz (Bruker AVANCE, Billerica, Massachusetts, US) or 500 MHz (Varian Inova, Palo Alto, California, US). The chemical shifts δ are reported in parts per million (ppm).Crystallization, data collection and refinement
RNase A and HEWL crystals were grown at 298 K using the hanging drop vapor diffusion method. RNase A crystals were obtained using a protein concentration of 22 mg mL−1 and a reservoir consisting of 22% PEG 4 K, 0.01 M sodium citrate pH 5.1.HEWL crystals were grown in 0.10 M HEPES pH 7.5 and 2.00 M sodium formate using protein in concentration 18 mg mL−1.
Crystals of the adducts formed upon reaction of the two proteins with [cis-Rh2(OAc)2(tfa)2] were obtained exposing metal-free protein crystals to a reservoir solution containing the metal complex, using the soaking method,30 as described in the Procedure section of Table 1.
| Protein | RNase A | RNase A | RNase A | RNase A | HEWL |
|---|---|---|---|---|---|
| Crystal 1 | Crystal 2 | Crystal 3 | Crystal 4 | ||
| PDB code | 7QPW | 7QQ0 | 7QPY | 7QPZ | 7QQ1 |
| a Rmerge = ∑hΣi |I(h,i)− < I(h)>|/∑hΣi I(h,i), where I(h,i) is the intensity of the ith measurement of reflection h and <I(h)> is the mean value of the intensity of reflection h. b As expected, Rh atoms bound to His residues have lower B-factor than their Rh partner. | |||||
| Procedure | Soaking with a saturated solution of [cis-Rh2(OAc)2(TFA)2] | Soaking with a 5 mM solution of [cis-Rh2(OAc)2(TFA)2] | Soaking with a 8 mM solution of [cis-Rh2(OAc)2(TFA)2] | Soaking with a saturated solution of [cis-Rh2(OAc)2(TFA)2] | Soaking with a 5 mM solution of [cis-Rh2(OAc)2(TFA)2] |
| Soaking time | 3 days | 4 days | 3 days | 3 days (freezed just a few hours later than Crystal1) | 4 days |
| Crystallization conditions | 22% PEG 4 K, 10 mM sodium citrate at pH 5.1 | 10 mM HEPES pH 7.5 and 2.00 M sodium formate | |||
| Data collection | |||||
| Space group | C2 | C2 | C2 | C2 | P43212 |
| a (Å) | 100.35 | 100.21 | 100.39 | 101.18 | 77.59 |
| b (Å) | 32.87 | 32.57 | 32.54 | 32.95 | 77.59 |
| c (Å) | 72.97 | 72.61 | 72.51 | 73.28 | 37.94 |
| β (°) | 90.16 | 90.30 | 90.43 | 90.17 | 90.00 |
| Resolution range (Å) | 73.06–1.15 | 41.14–1.32 | 50.11–1.42 | 50.86–1.45 | 54.86–1.48 |
| (1.15–1.17) | (1.35–1.32) | (1.44–1.42) | (1.48–1.45) | (1.50–1.48) | |
| Unique reflections | 84![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 723 (4212) 723 (4212) |
54908 (2752) |
44454 (2037) |
43428 (2151) |
19342 (918) |
| Completeness (%) | 99.6 (100.0) | 99.5 (100.0) | 99.1 (92.8) | 99.9 (100.0) | 96.3 (93.0) |
| Redundancy | 6.1 (5.4) | 5.5 (5.8) | 6.0 (4.4) | 5.2 (5.6) | 23.3 (24.9) |
| Rmerge (%)a | 0.078 (0.699) | 0.138 (0.862) | 0.076 (0.523) | 0.185 (0.796) | 0.058 (1.33) |
| Rpim | 0.051 (0.479) | 0.100 (0.570) | 0.050 (0.406) | 0.135 (0.557) | 0.017 (0.380) |
| Average I/σ(I) | 10.3 (2.2) | 10.8 (2.2) | 12.7 (2.3) | 11.8 (3.1) | 29.2 (2.7) |
| CC 1/2 | 0.997 (0.738) | 0.996 (0.704) | 0.996 (0.790) | 0.988 (0.670) | 0.999 (0.950) |
| Anom. completeness (%) | 98.6 (97.2) | 98.3 (96.3) | 98.8 (91.1) | 97.9 (99.4) | 97.1 (93.3) |
| Anom. Redundancy | 3.1 (2.8) | 2.8 (3.0) | 3.1 (2.3) | 2.7 (2.9) | 12.7 (13.1) |
| Refinement | |||||
| Asymmetric unit content | Two protein molecules | Two protein molecules | Two protein molecules | Two protein molecules | One protein molecule |
| Resolution range (Å) | 73.06–1.15 | 41.14–1.32 | 50.11–1.42 | 50.86–1.45 | 39.05–1.47 |
| N. of reflections | 80792 |
52632 |
41857 |
40445 |
18518 |
| N. of reflections (test set) | 5840 | 3855 | 2955 | 2936 | 1225 |
| R-factor/R-free (%) | 13.6/16.5 | 20.9/24.3 | 17.7/21.2 | 16.2/20.2 | 18.6/22.4 |
| N. of atoms | 2598 | 2450 | 2355 | 2499 | 1250 |
| Average B-factors (Å2) | |||||
| All atoms | 18.0 | 15.9 | 20.7 | 16.1 | 21.4 |
| Rh atomsb | 15.8/22.7/19.3/16.8 | 18.4/24.3/22.3 | 17.7/22.5/16.5/19.7/17.7 | 11.8/16.7/13.0/16.7 | 54.4/73.5/59.8 |
| 20.9/21.7/20.1/30.4 | 26.8/25.2/36.8 | 21.2/21.5/38.8/17.7/22.5 | 13.7/18.5/13.5/21.3 | ||
| Rh occupancy | 0.55/0.55/0.20/0.20 | 0.40/0.40/0.40 | 0.80/0.80/0.30/0.30/0.30 | 0.75/0.75/0.30/0.30 | 0.50/0.50/0.30 |
| 0.20/0.20/0.40/0.40 | 0.40/0.40/0.40 | 0.30/0.70/0.70/0.40/0.40 | 0.30/0.30/0.55/0.55 | ||
| R.m.s. deviations | |||||
| Bond lengths (Å) | 0.018 | 0.013 | 0.013 | 0.013 | 0.012 |
| Bond angles (°) | 3.40 | 3.10 | 3.69 | 2.80 | 1.92 |
| Ramachandran statistics (Coot analysis) | |||||
| N. of residues in | |||||
| Allowed/disallowed regions | 4/2 | 8/0 | 6/1 | 6/2 | 4/0 |
Crystals from each soaking condition were scooped into a loop using 20% glycerol as a cryoprotectant and then frozen with liquid N2.
X-ray diffraction data were measured on four different crystals of RNase A exposed to [cis-Rh2(OAc)2(tfa)2] and one crystal of HEWL exposed to [cis-Rh2(OAc)2(tfa)2] at the XRD2 beamline of Elettra synchrotron in Trieste, Italy. Data sets were processed and scaled through Global Phasing autoPROC pipeline.31 Data collection statistics are reported in Table 1.
The crystal structures were solved with Phaser32 using the previously reported molecular structures of metal-free RNase A with PDB code 1JVT (chain A without waters)33 and of metal-free HEWL (PDB code 193L)34 as molecular search models. Model building and visualization of the electron density maps were carried out using Coot.35 The final structures converged to Rfactors within the range 0.136–0.209 and to Rfree within the range 0.165–0.243. Refinements have been carried out using Refmac5;36 refinement statistics are reported in Table 1. Structure 1 was refined anisotropically, the other structures were refined isotropically. Structure validations were carried out using the PDB validation server.37 In all the structures here reported the presence of Rh centre has been confirmed through anomalous difference electron density maps. Although we have done our best to interpret the electron density maps, peaks >5 sigma level can be found in the final 2Fo–Fc maps. It is hard in our experience to get a ‘perfect’ difference electron density map when metals are bound to proteins in a crystal structure.30 A list of these peaks and the reasons why we did not interpret them are reported in Table S1,† together with the clashscore of each structure from PDB validation report, as recently done by Helliwell.38 In structures 3 and 4, positive electron density peaks in the Fourier difference Fo–Fc electron density maps remain also very close to some Rh centers or close to some Rh ligands. Occupancy of Rh atoms and their ligands were fixed to the same values. Occupancy values were evaluated trying to minimize the positive and negative peaks on Rh centres in the Fourier difference Fo–Fc electron density maps. Our occupancy assignment has been further validated reporting the Rfactor and Rfree values of the structures as function of dirhodium centre occupancy. Our occupancy assignments are those corresponding to the structures that refine with the best Rfactor and Rfree values, retaining a reasonable value of the B-factors of Rh centres. In the structures of the adducts with RNase A, residues 16–22 of the two molecules in the a.u. are not well defined in the electron density maps. The presence of tfa ligand close to dirhodium centre in structure 1 was further validated refining the structure with Phenix.refine, as suggested by other authors.39
In the structure of HEWL, radiation damage was observed. Coordinates and structure factors were deposited in the Protein Data Bank (PDB codes 7QPW, 7QQ0, 7QPY and 7QPZ for the Rh/RNase A adducts, 7QQ1 for the adduct with HEWL). Figures were prepared using Pymol (http://www.pymol.org).
UV-vis absorption spectroscopy and far UV-CD spectroscopy
UV–vis absorption spectra of [cis-Rh2(OAc)2(tfa)2] were recorded at room temperature using a Jasco V750 UV–vis spectrophotometer in the 340–700 nm range. Spectra were recorded in 10.0 mM sodium citrate pH 5.1 and in 5.0 mM HEPES pH 7.5 using 0.5 mM of the metal complex in the absence and in the presence of RNase A and HEWL, respectively (protein to metal molar ratio 1:3), every 1 nm at a scan rate of 200 nm min−1.
Circular dichroism (CD) spectra of the adducts formed upon reaction of the two proteins with the metal compound upon 24 h incubation were collected in the far-UV region (from 200 to 250 nm) using a Jasco J-810 spectropolarimeter (JASCO Corp., Milan, Italy) at 25 °C. Each spectrum was obtained using a quartz cell with path length of 0.1 cm, averaging three scans, subtracting contributions from the corresponding references and converting the signal to mean residue ellipticity in units of deg cm−2 dmol−1. Spectra were registered using protein samples obtained upon incubation of the proteins with [cis-Rh2(OAc)2(tfa)2] at a 1:3 molar ratio in 10.0 mM sodium citrate buffer at pH 5.1 and in 5.0 mM HEPES pH 7.5. Other experimental settings were: 50 nm min−1 scan speed, 2.0 nm bandwidth, 0.2 nm resolution, 50 mdeg sensitivity, and 4 s response.
19F nuclear magnetic resonance
19F NMR spectra were recorded at 25 °C using Bruker AVANCE spectrometer (Billerica, Massachusetts, US) operating at 376 MHz with TOPSPIN using autolocking and auto shimming. Spectra were acquired in 10.0 mM sodium citrate pH 5.1 and in 5.0 mM HEPES pH 7.5 (10% D2O) using 0.5 mM of the metal complex in the absence and in the presence of RNase A and HEWL, respectively (protein to metal molar ratio 1:1). The spectra were referenced with pure TFA in the same buffer solutions (10.0 mM sodium citrate 10% D2O at pH 5.1 and 5.0 mM HEPES 10% D2O at pH 7.5).
Results
Synthesis and characterization of [cis-Rh2(OAc)2(tfa)2]
[cis-Rh2(OAc)2(tfa)2] was synthesized as described previously.29 Accordingly, the treatment of [Rh2(OAc)4] with an excess of TFA at room temperature provided after 2 h the desired [cis-Rh2(OAc)2(tfa)2] in a very good yield (64%). 1H and 19F NMR data well agree with those reported elsewhere (see ESI† for further details).29Stability of [cis-Rh2(OAc)2(tfa)2] in aqueous solution and in solution reactivity with proteins
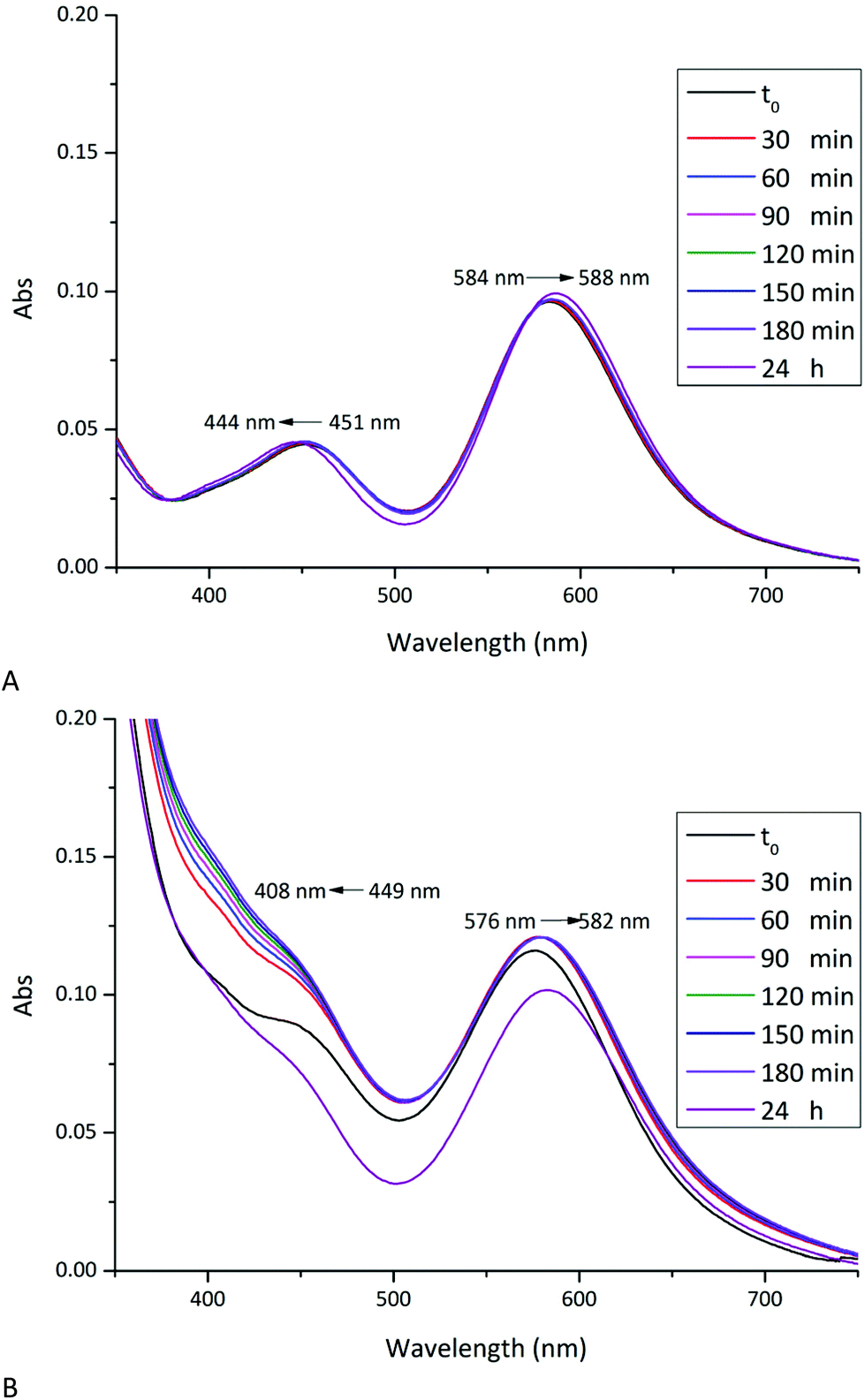
The stability of [cis-Rh2(Oac)2(tfa)2] in aqueous solvents and their reactivity with RNase A and HEWL has been investigated in solution by UV-vis absorption spectroscopy, 19F NMR spectroscopy and circular dichroism under the conditions used to obtain crystals of the two proteins (10.0 mM sodium citrate buffer pH 5.1 and 5.0 mM HEPES solution pH 7.5).UV-vis absorption spectra of [cis-Rh2(OAc)2(tfa)2] as function of time under the two different experimental conditions are reported in Fig. 2A and 3A. The spectra have been compared with those registered in the presence of RNase A (Fig. 2B) and HEWL (Fig. 3B). The spectral profiles of [cis-Rh2(OAc)2(tfa)2] in sodium citrate at pH 5.1 show two peaks in the visible region, at λmax = 451 and 584 nm, respectively (Fig. 2A). Following literature data on Rh2(OAc)421 and references therein, the band at 451 nm has been assigned to Rh2(π*) → Rh–O(σ*) transitions; while the band at 584 nm has been assigned to Rh2(π*) → Rh2(σ*) transition of the metal–metal single bond. The peak at λmax = 584 nm remains unaltered for three hours. After 24 h, a small shift of λmax from 584 to 588 nm has been observed. This suggests that the dimetallic centre is preserved for 24 h under this condition, in agreement with what was found for [Rh2(OAc)2].21 Analogously, the peak at λmax = 451 nm remains unaltered for three hours, while a blue shift of λmax from 451 nm up 444 nm has been observed upon 24 h. This shift can be attributed to the exchange process of dirhodium ligands with solvent molecules. Indeed, tfa is a labile ligand and can be rapidly lost in solution.
 | ||
| Fig. 2 Time course UV-vis spectra of [cis-Rh2(OAc)2(tfa)2] in 10.0 mM sodium citrate buffer pH 5.1 monitored over time (up to 24 h) in the absence (panel A) and in the presence (panel B) of RNase A (protein to metal molar ratio 1:3). | ||
 | ||
| Fig. 3 Time course UV-vis spectra of [cis-Rh2(OAc)2(tfa)2] in 5.0 mM HEPES solution pH 7.5, monitored over time (up to 24 h) in the absence (panel A) and in the presence (panel B) of HEWL (protein to metal molar ratio 1:3). | ||
In the presence of RNase A (Fig. 2B), the UV-vis spectrum of [cis-Rh2(OAc)2(tfa)2] is different when compared to that observed in the absence of the protein. Even in this spectrum two peaks in the visible region have been observed, at λmax = 449 nm and 576 nm. The spectrum significantly changes with time and, already after 30 minutes, a blue-shift of the peak at 449 nm is found. This peak almost disappears upon 24 h. This variation is associated with a red-shift of the peak from λmax = 576 up to 582 nm.
These data demonstrate that in the presence of RNase A the complex exchanges its tfa ligands even more rapidly than in the absence of the protein. This finding suggests a possible interaction of one or more residue side chains with the dirhodium compound. Data also indicate that in the presence of RNase A the Rh–Rh bond is preserved over time, suggesting that the interaction of the compound with the protein does not affect the dimetallic bond.
UV-vis spectrum of [cis-Rh2(OAc)2(tfa)2] in 5.0 mM HEPES buffer pH 7.5 shows two peaks at λmax = 452 and 580 nm (Fig. 3A). Under this experimental condition, significant variations of the spectrum profile are observed just after 30 min. Over time, a blue-shift of the first peak from 452 nm to 408 nm and a concomintant increase in the absorbance are observed. A blue-shift of the second peak from λmax = 580 nm to 576 nm, associated with a decrease of the absorbance, is also found. Superimposition of the spectra indicates the presence of an isosbestic point at 481 nm, which suggests the existence of an equilibrium between two species in solution. Extensive variations of the spectral profiles, as found in this case, suggest substantial structural variations for the [cis-Rh2(OAc)2(tfa)2] complex, which should maintain the Rh-Rh bond at least for three hours, but should degrade over time. In this respect, it is interesting to note that Rh2(OAc)4 has proved to be stable for 24 h under the same conditions.21
In the presence of HEWL (Fig. 3B), UV-vis spectrum of [cis-Rh2(OAc)2(tfa)2] is different from that of the compound alone, registered under the same experimental condition. At t0, two peaks at λmax = 446 and 577 nm are found. Also in this case, significant variations of the spectra are observed over time: a red-shift of λmax from 577 nm to 595 nm and an increase in the absorbance of the peak at 446 nm is found; with the peak at 446 nm that disappears over time. Superimposition of the spectra shows, also in this case, the presence of an isosbestic point at 621 nm. These large spectral variations suggest that in the presence of the protein the metal compound can degrade and that the Rh–Rh bond can be broken, as observed in the structure of the adduct formed upon reaction of HEWL with Rh2(OAc)4.21 The differences in the spectra of the compound collected in the absence and in the presence of the protein in 5.0 mM HEPES pH 7.5 suggest the existence of an interaction between [cis-Rh2(OAc)2(tfa)2] and HEWL.
To further validate these results we have taken advantages by the finding that the 19F nucleus is an excellent probe for the identification and quantification of different chemical environments. Thus, the exchange of dirhodium ligands has been evaluated by collecting 19F NMR spectra as function of time. Spectra of [cis-Rh2(OAc)2(tfa)2] alone and in presence of RNase A (Fig. 4A) and HEWL (Fig. 4B) were recorded under the two different experimental conditions (10.0 mM sodium citrate pH 5.1 and 5.0 mM HEPES pH 7.5 respectively) and compared with pure TFA in the same buffer solutions. Data reveal that tfa ligands are exchanged by solvent molecules over the time. Particularly, considering the spectra of the compounds alone, a complete release of tfa ligands in solution is observed for both conditions after 24 h. On the other hand, in presence of RNase A and HEWL, tfa exchange is faster with a complete release of tfa ligands after only 5 minutes (Fig. 4A) and 2 h (Fig. 4B) respectively. These data are consistent with those obtained by UV-vis absorption spectroscopy suggesting a possible interaction of dirhodium compounds with the proteins.
 | ||
| Fig. 4 Time course 19F NMR spectra of [cis-Rh2(OAc)2(tfa)2] monitored over time (up to 24 h) in 10.0 mM sodium citrate buffer pH 5.1 (10% D2O) in the absence and in the presence of RNase A (protein to metal ratio 1:1) (panel A) and in 5.0 mM HEPES buffer pH 7.5 (10% D2O) in the absence and in the presence of HEWL (protein to metal molar ratio 1:1)(panel B). | ||
CD spectra of RNase A and HEWL incubated for 24 h with [cis-Rh2(OAc)2(tfa)2] in 1:3 protein/metal compound molar ratio have been then collected to evaluate if the binding of the compounds in solution could affect the secondary structure of the proteins (Fig. S1†). CD spectra of RNase A and HEWL in the presence of the compound show a slight change of molar ellipticity, although the overall features of the spectra suggest that the proteins remain folded in the presence of the metal compound.
X-ray structures of the adduct with RNase A
Subsequently, four different structures of the adducts formed upon reaction of [cis-Rh2(OAc)2(tfa)2] with RNase A have been solved by X-ray crystallography. Each structure contains two independent protein molecules in the asymmetric unit (molecules A and B). The structures have been obtained from RNase A crystals treated with [cis-Rh2(OAc)2(tfa)2] for a different soaking time and using slightly different procedures (Table 1). X-ray diffraction data have been collected at high resolution (from 1.15 to 1.45 Å resolution) at the XRD2 beamline of Elettra synchrotron in Trieste, Italy. Data collection and refinement statistics are reported in Table 1. The overall structures of RNase A in the adduct with [cis-Rh2(OAc)2(tfa)2] are very similar to each other and superimpose well to that of the metal-free protein and to that in the adduct with [Rh2(OAc)4] (Fig. 5). Root mean square deviation (rmsd) of Cα atoms from the structure of the metal-free protein (PDB code 1JVT)33 is within the range 0.19–0.45 Å; rmsd from the structure of the adduct with [Rh2(OAc)4] is within the range 0.07–0.45 Å (Table S2†), suggesting that no significant structural change is induced by metal coordination.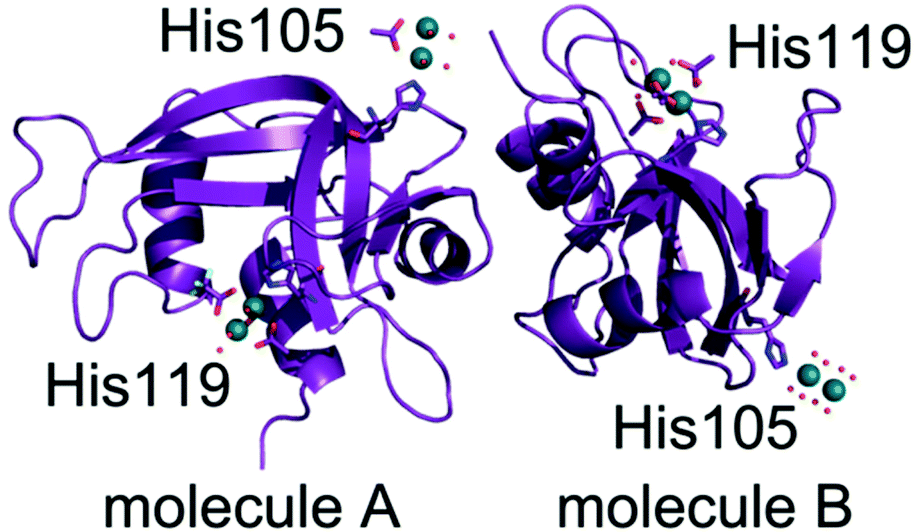 | ||
| Fig. 5 Overall structure of the two independent protein molecules (molecules A and B) in the asymmetric unit of the crystal of the adduct formed in the reaction of [cis-Rh2(OAc)2(tfa)2] with RNase A solved at 1.42 Å resolution (structure 3 in Table 1). Rh atoms are in green. | ||
In the majority of the structures, consistently with 19F NMR spectra, diffraction data indicate that dirhodium cores lose the tfa ligands.
In three out of the four structures of the Rh/protein adduct formed upon reaction of RNase A with [cis-Rh2(OAc)2(tfa)2] (structures 1, 2 and 4 in Table 1), dirhodium binding sites have been found at level of the side chains of His105 of molecule A and His119 of both protein molecules A and B of the asymmetric unit. In the other structure (structure 3 in Table 1), His105 and His119 of both protein molecules in the asymmetric unit are metalated.
In the structures 1 and 2 His119 of molecule A adopts two distinct conformations and one of the two conformations is bound to a dirhodium centre. In the structure 1 the dirhodium centre has two acetate ions, one tfa and two water molecules as equatorial ligands and a water molecule as axial ligand (occupancy = 0.55) (Fig. 6A). The same dirhodium fragment with highest occupancy (occupancy = 0.80 and 0.75, respectively) has been found in the structures 3 and 4, where a single conformation of the His has been observed (Fig. S2A and S2B†). In the structure 2, in this site, the dirhodium centre coordination sphere is completed by one acetate and six water molecules as equatorial ligands (Fig. S2C†).
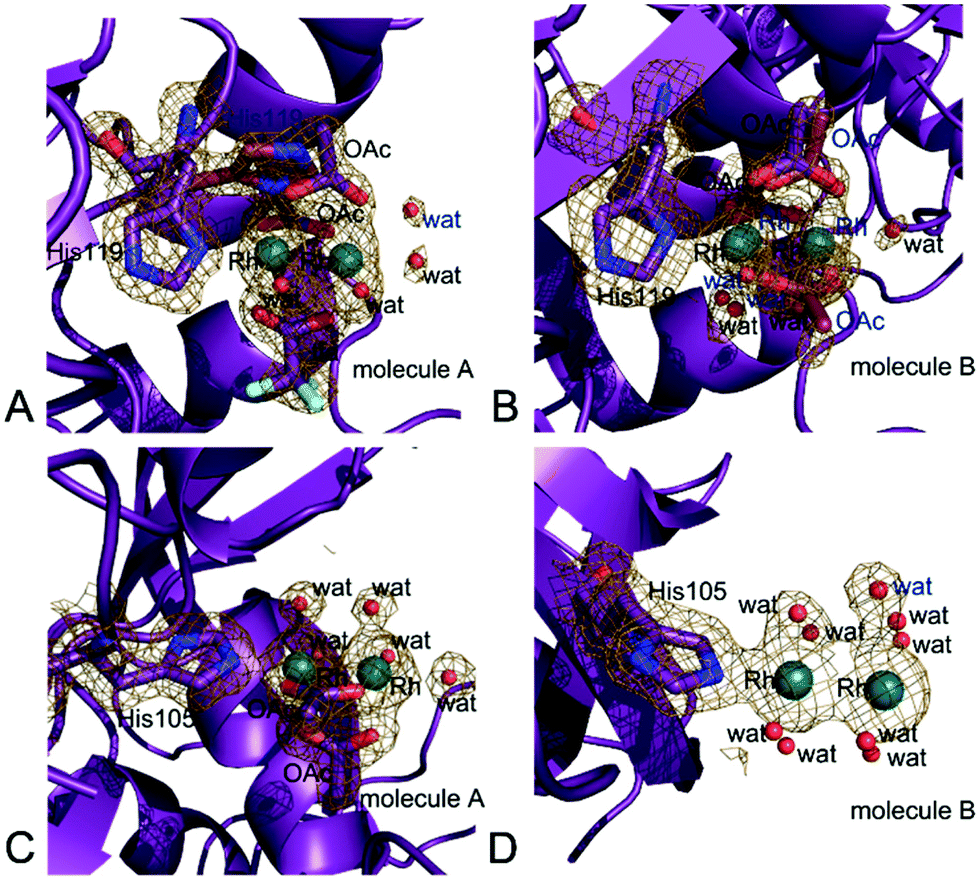 | ||
| Fig. 6 Details of the Rh binding site close to (A) His119 of molecule A in structure 1, (B) His119 of molecule B in structure 1. (C) His105 of molecule A in structure 4, (D) His105 of molecule B in structure 3. In panel A, the two conformations of His119 are shown. 2Fo–Fc electron density maps are contoured at 0.7σ. | ||
In molecule B, His119 adopts a single conformation in all the structures. In the structures 1 (Fig. 6B), 3 (Fig. S3A†) and 4 (Fig. S3B†) the dirhodium centre in this site adopts two alternative conformations and have two acetate ions, and four water molecules as equatorial ligands. In the structure 2, in this site, the dirhodium core has one acetate and six water molecules as equatorial ligands (Fig. S3C†).
Close to His105 of molecule A different dirhodium centres have been observed: in the structures 1 (Fig. S4A†) and 3 (Fig. S4B†), the dirhodium centres have one acetate and six water molecules as equatorial ligands, in the structure 2 (Fig. S4C†) only water molecules are found close to the Rh atoms, while in the structure 4 dirhodium unit has two acetate ions and four water molecules as equatorial ligands (Fig. 6C).
His105 of molecule B is metalated only in structure 3. At this site, only water molecules are observed (Fig. 6D).
A summary of the dirhodium ligands and of geometries of the dirhodium compound fragments observed in these structures is reported in Tables S3 and S4.† The values well agree with those obtained by a search in the Cambridge Structure Database on tetrakis(2-R-carboxylato-O,O′)-di-rhodium(II) complexes (average Rh–Rh distance = 2.402(24) Å, average Rh–O distance = 2.036(13) Å, average Rh–N distance = 2.256(69) Å, average Rh–X distance = 2.31(13) Å (query on CSD Version 5.43 – November 2021).
Overall the Rh binding sites on the RNase A structure are those that have been found in the previously solved structure of the adduct of the protein with Rh2(OAc)4,21 although in this latter case acetate ligands can be fully retained in the dirhodium binding sites.
X-ray structure of the adduct with HEWL
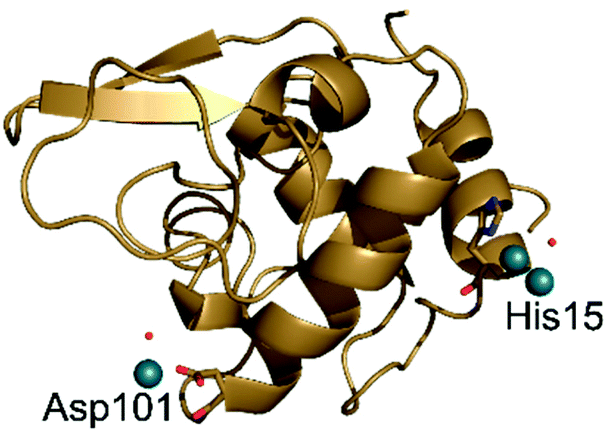
When [Rh2(OAc)4] reacts with HEWL it degrades under a variety of experimental conditions.24 In crystals of HEWL exposed to [Rh2(OAc)4.] in 10 mM HEPES at pH 7.5 and 2.00 M sodium formate, monometallic Rh centres are found close to the side chains of Arg14/His15, Lys13 and C-terminal carboxylate, Asp101, Asn93 and Lys96, Lys33.24The structure of HEWL derived from crystals soaked in a solution of [cis-Rh2(OAc)2(tfa)2] in 10 mM HEPES at pH 7.5 and 2.00 M sodium formate was refined at 1.48 Å resolution. The overall structure of HEWL in this crystal is almost identical to that of metal free protein (Fig. 7). Rmsd from the structure with PDB code 193L is as low as 0.15 Å. The structure of the protein is also very similar to that observed in the adduct with [Rh2(OAc)4] obtained under the same experimental conditions24 (rmsd = 0.13 Å).
 | ||
| Fig. 7 Overall structure of the adduct formed upon reaction of [cis-Rh2(OAc)2(tfa)2] with HEWL. Rh atoms are in green. | ||
However, in the adduct of HEWL with [cis-Rh2(OAc)2(tfa)2], Rh-containing fragments bound to the protein are different when compared to those found in the HEWL adduct with [Rh2(OAc)4].24 In the structure of HEWL in the presence of [cis-Rh2(OAc)2(tfa)2], a dirhodium core is coordinated to the ND1 atom of His15, with occupancy equal to 0.50 (Fig. 8A), while only monometallic fragments were found upon reaction of the protein with [Rh2(OAc)4].24 However it is possible that at His15 site dirhodium can degrade over time, leaving only monometallic fragments bound to the His side chain. This hypothesis is in line with the finding that at this site the coordination sphere of the dirhodium centre is not well defined in the electron density map. In our model, only a water molecule has been modelled close to the dirhodium center, but it is highly probable that the other ligands are not observed in the electron density map due to conformation disorder. Additional support to the idea that the dirhodium core can degrade in the crystal of the adduct formed upon reaction of HEWL with [cis-Rh2(OAc)2(tfa)2] comes from the presence of a monometallic Rh centre site in this structure, close to the side chain of Asp101 (occupancy = 0.30) (Fig. 8B).
 | ||
| Fig. 8 Details of the Rh binding site close to (A) His15 and (B) Asp101 in the structure of the Rh/HEWL adduct obtained upon reaction of the protein with [cis-Rh2(OAc)2(tfa)2]. 2Fo–Fc electron density maps are contoured at 0.7σ. | ||
Refinements with higher occupancy value at this Rh binding site produce structures with a slightly lower Rfactor value, but unrealistic B-factors (>90 Å2). This value is far from that of the OD2 atom of the Asp101 that coordinates the Rh centre (about 34 Å2).
Conclusions
Dirhodium paddlewheel compounds have been used in a variety of fields ranging from homogeneous catalysis to biological chemistry.1–12 The replacement of equatorial carboxylate ligands surrounding the dirhodium core strongly affects the properties of the dimetallic compounds. It has been shown that [cis-Rh2(OAc)2(tfa)2] can be used to build dirhodium/peptide adducts, inducing the formation of helical structures.17 Furthermore, it has been demonstrated that changing one bridging ligand on compounds with the general formula Rh2(μ-L)-(HNOCCF3)3 alters the rate of DNA-binding by greater than 100-fold with μ-L = tfa > OAc > trifluoroacetamidate.19 Thus, systematic changes of equatorial ligands of the dirhodium centre can tune the reactivity of these complexes towards biological macromolecules, leading to the potential development of more selective biological targeting agents.Despite several studies have been published on the cytotoxicity of dirhodium complexes as well as the reactivity of this class of molecules with DNA,14–16,18 little is known on the interaction of tfa-containing dirhodium compounds with proteins.
Here, the interaction of [cis-Rh2(OAc)2(tfa)2] with the model proteins RNase A and HEWL has been investigated by using a combination of experimental techniques. UV-vis absorption and 19F NMR spectra of the compound, in the absence and in the presence of the two proteins, indicate that the ligand exchange is faster when the compound is treated with HEWL and RNase A, thus suggesting that a reaction between the proteins and the metal compound occurs in solution. Notably, 19F NMR spectra reveal that [cis-Rh2(OAc)2(tfa)2] loses its tfa ligands after just a few minutes in the presence of RNase A and after 2 hours in the presence of HEWL. Structural analyses of five new crystal structures of adducts obtained upon reaction of the Rh compound with RNase A and HEWL allow us to delineate some general trends in the reactivity of this compound with proteins. The complex binds proteins without altering their overall conformation, as observed in the formation of all the known Rh/protein adduct23 and, more generally, in the case of the adducts formed upon reaction of metallodrugs with proteins.40,41 The dirhodium carboxylate compound reacts with proteins similarly to [Rh2(OAc)4]: dirhodium centres preferentially bind the side chains of His residues at the axial coordination site, but they can also bind Asp side chains. However, while in the final Rh/protein adducts obtained upon reaction of proteins with [Rh2(OAc)4] the dimetallic compound can retain its OAc ligands,21 tfa moieties are lost after incubation of proteins with [cis-Rh2(OAc)2(tfa)2]. Furthermore, depending on the experimental conditions both compounds can degrade.
Axial binding of protein residue side chain to dirhodium core can significantly alter the reactivity and catalytic activity of the metal compounds. In this respect it should be recalled that it has been already shown that dirhodium carbene reactivity is “tunable” by axial ligation opposite to the Rh–C bond42 and that dirhodium complexes with improved catalytic properties have been designed by incorporating ligands that shield the Rh core from axial coordination by external molecules.43 The origin of the effects of axial ligand coordination to dirhodium centre is related to structural and electronic changes of the Rh–Rh core arising from axial ligation.44
Overall, these results demonstrate that tfa-containing dirhodium tetracarboxylates can react with proteins similarly to their OAc-containing analogues although they can form different products of reaction, i.e. protein adducts with different Rh ligands and thus different potential reactivity. These data are important for the design of experiments devoted to the preparation of artificial metalloenzymes based on the reaction of peptide/protein adducts with dirhodium compounds. Our data also enrich the repertoire of structures of protein with Rh-containing compounds that are rarely reported in literature, as recently evidenced by a systematic review on this topic.23 Indeed, data also suggest that proteins and protein crystals can be used as immobilization platforms of dirhodium centres for heterogeneous catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Elettra staff for technical assistance.Notes and references
- (a) R. Hrdina, Dirhodium(II,II) Paddlewheel Complexes, Eur. J. Inorg. Chem., 2021, 6, 501–528 CrossRef; (b) R. Paulissenen, H. Reimlinger, E. Hayez, A. J. Hubert and P. Teyssie, Transition metal catalysed reactions of diazocompounds-II insertion in the hydroxylic bond, Tetrahedron Lett., 1973, 14, 2233–2236 CrossRef.
- (a) N. R. Candeias, C. A. M. Afonso and P. M. P. Gois, Making Expensive Dirhodium(II) Catalysts Cheaper: Rh(II) Recycling Method, Org. Biomol. Chem., 2012, 10, 3357–3378 RSC; (b) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Catalytic Carbene Insertion into C–H Bonds, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed.
- H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Stereoselective Cyclopropanation Reactions, Chem. Rev., 2003, 103(4), 977–1050 CrossRef CAS PubMed.
- (a) M. P. Doyle, K. G. High, C. L. Nesloney, T. W. Clayton Jr. and J. Lin, Rhodium(II) Perfluorobutyrate Catalyzed Hydrosilylation of 1-alkynes. Trans Addition and Rearrangement to Allylsilanes, Organometallics, 1991, 10, 1225–1126 CrossRef CAS; (b) M. P. Doyle, G. A. Devora, A. O. Nefedov and K. G. High, Addition/elimination in the Rhodium(II) Perfluorobutyrate Catalyzed Hydrosilylation of 1-Alkenes. Rhodium Hydride Promoted Isomerization and Hydrogenation, Organometallics, 1992, 11, 549–555 CrossRef CAS.
- P. Müller and C. Fruit, Enantioselective Catalytic Aziridinations and Asymmetric Nitrene Insertions into CH Bonds, Chem. Rev., 2003, 103, 2905–2919 CrossRef PubMed.
- W. Zeghida, C. Besnard and J. Lacour, Rhodium(II)-Catalyzed One-Pot Four-Component Synthesis of Functionalized Polyether Macrocycles at High Concentration, Angew. Chem., 2010, 122, 7411–7414 CrossRef.
- (a) J. Huang, C. J. Gallucci and C. Turro, Panchromatic Dirhodium Photocatalysts for Dihydrogen Generation with Red Light, Chem. Sci., 2020, 11, 9775–9783 RSC; (b) H. J. Sayre, A. Millet, K. R. Dunbar and C. Turro, Photocatalytic H2 Production by Dirhodium(II,II) Photosensitizers with Red Light, Chem. Commun., 2018, 54, 8332–8334 RSC; (c) J. Xie, C. Li, Q. Zhou, W. Wang, Y. Hou, B. Zhang and X. Wang, Large Improvement in the Catalytic Activity Due to Small Changes in the Diimine Ligands: New Mechanistic Insight into the Dirhodium(II,II) Complex-Based Photocatalytic H2 Production, Inorg. Chem., 2012, 51(11), 6376–6384 CrossRef CAS PubMed.
- (a) H. D. Manamperi, S. E. Witt and C. Turro, Selective Electrocatalytic Conversion of CO2 to HCOOH by a Cationic Rh2(II,II) Complex, ACS Appl. Energy Mater., 2019, 2, 7306–7314 CrossRef CAS; (b) S. E. Witt, T. A. White, Z. Li, K. R. Dunbar and C. Turro, Cationic Dirhodium(II,II) Complexes for the Electrocatalytic Reduction of CO2 to HCOOH, Chem. Commun., 2016, 52, 12175–12178 RSC.
- S. Pramodini Devi, R. K. Hemakumar Singh, W. Sujata and D. D. Joshi, Synthesis, DNA binding and antimicrobial studies on rhodium(II) complexes of dicyandiamide, Nucleosides, Nucleotides Nucleic Acids, 2020, 39(6), 923–942 CrossRef PubMed.
- A. Erck, L. Rainen, J. Whileyman, I.-M. Chang, A. P. Kimball and J. Bear, Studies of rhodium(II) carboxylates as potential antitumor agents, Proc. Soc. Exp. Biol. Med., 1974, 145(4), 1278–1283 CrossRef CAS PubMed.
- R. A. Howard, A. P. Kimball and J. L. Bear, Mechanism of action of tetra-μ-carboxylatodirhodium(II) in L1210 tumor suspension culture, Cancer Res., 1979, 39, 2568–2573 CAS.
- J. L. Bear., Rhodium compounds for antitumor use. In Precious Met. Proc. Jnt. Precious Met. Inst., Conf, 1986, pp. 337–344 Search PubMed.
- K. Sorasaenee, P. K.-L. Fu, A. M. Angeles-Boza, K. R. Dunbar and C. Turro, Inhibition of Transcription in Vitro by Anticancer Active Dirhodium(II) Complexes, Inorg. Chem., 2003, 42, 1267–1271 CrossRef CAS PubMed.
- H. T. Chifotides and K. R. Dunbar, Interactions of Metal-Metal-Bonded Antitumor Active Complexes with DNA Fragments and DNA, Acc. Chem. Res., 2005, 38, 146–156 CrossRef CAS PubMed.
- (a) M. Kang, H. T. Chifotides and K. R. Dunbar, D NMR Study of the DNA duplex d(CTCTC*A*ACTTCC), d(GGAAGTTGAGAG) cross-linked by the antitumor-active dirhodium(II,II) unit at the cytosine adenine step, Biochemistry, 2008, 47, 2265–2276 CrossRef CAS PubMed; (b) S. U. Dunham, H. T. Chifotides, S. Mikulski, A. E. Burr and K. R. Dunbar, Covalent Binding and Interstrand Cross-Linking of Duplex DNA by Dirhodium(II,II) Carboxylate Compounds, Biochemistry, 2005, 44, 996–1003 CrossRef CAS PubMed.
- J. D. Aguirre, A. M. Angeles-Boza, A. Chouai, J. P. Pellois, C. Turro and K. R. Dunbar, Live Cell Cytotoxicity Studies: Documentation of the Interactions of Antitumor Active Dirhodium Compounds with Nuclear DNA, J. Am. Chem. Soc., 2009, 131, 11353–11360 CrossRef CAS PubMed.
- A. N. Zaykov and Z. T. Ball, A General Synthesis of Dirhodium Metallopeptides as MDM2 Ligands, Chem. Commun., 2011, 47, 10927–10929 RSC.
- T. Chifotides, J. M. Koomen, M. Kang, S. E. Tichy, K. R. Dunbar and D. H. Russell, Binding of DNA Purine Sites to Dirhodium Compounds Probed by mass spectrometry, Inorg. Chem., 2004, 43, 6177–6187 CrossRef PubMed.
- S. U. Dunham, T. S. Remaley, B. S. Moore, D. L. Evans and S. U. Dunham Isolation, Characterizationand DNA Binding Kinetics of Three dirhodium(II,II) carboxyamidate complexes: Rh2(μ-L)(HNOCCF3)3 where L = [OOCCH3]-, [OOCCF3]-, or [HNOCCF3]-, Inorg. Chem., 2011, 50, 3458–3463 CrossRef CAS PubMed.
- A. M. Angeles-Boza, H. T. Chifotides, J. D. Aguirre, A. Chouai, P. K.-L. Fu, K. R. Dunbar and C. Turro, Dirhodium(II,II) Complexes: Molecular Characteristics that Affect in Vitro Activity, J. Med. Chem., 2006, 49, 6841–6847 CrossRef CAS PubMed.
- G. Ferraro, A. Pratesi, L. Messori and A. Merlino, Protein Interactions of Dirhodium Tetraacetate: a Structural Study, Dalton Trans., 2020, 49(8), 2412–2416 RSC.
- (a) L. Trynda and F. Pruchnik, Interactions of Tetra-Ì-acetatodirhodium(II) with Human Serum Albumin, J. Inorg. Biochem., 1995, 58, 69–77 CrossRef CAS PubMed; (b) B. P. Esposito, E. de Oliveira, S. B. Zyngier and R. Najjar, Effects of Serum Albumin in some Biological Properties of Rhodium(II) Complexes, J. Braz. Chem. Soc., 2000, 11, 447–452 CrossRef CAS.
- D. Loreto and A. Merlino, The Interaction of Rhodium Compounds with Proteins: A Structural Overview, Coord. Chem. Rev., 2021, 442, 213999 CrossRef CAS.
- D. Loreto, G. Ferraro and A. Merlino, Unusual Structural Features in the Adduct of Dirhodium Tetraacetate with Lysozyme, Int. J. Mol. Sci., 2021, 22(3), 1496 CrossRef CAS PubMed.
- L. Messori, T. Marzo, R. N. Fernandes Sanches, H.-U. Rehman, D. de Oliveira Silva and A. Merlino, Unusual Structural Features in the Lysozyme Derivative of the Tetrakis(acetato)chlorido Diruthenium(II,III) Complex, Angew. Chem., Int. Ed. Engl., 2014, 53(24), 6172–6175 CrossRef CAS PubMed.
- Z. T. Ball, Designing Enzyme-like Catalysts: a Rhodium(II) Metallopeptide Case Study, Acc. Chem. Res., 2013, 46(2), 560–570 CrossRef CAS PubMed.
- J. Liu, P. B. Groszewicz, Q. Wen, A. S. L. Thankamony, B. Zhang, U. Kunz, G. Sauer, Y. Xu, T. Gutmann and G. Buntkowsky, Revealing Structure Reactivity Relationship in Heterogenized Dirhodium Catalysts by Solid-State NMR Techniques, J. Phys. Chem. C, 2017, 121(32), 17409–17416 CrossRef CAS.
- J. Liu, A. Plog, P. Groszewicz, L. Zhao, Y. Xu, H. Breitzke, A. Stark, R. Hoffmann, T. Gutmann and K. Zhang, Design of a Heterogeneous Catalyst Based on Cellulose Nanocrystals for Cyclopropanation: Synthesis and Solid-State NMR Characterization, Chem. – Eur. J., 2015, 21, 12414–12420 CrossRef CAS PubMed.
- Y. Lou, T. P. Remarchuk and E. J. Corey, Catalysis of Enantioselective [2 + 1]-Cycloaddition Reactions of Ethyl Diazoacetate and Terminal Acettylenes Using Mixed-Ligand Complexes of the Series Rh2(RCO2)n(L*4−n). Stereochemical Heuristics for Ligand Exchange and Catalyst Synthesis, J. Am. Chem. Soc., 2005, 127, 14223–14230 CrossRef CAS PubMed.
- I. Russo Krauss, G. Ferraro, A. Pica, J. A. Márquez, J. R. Helliwell and A. Merlino, Principles and methods used to grow and optimize crystals of protein–metallodrug adducts, to determine metal binding sites and to assign metal ligands, Metallomics, 2017, 9, 1534–1547 CrossRef CAS PubMed.
- C. Vonrhein, C. Flensburg, P. Keller, A. Sharff, O. Smart, W. Paciorek, T. Womack and G. Bricogne, Data Processing and Analysis with the autoPROC Toolbox, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 293–302 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, Phaser Crystallographic Software, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- L. Vitagliano, A. Merlino, A. Zagari and L. Mazzarella, Reversible Substrate-Induced Domain Motions in Ribonuclease A, Proteins, 2002, 46, 97–104 CrossRef CAS PubMed.
- J. M. C. Vaney, S. Maignan, M. Ries-Kautt and A. Ducriux, High-resolution Structure (1.33(A) of a HEW Lysozyme Tetragonal Crystal Grown in the APCF Apparatus. Data and Structural Comparison with a Crystal Grown Under Microgravity from SpaceHab-01 Mission, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1996, 52, 505–517 CrossRef PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Features and Development of Coot, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- G. N. Murshudov, P. Skubák, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, REFMAC5 for the Refinement of Macromolecular Crystal Structures, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 355–367 CrossRef CAS PubMed.
- H. Berman, K. Henrickm and H. Nakamura, Announcing the Worldwide Protein Data Bank, Nat. Struct. Biol., 2003, 10, 980R CrossRef PubMed.
- A. Brink and J. R. Helliwell, Formation of a highly dense tetra-rhenium cluster in a protein crystal and its implications in medical imaging, IUCrJ, 2019, 6, 695–702 CrossRef CAS PubMed.
- J. R. Helliwell, The crystal structures of the enzyme hydroxymethylbilane synthase, also known as porphobilinogen deaminase, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2021, 77, 388–398 CrossRef CAS PubMed.
- D. Loreto, G. Ferraro and A. Merlino, Protein-metallodrugs interactions: Effects on the overall protein structure and characterization of AuRu and Pt binding sites, Int. J. Biol. Macromol., 2020, 163, 970–976 CrossRef CAS PubMed.
- A. Merlino, Recent advances in protein metalation: structural studies, Chem. Commun., 2021, 57(11), 1295–1307 RSC.
- A. F. Trindade, J. A. S. Coelho, C. A. M. Afonso, L. F. Veiros and P. M. P. Gois, Fine Tuning of Dirhodium(II) Complexes: Exploring the Axial Modification, ACS Catal., 2012, 2, 370–383 CrossRef CAS.
- R. Ohnishi, H. Ohta, S. Mori and M. Hayashi, Cationic dirhodium complexes Bridged by 2-Phosphinopyridines Having an exquisitely Positioned Axial Shielding Group: A Molecular Design for Enhancing the Catalytic Activity of the Dirhodium Core, Organometallics, 2021, 40, 2678–2690 CrossRef CAS.
- C. J. Laconsay, A. Pla-Quintana and D. J. Tantillo, Effects of Axial Solvent Coordination to Dirhodium Complexes on the Reactivity and Selectivity in C−H Insertion Reactions: A Computational Study, Organometallics, 2021, 40, 4120–4132 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2dt00082b |
| This journal is © The Royal Society of Chemistry 2022 |