
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand exchange among iodine(I) complexes†
Shilin
Yu
 * and
Jas S.
Ward
* and
Jas S.
Ward
Department of Chemistry, University of Jyvaskyla, 40014, Jyväskylä, Finland. E-mail: shandianyu1989@163.com
First published on 17th February 2022
Abstract
A detailed investigation of ligand exchange between iodine(I) ions in [N⋯I⋯N]+ halogen-bonded complexes is presented. Ligand exchange reactions were conducted to successfully confirm whether iodine(I) complex formation, via the classical [N⋯Ag⋯N]+ to [N⋯I⋯N]+ cation exchange reaction from their analogous Ag+ complexes, could be determined solely by using 1H NMR spectroscopy. In instances where the formation of the iodine(I) complex was unclear or in low yield by the traditional cation exchange reaction, a ligand exchange reaction was used to form the desired iodine(I) complexes in a quantitative manner. Mixing two homoleptic [N⋯I⋯N]+ iodine(I) complexes in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio was found to undergo a statistical ligand exchange, with 1H NMR studies showing that the preferred formation of the relative heteroleptic [N1⋯I⋯N2]+ complexes increases with greater differences in the Lewis basicities of two XB acceptors of the complexes involved.
:1 ratio was found to undergo a statistical ligand exchange, with 1H NMR studies showing that the preferred formation of the relative heteroleptic [N1⋯I⋯N2]+ complexes increases with greater differences in the Lewis basicities of two XB acceptors of the complexes involved.
Introduction
The halogen-bonding (XB) interaction, a type of non-covalent interaction, only started to draw attention in the 1990s due to the work by the groups of Metrangolo and Resnati,1,2 and Legon3,4 among others.5,6 Nowadays, because of the unique features of halogen bonding, such as directionality, a tuneable interaction strength, and an understanding of the utility of XB, it is becoming a booming topic in many fields, for instance supramolecular chemistry,7–9 medicinal chemistry,10–13 materials sciences,14–17 and organic synthesis and catalysis.18–22The halogen(I) complex, formed with a halenium ion (X+, where X = Cl, Br, I) and two Lewis bases in a linear fashion [L⋯X⋯L]+, is considered as one of the strongest halogen-bond complexes, along with those XB complexes using perfluorocarbons (PFHCs)1,2,23 or N-halosaccharin24,25 as XB donors. Barluenga's eponymous reagent, [I(Py)2][BF4] (Py = pyridine),26 which can be viewed as the archetypal halogen(I) complex, is known in synthetic chemistry as an iodinating and oxidising agent.27–30 Among the five halogens, iodine(I), bromine(I), and chlorine(I) have been reported to form three-centre-four-electron (3c-4e) bonds as [L⋯X⋯L]+ complexes, whereas fluorine(I) has been studied and was found to prefer conventional halogen bonds of the form L+–F⋯L,31 and astatine has been found to behave as a stronger halogen-bond donor than iodine both experimentally and computationally.32 The iodine(I) and bromine(I) complexes, predominantly of the [N⋯X⋯N]+ motif (X = Br, I),33 are stable at ambient conditions and can be prepared by the classical [N⋯Ag⋯N]+ to [N⋯I⋯N]+ cation exchange reaction from their analogous Ag+ complexes (Scheme 1a). The geometries of homoleptic [N⋯X⋯N]+ complexes (X = Cl, Br, I) have been extensively studied by Erdélyi and coworkers.31,34–36 Considering the halogen(I) complexes possible from the myriad of potential ligands, the reported halogen(I) complexes in the CSD database are still limited.37–42 The difficulty in obtaining single crystals of halogen(I) complexes sometimes makes it challenging to prove their formation, unless more definitive methods were available in solution. It has been reported that the 15N NMR chemical shift change of the coordinated nitrogen can indicate the formation of an [N⋯I⋯N]+ complex,43 though not always conclusively, as the 15N NMR chemical shift changes of the nitrogen atom in some protonated ligands also fall within similar expected ranges.44 Whilst the [N⋯Ag⋯N]+ to [N⋯I⋯N]+ cation exchange reaction is reliable for many substituted pyridines,42 it is not effective for all ligand types. With this classic method of preparation, the [N⋯I⋯N]+ complexes of some ligands can be afforded in low yield, but in conjunction with the protonated ligands as side products. These side products further complicate attempts to identify them solely based on the 1H NMR data, especially when the formed protonated ligands are often more amenable to recrystallisation, which can be a false indicator that the reactions have failed to produce the desired [N⋯I⋯N]+ complexes.
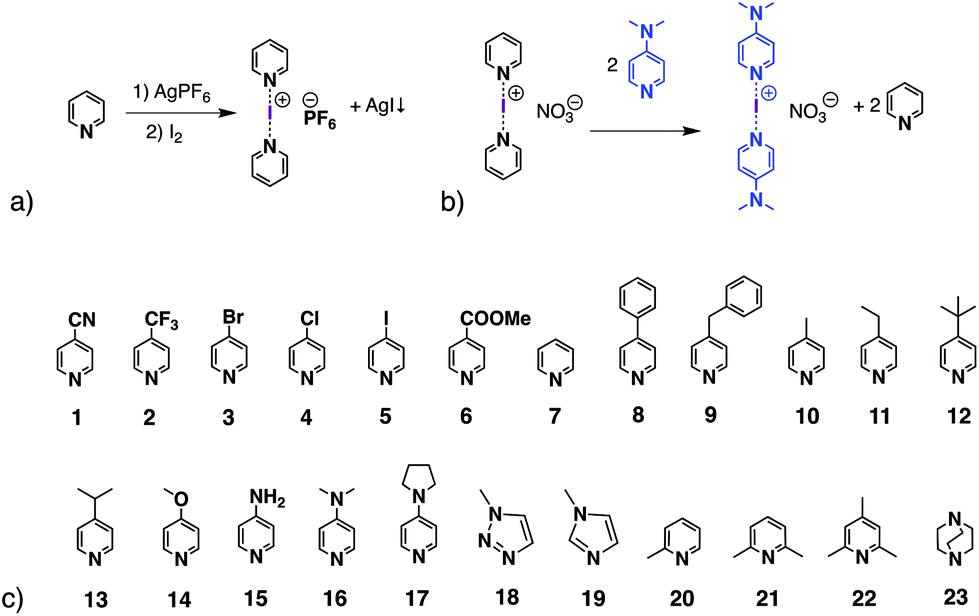 | ||
| Scheme 1 (a) Synthesis of iodine(I)–nitrogen, [N⋯I⋯N]+, complexes, using pyridine as a general example; (b) ligand exchange reaction of pyridine for DMAP about an [I]+, reported by Dutton et al.; (c) list of ligands 1–23 used in this work. | ||
In 2013 Dutton and co-workers reported the formation of the [I(DMAP)2][NO3] iodine(I) complex (DMAP = 4-dimethylaminopyridine; Scheme 1b) in a 71% yield, achieved by adding the free DMAP ligand into a solution of [I(Py)2][NO3].45 Furthermore, the UV-Vis kinetics of the dissociation of several pyridine-iodine(I) complexes in the presence of DMAP have been studied by Erdélyi and co-workers.36 Using the strategy of molecular clefts, heteroleptic halogen(I) complexes have been prepared,46 however, only very recently was the first unrestrained heteroleptic iodine(I) complex confirmed in the solid state,38 and following that was further studied in solution by the Erdélyi group.47 They concluded that the preference of iodine(I) was to form complexes with the more basic pyridine, which proceeded at slower ligand exchange rates; the mixing of two homoleptic halogen-bonded iodine(I) complexes leads to a statistical distribution of the pyridine ligands across the iodine(I) ions. Recently the utility of ligand exchange of discrete iodine(I) complexes has been demonstrated as an effective means to synthesise rare examples of iodine(I) macrocycles.48
Herein we present a comprehensive investigation of the ligand exchange reactions in iodine(I) complexes, the identity of which can be confirmed solely by 1H NMR spectroscopy using a strong Lewis base (e.g., DMAP). The exchange was determined to be quantitative in the conversion, despite iodine(I) complexes being used as the electrophilic reagent. The efficacy of the ligand exchange process was found to be strongly dependent on the difference of Lewis basicities between the two ligands involved. In some cases, the ligand exchange yields a more informative synthetic methodology when compared with the traditional [N⋯Ag⋯N]+ to [N⋯I⋯N]+ cation exchange reaction, as the ligand exchange process is more readily monitored. In addition, it was found that when the difference of the XB acceptor basicities in the iodine(I) system increases, a higher percentage of the heteroleptic iodine(I) complexes were formed, with greater covalent bond character between the iodine(I) and the XB acceptor possessing the higher Lewis basicity.
Results and discussion
Ligand exchange of iodine(I) complexes
These studies tested 23 ligands (Scheme 1c) to thoroughly investigate the ligand exchange behaviour of iodine(I) complexes. The free ligand DMAP (16) and iodine(I) complex [I(7)2]PF6 were chosen to represent the ligand exchange behaviour in the solution state, as the 1H NMR chemical shifts of complexes [I(16)2]PF6, [I(7)2]PF6 and [I(7)(16)]PF6 are known,38 and the formation of [I(16)2]NO3 through a ligand exchange reaction of [I(7)2]NO3 and 16 was already performed by Dutton and co-workers.45 The symmetrical iodine(I) complex [I(7)2]PF6 itself was prepared via the traditional [N⋯Ag⋯N]+ to [N⋯I⋯N]+ cation exchange reaction (Fig. 1a). As shown in Fig. 1b–e, with the addition of the free ligand DMAP (16; 0.5, 1.0, 1.5, and 2.0 equivalents) into the solution of [I(7)2]+, the 1H NMR spectra showed the gradual decrease of the peaks of [I(7)2]+ until it completely disappeared. Concomitantly a new set of signals for 7 appeared (Fig. 1b), increased (Fig. 1c), decreased (Fig. 1d), and finally disappeared (Fig. 1e), with the chemical shifts in agreement with the previously reported values for asymmetrical complex [I(7)(16)]+ (green trace).38 The green peaks of 16 in [I(7)(16)]+, which follows the same trend as the green 7, also supports that assignment. The signals for [I(16)2]+ (blue trace) appeared later than the [I(7)(16)]+ signals, and kept increasing until the complete consumption of the [I(7)(16)]+ species (green trace). The signals of 7 (black trace) increased and moved towards the chemical shifts observed for 7 as a free ligand, due to the increase of 7 being released in the reaction caused by the ligand exchange (for more detailed information, see Fig. S18†). When the ligand exchange reaches completion, the exchange yield of iodine(I) cation is 100%. From the above analysis, it is clear that before the full ligand exchange, the heteroleptic iodine(I) complex was first formed as the intermediate.38,47,49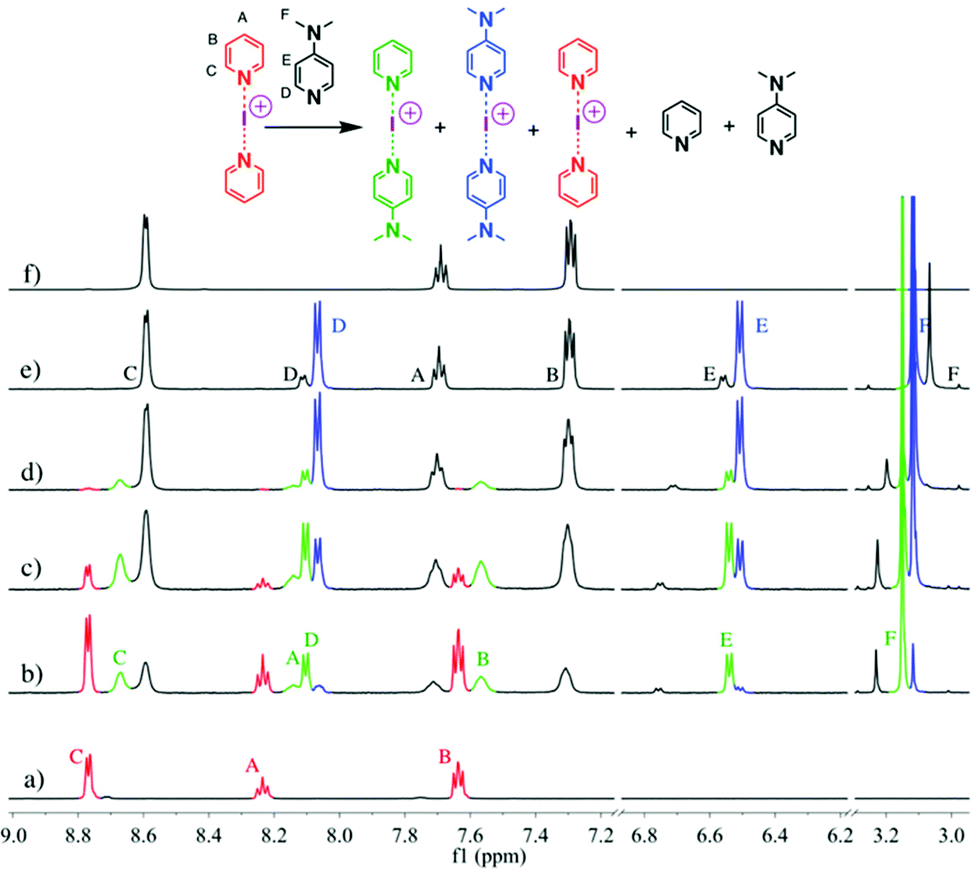 | ||
| Fig. 1 1H NMR spectra (500 MHz, CD2Cl2, 30 °C) of pure [I(7)2]PF6 (a) and [I(7)2]PF6 in the presence of 0.5 (b), 1.0 (c), 1.5 (d), and 2.0 equivalents (e) of 16, and of pure 7 (f). | ||
The signals of 7 in [I(7)(16)]+ (green trace) has shifted towards upfield when compared to [I(7)2]+; whilst the signals of 16 in [I(7)(16)]+ (green trace) has shifted downfield when compared to [I(16)2]+. This can be explained as the asymmetry of the N−I−N bond in [I(7)(16)]+ that causes a different extent of electron donation from 7 in [I(7)2]+ and 16 in [I(16)2]+ as compared to [I(7)(16)]+. As the iodine(I) cation gets more electrons from 16, it will pull less from 7.
Ligands 1–6, 8–14 and 18–23 were also used to prepare the relevant iodine(I) complexes via the [N⋯Ag⋯N]+ to [N⋯I⋯N]+ cation exchange reaction (see Section 2 in ESI†). The formation of [I(L)2]+ (L = 8–14, 18–19) are clean and were further confirmed through ligand exchange reactions. The reactions of [I(L)2]+ (L = 1–6, 20) were not particularly clean, but the ligand exchange process can monitor what components are present and which parts belong to the desired iodine(I) complexes. [I(10)2]PF6 was used to form iodine(I) complexes [I(15)2]+ and [I(17)2]+via ligand exchange reactions.
As shown in Fig. S45, S50† and Fig. 2a, the reactions of [I(L)2]+ (L = 21, 22, 23) from the traditional [N⋯Ag⋯N]+ to [N⋯I⋯N]+ cation exchange method did not proceed cleanly, while via the ligand exchange reaction between ligand 21 or 22 and the complex [I(18)2]+, which had a less basic ligand than 21 and 22, clean complexes of [I(21)2]+ and [I(22)2]+ were readily afforded (Fig. S46, S48, S51 and S53†). Additionally, using the same method, the complex of 23, [I(23)2]+, was also obtained (Fig. 2b, S55 and S56†), and further confirmed by the single crystal X-ray structure analysis (Fig. S67†).
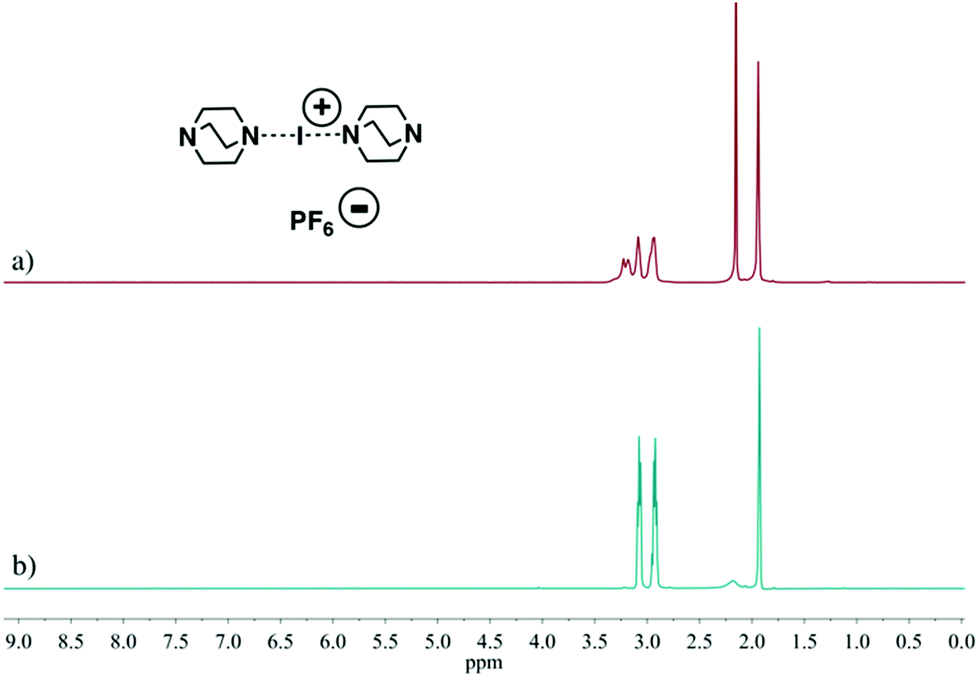 | ||
| Fig. 2 The 1H NMR spectra (500 MHz, CD3CN, 30 °C) of the crude mixture of reaction [I(23)2]PF6 by the traditional [N⋯Ag⋯N]+ to [N⋯I⋯N]+ cation exchange reaction (a) and the pure iodine(I) complex [I(23)2]PF6 prepared from the ligand exchange reaction between [I(18)2]PF6 and free ligand 23 (b). | ||
The efficacy of the iodine(I) ion transfer strongly depends on the Lewis basicities of the two involved ligands, as well as the stoichiometries of the iodine(I) complex and the free ligand. For example, with 20 eq. of 10 being introduced to 1 eq. of [I(16)2]PF6, no trace of [I(10)2]PF6 was observed. However, the addition of 2 eq. of free ligand 16 into 1 eq. of [I(10)2]PF6 causes the full iodine(I) ion transfer, whilst 3 eq. of the free ligand 16 were required with 1 eq. of [I(14)2]PF6 to exclusively yield [I(16)2]PF6.
It is noteworthy that, despite its reactive nature, the iodine(I) ion transfer was quantitative among all cases studied in this work, indicating the strong coordinative character of the iodine(I) ion. Among all of the above cases, the chemical shift of 16 in the homoleptic and multiple heteroleptic complexes, in each individual case, does not change during the whole process of ligand exchange when the ratios of the different species are changing. This is due to the slow exchange rate of ligand 16 in the iodine(I) complexes, as well as other experimental factors.47
Homoleptic versus heteroleptic preference in iodine(I) complexes
In a previous study,47 two homoleptic iodine(I) complexes, [I(2)2]+ and [I(7)2]+, were mixed together in different ratios to evaluate the resulting ratios of [I(2)2]+, [I(7)2]+, and freshly generated [I(2)(7)]+, by integration of the corresponding 1H NMR signals. From this they concluded that the ligand exchange proceeds due to a dominant entropic contribution. Herein, the difference of basicities of the two XB acceptors involved were investigated to see if they have an influence on the distribution of the homoleptic and heteroleptic species observed. The molar ratios of [I(L)2]+, [I(16)2]+, and [I(L)(16)]+ in solution were analysed by mixing complexes of [I(16)2]+ and [I(L)2]+ (L = 1, 7, 12, 15, 18) in a 1:1 ratio (Table 1). Surprisingly, as the Lewis basicities of ligand L decreases in the order of 15 (4-NH2Py) > 12 (4-tBuPy) > 7 (Py) > 18 (triazole) > 1 (4-CNPy), the percentage of the heteroleptic species [I(L)(16)]+ among the iodine(I) complexes was found to increase. Meanwhile, the signals of the free ligands L and 16 start to appear and increase for L = 7, 18, 1, suggesting that the heteroleptic complexes [I(L)(16)]+ were getting less stable upon increasing the difference of basicities of the two Lewis bases involved. The slightly pink colours of those reactions also support that, as elemental iodine is generated by the reaction.
:1 complexes [I(16)2]+ and [I(L)2]+ (L = 1, 7, 12, 15, 18); the comparisons of the 15N NMR chemical shift values of pyridinic nitrogen in 16 for the homoleptic complex [I(16)2]+, and the heteroleptic complexes [I(L)(16)]+ (L = 1, 7, 12, 15, 18)
| Comp. | [I(L)2]+:[I(L)(16)]+:[I(16)2]+ |
Δδa | ΔΔδb |
|---|---|---|---|
| a The 15N NMR chemical shift change of pyridinic nitrogen of DMAP upon complexation. b ΔΔδ = Δδ([I(L)(16)]+) − Δδ([(16)2]+). c Performed in CD3CN. d Protonated or free ligands excluded. | |||
| [I(16)2]+ | — | −107.6 (−110.7)c | 0 |
| [I(15)(16)]+ | 1:2.2:1 (52.4%)c |
−112.7c | −2.0c |
| [I(12)(16)]+ | 1:3.2:1 (61.5%) |
−123.6 | −16.3 |
| [I(7)(16)]+ | 1:4.8:1 (70.6%) |
−128.3 | −21.0 |
| [I(18)(16)]+ | 1:9.5:1 (82.6%) |
−135.0 | −27.4 |
| [I(1)(16)]+ | 1:23.5:1.2 (91.4%)d |
−143.4 | −35.8 |
The 15N NMR chemical shift values based on the 1H–15N HMBC measurements of the pyridinic nitrogen of DMAP (16) in the heteroleptic complexes [I(L)(16)]+ (L = 1, 7, 12, 15, 18) are also summarised in Table 1. The complexation-induced chemical shift change, ΔδN, defined as δ(15Ncomplex) − δ(15NDMAP), gets larger when decreasing the Lewis basicity of ligand L in the same order as previously described (15 > 12 > 7 > 18 > 1; vide supra), indicating the increasing electron density around the pyridinic nitrogen of DMAP (16). Even larger ΔδN values, up to −135.0 ppm were observed for [I(18)(16)]+ (cf. [I(1)(16)]+ = −143.4 ppm), than those previously reported for the stabilised O−I−N complex F3CC(O)(OI–DMAP), which had a value of −129 ppm.44 The Δδ values of the pyridinic nitrogen atoms of the weaker Lewis bases of the heteroleptic complexes [I(15)(16)]+, [I(12)(16)]+ and [I(7)(16)]+ were also recorded (Table S1†). The deshielding of the 15N NMR signals of weaker Lewis bases in [I(15)(16)]+, [I(12)(16)]+, and [I(7)(16)]+ as compared to the respective homoleptic complexes, [I(15)2]+, [I(12)2]+ and [I(7)2]+, were as expected. The values for [I(18)(16)]+ and [I(1)(16)]+ were unable to be determined due to the signal coalescence (Fig. S64 and S66†).
The single crystal of [I(18)(16)]PF6 was successfully obtained from dichloromethane upon slow evaporation (Fig. 3), only the second solid-state example of an unrestrained heteroleptic [N1⋯I⋯N2]+ complex, which had two crystallographically independent molecules in the asymmetric unit cell. A comparison of the I–N bond lengths between [I(18)(16)]+ (2.335(8)/2.340 Å for N(18)–I, 2.187(8)/2.163(8) Å for I–N(16)), and the symmetric analogues [I(18)2]+ (2.238(2) Å)50 and [I(16)2]+ (2.236(3)/2.251(3) Å),38 reveal that the I–N bond length in [I(18)(16)]+ is shortened. Consistent with the strictly linear geometry of the homoleptic 3c-4e [N⋯I⋯N]+ halogen bonds, the angle of the N–I–N in [I(18)(16)]+ falls within the same range with values of 178.1(4)° and 177.0(4)°.37,38 However, the single crystal structure of the only other reported heteroleptic [N1⋯I⋯N2]+ complex, [I(7)(16)]+, showed the reversed I–N bond lengths changes.38 Compared with I–N bond lengths in [I(7)2]+ and [I(16)2]+, the I–N bond length of 16 in [I(7)(16)]+ was slightly elongated, and the I–N bond length of 7 in [I(7)(16)]+ was slightly shortened in the asymmetric complex. This is probably due to the smaller difference of Lewis basicities between 7 and 16, as well as the influence of the unique packing interactions.
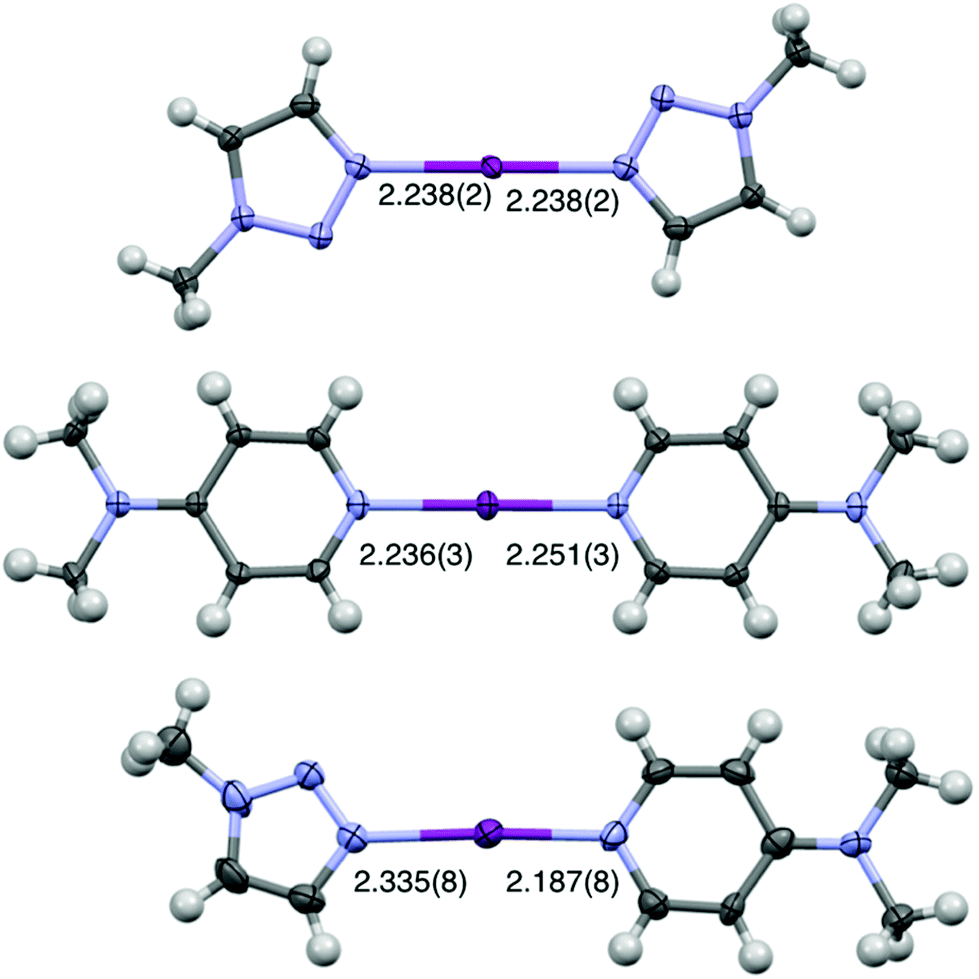 | ||
| Fig. 3 The X-ray crystal structures of (from top to bottom) [I(18)2]PF6, [I(16)2]PF6, and [I(18)(16)]PF6 (PF6 anions omitted for clarity; all lengths in Å; thermal displacement parameters at 50% probability).38,50 The second crystallographically independent molecule of [I(18)(16)]PF6 has been omitted for clarity (I–N(16) = 2.163(8) Å, I–N(18) = 2.340(8) Å). | ||
The DFT geometries of [I(L)(16)]+ (L = 1, 7, 12, 15, 16, 18) (Fig. 4) were calculated using the experimentally determined coordinates for the purpose of comparison, as had been reported previously for O−I−N halogen-bonded complexes.44,51 These calculations were performed to the M06-2X (def2-TZVP) level of theory, including a non-polar (DCM) solvent model, using the Spartan20 software package.52 The calculated models were all in good agreement with the experimentally determined structures obtained by X-ray crystallography, including the one for [I(18)(16)]+ (Fig. 3 and S70†).44,51 As shown in Fig. 4, upon decreasing the Lewis basicity of ligand L in the order of 16 (DMAP) > 15 (4-NH2Py) > 12 (4-tBuPy) > 7 (Py) > 18 (triazole) > 1 (4-CNPy), the N(L)–I bond length increases and the I–N(16) bond length decreases. This is consistent with the solution results (1H and 15N NMR chemical shift changes of ligand 16 in the relative complexes [I(L)(16)]+; Table 1). It should be noted that despite the increasing asymmetry of the N1–I–N2 bond, the N1–I–N2 angle remains strictly linear (Table S2†) even for [I(1)(16)]+, enforced by the nature of the 3c-4e [N1⋯I⋯N2]+ halogen bond in all halogen(I) complexes.36–40,42,50,53
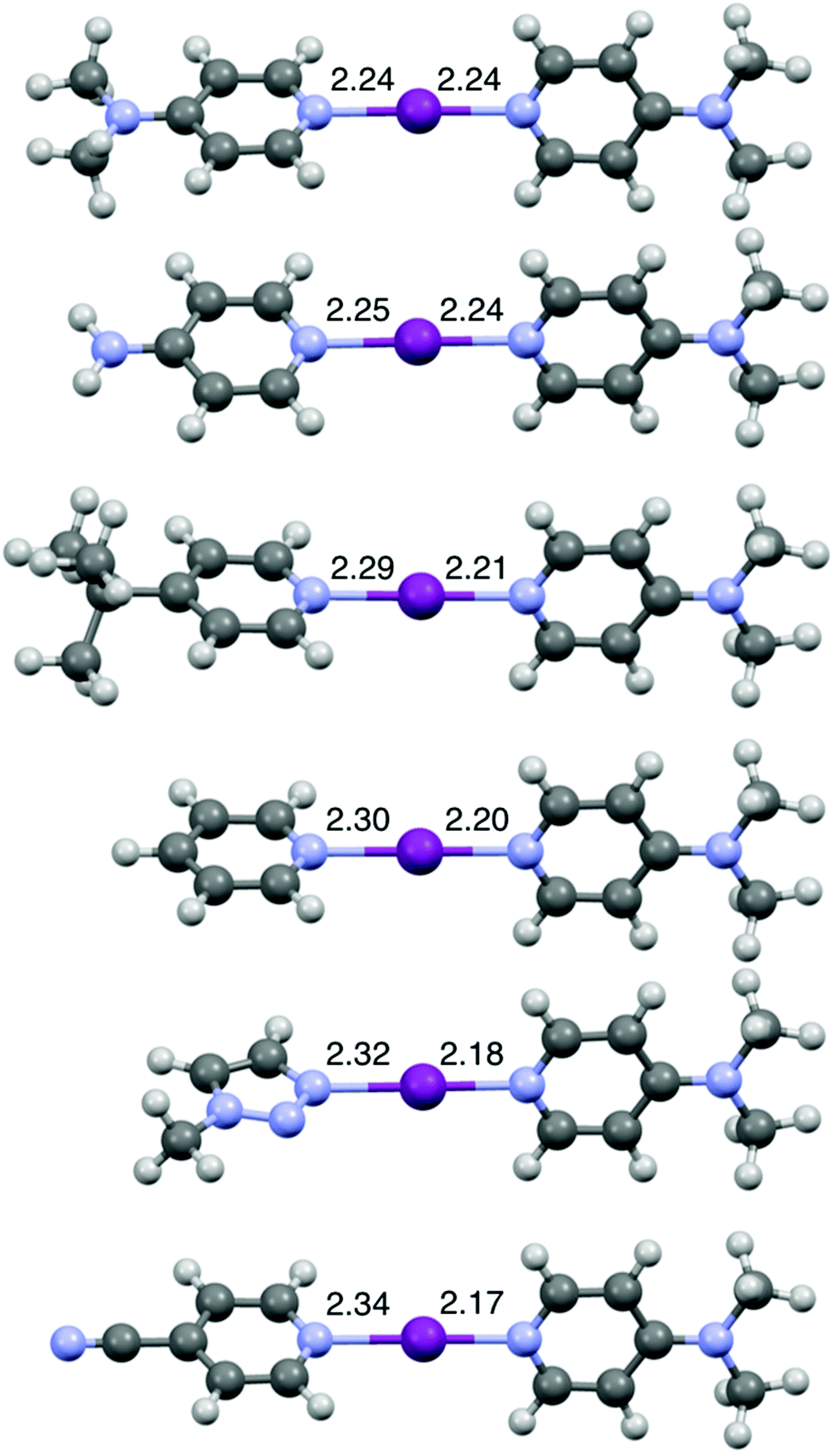 | ||
| Fig. 4 The computationally generated structures for [I(16)2]+ and [I(L)(16)]+ (L = 15, 12, 7, 18, 1) (in order of decreasing Lewis basicity from top to bottom), with the annotated N–I lengths in Å. | ||
Mechanism of ligand exchange
Similar to proposed mechanisms for halogen(I) transfer using three-centre iodine(I) complexes in organic synthesis,54–59 the ligand exchange process in iodine(I) complexes can be addressed (Scheme 2). The reversible dissociation of the three-centre complex (e.g., [I(7)2]+) causes the formation of free ligand 7 and an N-iodopyridinium cation [I-7]+ as the intermediate, which is subsequently captured by the strong XB acceptor (e.g., 16), leading to the formation of a heteroleptic halogen-bonded complex, viz. [I(7)(16)]+. Following this reasoning, [I(7)(16)]+ reversibly dissociates into free ligand 7 and an N-iodopyridinium cation [I-16]+, which interacts with another XB acceptor 16, forming a new homoleptic iodine complex [I(16)2]+. It should be noted that the dynamic character of the bis(acyloxy)iodates(I)-related axles in rotaxanes was also studied by the group of Šindelář, in which the carbonyl hypoiodite was analogously suggested to be the key intermediate in the transfer of the iodine(I) ion among different axles.60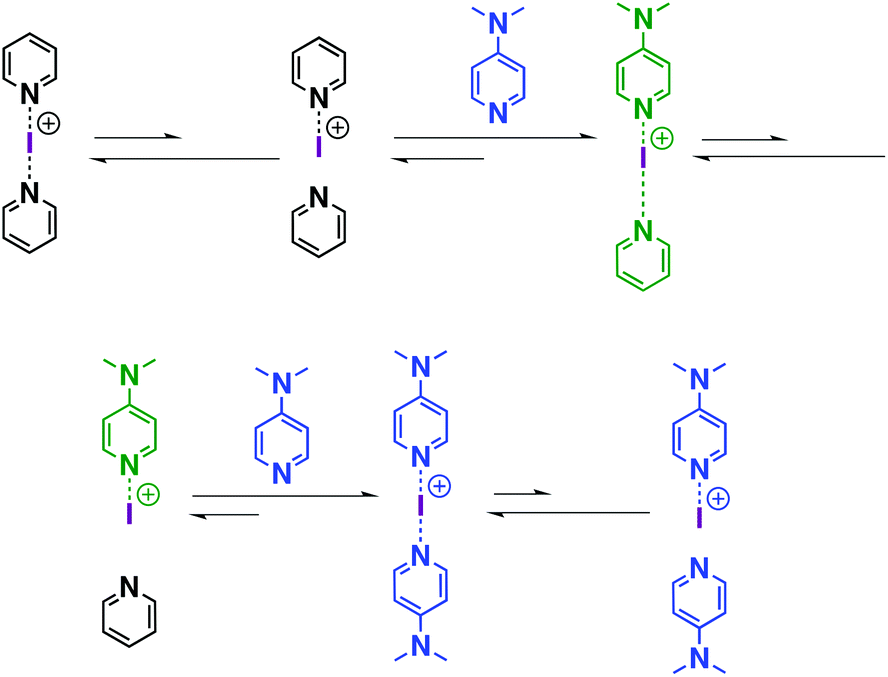 | ||
| Scheme 2 The proposed mechanism of the ligand exchange process. | ||
Conclusions
In this work, the iodine(I) cation was found to be an extraordinarily strong halogen bond donor that can be transferred among different ligands in a quantitative manner. A straightforward ligand exchange approach to synthesise and distinguish the formation of iodine(I) complexes exclusively by 1H NMR spectroscopy has been developed. This ligand exchange method can also be used to produce iodine(I) complexes that are not viable through the traditional [N⋯Ag⋯N]+ to [N⋯I⋯N]+ cation exchange reaction. The ligand exchange process represents a milder synthetic approach toward the synthesis of iodine(I) complexes when compared to the now ubiquitous cation exchange process. In stoichiometric mixtures of two homoleptic iodine(I) complexes, as the difference in basicities of the XB acceptors increases, the preference toward heteroleptic products increases, along with the increasing asymmetry of the [N1⋯I⋯N2]+ bond. The extensive studies of the ligand exchange process described in this work provides more information on the coordinative character of the iodine(I) ion, and will definitely aid in the construction of iodine(I)-based supramolecular structures, a field which is just beginning to gain traction, and is now picking up speed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Professor Kari Rissanen (University of Jyvaskyla, Finland) and Máté Erdélyi (Uppsala University, Sweden) for useful discussions, as well as the Magnus Ehrnrooth Foundation (J. S. W.) and the University of Jyvaskyla, Finland for financial support.Notes and references
- V. Amico, S. V. Meille, E. Corradi, M. T. Messina and G. Resnati, J. Am. Chem. Soc., 1998, 120, 8261–8262 CrossRef CAS.
- A. Farina, S. V. Meille, M. T. Messina, P. Metrangolo, G. Resnati and G. Vecchio, Angew. Chem., Int. Ed., 1999, 38, 2433–2436 CrossRef CAS PubMed.
- A. C. Legon, Angew. Chem., Int. Ed., 1999, 38, 2686–2714 CrossRef PubMed.
- A. C. Legon, in The Interaction of Dihalogens and Hydrogen Halides with Lewis Bases in the GasPhase: An Experimental Comparison of the Halogen Bond and the Hydrogen Bond BT – Halogen Bonding: Fundamentals and Applications, ed. P. Metrangolo and G. Resnati, Springer Berlin Heidelberg, Berlin, Heidelberg, 2008, pp. 17–64 Search PubMed.
- M. Fourmigué and P. Batail, Chem. Rev., 2004, 104, 5379–5418 CrossRef PubMed.
- K. Rissanen, CrystEngComm, 2008, 10, 1107–1113 RSC.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, Angew. Chem., Int. Ed., 2016, 55, 14033–14036 CrossRef CAS PubMed.
- L. Turunen, U. Warzok, C. A. Schalley and K. Rissanen, Chem, 2017, 3, 861–869 CAS.
- L. A. Hardegger, B. Kuhn, B. Spinnler, L. Anselm, R. Ecabert, M. Stihle, B. Gsell, R. Thoma, J. Diez, J. Benz, J.-M. Plancher, G. Hartmann, D. W. Banner, W. Haap and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 314–318 CrossRef CAS PubMed.
- Q. Zhang, Z. Xu and W. Zhu, J. Chem. Inf. Model., 2017, 57, 22–26 CrossRef CAS PubMed.
- A. M. S. Riel, R. K. Rowe, E. N. Ho, A.-C. C. Carlsson, A. K. Rappé, O. B. Berryman and P. S. Ho, Acc. Chem. Res., 2019, 52, 2870–2880 CrossRef CAS PubMed.
- W. Runguphan, X. Qu and S. E. O'Connor, Nature, 2010, 468, 461–464 CrossRef CAS PubMed.
- A. Sun, J. W. Lauher and N. S. Goroff, Science, 2006, 312, 1030LP–1034LP CrossRef PubMed.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed.
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2013, 5, 42–47 CrossRef CAS PubMed.
- H. Wang, H. K. Bisoyi, L. Wang, A. M. Urbas, T. J. Bunning and Q. Li, Angew. Chem., Int. Ed., 2018, 57, 1627–1631 CrossRef CAS PubMed.
- S. Schindler and S. M. Huber, in Halogen Bonds in Organic Synthesis and Organocatalysis BT – Halogen Bonding II: Impact on Materials Chemistry and Life Sciences, ed. P. Metrangolo and G. Resnati, Springer International Publishing, Cham, 2015, pp. 167–203 Search PubMed.
- D. A. Petrone, J. Ye and M. Lautens, Chem. Rev., 2016, 116, 8003–8104 CrossRef CAS PubMed.
- J. Wolf, F. Huber, N. Erochok, F. Heinen, V. Guérin, C. Y. Legault, S. F. Kirsch and S. M. Huber, Angew. Chem., Int. Ed., 2020, 59, 16496–16500 CrossRef CAS PubMed.
- R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS.
- R. L. Sutar, E. Engelage, R. Stoll and S. M. Huber, Angew. Chem., Int. Ed., 2020, 59, 6806–6810 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- O. Makhotkina, J. Lieffrig, O. Jeannin, M. Fourmigué, E. Aubert and E. Espinosa, Cryst. Growth Des., 2015, 15, 3464–3473 CrossRef CAS.
- R. Puttreddy, J. M. Rautiainen, T. Mäkelä and K. Rissanen, Angew. Chem., Int. Ed., 2019, 58, 18610–18618 CrossRef CAS PubMed.
- J. Barluenga, M. Trincado, E. Rubio and J. M. González, Angew. Chem., Int. Ed., 2003, 42, 2406–2409 CrossRef CAS PubMed.
- J. Barluenga, J. M. González, M. A. Garcia-Martin, P. J. Campos and G. Asensio, J. Chem. Soc., Chem. Commun., 1992, 1016–1017 RSC.
- J. Ezquerra, C. Pedregal, C. Lamas, J. Barluenga, M. Pérez, M. A. García-Martín and J. M. González, J. Org. Chem., 1996, 61, 5804–5812 CrossRef CAS.
- G. Espuña, G. Arsequell, G. Valencia, J. Barluenga, M. Pérez and J. M. González, Chem. Commun., 2000, 1307–1308 RSC.
- J. Barluenga, F. González-Bobes, M. C. Murguía, S. R. Ananthoju and J. M. González, Chem. – Eur. J., 2004, 10, 4206–4213 CrossRef CAS PubMed.
- A. Karim, M. Reitti, A.-C. C. Carlsson, J. Gräfenstein and M. Erdélyi, Chem. Sci., 2014, 5, 3226–3233 RSC.
- N. Guo, R. Maurice, D. Teze, J. Graton, J. Champion, G. Montavon and N. Galland, Nat. Chem., 2018, 10, 428–434 CrossRef CAS PubMed.
- K. Rissanen and M. Haukka, in Halonium Ions as Halogen Bond Donors in the Solid State [XL2]Y Complexes BT – Halogen Bonding II: Impact on Materials Chemistry and Life Sciences, ed. P. Metrangolo and G. Resnati, Springer International Publishing, Cham, 2015, pp. 77–90 Search PubMed.
- A.-C. C. Carlsson, J. Gräfenstein, J. L. Laurila, J. Bergquist and M. Erdélyi, Chem. Commun., 2012, 48, 1458–1460 RSC.
- A.-C. C. Carlsson, J. Gräfenstein, A. Budnjo, J. L. Laurila, J. Bergquist, A. Karim, R. Kleinmaier, U. Brath and M. Erdélyi, J. Am. Chem. Soc., 2012, 134, 5706–5715 CrossRef CAS PubMed.
- A.-C. C. Carlsson, K. Mehmeti, M. Uhrbom, A. Karim, M. Bedin, R. Puttreddy, R. Kleinmaier, A. A. Neverov, B. Nekoueishahraki, J. Gräfenstein, K. Rissanen and M. Erdélyi, J. Am. Chem. Soc., 2016, 138, 9853–9863 CrossRef CAS PubMed.
- M. Bedin, A. Karim, M. Reitti, A.-C. C. Carlsson, F. Topić, M. Cetina, F. Pan, V. Havel, F. Al-Ameri, V. Sindelar, K. Rissanen, J. Gräfenstein and M. Erdélyi, Chem. Sci., 2015, 6, 3746–3756 RSC.
- J. S. Ward, G. Fiorini, A. Frontera and K. Rissanen, Chem. Commun., 2020, 56, 8428–8431 RSC.
- J. S. Ward, A. Frontera and K. Rissanen, Dalton Trans., 2021, 50, 8297–8301 RSC.
- E. Taipale, M. Siepmann, K.-N. Truong and K. Rissanen, Chem. – Eur. J., 2021, 27, 17412–17419 CrossRef CAS PubMed.
- J. S. Ward, A. Frontera and K. Rissanen, Inorg. Chem., 2021, 60, 5383–5390 CrossRef CAS PubMed.
- J. S. Ward, A. Frontera and K. Rissanen, Chem. Commun., 2021, 57, 5094–5097 RSC.
- S. B. Hakkert, J. Gräfenstein and M. Erdelyi, Faraday Discuss., 2017, 203, 333–346 RSC.
- S. Yu, J. S. Ward, K.-N. Truong and K. Rissanen, Angew. Chem., Int. Ed., 2021, 60, 20739–20743 CrossRef CAS PubMed.
- D. C. Georgiou, P. Butler, E. C. Browne, D. J. D. Wilson and J. L. Dutton, Aust. J. Chem., 2013, 66, 1179–1188 CrossRef CAS.
- S. Lindblad, K. Mehmeti, A. X. Veiga, B. Nekoueishahraki, J. Gräfenstein and M. Erdélyi, J. Am. Chem. Soc., 2018, 140, 13503–13513 CrossRef CAS PubMed.
- D. von der Heiden, K. Rissanen and M. Erdélyi, Chem. Commun., 2020, 56, 14431–14434 RSC.
- S. Yu, E. Kalenius, A. Frontera and K. Rissanen, Chem. Commun., 2021, 57, 12464–12467 RSC.
- A.-C. C. Carlsson, M. Uhrbom, A. Karim, U. Brath, J. Gräfenstein and M. Erdélyi, CrystEngComm, 2013, 15, 3087–3092 RSC.
- S. Yu, P. Kumar, J. S. Ward, A. Frontera and K. Rissanen, Chem, 2021, 7, 948–958 CAS.
- E. Kramer, S. Yu, J. S. Ward and K. Rissanen, Dalton Trans., 2021, 50, 14990–14993 RSC.
- Spartan'20, Wavefunction Inc., Irvine CA, USA Search PubMed.
- L. Turunen and M. Erdélyi, Chem. Soc. Rev., 2020, 49, 2688–2700 RSC.
- T. Okitsu, D. Nakazawa, R. Taniguchi and A. Wada, Org. Lett., 2008, 10, 4967–4970 CrossRef CAS PubMed.
- R. S. Brown, R. W. Nagorski, A. J. Bennet, R. E. D. McClung, G. H. M. Aarts, M. Klobukowski, R. McDonald and B. D. Santarsiero, J. Am. Chem. Soc., 1994, 116, 2448–2456 CrossRef CAS.
- A. A. Neverov and R. S. Brown, J. Org. Chem., 1998, 63, 5977–5982 CrossRef CAS PubMed.
- X.-L. Cui and R. S. Brown, J. Org. Chem., 2000, 65, 5653–5658 CrossRef CAS PubMed.
- A. A. Neverov, H. X. Feng, K. Hamilton and R. S. Brown, J. Org. Chem., 2003, 68, 3802–3810 CrossRef CAS PubMed.
- S. Lindblad, F. Boróka Németh, T. Földes, D. von der Heiden, H. G. Vang, Z. L. Driscoll, E. R. Gonnering, I. Pápai, N. Bowling and M. Erdelyi, Chem. – Eur. J., 2021, 27, 13748–13756 CrossRef CAS PubMed.
- M. Kandrnálová, Z. Kokan, V. Havel, M. Nečas and V. Šindelář, Angew. Chem., Int. Ed., 2019, 58, 18182–18185 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compounds characterization, DFT calculations. CCDC 2127227 and 2127228. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt04309a |
| This journal is © The Royal Society of Chemistry 2022 |