
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimple conversion of trisodium phosphide, Na3P, into silyl- and cyanophosphides and the structure of a terminal silver phosphide†
Grégoire
Le Corre
 and
Hansjörg
Grützmacher
*
and
Hansjörg
Grützmacher
*
Department of Chemistry and Applied Biosciences ETH Zurich, Vladimir-Prelog-Weg 1, CH-8049 Zurich, Switzerland. E-mail: hgruetzmacher@ethz.ch
First published on 19th January 2022
Abstract
A reaction of trisodium phosphide (Na3P) with chlorosilanes allows for simple derivatization into silyl- and cyano-substituted phosphanide species which were compared with each other. The recently discovered cyano(triphenylsilyl)phosphanide shows unique coordination properties compared to bis(silyl)phosphides.
Silylated phosphanes and phosphides are useful building blocks in phosphorus chemistry1 and are soluble in organic solvents and suited to replace PH3 or M[PH2] which are mostly insoluble and difficult to handle.2 Particularly, tris(trimethylsilyl)phosphane P(TMS)3
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31 (TMS = SiMe3) and the alkali metal salts M[P(TMS)2] M[2] (M = Li, Na, K) are highly popular4 but their preparation requires a sodium-potassium alloy5 or phosphine gas which are hazardous materials.6 Tris(triphenylsilyl)phosphane 37 and the phosphides M[P(SiPh3)2] M[4]8 were prepared via silylation of a metal phosphide produced from elemental phosphorus and a Na/K alloy but their chemistry remains unexplored (Scheme 1).9
31 (TMS = SiMe3) and the alkali metal salts M[P(TMS)2] M[2] (M = Li, Na, K) are highly popular4 but their preparation requires a sodium-potassium alloy5 or phosphine gas which are hazardous materials.6 Tris(triphenylsilyl)phosphane 37 and the phosphides M[P(SiPh3)2] M[4]8 were prepared via silylation of a metal phosphide produced from elemental phosphorus and a Na/K alloy but their chemistry remains unexplored (Scheme 1).9
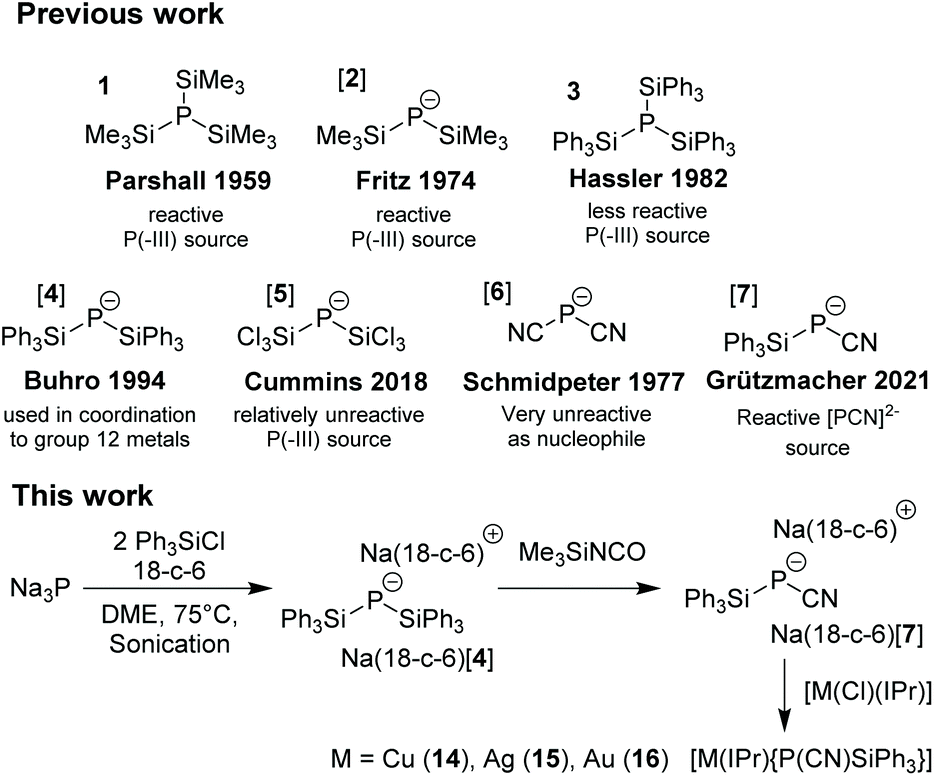 | ||
| Scheme 1 Existing phosphide species (top); direct synthesis of bis(silyl)phosphide from sodium phosphide (bottom). IPr = 1,3-Bis-(2,6-diisopropylphenyl)-imidazol-2-ylidene. | ||
In 2018, Cummins et al. reported the tetrabutylammonium salt of bis(trichlorosilyl)phosphide Bu4N[P(SiCl3)2] (Bu4N[5]) which was prepared by reduction of a phosphate derivative.10 This synthesis of a P3− silyl derivative directly from a P(V) precursor is ground-breaking, although the use of ten equivalents of trichlorosilane (Cl3SiH) per phosphorus atom is required. Therefore, a classical protocol which employs elemental phosphorus and a reducing agent, 12 M + P4 → 4 M3P, followed by a silylation reaction M3P + 3 R3SiX → P(SiR3)3 + 3 MX is still a competitive process.11
The phosphide [P(SiCl3)2]− can be used as a nucleophile but rather harsh reaction conditions are required. This indicates low nucleophilicity due to efficient depletion of the electron density from the P atom into the adjacent Si–Cl bonds via p(P) → σ*(SiCl) negative-hyperconjugation. Therefore [5]− is related to the bis(cyano)phosphide anion [6]−, first isolated as a sodium salt by Schmidpeter et al. more than 40 years ago.12 In this anion the electron density at P is delocalized into the π*(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) orbitals to such an extent that [P(CN)2]− even behaves as an electrophile and nucleophilic attack occurs under the displacement of cyanide, [P(CN)2]− + Nuc− → [P(CN)(Nuc)]− + CN−.13
N) orbitals to such an extent that [P(CN)2]− even behaves as an electrophile and nucleophilic attack occurs under the displacement of cyanide, [P(CN)2]− + Nuc− → [P(CN)(Nuc)]− + CN−.13
Recently, we have shown the synthesis of a mixed cyano(silyl)phosphanide from Na(OCP) which shows unique reactivity towards electrophiles.14
In this study, we set out to find a more convenient method for the preparation of phosphides by reactions of sodium phosphide and chlorosilanes in 1,2-dimethoxyethane (DME) as the solvent.
The sodium salt Na[P(SiCl3)2] (Na[5]) was prepared by a one-pot procedure by reacting red phosphorus with sodium metal in dimethoxyethane (DME) in the presence of 10 mol% naphthalene as an electron transfer reagent to give Na3P followed by the addition of SiCl4 at room temperature15 (Scheme 2(a)).
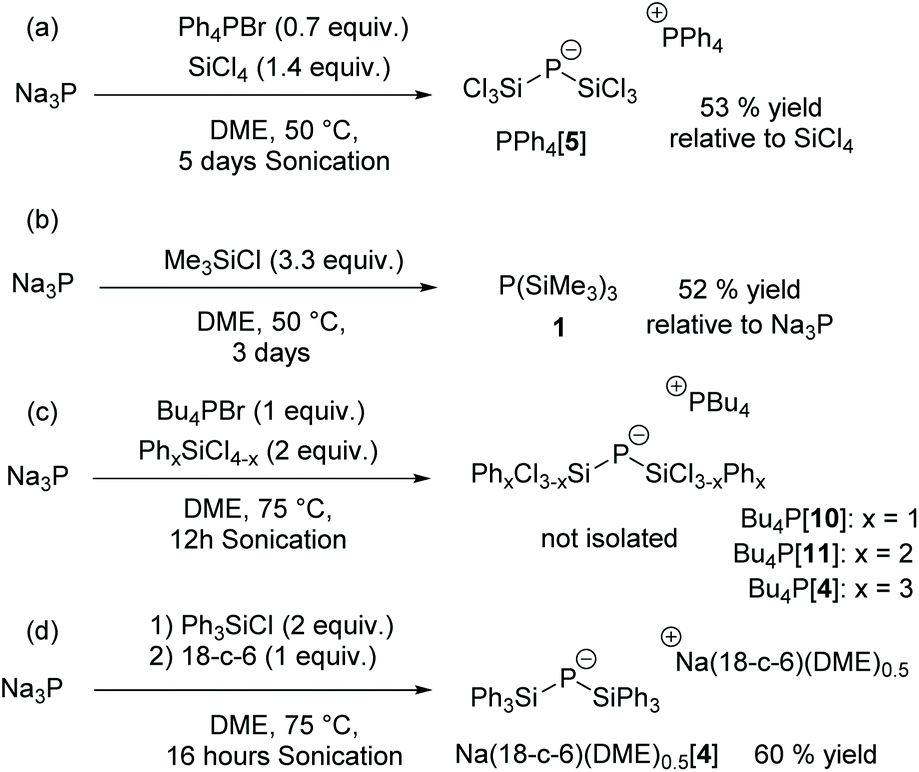 | ||
| Scheme 2 Reactions of Na3P with chlorosilanes. 18-c-6 = 18-crown-6. | ||
This reaction is slow but in the presence of nBu4NBr or Ph4PBr and under sonication, the reaction time is shortened to 5 days and [P(SiCl3)2]− can be obtained as tetraphenylphosphonium salt [δ(31P) = −171 ppm] in 53% yield (with respect to SiCl4).
The structure of Ph4P[P(SiCl3)2], Ph4P[5], was determined using single crystal X-ray diffraction (XRD). The resulting metric parameters are very similar to those of Bu4N[5] (see ESI Fig. S2†).10
With methylchlorosilanes Me4−xSiClx (x = 1, 2, 3) as reagents, complex product mixtures are obtained and only with 3.3 equivalents of Me3SiCl (TMSCl) a fair yield of P(TMS)3 (52%) is achieved after distillation, consistent with previous work (Scheme 2(b)).18 Notably, this reaction proceeds without the addition of phosphonium or ammonium salts.
The reaction with 2 equivalents of phenylchlorosilanes PhxSiCl4−x (x = 1, 2) in the presence of one equivalent of the phosphonium salt nBu4PBr is more selective and after 16 h at 75 °C under sonication, the bis(silyl)phosphides nBu4P[P(SiCl2Ph)2] [8] and nBu4P[P(SiClPh2)2] [9] could be characterized by NMR spectroscopy (Scheme 2(c)).
Bis(triphenylsilyl)phosphide was prepared by sonication of Na3P with Ph3SiCl at 75 °C in DME in the absence of phosphonium salts, followed by precipitation with 18-crown-6 to afford [Na(18-c-6)(dme)0.5]+[P(SiPh3)2]− [4] in 60% yield (Scheme 2(d)). Note that the formation of tris(triphenylsilyl)phosphane 37 is not observed in the 31P NMR spectrum of the reaction mixture even when the reaction was performed in dimethoxyethane at 115 °C in the presence of 10 mol% tetrabutylphosphonium bromide. This is in contrast to the reported synthesis of 3 from sodium–potassium phosphide and Ph3SiCl in boiling DME.7
In the structure of Na(18-crown-6)(DME)0.5[4] (ESI Fig. S1†), no close cation–anion interaction is observed and in the separated ion pair the P–Si distance is 2.19 Å and the Si–P–Si angle is 105° (similar to the corresponding data of [2]−).16
Although the isolation in the pure form was not possible for all derivatives, the 31P chemical shifts and the 1JPSi coupling constants of the complete series of chloro organylsilyl phosphides [P(SiPhxCl3−x)2]− [5], [8], [9], and [4] (x = 0, 1, 2, and 3, respectively) were obtained and allow a comparison with the data of [P(SiMe3)2]− [2], [P(CN)2]− [6] and [P(CN)SiPh3]− [7]. Clear trends are observed (Table 1): the higher the degree of chlorine substitution, the larger is 1JPSi and the more the 31P NMR signal is shifted to higher frequencies (less negative δ(31P)). Chlorine substitution appears to lead to shortening of the P–Si distance and narrowing of the Si–P–Si angle, which is verified experimentally for [4]− and [5]− and coherent with the NMR data of [8]− and [9]−. These effects are rationalized by the assumption that with increasing chlorine substitution negative hyperconjugation between the p-type lone-pair at phosphorus and the σ*(Si–Cl)-orbitals increases, provoking a shortening of the phosphorus–silicon bond which in turn leads to an increase of the 1JPSi scalar coupling. Concomitantly, the electron density at the 31P nucleus is depleted, leading to a high-frequency shift of the 31P chemical shift. Both effects overcompensate the narrowing of the Si–P–Si angle which is expected to cause a low-frequency shift.19
Noteworthy is the structural and spectroscopic similarity between the anions [P(SiCl3)2]− ([5]−, entry 2) and [P(CN)2]− ([6]−, entry 6) which suggests that the former is also a poor nucleophile. Indeed, Ph4P[5] does not react with SiCl4 in excess or with CH2Cl2. On the other hand, the similarities between [P(SiMe3)2]−16 ([2]−) and [P(SiPh3)2]− ([4]−) prompted us to investigate the reactivity of [4]− as a nucleophile in more detail.
As expected, the reaction of [4]− with dimethyl carbonate afforded Na(OCP) [δ(31P) = −394 ppm] comparable to the original synthesis of Li(OCP) by Becker20 using Li[P(SiMe3)2] as a reagent. Furthermore, [4]− was reacted with pivaloyl or mesitoyl chloride to afford mixtures of E- and Z-isomers (about 3:1 and 1:1 ratio, respectively) of the phosphaalkenes 10 and 11 (Scheme 3(a)) in analogy to the E/Z mixtures when the TMS-substituted derivatives TMS-P = C(OTMS)R (R = Mes, tBu) were prepared from P(TMS)31 or [P(TMS)2]− [2]−.1d,21
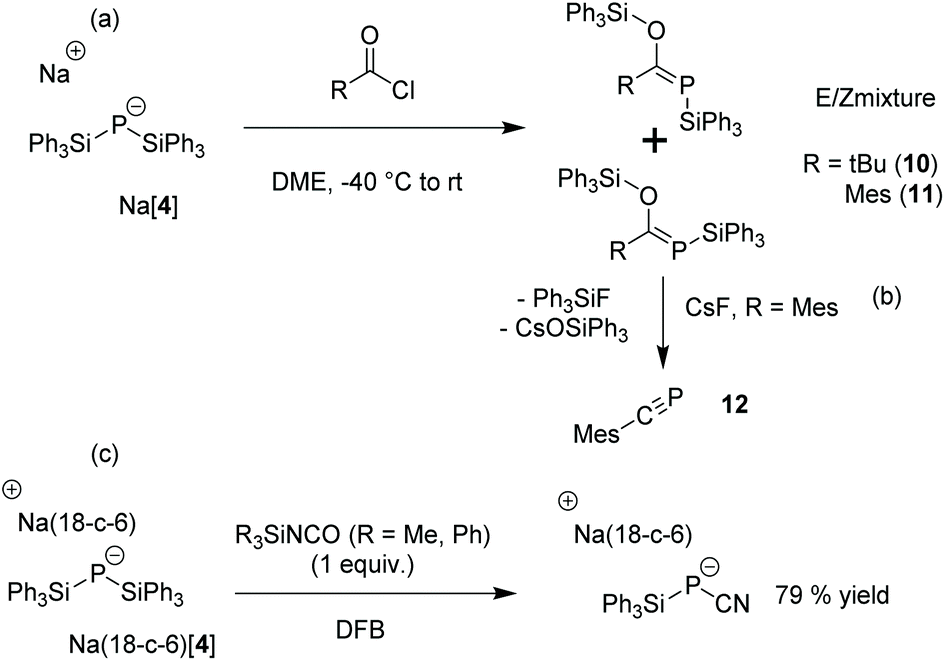 | ||
| Scheme 3 Reactivity of Na[4]. Mes = Mesityl. | ||
Only very few P-silyl phosphaalkenes TMS-P = C(OTMS)R (R = ferrocenyl or bicyclo[2.2.2]octanediyl) have been investigated by X-ray diffraction (XRD). Compounds with R = Mes, tBu are liquids at room temperature.22 Single crystals of the E-isomer of 10 and the Z-isomer of 11 can be easily grown by slow evaporation of a concentrated solution in hexane and were subjected to XRD experiments.
In both structures (Fig. 1), the phosphorus–carbon bond is slightly longer (1.71 Å for E-10 and 1.70 Å for Z-11) than the average P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond length in previously reported phosphaalkenes (1.66 Å).23 The phosphorus–silicon bonds (E-10: 2.27 Å; Z-11: 2.26 Å) are considerably elongated in comparison to [4]−. Both compounds have an almost planar Si–P–C–O unit (Si–P–C–O dihedral angles: Z-11: 6.1°; E-10 13.6°). The atoms Si1, C2, and P1 around the C–O unit are arranged in an almost planar fashion in Z-11 (Si–O–C–P torsion angle φ = 6.2°), while in E-10 this angle is φ = 37°, likely because the triphenylsilyl group is both repelled by the bulky tert-butyl moiety and by the electron density at phosphorus.
C bond length in previously reported phosphaalkenes (1.66 Å).23 The phosphorus–silicon bonds (E-10: 2.27 Å; Z-11: 2.26 Å) are considerably elongated in comparison to [4]−. Both compounds have an almost planar Si–P–C–O unit (Si–P–C–O dihedral angles: Z-11: 6.1°; E-10 13.6°). The atoms Si1, C2, and P1 around the C–O unit are arranged in an almost planar fashion in Z-11 (Si–O–C–P torsion angle φ = 6.2°), while in E-10 this angle is φ = 37°, likely because the triphenylsilyl group is both repelled by the bulky tert-butyl moiety and by the electron density at phosphorus.
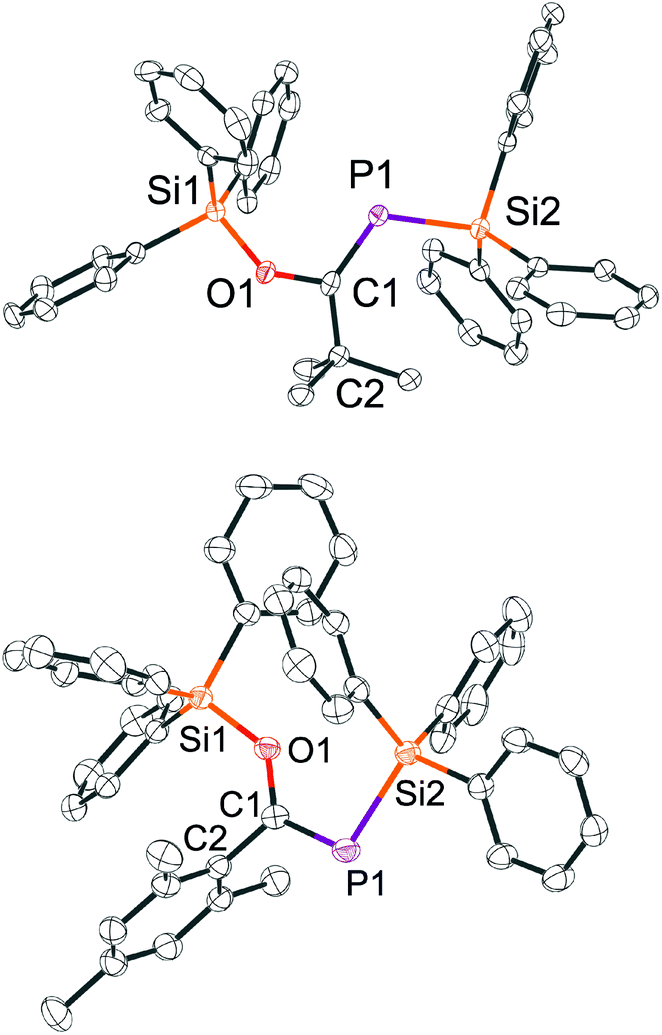 | ||
| Fig. 1 Structure of E-10 (top) and Z-11 (bottom) in the crystal (hydrogens omitted for clarity). Selected bond distances [Å] and angles [°] for E-10: Si1–O1 1.6871(17); O1–C1 1.354(3); P1–C1 1.704(2); Si2–P1 2.2604(9); C1–C2 1.497(3); C2–C1–P1 117.33(17); O1–C1–P1 124.47(18); C1 P1 Si2 102.34(9) Si2–P1–C1–O1 6.1(2) P1–C1–O1–Si1 12.73(13); for Z-11: Si1–O1 1.6688(19); O1–C1 1.372(3); P1–C1 1.718(3); Si2–P1 2.2696(10); C1–C2 1.528(4); C2–C1–P1 137.92(19); O1–C1–P1 114.02(19); O1–C1–C2 108.1(2); C1–P1–Si2 113.82(9); Si2–P1–C1–O1 12.58(15) P1–C1–O1–Si1 37.6(3). | ||
It has been shown that TMS-P = C(OTMS)R can be used as precursors for the corresponding phosphaalkynes, R-CP, via thermal elimination of hexamethyldisiloxane1d,24 or a reaction with AlCl3 at lower temperatures.21,25 Upon mixing 11 with one equivalent of cesium fluoride in order to initiate Ph3SiF and Cs(OSiPh3) elimination at room temperature in tetrahydrofuran (THF), the selective formation of phosphaalkyne 12 was observed by NMR [δ(31P) = +1 ppm and δ(13C) = 163.5 ppm (1JCP = 45 Hz)] (Scheme 3(b), ESI Fig. S32 and S33†). 10 does not react using the same reaction conditions, presumably because the steric hindrance due to the t-butyl group prevents 10 from achieving a conformation suitable for Ph3SiF and Cs(OSiPh3) elimination. Heating alone or treatment with AlCl3 did not lead to the formation of the corresponding phosphaalkynes R-CP (R = tBu, Mes).
[4]− reacts with two equivalents of cyanogen bromide to generate bis(cyano)phosphide [6]−. Since bromotriphenylsilane is generated as a byproduct in this reaction, we hypothesized that Ph3SiCl could be used catalytically in a reaction between Na3P and cyanogen bromide. While [6]− was not detected by 31P NMR after the sonication of Na3P for 16 h with two equivalents of BrCN in DME at 50 °C, it is formed as the major product when 20 mol% Ph3SiCl is added to the suspension. Na(18-crown-6)[6] was isolated in 43% yield from the reaction mixture. This provides a relatively efficient way to prepare Na+[P(CN)2]−, Na+[6]−, as other syntheses require a tenfold excess of P426 or the use of Ag salts or volatile HCN.12c
The reaction of [4]− with one equivalent of phenyl cyanate in DME gave a mixture of [6]− and cyano(triphenylsilyl)phosphide [7]− [δ(31P) = −282 ppm]. When one equivalent of triphenylsilyl- or trimethylsilyl isocyanate is used as a milder cyanation agent, only [7]− was observed in the NMR spectrum, allowing us to isolate Na(18-crown-6)[7] in 79% yield.
We investigated whether a trimethylsilyl-substituted equivalent of [7]− would be accessible via the reaction of Na[2] with trimethylsilyl isocyanate in THF. The product of this reaction was identified as P(TMS)31via31P- and 13C-NMR. Moreover, the reaction of [4]− with Me3Si-NCO in a DME/DFB mixture in the absence of 18-crown-6 afforded P(SiPh3)2(SiMe3) 1311 as the major product [δ(31P) = −255 ppm] along with minor amounts of [7]−.
From these two reactions, it can be concluded that both steric bulk on bis(silyl)phosphide and the complexation of sodium are essential in order to prevent salt metathesis with silyl isocyanates and favour the elimination of a disiloxane in the reaction of a bis(silyl)phosphide with a silyl isocyanate.
While terminally bonded bis(silyl) phosphide complexes of d10 valence electron configured noble metals Cu+ and Au+ stabilised with N-heterocyclic carbenes are known, Ag+ analogues remain elusive, as attempts to form such compounds typically result in the formation of Ag(I) phosphido clusters.27 Therefore, the push–pull substituted Na(18-crown-6)[7] was reacted with a series of coinage metal complexes [M(Cl)(IPr)] (IPr = 1,3-bis-(2,6-diisopropylphenyl)-imidazol-2-ylidene; M = Cu, Ag, Au) which allowed us to isolate the homologous series of complexes [M(IPr){P(CN)(SiPh3)}] 14 (M = Cu), 15 (M = Ag), and 16 (M = Au). These species have very similar conformations with a phosphorus atom in a pyramidal coordination sphere (the average sum of angles around phosphorus is 301°); see a plot of 15 in Fig. 2 as an example and the plots of 13 and 15 in Fig. S3 and S4,† respectively.
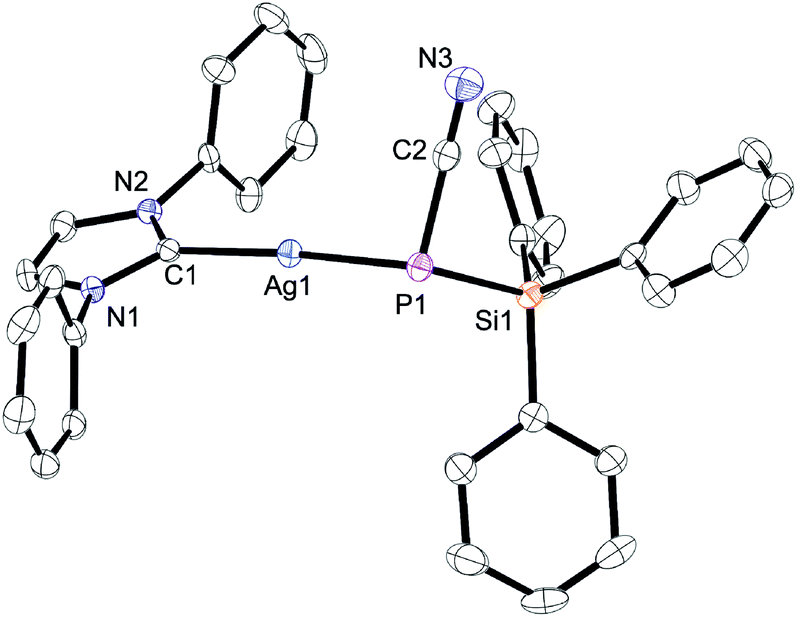 | ||
| Fig. 2 Structure of 15 in the crystal (hydrogens and isopropyl groups omitted for clarity). Selected bond distances [Å] and angles [°]: P1–Ag1 2.3757(8) Ag1–C1 2.109(3); P1–C2 1.778(3); C2–N3 1.150(4); P1–Si1 2.2392(11); Ag1–P1–C2 101.27(10); Si1–P1–C2 83.59(10); Si1–P1–Ag1 107.30(4); Ag1–P1–C1 5.32(8). | ||
Compared to Na(18-c-6)[7], the P–C and P–Si bond distances in the coinage metal complexes are longer while the C–N bond distances are shorter (ESI Table S1†). This is expected because the dative P → M bond causes a depletion of electron density at P which in turn weakens the donation into the σ*(Si–Ph) bonds (negative hyperconjugation) and π*-orbitals of the CN group. The lower magnitude of 1JP,C and 1JP,Si couplings and the higher CN stretching frequencies in 14–16 reflect these changes (Table S1†). In [Au(IPr)X]-type complexes, the 13C-NMR chemical shift of the carbenic carbon correlates with the σ-acidity of the [AuX] fragment.28 In 16, this carbon resonates at 195.8 ppm and at 188.6 ppm for [Au(IPr){P(SiMe3)2}].27b In 16, there is a relatively short distance between the Au atom and the carbon of the CN group (Au–C2 = 3.25 Å; sum of the van der Waals radii of Au and C: 3.8 Å).29 This secondary interaction is also found in 15 (Ag–C distance 3.23 Å; sum of van der Waals radii 3.8 Å) which may enhance the σ-acidity of the [M[7]] fragments thereby contributing to the overall stability. It also suggests that complexes of [7]− in an η2 or η3 binding mode may exist, as has been found in some complexes of related [X = C = P]− anions.30
In conclusion, simple silylation of trisodium phosphide gives access to a variety of silyl-substituted phosphanides. Particularly, the salts of [P(SiCl3)2]− and [P(SiPh3)2]− could be easily prepared using one-pot procedures by first mixing sodium and red phosphorus to give Na3P which was subsequently reacted with SiCl4 or Ph3SiCl. While [P(SiCl3)2]− is a weak nucleophile (comparable to [P(CN)2]−) which limits somewhat its use as a reagent, [P(SiPh3)2]− is a versatile reagent which allows us to access a variety of organophosphorus compounds such as phosphaalkenes or the push–pull cyano(silyl)phosphide [P(CN)(SiPh3)]−. The latter allowed us to access the first fully characterized example of a terminal Ag(I) phosphide, which highlights that this phosphide possesses unique properties. We hope to demonstrate by future work that especially [P(CN)(SiPh3)]− will allow the synthesis of new metal phosphides (for example via elimination of Ph3SiCN) which are currently highly sought after catalytic materials.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- (a) M. D. Healy, P. E. Laibinis, P. D. Stupik and A. R. Barron, J. Chem. Soc., Chem. Commun., 1989, 359–360 RSC; (b) R. Appel, D. Gudat, E. Niecke, M. Nieger, C. Porz and H. Westermann, Z. Naturforsch., B: Chem. Sci., 1991, 46, 865 CrossRef CAS; (c) G. Becker, W. Schwarz, N. Seidler and M. Westerhausen, Z. Anorg. Allg. Chem., 1992, 612, 72–82 CrossRef CAS; (d) G. Becker, G. Gresser and W. Uhl, Z. Naturforsch., B: Chem. Sci., 1981, 36, 16–19 CrossRef.
- G. Becker, G. Ditten, K. Hübler, U. Hübler, K. Merz, M. Niemeyer, N. Seidler, M. Westerhausen and Z. Zheng, Organosilicon Chemistry Set: From Molecules to Materials, 2005, pp. 161–186 Search PubMed.
- G. W. Parshall and R. V. Lindsey, J. Am. Chem. Soc., 1959, 81, 6273–6275 CrossRef CAS.
- (a) G. Fritz, H. Schäfer and W. Hölderich, Z. Anorg. Allg. Chem., 1974, 407, 266–286 CrossRef CAS; (b) F. Uhlig and R. Hummeltenberg, J. Organomet. Chem., 1993, 452, C9–C10 CrossRef CAS; (c) E. P. O. Fuchs, H. Heydt, M. Regitz, W. W. Schoeller and T. Busch, Tetrahedron Lett., 1989, 30, 5111–5114 CrossRef CAS; (d) A. R. Barron, A. H. Cowley and S. W. Hall, J. Chem. Soc., Chem. Commun., 1987, 980–981 RSC.
- G. Becker and W. Hölderich, Chem. Ber., 1975, 108, 2484–2485 CrossRef CAS.
- Y. J. Shin, S. W. Jun and J. H. Park, US Patent2017/226138, 2017 Search PubMed.
- K. Hassler, Monatsh. Chem., 1982, 113, 421–425 CrossRef CAS.
- M. A. Matchett, M. Y. Chiang and W. E. Buhro, Inorg. Chem., 1994, 33, 1109–1114 CrossRef CAS.
- D. C. Gary, B. A. Glassy and B. M. Cossairt, Chem. Mater., 2014, 26, 1734–1744 CrossRef CAS.
- M. B. Geeson and C. C. Cummins, Science, 2018, 359, 1383 CrossRef CAS PubMed.
- (a) H. Diskowski and T. Hofmann, Phosphorus, in Ullmann's Encyclopedia of Industrial Chemistry, 2000 Search PubMed; (b) W. Zulehner, B. Neuer and G. Rau, Silicon, in Ullmann's Encyclopedia of Industrial Chemistry, 2000 Search PubMed.
- (a) A. Schmidpeter and F. Zwaschka, Angew. Chem., Int. Ed. Engl., 1977, 16, 704–705 CrossRef; (b) A. Schmidpeter, W. Gebler, F. Zwaschka and W. S. Sheldrick, Angew. Chem., Int. Ed. Engl., 1980, 19, 722–723 CrossRef; (c) P. Panne, Ph.D. Thesis, University of Köln, 2004 Search PubMed; (d) J. F. Binder, S. C. Kosnik, P. B. J. St Onge and C. L. B. Macdonald, Chem. – Eur. J., 2018, 24, 14644–14648 CrossRef CAS PubMed.
- A. Schmidpeter and G. Burget, Z. Naturforsch., B: Chem. Sci., 1985, 40, 1306–1313 CrossRef.
- G. Le Corre, J. J. Gamboa-Carballo, Z. Li and H. Grützmacher, Angew. Chem., Int. Ed., 2021, 60(47), 24817–24822 CrossRef CAS PubMed.
- (a) M. C. J. M. Van Hooijdonk, G. Gerritsen and L. Brandsma, Phosphorus, Sulfur Silicon Relat. Elem., 2000, 162, 39–49 CrossRef CAS; (b) D. Stein, T. Ott and H. Grützmacher, Z. Anorg. Allg. Chem., 2009, 635, 682–686 CrossRef CAS.
- E. Hey, P. B. Hitchcock, M. F. Lappert and A. K. Rai, J. Organomet. Chem., 1987, 325, 1–12 CrossRef CAS.
- W. S. Sheldrick, J. Kroner, F. Zwaschka and A. Schmidpeter, Angew. Chem., Int. Ed. Engl., 1979, 18, 934–935 CrossRef.
- G. P. Weinberger, F. Sommer, A. Torvisco, R. C. Fischer and M. Flock, Eur. J. Inorg. Chem., 2020, 2020, 3778–3785 CrossRef CAS.
- J. H. Letcher and J. R. V. Wazer, J. Chem. Phys., 1966, 44, 815–829 CrossRef CAS.
- G. Becker, H. M. Hartmann and W. Schwarz, Z. Anorg. Allg. Chem., 1989, 577, 9–22 CrossRef CAS.
- A. Mack, E. Pierron, T. Allspach, U. Bergsträßer and M. Regitz, Synthesis, 1998, 1305–1313 CrossRef CAS.
- (a) A. Grünhagen, U. Pieper, T. Kottke and H. W. Roesky, Z. Anorg. Allg. Chem., 1994, 620, 716–722 CrossRef; (b) F. Brodkorb, M. Brym, C. Jones and C. Schulten, J. Organomet. Chem., 2006, 691, 1025–1029 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- (a) W. Rösch, U. Hees and M. Regitz, Chem. Ber., 1987, 120, 1645–1652 CrossRef; (b) W.-G. Veeck and M. Regitz, in Comprehensive Organic Functional Group Transformations, ed. A. R. Katritzky, O. Meth-Cohn and C. W. Rees, Elsevier Science, Oxford, 1995, pp. 1151–1160 Search PubMed.
- B. Breit and M. Regitz, Chem. Ber., 1996, 129, 489–494 CrossRef CAS.
- A. Schmidpeter, G. Burget, F. Zwaschka and W. S. Sheldrick, Z. Anorg. Allg. Chem., 1985, 527, 17–32 CrossRef CAS.
- (a) B. Khalili Najafabadi and J. F. Corrigan, J. Chem. Soc., Dalton Trans., 2015, 14235–14241 RSC; (b) B. Khalili Najafabadi and J. F. Corrigan, Can. J. Chem., 2016, 94, 593–598 CrossRef CAS; (c) B. Khalili Najafabadi and J. F. Corrigan, J. Chem. Soc., Chem. Commun., 2015, 51, 665–667 RSC.
- D. Marchione, M. A. Izquierdo, G. Bistoni, R. W. A. Havenith, A. Macchioni, D. Zuccaccia, F. Tarantelli and L. Belpassi, Chem. – Eur. J., 2017, 23, 2722–2728 CrossRef CAS PubMed.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
- (a) F. A. Watt, L. Burkhardt, R. Schoch, S. Mitzinger, M. Bauer, F. Weigend, J. M. Goicoechea, F. Tambornino and S. Hohloch, Angew. Chem., Int. Ed., 2021, 60, 9534–9539 CrossRef CAS PubMed; (b) L. Liu, D. A. Ruiz, F. Dahcheh, G. Bertrand, R. Suter, A. M. Tondreau and H. Grützmacher, Chem. Sci., 2016, 7, 2335–2341 RSC; (c) M. M. D. Roy, M. J. Ferguson, R. McDonald and E. Rivard, J. Chem. Soc., Chem. Commun., 2018, 54, 483–486 RSC; (d) A. Reinholdt, M. G. Jafari, C. Sandoval-Pauker, E. Ballestero-Martínez, M. R. Gau, M. Driess, B. Pinter and D. J. Mindiola, Angew. Chem., Int. Ed., 2021, 60, 17595–17600 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2126614–2126620. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt04223h |
| This journal is © The Royal Society of Chemistry 2022 |