
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeminal C–Cl and Si–Cl bond activation of chloromethanes and chlorosilanes by gallanediyl LGa†
Christoph
Helling
a,
Chelladurai
Ganesamoorthy
a,
Christoph
Wölper
a and
Stephan
Schulz
 *ab
*ab
aFaculty of Chemistry, University of Duisburg-Essen, Universitätsstr. 5-7, S07 S03 C30, D-45117 Essen, Germany. E-mail: stephan.schulz@uni-due.de; Fax: (+49) 201-1833830; Web: https://www.uni-due.de/ak_schulz/index_en.php
bCenter for NanoIntegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, 47057 Duisburg, Germany
First published on 12th January 2022
Abstract
The activation of relatively inert E–X σ-bonds by low-valent main group metal complexes is receiving increasing interest. We here confirm the promising potential of gallanediyl LGa (L = HC[C(Me)N(Dip)]2, Dip = 2,6-i-Pr2C6H3) to activate E–Cl (E = C, Si) σ-bonds of group 14 element compounds. Equimolar reactions of LGa with chloromethanes and chlorosilanes EHxCl4−x (E = C, x = 0–2; E = Si, x = 0, 1) occurred with E–Cl bond insertion and formation of gallylmethanes and -silanes L(Cl)GaEHxCl3−x (E = C, x = 2 (1), 1 (2), 0 (3); E = Si, x = 1 (4)). In contrast, consecutive insertion into a geminal E–Cl bond was observed with two equivalents of LGa, yielding digallyl complexes [L(Cl)Ga]2EHxCl2−x (E = C, x = 2 (5); E = Si, x = 1 (6), 0 (7)). Compounds 1–7 were characterized by heteronuclear NMR (1H, 13C, 29Si (4, 6)), IR spectroscopy and elemental analysis, and their solid-state structures were determined by single-crystal X-ray diffraction (sc-XRD).
Introduction
Bond activation reactions mediated by low-valent main group element complexes gained increasing interest within the past two decades due to the potential of establishing main group element-based catalytic transformations.1 In this light, the reactivity of monovalent group 13 β-diketiminates LM (M = Al, Ga, In; L = HC[C(Me)N(Dip)]2, Dip = 2,6-i-Pr2C6H3) in the formal oxidation state +I has been extensively investigated.2 Typical reaction patterns include the formation of Lewis acid–base adducts,3 oxidative addition reactions of σ- and π-bonds,4 and two-electron reduction reactions, which can also be utilized consecutively by optimizing the reaction conditions.5 A key step in transition metal-catalyzed C–C cross coupling reactions is the initial oxidative addition of C–X (X = F, Cl, Br, I) bonds to the metal center.6 Remarkably, group 13 diyls LM have been found to be capable of C–X oxidative addition. Indanediyl LIn was shown to react with alkyl bromides and iodides to yield the corresponding bond insertion products L(X)InR (R = Me, i-Pr, t-Bu; X = Br, I), whereas it was found unreactive towards alkyl chlorides and aryl iodides due to the stronger C–Cl and C(sp2)–I bonds, respectively.7 In contrast, stronger reducing gallanediyl LGa was found to activate the C–Cl σ-bond in t-BuCl and even the C–F σ-bonds in C6F6 and C6F5H under forcing conditions, yielding L(Cl)Gat-Bu (I)8 and L(F)GaArF (ArF = C6F5II, C6F4H III),9 respectively. The lightest and most reactive alanediyl LAl readily activated strong C(sp3)–F and C(sp2)–F bonds in alkyl, vinyl, and aryl fluorides via oxidative addition, α-fluoride elimination, and β-fluoride elimination mechanisms with formation of alanes L(F)AlR.10 Furthermore, LGa reacted with SnCl2 in reduction and bond insertion cascades to metalloid Sn clusters, [L(Cl)Ga]2Sn7 and [L(Cl)Ga]4Sn17,11 while intermetallic compounds [LGa]2Ge2 and [LGa]2Ge4 were obtained from reactions with (Cy3P)GeCl2/KC8 and (NHC)GeCl2, respectively.12 Oxidative additions of Sn–H and Pb–Cl bonds were observed with Ph3SnH and Me3PbCl, yielding L(H)GaSnPh3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4a and L(Cl)GaPbMe3,13 respectively, and two-fold bond insertion reactions produced [L(Cl)Ga]2SnCl28 and plumbylene [L(F3C(O)2SO)Ga]2Pb(THF).13
4a and L(Cl)GaPbMe3,13 respectively, and two-fold bond insertion reactions produced [L(Cl)Ga]2SnCl28 and plumbylene [L(F3C(O)2SO)Ga]2Pb(THF).13
In case of Si, reactions of LGa with SiX4 gave L(X)GaSiX3 (X = Cl IV, Br V),8,14 and compound V reacted with another equivalent of LGa to [L(Br)Ga]2SiBr2 (VI) via consecutive Si–Br bond insertion. Most remarkably, the reaction of VI with LGa under CO atmosphere produced the stable Si–carbonyl complex [L(Br)Ga]2Si(CO) (VII), whereas in the absence of CO transient silylene [L(Br)Ga]2Si was found to activate a C–C bond of the ligand backbone to form compound VIII (Fig. 1).14 In contrast, no further reactivity with LGa was observed for IV.8
 | ||
| Fig. 1 Selected compounds derived from LGa relevant to this study. | ||
We have reported in recent years on σ-bond activation reactions of group 13 diyls LM (M = Al, Ga, In) with main group element complexes of group 13,15 15,16 and 16,17 respectively, resulting in the formation of a large variety of complexes with unusual bonding properties and electronic structures including double bonded species, cluster-type complexes and main group metal centered radicals. We now became interested to study the general reactivity of LGa towards chloromethanes and chlorosilanes with geminal E–Cl bonds (E = C, Si), and report herein on the synthesis and characterization of mono- and bis-insertion products.
Results and discussion
Synthesis and properties
We initially studied reactions of LGa with dichloromethane (DCM, CH2Cl2), chloroform (CHCl3), and carbon tetrachloride (CCl4). Dissolution of LGa in both DCM and chloroform at ambient temperature immediately resulted in the formation of colorless solutions containing the C–Cl bond activation products L(Cl)GaCH2Cl (1) and L(Cl)GaCHCl2 (2), which were isolated as colorless crystalline solids in good yields after removal of the solvent and crystallization from n-pentane (1) and n-hexane (2), respectively (Scheme 1). | ||
| Scheme 1 Synthesis of compounds 1–4. | ||
In situ 1H NMR spectroscopic studies revealed the virtually quantitative formation of 1 and 2, and no other reaction products were detected in the reaction solutions. This observation contrasts those reported by Fischer et al., who observed subsequent decomposition reactions and formation of undefined products in reactions of LGa with DCM and chloroform.8 In contrast, dissolution of LGa in carbon tetrachloride at ambient temperature gave a new species together with considerable amounts of LGaCl218 as the major compounds in solution as determined by in situ1H NMR spectroscopy. The formation of LGaCl2 indicates the presence of a reduction pathway, which was successfully suppressed by addition of carbon tetrachloride to a toluene solution of LGa at low temperature. Pure L(Cl)GaCCl3 (3) was obtained as a colorless crystalline material in moderate yield after crystallization from DCM (Scheme 1). Under inert gas atmosphere, compounds 1 and 2 are stable in the solid-state and in solution even at elevated temperatures (80 °C), whereas 3 is stable in the solid-state but slowly decomposes in solution with formation of LGaCl2 and minor amounts of so far unidentified products, potentially arising from reactions with transient dichlorocarbene. The selective formation of compounds 1–3 is remarkable since only a few (low-valent) main group species were found to activate DCM and chloroform via simple C–Cl bond insertion, i.e. neutral stannylenes,19 germyliumylidene,20 phosphenium cations,21 and silylenes,22 while such reactivity was not yet observed towards carbon tetrachloride. With respect to group 13 elements, [In(L)]OSO2CF3 (L = 18-c-6, dibenzo-18-c-6) are the only low-oxidation state compounds reported to undergo distinct C–Cl bond insertion with DCM and chloroform.23
Since the formation of compound IV by Si–Cl oxidative addition of SiCl4 to LGa was previously reported,8 we became interested to study the selectivity of Si–H vs. Si–Cl bond insertion by reaction of LGa with HSiCl3. Addition of HSiCl3 to a solution of LGa in benzene afforded a colorless solution, from which the Si–Cl bond activation product L(Cl)GaSiHCl2 (4) was isolated as a colorless crystalline solid in good yield (Scheme 1). In situ1H NMR spectroscopy proved the selective formation of 4 with no observable signals corresponding to a potential Si–H bond activation product. Compound 4 is stable in the solid-state and in solution at ambient temperature in the absence of air and moisture even after prolonged storage. Similar Si–Cl bond activations of HSiCl3 were observed by a vinyl silylsilylene24 and a diamidosilylene,22b albeit in the latter case with low selectivity.
Intrigued by the consecutive Si–Br bond activation of compound V by reaction with LGa, the reactions of compounds 1–4 and IV with one equivalent of LGa were investigated. Compound 1 reacted with LGa at 100 °C within 7 hours via C–Cl bond insertion and formation of a rare Ga methylene complex, [L(Cl)Ga]2CH2 (5), which was isolated in moderate yield after recrystallization from n-hexane (Scheme 2). In contrast, compounds 2 and 3 reacted with LGa at 60 °C and ambient temperature, but the reactions only resulted in formation of LGaCl2 as the main product and several minor unidentified species as shown by in situ1H NMR spectroscopy. The large steric pressure in the hypothetically formed products [L(Cl)Ga]2C(X)Cl (X = H, Cl) in conjunction with the presence of a reactive C–Cl bond most likely induces the elimination of LGaCl2 concomitant with reduction of the carbon center, thus accounting for the several side products observed.
 | ||
| Scheme 2 Synthesis of compounds 5–7. | ||
Compound 5, which is exceptionally stable in both solid-state and solution under inert gas atmosphere, belongs to the very short list of structurally characterized group 13 element methylene complexes, which includes the parent Ga methylene [Ga8(μ-CH2)12],25 Al methylene [R2Al]2CH2 (R = Cl, CH(SiMe3)2, 2,4,6-i-Pr3C6H2, N(Me2CCH2)2CH2, N(SiMe3)2),26 and In methylene complexes [(L)X2In]2CH2 (X = Cl, Br; L = Me2NC2H4NMe2),27 respectively. In addition, several methylene complexes were obtained from two-fold C–Cl bond activation of DCM by silylenes,28 stannylenes,29 and a digermene,30 respectively.
Gallylsilanes 4 and II were found to react with LGa at 80 °C with Si–Cl bond insertion to afford digallylsilanes [L(Cl)Ga]2SiHCl (6) and [L(Cl)Ga]2SiCl2 (7) in good yields after recrystallization from benzene. In the reaction of LGa with 4, selective Si–Cl bond activation occurred and no indications for a Si–H activated product were observed. Fischer and co-workers8 only obtained compound IV even in the presence of excess LGa as conversion of IV to 7 requires thermal treatment to initiate the second bond insertion as was observed for V.14 Most conveniently, compounds 6 and 7 are synthesized by direct reactions of two equivalents of LGa with HSiCl3 and SiCl4, respectively. Both compounds, 6 and 7, are stable in the solid-state and in solution at ambient temperature in the absence of air and moisture. Apart from compound V,14 two-fold bond insertion into geminal Si-halogen bonds has only been observed by a cyclic digermene via initial Si–X addition to the double bond.31 Attempts to dehydrochlorinate 6 by addition of strong bases as well as further reduction of 7 using LGa or other reducing agents to obtain the corresponding silylene failed, showing the decisive influence of the Si–X bond strength.
Spectroscopic characterization
The 1H and 13C NMR spectra of compounds 1–5 and 7 feature the expected signals for the β-diketiminate ligand in reduced Cs symmetry due to the presence of two additional different substituents at each Ga center, e.g., two singlets (γ-CH, CCH3), two septets (CH(CH3)2), and four doublets (CH(CH3)2) were observed in the 1H NMR spectra. In contrast, compound 6 exhibits signals corresponding to a C1-symmetric β-diketiminate ligand, e.g., three singlets (γ-CH, CCH3), four septets (CH(CH3)2), and eight doublets (CH(CH3)2) in the 1H NMR spectrum, resulting from the heteroleptic substitution pattern at the central Si atom. The CH2Cl (2.62 ppm) and CHCl2 (5.31 ppm) resonances of 1 and 2, respectively, are observed at higher chemical shift compared to those of CH2Cl2 (4.27 ppm) and CHCl3 (6.15 ppm) in C6D6 due to the substitution of an electronegative Cl atom by an electropositive Ga atom. Virtually identical 1H NMR chemical shifts were observed for L′SiCH3−xClx complexes containing a related N,N′ chelating ligand L′ (x = 1, 2.68 ppm; x = 2, 5.34 ppm),22b whereas the shift to higher frequency is less pronounced in cationic Cl–In–CH3−xClx crown ether complexes (x = 1, 3.47, 4.37 ppm; x = 2, 5.87, 5.65 ppm).23 Similar higher chemical shifts compared to CH2Cl2 (53.5 ppm) and CHCl3 (77.8 ppm) are observed for the CH2Cl (25.4 ppm) and CHCl2 (59.6 ppm) 13C NMR resonances of 1 and 2, respectively, which could only be reliably located by the cross-peaks in the corresponding 2D-1H–13C-HSQC spectra due to their broadness. For the same reason, the CCl3 resonance of 3 was not observed. The CH2 (−1.02 ppm) and CH2 (−12.4 ppm) resonances of Ga methylene complex 5 appear at even higher frequency as a result of the high electron density at the central CH2 moiety imposed by the two electropositive gallyl substituents, and are comparable to those of THF and pyridine adducts of the parent Ga methylene [Ga8(μ-CH2)12] (δ1H 0.63–0.49 ppm)25 and [R2Al]2CH2 (δ1H R = CH(SiMe3)2 −0.50; 2,4,6-i-Pr3C6H2 −0.66; N(SiMe3)2 −0.67 ppm),26 while those of the double silylene insertion product {[H2CC(SiMe3)2]2(Cl)Si}2CH2 formed by reaction of two equivalents of silylene [H2CC(SiMe3)2]2Si with dichloromethane are found at lower frequency (δ1H 1.43 ppm; δ13C 20.2 ppm).28b In contrast to CH2Cl2, 1, and 5, the observed 1H NMR chemical shifts of the SiH protons in HSiCl3 (5.39 ppm), 4 (5.69 ppm), and 6 (4.30 ppm) show no distinct trend. However, the 1JHSi coupling constants measured from 29Si satellites gradually decrease from HSiCl3 (1JHSi = 371.0 Hz) over 4 (1JHSi = 244.0 Hz) to 6 (1JHSi = 178.0 Hz) most likely reflecting the change in hybridization: bonding to Ga will redirect the Si s character to that electropositive atom, which increases the p character in the Si–H bond and results in a smaller 1JHSi coupling constant.32 Moreover, these values are in accordance to those of Cl–Si–SiHCl2 complexes (5.34 ppm,22b 4.64 ppm (ref. 24)) formed in Si–Cl bond insertion of HSiCl3 with silylenes. Unfortunately, no meaningful 29Si NMR spectra of compound 7 was obtained due to its low solubility in organic solvents, whereas the 29Si spectra of 4 and 6 show resonces at 9.0 ppm (1JSiH = 244 Hz, 4) and −30.5 ppm (1JSi–H = 178 Hz, 6), respectively. The γ-CH resonances of compounds 1–7 remain largely unaffected by the different substituents at the Ga centers (δ1H 4.79–4.88; δ13C 97.7–98.6). The IR absorption bands corresponding to the Si–H stretching mode (νSi–H) are found at 2181 cm−1 and 2108 cm−1 for compounds 4 and 6, respectively.Single-crystal structure analysis
sc-XRD analyses confirmed the molecular structures of 1–7 in the solid state as one- and two-fold C–Cl and Si–Cl bond-activated complexes (Fig. 2, 3, S22 and S23†). Suitable crystals of 1–7 were obtained from saturated solutions in n-pentane (1), n-hexane (2, 5), benzene (4, 6, 7), and DCM/n-hexane (3). Compounds 2 and 4 as well as 3 and IV8 crystallize isomorphous in the orthorhombic space group Pnma and the monoclinic space group P21/m, respectively, while compound 1 crystallizes in the monoclinic space group P21/n with two independent yet similar molecules in the asymmetric unit, in which the CH2Cl and Ga(Cl)CH2Cl moieties are disordered over two positions, respectively. The SiHCl moiety in compound 6, which crystallized in the monoclinic space group P21/c, is disordered over two positions. Digallyl complexes 5 and 7 crystallize in the monoclinic space group C2/c and the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , respectively, and both incorporate co-crystallized solvent molecules (n-hexane 5, benzene 7). Selected bond lengths and angles are summarized in Table 1. The LGa moieties in the molecular structures of 1–7 feature the typical metrics observed in complexes of the type LGa(X)Y, i.e., the Ga atoms are located significantly out of the C3N2 planes of the ligand backbones. The Ga atoms adopt distorted tetrahedral coordination geometries with Cl–Ga–E bond angles ranging from 105.15(4)° to 117.90(3)°, which is slightly larger than the values observed in digallyl complexes 5–7. The Ga–C and Ga–Cl bond lengths of 1–3 are virtually identical to those of L(Cl)GaMe (Ga–C 1.956(2) Å, Ga–Cl 2.223(1) Å),33I (Ga–C 2.029(8) Å, Ga–Cl 2.251(2) Å),8 and L(Cl)GaCp (Ga–C 2.0006(17) Å, Ga–Cl 2.1887(5) Å),34 and the C–Cl bond lengths agree with those of related Si–CH2Cl (1.758 Å) and Si–C(Me)Cl2 (1.773 Å, 1.775 Å) complexes.22b The Ga–Cl bond lengths in 4 are similar to those of 1–3, and the Ga–Si and Si–Cl bonds lengths correspond to those of compounds IV (Ga–Si 2.3860(13) Å, Si–Cl 1.999(2) Å, 2.0135(15) Å)8 and V (Ga–Si 2.3992(14) Å).14 The [L(Cl)Ga] ligands in digallyl complexes 5–7 adopt twisted syn,syn conformations with respect to the Cl atoms, and the compounds feature Ga–E–Ga bond angles ranging from 123.01(6)° to 129.09(2)° comparable to those reported for other complexes of the type [L(X)Ga]2E (E = PBr,35 InEt,15 SnMe2,8 SiH2,14 SiBr2;14 Ga–E–Ga 120.01°–129.83°). Interestingly, the Ga–C bonds in 5 are considerably shorter than those in 1–3, while the Ga–Cl and Ga–N bonds are slightly elongated, which can be explained by a negative hyperconjugative interaction from the C–H bond orbital to the antibonding Ga–Cl/N orbitals (σC–H → σ*Ga–Cl/N) as were observed in related complexes containing electron-rich pnictogen centers instead of the CH2 group.16i,36 The Ga–C bond lengths in 5 are comparable to those of [Ga8(μ-CH2)12] (1.960(2) Å, 1.961(1) Å, 1.972(2) Å).25 The Ga–Si bond lengths in 7 agree with those of compound V (2.4247(5) Å, 2.4344(5) Å),14 while those of 6 significantly differ from each other (by 0.09 Å) with one longer and one shorter Ga–Si bond. The shorter Ga–Si bond in 6 is close to that of [L(Br)Ga]2SiH2 (2.3788(7) Å),14 which suggests uneven steric repulsion between the H and Cl, and the [L(Cl)Ga] ligands, respectively, leading to the observed distortions of bond lengths. The Ga–Cl and Si–Cl bonds in 6 and 7 are similar to those of compound 4 and IV (Ga–Cl 2.1980(11) Å, Si–Cl 1.999(2) Å, 2.0135(15) Å), respectively.8
, respectively, and both incorporate co-crystallized solvent molecules (n-hexane 5, benzene 7). Selected bond lengths and angles are summarized in Table 1. The LGa moieties in the molecular structures of 1–7 feature the typical metrics observed in complexes of the type LGa(X)Y, i.e., the Ga atoms are located significantly out of the C3N2 planes of the ligand backbones. The Ga atoms adopt distorted tetrahedral coordination geometries with Cl–Ga–E bond angles ranging from 105.15(4)° to 117.90(3)°, which is slightly larger than the values observed in digallyl complexes 5–7. The Ga–C and Ga–Cl bond lengths of 1–3 are virtually identical to those of L(Cl)GaMe (Ga–C 1.956(2) Å, Ga–Cl 2.223(1) Å),33I (Ga–C 2.029(8) Å, Ga–Cl 2.251(2) Å),8 and L(Cl)GaCp (Ga–C 2.0006(17) Å, Ga–Cl 2.1887(5) Å),34 and the C–Cl bond lengths agree with those of related Si–CH2Cl (1.758 Å) and Si–C(Me)Cl2 (1.773 Å, 1.775 Å) complexes.22b The Ga–Cl bond lengths in 4 are similar to those of 1–3, and the Ga–Si and Si–Cl bonds lengths correspond to those of compounds IV (Ga–Si 2.3860(13) Å, Si–Cl 1.999(2) Å, 2.0135(15) Å)8 and V (Ga–Si 2.3992(14) Å).14 The [L(Cl)Ga] ligands in digallyl complexes 5–7 adopt twisted syn,syn conformations with respect to the Cl atoms, and the compounds feature Ga–E–Ga bond angles ranging from 123.01(6)° to 129.09(2)° comparable to those reported for other complexes of the type [L(X)Ga]2E (E = PBr,35 InEt,15 SnMe2,8 SiH2,14 SiBr2;14 Ga–E–Ga 120.01°–129.83°). Interestingly, the Ga–C bonds in 5 are considerably shorter than those in 1–3, while the Ga–Cl and Ga–N bonds are slightly elongated, which can be explained by a negative hyperconjugative interaction from the C–H bond orbital to the antibonding Ga–Cl/N orbitals (σC–H → σ*Ga–Cl/N) as were observed in related complexes containing electron-rich pnictogen centers instead of the CH2 group.16i,36 The Ga–C bond lengths in 5 are comparable to those of [Ga8(μ-CH2)12] (1.960(2) Å, 1.961(1) Å, 1.972(2) Å).25 The Ga–Si bond lengths in 7 agree with those of compound V (2.4247(5) Å, 2.4344(5) Å),14 while those of 6 significantly differ from each other (by 0.09 Å) with one longer and one shorter Ga–Si bond. The shorter Ga–Si bond in 6 is close to that of [L(Br)Ga]2SiH2 (2.3788(7) Å),14 which suggests uneven steric repulsion between the H and Cl, and the [L(Cl)Ga] ligands, respectively, leading to the observed distortions of bond lengths. The Ga–Cl and Si–Cl bonds in 6 and 7 are similar to those of compound 4 and IV (Ga–Cl 2.1980(11) Å, Si–Cl 1.999(2) Å, 2.0135(15) Å), respectively.8
 | ||
| Fig. 2 Molecular structures of 1, 2, and 3 in the solid-state. Only one of the two independent molecules of 1 is shown. Hydrogen atoms (except CH2Cl and CHCl2) and the minor component of the disordered CH2Cl moiety were omitted for clarity. Displacement ellipsoids are drawn at 50% probability level, whereas hydrogen atoms are displayed as spheres of arbitrary radius. Symmetry generated parts in 2 and 3 are depicted with pale inner lines. | ||
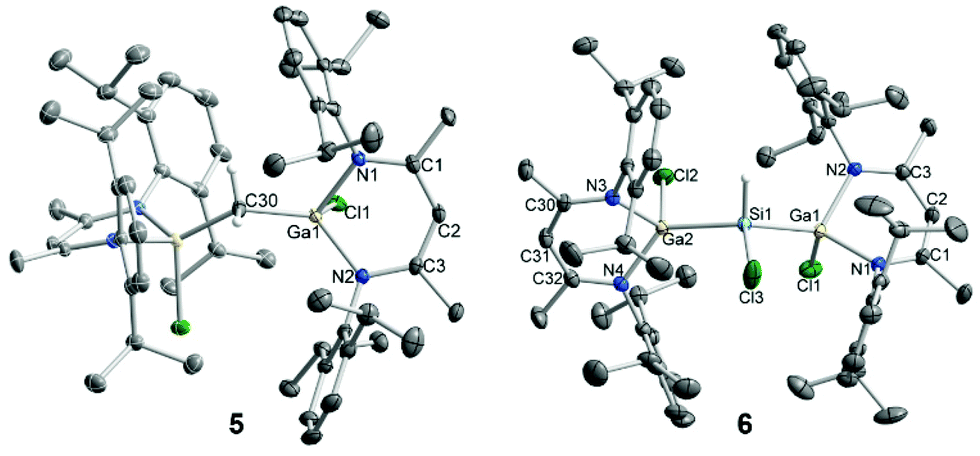 | ||
| Fig. 3 Molecular structures of 5 and 6 in the solid-state. Hydrogen atoms (except CH2 and SiHCl), the minor component of the disordered SiHCl moiety, and co-crystallized solvent molecules were omitted for clarity. Displacement ellipsoids are drawn at 50% probability level, whereas hydrogen atoms are displayed as spheres of arbitrary radius. Symmetry generated parts in 5 are depicted with pale inner lines. | ||
| 1 , | 2 | 3 | 4 | 5 | 6b | 7 | |
|---|---|---|---|---|---|---|---|
| a Average values of the two independent molecules. b Values given for the major component of disorder only. | |||||||
| Ga–Cl | 2.230(14) | 2.1933(5) | 2.1928(5) | 2.2016(5) | 2.2264(4) | 2.2131(4) | 2.2171(3) |
| 2.2173(5) | 2.2249(4) | ||||||
| Ga–E | 2.035(3) | 2.0172(19) | 2.0150(17) | 2.3809(6) | 1.9413(8) | 2.4540(14) | 2.4240(4) |
| 2.3632(14) | 2.4168(4) | ||||||
| Ga–N | 1.938(3) | 1.9283(9) | 1.9233(9) | 1.9281(9) | 1.9628(10) | 1.9548(12) | 1.9565(9) |
| 1.899(3) | 1.9481(10) | 1.9556(12) | 1.9537(9) | ||||
| 1.9546(12) | 1.9480(9) | ||||||
| 1.9519(12) | 1.9494(10) | ||||||
| E–Cl | 1.753(2) | 1.7731(12) | 1.7617(19) | 2.0528(5) | — | 2.0948(17) | 2.0807(5) |
| 1.7827(11) | 2.0775(5) | ||||||
| Cl–Ga–E | 114.2(7) | 110.58(6) | 106.84(6) | 109.56(2) | 117.90(3) | 116.20(4) | 115.11(2) |
| 105.15(4) | 113.69(2) | ||||||
| N–Ga–E | 113.0(8) | 115.04(4) | 118.10(4) | 117.00(3) | 109.96(5) | 117.81(5) | 122.68(3) |
| 110.8(7) | 124.78(3) | 118.18(5) | 114.65(3) | ||||
| 112.90(5) | 116.68(3) | ||||||
| 131.26(5) | 121.50(3) | ||||||
| Ga–E–Ga | — | — | — | — | 127.16(9) | 123.01(6) | 129.09(2) |
Conclusions
Gallanediyl LGa reacts with chloromethane solvents and chlorosilanes selectively with E–Cl bond activation, yielding oxidative addition products L(Cl)GaCHxCl3−x (x = 0 (3), 1 (2), 2 (1)) and L(Cl)GaSiHxCl3−x (x = 0 (IV), 1 (4)). Consecutive insertion of LGa into a geminal E–Cl bond furnished digallyl complexes [L(Cl)Ga]2CH2 (5), [L(Cl)Ga]2SiHCl (6), and [L(Cl)Ga]2SiCl2 (7), with 5 representing a rare Ga methylene complex. Hence, the scope of C–X (X = F, Cl, Br, I) bond activation reactions mediated by Ga(I) complexes was extended to compounds containing geminal C–X bonds. Moreover, the influence of the Si–X bond strength was disclosed as [L(Cl)Ga]2SiCl2 failed to give a silylene carbonyl complex analogous to V. Further studies on the C–X bond activation of polyhaloalkanes by Ga(I) as well as the utilization of carbenoids 1–3 as precursors for the release of halogenated and functionalized carbenes, and the use of 5 as methylene transfer reagent in synthetic transformations are under current investigation in our laboratory.Experimental
General procedures, materials, and instrumentation
All manipulations were carried out under an atmosphere of purified argon using standard Schlenk and glovebox techniques. Toluene, n-hexane, and n-pentane were dried with a MBraun Solvent Purification System (SPS), and benzene was distilled from Na/K alloy. Dichloromethane and chloroform were distilled from CaH2 and CaCl2, respectively, while carbon tetrachloride was dried with molecular sieves (4 Å) and degassed. Deuterated benzene was dried over activated molecular sieves (4 Å) and degassed prior to use. LGa37 was prepared according to literature procedures. SiCl4 and HSiCl3 were obtained from commercial sources and used as received. NMR spectra (δ in ppm) were recorded using a Bruker Avance DPX 300 (1H 300.1 MHz, 13C{1H} 75.5 MHz) or a Bruker Avance Neo 400 (1H 400.1 MHz, 13C{1H} 100.6 MHz) spectrometer and were referenced to internal C6D5H (1H δ = 7.16, 13C δ = 128.06). IR spectra were recorded in a glovebox with an ALPHA-T FT-IR spectrometer equipped with a single-reflection ATR sampling module. Microanalyses were performed at the Elemental Analysis Laboratory of the University of Duisburg-Essen. Melting points were measured in wax-sealed glass capillaries under argon atmosphere using a Thermo Scientific 9300 apparatus and are uncorrected.Synthesis of L(Cl)GaCH2Cl (1)
LGa (50 mg, 0.1026 mmol) was dissolved in CH2Cl2 (2 mL) yielding a colorless solution. Volatiles were removed in vacuo and the residue was dissolved in n-pentane (0.8 mL). Storage of the solution at ambient temperature overnight afforded colorless analytically pure crystals of 1. Yield: 38 mg (0.0664 mmol, 65%). Mp: 177 °C (dec.). Anal. calcd for C30H43Cl2GaN2: C, 62.96; H, 7.57; N, 4.99. Found: C, 63.1; H, 7.58; N, 5.17. 1H NMR (400.1 MHz, C6D6): δ 7.13 (m, 4 H, C6H3(i-Pr)2), 7.03 (m, 2 H, C6H3(i-Pr)2), 4.83 (s, 1 H, γ-CH), 3.74 (sept, 3JHH = 6.7 Hz, 2 H, CH(CH3)2), 3.18 (sept, 3JHH = 6.8 Hz, 2 H, CH(CH3)2), 2.62 (s, 2 H, CH2Cl), 1.53 (s, 6 H, CCH3), 1.50 (d, 3JHH = 6.8 Hz, 6 H, CH(CH3)2), 1.25 (d, 3JHH = 6.8 Hz, 6 H, CH(CH3)2), 1.19 (d, 3JHH = 6.7 Hz, 6 H, CH(CH3)2), 1.03 (d, 3JHH = 6.7 Hz, 6 H, CH(CH3)2). 13C{1H} NMR (100.6 MHz, C6D6): δ 170.4 (CCH3), 145.8 (C6H3(i-Pr)2), 143.1 (C6H3(i-Pr)2), 140.0 (C6H3(i-Pr)2), 127.9 (C6H3(i-Pr)2), 125.5 (C6H3(i-Pr)2), 124.0 (C6H3(i-Pr)2), 97.8 (γ-CH), 29.0 (CH(CH3)2), 28.3 (CH(CH3)2), 26.6 (CH(CH3)2), 25.4 (CH2Cl), 25.0 (CH(CH3)2), 24.5 (CH(CH3)2), 24.0 (CH(CH3)2), 23.4 (CCH3). IR (neat): ν 2964, 2962, 2867, 1526, 1438, 1383, 1316, 1260, 1178, 1021, 935, 872, 800, 758, 553, 450 cm−1.Synthesis of L(Cl)GaCHCl2 (2)
LGa (50 mg, 0.1026 mmol) was dissolved in CHCl3 (2 mL) yielding a colorless solution. Volatiles were removed in vacuo and the residue was dissolved in hot n-hexane (1 mL). Storage of the solution at ambient temperature overnight afforded colorless analytically pure crystals of 2. Yield: 48 mg (0.0791 mmol, 77%). Mp: 241 °C (dec.). Anal. calcd for C30H42Cl3GaN2: C, 59.39; H, 6.98; N, 4.62. Found: C, 59.3; H, 6.89; N, 4.79. 1H NMR (400.1 MHz, C6D6): δ 7.13 (m, 4 H, C6H3(i-Pr)2), 7.01 (m, 2 H, C6H3(i-Pr)2), 5.31 (s, 1 H, CHCl2), 4.79 (s, 1 H, γ-CH), 3.71 (sept, 3JHH = 6.7 Hz, 2 H, CH(CH3)2), 3.09 (sept, 3JHH = 6.8 Hz, 2 H, CH(CH3)2), 1.55 (d, 3JHH = 6.8 Hz, 6 H, CH(CH3)2), 1.51 (s, 6 H, CCH3), 1.26 (d, 3JHH = 6.8 Hz, 6 H, CH(CH3)2), 1.19 (d, 3JHH = 6.7 Hz, 6 H, CH(CH3)2), 0.99 (d, 3JHH = 6.7 Hz, 6 H, CH(CH3)2). 13C{1H} NMR (100.6 MHz, C6D6): δ 170.9 (CCH3), 145.7 (C6H3(i-Pr)2), 142.6 (C6H3(i-Pr)2), 140.1 (C6H3(i-Pr)2), 125.6 (C6H3(i-Pr)2), 123.8 (C6H3(i-Pr)2), 97.7 (γ-CH), 59.6 (CHCl2), 29.4 (CH(CH3)2), 28.2 (CH(CH3)2), 26.4 (CH(CH3)2), 25.0 (CH(CH3)2), 24.6 (CH(CH3)2), 23.7 (CH(CH3)2), 23.4 (CCH3). IR (neat): ν 3060, 2965, 2926, 2867, 1528, 1439, 1378, 1314, 1254, 1178, 1021, 934, 873, 799, 760, 684, 519, 453 cm−1.Synthesis of L(Cl)GaCCl3 (3)
An excess of CCl4 (0.1 mL) was added to a solution of LGa (200 mg, 0.4104 mmol) in toluene (5 mL) at −40 °C. The solution was warmed to ambient temperature and volatiles were removed in vacuo. The residue was dissolved in warm dichloromethane and stored at −30 °C overnight to afford colorless analytically pure crystals of 3. Yield: 95 mg (0.1481 mmol, 36%). Mp: 158 °C (dec.). Anal. calcd for C30H41Cl4GaN2: C, 56.20; H, 6.45; N, 4.37. Found: C, 56.35; H, 6.31; N, 4.46. 1H NMR (400.1 MHz, C6D6): 7.12 (m, 4 H, C6H3(i-Pr)2), 7.04 (m, 2 H, C6H3(i-Pr)2), 4.88 (s, 1 H, γ-CH), 3.80 (sept, 3JHH = 6.7 Hz, 2 H, CH(CH3)2), 3.40 (sept, 3JHH = 6.8 Hz, 2 H, CH(CH3)2), 1.56 (s, 6 H, CCH3), 1.52 (d, 3JHH = 6.8 Hz, 6 H, CH(CH3)2), 1.30 (d, 3JHH = 6.8 Hz, 6 H, CH(CH3)2), 1.18 (d, 3JHH = 6.7 Hz, 6 H, CH(CH3)2), 1.00 (d, 3JHH = 6.7 Hz, 6 H, CH(CH3)2). 13C{1H} NMR (100.6 MHz, C6D6): δ 172.0 (CCH3), 145.7 (C6H3(i-Pr)2), 143.2 (C6H3(i-Pr)2), 140.5 (C6H3(i-Pr)2), 125.5 (C6H3(i-Pr)2), 124.0 (C6H3(i-Pr)2), 98.5 (γ-CH), 29.8 (CH(CH3)2), 28.3 (CH(CH3)2), 25.6 (CH(CH3)2), 23.8 (CH(CH3)2), 25.0 (CH(CH3)2), 24.0 (CCH3) (CCl3 not observed). IR (neat): ν 3050, 2954, 2917, 2857, 1521, 1430, 1378, 1310, 1255, 1174, 1016, 933, 872, 793, 754, 710, 687, 635, 530, 441 cm−1.Synthesis of L(Cl)GaSiHCl2 (4)
HSiCl3 (27 mg, 0.1990 mmol, 20 μL) was added to a solution of LGa (97 mg, 0.1990 mmol) in benzene (1 mL) and the solution was stirred for 1 h at ambient temperature. Storage of the solution at 8 °C overnight afforded colorless analytically pure crystals of 4. Yield: 80 mg (0.1280 mmol, 65%). Mp: 190 °C (dec.). Anal. calcd for C29H42Cl3GaN2Si: C, 55.93; H, 6.80; N, 4.50. Found: C, 56.0; H, 6.75; N, 4.53. 1H NMR (300.1 MHz, C6D6): δ 7.18–7.03 (m, 6 H, C6H3(i-Pr)2), 5.69 (s, satellite: 1JHSi = 244.0 Hz, 1 H, SiH), 4.85 (s, 1 H, γ-CH), 3.73 (sept, 3JHH = 6.6 Hz, 2 H, CH(CH3)2), 3.16 (sept, 3JHH = 6.9 Hz, 2 H, CH(CH3)2), 1.54 (s, 6 H, CCH3), 1.51 (d, 3JHH = 6.6 Hz, 6 H, CH(CH3)2), 1.33 (d, 3JHH = 6.9 Hz, 6 H, CH(CH3)2), 1.18 (d, 3JHH = 6.9 Hz, 6 H, CH(CH3)2), 1.00 (d, 3JHH = 6.9 Hz, 6 H, CH(CH3)2). 13C{1H} NMR (C6D6, 75.5 MHz): δ 170.2 (CCH3), 146.1 (C6H3(i-Pr)2), 142.3 (C6H3(i-Pr)2), 140.2 (C6H3(i-Pr)2), 128.2 (C6H3(i-Pr)2), 125.8 (C6H3(i-Pr)2), 124.1 (C6H3(i-Pr)2), 98.0 (γ-CH), 29.8 (CH(CH3)2), 28.2 (CH(CH3)2), 26.8 (CH(CH3)2), 24.9 (CH(CH3)2), 24.5 (CH(CH3)2), 23.8 (CH(CH3)2), 23.2 (CCH3). 29Si NMR (119 MHz, C6D6, DEPT90): 9.0 (1JHSi = 244.0 Hz). IR (neat): ν 2966, 2923, 2864, 2181, 1525, 1462, 1435, 1380, 1314, 1252, 1177, 1099, 1021, 932, 873, 794, 756, 638, 592, 541, 513, 435 cm−1.Synthesis of [L(Cl)Ga]2CH2 (5)
A solution of LGa (90 mg, 0.1573 mmol) and 1 (77 mg, 0.1573 mmol) in benzene (1 mL) was heated to 100 °C for 7 h. Volatiles were removed in vacuo and the residue was dissolved in hot n-hexane (5 mL). Storage of the solution at −30 °C overnight afforded colorless analytically pure crystals of 5. Yield: 80 mg (0.0755 mmol, 48%). Mp: 358 °C. Anal. calcd for C59H84Cl2Ga2N4·C6H14: C, 68.13; H, 8.62; N, 4.89. Found: C, 68.1; H, 8.37; N, 4.86. 1H NMR (400.1 MHz, C6D6): δ 7.19 (dd, JHH = 7.6, 1.6 Hz, 4 H, C6H3(i-Pr)2), 7.13 (t, 3JHH = 7.6 Hz, 4 H, C6H3(i-Pr)2), 6.98 (dd, JHH = 7.6, 1.6 Hz, 4 H, C6H3(i-Pr)2), 4.88 (s, 2 H, γ-CH), 3.65 (sept, 3JHH = 6.7 Hz, 4 H, CH(CH3)2), 2.93 (sept, 3JHH = 6.7 Hz, 4 H, CH(CH3)2), 1.45 (s, 12 H, CCH3), 1.25 (d, 3JHH = 6.7 Hz, 12 H, CH(CH3)2), 1.20 (d, 3JHH = 6.7 Hz, 12 H, CH(CH3)2), 0.98 (d, 3JHH = 6.7 Hz, 12 H, CH(CH3)2), 0.94 (d, 3JHH = 6.7 Hz, 12 H, CH(CH3)2), −1.02 (s, 2 H, CH2). 13C{1H} NMR (C6D6, 100.6 MHz): δ 168.9 (CCH3), 145.9 (C6H3(i-Pr)2), 142.1 (C6H3(i-Pr)2), 142.0 (C6H3(i-Pr)2), 127.0 (C6H3(i-Pr)2), 125.5 (C6H3(i-Pr)2), 123.9 (C6H3(i-Pr)2), 97.9 (γ-CH), 28.9 (CH(CH3)2), 28.4 (CH(CH3)2), 27.4 (CH(CH3)2), 25.1 (CH(CH3)2), 24.7 (CH(CH3)2), 24.6 (CH(CH3)2), 24.2 (CCH3), −12.4 (CH2). IR (neat): ν 2953, 2916, 2857, 1520, 1430, 1377, 1312, 1254, 1174, 1095, 1018, 993, 933, 862, 793, 757, 684, 550, 497, 449 cm−1.Synthesis of [L(Cl)Ga]2SiHCl (6)
HSiCl3 (14 mg, 0.0990 mmol, 10 μL) was added to a solution of LGa (97 mg, 0.1990 mmol) in benzene (1 mL) and the solution was heated to 80 °C for 2 days. Storage of the solution at 8 °C overnight afforded colorless analytically pure crystals of 6. Yield: 67 mg (0.0600 mmol, 61%). Mp: 244 °C (dec.). Anal. calcd for C58H83Cl3Ga2N4Si: C, 62.75; H, 7.54; N, 5.05. Found: C, 62.78; H, 7.57; N, 5.15. 1H NMR (300.1 MHz, C6D6): δ 7.19–7.02 (m, 12 H, C6H3(i-Pr)2), 4.88 (s, 2 H, γ-CH), 4.30 (s, satellite: 1JHSi = 178.0 Hz, 1 H, SiH), 3.78 (sept, 3JHH = 6.9 Hz, 2 H, CH(CH3)2), 3.54 (sept, 3JHH = 6.6 Hz, 2 H, CH(CH3)2), 3.18 (m, 4 H, CH(CH3)2), 1.50 (s, 6 H, CCH3), 1.49 (s, 6 H, CCH3), 1.30 (d, 3JHH = 6.6 Hz, 6 H, CH(CH3)2), 1.29 (d, 3JHH = 6.9 Hz, 6 H, CH(CH3)2), 1.23 (d, 3JHH = 6.9 Hz, 6 H, CH(CH3)2), 1.21 (d, 3JHH = 6.9 Hz, 6 H, CH(CH3)2), 1.19 (d, 3JHH = 6.9 Hz, 6 H, CH(CH3)2), 1.15 (d, 3JHH = 6.6 Hz, 6 H, CH(CH3)2), 0.99 (d, 3JHH = 6.6 Hz, 6 H, CH(CH3)2), 0.95 (d, 3JHH = 6.6 Hz, 6 H, CH(CH3)2). 13C{1H} NMR (C6D6, 75.5 MHz): δ 169.6 (CCH3), 169.3 (CCH3), 146.2 (C6H3(i-Pr)2), 146.0 (C6H3(i-Pr)2), 143.2 (C6H3(i-Pr)2), 142.4 (C6H3(i-Pr)2), 142.3 (C6H3(i-Pr)2), 141.2 (C6H3(i-Pr)2), 127.7 (C6H3(i-Pr)2), 127.5 (C6H3(i-Pr)2), 125.3 (C6H3(i-Pr)2), 125.0 (C6H3(i-Pr)2), 123.7 (C6H3(i-Pr)2), 123.5 (C6H3(i-Pr)2), 98.6 (γ-CH), 29.8 (CH(CH3)2), 29.5 (CH(CH3)2), 28.3 (CH(CH3)2), 28.1 (CH(CH3)2), 27.9 (CH(CH3)2), 27.8 (CH(CH3)2), 25.1 (CH(CH3)2), 24.9 (CH(CH3)2), 24.4 (CH(CH3)2), 24.3 (CH(CH3)2), 24.1 (CH(CH3)2), 24.0 (CH(CH3)2), 23.6 (CCH3), 23.3 (CCH3). 29Si NMR (119 MHz, C6D6, DEPT90): −30.5 (1JHSi = 178.0 Hz). IR (neat): ν 2958, 2922, 2866, 2108, 1521, 1434, 1382, 1314, 1255, 1176, 1100, 1017, 937, 866, 799, 759, 735, 683, 636, 501, 441, 410 cm−1.Synthesis of [L(Cl)Ga]2SiCl2 (7)
SiCl4 (15 mg, 0.0870 mmol, 10 μL) was added to a solution of LGa (85 mg, 0.1740 mmol) in benzene (1 mL) and the solution was heated to 80 °C for 3 days. Storage of the solution at 8 °C overnight afforded colorless analytically pure crystals of 7. Yield: 78 mg (0.0680 mmol, 78%). Mp: 208 °C (dec.). Anal. calcd for C58H82Cl4Ga2N4Si: C, 60.86; H, 7.22; N, 4.89. Found: C, 60.83; H, 7.28; N, 4.81. 1H NMR (300.1 MHz, C6D6): δ 7.20–7.03 (m, 12 H, C6H3(i-Pr)2), 4.85 (s, 2 H, γ-CH), 3.67 (sept, 3JHH = 6.9 Hz, 4 H, CH(CH3)2), 3.23 (sept, 3JHH = 6.9 Hz, 4 H, CH(CH3)2), 1.46 (s, 12 H, CCH3), 1.33 (d, 3JHH = 6.6 Hz, 12 H, CH(CH3)2), 1.32 (d, 3JHH = 6.6 Hz, 12 H, CH(CH3)2), 1.23 (d, 3JHH = 6.9 Hz, 12 H, CH(CH3)2), 0.94 (d, 3JHH = 6.6 Hz, 12 H, CH(CH3)2). 13C{1H} NMR (C6D6, 75.5 MHz): δ 170.0 (CCH3), 145.9 (C6H3(i-Pr)2), 143.0 (C6H3(i-Pr)2), 142.4 (C6H3(i-Pr)2), 127.7 (C6H3(i-Pr)2), 125.2 (C6H3(i-Pr)2), 123.8 (C6H3(i-Pr)2), 98.6 (γ-CH), 29.6 (CH(CH3)2), 28.2 (CH(CH3)2), 27.9 (CH(CH3)2), 25.0 (CH(CH3)2), 24.6 (CH(CH3)2), 24.1 (CH(CH3)2), 24.0 (CCH3). A 29Si NMR spectrum of 7 could not be obtained due to its poor solubility. IR (neat): ν 2957, 2924, 2868, 1521, 1433, 1380, 1312, 1256, 1179, 1098, 1022, 937, 865, 796, 760, 711, 638, 496, 441 cm−1.Single-crystal X-ray diffraction
The crystals of 1–7 were mounted on nylon loops in inert oil. Crystallographic data of 1 was collected on a Bruker AXS D8 Venture diffractometer with Photon II detector (CuKα radiation, λ = 1.54178 Å, micro-focus source) at 102(2) K (Table S1†). Absorption corrections were performed semi-empirically from equivalent reflections on the basis of multiscans (Bruker AXS APEX2). Crystallographic data of 2–7 were collected on a Bruker D8 Kappa diffractometer with APEX2 detector (MoKα radiation, λ = 0.71073 Å) at 100(2) K and are summarized in Tables S1 and S2.† Absorption corrections were performed semi-empirically from equivalent reflections on the basis of multiscans (Bruker AXS APEX3). The structures were solved by direct methods (SHELXS-97)38 and refined anisotropically by full-matrix least-squares on F2 (SHELXL-2014).39 Hydrogen atoms were refined using a riding model or rigid methyl groups. In the crystal of 1, the central Ga(Cl)CH2Cl moiety in residue 1 and the CH2Cl group in residue 2 are disordered. Two alternate positions each were used for the modelling. In residue 1 a third orientation was identified yet strong parameter correlations made a refinement impossible thus the rather large anisotropic displacement parameters had to be accepted. All corresponding bond lengths of the disordered parts were restrained to be equal (SADI), additionally, the bond angles were restrained (SADI) in residue 1. RIGU restraints were applied to the anisotropic displacement parameters of the disordered atoms of residue 1, and C30_1 and C30′_1 were refined with common displacement parameters (EADP). The crystal of 2 was a non-merohedral twin and the model was refined against de-twinned HKLF4 data. In 4, one Dip group is disordered over two positions. Its bond lengths were restrained with SADI. In 5, an n-hexane molecule is disordered over two positions. All bond lengths and angles were restrained to be equal (SADI) and RIGU restraints were applied to the anisotropic displacement parameters. In 6, the central Si(H)Cl unit is disordered over two positions. The SiH hydrogen atoms were refined freely and the Si–H bond lengths were restrained to be equal (SADI). In 7, one of the four benzene molecules is disordered over two positions. The ADPs of the others suggest minor disorder. Any attempt to separate different orientations failed. All 1,2 and 1,3 distances of the solvent molecules were restraint to be equal (SADI) and the atoms were restrained to be on a common plane (FLAT). RIGU and ISOR restraints were applied to the atoms of the disorder model.CCDC-2127285 (1), 2127286 (2), 2127287 (3), 2127288 (4), 2127289 (5), 2127290 (6), 2127291 (7) contain the supplementary crystallographic data for this paper.†
Author contributions
C. H. and C. G. planned and executed all the experimental work and analysed the corresponding data. C. W. conducted the sc-XRD measurements and processed corresponding data. C. H. wrote the manuscript. S. S. supervised the project and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the University of Duisburg-Essen, the German Research Foundation DFG (grant no. SCHU 1069/26-1), and Evonik Industries (C. H., doctoral fellowship) is gratefully acknowledged. We thank J. Schoening for performing 29Si NMR measurements with compounds 4 and 6.Notes and references
- (a) T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608 CrossRef CAS PubMed; (b) P. P. Power, Nature, 2010, 463, 171 CrossRef CAS PubMed; (c) C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213 CrossRef CAS.
- M. Zhong, S. Sinhababu and H. W. Roesky, Dalton Trans., 2020, 49, 1351 RSC.
- (a) N. J. Hardman and P. P. Power, Chem. Commun., 2001, 1866 RSC; (b) C. Ganesamoorthy, M. Matthias, D. Bläser, C. Wölper and S. Schulz, Dalton Trans., 2016, 45, 11437 RSC.
- (a) A. Seifert, D. Scheid, G. Linti and T. Zessin, Chem. – Eur. J., 2009, 15, 12114 CrossRef CAS PubMed; (b) T. Chu, I. Korobkov and G. I. Nikonov, J. Am. Chem. Soc., 2014, 136, 9195 CrossRef CAS PubMed; (c) C. Bakewell, A. J. P. White and M. R. Crimmin, Chem. Sci., 2019, 10, 2452 RSC; (d) H. Zhu, J. Chai, H. Fan, H. W. Roesky, C. He, V. Jancik, H.-G. Schmidt, M. Noltemeyer, W. A. Merrill and P. P. Power, Angew. Chem., Int. Ed., 2005, 44, 5090 CrossRef CAS PubMed.
- (a) G. Prabusankar, C. Gemel, P. Parameswaran, C. Flener, G. Frenking and R. A. Fischer, Angew. Chem., Int. Ed., 2009, 48, 5526 CrossRef CAS PubMed; (b) L. Tuscher, C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, Angew. Chem., Int. Ed., 2015, 54, 10657 CrossRef CAS PubMed; (c) J. Krüger, C. Ganesamoorthy, L. John, C. Wölper and S. Schulz, Chem. – Eur. J., 2018, 24, 9157 CrossRef PubMed; (d) J. Krüger, C. Wölper, L. John, L. Song, P. R. Schreiner and S. Schulz, Eur. J. Inorg. Chem., 2019, 1669 CrossRef; (e) J. Krüger, C. Wölper and S. Schulz, Inorg. Chem., 2020, 59, 11142 CrossRef PubMed.
- K. M. Korch and D. A. Watson, Chem. Rev., 2019, 119, 8192 CrossRef CAS PubMed.
- M. S. Hill, P. B. Hitchcock and R. Pongtavornpinyo, Inorg. Chem., 2007, 46, 3783 CrossRef CAS PubMed.
- A. Kempter, C. Gemel and R. A. Fischer, Inorg. Chem., 2008, 47, 7279 CrossRef CAS PubMed.
- O. Kysliak, H. Görls and R. Kretschmer, J. Am. Chem. Soc., 2021, 143, 142 CrossRef CAS PubMed.
- (a) M. R. Crimmin, M. J. Butler and A. J. P. White, Chem. Commun., 2015, 51, 15994 RSC; (b) T. Chu, Y. Boyko, I. Korobkov and G. I. Nikonov, Organometallics, 2015, 34, 5363 CrossRef CAS; (c) C. Bakewell, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2018, 57, 6638 CrossRef CAS PubMed.
- G. Prabusankar, A. Kempter, C. Gemel, M.-K. Schröter and R. A. Fischer, Angew. Chem., Int. Ed., 2008, 47, 7234 CrossRef CAS PubMed.
- A. Doddi, C. Gemel, M. Winter, R. A. Fischer, C. Goedecke, H. S. Rzepa and G. Frenking, Angew. Chem., Int. Ed., 2013, 52, 450 CrossRef CAS PubMed.
- G. Prabusankar, C. Gemel, M. Winter, R. W. Seidel and R. A. Fischer, Chem. – Eur. J., 2010, 16, 6041 CrossRef CAS PubMed.
- C. Ganesamoorthy, J. Schoening, C. Wölper, L. Song, P. R. Schreiner and S. Schulz, Nat. Chem., 2020, 12, 608 CrossRef CAS PubMed.
- C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, Organometallics, 2015, 34, 2991 CrossRef CAS.
- (a) L. Tuscher, C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, Angew. Chem., Int. Ed., 2015, 54, 10657 CrossRef CAS PubMed; (b) L. Tuscher, C. Helling, C. Ganesamoorthy, J. Krüger, C. Wölper, W. Frank, A. S. Nizovtsev and S. Schulz, Chem. – Eur. J., 2017, 23, 12297 CrossRef CAS PubMed; (c) C. Ganesamoorthy, C. Helling, C. Wölper, W. Frank, E. Bill, G. E. Cutsail III and S. Schulz, Nat. Commun., 2018, 9, 87 CrossRef PubMed; (d) C. Helling, C. Wölper and S. Schulz, J. Am. Chem. Soc., 2018, 140, 5053 CrossRef CAS PubMed; (e) J. Krüger, C. Ganesamoorthy, L. John, C. Wölper and S. Schulz, Chem. – Eur. J., 2018, 24, 9157 CrossRef PubMed; (f) C. Helling, C. Wölper, Y. Schulte, G. Cutsail III and S. Schulz, Inorg. Chem., 2019, 58, 10323 CrossRef CAS PubMed; (g) J. Krüger, C. Wölper, L. John, L. Song, P. R. Schreiner and S. Schulz, Eur. J. Inorg. Chem., 2019, 1669 CrossRef; (h) J. Schoening, L. John, C. Wölper and S. Schulz, Dalton Trans., 2019, 48, 17729 RSC; (i) C. Helling, G. E. Cutsail III, H. Weinert, C. Wölper and S. Schulz, Angew. Chem., Int. Ed., 2020, 59, 7561 CrossRef CAS PubMed; (j) J. Krüger, C. Wölper and S. Schulz, Inorg. Chem., 2020, 59, 11142 CrossRef PubMed; (k) C. Helling, C. Wölper and S. Schulz, Dalton Trans., 2020, 49, 11835 RSC; (l) C. Helling, C. Wölper and S. Schulz, Eur. J. Inorg. Chem., 2020, 4225 CrossRef CAS; (m) B. Li, C. Wölper, G. Haberhauer and S. Schulz, Angew. Chem., Int. Ed., 2021, 60, 1986 CrossRef CAS PubMed; (n) B. Li, C. Wölper, G. Haberhauer and S. Schulz, Angew. Chem., Int. Ed., 2021, 60, 1986 CrossRef CAS PubMed; (o) M. K. Sharma, C. Wölper, G. Haberhauer and S. Schulz, Angew. Chem., Int. Ed., 2021, 60, 6784 CrossRef CAS PubMed; (p) H. M. Weinert, C. Wölper, J. Haak, G. E. Cutsail III and S. Schulz, Chem. Sci., 2021, 12, 14024 RSC.
- S. Schulz, C. Ganesamoorty, G. Bendt, D. Bläser and C. Wölper, Dalton Trans., 2015, 44, 5153 RSC.
- M. Stender, B. E. Eichler, N. J. Hardman, P. P. Power, J. Prust, M. Noltemeyer and H. W. Roesky, Inorg. Chem., 2001, 40, 2794 CrossRef CAS PubMed.
- (a) D. A. Dickie, P. T. K. Lee, O. A. Labeodan, G. Schatte, N. Weinberg, A. R. Lewis, G. M. Bernard, R. E. Wasylishen and J. A. C. Clyburne, Dalton Trans., 2007, 2862 RSC; (b) C. Drost, P. B. Hitchcock and M. F. Lappert, Organometallics, 2002, 21, 2095 CrossRef CAS.
- (a) A. Rit, R. Tirfoin and S. Aldridge, Angew. Chem., Int. Ed., 2016, 55, 378 CrossRef CAS PubMed; (b) M. M. D. Roy, S. Fujimori, M. J. Ferguson, R. McDonald, N. Tokitoh and E. Rivard, Chem. – Eur. J., 2018, 24, 14392 CrossRef CAS PubMed.
- (a) N. Burford, P. Losier, P. K. Bakshi and T. S. Cameron, Dalton Trans., 1993, 201 RSC; (b) N. Burford, P. Losier, C. Macdonald, V. Kyrimis, P. K. Bakshi and T. S. Cameron, Inorg. Chem., 1994, 33, 1434 CrossRef CAS.
- (a) D. C. H. Do, A. V. Protchenko, M. A. Fuentes, J. Hicks, P. Vasko and S. Aldridge, Chem. Commun., 2020, 56, 4684 RSC; (b) Y. Xiong, S. Yao and M. Driess, Organometallics, 2009, 28, 1927 CrossRef CAS.
- (a) C. G. Andrews and C. L. B. Macdonald, Angew. Chem., Int. Ed., 2005, 44, 7453 CrossRef CAS PubMed; (b) B. F. T. Cooper, C. G. Andrews and C. L. B. Macdonald, J. Organomet. Chem., 2007, 692, 2843 CrossRef CAS; (c) C. Peppe, D. G. Tuck, F. M. de Andrade, J. A. Nóbrega, M. A. Brown and R. A. Burrow, J. Organomet. Chem., 2005, 690, 925 CrossRef CAS.
- M. M. D. Roy, M. J. Ferguson, R. McDonald, Y. Zhou and E. Rivard, Chem. Sci., 2019, 10, 6476 RSC.
- M. Bonath, C. Maichle-Mössmer, P. Sirsch and R. Anwander, Angew. Chem., Int. Ed., 2019, 58, 8206 CrossRef CAS PubMed.
- (a) H. Lehmkuhl and R. Schäfer, Tetrahedron Lett., 1966, 7, 2315 CrossRef; (b) M. Layh and W. Uhl, Polyhedron, 1990, 9, 277 CrossRef CAS; (c) W. Uhl, C. Rösener, C. Stefaniak, T. Choy, B. Jasper-Peter, J. Kösters, M. Layh and A. Hepp, Z. Naturforsch., B: J. Chem. Sci., 2012, 67, 1081 CrossRef CAS; (d) K. Knabel, H. Nöth and T. Seifert, Z. Naturforsch., B: J. Chem. Sci., 2002, 57, 830 CrossRef CAS.
- M. A. Khan, C. Peppe and D. G. Tuck, Organometallics, 1986, 5, 525 CrossRef CAS.
- (a) B. Gehrhus and M. F. Lappert, J. Organomet. Chem., 2001, 617–618, 209 CrossRef CAS; (b) S. Ishida, T. Iwamoto, C. Kabuto and M. Kira, Chem. Lett., 2001, 30, 1102 CrossRef; (c) S. S. Sen, J. Hey, D. Kratzert, H. W. Roesky and D. Stalke, Organometallics, 2012, 31, 435 CrossRef CAS.
- M. J. S. Gynane, M. F. Lappert, S. J. Miles, A. J. Carty and N. J. Taylor, Dalton Trans., 1977, 2009 RSC.
- V. Y. Lee, H. Yasuda, M. Ichinohe and A. Sekiguchi, Angew. Chem., Int. Ed., 2005, 44, 6378 CrossRef CAS PubMed.
- T. Sugahara, T. Sasamori and N. Tokitoh, Angew. Chem., Int. Ed., 2017, 56, 9920 CrossRef CAS PubMed.
- (a) W. Scherer, P. Meixner, K. Batke, J. E. Barquera-Lozada, K. Ruhland, A. Fischer, G. Eickerling and K. Eichele, Angew. Chem., Int. Ed., 2016, 55, 11673 CrossRef CAS PubMed; (b) P. Meixner, K. Batke, A. Fischer, D. Schmitz, G. Eickerling, M. Kalter, K. Ruhland, K. Eichele, J. E. Barquera-Lozada, N. P. M. Casati, F. Montisci, P. Macchi and W. Scherer, J. Phys. Chem. A, 2017, 121, 7219 CrossRef CAS PubMed.
- S. Singh, H.-J. Ahn, A. Stasch, V. Jancik, H. W. Roesky, A. Pal, M. Biadene, R. Herbst-Irmer, M. Noltemeyer and H.-G. Schmidt, Inorg. Chem., 2006, 45, 1853 CrossRef CAS PubMed.
- C. Helling, C. Wölper, G. E. Cutsail III, G. Haberhauer and S. Schulz, Chem. – Eur. J., 2020, 26, 13390 CrossRef CAS PubMed.
- L. Tuscher, C. Helling, C. Wölper, W. Frank, A. S. Nizovtsev and S. Schulz, Chem. – Eur. J., 2018, 24, 3241 CrossRef CAS PubMed.
- (a) C. Helling, C. Wölper and S. Schulz, Dalton Trans., 2020, 49, 11835 RSC; (b) J. Krüger, C. Wölper and S. Schulz, Angew. Chem., Int. Ed., 2021, 60, 3572 CrossRef PubMed.
- N. J. Hardman, B. E. Eichler and P. P. Power, Chem. Commun., 2000, 1991 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
- (a) G. M. Sheldrick, SHELXL-2014, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 2014. See also Search PubMedG. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed; (b) shelXle, A Qt GUI for SHELXL: C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic (NMR and IR) and crystallographic data. CCDC 2127285–2127291. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt04192d |
| This journal is © The Royal Society of Chemistry 2022 |