
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tungsten(VI) selenide tetrachloride, WSeCl4 – synthesis, properties, coordination complexes and application of [WSeCl4(SenBu2)] for CVD growth of WSe2 thin films†
Victoria K.
Greenacre
 *a,
Andrew L.
Hector
a,
Ruomeng
Huang
b,
William
Levason
a,
Vikesh
Sethi
b and
Gillian
Reid
*a
*a,
Andrew L.
Hector
a,
Ruomeng
Huang
b,
William
Levason
a,
Vikesh
Sethi
b and
Gillian
Reid
*a
aSchool of Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: V.K.Greenacre@soton.ac.uk; G.Reid@soton.ac.uk
bSchool of Electronics and Computer Science, University of Southampton, Southampton SO17 1BJ, UK
First published on 11th January 2022
Abstract
WSeCl4 was obtained in good yield by heating WCl6 and Sb2Se3in vacuo. Green crystals grown by sublimation were shown by single crystal X-ray structure analysis to contain square pyramidal monomers with apical W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Se, and powder X-ray diffraction (PXRD) analysis confirmed this to be the only form present in the bulk sample. Density functional theory (DFT) calculations using the B3LYP-D3 functional replicated the structure, identified the key bonding orbitals, and were used to aid assignment of the IR spectrum of WSeCl4. Reaction of WSeCl4 with ligands L gave [WSeCl4(L)] (L = MeCN, DMF, thf, py, OPPh3, 2,2′-bipy, SeMe2, SenBu2), whilst the dimers [(WSeCl4)2(μ-L–L)] were formed with L–L = Ph2P(O)CH2P(O)Ph2, 1,4-dioxane and 4,4′-bipyridyl. The complexes were characterised by microanalysis, IR and 1H NMR spectroscopy, and single crystal X-ray structures determined for [WSeCl4(L)] (L = OPPh3, MeCN, DMF) and [(WSeCl4)2(μ-L–L)] (L–L = 1,4-dioxane, 4,4′-bipyridyl). All except the 2,2′-bipy complex, which is probably seven-coordinate, contain six-coordinate tungsten with the neutral donor trans to WSe. Alkylphosphines, including PMe3 and PEt3, decompose WSeCl4 upon contact, forming phosphine selenides (SePR3). In contrast, the selenoether complexes [WSeCl4(SeMe2)] and [WSeCl4(SenBu2)] were isolated and characterised. The crystal structure of the minor W(VI) by-product, [(WSeCl4)2(μ-SeMe2)], was determined and using SMe2, a few crystals of the W(V) species, [{WCl3(SMe2)}2(μ-Se)(μ-Se2)], were obtained and structurally characterised. The isolated W(VI) complexes are compared with those of WOCl4 and WSCl4 and the combination of experimental and computational data are consistent with WSeCl4 being a weaker Lewis acid and its complexes significantly less stable than those of the lighter analogues, especially in solution. Low pressure chemical vapour deposition (LPCVD) using [WSeCl4(SenBu2)] in the range 660–700 °C (0.1 mmHg) produced highly reflective thin films, which were identified to be WSe2 by grazing incidence X-ray diffraction (XRD), scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy. XRD analysis of the thinner films revealed them to be highly oriented in the <00l> direction.
Se, and powder X-ray diffraction (PXRD) analysis confirmed this to be the only form present in the bulk sample. Density functional theory (DFT) calculations using the B3LYP-D3 functional replicated the structure, identified the key bonding orbitals, and were used to aid assignment of the IR spectrum of WSeCl4. Reaction of WSeCl4 with ligands L gave [WSeCl4(L)] (L = MeCN, DMF, thf, py, OPPh3, 2,2′-bipy, SeMe2, SenBu2), whilst the dimers [(WSeCl4)2(μ-L–L)] were formed with L–L = Ph2P(O)CH2P(O)Ph2, 1,4-dioxane and 4,4′-bipyridyl. The complexes were characterised by microanalysis, IR and 1H NMR spectroscopy, and single crystal X-ray structures determined for [WSeCl4(L)] (L = OPPh3, MeCN, DMF) and [(WSeCl4)2(μ-L–L)] (L–L = 1,4-dioxane, 4,4′-bipyridyl). All except the 2,2′-bipy complex, which is probably seven-coordinate, contain six-coordinate tungsten with the neutral donor trans to WSe. Alkylphosphines, including PMe3 and PEt3, decompose WSeCl4 upon contact, forming phosphine selenides (SePR3). In contrast, the selenoether complexes [WSeCl4(SeMe2)] and [WSeCl4(SenBu2)] were isolated and characterised. The crystal structure of the minor W(VI) by-product, [(WSeCl4)2(μ-SeMe2)], was determined and using SMe2, a few crystals of the W(V) species, [{WCl3(SMe2)}2(μ-Se)(μ-Se2)], were obtained and structurally characterised. The isolated W(VI) complexes are compared with those of WOCl4 and WSCl4 and the combination of experimental and computational data are consistent with WSeCl4 being a weaker Lewis acid and its complexes significantly less stable than those of the lighter analogues, especially in solution. Low pressure chemical vapour deposition (LPCVD) using [WSeCl4(SenBu2)] in the range 660–700 °C (0.1 mmHg) produced highly reflective thin films, which were identified to be WSe2 by grazing incidence X-ray diffraction (XRD), scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy. XRD analysis of the thinner films revealed them to be highly oriented in the <00l> direction.
Introduction
Transition metal oxide halides have played a major role in the development of coordination and organometallic chemistry and in the applications of early d-block elements.1–3 In contrast, the heavier analogues, the sulfide and selenide halides, are little known and have been much less studied.4,5 There are no examples of molecular chalcogenide iodides, and whilst chalcogenide fluorides exist for Mo and W, their coordination chemistry is unexplored.6 The synthesis and chemistry of MEX3 (M = Nb, Ta; E = S, Se; X = Cl, Br) and WEX4 were developed in the period 1965–1985, much of it by the groups of Fowles, Rice and Drew.4,5 We have recently undertaken re-investigations and comparisons of the coordination chemistry of WSCl4 and WSCl3 with those of WOCl4 and WOCl3 with a range of N- and O-donor ligands,7 as well as with the softer phosphines and arsines,8 and thioethers.9 Comparisons of the chemistries of TaOCl3 and TaSCl3 have also been reported.10,11 Thioether complexes of NbSCl3, of type [{NbSCl3(SR2)}2] (R = Me, nBu) are obtained from reaction of NbCl5, SR2 and S(SiMe3)2 in CH2Cl2, whilst [NbSCl3(L–L)] (L–L = MeS(CH2)2SMe, MeS(CH2)3SMe, iPrS(CH2)2SiPr and nBuS(CH2)3SnBu) are made from reaction of L–L with pre-formed [NbSCl3(MeCN)2].12 Selected examples can function as single source precursors for low pressure chemical vapour deposition (LPCVD) of thin films of 3R-NbS2.12 [WSCl4(SMe2)] and [(WSCl4)2{μ-RS(CH2)2SR}] (R = Me, Ph, iPr) and [(WSCl4)2{μ-MeS(CH2)3SMe}] have been made by direct reaction of WSCl4 with the thioether in anhydrous CH2Cl2 solution.9 We also showed that [(WSCl4)2{μ-iPrS(CH2)2SiPr}] functions as a single source precursor to deposit 2H-WS2 thin films by low pressure chemical vapour deposition (LPCVD),9 providing further motivation to develop the chemistry of WSeCl4 and its complexes.Attempts to prepare dithio- or diseleno-ether complexes of NbSeCl3 were unsuccessful, but reaction of NbCl5, SenBu2 and (Me3Si)2Se in CH2Cl2 gave black [NbSe2Cl3(SenBu2)].12 The structure of the latter is unknown, but may contain a diselenide unit ([Se2]2−). This compound deposits 2H-NbSe2 films by LPCVD at 600–700 °C, 0.05 mmHg.12
The early transition metal dichalcogenides ME2 (M = Nb, Ta, Mo, V, W, etc., E = S, Se or Te) are layered semiconductors with good stabilities, and their properties can be tuned through the choice of metal and chalcogen.13 This versatility gives rise to a plethora of applications in (opto)electronics, magnetic materials, spintronics, sensors and electrocatalysis, which are very active areas of research.13–16 Production of the materials as thin films maximises the anisotropy of their magnetic or electronic properties, hence techniques to deposit 2D layers are of particular interest. Chemical vapour deposition (CVD) is widely used in industry as a low-cost, scalable and very versatile method to deposit such films using either dual or single source precursors.13–19 The latter can offer better control of film stoichiometry, cost-effective use of reagents, as well as the ability to selectively deposit the metal chalcogenide film onto lithographically patterned substrates.18 The application of molecular transition metal complexes as single source CVD precursors for the growth of metal chalcogenide thin films has been reviewed recently.20
Although WSeCl4 was first described in 1974,21 the few coordination complexes known are described in papers focussed rather on WSCl4 adducts,5,22–24 and a more detailed investigation of the selenide chloride is required to develop its chemistry and to determine if it could be the basis of a single source low pressure CVD route to WSe2. A small number of dual source CVD routes to WSe2 films have been described,19,20 including using WCl6 and SeEt2 in atmospheric pressure CVD,25 however, to the best of our knowledge there are no single source CVD reagents for tungsten diselenide films.
Here we report a detailed study of the synthesis, single crystal and powder X-ray structure and properties of WSeCl4, its coordination chemistry with a range of neutral N-, O- and Se-donor ligands, and demonstrate that the new complex, [WSeCl4(SenBu2)], can function as a single source precursor for the CVD growth of continuously interconnected WSe2 thin films.
Results and discussion
WSeCl4
WSeCl4 was prepared by heating together a finely ground mixture of WCl6 and Sb2Se3 in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in vacuo, according to the general method of Britnell et al.21 Obtaining good yields of the pure product required very careful control of the conditions, and hence the preparation and apparatus used in this work are described in detail in the Experimental section. The green powdered WSeCl4 is moisture sensitive, but can be stored in a sealed container in the glove box indefinitely. Attempts to obtain WSeCl4 by reaction of WCl6 and Se(SiMe3)2 in a variety of organic solvents, including CH2Cl2, toluene or MeCN, and at various temperatures from ambient to −78 °C, failed. This contrasts with the ready synthesis of WSCl4 from WCl6 and S(SiMe3)2, and WOCl4 from WCl6 and O(SiMe3)2.7–9,26 Refluxing WSeCl4 with Cl2CCCl2 also failed to produce WSeCl3, whereas the similar reaction of WSCl4 and Cl2CCCl2 forms WSCl3.27
:1 molar ratio in vacuo, according to the general method of Britnell et al.21 Obtaining good yields of the pure product required very careful control of the conditions, and hence the preparation and apparatus used in this work are described in detail in the Experimental section. The green powdered WSeCl4 is moisture sensitive, but can be stored in a sealed container in the glove box indefinitely. Attempts to obtain WSeCl4 by reaction of WCl6 and Se(SiMe3)2 in a variety of organic solvents, including CH2Cl2, toluene or MeCN, and at various temperatures from ambient to −78 °C, failed. This contrasts with the ready synthesis of WSCl4 from WCl6 and S(SiMe3)2, and WOCl4 from WCl6 and O(SiMe3)2.7–9,26 Refluxing WSeCl4 with Cl2CCCl2 also failed to produce WSeCl3, whereas the similar reaction of WSCl4 and Cl2CCCl2 forms WSCl3.27
Green single crystals of WSeCl4, which were obtained during the synthesis by sublimation, contained two independent molecules in the asymmetric unit, both monomeric square pyramids, with similar bond lengths and angles (Fig. 1). In contrast, WSCl4 adopts two structurally distinct polymorphs, a dimer, [SCl3W(μ-Cl)2WCl3S],28 and a tetramer, [Cl4SW(μ-Cl)SCl2W(μ-Cl)2WCl2S(μ-Cl)WSCl4].29 WSCl4 monomers are present in WSCl4·S8, which seems to be best described as co-crystallised WSCl4 and cyclo-octasulfur, rather than an adduct.30
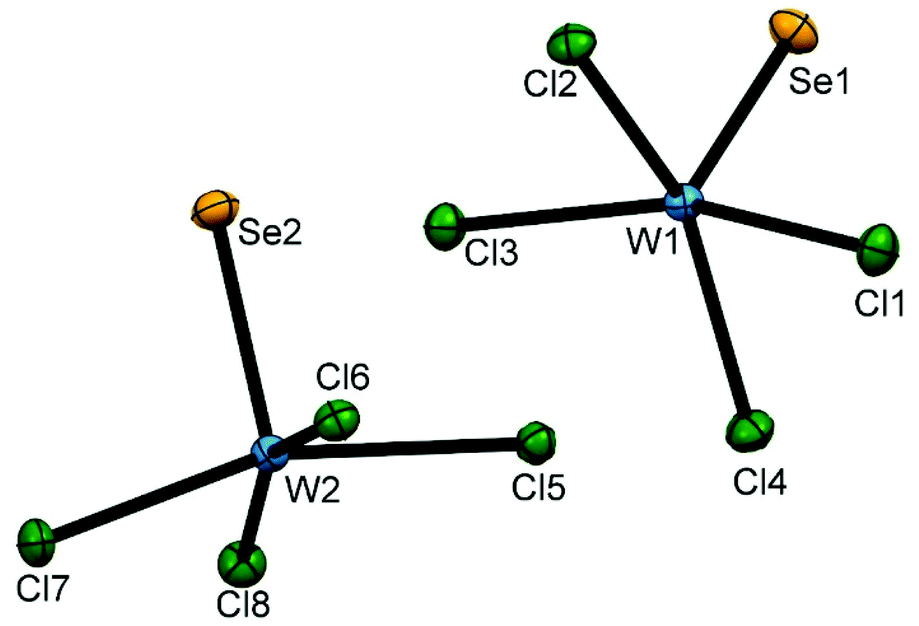 | ||
| Fig. 1 The crystal structure of the two independent molecules of WSeCl4 in the asymmetric unit showing the atom numbering scheme and with the ellipsoids drawn at the 50% probability level. Selected bond lengths (Å) and angles (°) are: W1–Se1 = 2.2099(4), W1–Cl3 = 2.2944(9), W1–Cl2 = 2.3048(8), W1–Cl4 = 2.3045(8), W1–Cl1 = 2.3038(9), Se1–W1–Cl3 = 101.46(2), Se1–W1–Cl2 = 101.15(2), Se1–W1–Cl4 = 101.98(2), Se1–W1–Cl1 = 101.66(3), Cl3–W1–Cl2 = 87.74(3), Cl3–W1–Cl4 = 87.68(3), Cl1–W1–Cl2 = 88.06(3), Cl1–W1–Cl4 = 87.30(3), W2–Se2 = 2.2128(4), W2–Cl5 = 2.3419(8), W2–Cl7 = 2.2762(9), W2–Cl6 = 2.3949(8), W2–Cl8 = 2.2689(8), Se2–W2–Cl5 = 98.24(2), Se2–W2–Cl7 = 101.18(2), Se2–W2–Cl6 = 97.65(2), Se2–W2–Cl8 = 101.49(2), Cl5–W2–Cl6 = 85.15(3), Cl7–W2–Cl6 = 88.32(3), Cl8–W2–Cl5 = 88.58(3), Cl8–W2–Cl7 = 91.45(3). | ||
Powder X-ray diffraction (PXRD) data from the powdered bulk WSeCl4 (ESI Fig. S1†) shows the same polymorph as identified in the single crystal structure, with no evidence for a second form. This solid state structure is very similar to that reported for the compound from vapour phase electron diffraction.31
The IR spectrum of WSeCl4 (Nujol mull) shows (ESI. Fig. S2†) a medium feature at 387 cm−1 assigned as ν(WSe) and intense features at 353 and 367 cm−1 assigned as ν(W–Cl). The ν(WSe) is in good agreement with literature data, viz. ν(WSe) 388,31 or 396 cm−1,21 and is supported by DFT calculations (below). The IR spectrum of WSeCl4 vapour, isolated in a N2 matrix at 10 K showed vibrations at 399 and 369 cm−1, both tentatively assigned as ν(W–Cl), while ν(WSe) was not identified.32 The reassignment of the band at 399 cm−1 as ν(WSe) seems merited in the light of these new results.
Density functional theory (DFT) calculations
To further explore the structure of WSeCl4, to investigate the frontier orbitals and to complement the experimental data with IR spectroscopic assignments, the ground state geometry of WSeCl4 was optimised using DFT methods, starting from the solid-state structure. These calculations were carried out with the B3LYP-D3 functional,33 (lanl2dz on W; 6-311G(d) on Se and Cl atoms) and the geometrical parameters were found to be in good agreement with the solid-state values (Fig. 2 and ESI Table S2†). The predicted IR harmonic wavenumbers in Table 1 confirm the assignment of the ν(WSe) peak in the experimental IR spectrum at a slightly higher frequency than ν(W–Cl).
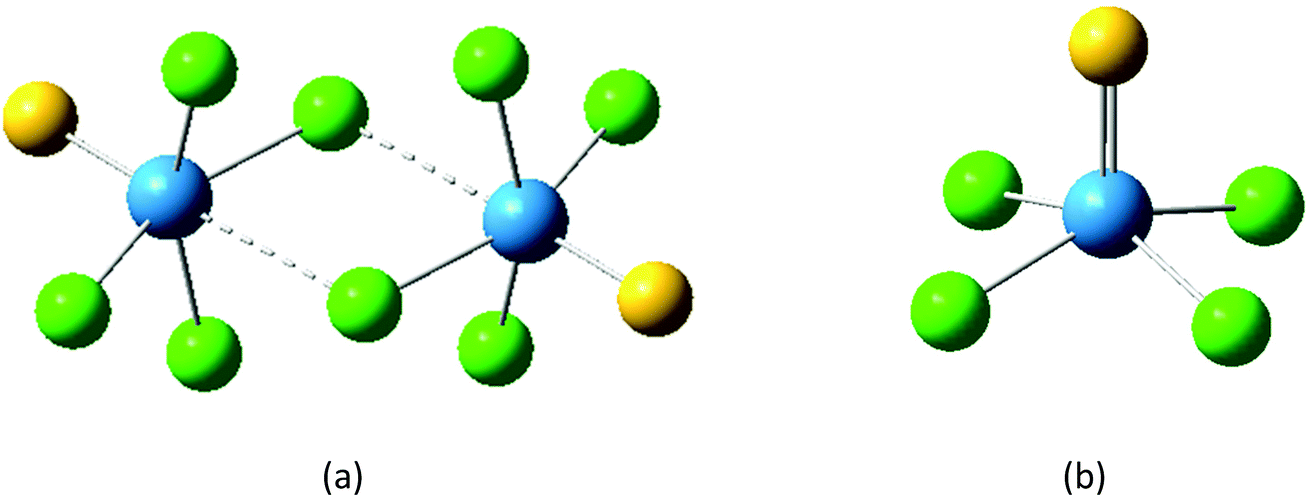 | ||
| Fig. 2 Schematic showing the DFT calculated structures for (a) WECl4 and (b) [ECl3W(μ-Cl)2WCl3E] (E = S, Se). | ||
As discussed above, the known polymorphs of WSCl4 in the solid state are a dimer or tetramer, while the crystals obtained of WSeCl4 show this to be a monomer. To further explore this difference, DFT calculations were undertaken to compare the energy of formation of the optimised selenide and sulfide dimer (with bridging Cl), i.e. the energy difference upon going from 2 WECl4 to [ECl3(W(μ-Cl)2WCl3E]. This showed the energy of formation of the sulfide-bridged dimer (ΔE) to be more negative (−78.4 kJ mol−1) than the selenide bridged dimer (−76.3 kJ mol−1), supporting the conclusion that for WSCl4 the dimer is more stable. This is further evidenced by examining the molecular orbitals of WSCl4 and WSeCl4 (ESI Fig. S30†), particularly the HOMO and HOMO−1 orbitals, which show more extensive contribution from the chlorides in WSCl4 compared to the WSeCl4.
Finally, calculations were undertaken to explore the relative Lewis acidities of the WECl4 species (E = S, Se) by considering the following reaction:
| WECl4 + OPPh3 → WECl4(OPPh3) (E = S, Se); |
where ΔE = E([WSCl4(OPPh3)]) − E(OPPh3) − E(WSCl4).
These showed that the ΔE for E = S (−161.2 kJ mol−1) was slightly more negative than for E = Se (−156.5 kJ mol−1), consistent with WSCl4 being the more Lewis acidic of the pair.
Complexes with N- and O-donor ligands
Despite the lower Lewis acidity of WSeCl4, the syntheses of a range of its complexes with neutral Group 15 and 16 donor ligands was undertaken (Scheme 1), and their properties compared with those of the WOCl4 and WSCl4 analogues. The reactions with ligands occurred readily, although the products were often unstable in solution and sometimes also in the solid state. Successful syntheses used carefully chosen (low polarity, weakly-coordinating, volatile) solvents and required rapid isolation of the products. Attempted recrystallisation of the complexes often resulted in significant decomposition rather than purification. The solution spectra also needed to be recorded from freshly prepared solutions to minimise significant decomposition. | ||
| Scheme 1 Synthesis routes to WSeCl4 and its complexes. | ||
The reaction of WSeCl4 with one molar equivalent of DMF, thf, pyridine, OPPh3 and 2,2′-bipy, as described in the Experimental section, resulted in green powders [WSeCl4(L)]; often the choice of solvent was key to obtaining the target complex in a pure form. In contrast, [WSeCl4(MeCN)] could be obtained from a range of solvents, as well as from neat MeCN, and could be used as a synthon for other complexes, although it proved to be little different from using WSeCl4 itself in most cases. The bidentates, Ph2P(O)CH2P(O)Ph2 (dppmO2), 1,4-dioxane and 4,4′-bipyridyl (L–L), reacted with two equivalents of WSeCl4 to form the ligand-bridged dimers, [(WSeCl4)2(μ-L–L)]. With one exception, ([WSeCl4(2,2′-bipy)] below), the spectroscopic data show the complexes contain six-coordinate distorted octahedral tungsten environments, which was confirmed by X-ray crystal structures of five examples (Fig. 3–7), showing the neutral ligand occupying the vacant site trans to WSe in WSeCl4. As is found in other complexes of this type, the tungsten lies out of the Cl4 plane towards the selenium. The effect of coordination on the WSe and W–Cl bond lengths is small, with only slightly longer bonds in the six-coordinate complexes. The 1H and (where appropriate) 31P{1H} NMR spectra confirm the presence of the coordinated neutral ligand, with chemical shifts generally very similar to those in the WOCl4 and WSCl4 analogues.7 The poor solubility and solution instability generally precluded using 77Se NMR spectroscopy (77Se: 7.58%, I = ½, Ξ = 19.09 MHz, Dc = 2.98) for their characterisation.
 | ||
| Fig. 3 The structure of [WSeCl4(OPPh3)] showing the atom numbering scheme and ellipsoids are shown at the 50% probability level. H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) are: W1–Se1 = 2.2321(2), W1–Cl1 = 2.3514(5), W1–Cl2 = 2.3577(5), W1–Cl3 = 2.3217(5), W1–Cl4 = 2.3218(5), W1–O1 = 2.1669(14), P1–O1 = 1.5066(15), Se1–W1–Cl1 = 97.836(15), Se1–W1–Cl2 = 94.628(14), Se1–W1–Cl3 = 96.375(15), Se1–W1–Cl4 = 97.047(15), Cl1–W1–Cl2 = 87.552(17), Cl3–W1–Cl2 = 88.861(18), Cl3–W1–Cl4 = 91.204(19), Cl4–W1–Cl1 = 89.494(18), O1–W1–Cl1 = 82.53(4), O1–W1–Cl2 = 84.52(4), O1–W1–Cl3 = 83.23(4), O1–W1–Cl4 = 83.81(4), P1–O1–W1 = 167.82(10). | ||
 | ||
| Fig. 4 The structure of [WSeCl4(DMF)] showing the atom numbering scheme and ellipsoids are shown at the 50% probability level. H atoms and a C6H6 solvent molecule are omitted for clarity. Note that the disorder in the structure was modelled using split atom occupancies and only the major form is shown here. Selected bond lengths (Å) and angles (°) are: W1–Se1 = 2.2478(7), W1–Cl1 = 2.3283(15), W1–Cl2 = 2.3252(15), W1–Cl3 = 2.3422(14), W1–Cl4 = 2.3466(14), W1–O1 = 2.173(4), Se1–W1–Cl1 = 98.79(4), Se1–W1–Cl2 = 98.22(4), Se1–W1–Cl3 = 95.60(4), Se1–W1–Cl4 = 96.22(4), Cl1–W1–Cl4 = 88.69(5), Cl2–W1–Cl1 = 89.50(6), Cl2–W1–Cl3 = 89.44(6), Cl3–W1–Cl4 = 88.75(5), O1–W1–Cl1 = 83.06(12), O1–W1–Cl2 = 81.74(13), O1–W1–Cl3 = 82.55(12), O1–W1–Cl4 = 83.82(13). | ||
 | ||
| Fig. 5 The structure of [WSeCl4(MeCN)] showing the atom numbering scheme. Ellipsoids are shown at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) are W1–Se1 = 2.2312(9), W1–Cl1 = 2.3248(17), W1–Cl2 = 2.3208(17), W1–N1 = 2.330(7), Se1–W1–Cl1 = 97.83(4), Se1–W1–Cl2 = 99.32(4), Cl1–W1–Cl1 = 90.30(9), Cl1–W1–N1 = 81.05(13), Cl2–W1–Cl1 = 88.83(6), Cl2–W1–Cl2 = 86.95(9), Cl2–W1–N1 = 81.83(14). | ||
 | ||
| Fig. 6 The structure of [(WSeCl4)2(μ-4,4′-bipy)] showing the atom numbering scheme. Ellipsoids are shown at the 50% probability level and H atoms and benzene solvent molecules are omitted for clarity. The asymmetric unit consists of two of these molecules, only one of which is shown. Selected bond lengths (Å) and angles (°) are: W1–Se1 = 2.2424(7), W1–Cl4 = 2.3292(17), W1–Cl1 = 2.3150(16), W1–Cl3 = 2.3113(17), W1–Cl2 = 2.3214(18), W1–N1 = 2.391(5), Se1–W1–Cl4 = 97.47(4), Se1–W1–Cl1 = 98.68(5), Se1–W1–Cl3 = 98.66(5), Se1–W1–Cl2 = 98.08(5), Cl4–W1–N1 = 81.40(14), Cl1–W1–Cl4 = 88.40(6), Cl1–W1–Cl2 = 88.92(7), Cl1–W1–N1 = 81.42(13), Cl3–W1–Cl4 = 88.66(7), Cl3–W1–Cl2 = 89.34(7), Cl3–W1–N1 = 81.22(13), Cl2–W1–N1 = 83.05(14). | ||
 | ||
| Fig. 7 The structure of [{WSeCl4}2(1,4-dioxane)] showing the atom numbering scheme. Ellipsoids are shown at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) are: W1–Se1 = 2.2285(4), W1–Cl1 = 2.3138(8), W1–Cl2 = 2.3273(8), W1–Cl3 = 2.3069(8), W1–Cl4 = 2.3081(8), W1–O1 = 2.398(2), Se1–W1–Cl1 = 98.44(2), Se1–W1–Cl2 = 98.97(2), Se1–W1–Cl3 = 99.60(2), Se1–W1–Cl4 = 98.73(2), Cl1–W1–Cl2 = 88.20(3), Cl1–W1–O1 = 80.83(6), Cl2–W1–O1 = 81.40(6), Cl3–W1–Cl2 = 88.80(3), Cl3–W1–Cl4 = 89.51(3), Cl3–W1–O1 = 81.12(6), Cl4–W1–Cl1 = 87.95(3), Cl4–W1–O1 = 80.88(6). | ||
In the IR spectra, the ν(W–Cl) occurred as two strong overlapping bands in the region 350–310 cm−1 and ν(WSe) as a weak or medium intensity band in the range 380–350 cm−1, somewhat lower frequency than in WSeCl4 itself, in accord with the increase in the coordination number.
Once isolated, the [WSeCl4(2,2′-bipy)] proved to very poorly soluble in suitable solvents, and X-ray quality crystals could not be obtained. However, the 1H NMR spectrum showed equivalent pyridyl rings, whilst the ν(W–Cl) = 318, 304 cm−1 and ν(WSe) = 345 cm−1 are lower than in the six-coordinate complexes, providing supporting evidence that [WSeCl4(2,2′-bipy)] is probably seven-coordinate and pentagonal bipyramidal. The corresponding [WOF4(2,2′-bipy)], [WOCl4(2,2′-bipy)] and [WSCl4(2,2′-bipy)] are also thought to be seven-coordinate based upon spectroscopic data, and [WOF4{o-C6H4(PMe2)2}], [WOCl4{o-C6H4(AsMe2)2}] and [WSCl4{o-C6H4(PMe2)2}] have been established as pentagonal bipyramids by single crystal X-ray analysis.8,35 Rather surprisingly, attempts to isolate the (unknown) anion, [WSeCl5]− with a range of cations, including [PPh4]+ and [NEt4]+, have been unsuccessful thus far, whereas the [WSCl5]− and [WS2Cl4]2− ions are well known.36,37
Reactions with soft P- and Se-donor ligands
The addition of PMe3 or PEt3 to a solution of WSeCl4 in CH2Cl2 immediately produced orange-brown solutions in which the only species evident in the 31P{1H} NMR spectra, in addition to small amounts of the free phosphine, were the corresponding phosphine selenides SePMe3 (δP = +9.4)38 and SePEt3 (δP = +45.2, 1JPSe = 680 Hz).39 The reaction of WECl4 (E = O, S, Se) with a large excess of PPh3 over several days in toluene was reported to form [WCl4(PPh3)2] and EPPh3.40 Similar, but much faster abstraction of the selenium occurs on contact with alkyl phosphines. Similarly, reaction of o-C6H4(PMe2)2 with WSeCl4 in CH2Cl2 instantly produced a green solution and a brown precipitate, and the 31P{1H} NMR spectrum showed several species present in the solution, with the major resonances at δ = +20 and +33, which may be due to phosphine selenides. This contrasts with the reactions of o-C6H4(PMe2)2 with WOCl4 and WSCl4, which gave high yields of the seven-coordinate complexes [WECl4{o-C6H4(PMe2)2}] (E = O, S), both of which were characterised, including the X-ray structure of the latter (as noted earlier).8 Decomposition of [WECl4{o-C6H4(PMe2)2}] in CH2Cl2 solution over a few days gave crystals of a number of tungsten(V) species including [WOCl3{o-C6H4(PMe2)2}] and [WCl4{o-C6H4(PMe2)2}2]+.8 This contrasts with the immediate reduction of the metal in the WSeCl4 systems in the present study. A few crystals obtained from the mother liquor of the o-C6H4(PMe2)2/WSeCl4 reaction were identified by X-ray analysis as [WCl4{o-C6H4(PMe2)2}2]Cl; the same eight-coordinate cation was found among the decomposition products of [WSCl4{o-C6H4(PMe2)2}] in CH2Cl2.8The reaction of WSeCl4 with SeMe2 in benzene resulted in green [WSeCl4(SeMe2)], identified by microanalysis and its IR spectrum, which showed ν(WSe) at 387 cm−1 and ν(W–Cl) at 343 cm−1. Use of SenBu2 in this reaction gave a dark red oil identified as [WSeCl4(SenBu2)] from its IR, 1H and 77Se{1H} NMR spectra, but attempts to isolate complexes with diselenoethers such as [(WSeCl4)2{μ-RSe(CH2)2SeR}] were unsuccessful. Similarly, attempts to prepare [(WSCl4)2{μ-MeSe(CH2)2SeMe}] were unsuccessful, although the [WSCl4(SeMe2)] analogue is well characterised.9 A few dark green crystals isolated from a solution of the [WSeCl4(SeMe2)] monomer in C6D6 unexpectedly proved to be the dinuclear [(WSeCl4)2(μ-SeMe2)] (Fig. 8), which shows two square pyramidal WSeCl4 groups linked by a bridging SeMe2 ligand. The d(WSe) and d(W–Cl) are similar to those in the complexes with hard donor ligands (above), while the d(W–SeMe2) is long at ∼3.0 Å. Selenoethers bridging between two metal centres and formally using both lone pairs are known for many d-block metals,41 although it was unexpected here given the modest acceptor ability of WSeCl4. This dimer presumably crystallised as the least soluble species in a partially dissociated solution of [WSeCl4(SeMe2)].
 | ||
| Fig. 8 The structure of [(WSeCl4)2(μ-SeMe2)] showing the atom numbering scheme. Ellipsoids are shown at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) are W1–Se1 = 2.2374(8), W2–Se3 = 2.2255(9), W1–Se2 = 3.0107(8), W2–Se2 = 3.0814(8), W1–Cl = 2.2871(19)–2.3311(19), W2–Cl = 2.3006(18)–2.3130(18), Se1–W1–Se2 = 178.42(3), Se3–W2–Se2 = 176.28(3), W1–Se2–W2 = 127.82(3), Se1–W1–Cl = 98.42(5) – 100.95(5), Se2–W1–Cl = 80.03(5) – 81.91(5), Se3–W2–Cl = 98.86(6) – 101.89(6), Se2–W2–Cl = 76.67(5) – 81.78(5). | ||
Similar reaction of WSeCl4 with SMe2 did not yield a pure product, however, a few crystals obtained by slow evaporation of CH2Cl2 from the reaction mixture were shown by X-ray crystallographic analysis to contain the W(V) dimer, [{WCl3(SMe2)}2(μ-Se)(μ-Se2)] (Fig. 9). Previous work showed that decomposition of several WSCl4 thioether complexes (or reduction with excess thioether) affords the corresponding W(V) complexes.5,9 However, decomposition of the WSeCl4 complexes prepared in the present study typically produced dark intractable solids, that were insoluble in organic solvents and could not be identified. Structurally characterised niobium(IV) complexes similar to [Cl3(SMe2)W(μ-Se)(μ-Se2)WCl3(SMe2)] include [(tht)Br2Nb(μ-Se)(μ-Se2)NbBr2(tht)] (tht = tetrahydrothiophene) and [(SMe2)Cl2Nb(μ-Se2)2NbCl2(SMe2)].5,21,42,43 In fact, very little is known about WSeCl3 and no complexes with neutral ligands have been described.5
 | ||
| Fig. 9 The structure of [{WCl3(SMe2)}2(μ-Se)(μ-Se2)] showing the atom numbering scheme. Ellipsoids are shown at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): W1–W1 = 2.8021(6), W1–Se1 = 2.4310(9), W1–Se2 = 2.5687(9), W1–Se3 = 2.5589(9), Se2–Se3 = 2.2633(14), W1–Cl1 = 2.4259(16), W1–Cl2 = 2.3944(16), W1–Cl3 = 2.3791(16), W1–S1 = 2.6017(18), W1–Se1–W1 = 70.39(3), W1–Se2–W1 = 66.11(3), W1–Se3–W1 = 66.40(3), Cl1–W1–S1 = 71.16(6), Cl2–W1–S1 = 76.35(6), Cl3–W1–S1 = 82.26(6), Cl3–W1–Cl1 = 153.31(6), Cl3–W1–Cl2 = 88.76(6), Cl2–W1–Cl1 = 87.33(6). | ||
Growth of WSe2 thin films via low pressure CVD
The isolation of [WSeCl4(SenBu2)] as an oil, containing a 1:2 ratio of tungsten to selenium, and with the n-butyl groups on the selenoether providing a potential low energy thermal decomposition pathway (e.g. via β-hydride elimination), raised the prospect that this might serve as a single source CVD precursor for the growth of tungsten diselenide thin films onto fused silica substrates, in a similar manner to [(WSCl4)2{μ-iPrS(CH2)2SiPr}], which deposits WS2 films.9 LPCVD using [WSeCl4(SenBu2)] produced highly reflective, silvery films of WSe2 at 0.1 mmHg and 660–700 °C.
Grazing incidence XRD analysis of the films obtained from [WSeCl4(SenBu2)] (Fig. 10 and S29†) are consistent with polycrystalline 4H WSe2 in space group P63/mmc. Lattice parameters were refined as: a = 3.2829(11), c = 13.130(6) (Rwp = 7.82%, literature: a = 3.285(1); c = 12.961(1) Å from the XRD pattern in Fig. S29a†).44 The grazing incidence diffraction patterns of the thinner films, for example, Fig. 10, are almost completely dominated by the 002 reflection, consistent with significant preferred orientation in the <00l> direction. This orientation is not unusual, as these layered materials typically grow initially with the c axis normal to the substrate, and suggests that the majority of the platelet crystals have grown in that orientation, with the platelets flat to the substrate. The average crystallite size in the WSe2 film was determined to be 9.4(14) nm based on the grazing incidence XRD data in Fig. S29a,† using the Williamson-Hall method.
 | ||
| Fig. 10 Grazing incidence XRD pattern (top) from a WSe2 thin film deposited by low pressure CVD using [WSeCl4(SenBu2)]. The broad feature at 2θ = 20–25° is from the SiO2 substrate. XRD pattern for bulk WSe2 (bottom).44 | ||
Fig. 11 presents top-view SEM images of the as-deposited WSe2 thin film. The film displays a smooth underlying interconnected polycrystalline layer with some more randomly oriented crystallites on top. The underlying layer comprises of hexagonal plate-like crystallites oriented with the c-axis normal to the substrate. This is supported by the preferential growth observed in the XRD patterns in Fig. 10 and Fig. S29b.†
 | ||
| Fig. 11 Morphological characterisation via SEM imaging for an as-deposited WSe2 film produced via low pressure CVD using [WSeCl4(SenBu2)], taken at magnifications (a) ×10000 and (b) ×40000. | ||
The Raman spectrum for the WSe2 film deposited via LPCVD is presented in Fig. 12. The two significant peaks located at 248.7 cm−1 and 258.5 cm−1 are commonly known as the E2g1 (green) and A1g (orange) vibrational modes, respectively, and are in good agreement with current literature.45 The inset is a schematic of the E2g1 and the A1g modes, which represent the in-plane and out-of-plane vibrational modes, respectively.46 The relative intensities of the E2g1 and the A1g modes suggest the suppression of the out-of-plane A1g mode being due to the thin nature of the film.46,47 The peaks labelled 2M are likely to be related to the combinational second order Raman modes.46–48 The origin of peak x located at 307.3 cm−1 is likely to be a combinational mode of low frequency modes due to its relatively large frequency. It has been suggested that this peak is linked to coupling of the E2g1 mode and an intra-layer shear force, due to reports that suggest suppression of this peak in monolayered samples.46,49,50
 | ||
| Fig. 12 Raman spectrum of the WSe2 film obtained using [WSeCl4(SenBu2)]. | ||
The film composition was investigated by X-ray photoelectron spectroscopy (XPS). Fig. 13a presents the XPS survey spectrum of the film. Both the W and Se peaks are clearly presented, with no impurities (e.g. C, Cl) observed, suggesting the WSe2 film is of high quality. Elemental scans were conducted of both the W 4f and Se 3d orbitals (shown in Fig. 13b–c). From the elemental scan of the W 4f orbital, two significant peaks positioned at 32.2 eV and 34.3 eV were observed. These peaks are in good agreement with literature and represent electron emission from the 4f7/2 and 4f5/2 atomic orbitals.51 An additional peak located at 37.5 eV was noted in the W scan, which has been suggested to be related to W 5p3/2.52 The Se scan in Fig. 13 displays two significant peaks at 54.6 eV and 55.7 eV related to the 3d5/2 and 3d3/2 orbitals respectively.52 From the spectra shown in Fig. 13, a WSe2.1 composition for the deposited films has been derived (i.e. W:Se = 32.1%:67.9%).
 | ||
| Fig. 13 XPS characterisation of an as-deposited WSe2 film produced via low pressure CVD using [WSeCl4(SenBu2)]. (a) XPS survey scan of binding energies in the range of 0 eV to 650 eV, with peaks related to the atomic orbitals of W (blue) and Se (orange) labelled. Elemental scans for the as-deposited WSe2 film showing the peaks associated with W (b) and Se (c). | ||
Experimental
Syntheses were performed using standard Schlenk and glovebox techniques under a dry N2 atmosphere. Solvents and ligands were dried by distillation from CaH2 (CH2Cl2, MeCN, DMF,) or Na/benzophenone ketyl (toluene, n-hexane, diethyl ether, thf, 1,4-dioxane, py). WCl6, PMe3, PEt3, SMe2, SeMe2, 2,2′-bipy and 4,4′-bipy were obtained from Sigma-Aldrich. Ph2P(O)CH2P(O)Ph2 was obtained by air oxidation of the corresponding diphosphine in CH2Cl2, catalysed by SnI4.52 Solid ligands were dried in vacuo for several hours immediately before use.Infrared spectra were recorded on a PerkinElmer Spectrum 100 spectrometer in the range 4000–200 cm−1, with samples prepared as Nujol mulls between CsI plates. 1H and 31P{1H} NMR spectra were recorded using a Bruker AV 400 spectrometer and referenced to the residual protio-resonance of the solvent and 85% H3PO4. Microanalyses on new compounds were undertaken by London Metropolitan University or Medac Ltd. Note that the complexes were generally very unstable, readily decomposing within a few hours in some cases. This hindered attempts to obtain satisfactory microanalyses on some samples from outsourced measurements – as noted below.
X-ray experimental
Crystals were grown from slow evaporation of saturated solutions in CH2Cl2 or by liquid–liquid diffusion using CH2Cl2 and hexane. Data collections used either a Rigaku OD UG2 goniometer equipped with a HyPix 6000HE hybrid pixel array detector mounted at the window of an FR-E + SuperBright molybdenum (λ = 0.71073 Å) rotating anode generator with VHF Varimax optics (70 micron focus) with the crystal held at 100 K (N2 cryostream), or a Rigaku OD UG2 goniometer equipped with a HyPix 6000HE hybrid pixel array detector mounted at the window of a MicroMax 007HF copper (λ = 1.54184 Å) rotating anode generator with VHF Varimax optics (70 micron focus) with the crystal held at 100 K (N2 cryostream). Crystallographic parameters are presented in Table S1.† Structure solution and refinement were performed using SHELX(T)-2018/2, SHELX-2018/3 through Olex253 and were mostly straightforward. H atoms were added and refined with a riding model. Where additional restraints were required, details are provided in the cif file for each structure found on CCDC.†Computational experimental
Geometry optimisation and frequency calculations were performed on WSeCl4 with the B3LYP hybrid functional, with the Grimme correction for dispersion (D3(BJ)),33 using Gaussian 16.54 A 6-311G(d) basis set was used on Se and Cl atoms while the RECP basis set LANL2DZ was used on the W atom. Calculations on WSCl4 were based on the crystal structure for WSeCl4, while the starting geometries for the dimers ([ECl3W(μ-Cl)2WCl3E]) (E = S, Se) were based on the structure for E = S reported in the literature.28For the complexes, the starting geometries for [WSeCl4(OPPh3)] and [WSCl4(OPPh3)]9 came from their respective crystal structures with the same functional and basis set as used for WSeCl4. Energy minima were confirmed by the absence of any imaginary frequencies using a frequency calculation and calculated frequencies were adjusted by standard scaling factors.34
Following the method of Britnell et al.,21 freshly ground WCl6 (6.19 g, 15.6 mmol) and Sb2Se3 (2.5 g, 5.2 mmol) were mixed in a glove box and added to a glass reaction tube (Fig. 14) under a dinitrogen atmosphere. The glass tube was then placed under vacuum and sealed with a gas torch at point (1). Section A was placed in a tube furnace at 120 °C for 4 days, with section C at room temperature, to allow the side products to deposit in the cooler section. This was then removed by sealing the glass at point (2). Section A was placed back in the furnace, with section B at room temperature for 4 days until more side product had collected in section B. At this point, joint (3) was broken in a glovebox and the solid remaining in section A removed and ground up, and the resulting green powder placed in a glass tube and heated at 170 °C for 6 h in vacuo. Any remaining SbCl3 pumped away and a grey solid had sublimed in the glass tube, leaving a brown residue which was discarded. The grey solid was removed in a glovebox and ground up to afford a bright green solid. Yield: 4.3 g, 68%. IR spectrum (Nujol, cm−1): 377 WSe, 367, 353, 336 W–Cl.
 | ||
| Fig. 14 (a) A borosilicate glass tube with three 1 cm constrictions, and a B14 joint, used for the solid-state reaction of WCl6 and Sb2Se3 to form WSeCl4; (b) grey-green solid remaining in the reaction tube after heating. | ||
N, 2281, ν(C–C) + δ(CH3), 373 WSe, 347, 332 W–Cl. 1H NMR (CD2Cl2): δ = 2.30 (s). The same complex can be obtained using CH2Cl2 or C6H6 as solvent or neat MeCN.
Se, 344, 330 W–Cl. 1H NMR (CD2Cl2): δ = 4.08 (s, [4H]), 1.96 (s, [4H]).
O, 360 WSe, 337, 309 W–Cl. 1H NMR (CD2Cl2): δ = 7.77 (m, [6H]), 7.67 (m, [3H]), 7.63 (m, [6H]). 31P{1H} NMR (CD2Cl2): δ = +45.7 (s). The same complex was obtained using C6H6, toluene or CS2 as solvent or by reaction of OPPh3 with [WSeCl4(MeCN)] in a 1:1 molar ratio.
O, 388 WSe, 367, 331 W–Cl. 1H NMR (C6D6): δ = 7.57 (s, [H]), 1.89 (s, [3H]), 1.35 (s, [3H]).
Se, 313 W–Cl. 1H NMR (CD2Cl2): δ = 8.92 (d, [2H], JHH = 4.7 Hz), 8.89 (d, [2H], JHH = 8.2 Hz), 8.28 (dt, [2H], JHH = 1.7, 7.8 Hz), 7.75 (dt, [2H], JHH = 1.0, 6.8 Hz).
Se, 327 W–Cl. 1H NMR (CD2Cl2): δ = 8.65 (br [2H]), 8.25 (br, [1H]), 7.82 (br, [2H]).
Se, 333 W–Cl. 1H NMR (CD2Cl2): δ = 9.20 (m, [4H]), 7.74 (m, [4H]).
Se, 361, 333 W–Cl. 1H NMR (C6D6): δ = 3.47 (s).
O, 366 WSe, 338, 337 W–Cl. 1H NMR (C6D6): δ = 7.80 (m, [8H]), 7.61 (m, [4H]), 6.90 (m, [8H]), 4.64 (t, [2H], 2JH–P = 15.6 Hz). 31P{1H} NMR (C6D6): δ = +45.9 (s). The product was very poorly soluble and the dilute solution showed significant decomposition, therefore solution data were collected in situ, but also show some decomposition.
Se, 344 W–Cl. 1H NMR (CD2Cl2): 1.41 (br s, [6H]).
Se, 352, 321 W–Cl. 1H NMR (in situ in C6D6): 2.48 (t, J = 7.6 Hz, [2H] CH2), 1.46 (m, [2H], CH2), 1.17 (m, [2H] CH2), 0.73 (t, J = 7.3 Hz, [3], CH3). 77Se{1H} NMR (CD2Cl2, 295 K): δ = 292; (183 K): δ = 295.
LPCVD growth of WSe2 films using [WSeCl4(SenBu2)]
The precursor (50–60 mg) was loaded into the precursor bulb at the closed end of a silica tube in a N2 purged glove box, silica substrates (ca. 1 × 8 × 20 mm3) were then positioned end-to-end lengthways along the tube away from the precursor. The tube was then set horizontally in a furnace so that the substrates were in the hot zone and the precursor protruded ca. 4 cm beyond the end of the tube furnace. The tube was evacuated to 0.1 mm Hg and the furnace heated to 700 °C and left for 15 min to allow the temperature to stabilise. The tube was gradually moved towards the hot zone until evaporation of the precursor began and the position was maintained until no further evaporation occurred, leaving a small amount of dark sticky residue in the precursor bulb. After ca. 10 min the tube was cooled to room temperature and the substrates were removed and stored for characterisation. Uniformly shiny silver films were observed on the substrates from the hot zone at temperatures between 650–700 °C (determined by temperature profiling), i.e. 8–18 cm away from the precursor.Film characterisation
X-ray diffraction (XRD) patterns were collected in grazing incidence mode (θ1 = 1°) using a Rigaku SmartLab diffractometer (Cu-Kα, λ = 1.5418 Å) with parallel X-ray beam and a Hypix detector operated in 1D mode. Phase matching and lattice parameter calculations (WS2) used the PDXL2 software package55 and diffraction patterns from ICSD.56 High resolution scanning electron microscopy (SEM) measurements were carried out with a field emission SEM (Jeol JSM 7500F) at an accelerating voltage of 2 kV. X-ray photoelectron spectroscopy (XPS) data were obtained using a ThermoScientific Theta Probe system with Al-Kα radiation (photon energy = 1486.6 eV). All peaks are calibrated against the adventitious C 1s peak at 284.6 eV. Raman spectra of the deposited films were measured at room temperature on a Renishaw InVia Micro Raman Spectrometer using 532 nm excitation. The incident laser power was adjusted to 0.1 mW for all samples.Conclusions
Single crystal and powder XRD data for WSeCl4 reveal discrete square pyramidal monomers, which contrasts with the weakly chloride bridged dimer and tetramer units in WSCl4, while WOCl4 is a strongly oxido-bridged polymer.3,57 The weaker Lewis acidity of WSeCl4 that this implies, is supported by DFT calculations and is also manifest in its coordination chemistry with neutral ligands. The synthesis and spectroscopic characterisation of the first extended series of complexes with neutral N-, O- and Se- donor ligands are described, which, with the exception of the seven-coordinate [WSeCl4(2,2′-bipy)], contain six-coordinate tungsten with the neutral donor trans to the WSe. The structures were confirmed by six single crystal X-ray studies, the first on WSeCl4 complexes. In contrast to the behaviour with WSCl4 and WOCl4,8 alkyl phosphines abstract Se from WSeCl4 upon contact. The complexes of WSeCl4 are less robust both in solution and in the solid state compared to those of WSCl4 and WOCl4, with some decomposing rapidly in solution.
Notably, despite its limited solution stability, [WSeCl4(SenBu2)] has been shown to function as a very effective LPCVD reagent for the deposition of thin films of the key WSe2 semiconductor; the first example of a single source precursor for this technologically important material. The resulting films were characterised by grazing incidence X-ray diffraction and scanning electron microscopy, both of which indicate very strong preferred growth in the <00l> direction, together with XPS and Raman spectroscopy. Further work focusing on optimising the film deposition and to explore the electrical properties of these CVD grown films is underway.
In contrast to the behaviour of [WSCl4(L)] analogues, the [WSeCl4(L)] generally do not lead to complexes of W(V), [WSeCl3(L)], on standing either at ambient temperature, by heating in solution, or by reaction with excess L. A few crystals of one W(V) example, [{WCl3(SMe2)}2(μ-Se)(μ-Se2)], were isolated from the attempted preparation of [WSeCl4(SMe2)].
Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
We thank EPSRC for support through EP/P025137/1 and the ADEPT Programme Grant (EP/N035437/1). We would also like to thank Prof. J. M. Dyke for helpful discussions regarding the DFT calculations reported herein. We wish to acknowledge the use of the EPSRC funded Physical Sciences Data-science Service hosted by the University of Southampton and STFC under grant number EP/S020357/1.References
- Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon, Oxford, 1987, vol. 3 Search PubMed.
- Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2003, vol. 4 Search PubMed.
- R. A. Walton, Prog. Inorg. Chem., 1972, 16, 1 CAS.
- D. A. Rice, Coord. Chem. Rev., 1978, 25, 199 CrossRef CAS.
- V. K. Greenacre, W. Levason, G. Reid and D. E. Smith, Coord. Chem. Rev., 2020, 424, 213512 CrossRef CAS.
- W. Levason, F. M. Monzittu and G. Reid, Coord. Chem. Rev., 2019, 391, 90 CrossRef CAS.
- V. K. Greenacre, A. L. Hector, W. Levason, G. Reid, D. E. Smith and L. Sutcliffe, Polyhedron, 2019, 162, 14 CrossRef CAS.
- W. Levason, G. Reid, D. E. Smith and W. Zhang, Polyhedron, 2020, 179, 114372 CrossRef CAS.
- D. E. Smith, V. K. Greenacre, A. L. Hector, W. Levason, G. Reid, F. Robinson and S. Thomas, Dalton Trans., 2020, 49, 2496 RSC.
- R. D. Bannister, W. Levason, M. E. Light, G. Reid and W. Zhang, Polyhedron, 2019, 167, 1 CrossRef CAS.
- R. D. Bannister, W. Levason, G. Reid and F. Robinson, Polyhedron, 2019, 169, 129 CrossRef CAS.
- Y.-P. Chang, A. L. Hector, W. Levason and G. Reid, Dalton Trans., 2017, 46, 9824 RSC.
- Chemical Vapour Deposition: Precursors, Processes and Applications, ed. A. C. Jones and M. L. Hitchman, The Royal Society of Chemistry, 2009 Search PubMed.
- M. Chhowalla, Z. Liu and H. Zhang, Chem. Soc. Rev., 2015, 44, 2584 RSC (ed. – themed issue on metal dichalcogenides).
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263 CrossRef CAS PubMed; J. Liu and X.-W. Liu, Adv. Mater., 2012, 24, 4097 CrossRef PubMed.
- J. R. Brent, N. Savjani and P. O'Brien, Prog. Mater. Sci., 2017, 89, 411 CrossRef CAS.
- Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575 CrossRef CAS PubMed; K. Lee, R. Gatensby, N. McEvoy, T. Hallam and G. S. Duesberg, Adv. Mater., 2013, 25, 6699 CrossRef PubMed; K. Xu, P. Chen, X. Li, C. Wu, Y. Guo, J. Zhao, X. Wu and Y. Xie, Angew. Chem., Int. Ed., 2013, 52, 10477 CrossRef PubMed; Z. Yan, C. Jiang, T. R. Pope, C. F. Tsang, J. L. Stickney, P. Goli, J. Renteria, T. T. Salguero and A. A. Balandin, J. Appl. Phys., 2013, 114, 20430 Search PubMed.
- S. L. Benjamin, C. H. de Groot, C. Gurnani, A. L. Hector, R. Huang, K. Ignatyev, W. Levason, S. J. Pearce, F. Thomas and G. Reid, Chem. Mater., 2013, 25, 4719 CrossRef CAS PubMed.
- M. D. Khan, M. A. Malik and N. Revaprasadu, Coord. Chem. Rev., 2019, 388, 24 CrossRef CAS.
- V. Brune, M. Grisch, R. Weißing, F. Hartle, M. Frank, S. Mishra and S. Mathur, Dalton Trans., 2021, 50, 12365 RSC.
- D. Britnell, G. W. A. Fowles and D. A. Rice, J. Chem. Soc., Dalton Trans., 1974, 2191 RSC.
- M. G. B. Drew, E. M. Page and D. A. Rice, J. Chem. Soc., Dalton Trans., 1983, 61 RSC.
- D. Britnell, G. W. A. Fowles and D. A. Rice, J. Chem. Soc., Dalton Trans., 1975, 213 RSC.
- G. W. A. Fowles, D. A. Rice and K. J. Shanton, J. Chem. Soc., Dalton Trans., 1978, 1658 RSC.
- N. D. Boscher, C. J. Carmalt and I. P. Parkin, J. Mater. Chem., 2006, 16, 122 RSC.
- V. C. Gibson, T. P. Kee and A. Shaw, Polyhedron, 1990, 9, 2293 CrossRef CAS.
- U. Müller and P. Klingelhöfer, Z. Anorg. Allg. Chem., 1984, 510, 109 CrossRef.
- M. G. B. Drew and R. Mandyczewky, J. Chem. Soc. A, 1970, 2815 RSC.
- F. A. Cotton, P. A. Kibala and R. B. W. Sandor, Inorg. Chem., 1989, 28, 2485 CrossRef CAS.
- D. L. Hughes, J. D. Lane and R. L. Richards, J. Chem. Soc., Dalton Trans., 1991, 1627 RSC.
- E. M. Page, D. A. Rice, K. Hagen, L. Hedberg and K. Hedberg, Inorg. Chem., 1982, 21, 3280 CrossRef CAS.
- P. J. Jones, W. Levason, J. S. Ogden, J. W. Turff, E. M. Page and D. A. Rice, J. Chem. Soc., Dalton Trans., 1983, 2625 RSC.
- (a) S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed; (c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed; (d) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- From https://cccbdb.nist.gov/vibscalejust.asp, Computational Chemistry Comparison and Benchmark Database.
- J. W. Emsley, W. Levason, G. Reid, W. Zhang and G. De Luca, J. Fluorine Chem., 2017, 197, 74 CrossRef CAS.
- M. G. B. Drew, G. W. A. Fowles, E. M. Page and D. A. Rice, J. Chem. Soc., Dalton Trans., 1981, 2409 RSC.
- P. Klingelhöfer and U. Müller, Z. Anorg. Allg. Chem., 1988, 536, 70 CrossRef.
- W. McFarlane and D. S. Rycroft, J. Chem. Soc., Dalton Trans., 1973, 2162 RSC.
- N. Muller, P. C. Lauterbur and J. Goldenson, J. Am. Chem. Soc., 1956, 78, 3556 Search PubMed.
- M. G. B. Drew, E. M. Page and D. A. Rice, J. Chem. Soc., Dalton Trans., 1983, 61 RSC.
- W. Levason, S. D. Orchard and G. Reid, Coord. Chem. Rev., 2002, 225, 159 CrossRef CAS.
- M. G. B. Drew, D. A. Rice and D. M. Williams, J. Chem. Soc., Dalton Trans., 1983, 2251 RSC.
- M. G. B. Drew, D. A. Rice and D. M. Williams, J. Chem. Soc., Dalton Trans., 1984, 1087 RSC.
- J. A. Champion, Br. J. Appl. Phys., 1965, 16, 1035 CrossRef CAS.
- W. Zhao, Z. Ghorannevis, K. K. Amara, J. R. Pang, M. Toh, X. Zhang, C. Kloc, P. H. Tan and G. Eda, Nanoscale, 2013, 5, 9677 RSC.
- H. Zeng, G.-B. Liu, J. Dai, Y. Yan, B. Zhu, R. He, L. Xie, S. Xu, X. Chen, W. Yao and X. Cui, Sci. Rep., 2013, 3, 1608 CrossRef PubMed.
- Y. Q. Ma, B. L. Liu, A. Y. Zhang, L. Chen, M. Fathi, A. Abbas, M. Y. Ge, C. F. Shen and C. W. Zhou, ACS Nano, 2015, 9, 7383 CrossRef CAS PubMed.
- X. Wang, X. Chen, Y. Zhou, C. Park, C. An, Y. Zhou, R. Zhang, C. Gu, W. Yang and Z. Yang, Sci. Rep., 2017, 46694 CrossRef CAS PubMed.
- X. Zhang, W. P. Han, J. B. Wu, S. Milana, Y. Lu, Q. Q. Li, A. C. Ferrari and P. H. Tan, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 115413 CrossRef.
- Y. Zhao, X. Luo, H. Li, J. Zhang, P. T. Araujo, C. K. Gan, J. Wu, H. Zhang, S. Y. Quek, M. S. Dresselhaus and Q. Xiong, Nano Lett., 2013, 13, 1007 CrossRef CAS PubMed.
- R. Zhang, D. Drysdale, V. Koutsos and R. Cheung, Adv. Funct. Mater., 2017, 1702455 CrossRef.
- W. Levason, R. Patel and G. Reid, J. Organomet. Chem., 2003, 688, 280 CrossRef CAS.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed; (c) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- S. Grazulis, D. Chateigner, R. T. Downs, A. F. Yokochi, M. Quiros, L. Lutterotti, E. Manakova, J. Butkus, P. Moeck and A. Le Bail, J. Appl. Crystallogr., 2009, 42, 726 CrossRef CAS PubMed.
- ICSD: Inorganic Crystal Structure Database (ICSD) accessed via the EPSRC funded Physical Sciences Data-science Service hosted by the University of Southampton and STFC.
- H. Hess and H. Hartung, Z. Anorg. Allg. Chem., 1966, 344, 157 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic parameters (Table S1), IR and NMR spectra for the new complexes (Fig. S2–S28), grazing incidence XRD patterns for further WSe2 films obtained in this work (Fig. S29), together with computational data from DFT calculations (Tables S2–S5 and Fig. S30). CCDC 2122137 (WSeCl4), 2122138 ([WSeCl4(OPPh3)]), 2122139 ([WSeCl4(DMF)]), 2122140 ([WSeCl4(MeCN)]), 2122141 ([(WSeCl4)2(4,4′-bipy)]), 2122142 ([WSeCl4(dioxane)]), 2122143 ([(WSeCl4)2(μ-SeMe2)]), 2122144 ([(WSeCl4)2(μ-Se)(μ-Se2)]). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03980f |
| This journal is © The Royal Society of Chemistry 2022 |