
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, solid-state, solution, and theoretical characterization of an “in-cage” scandium-NOTA complex†
Kelly E.
Aldrich
a,
Ivan A.
Popov
 ab,
Harrison D.
Root
a,
Enrique R.
Batista
*a,
Samuel M.
Greer
a,
Stosh A.
Kozimor
*a,
Laura M.
Lilley
a,
Maksim Y.
Livshits
a,
Veronika
Mocko
a,
Michael T.
Janicke
a,
Brian L.
Scott
a,
Benjamin W.
Stein
a and
Ping
Yang
*a
ab,
Harrison D.
Root
a,
Enrique R.
Batista
*a,
Samuel M.
Greer
a,
Stosh A.
Kozimor
*a,
Laura M.
Lilley
a,
Maksim Y.
Livshits
a,
Veronika
Mocko
a,
Michael T.
Janicke
a,
Brian L.
Scott
a,
Benjamin W.
Stein
a and
Ping
Yang
*a
aLos Alamos National Laboratory, Los Alamos, NM, USA. E-mail: erb@lanl.gov; stosh@lanl.gov; pyang@lanl.gov
bDepartment of Chemistry, The University of Akron, Akron, Ohio 44325-3601, USA
First published on 24th June 2022
Abstract
Developing chelators that strongly and selectively bind rare-earth elements (Sc, Y, La, and lanthanides) represents a longstanding fundamental challenge in inorganic chemistry. Solving these challenges is becoming more important because of increasing use of rare-earth elements in numerous technologies, ranging from paramagnets to luminescent materials. Within this context, we interrogated the complexation chemistry of the scandium(III) (Sc3+) trication with the hexadentate 1,4,7-triazacyclononane-1,4,7-triacetic acid (H3NOTA) chelator. This H3NOTA chelator is often regarded as an underperformer for complexing Sc3+. A common assumption is that metalation does not fully encapsulate Sc3+ within the NOTA3− macrocycle, leaving Sc3+ on the periphery of the chelate and susceptible to demetalation. Herein, we developed a synthetic approach that contradicted those assumptions. We confirmed that our procedure forced Sc3+ into the NOTA3− binding pocket by using single crystal X-ray diffraction to determine the Na[Sc(NOTA)(OOCCH3)] structure. Density functional theory (DFT) and 45Sc nuclear magnetic resonance (NMR) spectroscopy showed Sc3+ encapsulation was retained when the crystals were dissolved. Solution-phase and DFT studies revealed that [Sc(NOTA)(OOCCH3)]1− could accommodate an additional H2O capping ligand. Thermodynamic properties associated with the Sc-OOCCH3 and Sc-H2O capping ligand interactions demonstrated that these capping ligands occupied critical roles in stabilizing the [Sc(NOTA)] chelation complex.
Introduction
Recent advances in inorganic chemistry have supported development of a wide range of technologies based on rare-earth elements (Sc, Y, La, and lanthanides).1–6 This application space spans from superconducting materials and paramagnets7–9 to luminescent technologies10–12 as well as radiotherapeutics and radiodiagnostics.13–24 To advance the state-of-the-art in the aforementioned rare earth technologies, there is need to develop a better understanding of rare-earth element chemistry, in general. One area in need of attention is associated with rare-earth chelation. On the fundamental side, identifying chelators that bind strong and selective to Sc3+, Y3+, Lu3+, and the rest of the 4f-elements represents a longstanding challenge in inorganic chemistry. On the applied front, rare-earth chelation chemistry is important for purifying rare-earth elements in large-scale processing, remediating environmental contamination areas, and quantitatively analyzing for rare-earth elements in analytical samples.Breakthroughs in the radiopharmaceutical field have ignited recent interest in complexing one particular rare-earth element, namely Sc3+. The 44Sc and 43Sc isotopes are promising agents for positron emission tomography (PET) imaging and 47Sc is a potential therapeutic that emits low energy Auger electrons.13,25–33 Pharmaceutically relevant scandium chelators need to bind Sc3+ rapidly, irreversibly, under mild conditions, and in high yield.34 Most Sc3+ chelator design and metalation strategies are based on successful lanthanide chelation chemistry because Sc3+ shares many physical properties with lanthanide(III) cations. These characteristics include a highly stable +3 oxidation state, oxophilicity, and strong Lewis acidity. From this perspective, it is not surprising that many previous studies repurposed common lanthanide chelators for application with Sc3+. Some representative examples are provided in Chart 1; e.g. diethylenetriaminepentaacetic acid, H5DTPA, 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid, H4DOTA, and AAZTA.35–38
 | ||
| Chart 1 (a) Examples of some common chelates previously considered as Sc3+ binding agents; (b) an illustration of Sc3+ bound “in-cage”, with full chelation by the macrocycle, vs. “out-of-cage” with interactions between only the metal and pendant acetate arms. | ||
Despite strong similarities between Sc3+ and the other rare-earth elements, scandium is a first-row transition metal, which leads to significant differences compared to the lanthanides. It has valence 3d-orbitals, not 4f- and 5d-orbitals. It is also much smaller than the 4f-elements and, consequently, more Lewis acidic. For example, the eight-coordinate ionic radius for Sc3+ (0.87 Å) is over 0.3 Å smaller than the largest lanthanide (La3+ at 1.18 Å) and 0.1 Å smaller than the smallest lanthanide (Lu3+ at 0.97 Å).39 We propose that these distinctions should endow Sc3+ with chemical characteristics that can be tailored specifically to Sc3+ chelation and worry that treating Sc3+ as a small lanthanide is an oversimplification that limits innovation in Sc3+ chelator design. Within this context, we questioned why the small, hexadentate chelate H3NOTA (1,4,7-triazacyclononane-1,4,7-triacetic acid) was not more routinely used in Sc3+ chemistry. Reviewing the literature suggested NOTA3− often underperforms in Sc3+ binding, especially when compared to the larger DOTA4− chelate.35–38 This underperformance has been attributed to NOTA3− binding Sc3+ in a labile fashion, which is correlated with NOTA3− failing to encapsulated Sc3+ into its binding pocket where Sc3+ can interact with both the pendent acetate functional groups and the triaza-macrocyclic backbone (Chart 1). In the alternative and undesirable “out-of-cage” binding, Sc3+ only interacts with the acetate functional groups.36,38,40,41 Only by modifying NOTA's acetate substituents (e.g. longer linkers or alternate metal binding functionality) has “in-cage”, stable and robust Sc3+ binding been consistently realized with the 1,4,7-triaazacylcononane backbone of NOTA.34,42–44
Herein, we undertook a coordination chemistry study focused on better understanding Sc3+ complexation chemistry with NOTA3−. We synthesized an “in-cage” complex and used numerous characterization techniques to probe the interactions between Sc3+ and NOTA3−. For example, the solid-state structure of [Sc(NOTA)(OOCCH3)]1− was determined, for the first time, using single crystal X-ray diffraction. Subsequently, we showed that “in-cage” Sc3+ binding by NOTA3− was preserved in aqueous solution, using 45Sc NMR spectroscopy. Interpretation of the solution phase data was guided by theoretical studies that, when combined with experimental results, provided insight into the behavior of the [Sc(NOTA)] complex in aqueous solutions. These studies also highlighted the critical role of capping ligand(s), defined here as ligands occupying vacant coordination sites after metalating a chelator. For example, we observed that the capping acetate and H2O ligands were more than arbitrary ancillary binding agents that filled out Sc3+'s first coordination sphere. Instead, coordination of the capping ligands, specifically acetate, contributed substantially to the overall stability of the “in-cage” [Sc(NOTA)] chelation complex.
Results and discussion
Synthesis of Na[Sc(NOTA)(OOCCH3)]
Sodium scandium(III) 1,4,7-triazanonane-1,4,7-triacetate κ2-acetate, Na[Sc(NOTA)(OOCCH3)], was prepared from a reaction that fully encapsulated Sc3+ into 1,4,7-triazacyclononane-1,4,7-triacetate (NOTA3−). The resulting complex contained both Sc–ONOTA and Sc–NNOTA bonds (Scheme 1). Our experimental procedure was carried out under mild conditions. Metalation was achieved in aqueous media, under ambient atmosphere (air), using low temperatures, and in an aqueous solution that had been adjusted to a slightly acidic pH [acetate buffer, HOOCCH3/NaOOCCH3(aq); 0.1 M; pH of 5]. Under these conditions H3NOTA·3 HCl (1 equivalent) was combined with anhydrous scandium(III) triflate, Sc(OTf)3 (1 equivalent), and heated mildly (at 50 °C) for several hours to drive the reaction as far as possible toward complete complexation of Sc3+ by NOTA3−. This approach achieved high-yielding (∼70% isolated) and complete Sc3+ complexation with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand to metal ratio.
:1 ligand to metal ratio.
 | ||
| Scheme 1 Synthesis of Na[Sc(NOTA)(OOCCH3)] complex in acetate buffered solution. | ||
After heating, a small amount of free Sc3+(aq) (not complexed) was easily removed from the reaction mixture. This was achieved by precipitation of the uncomplexed Sc3+, which occurred rapidly when the pH was raised to 8 by addition of aqueous sodium hydroxide [NaOH(aq), 1 M, Ksp for Sc(OH)3 = 2.22 × 10−31].39 The resultant fine, white precipitate was removed via filtration and the filtrate was collected. Subsequent removal of the volatiles in vacuo left a white residue that contained a mixture of leftover reagents (NaOOCCH3, NaOH), byproducts (NaOTf and NaCl), and the Na[Sc(NOTA)(OOCCH3)] target compound. Purification of the Sc3+ complex from these salts required extraction of the reaction residue with a solvent mixture of methanol and water (1:1) followed by crystallization at reduced temperature (10 °C, 1 week), with acetone added as an antisolvent. This crystallization method yielded plate-like, colorless single crystals.
Solid-state characterization with single crystal X-Ray diffraction
Analysis of the colorless plates by single crystal X-ray diffraction confirmed formation of Na[Sc(NOTA)(OOCCH3)]. The crystallographic data were modeled in a C-centered, monoclinic crystal system in the C2/c space group. This model showed two [Sc(NOTA)(OOCCH3)]1− anions within the asymmetric unit (Fig. 1) that had nearly identical structures (see ESI† for more discussion on this topic). They each contained a single Sc3+ cation bound by the hexadentate NOTA3− chelate. There were three Sc–NNOTA bonds associated with the cyclic aza-macrocycle and three Sc–ONOTA bonds from the acetate arms. Although this binding motif is prototypical of complete metal binding by polyaza acetate chelators,38,42,45–47 it has not been previously observed for NOTA3− binding of Sc3+. The three acetate arms and their corresponding nitrogen atoms (from the macrocycle) were eclipsed, rather than twisted.48–51 Each [Sc(NOTA)] fragment was additionally capped by an acetate ligand (κ2-OOCCH31−) bound through both acetate oxygen atoms. This ligand environment around Sc3+ provided a total coordination number of eight, a common aqueous coordination number for Sc3+. Constraints imposed by the alkyl linkages of the hexadentate chelator (NOTA3−) and the capping ligand (κ2-OOCCH31−) prevented adaptation of an idealized 8-coordinate geometry for Sc3+ bound O and N atoms. Instead, the Sc3+ inner coordination sphere contained five O and three N atoms arranged somewhere between a bicapped trigonal antiprism (C2v) and a dodecahedron (D2d). These restrictions imparted low molecular symmetry. We identified only one pseudo-reflection plane, which contained the Sc3+ cation and the planar κ2-OOCCH31− ligand, and no rotational axes within each anionic fragment. Based on these findings, it seemed most appropriate to describe the [Sc(NOTA)(OOCCH3)]1− anion fragment using the Cs point group. | ||
| Fig. 1 Single crystal X-ray structure of Na[Sc(NOTA)(OOCCH3)]; Sc = orange, Na = aquamarine, N = blue, O = red, C = grey. Thermal ellipsoids are shown at 50% probability. Calculated H-atoms and disordered –CH2– groups on the NOTA3− backbone were omitted for clarity. Top – Asymmetric unit contents are shown, with two full [Sc(NOTA)(OOCCH3)]1− units bridged by Na1+ cations via carbonyl oxygens of the NOTA3− ligand and OOCCH31−. Bottom left – An overlay showing that the geometries for Sc1 are similar to Sc2 within the [Sc(NOTA)(OOCCH3)]1− substructure. Bottom right – The geometry of Sc1 in [Sc(NOTA)(OOCCH3)]1−. Data is from solution 1. | ||
Looking beyond the first few coordination spheres revealed that the [Sc(NOTA)(OOCCH3)]1− anions were arranged in two-dimensional sheets that extend parallel to the bc-plane (Fig. 2). Within each sheet, [Sc(NOTA)(OOCCH3)]1− anions were linked through a multifaceted Sc–O⋯Na⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O–Sc network that sandwiched the Na1+ cations between [Sc(NOTA)(OOCCH3)]1− anions. This arrangement caused the boundary of each sheet along the bc-plane to consist of hydrophobic CH2 groups from the macrocyclic NOTA-backbone (polyaza ring). Within the sandwich, the Na1+ cations were bridged by NOTA3− oxygen atoms that were not directly bound to Sc3+ (terminal carbonyl-like oxygens), as well as linkages involving both of the acetate oxygen atoms bound to Sc3+ in each anion fragment. Although the sum-total Na1+ coordination number (for each Na1+ atom) was six, the geometries around each Na1+ cation were not regular nor did they fall into idealized six-coordinate geometries (i.e. not perfectly octahedral or trigonal anitprismatic).
C–O–Sc network that sandwiched the Na1+ cations between [Sc(NOTA)(OOCCH3)]1− anions. This arrangement caused the boundary of each sheet along the bc-plane to consist of hydrophobic CH2 groups from the macrocyclic NOTA-backbone (polyaza ring). Within the sandwich, the Na1+ cations were bridged by NOTA3− oxygen atoms that were not directly bound to Sc3+ (terminal carbonyl-like oxygens), as well as linkages involving both of the acetate oxygen atoms bound to Sc3+ in each anion fragment. Although the sum-total Na1+ coordination number (for each Na1+ atom) was six, the geometries around each Na1+ cation were not regular nor did they fall into idealized six-coordinate geometries (i.e. not perfectly octahedral or trigonal anitprismatic).
 | ||
| Fig. 2 Extended structure of Na[Sc(NOTA)(OOCCH3)] with sheets of [Sc(NOTA)(OOCCH3)]x1− linked through Na1+ cations; Sc = orange, Na = aqua, N = blue, O = red, C = grey. The unit cell is shown and first-coordination sphere for Sc3+ and Na1+ have been represented as orange and aqua polyhedra, respectively. Calculated H-atoms were omitted for clarity. | ||
Many molecules of water (H2O) filled the void spaces between consecutive Na[Sc(NOTA)(OOCCH3)] layers. Exact numbers and positions of H2O molecules were difficult to determine. Hence, we investigated two crystallographic solutions. In the first model, solution 1, the electron density within the void space was treated using the “squeeze” function in Olex2 refinement software (with Platon, running ShelX refinement package).52–54 In the second model, solution 2, the residual void space electron density was modeled with discrete H2O molecules. Interatomic distances and angles for Na[Sc(NOTA)(OOCCH3)] from the two different models were essentially indistinguishable and largely fell within the uncertainty of the solution statistics. Solution 1 gave a slightly lower R-value compared to solution 2, R1 = 0.0427 vs. 0.0608. Based on this metric, we only included bond distances and angles from solution 1 herein. Related to this topic is a subtle detail associated with the Na[Sc(NOTA)(OOCCH3)] structure. Three Na1+ cations co-crystalized alongside two Sc(NOTA)(OOCCH3)1− anions. The system charge balances with one of the interstitial “water” molecules likely being a hydroxide (OH1−). Given the quality of the data, no attempt was made to model the position of the OH1− or the accompanying and likely proton disorder.
Investigation of solution structure with 45Sc NMR
A series of scandium-45 (45Sc) nuclear magnetic resonance (NMR) measurements were made to determine if the solid-state [Sc(NOTA)(OOCCH3)]1− structure described above persisted in solution. These studies exploited the unique accessibility of 45Sc NMR spectroscopy. First, the nuclear properties of naturally occurring 45Sc made it well suited for NMR spectroscopy: 100% abundant, nuclear spin of 7/2, quadrupole moment of −0.22b, and a magnetogyric ratio (γ45-Sc) of 6.5081 × 107 rad T−1 s−1 (0.3γ1-H = γ45-Sc).55,56 Second, the 45Sc NMR experiment could be conducted with common instrumentation because the 45Sc NMR excitation frequency is very close to that of the more commonly probed 13C nuclide (24.97% vs. 25.14% frequency ratios, respectively). This property makes standard broadband probes that have low frequency channels for 13C data acquisition compatible with 45Sc NMR experiments.The 45Sc NMR spectrum from single crystals of Na[Sc(NOTA)(OOCCH3)] dissolved in deuterated water (D2O) showed an intense resonance at 88.8 ppm (top, Fig. 3). This chemical shift was comparable to those reported previously from other examples of Sc3+ cations completely encapsulated by related chelating agents, e.g. Sc(DOTA) at 90 ppm, Sc(DTPA) at 79 ppm, Sc(DO3AP) at 100 ppm, and Sc(AAZTA) at 80 ppm.38,41,42 This good agreement suggested that upon dissolution, the Sc3+ cation remained encapsulated by the NOTA3− chelate, like the “in-cage” solid-state structure described above and depicted in Scheme 1. Consistent with this observation was the mismatch between our 45Sc chemical shifts and those reported for the “out-of-cage” complexation of Sc3+ by polyaza acetic acid ligands. Of specific relevance were the observations made by Huclier-Markai and co-workers.36 Those researchers observed [Sc(NOTA)] as an intermediate to “out-of-cage” complex, which had a diagnostic resonance shifted far up-field, near 20 ppm. That shift was similar, within 10 ppm, to Sc(oxalate)45− and the related “out-of-cage” Sc(DO3AP).38,41 Hence, the complexation method used by Huclier-Markai and co-workers led to “out-of-cage” Sc3+ binding, while the method used in the present work leads to “in-cage” Sc3+ binding.
 | ||
| Fig. 3 45Sc NMR spectra from single crystals of Na[Sc(NOTA)(OOCCH3)] dissolved in D2O (top), D2O with H217O (middle), and DMSO-d6 (bottom). Spectra were collected at 20 °C with an operating frequency of 97 MHz. For additional discussion of the DMSO-d6 spectrum, see ESI.† | ||
We attributed the resonance at 88.8 ppm to the [Sc(NOTA)(OOCCH3)]1− compound. However, the small downfield shoulder at 99.8 ppm clearly indicated that there was an additional 45Sc complex present in solution, one also encapsulated “in-cage” by the NOTA3− chelate (as indicated by the chemical shift). These signals were successfully deconvoluted using Gaussian fitting (in MestreNova V.14.1), which showed the 88.8 to 99.8 ppm peak intensity ratio was about 1.5 to 1 at 20 °C. The 45Sc spectrum also showed dependence on temperature (see Fig. S5–7† and discussion below). Increasing the temperature from 20 to 80 °C shifted the peak positions determined by Gaussian deconvolution slightly (Δ < 1 ppm). More significantly, increasing temperature also caused the peak at 99.8 ppm to decrease in intensity relative to the peak at 88.8 ppm. This behavior suggested the Sc3+ species associated with these two features were related by a dynamic exchange process in solution. We speculated that this exchange subtly altered the first coordination sphere of [Sc(NOTA)(OOCCH3)]1− (responsible for the peak at 88.8 ppm). Given that both resonances were consistent with Sc3+ encapsulation by NOTA3−, this first coordination sphere change was reasonably associated with the capping ligand. Likely processes responsible for the two peaks were (1) slipping the acetate from bidentate κ2-OOCCH31− to monodentate κ1-OOCCH31−, (2) hydration to add Sc-(H2O) bonding interactions (Fig. 4a), or (3) substitution of the capping acetate ligand with water (Fig. 4b). The following experiments and computational studies (vide infra) suggested that contributions from scenarios 1 and 2 were most likely responsible for the observed exchange behavior (Fig. 4a), while scenario 3 was significantly unfavorable (Fig. 4b).
 | ||
| Fig. 4 Two possible interpretations of the equilibrium process observed by 45Sc NMR spectroscopy of Na[Sc(NOTA)(OOCCH3)] crystals dissolved in D2O. The aqueous environment gives rise to two species of varying concentrations in solution; (a) coordination of water without loss of the acetate ligand vs. (b) exchange of the capping acetate ligand for water. Note, our analyses suggest that process (a) is the primary exchange process accessible in aqueous solutions. | ||
To more precisely characterize the origin of the two 45Sc resonances, two additional experiments were carried out. First, the NMR solvent was changed from D2O to DMSO-d6. The 45Sc NMR spectrum obtained from single crystals of Na[Sc(NOTA)(OOCCH3)] dissolved in DMSO-d6 showed a single broad feature at 99.47 ppm. We attributed this feature to the anhydrous [Sc(NOTA)(OOCCH3)]1− complex (see ESI†). This observation reinforced our conclusion that dissolving the crystalline material generated a solution that contained a single 45Sc species of the general formal [Sc(NOTA)(OOCCH3)]1−; however, we acknowledged that a DMSO adduct was also possible. The second 45Sc NMR experiment resembled the original measurement shown in Fig. 3 in that Na[Sc(NOTA)(OOCCH3)] crystals were dissolved in D2O. However, this solution was spiked with 17O isotopically enriched H217O (10 μL at 90% enrichment). The quadrupolar 17O nucleus (spin 5/2) provided an opportunity to identify if Sc-(H2O) bonds formed in solution. Close contact between the quadrupolar 17O and 45Sc nuclei through Sc-(H2O) interactions should enhance spin–spin relaxation, shorten the nuclear relaxation times (T2) for interacting 17O and 45Sc nuclei during the NMR experiment, and dramatically broaden the observed 45Sc NMR resonance.55,56 As demonstrated in Fig. 3, this prediction aligned well with the changes observed spectroscopically upon inclusion of H217O. The 45Sc NMR spectrum in D2O spiked with H217O showed a single resonance at 87.6 ppm. Meanwhile, the downfield shoulder at 99.8 ppm vanished into the baseline, likely owing to formation of Sc–17OH2O bonds. Reasonable attempts to deconvolute the spectrum with two Gaussians failed, indicating that the remaining signal intensity at 87.6 ppm originated from a single 45Sc species.
Based on the above experimental results (and the calculations described below), we concluded that the 45Sc NMR spectrum showed a combination of two species that coexisted in H2O solutions on the 45Sc NMR time scale. Two possible scenarios for this exchange process were presented in Fig. 4. The major contributor to the spectrum at room temperature was the anhydrous [Sc(NOTA)(OOCCH3)]1− that exhibited the 45Sc resonance near 88 ppm. There was also a minor species that exhibited a 45Sc resonance near 100 ppm, namely the hydrated [Sc(NOTA)(OOCCH3)(H2O)]1− (Fig. 4a). The data showed the Sc3+ cation in both [Sc(NOTA)(OOCCH3)(H2O)]1− and [Sc(NOTA)(OOCCH3)]1− resided in the binding pocket of NOTA3−, akin to the solid-state structure (vide supra), as evident from the dramatic downfield shifts of both species relative to the Sc(NO3)3(H2O)x standard (0 ppm). The only observable difference between the two coordination complexes was the presence of an H2O ligand in [Sc(NOTA)(OOCCH3)(H2O)]1− (Fig. 4a). The experimental data did not provide insight into the Sc-OOCCH3 binding mode and 1H and 13C NMR spectra did not generate additional insight. Hence, we have refrained from speculating on the degree of mono- vs. bidentate character associated with the Sc-OOCCH3 interaction (in either species) and simply acknowledge that both are possible in solution.
Experimental approximation of H2O coordination thermodynamics
Characterizing the solution-phase exchange process between [Sc(NOTA)(OOCCH3)]1− (88.8 ppm, linewidth = 450 Hz) and [Sc(NOTA)(OOCCH3)(H2O)]1− (99.8 ppm, linewidth = 880 Hz) by variable temperature 45Sc NMR spectroscopy (in D2O) provided an estimate of the thermodynamic properties associated with the H2O coordination process (note, chemical shifts dependence on temperature were reversible). This was achieved after assuming that these two 45Sc species had similar quadrupolar relaxations, which seemed reasonable owing to similar chemical shifts, linewidths, and coordination environments. Hence, a Van't Hoff approximation for the thermodynamic parameters associated with the H2O coordination process (Fig. 4a) was generated from the following equations: | (1) |
 | (2) |
Here, Keq was the equilibrium constant, R was the ideal gas constant (1.987 × 10−3 kcal mol−1 K−1), T was temperature (Kelvin), and ΔH and ΔS were the enthalpy and entropy changes for the exchange process, respectively. Plotting the dependence of Keq on  showed a linear relationship over the temperature region probed (Fig. 5). A linear fit of the data gave a ΔH = −1.7 ± 0.2 kcal mol−1 as described in eqn (1) and ΔS = −0.0064 ± 0.0005 kcal mol−1 K−1. As expected, these data suggested ΔS favored formation of reactants, [Sc(NOTA)(OOCCH3)]1− and H2O. This value was offset slightly by enthalpic stability that favored the [Sc(NOTA)(OOCCH3)(H2O)]1− product, which we attributed to formation of the Sc-H2O bond. The entire process was close to thermoneutral at room temperature, with a calculated ΔG of +0.2 ± 0.2 kcal mol−1 at T = 293.15 K. Overall, these values match the qualitative observation of more [Sc(NOTA)(OOCCH3)]1− in solution compared to [Sc(NOTA)(OOCCH3)(H2O)]1− (roughly 1.5:1).
showed a linear relationship over the temperature region probed (Fig. 5). A linear fit of the data gave a ΔH = −1.7 ± 0.2 kcal mol−1 as described in eqn (1) and ΔS = −0.0064 ± 0.0005 kcal mol−1 K−1. As expected, these data suggested ΔS favored formation of reactants, [Sc(NOTA)(OOCCH3)]1− and H2O. This value was offset slightly by enthalpic stability that favored the [Sc(NOTA)(OOCCH3)(H2O)]1− product, which we attributed to formation of the Sc-H2O bond. The entire process was close to thermoneutral at room temperature, with a calculated ΔG of +0.2 ± 0.2 kcal mol−1 at T = 293.15 K. Overall, these values match the qualitative observation of more [Sc(NOTA)(OOCCH3)]1− in solution compared to [Sc(NOTA)(OOCCH3)(H2O)]1− (roughly 1.5:1).
 | ||
| Fig. 5 Top: A Van't Hoff analysis of the equilibrium constant (Keq) that relates conversion of [Sc(NOTA)(OOCCH3)]1− to [Sc(NOTA)(OOCCH3)(H2O)]1−. Metrics from this plot were used to solve eqn (1) and (2). Bottom: Normalized distribution of the minor [Sc(NOTA)(OOCCH3)(H2O)]1− and major [Sc(NOTA)(OOCCH3)]1− peaks observed by 45Sc NMR spectroscopy from Na[Sc(NOTA)(OOCCH3)] crystals dissolved in D2O as a function of temperature. Peak deconvolution was performed using the “peak fitting” tool in MestreNova software V 14.1. The total peak area, [Sc(NOTA)(OOCCH3)]1− + [Sc(NOTA)(OOCCH3)(H2O)1−], was normalized to 1.0 (100%), see ESI† for more details. | ||
Calculated [Sc(NOTA)(OOCCH3)]1− and [Sc(NOTA)(OOCCH3)(H2O)]1− coordination thermodynamics
To guide the interpretation of [Sc(NOTA)(OOCCH3)]1− solution phase behavior, particularly in terms of interpreting the 45Sc NMR results discussed above, density functional theory (DFT) calculations were performed. Emphasis was placed on assessing computationally determined vs. experimentally derived Gibbs free energy (ΔG) parameters associated with conversion of Na[Sc(NOTA)(OOCCH3)] and H2O to Na[Sc(NOTA)(OOCCH3)(H2O)]. Note, these calculations included the [Sc(NOTA)(OOCCH3)]1− and the Na1+ counter cation. Calculations were also made that accounted for potential coordination with DMSO. The calculated structure for Na[Sc(NOTA)(OOCCH3)] agreed well with the experimental data (Table 1 and in the ESI†). For instance, the calculations showed stable “in-cage” Sc3+ binding by NOTA3− with bidentate binding of OOCCH31−. The calculated Sc–ligand distances were similar to those observed experimentally. One slight deviation was associated with the Sc–NNOTA bond distances of the NOTA3− macrocyclic backbone. Here, theoretically predicted Sc–NNOTA distances were slightly longer than the single crystal X-ray diffraction data by about +1.4% (on average). Similar differences in M–N distances between DFT optimized structures and X-ray diffraction data have been observed previously, specifically with M(DOTP)5− (M = Ac, Am, Cm, La)57 and M(HOPO)1− (M = Am, Cm, Cf) complexes;58 DOTP is 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetra(methylene)phosphonic acid and HOPO is an octadentate hydroxypyridinone ligand. Usually, such discrepancies emerge when calculations focus on a single molecule and omit crystal packing effects from the extended solid, e.g. the presence of multiple counter cations surrounding each anion fragment (Fig. 2, see ESI†). It is also worth noting that the differences are relative to the static structure observed in the solid state, while the thermodynamic comparisons were made versus the solvated complex.| Experimental | Bond | Fragment 1 distance/angle (°/Å) | Bond | Fragment 2 distance/angle (°/Å) |
|---|---|---|---|---|
| Sc-NOTA | Sc1–N1 | 2.384(2) | Sc2–N4 | 2.376(2) |
| Sc1–N2 | 2.374(2) | Sc2–N5 | 2.368(2) | |
| Sc1–N3 | 2.393(2) | Sc2–N6 | 2.436(2) | |
| Sc1–O1 | 2.116(1) | Sc2–O9 | 2.131(1) | |
| Sc1–O3 | 2.165(1) | Sc2–O11 | 2.158(1) | |
| Sc1–O5 | 2.155(1) | Sc2–O13 | 2.158(1) | |
| Sc-OOCCH3 | Sc1–O7 | 2.377(1) | Sc2–O15 | 2.328(1) |
| Sc1–O8 | 2.213(1) | Sc2–O16 | 2.180(1) | |
| κ2-(OOCCH3) | O7–C13–O8 | 118.89(17) | O15–C27–O16 | 118.75(17) |
| C13–O7 | 1.258(2) | C27–O15 | 1.262(2) | |
| C13–O8 | 1.283(2) | C27–O16 | 1.278(2) |
With a reasonable and validated calculated structure for [Sc(NOTA)(OOCCH3)]1− in hand, two categories of reactions involving water with Na[Sc(NOTA)(OOCCH3)] were explored. We initially probed association of water to the complex to generate Na[Sc(NOTA)(OOCCH3)(H2O)] (Fig. 4a and S15b†) and then interrogated acetate substitution by water to form [Sc(NOTA)(H2O)] or [Sc(NOTA)(H2O)2] (Fig. 4b and S15b†). Acetate substitution by water was calculated to be highly unfavorable based on the large and positive ΔG values calculated for one (+18.17 kcal mol−1) and for two (+24.94 kcal mol−1) water molecules (see ESI†). In contrast, the calculations showed that water association was thermodynamically favorable with a small, slightly negative ΔG of −0.94 kcal mol−1. A similar value of −0.74 kcal mol−1 was obtained if the calculations took into account a 1st-shell-hydrated Na1+ cation that stabilized [Sc(NOTA)(OOCCH3)]1− complex, see ESI.† Both of these values were in exceptional agreement with the experimentally derived +0.2 ± 0.2 kcal mol−1 at T = 293.15 K, especially when uncertainty between the experimental and theoretically derived values were considered. These metrics suggested that this reaction was close to thermoneutral at room temperature. The computed ΔH (−4.58 kcal mol−1) and ΔS (−0.01 kcal mol−1 K−1) parameters were also in agreement with the values estimated from experiment (−1.7 ± 0.2 and −0.0064 ± 0.0005 kcal mol−1 K−1, respectively), suggesting that the reaction was enthalpically favored. The small (close-to-zero) Gibbs free energy suggested that the [Sc(NOTA)(OOCCH3)]1− and [Sc(NOTA)(OOCCH3)(H2O)]1− structures can coexist in solution. These computational results were consistent with the mixture of two species observed experimentally. In addition to the water addition/water-acetate substitution reactions, we also considered DMSO–acetate substitution and DMSO addition reactions with Na[Sc(NOTA)(OOCCH3)]. Our calculations show that they are less thermodynamically favorable (Fig. S17 and S18†), with significantly more positive ΔG values in the range of +7.81–17.46 kcal mol−1 (eqn (S8)–(S10) in the ESI†) than the reaction of water association with Na[Sc(NOTA)(OOCCH3)].
In the calculated water addition reaction, acetate slips from bidentate (κ2-OOCCH31−) to monodentate (κ1-OOCCH31−). This modification accommodated water occupation without increasing the Sc3+ coordination number beyond eight (Fig. 4, 6 and S12, S15a†). The calculated Sc–OH2O (O9) distance was 2.374 Å and compared reasonably well with other analogous Sc–OH2O experimental values.42 It seemed likely that this calculated structure was further stabilized by hydrogen bonding between the coordinated H2O ligand and the κ1-OOCCH31− (Fig. 6).
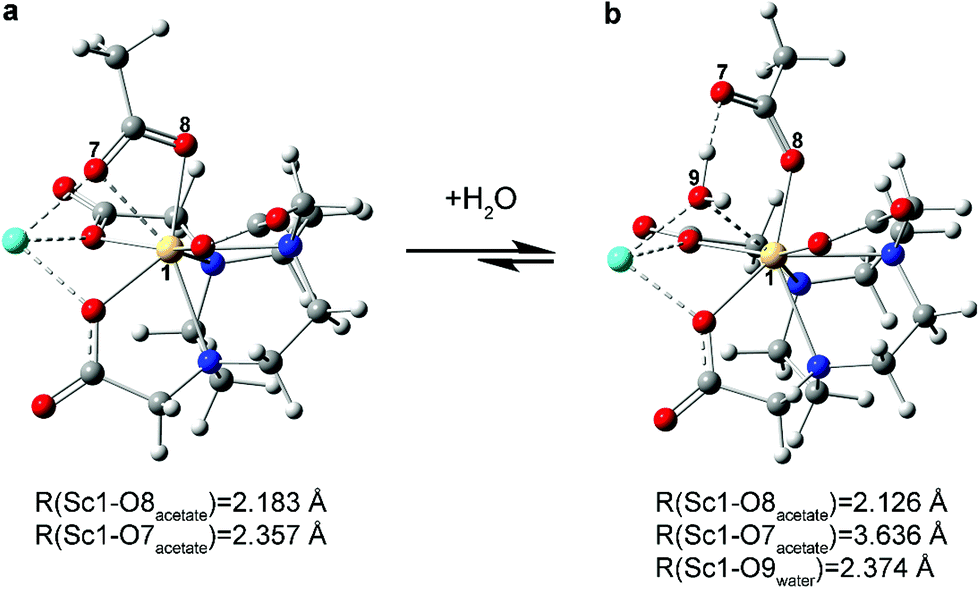 | ||
| Fig. 6 Association of a water molecule to Na[Sc(NOTA)(OOCCH3)]. Initially (a) the acetate is bound to Sc in a bidentate fashion (κ2-OOCCH31−); upon water coordination (b) the acetate slips to a monodentate binding mode (κ1-OOCCH31−). | ||
It was important to evaluate the challenge associated with computationally determining the absolute value of Gibbs free energy for the hydration reactions considered. This exercise put our calculated ΔG of −0.94 kcal mol−1 (−0.74 kcal mol−1 taking into account 1st-shell-hydrated Na1+ stabilizing [Sc(NOTA)(OOCCH3)]1− complex) for the hydration reaction into perspective. To approximate hydrogen bonding interactions that occur in solution, we used clusters of water molecules based on previously determined global minima from the gas-phase calculations.59 In this way, effects stemming from explicit waters of solvation were included, granted to a limited extent. The approach did not model dynamic hydrogen bonding interactions present in bulk water nor did it capture contributions from the outer sphere sodium cations. It did, however, provide a more reasonable alternative to ignoring bulk effects altogether (using a single water molecule on the reactant side), which is well documented to severely overestimate ΔG.57,58,60,61 It is worth noting that the calculated ΔG values fluctuated with sizes and shapes of water clusters considered in the hydration process for water association (see eqn (S4)–(7) in the ESI†). These results illustrate the challenge in determining the absolute values of ΔG for hydration reactions with high accuracy.
The DFT calculations provided guidance for interpreting what reactions were responsible for the exchange process observed experimentally (Fig. 3 and 4). These results attributed the two species observed by 45Sc NMR spectroscopy to a mixture of [Sc(NOTA)(OOCCH3)]1− and [Sc(NOTA)(OOCCH3)(H2O)]1−. The calculated thermodynamic parameters were on the same order of magnitude (within a few kcal mol−1) as the experimentally determined values. This interpretation was also self-consistent with the H217O labeled 45Sc NMR experiment and the 45Sc NMR experiment carried out in DMSO-d6. Alternative mixtures, e.g. [Sc(NOTA)(OOCCH3)]1− and [Sc(NOTA)(H2O)n] (n = 1, 2), and [Sc(NOTA)(OOCCH3)]1−/[Sc(NOTA)(DMSO)n] (n = 1, 2), were discarded based on the massively positive calculated ΔG values.
Outlook
We have established an alternate synthetic approach that fully encapsulated Sc3+ into the NOTA3− binding pocket, providing the Na[Sc(NOTA)(OOCCH3)] complex in 69% isolated yield. Conditions for Sc3+ complexation by NOTA3− were identified that could be carried out under relatively mild conditions. The procedure successfully proceeded in aqueous media, under ambient atmosphere (air), in acetate buffer solutions [HOOCCH3/NaOOCCH3(aq); 0.1 M] that were slightly acidic (pH = 5), and at relatively low temperatures (50 °C). Characterization in the solid-state showed that Sc3+ was bound “in-cage” by NOTA3−. Achieving this “in-cage” binding required a gentle synthetic hand. For example, previous studies showed the ease at which “out-of-cage” binding prevailed. Only by heating Sc3+ with H3NOTA, for longer reaction times, and in an acetic acid/acetate buffer solution did we generate an “in-cage” [Sc(NOTA)] complex.36,38 Our [Sc(NOTA)] species was capped with a bidentate κ2-OOCCH31− ligand, which provided stability (see below) and gave Sc3+ a coordination number of eight. Reaching steric saturation in aqueous media with only eight ligands also represented a distinction for Sc3+ in comparison to the larger lanthanide(III) cations, whose aqueous coordination numbers often range from 9–11.Solution phase 45Sc NMR studies demonstrated that the “in-cage” binding of Sc3+ by NOTA3− was preserved in aqueous solution. Computational results were consistent with this interpretation and suggested that in solution a mixture of [Sc(NOTA)(OOCCH3)]1− and [Sc(NOTA)(OOCCH3)(H2O)]1− coexisted. These calculations were supported by 45Sc NMR measurements made on Na[Sc(NOTA)(OOCCH3)] dissolved in water with H217O. The presence of H217O dramatically enhanced relaxation of the 45Sc resonance from [Sc(NOTA)(OOCCH3)(H217O)]1−, as a result of the close 45Sc–17OH2O interaction. We interpret our results as suggesting that [Sc(NOTA)(OOCCH3)]1− was the dominant species in solution and that the [Sc(NOTA)(OOCCH3)]1− to [Sc(NOTA)(OOCCH3)(H2O)]1− ratio was ∼1.5 to 1.
While characterizing the dynamic exchange behavior for conversion of [Sc(NOTA)(OOCCH3)]1− to [Sc(NOTA)(OOCCH3)(H2O)]1−, we identified the capping ligand(s) (OOCCH31− and H2O) substantially influenced thermodynamic stability of [Sc(NOTA)(L)x] (L = capping ligands). Both [Sc(NOTA)(OOCCH3)]1− and [Sc(NOTA)(OOCCH3)(H2O)]1− were calculated to be markedly more stable (by a ΔG of ∼20 kcal mol−1) than the simple hydrate, [Sc(NOTA)(H2O)n] (n = 1 or 2). These results highlight the importance of the capping ligands (L) in a coordination complex with the general formula M(chelator)(L)x; M being a metal like Sc3+ and chelator representing a binding agent like NOTA3−. Our results suggest that the capping ligand is more than a simple ancillary ligand and can be used to influence M(chelator) stability.
Taken as a whole, the synthetic and computational assessment of “in-cage” Sc3+ binding by NOTA3− provided insight into rare-earth complexation chemistry, particularly for Sc3+. The results highlighted how subtle variation for a given complexation method led to profoundly distinct outcomes, e.g. “in cage” and inert complexation vs. “out-of-cage” and labile binding. The data also piqued our interest in better defining the capping ligand's role in stabilizing rare-earth chelation complexes. Hence, current efforts are underway to further define Sc3+ speciation in aqueous media that contain a wider range of complexing agents and capping ligands. It is our aspiration that future studies carried out by us and others in the field will advance understanding of rare-earth complexation chemistry and contribute to developing the next generation of selective and strong binding rare-earth chelates. Success could impact rare-earth technologies broadly, and aid in solving 4f-element chelation changes.
Methods
General considerations
All reactions and manipulations were carried out in air at ambient pressures and temperatures. Anhydrous scandium(III) triflate [Sc(OTf)3, Sigma-Aldrich, 99%], scandium(III) nitrate hexahydrate [Sc(NO3)3·6 H2O, Sigma-Aldrich], 1,4,7-triazacyclononane-1,4,7-triacetic acid (H3NOTA·3 HCl, Macrocyclics), sodium acetate (NaOOCCH3, Fischer), and sodium hydroxide (NaOH, pellets, Fischer) were used as received without further purification. Deuterated water (D2O, Sigma-Aldrich) and deuterated dimethylsulfoxide (DMSO-d6, 99.9%, anhydrous, Sigma-Aldrich), used as NMR spectroscopy solvents, were also purchased and used without further purification. Oxygen-17 enriched water (H217O; estimated 90% 17O) was procured from legacy chemical inventories at LANL, and had poorly identified origins. Isotopic enrichment was determined experimentally prior to use. All pH measurements were made using Whatman pH paper (0–14 pH).Instrumentation
NMR experiments were performed on a Bruker AVANCE™ 400 MHz solution spectrometer equipped with a 5 mm broadband tunable probe for X nuclei. Infrared spectra were collected on a commercial ThermoFisher Scientific Nicolet iS5 ATR-FTIR. High resolution mass spectrometry was performed on an Agilent 6210 LC-TOF (ESI, APCI, APPI).Single crystal X-Ray diffraction
Data for Na[Sc(NOTA)(OOCCH3)] were collected on a Bruker D8 Quest diffractometer configured with a CPAD Photon II™ area detector and MoKα (λ = 0.71073 Å) IμS 3.0 micro source™. The crystal was cooled to 100 K employing an Oxford Cryostream 800™ liquid nitrogen cryostat. A hemisphere of data was collected using omega scans and 1.00° frame widths. Data collection and initial indexing and cell refinement were handled using APEX 3 software.62 Frame integration, including Lorentz-polarization corrections, and final cell parameter calculations were carried out using SAINT+ software.63 The data were corrected for absorption using redundant reflections and the SADABS program.64 The structure was solved using Intrinsic Phasing and difference Fourier techniques. All hydrogen atom positions were idealized, and rode on the atom to which they were attached. The final refinement included anisotropic temperature factors on all non-hydrogen atoms. Structure solution, refinement, graphics, and creation of publication materials were performed using SHELXTL.54 Hydrogen atom positions were idealized, and all non-hydrogen atoms were refined anisotropically. Cell indexing, data collection, integration, structure solution, and refinement were performed using Bruker and SHELXTL software. Additional details are included in the ESI.†CCDC deposit number: 2072300 and 2076191.†
Synthesis of sodium scandium(III) 1,4,7-triazacyclononane-1,4,7-triacetate κ2-acetate, Na[Sc(NOTA)(OOCCH3)]
A scintillation vial (20 mL) was loaded with Sc(OTf)3 (49 mg, 0.1 mmol), a Teflon stir bar, and an aqueous solution of NaOOCCH3 (8 mL; 0.1 M in NaOOCCH3; pH = 5; adjusted by addition of acetic acid). Note, all pH measurements were determined using pH paper. Separately, H3NOTA·3 HCl (41 mg, 0.1 mmol) was dissolved in an aqueous solution of NaOOCCH3 (3 mL; 0.1 M NaOOCCH3; pH = 4). This colorless aqueous solution of H3−nNOTAn− was added dropwise to the stirring Sc solution described above. Upon addition the reaction solution pH dropped and was subsequently adjusted to a pH of 5 by dropwise addition of NaOH (2 M). Because the homogeneity of this solution is quite sensitive to the NaOH(aq) addition [i.e. rapid addition can result in Sc(OH)xn− precipitate] the base was added slowly. The reaction solution was then heated to 50 °C and stirred overnight (∼12 h). The solution was cooled and aqueous NaOH (2 M) was added to adjust the pH to 8. This caused unreacted Sc3+ to precipitate as scandium(III) hydroxide [Sc(OH)x(H2O)9−x3−x] and slight opacity of the initially transparent solution. The precipitate represented a small percentage of dissolved Sc3+ and was easily removed by passing the solution through a filter stick packed with filter paper and Celite. Collecting the transparent and colorless filtrate into a clean scintillation vial and removing the solvent by rotary evaporation yielded a white residue. The residue was extracted with a mixture of H2O and MeOH (3 mL, 1:1, v:v). The extracts were filtered through Celite into a glass V-vial (10 mL) and the filtrate was carefully layered with acetone (1 mL). This solution was then stored at 10 °C for 1 week. From the crystallization solution, colorless plate-like crystals formed (18 mg, 41% yield) that were suitable for characterization by single crystal X-ray diffraction. Addition of more acetone (1.5 mL) to the equilibrated solution produced a second crop of crystals (12 mg, 28%). After drying the crystals, the total crystalline yield for Na[Sc(NOTA)(OOCCH3)] was 69%.
NMR: 1H (D2O, 20 °C, 400.13 MHz): δ 3.69 (br, s), 3.54 (br, s), 2.95 (br, m), 1.78 (s, OOCCH3). 1H (DMSO-d6, 20 °C, 400.13 MHz): δ 3.50 (br, s), 3.39 (s), 3.12 (br, m), 2.91 (br, m), 1.83 (s). 45Sc (D2O, 20 °C, 97.198 MHz): δ 99.8 ppm (fitted peak width, 840 Hz), 88.8 ppm (fitted peak width, 440 Hz). 45Sc (DMSO-d6, 20 °C, 97.198 MHz): δ 99.47 ppm (fitted peak width, 3744 Hz).
IR (cm −1 ): 3370 (br, m); 3130 (s); 3040 (s), 2860 (br, shoulder); 1630 (br, shoulder), 1570 (s); 1400 (s). See ESI† for spectra and additional details.
HRMS [Sc(NOTA)]: (ToF, positive ion mode) ScN3O6C12H19 (M + H)+ 346.0827, observed 346.0841; ScNaN3O6C12H18 (M + Na)+ 368.0658, observed 368.0647.
Computational details
All complexes were optimized without any constraints using self-consistent reaction field approach based on the integral equation formalism of the polarized continuum model (PCM)65,66 as implemented in Gaussian 16 software package (Version B.01).67 Water and dimethylsulfoxide (DMSO) were used as solvents in the respective reactions. Harmonic frequency calculations were performed to confirm that the optimized structures were stationary points on the potential energy surface. The initial geometry of the Na[Sc(NOTA)(OOCCH3)] complex was taken from the experimental XRD data. Energies and geometries were reported from results using the PBE068 hybrid DFT functional and def2-TZVP69 triple zeta valence basis set. Dispersion corrections (D3)70 were applied to account for possible intramolecular noncovalent interactions that can be important for the correct description of solvent-complex interaction. As reported previously for various transition-metal-based complexes,71 PBE0-D3 was found to be the best functional in the complete benchmark set relative to estimated CCSD(T)/CBS reference data, with a mean absolute deviation from the reference values of 1.1 kcal mol−1. Wavefunctions of the studied species were found to be stable, indicating that the calculations converged to the ground electronic state. Water clusters containing 16–19 molecules were used to more accurately calculate the energy of water binding/exchange between bulk water and complexes. Using only a single water molecule would neglect the energy from hydrogen bonding in water clusters and lead to less accurate ΔG values. The geometries of these water clusters were taken from the previously established global minima gas-phase structures,59 and were fully optimized in solvent medium using the PCM model in this study.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We acknowledge the US Department of Energy, Office of Science, Office of Basic Energy Sciences, Heavy Element Chemistry program (2020LANLE372) and LANL's LDRD-DR project (20180005DR) for support. Los Alamos National Laboratory is operated by Triad National Security, LLC, for the National Nuclear Security Administration of US Department of Energy (contract no. 89233218CNA000001). We are grateful for postdoctoral support provided (in-part) by the Glenn T. Seaborg Institute (KEA), by the LANL Director's Fellowship Program (SMG), and by the J. Robert Oppenheimer Distinguished Postdoctoral Fellowship (IAP). I. A. P. acknowledges the computational resources at the ARCC HPC cluster at the University of Akron. We would like to thank Mr Saman Shafaie and the Northwestern University IMSERC for performing the HRMS analyses.Notes and references
- Z. Guo and P. J. Sadler, Adv. Inorg. Chem., 2000, 49, 183–306 CrossRef CAS.
- N. Farrell, Coord. Chem. Rev., 2002, 232, 1–4 CrossRef CAS.
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS PubMed.
- J. Karges, ChemBioChem, 2020, 21, 3044–3046 CrossRef CAS PubMed.
- B. A. Corbin, A. C. Pollard, M. J. Allen and M. D. Pagel, Mol. Imaging Biol., 2019, 21, 193–199 CrossRef PubMed.
- K. J. Franz and N. Metzler-Nolte, Chem. Rev., 2019, 119, 727–729 CrossRef CAS PubMed.
- A. J. Dos santos-García, M. Á. Alario-Franco and R. Sáez-Puche, Lanthanides: Superconducting Materials, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, 2012, DOI:10.1002/9781119951438.eibc2039.
- J. E. McPeak, S. S. Eaton and G. R. Eaton, Electron paramagnetic resonance of lanthanides, Elsevier Inc., 1st edn, 2021, vol. 651 Search PubMed.
- D. Parker, E. A. Suturina, I. Kuprov and N. F. Chilton, Acc. Chem. Res., 2020, 53, 1520–1534 CrossRef CAS PubMed.
- J. C. G. Bünzli, Handb. Phys. Chem. Rare Earths, 2016, 50, 141–176 Search PubMed.
- D. Parker, J. D. Fradgley and K. L. Wong, Chem. Soc. Rev., 2021, 50, 8193–8213 RSC.
- S. V. Eliseeva and J. C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- C. Müller, K. A. Domnanich, C. A. Umbricht and N. P. Van Der Meulen, Br. J. Radiol., 2018, 91, 1–13 Search PubMed.
- R. F. Cavaier, F. Haddad, T. Sounalet, T. Stora and I. Zahi, Phys. Procedia, 2017, 90, 157–163 CrossRef.
- F. D. C. Guerra Liberal, A. A. S. Tavares and J. M. R. S. Tavares, Appl. Radiat. Isot., 2016, 110, 87–99 CrossRef CAS PubMed.
- S. J. Goldsmith, Semin. Nucl. Med., 2020, 50, 87–97 CrossRef PubMed.
- C. Müller, K. Zhernosekov, U. Köster, K. Johnston, H. Dorrer, A. Hohn, N. T. Van Der Walt, A. Türler and R. Schibli, J. Nucl. Med., 2012, 53, 1951–1959 CrossRef PubMed.
- R. P. Baum, A. Singh, M. Benešová, C. Vermeulen, S. Gnesin, U. Köster, K. Johnston, D. Müller, S. Senftleben, H. R. Kulkarni, A. Türler, R. Schibli, J. O. Prior, N. P. Van Der Meulen and C. Müller, Dalton Trans., 2017, 46, 14638–14646 RSC.
- C. Müller, C. A. Umbricht, N. Gracheva, V. J. Tschan, G. Pellegrini, P. Bernhardt, J. R. Zeevaart, U. Köster, R. Schibli and N. P. van der Meulen, Eur. J. Nucl. Med. Mol. Imaging, 2019, 46, 1919–1930 CrossRef PubMed.
- A. K. M. Rahman and A. Awal, J. Radioanal. Nucl. Chem., 2020, 323, 731–740 CrossRef CAS.
- C. Vermeulen, G. F. Steyn, F. Szelecsényi, Z. Kovács, K. Suzuki, K. Nagatsu, T. Fukumura, A. Hohn and T. N. Van Der Walt, Nucl. Instrum. Methods Phys. Res., Sect. B, 2012, 275, 24–32 CrossRef CAS.
- E. Hindie, P. Zanotti-Fregonara, M. A. Quinto, C. Morgat and C. Champion, J. Nucl. Med., 2016, 57, 759–764 CrossRef CAS PubMed.
- C. Champion, M. A. Quinto, C. Morgat, P. Zanotti-Fregonara and E. Hindié, Theranostics, 2016, 6, 1611–1618 CrossRef CAS PubMed.
- C. Müller, E. Fischer, M. Behe, U. Köster, H. Dorrer, J. Reber, S. Haller, S. Cohrs, A. Blanc, J. Grünberg, M. Bunka, K. Zhernosekov, N. van der Meulen, K. Johnston, A. Türler and R. Schibli, Nucl. Med. Biol., 2014, 41, e58–e65 CrossRef PubMed.
- C. Müller, M. Bunka, J. Reber, C. Fischer, K. Zhernosekov, A. Türler and R. Schibli, J. Nucl. Med., 2013, 54, 2168–2174 CrossRef PubMed.
- P. Santhanam, D. Taieb, L. Solnes, W. Marashdeh and P. W. Ladenson, Clin. Endocrinol., 2017, 86, 645–651 CrossRef CAS PubMed.
- S. Ferguson, H. S. Jans, M. Wuest, T. Riauka and F. Wuest, EJNMMI Phys., 2019, 6, 1–14 CrossRef PubMed.
- G. A. Wiseman, K. Pacak, M. S. O'Dorisio, D. R. Neumann, A. D. Waxman, D. A. Mankoff, S. I. Heiba, A. N. Serafini, S. S. Tumeh, N. Khutoryansky and A. F. Jacobson, J. Nucl. Med., 2009, 50, 1448–1454 CrossRef CAS PubMed.
- E. Eppard, A. de la Fuente, M. Benešová, A. Khawar, R. A. Bundschuh, F. C. Gärtner, B. Kreppel, K. Kopka, M. Essler and F. Rösch, Theranostics, 2017, 7, 4359–4369 CrossRef CAS PubMed.
- S. M. Qaim, B. Scholten and B. Neumaier, J. Radioanal. Nucl. Chem., 2018, 318, 1493–1509 CrossRef CAS.
- J. H. Turner, Br. J. Radiol., 2018, 91, 1–9 Search PubMed.
- C. S. Loveless, L. L. Radford, S. J. Ferran, S. L. Queern, M. R. Shepherd and S. E. Lapi, EJNMMI Res., 2019, 9, 1–10 CrossRef CAS PubMed.
- B. A. Vaughn, A. J. Koller and E. Boros, in Methods in Enzymology, Elsevier Inc., 1st edn, 2021, vol. 651, pp. 343–371 Search PubMed.
- B. A. Vaughn, S. H. Ahn, E. Aluicio-Sarduy, J. Devaraj, A. P. Olson, J. Engle and E. Boros, Chem. Sci., 2020, 11, 333–342 RSC.
- M. Połosak, A. Piotrowska, S. Krajewski and A. Bilewicz, J. Radioanal. Nucl. Chem., 2013, 295, 1867–1872 CrossRef PubMed.
- S. Huclier-Markai, A. Sabatie, S. Ribet, V. Kubíček, M. Paris, C. Vidaud, P. Hermann and C. S. Cutler, Radiochim. Acta, 2011, 99, 653–662 CrossRef CAS.
- A. Majkowska-Pilip and A. Bilewicz, J. Inorg. Biochem., 2011, 105, 313–320 CrossRef CAS PubMed.
- M. Pniok, V. Kubíček, J. Havlíčková, J. Kotek, A. Sabatie-Gogová, J. Plutnar, S. Huclier-Markai and P. Hermann, Chem. – Eur. J., 2014, 20, 7944–7955 CrossRef CAS PubMed.
- M. L. Williams, Occup. Environ. Med., 1996, 53, 504 CrossRef.
- S. Huclier-Markai, R. Kerdjoudj, C. Alliot, A. C. Bonraisin, N. Michel, F. Haddad and J. Barbet, Nucl. Med. Biol., 2014, 41, e36–e43 CrossRef CAS PubMed.
- R. Kerdjoudj, M. Pniok, C. Alliot, V. Kubíček, J. Havlíčková, F. Rösch, P. Hermann and S. Huclier-Markai, Dalton Trans., 2016, 45, 1398–1409 RSC.
- G. Nagy, D. Szikra, G. Trencsényi, A. Fekete, I. Garai, A. M. Giani, R. Negri, N. Masciocchi, A. Maiocchi, F. Uggeri, I. Tóth, S. Aime, G. B. Giovenzana and Z. Baranyai, Angew. Chem., Int. Ed., 2017, 56, 2118–2122 CrossRef CAS PubMed.
- J. Sinnes, J. Nagel and F. Rösch, EJNMMI Radiopharm. Chem., 2019, 4, 1–10 CrossRef PubMed.
- B. A. Vaughn, A. J. Koller, Z. Chen, S. H. Ahn, C. S. Loveless, S. J. Cingoranelli, Y. Yang, A. Cirri, C. J. Johnson, S. E. Lapi, K. W. Chapman and E. Boros, Bioconjugate Chem., 2021, 32, 1232–1241 CrossRef CAS PubMed.
- S. Aime, A. Barge, F. Benetollo, G. Bombieri, M. Botta and F. Uggeri, Inorg. Chem., 1997, 36, 4287–4289 CrossRef CAS.
- F. Benetollo, G. Bombieri, L. Calabi, S. Aime and M. Botta, Inorg. Chem., 2003, 42, 148–157 CrossRef CAS PubMed.
- P. Vojtíšek, P. Cígler, J. Kotek, J. Rudovský, P. Hermann and I. Lukeš, Inorg. Chem., 2005, 44, 5591–5599 CrossRef PubMed.
- F. Benetollo, G. Bombieri, L. Calabi, S. Aime and M. Botta, Inorg. Chem., 2003, 42, 148–157 CrossRef CAS PubMed.
- C. Kumas, W. S. Fernando, P. Zhao, M. Regueiro-Figueroa, G. E. Kiefer, A. F. Martins, C. Platas-Iglesias and A. D. Sherry, Inorg. Chem., 2016, 55, 9297–9305 CrossRef CAS PubMed.
- K. J. Miller, A. A. Saherwala, B. C. Webber, Y. Wu, A. D. Sherry and M. Woods, Inorg. Chem., 2010, 49, 8662–8664 CrossRef CAS PubMed.
- J. Blahut, P. Hermann, Z. Tošner and C. Platas-Iglesias, Phys. Chem. Chem. Phys., 2017, 19, 26662–26671 RSC.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, SHELXTL, University of Gottingen, Germany, 2016 Search PubMed.
- J. Mason, Multinuclear NMR, Plenum Press, New York, 6th edn, 1987 Search PubMed.
- A. Abragam, The Principles of Nuclear Magnetism, Clarendon Press, 1961 Search PubMed.
- B. W. Stein, A. Morgenstern, E. R. Batista, E. R. Birnbaum, S. E. Bone, S. K. Cary, M. G. Ferrier, K. D. John, J. L. Pacheco, S. A. Kozimor, V. Mocko, B. L. Scott and P. Yang, J. Am. Chem. Soc., 2019, 141, 19404–19414 CrossRef CAS PubMed.
- M. P. Kelley, G. J. P. Deblonde, J. Su, C. H. Booth, R. J. Abergel, E. R. Batista and P. Yang, Inorg. Chem., 2018, 57, 5352–5363 CrossRef CAS PubMed.
- J. T. Su, X. Xu and W. A. Goddard III, J. Phys. Chem. A, 2004, 108(47), 10518–10526 CrossRef CAS.
- M. P. Kelley, J. Su, M. Urban, M. Luckey, E. R. Batista, P. Yang and J. C. Shafer, J. Am. Chem. Soc., 2017, 139, 9901–9908 CrossRef CAS PubMed.
- X. Cao, D. Heidelberg, J. Ciupka and M. Dolg, Inorg. Chem., 2010, 49, 10307–10315 CrossRef CAS PubMed.
- APEX3, Bruker, AXS Inc., Madison, Wisconsin, 2019 Search PubMed.
- SAINT+, Bruker, AXS Inc., Madison, Wisconsin, 2013 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- B. Mennucci, E. Cancès and J. Tomasi, J. Phys. Chem. B, 1997, 101, 10506–10517 CrossRef CAS.
- E. Cancès and B. Mennucci, J. Math. Chem., 1998, 23, 309–326 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 132 CrossRef PubMed.
- M. Steinmetz and S. Grimme, ChemistryOpen, 2013, 2, 115–124 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra including fits for VT NMR; additional discussion of X-Ray diffraction data; additional information on the computed reactions and method details. CIF files for SOLUTION_1 of the solid-state single crystal structure. CCDC 2072300 and 2076191. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d1dt03887g |
| This journal is © The Royal Society of Chemistry 2022 |