
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of cysteine thiols on the catalytic and anticancer activity of Ru(II) sulfonyl-ethylenediamine complexes†
Feng
Chen
 ab,
Isolda
Romero-Canelón
ac,
Abraha
Habtemariam
a,
Ji-Inn
Song
a,
Samya
Banerjee
ad,
Guy J.
Clarkson
a,
Lijiang
Song
a,
Ivan
Prokes
a and
Peter J.
Sadler
*a
ab,
Isolda
Romero-Canelón
ac,
Abraha
Habtemariam
a,
Ji-Inn
Song
a,
Samya
Banerjee
ad,
Guy J.
Clarkson
a,
Lijiang
Song
a,
Ivan
Prokes
a and
Peter J.
Sadler
*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: P.J.Sadler@warwick.ac.uk
bSchool of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang 212013, PR China
cSchool of Pharmacy, University of Birmingham, Birmingham B15 2TT, UK
dDepartment of Chemistry, Indian Institute of Technology (BHU), Varanasi, UP-221005, India
First published on 28th February 2022
Abstract
We have synthesized a series of novel substituted sulfonyl ethylenediamine (en) RuII arene complexes 1–8 of [(η6-arene)Ru(R1-SO2-EnBz)X], where the arene is benzene, HO(CH2)2O-phenyl or biphenyl (biph), X = Cl or I, and R1 is phenyl, 4-Me-phenyl, 4-NO2-phenyl or dansyl. The ‘piano-stool’ structure of complex 3, [(η6-biph)Ru(4-Me-phenyl-SO2-EnBz)I], was confirmed by X-ray crystallography. The 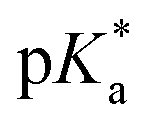 values of their aqua adducts were determined to be high (9.1 to 9.7). Complexes 1–8 have antiproliferative activity against human A2780 ovarian, and A549 lung cancer cells with IC50 values ranging from 4.1 to >50 μM, although, remarkably, complex 7 [(η6-biph)Ru(phenyl-SO2-EnBz)Cl] was inactive towards A2780 cells, but as potent as the clinical drug cisplatin towards A549 cells. All these complexes also showed catalytic activity in transfer hydrogenation (TH) of NAD+ to NADH with sodium formate as hydride donor, with TOFs in the range of 2.5–9.7 h−1. The complexes reacted rapidly with the thiols glutathione (GSH) and N-acetyl-L-cysteine (NAC), forming dinuclear bridged complexes [(η6-biph)2Ru2(GS)3]2− or [(η6-biph)2Ru2(NAC-H)3]2−, with the liberation of the diamine ligand which was detected by LC-MS. In addition, the switching on of fluorescence for complex 8 in aqueous solution confirmed release of the chelated DsEnBz ligand in reactions with these thiols. Reactions with GSH hampered the catalytic TH of NAD+ to NADH due to the decomposition of the complexes. Co-administration to cells of complex 2 [(η6-biph)Ru(4-Me-phenyl-SO2-EnBz)Cl] with L-buthionine sulfoximine (L-BSO), an inhibitor of GSH synthesis, partially restored the anticancer activity towards A2780 ovarian cancer cells. Complex 2 caused a concentration-dependent G1 phase cell cycle arrest, and induced a significant level of reactive oxygen species (ROS) in A2780 human ovarian cancer cells. The amount of induced ROS decreased with increase in GSH concentration, perhaps due to the formation of the dinuclear Ru-SG complex.
values of their aqua adducts were determined to be high (9.1 to 9.7). Complexes 1–8 have antiproliferative activity against human A2780 ovarian, and A549 lung cancer cells with IC50 values ranging from 4.1 to >50 μM, although, remarkably, complex 7 [(η6-biph)Ru(phenyl-SO2-EnBz)Cl] was inactive towards A2780 cells, but as potent as the clinical drug cisplatin towards A549 cells. All these complexes also showed catalytic activity in transfer hydrogenation (TH) of NAD+ to NADH with sodium formate as hydride donor, with TOFs in the range of 2.5–9.7 h−1. The complexes reacted rapidly with the thiols glutathione (GSH) and N-acetyl-L-cysteine (NAC), forming dinuclear bridged complexes [(η6-biph)2Ru2(GS)3]2− or [(η6-biph)2Ru2(NAC-H)3]2−, with the liberation of the diamine ligand which was detected by LC-MS. In addition, the switching on of fluorescence for complex 8 in aqueous solution confirmed release of the chelated DsEnBz ligand in reactions with these thiols. Reactions with GSH hampered the catalytic TH of NAD+ to NADH due to the decomposition of the complexes. Co-administration to cells of complex 2 [(η6-biph)Ru(4-Me-phenyl-SO2-EnBz)Cl] with L-buthionine sulfoximine (L-BSO), an inhibitor of GSH synthesis, partially restored the anticancer activity towards A2780 ovarian cancer cells. Complex 2 caused a concentration-dependent G1 phase cell cycle arrest, and induced a significant level of reactive oxygen species (ROS) in A2780 human ovarian cancer cells. The amount of induced ROS decreased with increase in GSH concentration, perhaps due to the formation of the dinuclear Ru-SG complex.
Introduction
Studies of the antiproliferative activity of organometallic complexes, especially the Ir,1–3 Os,4 Rh5,6 and Ru7,8 complexes, have been stimulated by the clinical success of platinum anticancer drugs such as cisplatin, carboplatin and oxaliplatin.9–11 The activity of organometallic half-sandwich complexes can be tuned by the choice of the arene, chelating and monodentate ligands.12,13 In recent years, the possibility of using synthetic organometallic complexes as catalysts for transfer hydrogenation (TH) reactions in cells has emerged as a potential way of modulating intracellular redox states and inducing cancer cell-killing by a mechanism which can circumvent resistance.14,15 Noyori-type RuII complex JS2 [(η6-p-cym)Ru(TsEn)Cl] exhibits potent catalytic TH activity in the reduction of co-enzyme NAD+ to NADH. Up to a 90% decrease in cancer cell viability was observed in the presence of non-toxic doses of sodium formate (Fig. 1), with the ratio of NAD+/NADH greatly decreasing in cancer cells.16 The analogue complex [(η6-p-cym)Ru(TsEnBz)Cl] shows enhanced activity in the TH of NAD+ with formate as hydride donor,17 however, the reaction appeared to be hampered by interaction of the RuII complex with thiol-containing molecules, including the tripeptide glutathione, γ-L-Glu-L-Cys-Gly (GSH, Fig. 1).17 We have therefore explored the role of GSH in mediating reactions of these half-sandwich organometallic catalysts in this work. | ||
| Fig. 1 Structures of Noyori-type RuII anticancer catalysts, glutathione (GSH) and N-acetyl-L-cysteine (NAC). | ||
GSH is an important tripeptide that exists ubiquitously in all eukaryotic cells (in mM concentrations); it can be oxidized, e.g. to GSSG, to protect cells from being damaged by reactive oxygen species (ROS, metabolic side products).18–20 GSH can also interact with metal complexes in cells, and many organometallic complexes are thiophilic.21 By taking advantage of such interactions, thiols have been used as switch-on probes to trigger luminescence or fluorescence in cells, by either reacting with a probe or by displacement of fluorescent ligands, which can be used to map their distributions in cells.22,23 We have found that the tethered RuII complex [Ru(η6-Ph(CH2)3-ethylenediamine-N-Ts)Cl] can rapidly react with GSH to form a Ru-SG adduct which can decompose slowly.24
In the present work, we have studied the effect of the amino acid cysteine and the tripeptide glutathione on the catalytic and anticancer activity of RuII sulfonyl ethylenediamine complexes 1–8 [(η6-arene)Ru(R1-SO2-EnBz)X], where the arene is benzene, HO(CH2)2O-phenyl, or biphenyl, and R1 is various sulfonyl substituents (Table 1). The catalytic TH reduction of NAD+ to NADH using sodium formate as hydride source was studied, as well as reactions of 1–8 with the tripeptide GSH and N-acetyl-L-cysteine (NAC) (Fig. 1), and the effect of GSH on the TH of NAD+. The anticancer activity of complexes 1–8 towards A2780 ovarian and A549 lung human cancer cells was determined together with the effect of coadministration with GSH, NAC, and the redox modulator L-buthionine sulfoximine (L-BSO), an inhibitor of GSH biosynthesis. Cell cycle arrest and the influence of GSH on induction of ROS were also investigated. The study revealed an interesting and unusual role for GSH in the biological activity of this class of organometallic transfer hydrogenation catalysts.
|
|
|||
|---|---|---|---|
| Complex | R | R1 | X |
| 1 | H | 4-Me-Ph | Cl |
| 2 | Ph | 4-Me-Ph | Cl |
| 3 | Ph | 4-Me-Ph | I |
| 4 | O(CH2)2OH | 4-Me-Ph | Cl |
| 5 | Ph | 4-Nitro-Ph | Cl |
| 6 | Ph | 4-F-Ph | Cl |
| 7 | Ph | Ph | Cl |
| 8 | Ph | Dansyl (Ds) | Cl |

Results
Synthesis and characterizations
The chelating diamine ligands [BzEn-SO2-R1], where R1 is phenyl, 4-Me-phenyl, 4-F-phenyl, 4-NO2-phenyl and Dansyl, were synthesized following a reported method,17 as were complexes 1–8 (Table 1).25 Triethylamine (4 mol equiv.) and ligand (2–2.1 mol equiv.) were added to the a solution of degassed isopropanol and RuII dimer [(η6-arene)RuCl2]2, and the reactions were then stirred under a N2 atmosphere at 365 K for 10 h. All the synthesized complexes were purified 2× by silica column chromatography (MeOH/DCM, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 v/v) and recrystallization, and characterized by NMR spectroscopy (1H, 13C and 19F, Fig. S1–S17 in the ESI†), high resolution mass spectrometry (HR-MS, Fig. S18–S25 in the ESI†), and elemental analysis (CHN).
:8 v/v) and recrystallization, and characterized by NMR spectroscopy (1H, 13C and 19F, Fig. S1–S17 in the ESI†), high resolution mass spectrometry (HR-MS, Fig. S18–S25 in the ESI†), and elemental analysis (CHN).
X-ray crystal structure
A crystal of complex 3 [(η6-biph)Ru(4-Me-phenyl-SO2-EnBz)I] suitable for X-ray crystallographic analysis was obtained by the slow diffusion of diethyl ether into a saturated methanol solution of complex 3 at ambient temperature. Selected bond lengths and angles for complex 3 are given in Table 2, crystallography data are listed in Tables S1 and S2,† and the molecular structure is shown in Fig. 2. Complex 3 adopts a pseudo-tetrahedral geometry with a η6-phenyl ring on one face of the metal centre occupying 3 coordination sites. The sulphonamide nitrogen of the ethylenediamine ligand is deprotonated and bound as a monoanionic bidentate ligand to Ru, together with an iodido ligand completing the coordination sphere of the complex.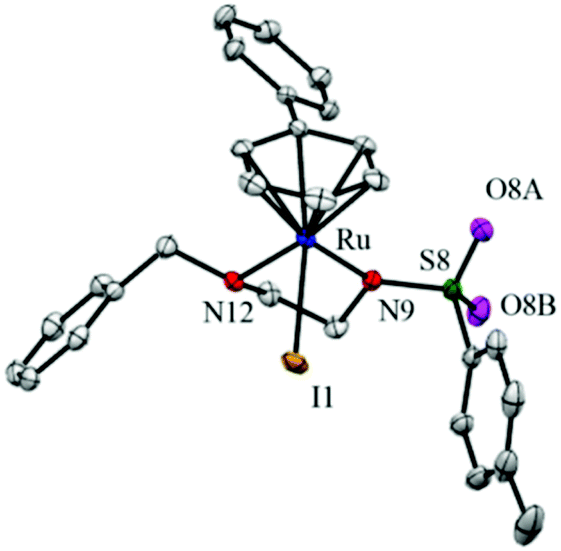 | ||
| Fig. 2 ORTEP diagram for complex 3 [(η6-biph)Ru(4-Me-phenyl-SO2EnBz)I]. Ellipsoids are shown at the 50% probability level. All hydrogen atoms have been omitted for clarity. | ||
| Bonds | Length (Å)/angle (°) |
|---|---|
| Ru–N9 | 2.123(3) |
| Ru–N12 | 2.174(3) |
| Ru–I1 | 2.7434(3) |
| Ru–arene (centroid) | 1.672 |
| N9–Ru–N12 | 78.60(11) |
| N9–Ru–I1 | 90.65(7) |
| N12–Ru–I1 | 83.90(8) |
 determination
determination
The 
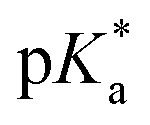 values (pKa determined in deuterated solvent) of the aqua adducts of complexes 1, 2 and 4–7 were determined by titration over the pH* range from 2 to 12. The 1H NMR chemical shifts of protons of the sulfonyl phenyls as a function pH were fitted to the Henderson–Hasselbalch equation (Fig. S26 in the ESI†). All the
values (pKa determined in deuterated solvent) of the aqua adducts of complexes 1, 2 and 4–7 were determined by titration over the pH* range from 2 to 12. The 1H NMR chemical shifts of protons of the sulfonyl phenyls as a function pH were fitted to the Henderson–Hasselbalch equation (Fig. S26 in the ESI†). All the  values of these aqua complexes are in the range of 9.10–9.75 (Table 3).
values of these aqua complexes are in the range of 9.10–9.75 (Table 3).
 values for the aqua adducts
values for the aqua adducts
| Complex | TOF (h−1) |
|
|---|---|---|
| 1 | 7.5 ± 0.3 | 9.67 ± 0.03 |
| 2 | 7.9 ± 0.4 | 9.3 ± 0.1 |
| 3 | 5.7 ± 1.4 | n. d. |
| 4 | 2.5 ± 0.1 | 9.71 ± 0.05 |
| 5 | 9.1 ± 0.5 | 9.1 ± 0.2 |
| 6 | 9.7 ± 0.1 | 9.12 ± 0.04 |
| 7 | 6.74 ± 0.04 | 9.34 ± 0.07 |
| 8 | 3.7 ± 0.6 | n. d. |

Nucleobase binding
The interaction of complex 2 with model nucleobase 9-ethylguanine (9-EtG) was studied by 1H NMR spectroscopy (Fig. S27 in the ESI†). The reactions were performed by titrating a solution of complex 2 (2 mM, 10% MeOD-d4 in D2O) with 9-EtG in D2O at 310 K, in 0.5 mol equiv. steps. Complex 2 reacted rapidly with 9-EtG, and binding appeared to be complete within 5 min. Adduct formation was confirmed by following a new set of peaks, with slow exchange on the NMR time scale between the aqua adduct of 2 and its 9-EtG adduct. Peaks for complex 2 disappeared when 1.0 mol equiv. of 9-EtG was added, indicating strong binding and completion of the reaction (Fig. S27 in the ESI†).Kinetics of transfer hydrogenation
Transfer hydrogenation (TH) of nicotinamide adenine dinucleotide (NAD+) to give NADH was studied in aqueous media using complexes 1–8 as catalysts and sodium formate as hydride source (MeOD-d4/D2O, 1:9 (v/v), pH* 7.2 ± 0.1, 310 K). All the kinetic experiments were monitored by 1H NMR with RuII, NAD+, and sodium formate in a mol ratio of 1:4:25. As shown in Table 3, the turnover frequencies (TOFs) for complexes 1–8 are in the range 2.5–9.7 h−1, Complex 6 gave the highest TOF (9.7 ± 0.1 h−1), while complex 4 gave the lowest (2.5 ± 0.1 h−1). Complexes 1 [(η6-benzene)Ru(4-Me-phenyl-SO2EnBz)Cl] and 2 [(η6-biph)Ru(4-Me-phenyl-SO2EnBz)Cl] bearing the 4-Me-phenyl-SO2EnBz ligand have similar TOF values (7.5 ± 0.3 and 7.9 ± 0.4 h−1, respectively), while complexes 5 and 6 with strong 4-NO2 and 4-F electron-withdrawing groups have higher catalytic efficiency than 2, with TOF values of 9.1 ± 0.5 and 9.7 ± 0.1 h−1, respectively; while complex 7 (phenyl) with the relatively weaker electron-withdrawing group gave a slightly lower TOF value (6.74 ± 0.04 h−1).
Reactions with glutathione
Reactions of complex 2 with GSH were investigated via a series of concentration-dependent experiments and monitored by 1H NMR, at pH* 7.2, (pH* = pH meter reading in deuterated solvent), 310 K. Complex 2 (2 mM) and GSH were mixed in the mol ratio of 1:X, where X = 1, 2, 5, 10, respectively, in the mixed solvent of MeOD-d4 and D2O, 1:9 (v/v). Reaction of complex 2 with 1.0 mol equiv. GSH led to the disappearance of 1H NMR peaks for 2 in the biphenyl ligand region of the spectrum within 10 min, and generated a new set of peaks shifted to lower field (Fig. S28 in the ESI†). No further change in this region was observed with further addition of GSH (2–10 mol equiv.). However, the products were difficult to identify based on the 1H NMR spectra alone (Fig. S28 in the ESI†).
Identification of GSH/NAC adducts by LC-MS
HPLC and LC-MS were used to elucidate the nature of the products from reactions of complex 2 with GSH. Similar reactions with N-acetyl-L-cysteine (NAC) were studied for comparison.Complex 2 (2 mM in MeOH/H2O, 1:9 (v/v)) and GSH or NAC (10 mol equiv., in H2O) were mixed in a vial and pre-incubated at 310 K for 24 h (pH 7.10 ± 0.1). As can be seen from Fig. S29,† the reactions proceeded with >95% and 100% conversions to form the RuII-SG and RuII-NAC adducts, respectively, as determined by HPLC. HPLC peak p4, assignable to complex 2, disappeared after 24 h co-incubation at 310 K, with two new peaks p1 and p2 emerging (Fig. S29 in the ESI†). Subsequently, reactions were studied by LC-MS using the same conditions. MS peaks for dinuclear complexes [(η6-biph)2Ru2(GS)3]2−2a and [(η6-biph)2Ru2(NAC-H)3]2−2b are assignable to HPLC peaks p1 and p2, respectively. Displaced free chelating TsEnBz ligand was detected as peak p3. The peak assignments are listed in Table S3 in the ESI.†
Next, the isolation of the dinuclear complexes 2a and 2b was attempted by HPLC using a ZORBAX Eclipse XDB-C18 Semi-preparative column (9.4 × 250 mm). Complex 2b was collected and characterized by 1H NMR (Fig. S30 in the ESI†). A high resolution MS peak at 715.6011 m/z was observed, which corresponds to [(η6-biph)2Ru2(GS)3+4H]2+ (2a in Fig. S31 in the ESI†), and the high resolution peak at 998.0339 m/z can be assigned to [(η6-biph)2Ru2(NAC)3]+ (2b in Fig. S32 in the ESI†).
Effect of GSH on transfer hydrogenation of NAD+
Given the high abundance of GSH in mammalian cells, the influence of GSH on the TH of NAD+ catalysed by complex 2 was investigated under similar conditions: 2 (1.4 mM), 10% MeOD-d4/90% D2O (v/v), with NAD+, GSH and sodium formate (D2O) in the mol ratios of 1:4:X:25, where X = 0.2, 0.5, 1 and 2, pH* 7.2, 310 K. 1H NMR spectra were recorded every 5 min. The catalytic efficiency of complex 2 was little affected, with the TOF decreasing slightly from 7.9 ± 0.4 h−1 to 6.29 ± 0.53 h−1 when 0.2 mol equiv. GSH was present. However, the TOF dropped dramatically to 0.91 ± 0.43 h−1 when 0.5 mol equiv. GSH was co-administered. The reaction totally stopped when 1 mol equiv. or more GSH was added, probably due to the completing reaction of 2 with GSH.
Fluorescence-detected thiol-triggered chelated ligand release from complex 8
The fluorescence of the chelated phenylsulfonyl ethylenediamine dansyl-ligand DsEnBz was fully quenched when it was present as a chelated ligand in complex 8 (Table 1). Complex 8 alone fluoresced only very weakly when dissolved in a mixed solvent of DMSO and H2O (1:9, v/v), Fig. 3. As found above, GSH can react rapidly with complex 8 to form [(η6-biph)2Ru2(GS)3]2−, accompanied by the release of the sulfonyl ethylenediamine ligand. Such a reaction should switch-on the fluorescence of DsEnBz through its release from complex 8. An immediate emission was observed when complex 8 (2 mM in DMSO/H2O, 2:8(v/v)) was treated with GSH or NAC (20 mM in H2O, Fig. 3).
 | ||
| Fig. 3 GSH- and NAC-triggered fluorescence of complex 8. (a) UV-vis detection of reactions of complex 8 with GSH or NAC; (b) solution excited at 350 nm; (c) solution excited at 405 nm; (d) A: complex 8 in DMSO, B: complex 8 in DMSO/H2O (1:9 (v/v)), C: complex 8 (0.1 mM, DMSO/H2O, 1:9 (v/v)) with GSH (2 mM in water) and D: complex 8 (0.1 mM, DMSO/H2O, 1:9 (v/v)) with NAC (2 mM in water) under UVA; (e) fluorescence from reactions of complex 8 with L-leucine (E, 10 mol equiv.), L-tryptophan (F, 10 mol equiv.) and 1-butanethiol (G, 10 mol equiv.) under UVA. | ||
The reaction was initially detected by UV-vis spectroscopy at 310 K, pH 7 (Fig. 3a). Next, a ca. 200-fold increase in emission intensity was observed on adding GSH (10 mol equiv.) to an aqueous solution of complex 8, pH 7, 310 K, on excitation at 350 nm (Fig. 3b). NAC induced a stronger increase in fluorescence under the same conditions (ca. 1.7-fold stronger than GSH). When excited at 405 nm, the emission intensity was relatively lower, only about 40-fold intensity for GSH and 60-fold for NAC compared to complex 8 alone, Fig. 3c. In order to confirm the importance of the thiol groups in these reactions, complex 8 was reacted with the amino acids L-leucine and L-tryptophan, and the thiol-containing molecule 1-butanethiol. No increase in fluorescence was observed when 8 was mixed with thiol-free amino acids; however, a relatively strong fluorescence emission was found when 1-butanethiol was added, indicating the key role of the thiol group.
Antiproliferative activity
The antiproliferative activity of complexes 1–8 towards A2780 human ovarian and A549 human lung cancer cells was determined (Table 4). The clinical drug cisplatin (CDDP) was studied as a comparison. As can be seen in Table 4, these complexes gave a broad range of IC50 values ranging from 3.57 to >50 μM and 4.1 to 38.5 μM against A2780 human ovarian and A549 human lung cancer cells, respectively. Complex 6 [(η6-biph)Ru(4-F-phenyl-SO2-EnBz)Cl] was most potent towards A2780 cancer cells (IC50, 3.57 ± 0.98 μM), towards which complex 7 [(η6-biph)Ru(phenyl-SO2-EnBz)Cl] was inactive. However, complex 7 was potent towards A549 lung cancer cells with an IC50, 4.1 ± 1.3 μM, comparable to CDDP (IC50, 3.1 ± 0.1 μM).| IC50a (μM) |
||
|---|---|---|
| Complex | A2780 | A549 |
| a Data are shown as mean ± standard deviation (STD), cell viability was assessed after 24 h incubation with RuII complexes and washing with PBS. | ||
| 1 | 8.32 ± 0.54 | 28.8 ± 2.6 |
| 2 | 11.25 ± 0.08 | 13.5 ± 1.4 |
| 3 | 18.4 ± 1.2 | 32.2 ± 0.7 |
| 4 | 14.25 ± 0.06 | 16.1 ± 2.4 |
| 5 | 3.57 ± 0.98 | 29.8 ± 1.1 |
| 6 | 5.6 ± 0.5 | 13.7 ± 0.1 |
| 7 | >50 | 4.1 ± 1.3 |
| 8 | 39.4 ± 3.4 | 38.5 ± 1.9 |
| CDDP | 1.2 ± 0.02 | 3.1 ± 0.1 |
Effect of L-buthionine sulfoximine on antiproliferative activity
L-Buthionine sulfoximine (L-BSO) is a specific inhibitor of γ-glutamylcysteine synthetase,26 an enzyme involved in the biosynthesis of GSH. Treatment with L-BSO can scavenge the intracellular GSH levels up to 40%, which effectively hampers cellular GSH synthesis.27 The anticancer activity of complex 2 against A549 human lung cancer cells after co-incubation with L-BSO was determined, to provide insights into the role of GSH in antiproliferative activity. Complex 2 was coadministered with three different concentrations of L-BSO: 1, 5 and 50 μM. After 24 h co-incubation with L-BSO (at concentrations of 1 and 5 μM), the antiproliferative activity of 2 (IC50, ca. 13 μM) remained unchanged; but increased by 61% to 8.3 ± 0.5 μM when co-treated with 50 μM L-BSO.Effect of GSH and NAC on anticancer activity
GSH often acts as a detoxification agent for metal-based drugs in cells, and some drug-resistant cancer cells are capable of generating higher levels of GSH to circumvent damage.21N-Acetyl-L-cysteine (NAC) is scavenger of reactive oxygen species (ROS), which can block cisplatin related caspase-3 activation and cell apoptosis.28Since complex 2 can react rapidly with GSH to form the dimer 2a [(η6-biph)2Ru2(GS)3]2−, co-administration of complex 2 with GSH (5, 10 and 50 μM) was studied, to investigate the effect of GSH on the antiproliferative activity in A2780 human ovarian carcinoma cells. Cells were incubated with three concentrations of GSH (5, 10 and 50 μM) as controls. The results indicated that GSH exposure only is not toxic towards A2780 cancer cells, Table S4 (in the ESI†). After 72 h of recovery time in drug-free medium, cell survival was evaluated using the sulforhodamine B colorimetric assay. As shown in Fig. 4, the antiproliferative activity decreased gradually with increase of GSH concentration, giving IC50 values of 22.41 ± 1.25, 29.9 ± 2.1 and >50 μM towards A2780 cells and, 27.33 ± 0.54, 43.93 ± 3.54 and >50 μM towards A549 cancer cells, for GSH concentrations of 5, 10, and 50 μM, respectively.
 | ||
| Fig. 4 Effect of co-administration with GSH (5, 10 or 50 μM) on the antiproliferative activity of complex 2 against human A2780 ovarian and A549 lung cancer cells. Data are presented in mean ± standard deviation (STD). | ||
Co-treatment with complex 2 and NAC gave rise to a similar trend, in which the anticancer activity decreased with an increase in NAC concentration (under similar conditions as those for GSH above), IC50 values are 25.8 ± 0.9, 39.9 ± 0.9 and >50 μM, for NAC concentrations of 5, 10 and 50 μM, respectively.
Cell cycle arrest
A cell cycle analysis for A2780 human ovarian cancer cells treated with the representative complex 2 was performed using propidium iodide staining and flow cytometry. A2780 cancer cells were incubated with IC50 or 2 × IC50 concentrations of complex 2 for 24 h. In comparison to negative control population, complex 2 increased cell cycle arrest at the G1 phase when the drug concentration increased from IC50 to 2 × IC50, Fig. 5. | ||
| Fig. 5 Cell cycle analysis for A2780 human ovarian cancer cells after 24 h exposure to complex 2 at 310 K at IC50 and 2 × IC50 concentrations. Cell staining for flow cytometry was carried out using PI/RNase. The percentage of cell populations in each cell cycle phase for the negative control and complex 2 are compared. p-Values were calculated after a t-test against the negative control data, *p < 0.05, **p < 0.01. | ||
Reactive oxygen species (ROS)
The level of reactive oxygen species (ROS) induced by complex 2 and GSH in combination with 2 in A2780 human ovarian cancer cells was determined at the IC50 concentration of the complex by flow cytometry fluorescence analysis (Fig. 6). These experiments were carried out following previously described protocols.29 A2780 cancer cells were treated at a fixed IC50 concentration of 2 and GSH (0.5 or 5 μM) without any recovery time. The total level of oxidative stress (including H2O2, peroxy and hydroxyl radicals, peroxynitrite, and NO) in the FITC-A channel and superoxide production in PE-A channel were monitored. ROS levels were detected in more than 70% of A2780 cancer cells. The populations of A2780 cells showing high fluorescence in FITC-A channel (ca. 73.1 ± 1.9%, Table S5 in the ESI†) for complex 2 alone and low fluorescence in channel PE-A (ca. 26.8 ± 2.0%), indicate a major induction of oxidative stress in A2780 cancer cells. Interestingly, cell populations in FITC-A-/PE-A+ and FITC-A+/PE-A+ channels decreased with increase of GSH concentration, from ca. 16.8 ± 0.8% to ca. 5.9 ± 0.5%. However, inversely, cell populations in FITC-A+/PE-A-channels increased when 5 μM GSH was co-administered with 2, from 71.7 ± 1.7% to 86.3 ± 0.5%, suggesting a higher level of oxidative stress (Table S5 in the ESI†). | ||
| Fig. 6 ROS induction in A2780 cancer cells exposed to complex 2 alone, 2 with 0.5 μM GSH, and 2 with 5 μM GSH. FITC-A channel detects total oxidative stress, and PE-A channel detects production of superoxide. p-Values were calculated after two-tailed Welch's t-tests to determine the significance of variations, ap > 0.05, *p < 0.05, **p < 0.01 and ***p < 0.001. | ||
Discussion
The X-ray crystal structure of complex 3 [(η6-biph)Ru(4-Me-phenyl-SO2-EnBz)I] shows that it has the typical half-sandwich ‘piano-stool’ geometry (Fig. 2), similar to related RuII complexes.25,30 The Ru–N bond lengths of 2.123 Å (Ru–N(−)) and 2.174 Å (Ru–N(H)), are close to those reported for [(η6-p-cym)Ru(TsEnEt)Cl] (2.126(9) Å and 2.1702(11) Å, respectively),25 but the Ru–N12 bond length is 0.08 Å longer than for the related complex [(η6-biph)Ru(TsEn)Cl] (2.096(4) Å). As expected, the Ru–I bond length is much longer than that of the Ru–Cl bond in [(η6-biph)Ru(TsEn)Cl] (2.7434(3) Å versus 2.4444(11) Å), and close to that in the Os complex [(η6-p-cym)Os(Impy-OH)I]+ (2.7247(4) Å).26 The N12–Ru–I bond angle of 83.90° is smaller than that of [(η6-p-cym)Ru(EtTsEn)Cl] (87.55°). The remaining bond lengths and angles are similar to those of related RuII complexes.In aqueous solution the ability of these halido sulfonylethylenediamine complexes to undergo hydrolysis and generate aqua adducts,17,25,26 decreases with the halido ligand in the order of Cl ≈ Br > I. Halido complexes with strong δ-donor ligands like en (ethylenediamine) and acac (acetylacetone), hydrolyse quickly and produce basic hydroxido adducts (pKa > 7), while the π-acceptor ligands like azopyridine, hydrolyse slowly and give more acidic aqua complexes.26,31 The aqua adducts of the catalysts studied here with high  values of 9.1–9.7 (Table 3), would be predominantly protonated in physiological media (pH 7.4). Aqua adducts are usually much more reactive than the corresponding hydroxido adducts and more readily undergo substitution reactions, which is favourable for a catalytic centre.
values of 9.1–9.7 (Table 3), would be predominantly protonated in physiological media (pH 7.4). Aqua adducts are usually much more reactive than the corresponding hydroxido adducts and more readily undergo substitution reactions, which is favourable for a catalytic centre.
RuII sulfonyl ethylenediamine complexes show potent catalytic activity in (sometimes asymmetric) transfer hydrogenation (TH) reactions with ketones, imines, and importantly cellular coenzyme nicotinamide adenine dinucleotide, NAD+.25,32–36 The efficiency for catalysis of transfer hydrogenation of NAD+ by complex 1 [(η6-benzene)Ru(TsEnBz)Cl] and 2 [(η6-biph)Ru(TsEnBz)Cl] was similar (TOFs of 7.5 ± 0.3 h−1 and 7.9 ± 0.4 h−1, respectively), in comparison to [(η6-p-cym)Ru(TsEnBz)Cl] (7.4 ± 0.1 h−1), but decreased with change in arene in the order: biph (2) > benzene (7) > p-cym, which is a slightly different arene order from the previous observations: benzene > biph > p-cym.25 Complex 4 [(η6-HO(CH2)2O-phenyl)Ru(TsEnBz)Cl] with hydrophilic side group HO(CH2)2O- on the phenyl arene has significantly enhanced the water solubility (up to 10 mg mL−1 in aqueous solution), while it gave the lowest TOF (2.5 ± 0.1 h−1); this may be because the terminal –OH group reversibly triggers rapid tethered ring formation and deformation by binding of the pendant alcohol–oxygen to the metal centre in aqueous solution, thereby hindering NAD+ approach to the Ru centre.37–39
Interestingly, Süss-Fink et al. have reported a series of dinuclear dithiolato and trithiolato RuII complexes [(η6-p-cym)2Ru2(SR)2]2+ or [η6-(p-cym)2Ru2(SR)3]+ where R is an aromatic group, which exhibit sub-micromolar anticancer activity against both A2780 and A2780 cisplatin-resistant human ovarian cancer cells.40–42 Catalytic oxidation of GSH to GSSG by such complexes suggested they can regulate GSH levels in cells.43 Aldrich-Wright et al. have reported the slow degradation of the PtII complex [(5,6-dimethyl-1,10-phenanthroline)(1S,2S-diaminocyclohexane)-Pt(II)] by GSH to form the [(5,6-dimethyl-1,10-phenanthroline)2Pt2(SG)2]2+, as a GS-bridged dimer.44 Complex 2 reacted with GSH or NAC to form an Ru-SG/NAC-bridged dimer, that significantly hampered the catalytic TH of NAD+, while retriggering DsEnBz ligand fluorescence of complex 8, showing that this complex has potential for tracking such ligand dissociation in cells. A recent publication by Briš et al. described reactions of organo-ruthenium complexes with cysteine and its analogues studied by mass spectrometry.45 A ruthenium complex with 1-(4-chlorophenyl)-4,4,4-trifluorobutane-1,3-dione as ligand underwent solvolysis in water to form a [Ru2(p-cym)2(OH)3]+ hydroxido-bridged dimer; subsequently hydroxide ligands were displaced by deprotonated cysteine to give the cysteine-bridged dimer similar to complex 2b in this work.
As an inhibitor of the enzyme γ-glutamyl cysteine synthetase, L-BSO can limit the cellular synthesis of GSH, and enhance ROS levels to induce cell apoptosis. Co-treatment of organometallic RuII or OsII complexes with L-BSO has been developed as a strategy to overcome GSH mediated detoxification of drugs.46,47 For example, L-BSO can restore CDDP activity against several CDDP-resistant cancer cell lines. L-BSO has been shown to cause a significant reduction in A2780 cellular GSH levels (ca. 50% with 5 μM L-BSO) and a significant enhancement of anticancer activity towards ovarian cancer cells upon co-administration of organo-Os complex [Os(η6-p-cym)(p-NMe2-Azpy)I]PF6 with L-BSO (5 μM dose), with 87% improvement in anticancer activity at an equipotent 2 × IC50 concentration of the complex.27,28 However, such restoration of antiproliferative activity by L-BSO only occurs when a complex is already biologically active.27 In the present work, enhancement of the anticancer activity against A549 cancer cells was observed at an L-BSO concentration of 50 μM with IC50 decreasing from ca. 13 to 8.3 μM; with levels of GSH reduced to ca. 61%.28 High L-BSO concentrations probably interfere with cellular GSH synthesis, while excessive GSH might react with the complex to destroy catalytic activity but promote the generation of biologically active dinuclear Ru(II) species.42
The cell cycle arrest study of complex 2 in A2780 cancer cells revealed a dose-dependent cell population increase in G1 phase (66.7 ± 1.5% to 75.2 ± 0.2% and 80 ± 2% at IC50 and 2 × IC50 concentrations), but a cell population depletion in G2/M and S phase, which implies that complex 2 is less likely to have DNA as a target site, in agreement with the previous study.16 DNA-targeted compounds normally cause cell accumulation in the S phase or G2/M phase, e.g. cisplatin.48
Reactive Oxygen Species (ROS) display important roles in cell metabolism. As respiratory side-products of mitochondria, over-production from ROS damages proteins or causes oxidation of DNA nucleobases to induce cell apoptosis, and ROS-mediated apoptotic signalling is usually associated with reduction of cytosol or mitochondrial GSH levels.49,50 Organo Ir, Os and Ru complexes have been widely reported as anticancer agents which can induce cell apoptosis via ROS-involving pathways.28,51–53 Complex 2 can induce significant amounts of superoxide in A2780 ovarian cancer cells (up to 16% of the cell population, Table S5 in the ESI†). Co-administration of complex 2 with GSH reduces both superoxide levels and antiproliferative activity against A2780 human ovarian cancer cells. The levels of superoxide showed an inverse relationship with the concentrations of GSH added (Table S5 in the ESI†), and a similar trend in antiproliferative activity with GSH concentration was also observed. This might imply that ROS play an important role in killing cancer cells for these complexes.
Conclusions
A series of RuII sulfonyl-substituted ethylenediamine complexes of the type [(η6-arene)Ru(R1-SO2-EnBz)X] (where the arene is benzene, biphenyl or HOCH2CH2O-phenyl, R1 is 4-Me-phenyl, phenyl, 4-F-phenyl, 4-NO2-phenyl or Dansyl, and X is halide) were synthesized and fully characterized. The half-sandwich structure of complex 3 was confirmed by X-ray crystallography. These complexes retained high potency in transfer hydrogenation of coenzyme NAD+ with formate as hydride donor. The introduction of the substituted arene HOCH2CH2O-phenyl (complex 4) significantly improved the water solubility of the complex, albeit with reduced catalytic activity.Complex 2 [(η6-biph)Ru(4-Me-phenyl-SO2-EnBz)Cl] bound strongly to 9-ethylguanine to form the 2-9-EtG-N7 adduct. However, complex 2 caused only G1 cell cycle arrest in a concentration-dependent manner, and is unlikely to target DNA in cells. Complex 2 exhibited a high affinity for GSH and NAC, to form the Ru-thiolate bridged dimers [(η6-biph)2Ru2(GS)3]2− and [(η6-biph)2Ru2(NAC-H)3]2−. Co-incubation of complex 2 with increasing GSH concentrations effectively reduced induction of reactive oxygen species in cells, and decreased the antiproliferative activity significantly. Such reactions with GSH lead to the release of the chelated diamine ligand, and hence trigger the fluorescence of the free dansyl ligand upon release from complex 8 [(η6-biph)Ru(DsEnBz)Cl], which might provide a basis for a study of such release in cells. Reactions of RuII complexes with GSH to form the dinuclear species may offer a new class of TH catalysts that can also form cytotoxic thiolate-bridged complexes in cells. Future work on strategies to control the rate and extent of reactions of these catalysts with GSH, might lead to new concepts in the design of this class of multi-targeting candidate metallodrugs.
Experimental section
Materials
Dansyl chloride was purchased from Sigma-Aldrich. The RuII-η6-arene precursor dimers were prepared following literature methods,25,54 as were the ligands (synthesis is presented in ESI†).17 The solvents used for NMR spectroscopy were purchased from Sigma-Aldrich and Cambridge Isotope Laboratories Inc. Non-dried solvents used in syntheses were obtained from Fisher Scientific. Glutathione and N-acetyl-L-cysteine were purchased from Fisher Scientific.Synthesis and characterizations
:9 (v/v)), to afford a red solid. Yield = 134.7 mg (65%). 1H NMR (400 MHz, CDCl3): δH 2.10–2.24 (m, 2H), 2.33 (s, 3H), 2.40–2.42 (m, 1H), 3.08 (dd, J = 3.1 Hz, 11.2 Hz, 1H), 3.75 (t, J = 10.1 Hz, 1H), 4.19 (q, J = 10.8 Hz, 13.2 Hz, 1H), 4.85 (dd, J = 10.1 Hz, 13.4 Hz, 1H), 5.70 (s, 6H), 7.14 (d, J = 8 Hz, 2H), 7.30–7.32 (m, 2H), 7.35–7.38 (m, 3H), 7.71 (d, J = 8.1 Hz, 2H); 13C NMR (125.73 MHz, CDCl3): δc 21.4, 48.3, 55.5, 62.2, 83.1, 127.3, 128.4, 128.7, 128.9, 129.3, 135.7, 140.1, 140.7; HR-MS: calcd for [C22H25N2O2SRu]+ 483.0680 m/z, found: 483.0683 m/z. Elemental analysis: calcd for [C22H25N2O2SRuCl]0.1(H2O): C, 50.83%; H, 4.89%; N, 5.39%. Found: C, 50.84%; H, 4.81%; N, 5.42%.
:9 (v/v)), giving an orange-red solid. Yield = 132.7 mg (73%). 1H NMR (400 MHz, CDCl3): δH 1.89–1.95 (m, 1H), 2.11–2.17 (m, 2H), 2.34 (s, 3H), 3.08 (dd, J = 3.6 Hz, 11.2 Hz, 1H), 3.59 (q, J = 10.4 Hz, 13.2 Hz, 1H), 3.73 (t, J = 11.5 Hz, 1H), 4.38 (dd, J = 3.9 Hz, 13.3 Hz, 1H), 5.47 (t, J = 7.7 Hz, 1H), 5.98–6.01 (m, 2H), 6.06 (t, J = 5.6 Hz, 1H), 6.57 (d, J = 5.4 Hz, 1H), 7.03–7.05 (m, 2H), 7.15 (d, J = 8.0 Hz, 2H), 7.28–7.31 (m, 4H), 7.53–7.56 (m, 2H), 7.72 (d, J = 8.1 Hz, 2H), 7.80–7.83 (m, 2H); 13C NMR (125.73 MHz, CDCl3): δC 21.4, 48.3, 53.9, 60.5, 78.7, 78.7, 86.4, 88.1, 89.0, 90.4, 127.4, 128.0, 128.2, 128.7, 129.1, 129.4, 129.6, 134.7, 135.7, 140.0, 140.8; HR-MS: calcd for [C28H29N2O2SRu]+ 559.0993 m/z, found: 559.0990 m/z. Elemental analysis: calcd for [C28H29N2O2SRuCl]0.3(H2O): C, 56.09%; H, 4.98%; N, 4.67%. Found: C, 56.02%; H, 5.01%; N, 4.73%.
:8 (v/v)). A red solid was obtained. Yield = 72.6 mg (54%). 1H NMR (400 MHz, CDCl3): δH 1.92–1.98 (m, 1H), 2.07–2.15 (m, 2H), 2.35 (s, 3H), 3.14–3.18 (m, 1H), 3.40 (q, J = 10.9 Hz, 13.2 Hz, 1H), 3.94 (t, J = 11.5 Hz, 1H), 4.32 (dd, J = 4.0 Hz, 13.4 Hz, 1H), 5.40 (t, J = 5.6 Hz, 1H), 5.88 (d, J = 5.8 Hz, 1H), 6.04 (t, J = 5.5 Hz, 1H), 6.34 (t, J = 5.7 Hz, 1H), 6.85 (d, J = 6.1 Hz, 1H), 7.00–7.01 (m, 2H), 7.16 (d, J = 8.2 Hz, 1H), 7.29–7.34 (m, 3H), 7.48–7.54 (m, 3H), 7.70 (d, J = 8.1 Hz, 2H), 7.88–7.91 (m, 2H); 13C NMR (125.73 MHz, CDCl3): δC 21.4, 49.4, 53.5, 60.5, 78.6, 85.8, 87.4, 90.3, 127.7, 127.8, 128.3, 128.6, 128.8, 129.1, 129.5, 129.7, 134.8, 135.7, 139.3, 140.7; HR-MS: calcd for [C28H29N2O2SRu]+ 559.0993 m/z, found: 559.0994 m/z. Elemental analysis: calcd for [C28H29N2O2SRuI]0.2(H2O): C, 48.80%; H, 4.30%; N, 4.06%. Found: C, 48.74%; H, 4.17%; N, 3.96%.
:9 (v/v)). A bright red solid was obtained. Yield = 104 mg (56%). 1H NMR (400 MHz, CDCl3): δH 2.12 (td, J = 2.8 Hz, 11.6 Hz, 1H), 2.22 (dd, J = 4.1 Hz, 11.8 Hz, 1H), 2.34 (s, 3H), 2.48 (d, J = 10.1 Hz, 1H), 3.00 (dd, J = 3.8 Hz, 11.4 Hz, 1H), 3.24 (s, broad, 1H), 3.76 (t, J = 11.3 Hz, 1H), 3.92–3.95 (m, 1H), 4.04–4.08 (m, 1H), 4.17 (q, J = 10.3 Hz, 13.3 Hz, 1H), 4.22–4.32 (m, 2H), 4.77 (dd, J = 4.1 Hz, 13.4 Hz, 1H), 5.04 (t, J = 5.2 Hz, 2H), 5.52 (t, J = 5.6 Hz, 1H), 5.56 (dd, J = 1.0 Hz, 5.9 Hz, 1H), 6.48 (t, J = 5.7 Hz, 1H), 7.15 (d, J = 8.0 Hz, 2H), 7.31 (d, J = 7.5 Hz, 2H), 7.35–7.40 (m, 3H), 7.75 (d, J = 8.2 Hz, 2H); 13C NMR (125.73 MHz, CDCl3): δC 21.4, 47.9, 55.9, 60.8, 61.6, 61.9, 66.5, 68.8, 71.5, 87.1, 90.1, 127.6, 128.4, 128.7, 128.8, 129.3, 134.4, 135.7, 139.9, 140.8; ESI-MS: calcd for [C24H29N2O4SRu]+ 543.0891 m/z, found: 543.0891 m/z. Elemental analysis: calcd for [C24H29N2O4SRuCl]: C, 49.87%; H, 5.06%; N, 4.85%. Found: C, 50.60%; H, 5.06%; N, 4.60%.
:9 (v/v)). A dark red solid was obtained. Yield = 90 mg (46%). 1H NMR (400 MHz, CDCl3): δH 1.96–2.02 (m, 1H), 2.09 (td, J = 3.2 Hz, 12.8 Hz, 1H), 2.24 (d, J = 10.4 Hz, 1H), 3.17 (dd, J = 3.6 Hz, 11.0 Hz, 1H), 3.65–3.70 (m, 2H), 4.41 (q, J = 9.4 Hz, 18.6 Hz, 1H), 5.48 (t, J = 5.7 Hz, 1H), 5.94 (t, J = 5.8 Hz, 1H), 6.04–6.07 (m, 2H), 6.51 (d, J = 5.7 Hz, 1H), 7.04–7.06 (m, 2H), 7.31–7.33 (m, 3H), 7.53–7.55 (m, 3H), 7.79–7.82 (m, 2H), 7.92 (d, J = 8.0 Hz, 2H), 8.15 (d, J = 8.0 Hz, 2H); 13C NMR (125.73 MHz, CDCl3): δC 48.2, 53.9, 60.7, 78.5, 78.9, 86.7, 88.2, 88.8, 90.9, 123.4, 127.9, 128.2, 128.4, 128.8, 129.2, 129.5, 129.9, 134.3, 135.4, 148.7; HR-MS: calcd for [C27H26N3O4SRu]+ 590.0688 m/z, found: 590.0688 m/z. Elemental analysis: calcd for [C27H26N3O4SRuCl]: C, 51.88%; H, 4.19%; N, 6.72%. Found: C, 51.70%; H, 4.22%; N, 6.69%.
:9 (v/v)). A dark red solid was obtained. Yield = 101 mg (54%). 1H NMR (400 MHz, CDCl3): δH 1.88–1.98 (m, 1H), 2.10 (dt, J = 3.2 Hz, 12.7 Hz, 1H), 2.17–2.20 (m, 1H), 3.08 (dd, J = 4.1 Hz, 11.6 Hz, 1H), 3.62 (dd, J = 10.3 Hz, 13.0 Hz, 1H), 3.69–3.76 (m, 1H), 4.39 (dd, J = 3.9 Hz, 13.0 Hz, 1H), 5.47 (t, J = 5.7 Hz, 1H), 5.97 (t, J = 5.7 Hz, 1H), 6.01 (d, J = 6.0 Hz, 1H), 6.05 (t, J = 5.7 Hz, 1H), 6.55 (d, J = 5.6 Hz, 1H), 7.00 (t, J = 8.8 Hz, 2H), 7.04–7.06 (m, 2H), 7.30–7.32 (m, 3H), 7.51–7.55 (m, 3H), 7.81–7.84 (m, 4H); 13C NMR (125.73 MHz, CDCl3): δC 48.2, 53.9, 60.6, 78.6, 78.8, 86.5, 88.1, 88.9, 90.6, 114.9, 115.1, 128.0, 128.2, 128.7, 129.2, 129.4, 129.7, 129.7, 129.8, 134.6, 135.6, 163.0, 165.0; 19F NMR (376.4 MHz, CDCl3, spectrum referenced to trifluoro-acetic acid at −76.55 ppm): δF −109.9. HR-MS: calcd for [C27H26FN2O2SRu]+ 563.0743 m/z, found: 563.0742 m/z. Elemental analysis: calcd for [C27H26FN2O2SRuCl]1.4(H2O): C, 52.03%; H, 4.66%; N, 4.49%. Found: C, 52.02%; H, 4.24%; N, 4.78%.
:9 (v/v)). A dark red solid was obtained. Yield = 60.3 mg (34%). 1H NMR (500 MHz, CDCl3): δH 1.88–1.95 (m, 1H), 2.11–2.18 (m, 2H), 3.11 (dd, J = 5.1 Hz, 9 Hz, 1H), 3.58 (dd, J = 10.4 Hz, 13.4 Hz, 1H), 3.71–3.76 (m, 1H), 4.38 (dd, J = 4.2 Hz, 13.4 Hz, 1H), 5.46 (t, J = 5.7 Hz, 1H), 5.98 (d, J = 6.0 Hz, 1H), 6.01 (d, J = 5.7 Hz, 1H), 6.05 (t, J = 5.7 Hz, 1H), 6.58 (d, J = 5.5 Hz, 1H), 7.03–7.05 (m, 2H), 7.29–7.30 (m, 3H), 7.33–7.40 (m, 4H), 7.52 (d, J = 7.5 Hz, 2H), 7.83 (t, J = 8.5 Hz, 4H); 13C NMR (125.73 MHz, CDCl3): δC 48.3, 53.9, 60.5, 78.7, 78.8, 86.4, 88.1, 88.9, 90.5, 127.3, 128.0, 128.1, 128.2, 128.3, 128.6, 129.1, 129.4, 129.6, 130.5, 134.7, 135.7, 142.9; HR-MS: calcd for [C27H27N2O2SRu]+ 545.0837 m/z, found: 545.0834 m/z. Elemental analysis: calcd for [C27H27N2O2SRuCl]0.4(H2O): C, 55.22%; H, 4.77%; N, 4.77%. Found: C, 55.14%; H, 4.62%; N, 4.86%.
:9 (v/v)). A dark red solid was obtained. Yield = 138 mg (67%). 1H NMR (400 MHz, CDCl3): δH 1.87–1.97 (m, 1H), 2.09–2.12 (m, 1H), 2.35 (dt, J = 2.7 Hz, 12.7 Hz, 1H), 2.84 (s, 6H), 3.10 (dd, J = 3.9 Hz, 12.7 Hz, 1H), 3.55 (dd, J = 10.5 Hz, 13.2 Hz, 1H), 3.92 (s, broad, 1H), 4.38 (dd, J = 4.0 Hz, 13.5 Hz, 1H), 5.63 (t, J = 5.6 Hz, 1H), 5.99–6.03 (m, 2H), 6.08 (t, J = 5.6 Hz, 1H), 6.52 (d, J = 5.4 Hz, 1H), 7.04–7.06 (m, 2H), 7.12 (d, J = 7.4 Hz, 1H), 7.29–7.32 (m, 3H), 7.39 (t, J = 7.8 Hz, 1H), 7.47–7.53 (m, 4H), 7.83–7.84 (m, 2H), 8.38 (dd, J = 8.6 Hz, 11.7 Hz, 2H), 8.73 (d, J = 8.6 Hz, 1H); 13C NMR (125.73 MHz, CDCl3): δC 45.5, 48.5, 54.36, 60.1, 79.7, 80.2, 85.5, 85.7, 86.5, 92.2, 114.6, 121.5, 123.5, 127.0, 127.1, 127.4, 127.8, 127.9, 128.1, 128.2, 128.3, 128.5, 128.8, 128.9, 129.4, 129.7, 130.0, 130.1, 130.6, 134.7, 135.9, 151.0; HR-MS: calcd for [C33H34N3O2SRu]+ 638.1415 m/z, found: 638.1419 m/z. Elemental analysis: calcd for [C33H34N3O2SRuCl]0.9(H2O): C, 57.49%; H, 5.23%; N, 6.09%. Found: C, 57.41%; H, 4.97%; N, 6.21%.
X-ray crystallography
Diffraction data for complex 3 were collected on an Oxford Diffraction Gemini four-circle system with an AtlasS2 CCD area detector. The structure of complex 3 was refined by full-matrix least-squares against F2 using Olex255 and was solved by with the ShelXT56 structure solution program using Intrinsic Phasing and refined with the ShelXL57 refinement package using Least Squares minimisation. The atoms from the sulphonamide nitrogen to the end of the chain (C10 C11 N12 C13) were modelled as disordered over two positions related by a small ruffle in the chain. The occupancy of the two positions was linked to a free variable which refined to 86:14. The minor component was refined isotopically. The NH of the major component was located in a difference map though both it and the NH of the minor position were placed at calculated positions for the rest of the refinement. The data were processed by the modelling program Mercury 3.8. X-ray crystallographic data for complex 3 have been deposited in the Cambridge Crystallographic Data Centre under the accession number CCDC 2117792.†
In vitro growth inhibition assays
The antiproliferative activity of complexes 1–8 was determined against A2780 human ovarian and A549 human lung cancer cells. Briefly, 5000 cells per well were seeded in 96-well plates. The plates were left to pre-incubate with drug-free medium at 310 K for 48 h before adding different concentrations of the tested compounds. Exact drug (Ru) concentrations were determined by ICP-OES. A drug exposure period of 24 h was allowed. After this, supernatants were removed by suction and each well was washed with PBS. A further 72 h were allowed for the cells to recover in drug-free medium at 310 K. The Sulforhodamine B (SRB) assay was used to determine cell viability. IC50 values, as the concentration that causes 50% cell death, were determined as duplicates of triplicates in two independent sets of experiments and their standard deviations were calculated.For cell growth inhibition by GSH/NAC with complex 2, GSH/NAC at concentrations of 5, 10 and 50 μM were added to the cells first, followed by complex 2 (within 10 min), and the cytotoxicity was monitored using the SRB assay described above. After 24 h co-incubation with complex 2 and GSH, cancer cell viability was assessed after washing the cells with PBS.
Cell cycle arrest
Approximately 1.5 × 106 per well of A2780 human ovarian cancer cells were cultured in a six-well plate and pre-incubated in drug-free media at 310 K for 24 h, after which complex 2 at equipotent IC50 concentration were added. After drug exposure for 24 h, supernatants were removed by suction and cells were washed with PBS. Then A2780 cells were harvested using trypsin-EDTA and fixed with cold 70% ethanol for 2 h. DNA staining was obtained by re-suspension of cell pellets in PBS (containing propidium iodide (PI) and RNase). Cell pellets were washed and re-suspended in PBS before being analysed in a Becton Dickinson FACScan flow cytometer using excitation of DNA-bound PI at 536 nm, with emission at 617 nm. Data were processed with Flowjo software.ROS determination
ROS/superoxide induction in A2780 cells induced by complex 2 was determined using the Total ROS/Superoxide detection kit (Enzo-Life Sciences) according to the instructions. The analysis was performed via Flow cytometry. Generally, 1.0 × 106 of A2780 cells per well were seeded in the six-well plate and then pre-incubated in drug-free media at 310 K for 24 h (under 5% CO2 humidified conditions), and then drugs were added to triplicates wells at equipotent IC50 concentration. After 24 h of drug exposure, supernatants were removed by suction and cells were washed with PBS and harvested. Cell pellets were then re-suspended in PBS buffer containing the orange/green fluorescent reagents to achieve cell staining. Cells were analysed in a BD LSR II flow cytometer (488 nm laser) using FITC-A channel: 575/26 nm for the oxidative stress and PE-A channel: 530/30 nm for superoxide detection. Data were gated using positive-stained (pyocyanin positive control), untreated-stained and untreated-unstained control samples, acquired as instrumental triplicates by using Flowjo V10 for Windows software. All samples were kept under dark conditions to avoid light-induced ROS production.Author contributions
F. C., S. B., A. H., and P. J. S. designed the project. F. C. carried out the synthesis and characterisation of ligands and complexes, determined the values, UV-vis spectra and turnover frequencies. I. R.-C. and J.-I. S. carried out the antiproliferative cell studies and related biochemical assays. G. J. C. carried out the X-ray crystallography. L. S. the high resolution mass spectra work, and I. P. the NMR work. All authors contributed to the writing of the paper.
values, UV-vis spectra and turnover frequencies. I. R.-C. and J.-I. S. carried out the antiproliferative cell studies and related biochemical assays. G. J. C. carried out the X-ray crystallography. L. S. the high resolution mass spectra work, and I. P. the NMR work. All authors contributed to the writing of the paper.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC (grants EP/F034210/1, EP/P030572/1, and EP/M027503/1), China Scholarship Council (CSC; scholarship for F. C.), and Royal Society (Newton-Bhahba International Fellowship, NF151429, for S. B.) for support.References
- Z. Liu and P. J. Sadler, Acc. Chem. Res., 2014, 47, 1174–1185 CrossRef CAS PubMed.
- W.-Y. Zhang, S. Banerjee, G. M. Hughes, H. E. Bridgewater, J.-I. Song, B. G. Breeze, G. J. Clarkson, J. P. C. Coverdale, C. Sanchez-Cano, F. Ponte, E. Sicilia and P. J. Sadler, Chem. Sci., 2020, 11, 5466–5480 RSC.
- S. Kuang, X. Liao, X. Zhang, T. W. Rees, R. Guan, K. Xiong, Y. Chen, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2020, 59, 3315–3321 CrossRef CAS PubMed.
- S. Banerjee and P. J. Sadler, RSC Chem. Biol., 2021, 2, 12–29 RSC.
- R. M. Lord, M. Zegke, A. M. Basri, C. M. Pask and P. C. McGowan, Inorg. Chem., 2021, 60, 2076–2086 CrossRef CAS PubMed.
- M. Hanif, J. Arshad, J. W. Astin, Z. Rana, A. Zafar, S. Movassaghi, E. Leung, K. Patel, T. Söhnel, J. Reynisson, V. Sarojini, R. J. Rosengren, S. M. F. Jamieson and C. G. Hartinger, Angew. Chem., Int. Ed., 2020, 59, 14609–14614 CrossRef CAS PubMed.
- J. M. Cross, T. R. Blower, A. D. H. Kingdon, R. Pal, D. M. Picton and J. W. Walton, Molecules, 2020, 25, 2383 CrossRef CAS PubMed.
- W.-J. Wang, X. Mu, C.-P. Tan, Y.-J. Wang, Y. Zhang, G. Li and Z.-W. Mao, J. Am. Chem. Soc., 2021, 143, 11370–11381 CrossRef CAS PubMed.
- B. Rosenberg, L. Vancamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed.
- X. Wang, X. Wang and Z. Guo, Acc. Chem. Res., 2015, 48, 2622–2631 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- S. K. Tripathy, A. C. Taviti, N. Dehury, A. Sahoo, S. Pal, T. K. Beuria and S. Patra, Dalton Trans., 2015, 44, 5114–5124 RSC.
- X. Wang, X. Wang, S. Jin, N. Muhammad and Z. Guo, Chem. Rev., 2019, 119, 1138–1192 CrossRef CAS PubMed.
- Y. K. Yan, M. Melchart, A. Habtemariam, A. F. A. Peacock and P. J. Sadler, J. Biol. Inorg. Chem., 2006, 11, 483–488 CrossRef CAS PubMed.
- Y. Fu, C. Sanchez-Cano, R. Soni, I. Romero-Canelón, J. M. Hearn, Z. Liu, M. Wills and P. J. Sadler, Dalton Trans., 2016, 45, 8367–8378 RSC.
- J. J. Soldevila-Barreda, I. Romero-Canelón, A. Habtemariam and P. J. Sadler, Nat. Commun., 2015, 6, 6582 CrossRef CAS PubMed.
- F. Chen, J. J. Soldevila-Barreda, I. Romero-Canelón, J. P. C. Coverdale, J.-I. Song, G. J. Clarkson, J. Kasparkova, A. Habtemariam, V. Brabec, J. A. Wolny, V. Schünemann and P. J. Sadler, Dalton Trans., 2018, 47, 7178–7189 RSC.
- A. T. Dharmaraja, J. Med. Chem., 2017, 60, 3221–3240 CrossRef CAS PubMed.
- Y. Kasherman, S. Sturup and D. Gibson, J. Med. Chem., 2009, 52, 4319–4328 CrossRef CAS PubMed.
- G. Garai-Ibabe, L. Saa and V. Pavlov, Anal. Chem., 2013, 85, 5542–5546 CrossRef CAS PubMed.
- M. Kartalou and J. M. Essigmann, Mutat. Res., 2001, 478, 23–43 CrossRef CAS PubMed.
- R. Zhang, X. Yu, Z. Ye, G. Wang, W. Zhang and J. Yuan, Inorg. Chem., 2010, 49, 7898–7903 CrossRef CAS PubMed.
- T. Zou, C. T. Lum, S. S.-Y. Chui and C.-M. Che, Angew. Chem., Int. Ed., 2013, 52, 2930–2933 CrossRef CAS PubMed.
- F. Chen, I. Romero-Canelón, J. J. Soldevila-Barreda, J.-I. Song, J. P. C. Coverdale, G. J. Clarkson, J. Kasparkova, A. Habtemariam, M. Wills, V. Brabec and P. J. Sadler, Organometallics, 2018, 37, 1555–1566 CrossRef CAS PubMed.
- J. J. Soldevila-Barreda, P. C. A. Bruijnincx, A. Habtemariam, G. J. Clarkson, R. J. Deeth and P. J. Sadler, Organometallics, 2012, 31, 5958–5967 CrossRef CAS.
- Y. Fu, M. J. Romero, A. Habtemariam, M. E. Snowden, L. Song, G. J. Clarkson, B. Qamar, A. M. Pizarro, P. R. Unwin and P. J. Sadler, Chem. Sci., 2012, 3, 2485–2494 RSC.
- I. Romero-Canelón, L. Salassa and P. J. Sadler, J. Med. Chem., 2013, 56, 1291–1300 CrossRef PubMed.
- Y. Fu, A. Habtemariam, A. M. Pizarro, S. H. van Rijt, D. J. Healey, P. A. Cooper, S. D. Shnyder, G. J. Clarkson and P. J. Sadler, J. Med. Chem., 2010, 53, 8192–8196 CrossRef CAS PubMed.
- I. Romero-Canelón, M. Mos and P. J. Sadler, J. Med. Chem., 2015, 58, 7874–7880 CrossRef PubMed.
- R. E. Morris, R. E. Aird, P. S. Murdoch, H. Chen, J. Cummings, N. D. Hughes, S. Parsons, A. Parkin, G. Boyd, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2001, 44, 3616–3621 CrossRef CAS PubMed.
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F.-X. Wang, W.-Y. Zhang and Z. Zhang, Chem. Sci., 2020, 11, 12888–12917 RSC.
- S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562–7563 CrossRef CAS.
- A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521–2522 CrossRef CAS.
- K. J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed. Engl., 1997, 36, 285–288 CrossRef CAS.
- X. Li, W. Chen, W. Hems, F. King and J. Xiao, Org. Lett., 2003, 5, 4559–4561 CrossRef CAS PubMed.
- X. Li, X. Wu, W. Chen, F. E. Hancock, F. King and J. Xiao, Org. Lett., 2004, 6, 3321–3324 CrossRef CAS PubMed.
- A. M. Pizarro, M. Melchart, A. Habtemariam, L. Salassa, F. P. A. Fabbiani, S. Parsons and P. J. Sadler, Inorg. Chem., 2010, 49, 3310–3319 CrossRef CAS PubMed.
- F. Martínez-Peña, S. Infante-Tadeo, A. Habtemariam and A. M. Pizarro, Inorg. Chem., 2018, 57, 5657–5668 CrossRef PubMed.
- S. Infante-Tadeo, V. Rodríguez-Fanjul, A. Habtemariam and A. M. Pizarro, Chem. Sci., 2021, 12, 9287–9297 RSC.
- A. F. Ibao, M. Gras, B. Therrien, G. Süss-Fink, O. Zava and P. J. Dyson, Eur. J. Inorg. Chem., 2012, 1531–1535 CrossRef CAS.
- F. Giannini, L. E. H. Paul, J. Furrer, B. Therrien and G. Süss-Fink, New J. Chem., 2013, 37, 3503–3511 RSC.
- F. Chérioux, B. Therrien and G. Süss-Fink, Inorg. Chim. Acta, 2004, 357, 834–838 CrossRef.
- F. Giannini, G. Süss-Fink and J. Furrer, Inorg. Chem., 2011, 50, 10552–10554 CrossRef CAS PubMed.
- S. Kemp, N. J. Wheate, M. J. Pisani and J. R. Aldrich-Wright, J. Med. Chem., 2008, 51, 2787–2794 CrossRef CAS PubMed.
- A. Briš, J. Jašík, I. Turel and J. Roithová, Dalton Trans., 2019, 48, 2626–2634 RSC.
- E. Păunescu, M. Soudani, P. Martin, R. Scopelliti, M. L. Bello and P. J. Dyson, Organometallics, 2017, 36, 3313–3321 CrossRef.
- I. Romero-Canelón and P. J. Sadler, Inorg. Chem., 2013, 52, 12276–12291 CrossRef PubMed.
- C. M. Sorenson, M. A. Barry and A. Eastman, J. Natl. Cancer Inst., 1990, 82, 749–755 CrossRef CAS PubMed.
- M. L. Circu and T. Y. Aw, Free Radicals Biol. Med., 2010, 48, 749–762 CrossRef CAS PubMed.
- M. P. Murphy, Biochem. J., 2009, 417, 1–13 CrossRef CAS PubMed.
- S. Kuang, X. Liao, X. Zhang, T. W. Rees, R. Guan, K. Xiong, Y. Chen, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2020, 59, 3315–3321 CrossRef CAS PubMed.
- M. J. Chow, C. Licona, D. Y. Q. Wong, G. Pastorin, C. Gaiddon and W. H. Ang, J. Med. Chem., 2014, 57, 6043–6059 CrossRef CAS PubMed.
- M. J. Chow, C. Licona, G. Pastorin, G. Mellitzer, W. H. Ang and C. Gaiddon, Chem. Sci., 2016, 7, 4117–4124 RSC.
- J. Soleimannejad and C. White, Organometallics, 2005, 24, 2538–2541 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2117792. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03856g |
| This journal is © The Royal Society of Chemistry 2022 |