
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancement of the catalytic activity of Mg/Al layered double hydroxide for glycerol oligomers production†
Fernando José Soares
Barros
 a,
Yue
Liu
b,
Clarissa Dantas
Paula
a,
Francisco Murilo Tavares
de Luna
a,
Enrique
Rodríguez-Castellón
*c and
Rodrigo
Silveira Vieira
a
a,
Yue
Liu
b,
Clarissa Dantas
Paula
a,
Francisco Murilo Tavares
de Luna
a,
Enrique
Rodríguez-Castellón
*c and
Rodrigo
Silveira Vieira
a
aUniversidade Federal do Ceará, Departamento de Engenharia Química, Grupo de Pesquisa em Separações por Adsorção – GPSA, Campus do Pici, 709, Fortaleza, CE 60.455-760, Brazil. E-mail: Rodrigo@gpsa.ufc.br
bTechnische Universität München, Department of Chemistry and Catalysis Research Center, Lichtenbergstrasse 4, D-84747 Garching, Germany. E-mail: yue.liu@tum.de
cUniversidad de Málaga, Departamento de Química Inorgánica, Mineralogía y Cristalografía (Unidad Asociada ICP-CSIC), Facultad de Ciencias, Campus de Teatinos s/n, 29071, Málaga, Spain
First published on 27th January 2022
Abstract
In this study, the impact of rehydration on the catalytic properties of Mg/Al layered double hydroxides (LDH) for glycerol oligomerization was assesed. Although previous works have employed other LDH derived materials in this reaction, little information on recyclability is published. After observing the initial results on how basicity and surface area were related to the catalytic activty, an LDH modification strategy was developed with the addition of acetic acid. Changes on the basic site distribution were noticed and consequently, selectivity to diglycerol was improved. The best catalytic performance (reaction with 4 wt% cat., at 240 °C for 8 hours) led to 64% of glycerol conversion (XGly) and 37% of diglycerol selectivity (Sdi). Aditionally, reciclying of modified LDH was better than the non acid treated material, presenting higher yield of diglycerol. Catalyst deactivation was related to the harsh reaction conditions and to the blockage of active species by impurities. Loss of metallic species by leaching to the reaction products was not oberseved, an advantage in comparison with previous works.
Introduction
Reduced global oil reserves are increasing the search for renewable energy sources, such as biodiesel. This fuel is typically produced by the transesterification of vegetable oils and fats with a short chain alcohol (methanol or ethanol). Its growing production drives an excess of by-product glycerol in the market.1 Besides the attractive price, this compound is versatile, non-toxic and biodegradable. Among the wide variety of transformation pathways for glycerol, the production of oligomers is an emerging topic, for instance diglycerols have numerous applications, from personal care formulations to polymer industry.2,3The main issue on the direct oligomerization of glycerol is to control the degree of polymerization and regioselectivity to form a well-defined oligomer.4 Selectivity control in most glycerol reactions is difficult, because it is highly functionalized and thus able to participate in several reaction pathways. In the case of glycerol etherification, the formation of higher oligomers and polymers are consecutive reactions, therefore control of the reaction conditions to avoid longer polymeric chains or undesired by-products, such as acrolein is essential.
Studies on glycerol oligomerization in homogeneous phase with acid catalysts, e.g. H2SO4, points to fast routes but low selectivity.5 In homogeneous base catalysis, a loss on diglycerol selectivity at complete glycerol conversion was observed for CsHCO3.6 The use of heterogeneous catalysts for this reaction is desirable due to advantages such as fewer purification steps and recyclability. Basic heterogeneous catalysts, such as alkaline earth oxides (CaO, SrO and BaO) and Cs supported on MCM-41, have been studied.7,8 Acidic heterogeneous catalysts were also tested by using materials such as zeolites.9,10
Recent studies have proposed new approaches to use heterogeneous catalysis to produce glycerol oligomers. However, most of them did not report recycling tests. Kirby et al. reported the use of colloidal Ca particles synthesized by dispersing CaO as nanoparticles or thin films on a carbon nanofiber support.11 Galy et al. evaluated this reaction in a continuous flow reactor using K2CO3 as homogeneous catalyst.12 The use of microwaves as heat source with Na2CO3 as catalyst was also investigated.13 Lee et al. recently addressed the catalytic activities of alkali metal acetates (LiOAc, NaOAc, and KOAc).14
In our previous works, low cost catalysts from calcium natural sources were used, such as waste eggshells and dolomite (Ca and Mg carbonate).15,16 Both materials displayed glycerol conversions around 80% and high yield of oligomers. However, the leaching of Ca species was expressive, while Mg had more stability. This fact has led us to choose Mg as bimetallic cation in the LDH prepared in this work.
Layered double hydroxides and their derivatives have been extensively investigated as catalysts in recent years. They have a wide range of surface area and their acidic/basic properties can be tuned by modifying their composition. LDH may serve as precursors, to form an oxyhydroxide or a double oxide by calcination.17,18 For glycerol oligomerization, mixed oxides from Mg/Al, Ca/Al and Mg/Fe LDHs have been used.19–21
LDH derived mixed oxides are able to recover the layered structure upon contact with water or anionic aqueous solutions, in a feature frequently referred as “memory effect”.22,23 Use of rehydration as a strategy to improve activity through the changes in the physicochemical properties has been reported for aldol condensation,24 isomerization of glucose25 and transesterification of glycerol.26
In this study we will discuss the changes in the properties and activity for glycerol oligomerization arising from the LDH modification through calcination followed by rehydration via mechanic stirring or ultrasound. Due to the lack of studies on catalyst reusability in glycerol oligomerization, special attention was paid to this feature. To understand LDH stability, characterization of fresh and spent catalysts was performed as well as leaching tests on reaction products.
Experimental
Catalyst preparation
The LDH with a Mg/Al molar ratio of 3 was synthesized by the co-precipitation method, following the procedure described elsewhere.27 An aqueous precipitating solution of NaOH/Na2CO3 was slowly added to the solution of Mg(NO3)2·6H2O and Al(NO3)3·9H2O. The mixture was aged under mechanical stirring for 30 minutes at room temperature. The precipitates were separated by centrifugation and then washed with deionized water (until pH around 7). The material was dried at 70 °C for 48 hours and labelled as MAS. Mixed oxides were obtained by calcination at 450 °C for 24 hours (MAC). Two different types of rehydration were used on the mixed oxides: assisted by ultrasound or mechanical agitation. For ultrasound tests, 0.6 g of sample were mixed with 100 mL of deionized water, 0.5 h or 1 h of agitation were employed, samples MAU05 and MAU1, respectively. In the mechanical stirring, the same ratio of samples/deionized water was stirred for one hour or 24 hours, MAM1 and MAM24, respectively.After the observation of catalytic application results, a new material was prepared by adding 20 mL of acetic acid to 1.4 g of MAU05, with magnetic agitation for half an hour. Prior reaction, the material was dried overnight at 120 °C and labeled as MAU05P.
Catalyst characterization
The contents of Mg and Al in the solids were determined by ICP-OES. 10 mg of sample was dissolved in 50 mL of a 2% HNO3 aqueous solution, and then filtrated through a syringe filter with a pore diameter of 0.45 μm. Next, 2 mL of this solution were diluted into 20 mL. The calibration curves were prepared with a multielemental standard (SpecSol) on a nitric acid aqueous solution.Lamellar structure formation as well as the changes after thermal and rehydration processes were evaluated by X-ray diffraction (XRD) on a Bruker D8 Advanced diffractometer, using Cu Kα (40 kV, 40 mA) radiation. The diffractograms were collected in 2θ = 2°–90° range with a step of 0.05° and a counting time of 1 s per step. Results were compared to Joint Committee on Powder Diffraction Standards (JCPDS). FTIR-ATR Spectroscopy (VERTEX70, BRUKER), was used to monitor changes in the functional groups, in the range of 600–4400 cm−1, with spectral resolution of 4 cm−1.
The nitrogen adsorption–desorption measurements were carried out at −196 °C in an ASAP 2420 equipment (Micrometrics). The samples were pretreated at 120 °C for 12 hours with N2. Pore diameter and pore volume were determined from the adsorption branch of the isotherms by the Barrett–Joyner–Hallenda (BJH) method. The surface area was determined with the Brunauer–Emmett–Teller (BET) method (SBET). The morphology of the samples was observed by transmission electronic microscopy (TEM), Morgagni 268D 100 kV (FEI). Thermogravimetric analysis/differential thermal analysis (TGA/DTA) was carried out on a STA 449 F3 – JUPITER/NETZSCH analyser operating under the following conditions: 20 mg of sample, nitrogen flow of 80 mL min−1; heating rate of 5 °C min−1; temperature range from40 to 800 °C.
Temperature-programmed desorption of CO2 was used to evaluate the basicity. Catalysts were analyzed in a self-constructed 6-fold TPD unit. The procedure consisted on filling one tube with 50 mg of NaHCO3 (Sigma Aldrich) as standard; the other tubes were filled with the same amount of samples to be measured and they all were pretreated in vacuum at 100 °C for 1 hour. The reaction temperature was lowered to 50 °C and pure CO2 stream was subsequently introduced into the reactor for 1 hour. Then, the thermal desorption of CO2 was conducted from 50 to 770 °C (10 °C min−1) and was registered using a mass spectrometer Pfeiffer QMS 200 (Pfeiffer Vacuum), that monitored the weight of CO2 during the experiment. The mass of samples was measured at the end of process, to calculate basicity.
Glycerol oligomerization tests
Glycerol oligomerization was carried out in a batch reactor (Parr) under nitrogen flow, typical tests used 50 g of glycerol. The autoclave was equipped with a system to collect the water formed. First, it was evaluated the influence of reaction parameters, using MAC calcinated at 450 °C. The effect of reaction temperature (210–240 °C) was assessed for reaction of 24 hours with 2 wt% of catalyst. Next, the influence of catalyst loading (1–4 wt%) was studied. Rehydrated LDH (MAU05, MAU1, MAM1 and MAM24) were tested over the best set of reaction conditions found. Product analysis was measured by gas chromatography with flame ionization detector GC-FID (Agilent) and a CP-SIL 5CB column (100% dimethylpolysiloxane, 25 m × 0.25 mm × 0.25 μm, Agilent). Samples were silylated with BSTFA (N,O-Bis(trimethylsilyl)trifluoroacetamide), using pyridine as solvent, following the methodology reported elsewhere.We have used the same equations of our previous work to calculate glycerol conversion XGly, selectivity to di and triglycerol (Sdi and Stri) and yield of oligomers (Ydi and Ytri) as presented in the following equations.15 Glycerol conversion (XGly) was calculated as:
 | (1) |
where, n0 is the initial amount of glycerol in mol and nf is the final amount of glycerol in mol.
The selectivity (Solig) was calculated using eqn (2).
 | (2) |
Where nolig is the amount of each oligomer (diglycerol or triglycerol) in mol. The Yield of each oligomer was calculated as.
 | (3) |
Oligoglycerols higher than triglycerol could not be resolved by this analytical procedure. Thus, once the percentage of glycerol and the selectivity to the oligomers (di- and triglycerol) were calculated, the missing fraction was assumed to be the selectivity to higher oligomers and other products (Sho).11 Yield of higher oligomers and other products (Yho) was calculated on the same way.
Acrolein detection was performed on the water collected in each reaction, using GC-FID with a DB-Wax column (polyethylene glycol stationary phase, 60 m × 0.32 mm × 0.25 μm, agilent), following a previously reported methodology.29 Hydroquinone and 1-butanol were used as polymerization inhibitor and internal standard, respectively.
Catalyst recycling
Reusability tests were performed over the best set of reaction conditions. Spent catalysts were recovered as follows: solids and reaction medium were diluted in deionized water and then separated by vacuum filtration on a porous plate. The recovered material was dried at 80 °C overnight prior the new reaction cycle.To observe changes related to the presence of adsorbed species, TGA/DTA of spent catalysts was carried out following the same procedure describe for fresh solids.
The surface of fresh and spent catalysts was studied by X-Ray Photoelectron Spectroscopy (XPS). The analysis was performed with a spectrometer (Physical Electronics PHI 5700) equipped with a non-monochromatic Mg Kα radiation (1253.6 eV) with a multichannel detector. Each spectrum was collected in the constant-pass energy mode at 29.35 eV, using a 720 mm diameter analysis area. Charge referencing was done against adventitious carbon (C 1s at 284.8 eV). Data acquisition and analysis was performed with PHI ACCESS ESCA-V6.0F software package. The signals were subtracted using a Shirley-type background and fitted with Gaussian–Lorentzian curves to determine more accurately the binding energies of the different element core levels. Samples were outgassed for 12 hours prior measurement.
To a better understanding of Mg/Al LDH stability in glycerol oligomerization, the reaction products were analyzed by ICP-OES to detect any trace of metal leached from catalysts to the reaction medium. For sample preparation, one mg of reaction product was diluted on 10 mL of ultrapure water and filtrated through a syringe filter with a pore diameter of 0.45 μm. Calibration curves were prepared under the same conditions of catalyst analysis.
Results and discussion
Catalyst characterization
The analyses of the materials as-synthesized (MAS), after calcination (MAC), rehydrated by ultrasound for 0.5 or 1 h (MAU05 and MAU1) or mechanical stirring for 1 or 24 h (MAM1and MAM24) are presented and discussed. First, ICP measurements (Table 1) confirmed that the as-synthesized material (MAS) had a Mg/Al molar ratio close to that used in synthesis, and that this value was not significantly changed across calcination and rehydration steps.| Sample | Mg/Al | d 003 | d 110 | a (Å) | c (Å) | IFS (Å) | I 006/I003 | FWHM110 | FWHM003 |
|---|---|---|---|---|---|---|---|---|---|
| *d003 and d110: d spacing; *a and c (Å): unit cell dimensions; *ISF (Å): interlayer free space; *I006/I003: relative intensities; *FWHM110 and FWHM003: full width at half maximum. The interlayer free space (IFS) was calculated using the expression ISF = (c/3) − 4.8, where the thickness of brucite-like layer is considered as 4.8 Å (ref. 28). | |||||||||
| MAS | 2.90 | 7.800 | 3.913 | 7.826 | 23.400 | 3.00 | 0.47 | 0.541 | 0.637 |
| MAC | 3.09 | — | — | — | — | — | — | — | — |
| MAU05 | 3.18 | 7.605 | 1.528 | 3.056 | 22.815 | 2.81 | 0.35 | 0.492 | 0.541 |
| MAU1 | 3.28 | 7.814 | 1.533 | 3.066 | 23.442 | 3.01 | 0.33 | 0.394 | 0.689 |
| MAM1 | 3.17 | 7.740 | 1.529 | 3.058 | 23.220 | 2.94 | 0.28 | 0.590 | 0.541 |
| MAM24 | 3.05 | 7.801 | 1.535 | 3.070 | 23.403 | 3.00 | 0.36 | 0.394 | 0.541 |
From XRD in Fig. 1a, MAS exhibits a single phase, which corresponds to the typical structure of a hydrotalcite (JCPDS 70-2151), with well-defined and symmetric d003, d006, d110 and d113 peaks and wide asymmetric d012, d015 and d018 peaks. MAC shows a mixed oxide of periclase type (JCPDS 45-0946), with the characteristic d200 and d220 reflections, pointing to the total collapse of the lamellar structure after calcination of MAS.30 All rehydrated samples (MAU05, MAU1, MAM1 and MAM24) show the typical patterns for LDH. Although lamellar structure is recovered on them, some differences can be noticed on their XRD profiles. MAU1 and MAM24 present slightly higher and better-defined peaks than MAS, whereas the d012, d015 and d018 peaks become more symmetrical for all rehydrated samples. Rehydrated materials displayed a reduction of relative intensities on the main diffraction peaks (I006/I003), presented in Table 1, ranging from 0.28 to 0.36 in comparison with MAS (0.47). This suggests lower crystallinity and presence of defects on rehydrated samples. The reduction of carbonate amount, as a result of the change on interlamellar composition, can be noticed by the decrease of the a lattice parameter. The interlayer free space (IFS) has reduced significantly on MAU05 and MAM1, in agreement with the findings of previous works, where calcination and rehydration steps are considered as aging procedures that improve crystallinity.30
 | ||
| Fig. 1 (a) XRD and (b) FTIR for MAS, MAC, MAU05, MAU1, MAM1 and MAM24. | ||
In the FTIR spectra, (Fig. 1b), the band assigned to the hydroxyl group present in the lamellae and interlamellar water, around 3500 cm−1, is on all samples except in the case of MAC. The spectrum of MAC exhibits a reduction of this band due to the elimination of OH− groups after calcination. The same trend was observed for the decreased band related to interlayer water deformation vibration at 1630 cm−1 on MAC. The bands related to the carbonate group are at around 1350 cm−1 and 630 cm−1,24 also with lower intensity but still partly remained in MAC, suggesting that the calcination did not eliminate completely hydroxyls and carbonates.
Since the FTIR results suggests differences in the water and carbonate contents in the synthesized materials, and the equipment resolution limits a more precise quantification, a thermogravimetric analysis was performed to compare their profiles. TGA/DTG (Fig. 2) shows that all materials, except MAC, present two weight loss steps. In the first loss, from room temperature to 200 °C, adsorbed and interlamellar water desorbs. The weight loss for this step is quite similar for MAU05 and MAM1 while higher on MAU1 and MAM24, Table S1 (ESI†), indicates that samples MAU05 and MAM1 have similar amounts of water molecules and longer time of rehydration makes MAU1 and MAM24 with more sorbed water. The second weight loss, from 200 to 450 °C, is related to the removal of condensed water molecules and carbon dioxide from carbonate anions present in the LDH interlayer space. The lower weight loss for sample MAC suggests that during calcination LDH was transformed into periclase, in accordance with XRD results.31,32
 | ||
| Fig. 2 (a) TGA and (b) DTG for MAS, MAC, MAU05, MAU1, MAM1 and MAM24. | ||
Textural properties are presented in Table 2, where it is possible to notice that the calcined material, MAC, exhibited similar properties to that of MAS. MAU05 and MAU1 both show lower BET area (SBET), pore volume (Vp) and pore diameter (Dp) compared to MAS. This decrease was more significant after one hour of sonication and was attributed to the formation and growth of mexinerite particles (a basic magnesium carbonate), as well as to the high surface tension of water, which may lead to the formation of a platelet agglomeration, provoking the closure of small mesopores.26 Contrary to MAU samples, MAM samples do not have a lower SBET compared to MAC. On MAM24, BET area, pore volume and diameter increased substantially. This is due to the fact that mechanical stirring in liquid phase can break and exfoliate the hydrotalcite-like platelets, increasing the specific surface area.24,32 The N2 adsorption isotherms are presented in Fig. S1 (ESI†). Mesoporous character of all samples was confirmed by their pore size distribution, the major pores were at diameter of 12 nm, Fig. S2 (ESI†).
| Sample | S BET (m2 g−1) | V P (cm3 g−1) | D P (nm) | Basic site concentration normalized to mass (mmol g−1) | Basic site concentration normalized to surface area (mmol m−2) | ||
|---|---|---|---|---|---|---|---|
| Medium | Strong | Total | |||||
| MAS | 58 | 0.13 | 19.57 | 8.68 | 4.72 | 13.40 | 0.23 |
| MAC | 62 | 0.17 | 19.42 | 7.16 | 2.61 | 9.77 | 0.16 |
| MAU05 | 44 | 0.04 | 3.30 | 4.33 | 2.66 | 6.99 | 0.16 |
| MAU1 | 26 | 0.03 | 3.31 | 3.99 | 2.16 | 6.15 | 0.24 |
| MAM1 | 56 | 0.05 | 3.29 | 5.24 | 3.34 | 8.58 | 0.15 |
| MAM24 | 112 | 0.33 | 16.40 | 4.90 | 3.12 | 8.02 | 0.07 |
The basicity of the materials was studied by temperature-programmed desorption of CO2. They all presented similar CO2-TPD profiles, with two desorption peaks at around 450 °C (medium strength) and 650 °C (strong basic sites) respectively (Fig. 3). The first peak is ascribed to the decomposition of bidentate carbonates and bicarbonate species.24 The second peak is attributed to monodentate carbonates. It has more pronounced intensity on MAS, MAU05, MAM1 and MAM24 than those on MAC and MAU1. The quantitative concentration of basic sites is compiled in Table 2 and shows the following sequence of basic site concentration normalized to surface area: MAU1 > MAS > MAC = MAU05 > MAM1 > MAM24.
 | ||
| Fig. 3 CO2-TPD profiles of MAS, MAC, MAU05, MAU1, MAM1 and MAM24. | ||
TEM micrographs of the materials are shown in Fig. 4, MAS exhibits an irregular morphology and the dark areas indicates the presence of dense agglomerations of particles. Such features are maintained in MAC. The rehydrated materials MAU05, MAU1 and MAM1 displayed several thin layered crystals, typical for LDHs. On MAU05, MAU1 and MAM1, the agglomeration of these thin platelets is responsible for their lower surface areas (44, 26 and 56 m2 g−1) in comparison to that of MAM24 (112 m2 g−1).28,30 MAM24 presented hexagonal like crystallites, characteristic of LDH structures, and fewer agglomerates of platelets. This result combined with XRD measurements supports that long time mechanical stirring improves crystallinity of LDH.
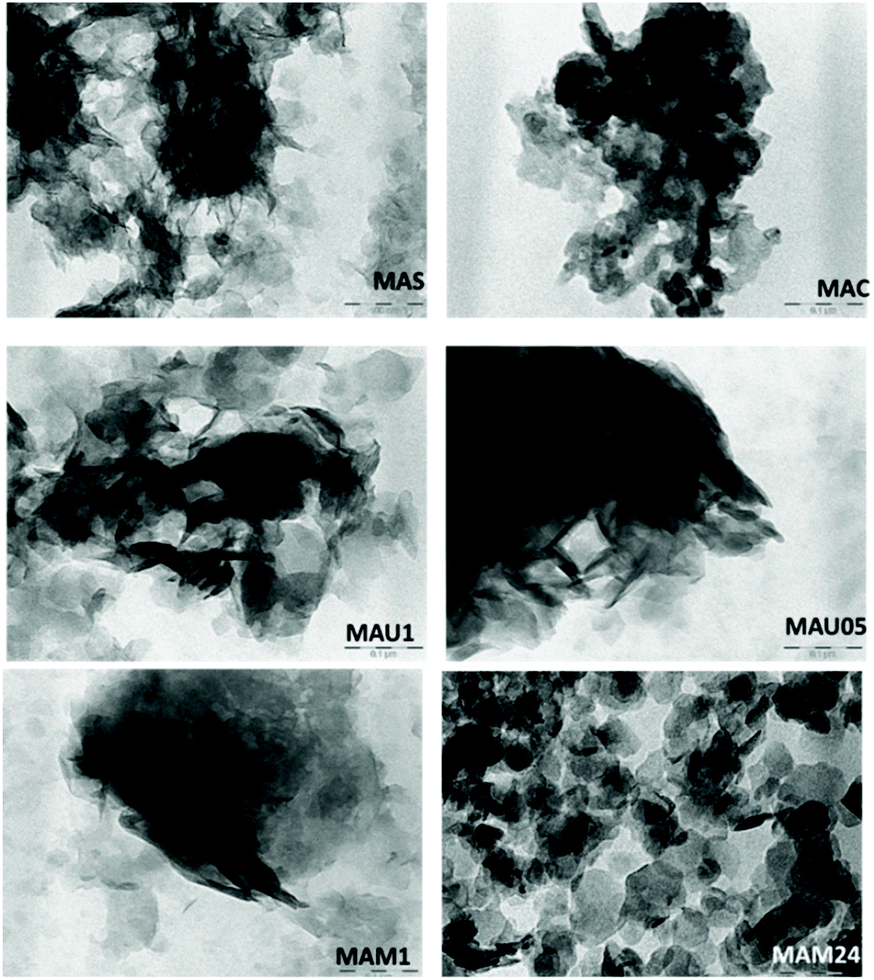 | ||
| Fig. 4 TEM images (scale bar 0.1 μm) for MAS, MAC MAU30, MAU60, MAM60 and MAM24. | ||
Catalytic application
Catalytic conversion of glycerol to oligomers was carried out on these LDHs. To screen and optimize reaction parameters, (Fig. 5a), MAC was tested in reaction temperatures from 210 to 240 °C, for 24 hours. The reaction at 210 °C led to very low glycerol conversion (XGly) and yield of diglycerol (Ydi), 3.68 and 1.40% respectively. By-products are mainly higher oligomers while monomolecular dehydration products (acrolein) are very low (Table S2 (ESI†). By increasing temperature to 240 °C, XGly reached 49% and the highest yield of oligomers was obtained, 19% Ydi and 13% Ytri. Thus, this temperature was used in the next tests. It is important to note here that the selectivity in this work is defined as percentage of each oligomer product (di or triglycerol) in the amount of converted glycerol, not the ratio of the respective product to the sum of products founded, which overestimates selectivity by ignoring missing by-products yet widely used in other works.20,33 Increasing catalyst loading of MAC from 2 to 3 wt%, an expressive increase of XGly (from 49 to 65%) was noticed. | ||
| Fig. 5 Influence in glycerol oligomerization of (a) reaction temperature (reaction conditions: 2 wt% of MAC and 24 h), (b) catalyst loading (240 °C and 24 h) (c) reaction time (4 wt% and 240 °C). In terms of glycerol conversion (XGly); selectivity to diglycerol (Sdi), triglycerol (Stri) and for high oligomers and other products (Sho); yield of diglycerol (Ydi) and triglycerol yield (Ytri). | ||
However, further increment to 4 wt% did not improve conversion but on the contrary led to an expressive drop in selectivity to diglycerol, from 29 to 4%, pointing to the increased consumption of diglycerol by its oligomerization making more high oligomers (Sho of 91%), in agreement with previous works.6,15,34 Decreasing the reaction time from 24 h to 8 h only lead to a drop of XGly by 10%, but largely improved the selectivity to di- and triglycerol selectivity from 4 to 24% and 5 to 7.6%, respectively. After these screening tests, the best reaction condition was chosen as 4 wt% catalyst loading, 240 °C and 8 h.
The catalytic results of rehydrated materials are shown in Fig. 6a. MAU05, MAM1 and MAM24 led to higher XGly than MAU1 (62, 64 and 73%, respectively, against 50%), in a correlation with their higher basicity. The higher conversion on more basic catalysts was also shown on a series of mixed oxides derived from Mg/Al LDH.19
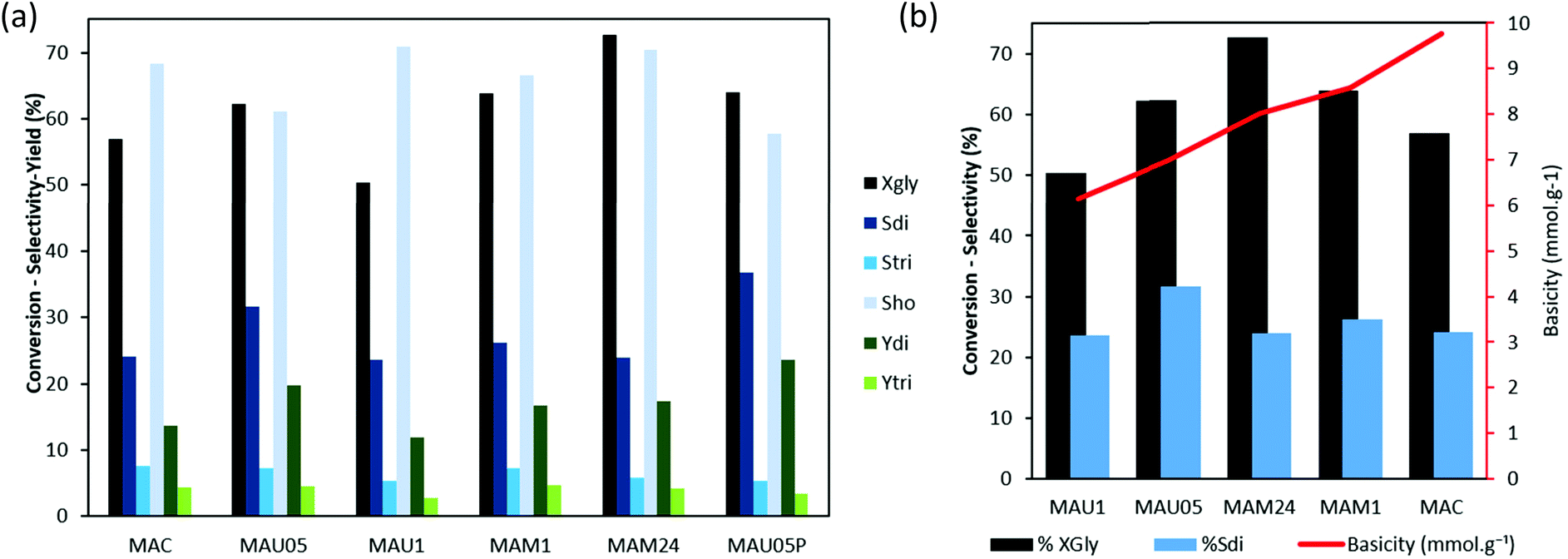 | ||
| Fig. 6 (a) Catalytic conversion of glycerol on rehydrated catalysts (reactions conditions: 4 wt% cat., 240 °C and 8 h). (b) Conversion of glycerol and selectivity of diglycerol as a function of basic site concentration normalized to mass on the rehydrated catalysts. | ||
The better activity presented by rehydrated catalysts in comparison to the calcined material, can be explained, considering a reaction mechanism (Scheme 1) where the first step of glycerol oligomerization is the proton extraction, and on the second step a coordinatively unsaturated metal site is able to activate a hydroxyl group of another glycerol molecule, which is attacked by the nucleophilic oxygen obtained in the first step.7,16 The surface hydroxyl groups of rehydrated LDH lattice acts as Brønsted basic centres in the first step, being more appropriate than the Lewis basic centres (O2−) present in the calcined material, MAC.
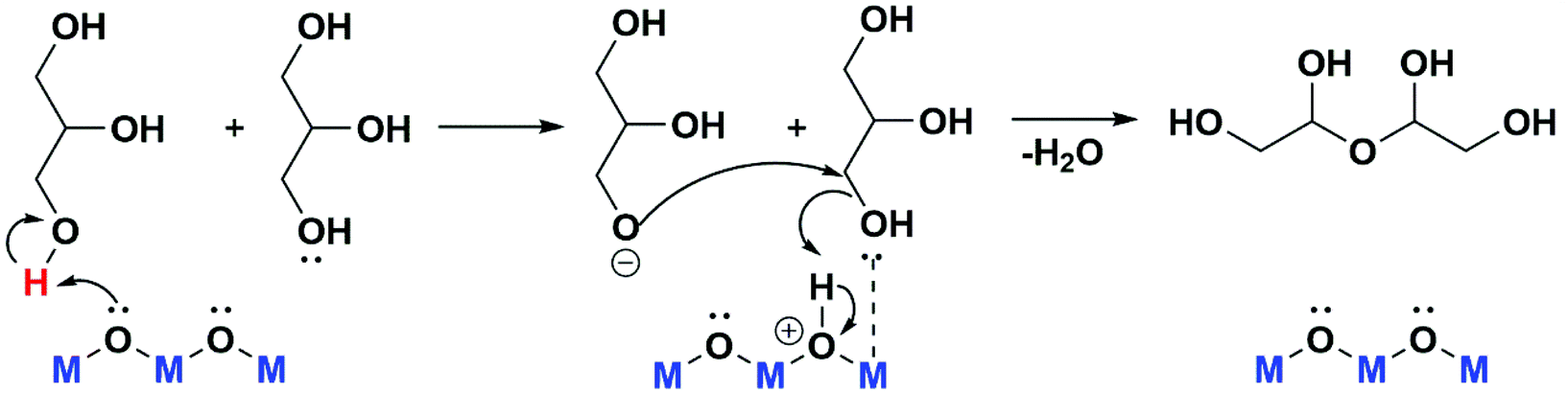 | ||
| Scheme 1 Representation of possible mechanism for oligomers formation from glycerol.7 | ||
Fig. 6b shows the role of the weight normalized basic site concentration of the catalysts on the glycerol conversion and diglycerol selectivity. With the increase of basic site concentration from 6 to 8 mmol g−1, the XGly is enhanced, while Sdi end ups on the same level (24% for MAM24 and MAU1). Important to notice that by using MAU05 a maximum on Sdi (32%) was obtained, and also that this catalyst has an average number of basic sites (6.99 mmol g−1) compared to the previous ones. This confirms that the possibility of tunning LDH properties with rehydration can be used to favored selectivity in glycerol oligomerization. Further increase of basicity from 8 mmol g−1 onwards reached similar XGly levels and reduced selectivity to the dimer, indicating that a higher basic site concentration promotes the reaction of glycerol but surpassing the optimal amount, it supports higher oligomerization and thus decrease selectivity to diglycerol as well as deactivate the catalysts.
A comparison between these catalysts led to the hypothesis of whether tuning these properties for an even lower total basicity and surface area could improve glycerol conversion and the selectivity to the oligomers. To verify this, we implemented an acetic acid modification on MAU05: it was dried overnight at 80 °C, then 15 mL of acetic acid were added to it and this new material was prepared via magnetic stirring for half an hour and labeled as MAU30P. The prepared MAU05P has 18% less medium strength basic sites than MAU05, based on CO2-TPD results (Fig. S4†).
The reaction with MAU05P, (Fig. 6a), led to XGly (64%) very similar to MAU05 (63%). Important growth of diglycerol yield was achieved, reaching 24% on MAU05P, against 19.5% on MAU05. In particular, MAU05P exhibited a better stability against catalyst deactivation than MAU05.
The catalytic performance of MAU05P is interesting in comparison with previous works using Mg/Al LDH or its derivatives for glycerol oligomerization, since it reached similar conversion in less time. It is important to notice that some of these works use different expressions to calculate selectivity. Mixed oxides from Mg/Al LDH coprecipitated with NaOH and Na2CO3 reported by García-Sancho et al. (2011) led to 51% of XGly (reactions at 220 °C for 24 hours), the selectivity was calculated as the weight ratio of the respective product to the sum of products formed, being 84.8% for Sdi and 15.2% for Stri;19 using this same approach in MAU05P data, Sdi and Stri would be 82.43 and 17.57% respectively. Anuar, Abdullah and Othman (2013) also used the previous expressions, and their calcined Mg/Al LDH led to 78% of XGly, and approximately 80% of Sdi and 20% of Stri (reaction at 240 °C for 16 hours).33 Whereas Pérez-Barrado et al. (2015) studied calcined LDH (coprecipitated with NH3/(NH4)2CO3) and had a 60% of XGly (reaction at 235 °C for 16 hours), in their work the selectivity term included a stoichiometric constant (2 for diglycerol and 3 for triglycerol), reaching 44% Sdi and 11% Stri.35 The application of this expression to the data in the present work results in 73.38% of Sdi and 17.27% of Stri.
Recycling test, (Fig. 7b), shows that MAU05 had an expressive drop on XGly already after the first reuse (from 63 to 21%). Besides, two reuses were possible, with a very low activity on the 3rd run (9% of XGly and Ydi 4%). For MAU05P, XGly reaches 31% in the second run, with 16% of Ydi. In this case, on the third run, there is an unexpected growing of XGly to 61%, with similar Ydi, 18%.
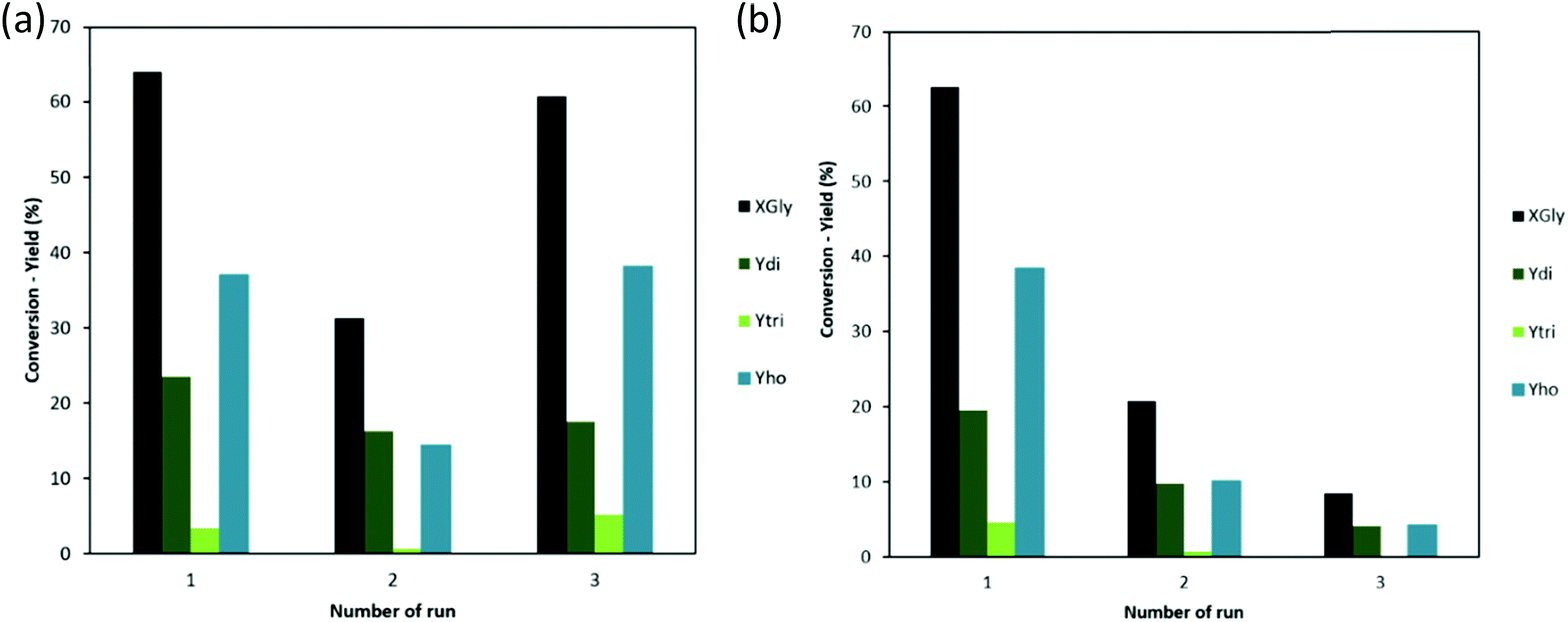 | ||
| Fig. 7 Reusability test (reaction conditions: 4 wt% cat., 240 °C and 8 h): (a) MAU05P (b) MAU05. | ||
The Literature on catalyst recycling in glycerol oligomerization is limited. However, in recent years new contributions are helping to clarify this issue. In our previous work,16 calcined dolomite was used, under optimal conditions (reaction with 2 wt% of dolomite for 24 h at 220 °C), showing a decrease in glycerol conversion from 80 to 35% after the first reuse and 40% after second reuse, with Sdi around 50%. The deactivation was explained by the expressive amount of Ca and Mg that leached into the reaction medium. Aloui et al. have worked with a series of mixed oxides derived from tetrametallic hydrotalcite, Mg2+/(Al3++Zr4+), with different Ni loadings.36Their recycling study was carried out using 2 wt% of catalyst for a rection at 220 °C for 24 h. Between each run, the catalyst was separated by decantation and washed with water. Glycerol conversion decreased from 55 to 35% after 4 runs. In all cases, the only product obtained was diglycerol. The work of Caputo et al. reported the use of lanthanum oxide supported on mesoporous silica gel KIT-6 for glycerol oligomerization. In this case, the reactions had 2 wt% of catalyst, at 240 °C for 24 h, leading to 72% of XGly in the first cycle. After each run, the catalyst was washed with methanol and calcinated at 700 °C to remove the organic phase. The catalyst was used for three times. In the second and third runs, conversion gradually decreased to 55% and 25% and formation of diglycerol was favored upon recycling. In comparison with the results on the present work, using MAU05P it was possible to increase the XGly in the first two cycles with only 8 hours of reaction, Fig. 7a. Hydroxyapatite, Ca10(PO4)6(OH)2, was used as catalyst in the work of Ebadipour et al., the optimal conditions were 0.5 mol% of catalyst used at 245 °C for 8 hours. In their reusability study, they compared the recovered catalyst when washed with water or ethanol. Ethanol had the best performance, allowing the catalyst to be used for three runs without loss in activity, with an XGLy of 15% and 100% selectivity to triglycerol.37
Several tests were performed in order to understand the catalyst deactivation. First, elemental analysis (Table S4 ESI†) of the reaction mixture showed the absence of leaching of Mg and Al from MAU05P. Next, the spent catalysts were submitted to thermogravimetric analysis and XPS.
It was reported in the literature that the presence of organic deposits, quantified by thermal analysis, in the recycling of rehydrated Mg/Al catalysts in aldol condensation38 and also by deposits, quantified by thermal analysis, in the recycling of rehydrated Mg/Al catalysts in aldol condensation38 and also by using their mixed oxides for glycerol carbonate production39 was a key factor to deactivation. These deposits modify the nature of the active centers, blocking their accessibility, therefore decreasing the catalytic activity. After the poisoning with acetic acid, TGA of MAU05P showed the characteristic two steps of weight loss, the first from room temperature to 200 °C and the second from 200 to 450 °C, Fig. S3e.† However, its values are 16.9% for the first step and 56.7% for the second, against 11.3% and 32.6% in MAU05 (Fig. 2), pointing to the presence of remaining acetic acid on the solid, as can be seen in FTIR results, Fig. S3b.† The thermograms of used MAU05P maintained the typical steps upon recycling, with weight losses under 100 °C of 6%, 9% and 4% from 1st to 3rd cycle, respectively (Fig. 8). While, from 200 to 500 °C the losses were of 40, 55 and 44%. This expressive variation on the second region can be assigned to the presence of adsorbed species such as unreacted glycerol and remaining reaction products (diglycerol, triglycerol and high oligomers). The spent catalyst from second run had a higher amount of water compared to the others, as can be seen in the peak below 100 °C, it has also featured a substantial weight loss peak at around 200 °C, Fig. 8b. By comparing the catalytic performance (Fig. 7), with the thermal analysis (Fig. 8), it is noticeable that higher amount of remaining sorbed species on used catalyst points to lower catalytic activity in that cycle. The third reaction cycle displayed improvement in glycerol conversion. Notice that the spent catalyst on this reaction had similar total weight loss to the first cycle. However, catalyst degradation over reaction cycles plus the effect of blockage of active sites affects the products distribution, with the reduction of diglycerol formation in comparison to the first cycle. TGA/DTG of MA05 spent form after the third reaction cycle is presented in Fig. S5,† the total weight loss was of 69% against 44% on its fresh form. Presence of unreacted glycerol and remaining reaction products has strongly reduced the XGly (9%), in contrast with MAU30P, 61% of XGly for a weight loss of 47% on the last reaction cycle.
 | ||
| Fig. 8 Thermogravimetric analysis of spent catalysts from reusability test of MAU05P: (a) TGA and (b) DTG. | ||
The surfaces of fresh and spent catalysts of MAU05 (spent from 3rd run) and MAU05P (spent from 1st run) were studied by XPS, Table 3, Fig. 9 and Fig. S6.† Firstly, it is possible to notice a difference on the surface atomic ratio of Mg/Al on fresh catalysts, the material after treatment with acetic acid presents a higher ratio than MAU05 (2.59 compared to 1.75), due to the concentration of Mg. Also, Surface Mg/Al molar ratios are lower than the ratio on the bulk determined by ICP-OES, Table 1, suggesting the presence of a higher proportion of Al species on the catalyst surface. This superficial enrichment of aluminium was also observed by García-Sancho et al. preparing Mg/Al LDH by coprecipitation.19 Spent catalysts had a reduction of such ratio, reaching 0.93 on MAU05 after 3 reaction cycles and 1.42 on MAU05P after the first cycle, as a consequence of the decrease of Mg atomic concentration. As Mg and Al amounts were not found the reaction products by ICP, one possible explanation is that these species were transferred to the bulk catalyst. The reduction on the exposed Mg on these catalysts seems to be corelated to the reduction on XGly observed in reusability tests (Fig. 7). Guerrero-Urbaneja et al. have found similar correlation between catalytic behavior and structural stability (inferred from the superficial MII/MIII atomic ratio) of mixed oxides derived of Mg/Fe LDH.
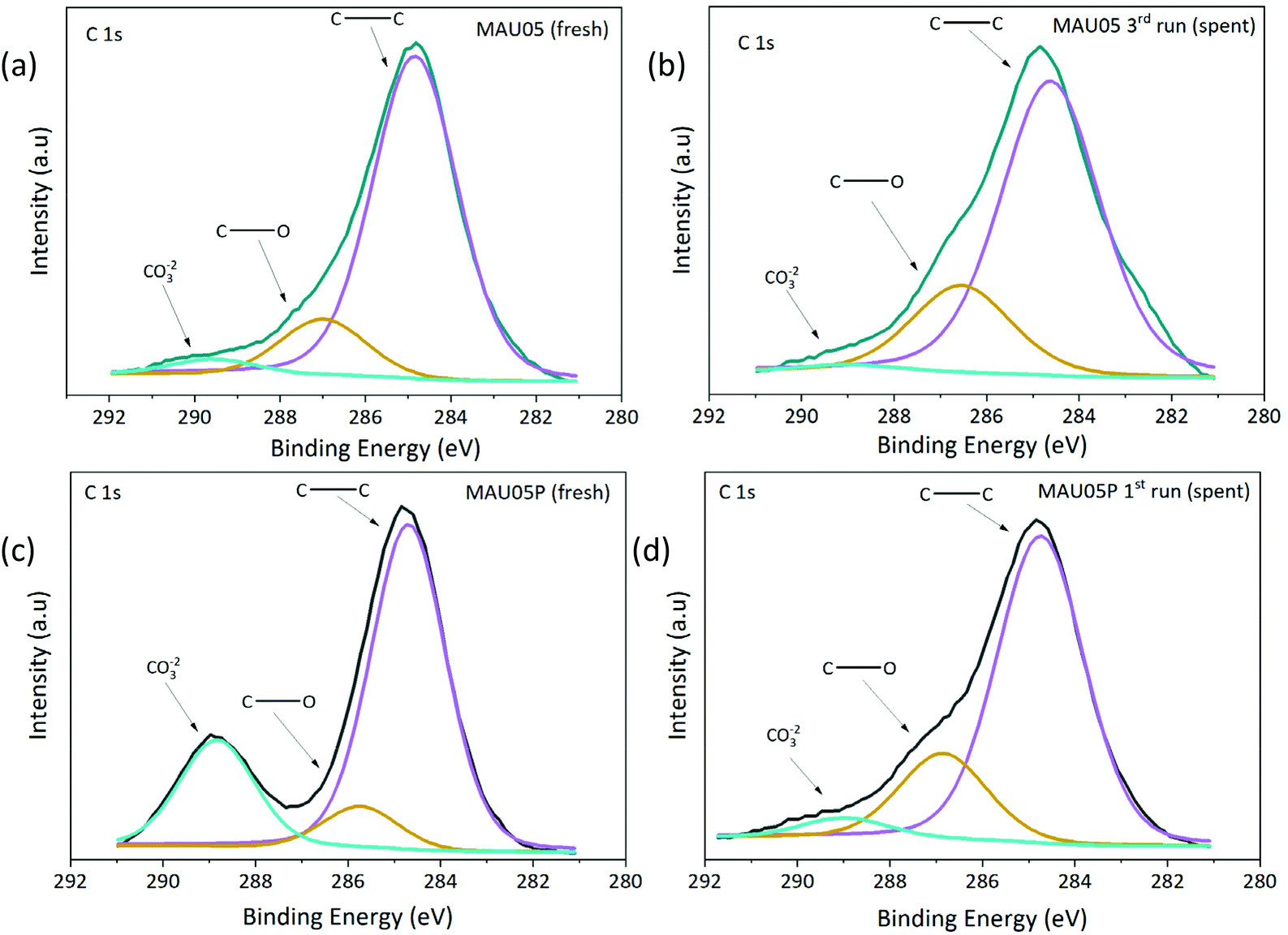 | ||
| Fig. 9 XPS spectra for C 1s region: (a) MAU05 fresh, (b) MAU05 spent, (c) MAU05P fresh (d) MAU05P spent. | ||
| Sample | Binding energy (eV) | Atomic concentration (%) | Molar ratio | ||||||
|---|---|---|---|---|---|---|---|---|---|
| C 1s | O 1s | Mg 2p | Al 2p | C | O | Mg | Al | Mg/Al | |
| MAU05 (fresh) | 284.8 (83%) | 532.5 | 50.7 | 75.0 | 54.30 | 33.46 | 6.75 | 4.29 | 1.75 |
| 287.0 (13%) | |||||||||
| 289.6 (4%) | |||||||||
| MAU05 3rd run (spent) | 284.6 (76%) | 530.0 (10%) | 50.5 | 74.6 | 56.04 | 30.43 | 5.06 | 6.01 | 0.99 |
| 286.6 (22%) | 532.2 (90%) | ||||||||
| 289 (2%) | |||||||||
| MAU05P (fresh) | 284.7 (69%) | 531.7 | 50.4 | 73.6 | 56.72 | 31.69 | 8.12 | 3.48 | 2.59 |
| 285.7 (9%) | |||||||||
| 288.8 (22%) | |||||||||
| MAU05P1st run (spent) | 284.7 (74%) | 532.5 | 50.7 | 75.0 | 50.02 | 36.55 | 7.18 | 5.62 | 1.42 |
| 286.7 (21%) | |||||||||
| 289.0 (5%) | |||||||||
The C 1s core level spectra of fresh and spent catalysts displays three contributions (Fig. 9). The main contribution at 284.8 eV and the second contribution at 286.7 eV are attributed to adventitious carbon (C–C and C–O, respectively). The third contribution, at 289.0 eV, is related to the presence of carbonate or carboxylate. Distribution of these bands are markedly different for MAU05 and MAU05P, the later having 22% of carbonate, which is probably arising from the treatment with acetic acid. The presence of these groups is in accordance with FTIR results (Fig. S3b†). After the first reaction cycle, MAU05P displays a reduction of carbonate band to 5%. The increase of the intensity of the contribution at 286.7 eV (C–O) is noticeable in the recycling of both catalysts, and can be related to unreacted glycerol or oligomers at the surface. The addition of acetic acid to MAU05 has changed its surface and improved the recyclability of MAU05P. Even after a reduction on surface carbonate content of spent catalyst from reuse (Fig. 9b), this material was able to present higher activity compared to MAU05 in the same cycle (Fig. 7).
Conclusions
This study shows that changing Mg/Al LDH properties via simple rehydration methods was able to improve its catalytic activity for glycerol oligomerization. Despite the recovery of lamellar structure either using ultrasound or mechanic stirring, the rehydrated materials have shown the presence of defects on the lamellae and changes on interlamellar composition, mainly reduction of carbonate and differences in water content. These new features were followed by changes in the basic sites and therefore in the activity for glycerol oligomerization. Such correlation indicates that tuning of catalyst basicity is crucial to obtain better selectivity in glycerol oligomerization.A simple modification based on the addition of acetic acid to the LDH reconstructed by ultrasound for 30 minutes was enough to adjust base site distribution and amount in order to successfully improve selectivity to diglycerol, reaching 37%, for XGly of 64%. The modification has also proven to have a positive effect on recyclability, since MAU05P insured lower deactivation and higher yield of the oligomers compared to the previous material.
The investigation of deactivation has shown that there was no leaching of metallic species from catalysts to the reaction products, an advance in comparison with previous efforts. In contrast, the increase of carbon on the surface as well as on the bulk spent catalyst, pointed to the contribution of unreacted glycerol and adsorbed products to reduce activity over reaction cycles. The good catalytic performance makes Mg/Al LDH catalyst a viable alternative for glycerol oligomerization.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are very grateful to Prof. J. A. Lercher and the Chair of Technical Chemistry II at Technische Unviersität München for the support and facilities provided for experiments. We are grateful to Smart2 Erasmus Program, CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico), Centro de Tecnologias Estratégicas do Nordeste [INT/MCT-FACEPE APQ-1015-3.06/14], Central Analítica – [UFC/CT-INFRA/MCTI-SISNANO/Pró- Equipamentos], Project CAPES/FUNCAP (Áreas Estratégicas) [Project E1-0079-0004301].Notes and references
- M. V. Sivaiah, S. Robles-Manuel, S. Valange and J. Barrault, Catal. Today, 2012, 198, 305–313 CrossRef CAS.
- A. Taghizadeh, P. Sarazin and B. D. Favis, J. Mater. Sci., 2013, 48, 1799–1811 CrossRef CAS.
- A. Martin and M. Richter, Eur. J. Lipid Sci. Technol., 2011, 113, 100–117 CrossRef CAS.
- M. Sutter, E. Da Silva, N. Duguet, Y. Raoul, E. Me and M. Lemaire, Chem. Rev., 2015, 115, 8609–8651 CrossRef CAS PubMed.
- M. A. Medeiros, M. H. Araujo, R. Augusti, L. C. A. De Oliveira and R. M. Lago, J. Braz. Chem. Soc., 2009, 20, 1667–1673 CrossRef CAS.
- M. Richter, Y. K. Krisnandi, R. Eckelt and A. Martin, Catal. Commun., 2008, 9, 2112–2116 CrossRef CAS.
- A. M. Ruppert, J. D. Meeldijk, B. W. M. Kuipers, B. H. Ernø and B. M. Weckhuysen, Chem. – Eur. J., 2008, 2016–2024 CrossRef CAS PubMed.
- J. Clacens, Y. Pouilloux and J. Barrault, Appl. Catal., A, 2022, 227, 181–190 CrossRef.
- J. I. Eshuis, J. A. Laan and R. P. Potman, Polymerization of glycerol using a zeolite catalyst, US Pat., 5635588, 1997 Search PubMed.
- K. Cottin, J.-M. Clacens, Y. Pouilloux and J. Barrault, Ol., Corps Gras, Lipides, 1998, 5, 405–412 Search PubMed.
- F. Kirby, A. Nieuwelink, B. W. M. Kuipers, A. Kaiser, P. C. A. Bruijnincx and B. M. Weckhuysen, Chem. – Eur. J., 2015, 21, 5101–5109 CrossRef CAS PubMed.
- N. Galy, R. Nguyen, P. Blach, S. Sambou, D. Luart and C. Len, J. Ind. Eng. Chem., 2017, 51, 312–318 CrossRef CAS.
- P. Bookong, S. Ruchirawat and S. Boonyarattanakalin, Chem. Eng. J., 2015, 275, 253–261 CrossRef CAS.
- J. H. Lee, S. K. Park, J. Ryu, H. Lee and J. S. Lee, Bull. Korean Chem. Soc., 2018, 39, 722–725 CrossRef CAS.
- F. J. S. Barros, R. Moreno-tost, J. A. Cecilia, A. L. Ledesma-mu, L. C. C. De Oliveira, F. M. T. Luna and R. S. Vieira, Mol. Catal., 2017, 433, 282–290 CrossRef CAS.
- F. J. S. Barros, J. A. Cecilia, R. Moreno-Tost, M. F. de Oliveira, E. Rodríguez-Castellón, F. M. T. Luna and R. S. Vieira, Waste Biomass Valorization, 2018, 1–14 CAS.
- E. L. Crepaldi and J. B. Valim, Quim. Nova, 1998, 21(3), 300–311 CrossRef CAS.
- X. Zhang, Z. Wang, Y. Tang, N. Qiao and Y. Li, Catal. Sci. Technol., 2015, 5, 4991–4999 RSC.
- C. García-Sancho, R. Moreno-tost, J. M. Mérida-robles, J. Santamaría-gonzález, A. Jiménez-lópez and P. M. Torres, Catal. Today, 2011, 167, 84–90 CrossRef.
- P. Guerrero-Urbaneja, C. García-Sancho, R. Moreno-Tost, J. Mérida-Robles, J. Santamaría-González, A. Jiménez-López and P. Maireles-Torres, Appl. Catal., A, 2014, 470, 199–207 CrossRef CAS.
- E. Pérez-Barrado, M. C. Pujol, M. Aguiló, J. Llorca, Y. Cesteros, F. Díaz, J. Pallarès, L. F. Marsal and P. Salagre, Chem. Eng. J., 2015, 264, 547–556 CrossRef.
- E. Lima, D. Jesu, R. Isabel and M. Vera, Inorg. Chem., 2012, 51, 7774–7781 CrossRef CAS PubMed.
- J. Pérez-ramírez, S. Abelló and N. M. Van Der Pers, Chem. – Eur. J., 2007, 13, 870–878 CrossRef PubMed.
- S. Abelló, F. Medina, D. Tichit, J. Pérez-Ramírez, J. C. Groen, J. E. Sueiras, P. Salagre and Y. Cesteros, Chem. – Eur. J., 2005, 11, 728–739 CrossRef PubMed.
- G. Lee, Y. Jeong, A. Takagaki and J. C. Jung, J. Mol. Catal. A: Chem., 2014, 393, 289–295 CrossRef CAS.
- M. G. Álvarez, R. J. Chimentão, F. Figueras and F. Medina, Appl. Clay Sci., 2012, 58, 16–24 CrossRef.
- D. C. Carvalho, N. A. Ferreira, J. M. Filho, O. P. Ferreira, J. M. Soares and A. C. Oliveira, Catal. Today, 2015, 250, 155–165 CrossRef CAS.
- O. D. Pavel, R. Bîrjega, M. Che, G. Costentin and E. Angelescu, Catal. Commun., 2008, 9, 1974–1978 CrossRef CAS.
- H. Atia, U. Armbruster and A. Martin, Appl. Catal., A, 2011, 393, 331–339 CrossRef CAS.
- E. Angelescu, O. D. Pavel, R. Bîrjega, M. Florea and R. Zăvoianu, Appl. Catal., A, 2008, 341, 50–57 CrossRef CAS.
- R. J. Chimentão, S. Abelló, F. Medina, J. Llorca, J. E. Sueiras, Y. Cesteros and P. Salagre, J. Catal., 2007, 252, 249–257 CrossRef.
- S. K. Sharma, P. A. Parikh and R. V. Jasra, Appl. Catal., A, 2010, 386, 34–42 CrossRef CAS.
- M. R. Anuar, A. Z. Abdullah and M. R. Othman, Catal. Commun., 2013, 32, 67–70 CrossRef CAS.
- Z. Gholami, A. Z. Abdullah and K. T. Lee, Chem. Eng. Commun., 2015, 202, 1397–1405 CrossRef CAS.
- E. Pérez-Barrado, M. C. Pujol, M. Aguiló, J. Llorca, Y. Cesteros, F. Díaz, J. Pallarès, L. F. Marsal and P. Salagre, Chem. Eng. J., 2015, 264, 547–556 CrossRef.
- M. Aloui, J. A. Cecilia, R. Moreno-Tost, S. B. Ghorbel, M. S. Zina and E. Rodríguez-Castellón, J. Sol-Gel Sci. Technol., 2021, 97, 351–364 CrossRef CAS.
- N. Ebadipour, S. Paul, B. Katryniok and F. Dumeignil, Catal. Today, 2021, 11, 1247 CAS.
- S. Abelló, D. Vijaya-Shankar and J. Pérez-Ramírez, Appl. Catal., A, 2008, 342, 119–125 CrossRef.
- G. M. Lari, A. B. L. De Moura, L. Weimann, S. Mitchell, C. Mondelli and J. Pérez-Ramírez, J. Mater. Chem. A, 2017, 5, 16200–16211 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization for catalysts: thermogravimetric analysis, nitrogen adsorption isotherms, BJH pore size distribution, XRD, CO2-TPD, TEM, FTIR and XPS. Selectivity to acrolein. See DOI: 10.1039/d1dt03817f |
| This journal is © The Royal Society of Chemistry 2022 |