
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLuminescent chromium(0) and manganese(I) complexes
Christina
Wegeberg
 and
Oliver S.
Wenger
*
and
Oliver S.
Wenger
*
Department of Chemistry, University of Basel, St. Johanns-Ring 19, 4056 Basel, Switzerland. E-mail: oliver.wenger@unibas.ch
First published on 15th December 2021
Abstract
In this Frontier article, recently discovered chromium(0) and manganese(I) complexes emitting from metal-to-ligand charge transfer (MLCT) excited states are highlighted. Chelating isocyanide ligands give access to this new class of 3d6 emitters with MLCT lifetimes in (or close to) the nanosecond regime in solution at room temperature. Although the so far achievable luminescence quantum yields in these open-shell complexes are yet comparatively low, the photophysical properties of the new chromium(0) and manganese(I) isocyanides are reminiscent of those of well-known ruthenium(II) polypyridines. Our findings provide insight into how undesired nonradiative MLCT deactivation in 3d6 complexes can be counteracted, and they seem therefore relevant for the further development of new luminescent first-row transition metal complexes based on iron(II) and cobalt(III) in addition to chromium(0) and manganese(I).
Introduction
Transition metal complexes of precious 4d6 and 5d6 ions such as ruthenium(II) and iridium(III) have extensively been explored in the fields of inorganic photophysics and photochemistry due to their favorable electronic structures leading to long-lived and emissive MLCT excited states with attractive redox properties. Consequently, these complexes have been widely used as luminophores, sensitizers, photoredox catalysts, and dyes in solar cells.1–7 The use of more earth-abundant metal elements is, however, an obvious goal to secure cheaper and more sustainable technologies. With their d6 valence electron configuration, cobalt(III), iron(II), manganese(I) and chromium(0) are in principle isoelectronic and earth-abundant possible alternatives to ruthenium(II) and iridium(III). The challenge with the use of first-row transition metals with an open-shell configuration is their weak ligand fields (compare Fig. 1a and b), which result in energetically low-lying metal-centered (MC) excited states that can typically enable fast nonradiative deactivation of the MLCT excited state back to the electronic ground state (compare Fig. 1c and d).8–10 In [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) the 3MLCT lifetime is for instance on the order of 500 ns, but in [Fe(bpy)3]2+ it is only ca. 50–100 fs.11,12 Recent work on new types of iron(II) compounds has led to MLCT lifetimes up to a few nanoseconds, but no MLCT photoluminescence was detectable.13–16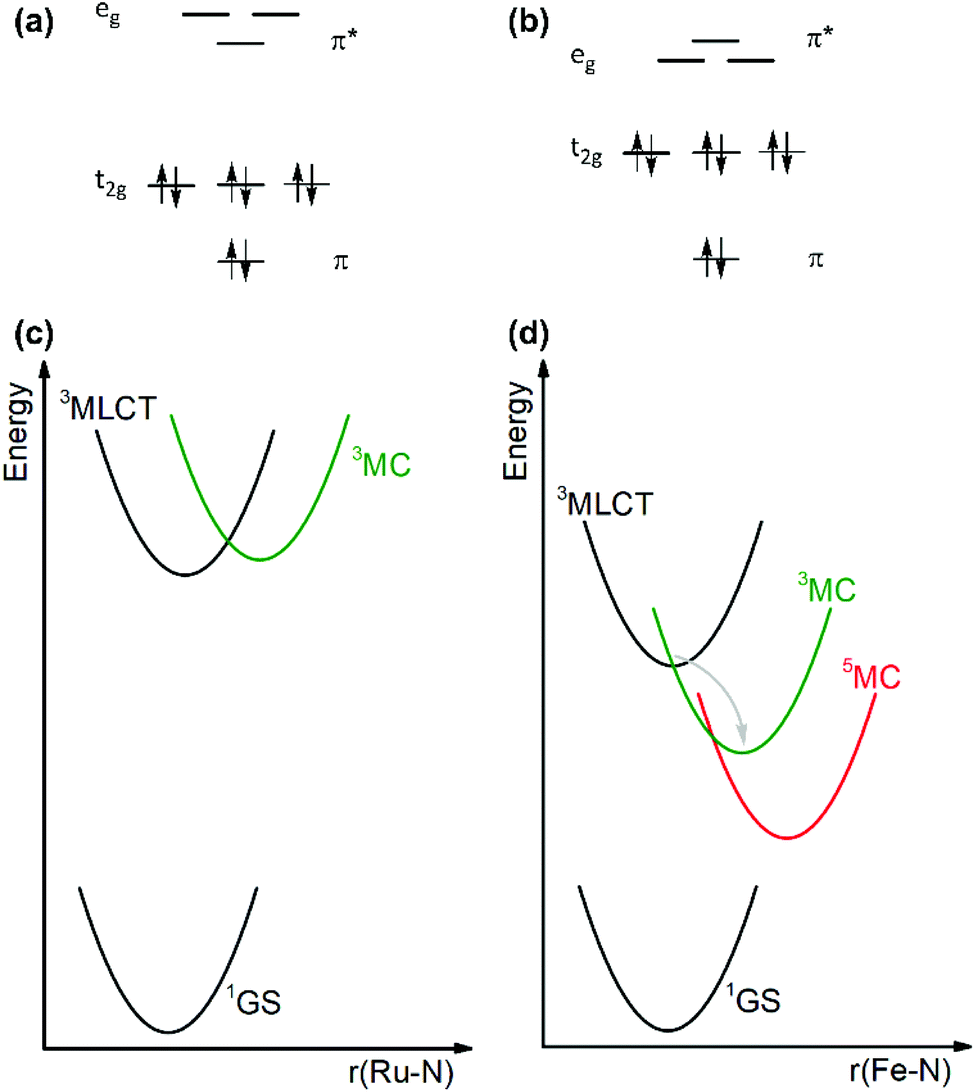 | ||
| Fig. 1 Schematic representation of the low-spin d6 electron configuration in Oh point symmetry, including ligand-based π/π* orbitals in addition to metal-based t2g and ee orbitals in either a strong (a) or weak (b) ligand field as encountered for example in [Ru(bpy)3]2+ and [Fe(bpy)3]2+, respectively. Simplified potential well diagrams with the key electronic states of (c) [Ru(bpy)3]2+ and (d) [Fe(bpy)3]2+. 1GS denotes the electronic ground state. The horizontal axes in (c) and (d) are nuclear coordinates, along which the complexes distort upon electronic excitation. Here, this has been oversimplified to the metal–ligand bond distances. | ||
Discussion
Inspired by early studies of group 6 metal complexes with monodentate isocyanide ligands17,18 and recent reports of luminescent tungsten(0) complexes with other types of monodentate isocyanides,22–24 our group discovered that chelating isocyanide ligands facilitate access to luminescent chromium(0) and manganese(I) complexes (Fig. 2).19–21 The collective σ-donating and π-accepting properties of the isocyanide ligands25 cause a strong ligand field, in which the abovementioned MC states are energetically less accessible than in typical iron(II) polypyridines,26 thereby allowing for emission from the lowest MLCT excited state. The chelating nature of our di- and triisocyanides imparts robustness, and possibly contributes to reducing the efficiency of (undesired) nonradiative excited-state relaxation processes as a consequence of enhanced rigidity.27 Owing to improved ligand design, we have now been able to make the important step from luminescent molybdenum(0)27–29 and tungsten(0)17,18,22–24,67 complexes to emissive compounds of the first-row of transition metals.19–21,30 | ||
| Fig. 2 (a) Molecular structures of the chelating diisocyanide ligands LH, Lpyr, Lbi and the triisocyanide ligand Ltri. (b) Generic structures of homoleptic tris(diisocyanide) (up) and bis(triisocyanide) (down) complexes. M = Cr0, n = 0; M = MnI, n = 1. | ||
The chelating bidentate ligand LH (Fig. 2) provided access to the complex [Cr(LH)3], which was our first example of a 3d6 complex emitting in solution at room temperature (Table 1 and Fig. 3a black).19 Prior to that, MLCT emission from 3d6 complexes under such conditions had been largely unknown, though an early study already reported on photoluminescence from [Cr(CO)4(bpy)], but this was a rather curious case of a presumed dual emission.31,32 Group 6 metal carbonyls typically undergo photo-dissociation of CO ligands,33,34 and against this background chelating isocyanides seem advantageous to avoid photo-degradation due to loss of ligands. The 3MLCT excited state lifetime of [Cr(LH)3] is 2.2 ns in deaerated THF, which is long enough for [Cr(LH)3] to engage as photosensitizer in a triplet–triplet annihilation upconversion process with anthracene.19
 | ||
| Fig. 3 (a) UV–Vis absorption spectra (solid) of [Cr(LH)3] in deaerated THF (black) and [Cr(Lpyr)3] in deaerated cyclooctane (green) as well as emission spectra (dashed) of [Cr(LH)3] in deaerated THF (black) and [Cr(Lpyr)3] in deaerated solutions of n-hexane (blue), cyclooctane (green) and toluene (red). [Cr(LH)3] and [Cr(Lpyr)3] were excited at 532 nm and 473 nm, respectively. (b) UV–Vis absorption spectrum (solid) and emission spectrum (dashed) of [Mn(Ltri)2]+ in deaerated acetonitrile. Excitation occurred at 410 nm. All data sets were obtained at 20 °C. | ||
| Complex | Solvent | λ abs [nm] | λ em [nm] |
λ
em![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a at 77 K [nm] a at 77 K [nm] |
τ 1, τ2, τ3 [ns] | τ avg [ns] | φ [%] | Ref |
|---|---|---|---|---|---|---|---|---|
| a Data obtained in 2-methyl-THF. λabs: 1MLCT absorption band maxima, λem: emission band maxima, τ1–3: luminescence lifetime decay components, τavg: weighted average luminescence lifetime. | ||||||||
| [Cr(LH)3] | THF | 475 | 630 | 645 | 2.2 (100%) | 2.2 | 0.001 | 19 |
| [Cr(Lpyr)3] | Cyclooctane | 475 | 682 | 692 | 4.05 ns (48%), 8.00 ns (52%) | 6.10 | 0.09 | 20 |
| [Mn(Lbi)3]+ | CH3CN | 385 | 485 | 480; 620 | 0.374 ns (79.2%), 1.84 ns (19.3%), 5.85 ns (1.5%) | 0.74 | 0.05 | 21 |
| [Mn(Ltri)2]+ | CH3CN | 395 | 525 | 480; 600 | 0.635 ns (56.4%), 2.07 ns (33.6%), 6.74 ns (10.0%) | 1.73 | 0.03 | 21 |
The luminescence quantum yield of [Cr(LH)3] is, however, barely above the detection limit (10−5), making further improvement of the ligand and complex design highly desirable. We have previously shown that modifications of the diisocyanide ligand backbone allow for adjustment of the photophysical properties of isoelectronic molybdenum(0) tris(diisocyanide) complexes,27–29 hence we reasoned that an improved ligand design could potentially enhance the photophysical properties of chromium(0) MLCT emitters.
Extension of the π-system of the α-diimine ligands of ruthenium(II) complexes has previously been identified as a useful strategy to improve the photophysical properties of 4d6 MLCT luminophores.35–38 This change in ligand design can have one of two advantageous effects. First, it can lead to a more delocalized MLCT excited state relative to complexes bearing ligands without extended π-system. Second, it can cause an electronic structure in which a ligand-centered (LC) excited state localized on the extended π-system becomes important for the overall photophysics. The first scenario is commonly referred to as the delocalization effect,35,36,39–45 and the latter is often referred to as the triplet reservoir effect.37,38,46–48 Which one of these two effects dominates is largely dependent on whether there is substantial electronic coupling between the metal core and the extended π-system, or whether these two parts are electronically decoupled from one another. For the triplet reservoir effect, it is furthermore necessary that the emissive 3MLCT excited state and the 3LC state are energetically close to each other, such that the typically very long-lived 3LC (dark) state over time can feed the more rapidly decaying luminescent 3MLCT excited state, hereby elongating the excited state lifetime of the d6 emitter. It seemed interesting to explore whether these concepts known from precious 4d6 complexes are also applicable to 3d6 congeners, and for that purpose we targeted the attachment of an extended π-system at the periphery of the LH ligand, and the subsequent coordination of the resulting new ligand to chromium(0). The energy of the emissive 3MLCT excited state of [Cr(LH)3] was estimated to 2.05 eV, which is close to the triplet energy of pyrene (2.10 eV),19,49 suggesting that pyrenyl substituents at LH might result in one of the two abovementioned effects.
Indeed, the luminescent compound [Cr(Lpyr)3] (Fig. 1) has substantially improved photophysical properties relative to the [Cr(LH)3] parent complex without anchored pyrene moieties (Table 1).20 Its (average) 3MLCT lifetime of 6.10 ns in deaerated cyclooctane at 20 °C is, to the best of our knowledge, the longest 3MLCT lifetime reported to date for any 3d6 complex under comparable conditions. Recent record 3MLCT lifetimes (of dark, non-emissive states) for isoelectronic FeII complexes are 528 ps and 2.6 ns.13,14 Remarkably, the luminescence quantum yield (φ) of 9 × 10−4 for [Cr(Lpyr)3] is 90 times greater than that of [Cr(LH)3], and this is despite a redshift of the emission by 1100 cm−1 for [Cr(Lpyr)3] relative to [Cr(LH)3] (Fig. 3a). Typically, luminescence decays of MLCT emitters follow the so-called energy gap law, according to which a lower excited-state energy enables more rapid nonradiative emission.50 Against this background, the much enhanced photoluminescence properties of the pyrene-decorated Cr0 complex are all the more remarkable. The photophysics of [Cr(Lpyr)3] are dominated by the delocalization effect, which is reflected in noticeable electronic coupling between the chromium(0) core and the pyrenyl in the excited 3MLCT state. Specifically, redshifted emission maxima of [Cr(Lpyr)3] relative to [Cr(LH)3] and a strong emission solvatochromic effect are detected for [Cr(Lpyr)3], which was not observed for [Cr(LH)3]. The emission band maximum for [Cr(Lpyr)3] shifts from 675 nm in n-hexane to 740 nm in toluene (Fig. 3a), whereas in [Cr(LH)3] the emission band maximum only shifts by 5 nm between these two solvents. Such a strong emission solvatochromism suggests that the change in dipole moment between the ground state and the emissive 3MLCT state is larger in [Cr(Lpyr)3] than in [Cr(LH)3]. This in turn is compatible with the view that the excited electron in the emissive 3MLCT state of [Cr(Lpyr)3] is delocalized over a larger portion of the ligand π-system (diisocyanide m-terphenyl backbone and attached pyrene unit) than in the 3MLCT state of [Cr(LH)3] (in which only the diisocyanide m-terphenyl backbone but no pyrene is present). As a consequence, the photoactive MLCT excited state in [Cr(Lpyr)3], likely becomes less distorted than in [Cr(LH)3], which leads to a smaller overlap between vibrational wavefunctions of the ground and excited state potential energy surfaces.39,42 This effect can be expected to decrease non-radiative excited-state decay rates and likely contributes substantially to the improved photophysical properties of [Cr(Lpyr)3] relative to [Cr(LH)3], though other (yet unidentified effects) could play important roles as well.
The MLCT luminescence lifetimes and quantum yields for [Cr(Lpyr)3] exhibit an unusual bell-shaped dependence on solvent polarity indicative of two counteracting effects governing the MLCT deactivation,20 a behavior that is not typically seen for precious metal-based 4d6 and 5d6 MLCT luminophores. Polar solvents stabilize the emissive 3MLCT state of [Cr(Lpyr)3] and thereby decrease the energy gap to the electronic ground state, which accelerates non-radiative relaxation. Conversely, apolar solvents raise the 3MLCT energy but presumably decrease the barrier for thermal deactivation via near-by higher lying MC excited states.51,52 The sweet spot with optimal luminescence lifetime and quantum yield of [Cr(Lpyr)3] is reached in cyclooctane.
Complexation of manganese(I) to the chelating bidentate isocyanide ligand Lbi and the tridentate isocyanide Ltri (Fig. 2a) resulted in the first examples of MLCT-luminescent manganese complexes in solution at room temperature.21 Previously, only a molecular complex of manganese(IV) with luminescence properties has been reported, however, this compound was reported to emit primarily in the solid state and not from an MLCT state.53 Both [Mn(Lbi)3]+ and [Mn(Ltri)2]+ (Fig. 3b) have a broad unstructured emission band around 500 nm, and the luminescence lifetimes approach the nanosecond regime in deaerated acetonitrile at room temperature (Table 1). The photoactive excited states have dominant MLCT character, as demonstrated by a combination of UV–Vis transient absorption and spectro-electrochemical studies. These MLCT states participate in electron transfer processes, as shown by a laser experiment, in which methyl viologen was reduced transiently from its initial dication to the radical monoanion form. Furthermore, the photoactive MLCT excited states of [Mn(Lbi)3]+ and [Mn(Ltri)2]+ engage in bimolecular triplet–triplet energy transfer reactions with polyaromatic hydrocarbons possessing triplet energies below 2.3 eV. For instance, in an UV–Vis transient absorption experiment performed in the presence of excess pyrene, the hydrocarbon triplet excited state was readily detectable following selective excitation of the MnI complexes. This experiment confirms that the energy-donating excited state has triplet spin multiplicity. Certain aspects of the MnI isocyanide photophysics have remained yet somewhat unclear, for example the role of a ligand-centered 3π–π* excited state, which will require further studies to comprehend in detail.
The manganese(I) isocyanide complexes are air stable, and their oxidation to the +II oxidation state is reversible. This opens perspectives for possible applications in photocatalysis, in which the 3MLCT-excited manganese(I) complexes could act as electron donors to various substrates. After photo-oxidation, the initial +I oxidation state of manganese can likely be regenerated with common sacrificial reagents such as tertiary amines, because the reduction of MnII to MnI is comparatively easy to accomplish in this class of compounds.17,54 In the case of chromium, the analogous d5 to d6 reduction of CrI to Cr0 is more difficult to achieve, mainly because of the lower (cationic) charge of CrI compared to MnII.55 Consequently, substantially stronger sacrificial electron donors than the commonly used tertiary amines would likely be required in photoredox reactions based on chromium(0) catalysts.
Conclusions
Our recent progress on luminescent chromium(0) and manganese(I) complexes demonstrates that with sophisticated ligand design, it is possible to obtain d6 emitters based on earth-abundant elements, which are essentially analogues of the extensively explored [Ru(bpy)3]2+ and its numerous 4d6 and 5d6 congeners. These luminescent 3d6 complexes have a rich photoreactivity, which include photoinduced electron transfer as well as triplet energy transfer, and furthermore they are amenable to photochemical (triplet–triplet annihilation) upconversion.Deactivation of the lowest MLCT state of isoelectronic iron(II) complexes often occurs on a very rapid time-scale due to low-lying MC excited states. In analogy, we found that low-lying excited MC states play an important role in the deactivation of the MLCT states in our isoelectronic chromium(0) and manganese(I) complexes. Even subtle changes in solvent polarity can significantly influence the radiative and non-radiative decay rates, which highlights some of the challenges associated with the comparatively weak ligand fields encountered in first-row transition metal complexes relative to those of the second or the third row of d-metals (compare Fig. 1a and b). The issue of low-lying MC states (Fig. 1d) is a key difference between the 3d6 compounds considered here and the well-known class of emissive CuI complexes, which have a completely filled 3d10 subshell.56,57 This makes it much more difficult to establish long-lived and luminescent MLCT excited states in the open-shell compounds, whilst recent studies of linear copper(I) complexes have reported outstanding luminescence properties.58–60 Similarly, newly designed CrIII (d3) complexes feature remarkable luminescence quantum yields and excited state lifetimes,61–63 but those are complexes emitting from so-called spin–flip (MC) states, which are very little distorted relative to the electronic ground state, and as such are not directly comparable to the MLCT excited states of d6 compounds. Another strategy to develop d6 emitters is to install luminescent MC states rather than MLCT states. This was recently accomplished for a CoIII complex with the use of a scorpionate carbene ligand.64 The same ligand has also proven useful in making an iron(III) (d5) complex emit from a ligand-to-metal charge transfer (LMCT) excited state, applicable to photocatalytic reactions.65,66,68 Thus, each d-electron configuration has its own photophysical characteristics, and further research in all directions seems highly desirable in the interest of broadening our understanding of photoactive excited states in first-row transition metal complexes.
Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. W. thanks the Independent Research Fund Denmark for an international postdoctoral grant (9059-00003B). O. S. W. acknowledges funding from the Swiss National Science Foundation through grant number 200021_178760.References
- M. S. Lowry and S. Bernhard, Chem. – Eur. J., 2006, 12, 7970–7977 CrossRef CAS PubMed.
- V. Balzani, G. Bergamini, F. Marchioni and P. Ceroni, Coord. Chem. Rev., 2006, 250, 1254–1266 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178–8211 CrossRef CAS PubMed.
- Y. You and W. Nam, Chem. Soc. Rev., 2012, 41, 7061–7084 RSC.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- O. S. Wenger, Chem. – Eur. J., 2019, 25, 6043–6052 CrossRef CAS PubMed.
- C. Förster and K. Heinze, Chem. Soc. Rev., 2020, 49, 1057–1070 RSC.
- J. K. McCusker, Science, 2019, 363, 484–488 CrossRef CAS PubMed.
- S. F. McClanahan and J. R. Kincaid, J. Am. Chem. Soc., 1986, 108, 3840–3841 CrossRef CAS.
- G. Auböck and M. Chergui, Nat. Chem., 2015, 7, 629–633 CrossRef PubMed.
- P. Chábera, K. S. Kjaer, O. Prakash, A. Honarfar, Y. Liu, L. A. Fredin, T. C. B. Harlang, S. Lidin, J. Uhlig, V. Sundström, R. Lomoth, P. Persson and K. Wärnmark, J. Phys. Chem. Lett., 2018, 9, 459–463 CrossRef PubMed.
- J. D. Braun, I. B. Lozada, C. Kolodziej, C. Burda, K. M. E. Newman, J. van Lierop, R. L. Davis and D. E. Herbert, Nat. Chem., 2019, 11, 1144–1150 CrossRef CAS PubMed.
- L. Liu, T. Duchanois, T. Etienne, A. Monari, M. Beley, X. Assfeld, S. Haacke and P. C. Gros, Phys. Chem. Chem. Phys., 2016, 18, 12550–12556 RSC.
- Y. Vukadinovic, L. Burkhardt, A. Päpcke, A. Miletic, L. Fritsch, B. Altenburger, R. Schoch, A. Neuba, S. Lochbrunner and M. Bauer, Inorg. Chem., 2020, 59, 8762–8774 CrossRef CAS PubMed.
- K. R. Mann, M. Cimolino, G. L. Geoffroy, G. S. Hammond, A. A. Orio, G. Albertin and H. B. Gray, Inorg. Chim. Acta, 1976, 16, 97–101 CrossRef CAS.
- K. R. Mann, H. B. Gray and G. S. Hammond, J. Am. Chem. Soc., 1977, 99, 306–307 CrossRef CAS.
- L. A. Büldt, X. Guo, R. Vogel, A. Prescimone and O. S. Wenger, J. Am. Chem. Soc., 2017, 139, 985–992 CrossRef PubMed.
- C. Wegeberg, D. Häussinger and O. S. Wenger, J. Am. Chem. Soc., 2021, 143, 15800–15811 CrossRef CAS PubMed.
- P. Herr, C. Kerzig, C. B. Larsen, D. Häussinger and O. S. Wenger, Nat. Chem., 2021, 13, 956–962 CrossRef CAS PubMed.
- W. Sattler, M. E. Ener, J. D. Blakemore, A. A. Rachford, P. J. LaBeaume, J. W. Thackeray, J. F. Cameron, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2013, 135, 10614–10617 CrossRef CAS PubMed.
- W. Sattler, L. M. Henling, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2015, 137, 1198–1205 CrossRef CAS PubMed.
- H. Kvapilová, W. Sattler, A. Sattler, I. V. Sazanovich, I. P. Clark, M. Towrie, H. B. Gray, S. Záliš and A. Vlček, Inorg. Chem., 2015, 54, 8518–8528 CrossRef PubMed.
- G. D. Sutton, M. E. Olumba, Y. H. Nguyen and T. S. Teets, Dalton Trans., 2021 10.1039/d1dt03312c.
- W. Zhang, R. Alonso-Mori, U. Bergmann, C. Bressler, M. Chollet, A. Galler, W. Gawelda, R. G. Hadt, R. W. Hartsock, T. Kroll, K. S. Kjær, K. Kubiček, H. T. Lemke, H. W. Liang, D. A. Meyer, M. M. Nielsen, C. Purser, J. S. Robinson, E. I. Solomon, Z. Sun, D. Sokaras, T. B. van Driel, G. Vankó, T.-C. Weng, D. Zhu and K. J. Gaffney, Nature, 2014, 509, 345–348 CrossRef CAS PubMed.
- P. Herr, F. Glaser, L. A. Büldt, C. B. Larsen and O. S. Wenger, J. Am. Chem. Soc., 2019, 141, 14394–14402 CrossRef CAS PubMed.
- L. A. Büldt, X. Guo, A. Prescimone and O. S. Wenger, Angew. Chem., Int. Ed., 2016, 55, 11247–11250 CrossRef PubMed.
- J. B. Bilger, C. Kerzig, C. B. Larsen and O. S. Wenger, J. Am. Chem. Soc., 2021, 143, 1651–1663 CrossRef CAS PubMed.
- C. Wegeberg and O. S. Wenger, JACS Au, 2021, 1, 1860–1876 CrossRef CAS PubMed.
- D. M. Manuta and A. J. Lees, Inorg. Chem., 1986, 25, 1354–1359 CrossRef CAS.
- A. J. Lees, Chem. Rev., 1987, 87, 711–743 CrossRef CAS.
- I. R. Farrell, P. Matousek, M. Towrie, A. W. Parker, D. C. Grills, M. W. George and A. Vlcek, Inorg. Chem., 2002, 41, 4318–4323 CrossRef CAS PubMed.
- W. C. Henke, C. J. Otolski, W. N. G. Moore, C. G. Elles and J. D. Blakemore, Inorg. Chem., 2020, 59, 2178–2187 CrossRef CAS PubMed.
- J. A. Treadway, B. Loeb, R. Lopez, P. A. Anderson, F. R. Keene and T. J. Meyer, Inorg. Chem., 1996, 35, 2242–2246 CrossRef CAS PubMed.
- N. H. Damrauer, T. R. Boussie, M. Devenney and J. K. McCusker, J. Am. Chem. Soc., 1997, 119, 8253–8268 CrossRef CAS.
- A. J. Howarth, M. B. Majewski and M. O. Wolf, Coord. Chem. Rev., 2015, 282–283, 139–149 CrossRef CAS.
- X. Wang, A. Del Guerzo and R. H. Schmehl, J. Photochem. Photobiol., C, 2004, 5, 55–77 CrossRef CAS.
- G. F. Strouse, J. R. Schoonover, R. Duesing, S. Boyde, W. E. J. Jones and T. J. Meyer, Inorg. Chem., 1995, 34, 473–487 CrossRef CAS.
- J. A. Treadway, G. F. Strouse, R. R. Ruminski and T. J. Meyer, Inorg. Chem., 2001, 40, 4508–4509 CrossRef CAS PubMed.
- V. Grosshenny, A. Harriman, F. M. Romero and R. Ziessel, J. Phys. Chem., 1996, 100, 17472–17484 CrossRef CAS.
- S. Boyde, G. F. Strouse, W. E. Jones and T. J. Meyer, J. Am. Chem. Soc., 1990, 112, 7395–7396 CrossRef CAS.
- C.-Y. Hung, T.-L. Wang, Y. Jang, W. Y. Kim, R. H. Schmehl and R. P. Thummel, Inorg. Chem., 1996, 35, 5953–5956 CrossRef CAS.
- E. M. Kober, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1984, 23, 2098–2104 CrossRef CAS.
- Y. Wang, S. Liu, M. R. Pinto, D. M. Dattelbaum, J. R. Schoonover and K. S. Schanze, J. Phys. Chem. A, 2001, 105, 11118–11127 CrossRef CAS.
- X. Zhang, Y. Hou, X. Xiao, X. Chen, M. Hu, X. Geng, Z. Wang and J. Zhao, Coord. Chem. Rev., 2020, 417, 213371 CrossRef CAS.
- C. Mongin, P. Moroz, M. Zamkov and F. N. Castellano, Nat. Chem., 2018, 10, 225–230 CrossRef CAS PubMed.
- N. D. McClenaghan, Y. Leydet, B. Maubert, M. T. Indelli and S. Campagna, Coord. Chem. Rev., 2005, 249, 1336–1350 CrossRef CAS.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook Of Photochemistry, CRC Press, Boca Raton, 3rd edn, 2006 Search PubMed.
- J. V. Caspar, E. M. Kober, B. P. Sullivan and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 630–632 CrossRef CAS.
- A. Soupart, F. Alary, J.-L. Heully, P. I. P. Elliott and I. M. Dixon, Inorg. Chem., 2018, 57, 3192–3196 CrossRef CAS PubMed.
- A. Soupart, F. Alary, J.-L. Heully, P. I. P. Elliott and I. M. Dixon, Coord. Chem. Rev., 2020, 408, 213184 CrossRef CAS.
- J. P. Harris, C. Reber, H. E. Colmer, T. A. Jackson, A. P. Forshaw, J. M. Smith, R. A. Kinney and J. Telser, Can. J. Chem., 2017, 95, 547–552 CrossRef.
- P. M. Treichel, D. W. Firsich and G. P. Essenmacher, Inorg. Chem., 1979, 18, 2405–2409 CrossRef CAS.
- P. M. Treichel and G. J. Essenmacher, Inorg. Chem., 1976, 15, 146–150 CrossRef CAS.
- N. Armaroli, Chem. Soc. Rev., 2001, 30, 113–124 RSC.
- M. S. Lazorski and F. N. Castellano, Polyhedron, 2014, 82, 57–70 CrossRef CAS.
- D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. H. Rivett, S. Jones, T. H. Thomas, M. Abdi Jalebi, R. H. Friend, M. Linnolahti, M. Bochmann and D. Credgington, Science, 2017, 356, 159–163 CrossRef CAS PubMed.
- R. Hamze, J. L. Peltier, D. Sylvinson, M. Jung, J. Cardenas, R. Haiges, M. Soleilhavoup, R. Jazzar, P. I. Djurovich, G. Bertrand and M. E. Thompson, Science, 2019, 363, 601–606 CrossRef CAS PubMed.
- M. Gernert, L. Balles-Wolf, F. Kerner, U. Müller, A. Schmiedel, M. Holzapfel, C. M. Marian, J. Pflaum, C. Lambert and A. Steffen, J. Am. Chem. Soc., 2020, 142, 8897–8909 CrossRef CAS PubMed.
- C. Wang, S. Otto, M. Dorn, E. Kreidt, J. Lebon, L. Sršan, P. Di Martino-Fumo, M. Gerhards, U. Resch-Genger, M. Seitz and K. Heinze, Angew. Chem., Int. Ed., 2018, 57, 1112–1116 CrossRef CAS PubMed.
- J.-R. Jiménez, B. Doistau, C. M. Cruz, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, J. Am. Chem. Soc., 2019, 141, 13244–13252 CrossRef PubMed.
- F. Reichenauer, C. Wang, C. Förster, P. Boden, N. Ugur, R. Báez-Cruz, J. Kalmbach, L. M. Carrella, E. Rentschler, C. Ramanan, G. Niedner-Schatteburg, M. Gerhards, M. Seitz, U. Resch-Genger and K. Heinze, J. Am. Chem. Soc., 2021, 143, 11843–11855 CrossRef CAS PubMed.
- S. Kaufhold, N. W. Rosemann, P. Chábera, L. Lindh, I. Bolaño Losada, J. Uhlig, T. Pascher, D. Strand, K. Wärnmark, A. Yartsev and P. Persson, J. Am. Chem. Soc., 2021, 143, 1307–1312 CrossRef CAS PubMed.
- K. S. Kjær, N. Kaul, O. Prakash, P. Chábera, N. W. Rosemann, A. Honarfar, O. Gordivska, L. A. Fredin, K.-E. Bergquist, L. Häggström, T. Ericsson, L. Lindh, A. Yartsev, S. Styring, P. Huang, J. Uhlig, J. Bendix, D. Strand, V. Sundström, P. Persson, R. Lomoth and K. Wärnmark, Science, 2019, 363, 249–253 CrossRef PubMed.
- A. Aydogan, R. E. Bangle, A. Cadranel, M. D. Turlington, D. T. Conroy, E. Cauët, M. L. Singleton, G. J. Meyer, R. N. Sampaio, B. Elias and L. Troian-Gautier, J. Am. Chem. Soc., 2021, 143, 15661–15673 CrossRef CAS PubMed.
- J. Fajardo Jr., A. T. Barth, M. Morales, M. K. Takase, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2021, 143, 19389–19398 CrossRef CAS PubMed.
- N. Kaul and R. Lomoth, J. Am. Chem. Soc., 2021, 143, 10816–10821 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |