DOI:
10.1039/D1DT03641F
(Paper)
Dalton Trans., 2022,
51, 1806-1818
Acid/base responsive assembly/dis-assembly of a family of zirconium(IV) clusters with a cyclic imide-dioxime ligand†
Received
27th October 2021
, Accepted 24th December 2021
First published on 24th December 2021
Abstract
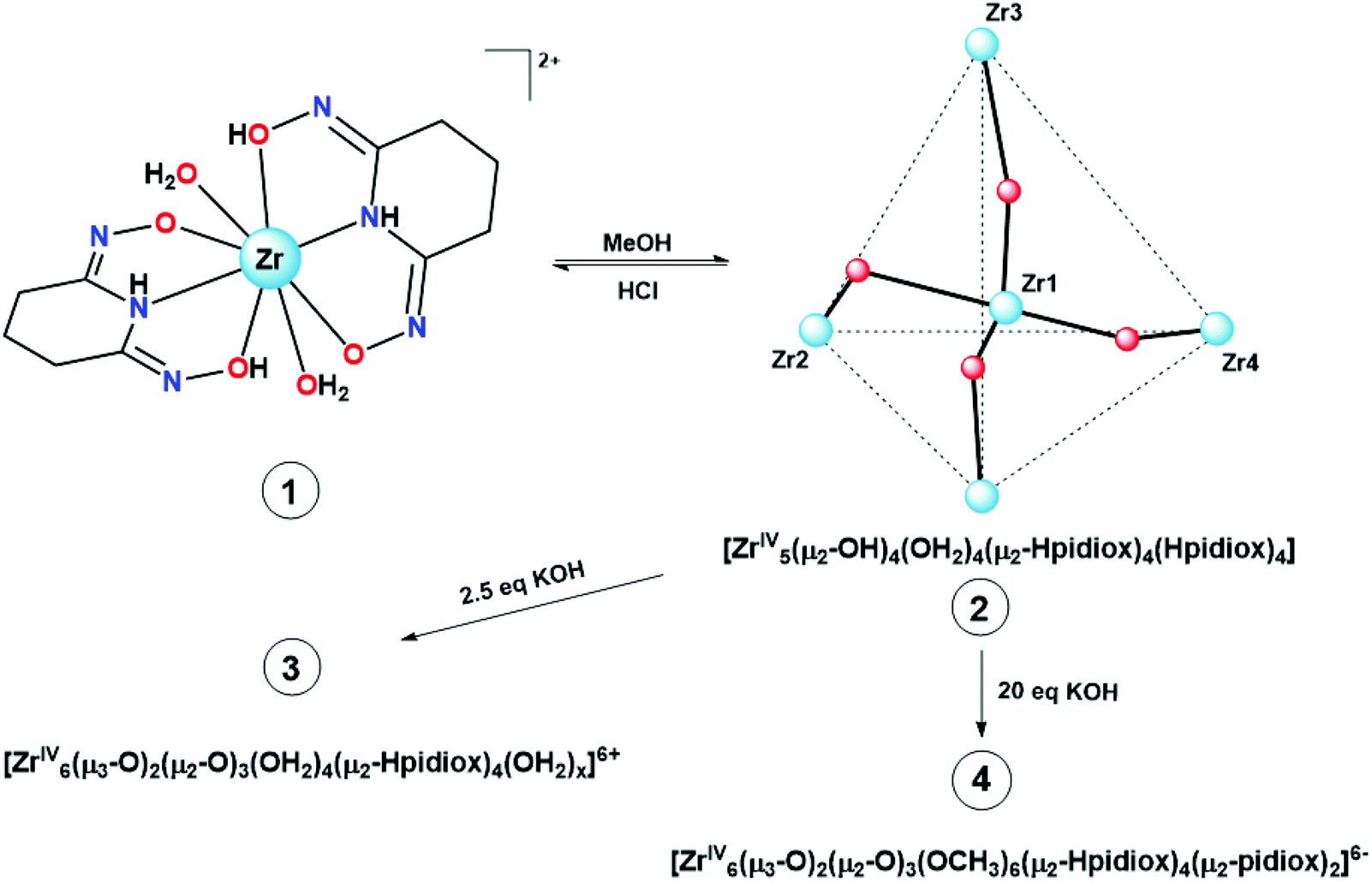
The hydrolytically stable dioxime ligand (2Z-6Z)-piperidine-2,6-dione (H3pidiox) acts as a strong chelator mainly with hard metals in high oxidation states, a pre-requisite for potential applications in metal sequestering processes from aqueous solutions. Reaction of ZrCl4 with H3pidiox in methanol gives the mononuclear compound [ZrIV(η1,η1,η2-H2pidiox-O,N,O′)2(OH2)2]Cl2·H2O·CH3OH (1), while the same reaction mixture in the presence of KOH gave the pentanuclear ZrOC [ZrIV5(μ2-OH)4(OH2)4(μ2–η1,η1,η2-Hpidiox-O,N,O′)4(η1,η1,η1-HpidioxO,N,O′)4]·5KCl·3CH3OH·8H2O (2). Compound 1 is formed at very acidic pH = 0, and the pentanuclear ZrOC 2 at higher pH values (pH = 2). Compounds 1 and 2 were characterized by single crystal X-ray structure analysis, multi-nuclear NMR spectroscopy and ESI-MS spectrometry. The single crystal X-ray structure analysis of 1 revealed a mononuclear zirconium(IV) compound containing an eight-coordinate zirconium atom bound to two singly deprotonated H2pidiox− ligands and two water molecules in a severely distorted bicapped octahedral geometry. The pentanuclear ZrOC 2 constitutes the second example of a Zr5 cluster to be reported and the first one in which the four zirconium atoms are arranged in a tetrahedral arrangement with the fifth occupying the center of the tetrahedron. 1D and 2D NMR spectroscopies of the acidic CD3OD solutions of complex 1 reveal a fast equilibrium between 1 and 2. Addition of KOH into a CH3OH solution of 2 results in the controlled fast transformation of 2 to an asymmetric hexanuclear ZrOC 3 as evidenced by the NMR and real-time ESI-MS solution studies. Further addition of KOH to the solution of 3 leads to the ZrOC 4, and on the basis of NMR and ESI-MS data and in comparison with the known hexanuclear titanium(IV)/H3pidiox cluster, it is concluded that the cluster 4 should have a hexanuclear structure. Electrospray ionization mass spectrometry (ESI-MS) demonstrated not only the structural stability 1 and 2 in solution, but also revealed the reversible pH driven dis-assembly/re-assembly process between the monomeric 1 and the pentanuclear ZrOC 2.
Introduction
Metal-oxo clusters (MOCs) are polynuclear complexes consisting of an inorganic core formed by metals in their highest oxidation state linked by oxygen atoms and stabilized by capping ligands. MOCs of group IV are a very promising class of compounds with many applications.1–9 In particular, the study of zirconium oxo clusters (ZrOCs) is of vital importance since they are processable molecular analogues of ZrO2 which has a wide range of applications in fuel cells,10 photocatalysis,11,12 and photochemical water splitting.13 Moreover, various ZrOCs have been reported as molecule-based building blocks in the construction of metal–organic frameworks (MOFs), with numerous applications in gas storage and separation,14–17 adsorption,18–21 catalysis,22–25 and drug delivery.26
The largest Zr oxo cluster, reported to date, is a {Zr70} cluster,27 while the majority of the reported clusters are mainly of lower nuclearity such as {Zr4}, {Zr6}, and {Zr12}.28 To the best of our knowledge, there is only one example of a {Zr5} oxo cluster ever reported with a square pyramidal arrangement of the five zirconium atoms.29 In general, the chemistry of the group IV metals remains underdeveloped.28
Oximes are strong binders to hard metals in their high oxidation states and protect them from hydrolysis. Reactivity studies of oximes with early transition metals and formation of relevant complexes is underexplored in comparison with the oximate derivatives of the later transition metals.30 The hydrolytically stable31 ligand (2Z-6Z)-piperidine-2,6-dione dioxime (H3pidiox, Scheme 1A) acts mainly as a tridentate chelator forming two fused five-membered chelate rings, with various degrees of deprotonation, in complexes of metals like Ti, V, U, and Fe32–36 (Scheme 1B) and as chelating–bridging ligand (Scheme 1C).32
 |
| | Scheme 1 The ligand used in this study (A) and the chelating (B) and the chelating–bridging coordination modes of H3pidiox (C). | |
Recently, our group reported the isolation of a new hexanuclear {Ti6O5} polyoxo-titanium cluster using H3pidiox as the ligand.32 In the present study, we report the synthesis of the mononuclear zirconium complex [ZrIV(η1,η1,η2-H2pidiox-O,N,O′)2(OH2)2]Cl2·H2O·CH3OH (1) and the pentanuclear ZrOC [ZrIV5(μ2-OH)4(OH2)4(μ2–η1,η1,η2-Hpidiox-O,N,O′)4(η1,η1,η1-Hpidiox-O,N,O′)4]·5KCl·3CH3OH·8H2O (2). Compound 1 was synthesized at very acidic conditions (pH = 0), while the cluster 2 at higher pH values (pH = 2). Interestingly, 1 is spontaneously converted to 2 in methanol solution and 2 to 1 upon addition of HCl to the methanol solution of 2 as it was evidenced by NMR spectroscopy and ESI-MS spectrometry. The cluster 2 is the second example of a pentanuclear ZrOC reported to date with four tetrahedrally arranged zirconium atoms with the fifth zirconium atom occupying the center of the tetrahedron.
Materials and methods
Experimental details
All chemicals and solvents were purchased from Sigma-Aldrich and Merck, were of reagent grade, and were used without further purification. H3pidiox was synthesized according to literature.32 C, H, and N analyses were conducted by the microanalytical service of the School of Chemistry, the University of Glasgow.
Synthesis of [ZrIV(η1,η1,η2-H2pidiox-O,N,O′)2(OH2)2]Cl2·H2O·CH3OH (1)
To a stirred moist methyl alcohol solution (4 ml) were successively added H3pidiox (122.8 mg, 0.858 mmol), and ZrCl4 (100 mg, 0.429 mmol). Then, the solution was filtered (pH = 0), and the colorless filtrate was kept at room temperature (∼20 °C) for 5–6 days, during which period 125 mg of white crystals of compound 1 were formed. The crystals were filtered off and dried at an ambient temperature (∼20 °C). (Yield: 55%, based on ZrCl4). Elemental anal. calc. for (C11H26N6O8Cl2Zr, Mr = 532.487 g mol−1): C, 24.81; H, 4.92; N, 15.78; found: C, 24.78; H, 4.82; N, 15.63.
Synthesis of [ZrIV5(μ2-OH)4(OH2)4(μ2–η1,η1,η2-Hpidiox-O,N,O′)4(η1,η1,η1-Hpidiox-O,N,O′)4]·5KCl·3CH3OH·8H2O (2)
To a stirred moist methyl alcohol solution (4 ml) were successively added H3pidiox (122.8 mg, 0.858 mmol), and ZrCl4 (100 mg, 0.429 mmol). Then, upon addition of solid KOH (48.1 mg, 0.858 mmol) in one portion a white precipitate was formed which was filtered off and the colorless filtrate (pH = 2) was kept at 4–5 °C for 9–10 days during which period 94.0 mg of white crystals of compound 2 along with other species (insoluble unidentified material) were formed. The crystals and the solid were filtered off and crystals of compound 2 were chosen under the microscope. Their identity was verified by single crystal X-ray structure analysis. Yield, 30 mg (15%, based on ZrCl4) Elemental anal. calc. for (C43H96N24O35Cl5K5Zr5, Mr = 2338.234 g mol−1): C, 22.08; H, 4.13; N, 14.37; found: C, 22.12; H, 4.07; N, 14.42.
X-ray crystallographic details
Suitable single crystals were selected and mounted onto a rubber loop using Fomblin oil. Single-crystal X-ray diffraction data of 1 was recorded on a Bruker Apex II Quazar CCD diffractometer (Bruker, Bremen, Germany) (λ (Mo Kα) = 0.71073 Å) at 150 K equipped with a graphite monochromator. Data collection and reduction were performed using the Apex2 software package. In the case of ZrOC 2, single crystal X-ray diffraction data of the compound was collected using an Xcalibur Oxford diffractometer equipped with a Sapphire 3 CCD detector and a 4-cycle Kappa geometry goniometer, using enhanced Mo Kα (λ = 0.71073 Å) at 150 K equipped with a graphite monochromator. Analytical absorption correction was applied using CrysAlis RED software. CrysAlis CCD and CrysAlis RED software were used for data collection and data reduction/cell refinement respectively. Structure solution and refinement were carried out with SHELXS-2014![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 45 and SHELXL-201446 using the WinGX software package.47 Corrections for the incident and diffracted beam absorption effects were applied using empirical absorption corrections.48 All the non-H atoms were refined anisotropically. Solvent molecule sites were found and included in the refinement of the structures. In the case of compound 2 the solvent area within the unit cell suffers from severe disorder issues. Thus, the unambiguous composition was determined by means of elemental analyses. Final unit cell data and refinement statistics for compounds 1 and 2 are collated in Table 1. The crystallographic data for compounds 1 and 2 (CCDC 1: 2113958;†2: 2113960†).
45 and SHELXL-201446 using the WinGX software package.47 Corrections for the incident and diffracted beam absorption effects were applied using empirical absorption corrections.48 All the non-H atoms were refined anisotropically. Solvent molecule sites were found and included in the refinement of the structures. In the case of compound 2 the solvent area within the unit cell suffers from severe disorder issues. Thus, the unambiguous composition was determined by means of elemental analyses. Final unit cell data and refinement statistics for compounds 1 and 2 are collated in Table 1. The crystallographic data for compounds 1 and 2 (CCDC 1: 2113958;†2: 2113960†).
Table 1 Crystal data and details of the structure determination and refinement for 1 and ZrOC 2
| |
1
|
2
|
| Formula |
C11H25.60Cl2N6O8Zr |
C85.85H112Cl9.60K8.60N48O69.94Zr10 |
| Formula weight |
532.09 g mol−1 |
4524.33 g mol−1 |
| Temperature |
174(2) K |
150(2) K |
| Wavelength |
71.073 pm |
71.073 pm |
| Crystal system |
Orthorhombic |
Monoclinic |
| Space group |
Pbca
|
P21/n |
| Unit cell dimensions |
a = 1590.10(6) pm |
a = 1445.6(4) pm |
|
b = 1421.21(5) pm |
b = 4026.8(10) pm |
|
c = 1768.33(9) pm |
c = 1468.2(4) pm |
|
α = β = γ 90° |
α = β = 90° |
|
|
β = 91.042(4)° |
| Volume |
3970.9(3) nm3 |
8545(4) nm3 |
|
Z
|
8 |
2 |
| Density (calculated) |
1.780 g cm−3 |
1.758 g cm−3 |
| Absorption coefficient |
0.876 mm−1 |
1.042 mm−1 |
|
F(000) |
2172.8 |
4499.0 |
| Crystal size |
0.100 × 0.080 × 0.070 mm3 |
0.200 × 0.180 × 0.150 mm3 |
| Theta range for data collection |
2.562 to 53.506° |
1.477 to 26.445° |
| Index ranges |
−26 < =h < = 30 |
−18 < =h < = 18 |
| −28 < =k < = 22 |
−50 < =k < = 50 |
| −36 < =l < = 27 |
−18 < =l < = 18 |
| Reflections collected |
46305 |
65170 |
| Independent reflections |
17472 |
16786 [R(int) = 0.0970] |
| Completeness to theta = 25.242° |
99.9% |
100.0% |
| Absorption correction |
Multi-Scan |
Empirical |
| Max. and min. transmission |
0.941 and 0.919 |
0.850 and 0.805 |
| Refinement method |
Full-matrix least-squares on F2 |
| Goodness-of-fit on F2 |
1.044 |
1.020 |
| Final R indices [I > 2sigma(I)] |
R
1 = 0.0345, wR2 = 0.0783 |
R
1 = 0.0581, wR2 = 0.1508 |
NMR experimental details
All NMR samples were prepared by the dissolution of the crystals of the complex in CD3OD at room temperature just prior to NMR spectrometric determinations. NMR spectra were recorded on a Bruker Avance III 300 MHz spectrometer. 2D {1H,13C} grHSQC and 2D {1H,13C} grHMQC spectra were obtained by using standard pulse sequences of Bruker Topspin 3.0 software. The spectra were acquired using, for 2D {1H} grHSQC 128 increments (with 16 scans each) covering 5.0 ppm at F2 dimension and 150 ppm at F1, and for 2D {1H,13C} grHMQC 128 increments (with 16 scans each) covering 5.0 ppm at F2 dimension and 200 ppm at F1. Standard DCl solutions (0.10 M) were prepared by adding concentrated HCl in CD3OD. Assignments of the peaks were confirmed also by spike experiments.
ESI-MS experimental details
All MS data were collected using a Q-trap, time-of-flight MS (Maxis Impact MS) instrument supplied by Bruker Daltonics Ltd. The detector was a time-of-flight, micro-channel plate detector and all data was processed using the Bruker Daltonics Data Analysis 4.1 software, whilst simulated isotope patterns were investigated using Bruker Isotope Pattern software and Molecular Weight Calculator 6.45. The calibration solution used was Agilent ES tuning mix solution, Recorder No. G2421A, enabling calibration between approximately 100 m/z and 2000 m/z. This solution was diluted 60:1 with MeCN. Samples were dissolved in MeOH and introduced into the MS via direct injection at 180 μL h−1. The ion polarity for all MS scans recorded was negative, at 180 °C, with the voltage of the capillary tip set at 4000 V, endplate offset at −500 V, funnel 1 RF at 300 Vpp and funnel 2 RF at 400 Vpp.
Results and discussion
Synthesis of 1 and 2
The synthesis of the zirconium(IV) compound 1 and ZrOC 2 takes place according to eqn (1) and (2). The almost quantitative transformation of 1 to 2 occurs spontaneously upon dissolution of 1 to methanol according to eqn (3) and vice versa upon addition of HCl to the methanol solution of ZrOC 2 (Scheme 2) according to the multi-nuclear NMR and ESI-MS measurements (vide infra). Thus, it is crystal-clear that at very low pH the mononuclear compound is formed containing two singly deprotonated ligands and at higher pH the pentanuclear ZrOC is assembled with a further deprotonation of the ligands. The formation of compound 1 at very low pH values means that the ligand H3pidiox could be used as a sequestering agent for zirconium.
 |
| | Scheme 2 The formation of the ZrOCs 2, 3, and 4 starting from 1. The molecular formulae for the hexanuclear ZrOCs 3 and 4 are suggested based on NMR and ESI-MS studies. | |
Sequential addition of 2.5 and 20 equivalents of KOH to the methanol solution of 2 results in the formation of the new hexanuclear ZrOCs (vide infra) 3 (see Fig. 11A) and 4 (see Fig. 11B; eqn (4)) respectively (Scheme 2). The molecular formulae of 3 and 4 (Scheme 2) were based on the ESI-MS and ESI-MS/NMR data respectively. Efforts to isolate the ZrOCs 3 and 4 have been unsuccessful thus far.
| | 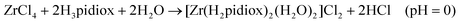 | (1) |
| |  | (2) |
| |  | (3) |
| |  | (4) |
Description of the structures
The molecular structure of the cation [ZrIV(η1,η1,η1-H2pidiox-O,N,O′)2(OH2)2]2+ of 1 is presented in Fig. 1. Interatomic distances and bond angles relevant to the Zr(1) coordination sphere are listed in Table 2. The six donor atoms of the two singly deprotonated H2pidiox− ligands and the two oxygen atoms of the two water molecules surrounding the zirconium(IV) atom are disposed in a bicapped severely distorted octahedral geometry (Fig. S1†). The two oxime O(7), O(8) oxygen atoms, the endocyclic nitrogen atom N(5) of one H2pidiox− ligand and the O(18) of a water molecule occupy the equatorial plane. The axial positions are defined from the oxime O(9) of the second chelate ligand and a water oxygen atom, O(17). The oxime oxygen atom O(10) and the endocyclic nitrogen atom N(2) are capping the trigonal faces O(8), O(17), O(18) and O(8), O(9), O(18) of the octahedron, respectively. Each of the two singly-deprotonated H2pidiox− ligands forms two five-membered fused chelate rings and is meridionally ligated to the zirconium(IV) center. The two H2pidiox− ligands in 1 are almost perpendicular to each other [89.21(8)°].
 |
| | Fig. 1 ORTEP plot (50% probability level) of the cation [ZrIV(η1,η1,η1-H2pidiox-O,N,O′)2(OH2)2]2+ of 1 with a partial labeling scheme. | |
Table 2 Interatomic distances (Å) and angles (°) relevant to the Zr(1) coordination sphere for the mononuclear complex 1
|
Bond distances
|
| Zr(1)–N(5) |
2.255(3) |
Zr(1)–O(9) |
2.182(2) |
| Zr(1)–N(2) |
2.270(2) |
Zr(1)–O(10) |
2.191(2) |
| Zr(1)–O(7) |
2.192(2) |
Zr(1)–O(17) |
2.212(3) |
| Zr(1)–O(8) |
2.146(3) |
Zr(1)–O(18) |
2.224(3) |
|
Bond angles
|
| O(8)–Zr(1)–O(9) |
93.17(11) |
O(17)–Zr(1)–O(18) |
93.54(11) |
| O(8)–Zr(1)–O(10) |
79.64(10) |
O(8)–Zr(1)–N(5) |
68.40(10) |
| O(9)–Zr(1)–O(10) |
135.21(8) |
O(9)–Zr(1)–N(5) |
76.19(9) |
| O(8)–Zr(1)–O(7) |
136.09(9) |
O(10)–Zr(1)–N(5) |
137.05(10) |
| O(9)–Zr(1)–O(7) |
81.53(9) |
O(7)–Zr(1)–N(5) |
68.00(9) |
| O(10)–Zr(1)–O(7) |
132.69(9) |
O(17)–Zr(1)–N(5) |
81.49(10) |
| O(8)–Zr(1)–O(17) |
89.47(12) |
O(18)–Zr(1)–N(5) |
135.72(9) |
| O(9)–Zr(1)–O(17) |
154.73(9) |
O(8)–Zr(1)–N(2) |
84.35(11) |
| O(10)–Zr(1)–O(17) |
69.96(9) |
O(9)–Zr(1)–N(2) |
67.17(8) |
| O(7)–Zr(1)–O(17) |
79.22(9) |
O(10)–Zr(1)–N(2) |
68.14(9) |
| O(8)–Zr(1)–O(18) |
155.86(9) |
O(7)–Zr(1)–N(2) |
130.89(9) |
| O(9)–Zr(1)–O(18) |
94.22(10) |
O(17)–Zr(1)–N(2) |
138.08(9) |
| O(10)–Zr(1)–O(18) |
78.94(9) |
O(18)–Zr(1)–N(2) |
77.45(10) |
| O(7)–Zr(1)–O(18) |
67.85(9) |
N(5)–Zr(1)–N(2) |
132.64(10) |
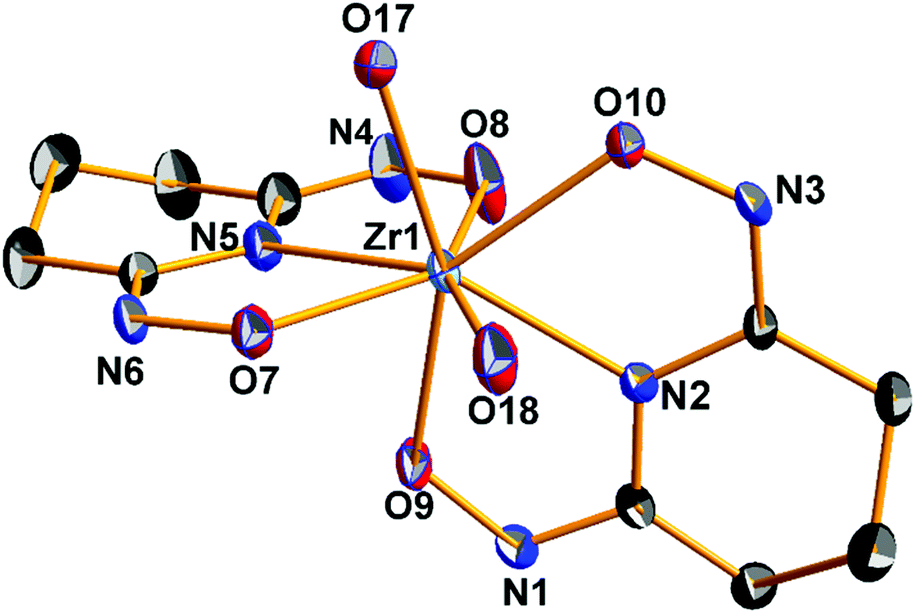
The single-crystal X-ray analysis of the ZrOC 2 reveals a pentanuclear molecular structure (Fig. 2) containing a {ZrIV5} cluster core. Selected bond lengths and angles are listed in Table 3. Bond valence sum calculation (BVS) for the μ2-O(17), O(19), O(23), and O(24) oxygen atoms indicate that are monoprotonated, while in the case of the terminal oxygen atoms O(18), O(20), O(22), and O(25) found to be doubly protonated (aqua ligands).
 |
| | Fig. 2 ORTEP plot (50% probability level) of the neutral pentanuclear ZrOC 2 with a partial labeling scheme. Hydrogen atoms and molecules of crystallization have been omitted for clarity. | |
Table 3 Interatomic distances (Å) and angles (°) relevant to the Zr(1/2) coordination spheres for the pentanuclear ZrOC 2
|
Bond distances
|
| Zr(1)–O(1) |
2.232(5) |
Zr(2)–N(2) |
2.279(7) |
| Zr(1)–O(10) |
2.250(6) |
Zr(2)–N(5) |
2.240(6) |
| Zr(1)–O(11) |
2.222(5) |
Zr(2)–O(7) |
2.162(6) |
| Zr(1)–O(17) |
2.160(6) |
Zr(2)–O(8) |
2.190(6) |
| Zr(1)–O(19) |
2.176(6) |
Zr(2)–O(9) |
2.161(6) |
| Zr(1)–O(21) |
2.217(5) |
Zr(2)–O(10) |
2.287(5) |
| Zr(1)–O(23) |
2.163(6) |
Zr(2)–O(17) |
2.142(6) |
| Zr(1)–O(24) |
2.155(6) |
Zr(2)–O(18) |
2.214(6) |
|
Bond angles
|
| O(1)–Zr(1)–O(10) |
93.6(2) |
N(2)–Zr(2)–N(5) |
132.4(2) |
| O(1)–Zr(1)–O(11) |
157.8(2) |
N(2)–Zr(2)–O(7) |
135.1(2) |
| O(1)–Zr(1)–O(17) |
82.6(2) |
N(2)–Zr(2)–O(8) |
48.4(2) |
| O(1)–Zr(1)–O(19) |
80.7(2) |
N(2)–Zr(2)–O(9) |
68.8(2) |
| O(1)–Zr(1)–O(21) |
89.8(2) |
N(2)–Zr(2)–O(10) |
66.5(2) |
| O(1)–Zr(1)–O(23) |
138.1(2) |
N(2)–Zr(2)–O(17) |
130.2(2) |
| O(1)–Zr(1)–O(24) |
63.7(2) |
N(2)–Zr(2)–O(18) |
80.3(2) |
| O(10)–Zr(1)–O(11) |
90.9(2) |
N(5)–Zr(2)–O(7) |
68.7(2) |
| O(10)–Zr(1)–O(17) |
64.2(2) |
N(5)–Zr(2)–O(8) |
68.4(2) |
| O(10)–Zr(1)–O(19) |
138.2(2) |
N(5)–Zr(2)–O(9) |
80.3(2) |
| O(10)–Zr(1)–O(21) |
157.9(2) |
N(5)–Zr(2)–O(10) |
131.8(2) |
| O(10)–Zr(1)–O(23) |
81.9(2) |
N(5)–Zr(2)–O(17) |
85.4(2) |
| O(10)–Zr(1)–O(24) |
80.0(2) |
N(5)–Zr(2)–O(18) |
139.7(2) |
| O(11)–Zr(1)–O(17) |
79.8(2) |
O(7)–Zr(2)–O(8) |
137.0(2) |
| O(11)–Zr(1)–O(19) |
81.5(2) |
O(7)–Zr(2)–O(9) |
80.1(2) |
| O(11)–Zr(1)–O(21) |
94.2(2) |
O(7)–Zr(2)–O(10) |
135.4(2) |
| O(11)–Zr(1)–O(23) |
64.0(2) |
O(7)–Zr(2)–O(17) |
83.0(2) |
| O(11)–Zr(1)–O(24) |
138.5(2) |
O(7)–Zr(2)–O(18) |
71.1(2) |
| O(17)–Zr(1)–O(19) |
74.0(2) |
O(8)–Zr(2)–O(9) |
94.1(2) |
| O(17)–Zr(1)–O(21) |
137.9(2) |
O(8)–Zr(2)–O(10) |
76.4(2) |
| O(17)–Zr(1)–O(23) |
129.7(2) |
O(8)–Zr(2)–O(17) |
92.3(2) |
| O(17)–Zr(1)–O(24) |
128.8(2) |
O(8)–Zr(2)–O(18) |
151.6(2) |
| O(19)–Zr(1)–O(21) |
63.9(2) |
O(9)–Zr(2)–O(10) |
135.3(2) |
| O(19)–Zr(1)–O(23) |
128.6(2) |
O(9)–Zr(2)–O(17) |
160.9(2) |
| O(19)–Zr(1)–O(24) |
130.6(2) |
O(9)–Zr(2)–O(18) |
95.5(2) |
| O(21)–Zr(1)–O(23) |
81.1(2) |
O(10)–Zr(2)–O(17) |
63.8(2) |
| O(21)–Zr(1)–O(24) |
82.0(2) |
O(10)–Zr(2)–O(18) |
78.0(2) |
| O(23)–Zr(1)–O(24) |
74.6(2) |
O(17)–Zr(2)–O(18) |
87.3(2) |
The four outer zirconium(IV) atoms adopt a distorted tetrahedral arrangement (Fig. 3) and in the center of the Zr4 distorted tetrahedron is located the fifth zirconium(IV) atom coordinated to four μ2-bridging oxime oxygen atoms and four μ2-OH− groups (Scheme 3) in a bicapped distorted octahedral O8 coordination (Fig. S2A†). The distorted octahedron is defined by the axial O(1), O(11) and the equatorial O(10), O(17), O(19), O(21) oxygen atoms. The trigonal faces of the octahedron O(10), O(11), O(21) and O(10), O(1), O(21) are capped by the oxygen atoms O(23) and O(24) respectively (Fig. S2A†). Surprisingly, the only other discrete pentanuclear ZrOC reported thus far is the compound {Zr5O4[(CH3)2BrCCO2]10(OnPr)2(nPrOH)4} in which the five zirconium(IV) atoms form a square pyramidal arrangement.27 The coordination sphere of the five zirconium(IV) atoms in 2 is not uniform; while the four outer zirconium(IV) atoms are coordinated by six oxygen and two endocyclic nitrogen atoms, the central Zr(1) is coordinated by eight oxygen atoms (Scheme 3).
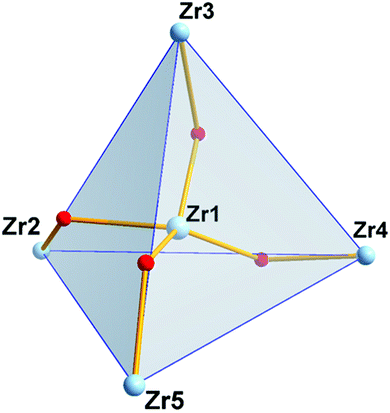 |
| | Fig. 3 The distorted tetrahedral arrangement of [Zr5(μ2-OH)4] structural unit. | |
 |
| | Scheme 3 The molecular drawing of the ZrOC [ZrIV5(μ2-OH)4(OH2)4(μ2–η1,η1,η2-Hpidiox-O,N,O′)4(η1,η1,η1-Hpidiox-O,N,O′)4]. | |
The central zirconium(1) polyhedron is edge shared with each one of the four peripheral zirconium(IV) polyhedra through a μ2-oxime oxygen atom and a μ2-OH−group (Fig. 4).
 |
| | Fig. 4 Polyhedral representation of the neutral pentanuclear ZrOC 2. | |
The central Zr(1) atom shows two sets of Zr–O bonds with mean Zr–O bond lengths of 2.164(6) Å for the bridging μ2-OH− groups and 2.230(8) Å for the bridging μ2–O− atoms of oximes. The outer Zr atoms are bonded to two di-deprotonated Hpidiox2− ligands, one of which acts as a tridentate-O,N,O chelate through the deprotonated oxime oxygens and the endocyclic nitrogen forming two five-membered fused chelate rings and the other one acts as a chelate-O,N,O′ bridging through the one oxime atom (Scheme 3) rendering the two Hpidiox2− ligands almost perpendicular to each other [89.9(1)°]. The coordination sphere of the outer zirconium atoms is completed by a water molecule and a μ2-OH− group leading to an eight-coordinate N2O6 coordination sphere with bicapped distorted octahedral geometry (Fig. S2B†). The distorted octahedron for Zr(2) is defined by the axial O(17), O(9) and the equatorial O(18), O(7), N(5), O(8) atoms. The O(18), O(9), O(8) and O(18), O(17), O(8) faces of octahedron are capped by the N(2) and O(10) atoms respectively (Fig. S2B†).
NMR spectroscopy
The 1H NMR spectrum of the pentanuclear ZrOC 2 in solution (CD3OD) is shown in Fig. 5(a), and its 13C and 1H NMR peaks are listed in Table 4.
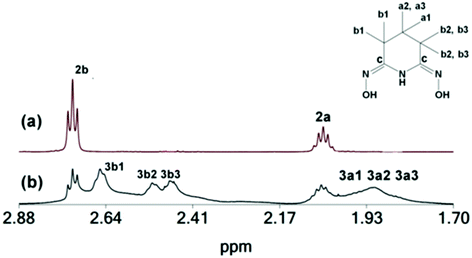 |
| | Fig. 5
1H NMR spectrum of (a) the crystals of 2, (b) of the product isolated from the reaction of ZrCl4, H3pidiox and KOH in CH3OH and the labelling of the carbon and hydrogen atoms of the ligand H3pidiox. | |
Table 4
13C and 1H (in parentheses) chemical shifts (ppm) of the ZrOCs 2, 3, and 4 in solution (CD3OD) and of the ligand H3pidiox
|
13C (1H)/ppm |
2
|
3
|
4
|
H3pidiox |
| C(a) |
18.10 (2.064) |
18.50 (2.070, 1.967, 1.861) |
20.30 (1.895, 1.662) |
19.02 (1.752) |
| C(b) |
20.35 (2.739) |
20.48, 20.49, 22.32 (2.657, 2.517, 2.502) |
23.28 (2.396) |
25.31 (2.368) |
| C(c) |
156.4 |
|
|
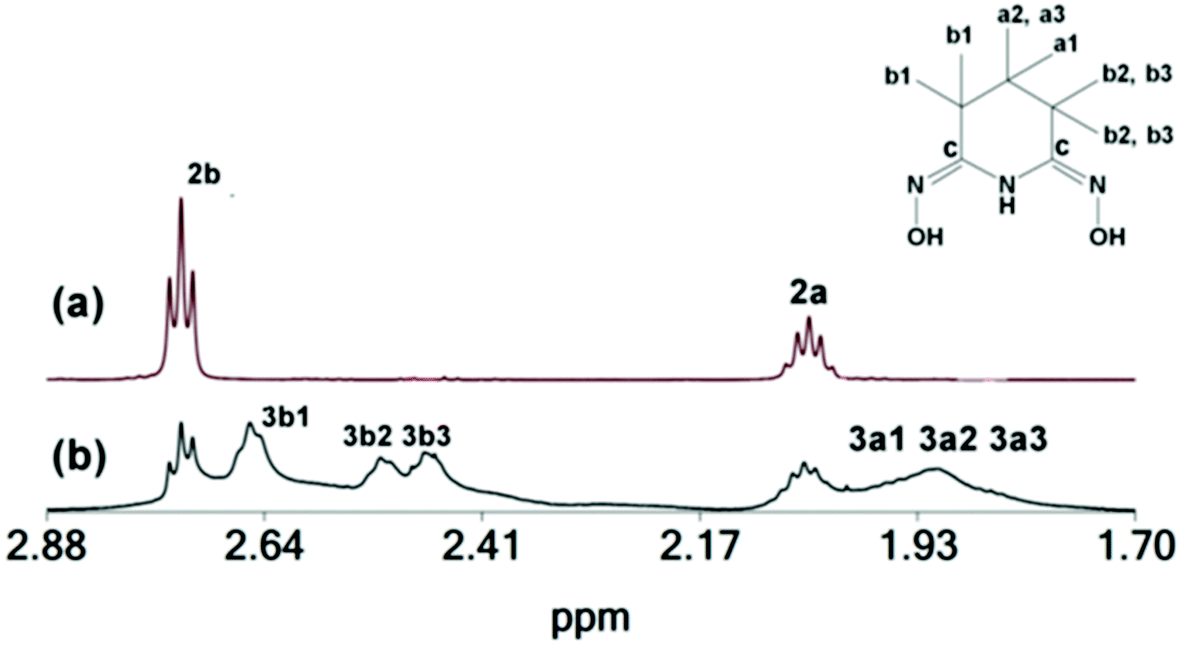
146.0 |
The solid isolated from the reaction of ZrCl4, H3pidiox and KOH in CH3OH is a mixture, of two species 2 and 3 [Fig. 5(b)], from which the single crystals of 2, were separated manually under a microscope and characterized in the solid state (X-ray, elemental analysis) and in solution [NMR, Fig. 5(a), and ESI-MS spectrometry]. The second component of the solid, complex 3 was characterized by 2D NMR spectra in solution (CD3OD) (Fig. S3–S6†). The spectra show that 3 contains two types of Hpidiox2− ligands. Each ligand has two sets of peaks for the Hb (3b1, 3b2 and 3b1, 3b3) and two sets of peaks for the Ha protons (3a1, 3a2 and 3a1, 3a3); see Fig. 5. The 2D{1H} grEXSY spectroscopy revealed that the 3 is fluctional and the environments of 3b1 protons exchange with those of 3b2 and 3b3. However, between the two Hpidiox2− ligands of 3 there is no exchange process supporting an intramolecular mechanism.
The 1H NMR spectrum of 1 in solution (CD3OD) gave the same peaks as 2 (Fig. 6). Apparently, dissolution of 1 in CH3OH results in the formation of 2 (in line with the MS measurements; vide infra), because the acidity of the CH3OH solution of 1 is less than the acidity in the reaction mixture during the synthesis of 1. However, according to eqn (3) (vide supra) conversion of 1 to 2 results in the release of two H3pidiox molecules, but the 1H NMR spectrum of 1 in CD3OD shows only the peaks of 2, suggesting that 2 and H3pidiox at this acidic condition are in fast exchange and the peaks of 2 and H3pidiox have been collapsed in one set of peaks. The hypothesis of the peaks’ overlap was confirmed by the 2D {1H,13C} grHSQC NMR spectroscopy (Fig. 6). The 2D {1H,13C} grHSQC spectrum of the CD3OD solution of 1 resolved the 1H NMR peaks to two major species assigned to 2 {(δ13C 20.71[H(b)] and 18.08[H(a)] ppm} and H3pidiox {δ13C 18.05[H(b)] and 20.21[H(a)] ppm}.
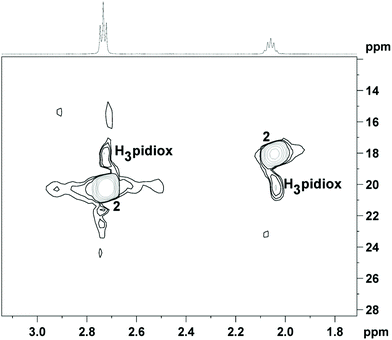 |
| | Fig. 6 2D {1H, 13C} grHSQC NMR spectrum of 1 in solution (CD3OD, 4.00 mM). | |
To further investigate the speciation of the zirconium(IV) species in CD3OD solution of 1, NMR experiments were carried out by adding various quantities of DCl to the solution of 1 (CD3OD) (Fig. 7). Addition of 1.00 mM DCl into the CD3OD solution of 1 results in an increase of the intensity of the 1H NMR peaks attributed to either an increase of the exchange rate of the couple H3pidiox/2 or a decrease of the energy difference (Δν) between the peaks of H3pidiox and 2 or both. Addition, of higher quantities of DCl up to 160 mM results in a shift of both Hb and Ha 1H NMR peaks to higher field and broadening, assigned to the formation of 1 which is in fast exchange with both 2 and H3pidiox. Addition of 240 mM of DCl into the CD3OH solution of 1 (4.00 mM) results in the dissociation of one of the H2pidiox− from 1. Addition of more than 1280 mM DCl results in full collapse of the 1H NMR spectra due to the fast exchange between the zirconium species and the free ligand.
 |
| | Fig. 7
1H NMR spectra of 1 in solution (CD3OD, 4.00 mM, first bottom spectrum) and after the addition of various quantities of DCl (1–1280 mM). | |
The changes of the 1H NMR spectra of 1 in solution (CD3OD, 4.00 mM) vs. time after the addition of more than 240 mM DCl are shown in Fig. S7.† Three hours after the addition of 1280 mM DCl into the CD3OD solution of 1 (4.00 mM), compound 1 has been decomposed to [ZrIV(H2pidiox)(H2O)X]3+ and H3pidiox. Fifteen hours later the peaks of the free ligand disappeared, and the spectrum shows peaks from four unknown molecules at higher field assigned to the decomposition products of H3pidiox.
The 1H NMR spectra of 1 in solution (CD3OD) after the addition of various quantities of KOD are shown in Fig. 8. Addition of 2.0 equivalents of KOD to a CD3OD solution of 1 results in the formation of the zirconium(IV) compound 3, which is the major species in the solution and further addition of ≥20 equivalents of KOD to it results in the formation of a new ZrOC 4. The 1H NMR pattern of 4 resembles the pattern of the previously reported hexanuclear titanium(IV) cluster32 [TiIV6(μ3-O)2(μ2-O)3(CH3O)6(μ2–η1,η1,η2-Hpidiox-O,N,O′)4(μ2–η1,η1,η2-pidiox-O,N,O′)2]6− (5) and the only difference is that the 1H NMR peaks of 4 are shifted towards higher (∼0.6 ppm) and 13C peaks to lower field (∼2.5 ppm) in comparison to its titanium(IV) analogue (Fig. S8†). The 2D {1H} grNOESY spectrum (Fig. S9†) shows strong NOE interactions between 4a1 and 4a2 and between 4b and 4a protons, supporting our interpretation that 4a1 and 4a2 are geminal protons with a different environment created by a structure like the hexanuclear cluster 5. However, conversion of 2 to 4 will result in the release of free H3pidiox ligands (eqn (4)), but the 1H NMR spectra do not show any peaks originating from the free ligand. A brown colour which is developed into the solution with the time after the addition of the base in the colourless solution of 1 suggests the decomposition of the ligand probably to species that cannot be detected easily by 1H NMR. Multiple peaks at 2.2 ppm [Fig. 8, solution of 1 (4.0 mM) + KOD (160 mM)] might be originated from the decomposition of the free ligand.
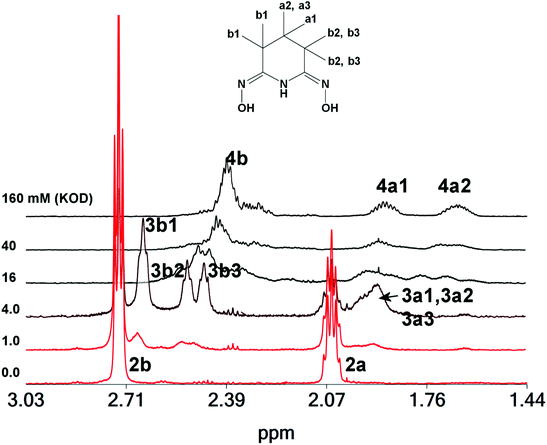 |
| | Fig. 8
1H NMR spectra of 1 in solution (CD3OD, 4.00 mM, first bottom spectrum) and after the addition of various quantities of KOD (1–160 mM). | |
We conclude that 4 has a similar structure to its hexanuclear titanium(IV) analogue 5. The differences in the chemical shifts of the 1H and 13C NMR peaks between 4 and 5 is probably due to the ligation of Hpidiox2− to ZrIV, a second row transition metal, in comparison to TiIV, which is a first row transition metal.
ESI-MS spectrometry
Electrospray ionization mass spectrometry facilitated not only the identification37–42 and confirmation of the structural stability of the species in solution but helped us to demonstrate the pH driven dis-assembly/re-assembly process between the monomeric 1 and pentanuclear ZrOC 2 reported in this work.
The ESI-MS studies were performed in methanol in positive ionization mode. The observation of the doubly charged higher m/z value distribution envelopes correspond to the pentanuclear moiety resulting from the variable number of protons, counterions and solvent molecules (see Table 5) as shown in Fig. 9. Upon dissolution of the compound 1 in methanol (less acidic environment) it reorganizes and re-assembles into the pentanuclear {Zr5} species. Additionally, transition metal clusters are generally susceptible to redox processes under the employed ionization conditions which can occasionally induce partial fragmentation of the species. This type of behavior is quite common in ESI-MS solution studies of compounds.40–44 In this case, the region of higher m/z values is populated by a series of + 2 charged distribution envelopes assigned to the intact {Zr5} ZrOC (Fig. 9). In this case, a group of distribution envelopes are clustered within the range of ca. 600–1200 m/z, while the envelope centered at 560.99 m/z corresponds to the (Zr4} fragment of 2. Furthermore, the isotopic distribution envelope centered at 1120.97 m/z can be assigned to the intact {ZrIV5(C5H7N3O2)8 O8Cl4H18(OH2)4(OHCH3)3}2+ cationic ZrOC (Table 5). In the range of ca. 200–400 m/z values, traces of the monomeric cluster 1 have been identified at 373.00 m/z and assigned as a singly charged [ZrIV(C5H7O2N3)2H]+ probably what has been left over following the in situ assembly of the {Zr5} moiety 2. At this point, it is worth noting that crystals of 2 gave the same mass spectrum to that shown in Fig. 9. Interestingly, upon acidification of the methanolic solution with two drops of concentrated HCl, induces the conversion of cluster 2 back to 1via a dis-assembly process. This assembly/dis-assembly process is fully reversible. Fig. 10 shows two envelopes centred at 301.87 and 372.98 m/z values which correspond to the intact mononuclear cluster 1, [ZrIV(C5H7O2N3)2H]+, and the fragmentation product formed during the ionisation process, [ZrIV(C5H7O2N3)(OH2)3(OH)]+.
 |
| | Fig. 9 Positive ion mass spectrum of 1 and its re-assembly in a methanol solution. The isotopic distribution envelopes correspond to doubly charged species centred at 1120.97, 933.98, 746.98, 676.44 and 560.99 m/z and can be attributed to the intact pentanuclear species and its fragments. Remainers from the structural transformations of the monomeric complex [ZrIV(C5H7O2N3)2H]+ can be observed from the left-over traces centered at 373.00 m/z. | |
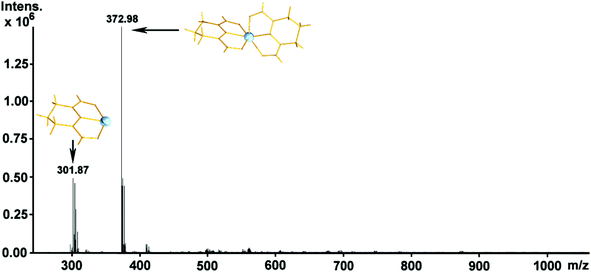 |
| | Fig. 10 Positive ion mass spectrum of 1 in CH3OH acidified with two drops of concentrated aqueous HCl which exhibits two characteristic singly charged isotopic distribution envelopes centered at 301.87 and 372.98 m/z with the formulae of [ZrIV(C5H7O2N3)(OH2)3(OH)]+ and [ZrIV(C5H7O2N3)2H]+ respectively. | |
Table 5 Representation of the experimentally identified and simulated m/z values of distribution envelopes of the {Zr5} ZrOC
| Exp. |
Theor. |
Charge |
Formula |
| 1120.97 |
1121.0 |
+2 |
{Zr5IV(C5H7N3O2)8O8Cl4H18(OH2)4(HOCH3)3}2+ |
| 933.98 |
933.99 |
+2 |
{Zr5IV(C5H7N3O2)8O7Cl3H15(OH2)(HOCH3)}2+ |
| 746.98 |
746.95 |
+2 |
{Zr5IV(C5H7N3O2)6O8H10(OH2)3}2+ |
| 676.44 |
676.45 |
+2 |
{Zr5IV(C5H7N3O2)5O4(HOCH3)4}2+ |
| 560.99 |
1155.92 |
+2 |
{Zr4IV(C5H7N3O2)4O4(HOCH3)4H2}2+ |
Fig. 11 shows the real time monitoring of the structural evolution process as a function of the added KOH to the solution (CH3OH) of {Zr5} cluster 2. More specifically, as a function of increased amount of KOH into the reaction mixture the pentanuclear species undergoes a major structural re-arrangement and re-assembles to the hexanuclear {Zr6} ZrOC as shown in Fig. 11B. Fig. 11A shows the growth stage where there is a mixture of incomplete intermediate {Zr6} cluster e.g. {ZrIII6O5 (C5H7N3O2)4(HOCH3)2(OH2)3H2}2+3 and the final product namely {ZrIV6O5(C5H7N3O2)6(HOCH3)5(OH2)5}2+ (Table 6). Interestingly, evidence of the {Zr5} ZrOC can be identified within the range of 400–500 m/z values. The hexanuclear {Zr6} ZrOC retains its integrity within a range of KOH concentrations (2.5–35 eq.) as shown in Fig. 11C where a higher number of distribution envelopes can still be identified as the {Zr6} ZrOC. Reaching basic enough conditions (>35 eq. of KOH) the cluster is not able to retain its integrity anymore and only species of decomposition can be identified (Fig. 11D).
 |
| | Fig. 11 Real time monitoring of the structural evolution process as a function of the added KOH to the solution of {Zr5} species 2. After addition of: (A) 2.5 eq. of KOH (intermediate compound 3); (B) 25 eq. of KOH; (compound 4) (C) 35 eq. (compound 4 still present) and (D) 45 eq. of KOH respectively. | |
Table 6 Representation of the experimentally identified and simulated m/z values of distribution envelopes observed during the addition of KOH to the solution (CH3OH) of 2
| Exp. |
Theor. |
Charge |
Formula |
| 410.98 |
411.05 |
+1 |
{ZrIVO(C5H7N3O2)2(OH2)H}+ |
| 448.90 |
449.00 |
+2 |
{Zr2IIIO3(C5H7N3O2)(HOCH3)3H10}2+ |
| 486.90 |
487.10 |
+2 |
{Zr2IVO(C5H7N3O2)4(HOCH3)6(OH2)H4}2+ |
| 655.93 |
655.86 |
+2 |
{Zr6IIIO5(C5H7N3O2)4(HOCH3)2(OH2)3H2}2+ |
| 674.91 |
675.34 |
+2 |
{Zr2IIIZr4IVO5(C5H7N3O2)5(OH2)}2+ |
| 692.90 |
693.35 |
+2 |
{Zr2IIIZr4IVO5(C5H7N3O2)5(OH2)3}2+ |
| 712.37 |
712.37 |
+2 |
{Zr4IIIZr2IVO5(C5H7N3O2)5(OH2)5H2}2+ |
| 822.96 |
822.94 |
+2 |
{Zr4IIIZr2IVO5(C5H7N3O2)6(HOCH3)3(OH2)4H4}2+ |
| 843.92 |
843.94 |
+2 |
{Zr4IIIZr2IVO5(C5H7N3O2)6(HOCH3)5(OH2)3}2+ |
| 861.90 |
861.95 |
+2 |
{Zr6IVO5(C5H7N3O2)6(HOCH3)5(OH2)5}2+ |
| 880.97 |
880.97 |
+2 |
{Zr2IIIZr4IVO5(C5H7N3O2)6(HOCH3)3(OH2)7H2}2+ |
| 898.88 |
898.98 |
+2 |
{Zr2IIIZr4IVO5(C5H7N3O2)6(HOCH3)3(OH2)9H2}2+ |
| 936.81 |
937.02 |
+2 |
{Zr6IIIO5(C5H7N3O2)6(HOCH3)5(OH2)13H6}2+ |
| 994.79 |
994.87 |
+1 |
{Zr4IIIO(C5H7N3O2)4(HOCH3)(OH2)H}3+ |
| 1032.75 |
1032.89 |
+1 |
{Zr4IIIO(C5H7N3O2)4(HOCH3)(OH2)3H}3+ |
Conclusions
In summary, the mononuclear (1) zirconium(IV) compound and pentanuclear ZrOC (2) were synthesized by reacting the ligand H3pidiox with ZrCl4 in methanol (pH = 0) for 1 and H3pidiox, ZrCl4 and KOH (pH = 2) for 2 respectively. The pH is a crucial factor for the synthesis and interconversion of either 1 or 2. Single crystal X-ray structure analysis revealed that the pentanuclear cluster 2 constitutes the second discrete {Zr5} to be reported and the first one with a distorted tetrahedral arrangement of the four zirconium atoms and the fifth zirconium atom in the centre of the tetrahedron.
Dissolution of 1 in methanol results in the assembly of ZrOC 2 in the solution, and strong acidification of it dis-assembles into 1. The dis-assembly/re-assembly process was revealed and monitored by NMR spectroscopy and ESI-MS spectrometry. Moreover, addition of a base, such as KOH, in the methanol solution of 2 results in the gradual formation of two additional ZrIV species. More specifically, addition of 1–2 eq. of KOH to the solution of 2 creates mainly an asymmetric intermediate zirconium(IV) species 3, during the dis-assembly/re-assembly stage where incomplete {Zr6} cluster co-exists in solution. Addition of 2.5–25 equivalents of KOH complete the formation of the hexanuclear ZrOC 4 as evidenced by the real-time ESI-MS monitoring and NMR data. Interestingly, the acidity and basicity of the methanol solution can be used as a switch to control the speciation and structural evolution process towards the formation of higher nuclearity ZrIV/H3pidiox-based compounds.
ESI-MS proved to be crucial for the verification of the structural stability of the species in solution and revealed an interesting acid/base driven dis-assembly/re-assembly process associated with this family of clusters.
The extraordinary stability of 1 under very acidic condition as it was demonstrated by NMR and ESI-MS studies implies that H3pidiox is an excellent sequestering agent for zirconium and the subsequent construction of multinuclear molecular materials.
Author contributions
Conceptualization, T. A. K., A. D. K. and H. N. M.; synthesis of the ligand H3pidiox and of the zirconium(IV) compounds 1 and 2 S. S. P.; ESI-MS spectrometry H. N. M.; crystallography, H. N. M. and A. D. K.; NMR spectroscopy, S. H., M. G. P. and A. D. K.; writing-original draft preparation, T. A. K., H. N. M., A. D. K., M. G. P. and S. S. P.; writing—review and editing, T. A. K., H. N. M. and A. D. K.; supervision of all contributions, T. A. K., H. N. M. and A. D. K. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The research work was supported by the Hellenic Foundation for Research and Innovation (HFRI) under the HFRI PhD Fellowship grant (Fellowship Number: 1553). This work was co-funded by the European Regional Development Fund and the Republic of Cyprus through the Research and Innovation Foundation (Project: EXCELLENCE/1216/0515). We would like to thank EPSRC (EP/R01308X/1) and the University of Glasgow for supporting this work.
References
- T. Xu, X. Hou, Y. Wang, J. Zhang, J. Zhang and B. Liu, Dalton Trans., 2017, 46, 10185–10188 RSC.
- S. S. Passadis, M. G. Papanikolaou, A. Elliott, C. G. Tsiafoulis, A. C. Tsipis, A. D. Keramidas, H. N. Miras and T. A. Kabanos, Inorg. Chem., 2020, 59, 18345–18357 CrossRef CAS PubMed.
- M. Y. Gao, S. Chen, L. X. Hu, L. Zhang and J. Zhang, Dalton Trans., 2017, 46, 10630–10634 RSC.
- C. Wang, C. Liu, L. J. Li and Z. M. Sun, Inorg. Chem., 2019, 58, 6312–6319 CrossRef CAS PubMed.
- J.-L. Hou, P. Huo, Z.-Z. Tang, L.-N. Cui, Q.-Y. Zhu and J. Dai, Inorg. Chem., 2018, 57, 7420–7427 CrossRef CAS PubMed.
- D.-H. Zou, L.-N. Cui, P.-Y. Liu, S. Yang, Q.-Y. Zhu and J. Dai, Inorg. Chem., 2019, 58(14), 9246–9252 CrossRef CAS PubMed.
- J. Moons, F. Azambuja, J. Mihailovic, K. Kozma, K. Smiljanic, M. Amiri, T. Cirkovic-Velickovic, M. Nyman and T. N. Parac-Vogt, Angew. Chem., 2020, 132(23), 9179–9186 CrossRef.
- M. Janek, A. Radtke, T. M. Muziol, M. Jerzykiewicz and P. Piszczek, Materials, 2018, 11, 1661 CrossRef PubMed.
- M. Janek, T. M. Muziol and P. Piszczek, Materials, 2019, 12, 3195 CrossRef CAS PubMed.
- B. Ksapabutr, T. Chalermkiti, S. Wongkasemjit and M. Panapoy, Thin Solid Films, 2010, 518, 6518–6521 CrossRef CAS.
- M. Ismael, Y. Wu and M. Wark, New J. Chem., 2019, 43, 4455–4462 RSC.
- Y. Zhao, Y. Zhang, J. Li and X. Du, Mater. Lett., 2014, 130, 139–142 CrossRef CAS.
- Y.-S. Lai, Y. H. Su and M. I. Lin, Dyes Pigm., 2014, 103, 76–81 CrossRef CAS.
- J. Zhu, P. M. Usoy, W. Xu, P. J. Celis-Salazar, S. Lin, M. C. Kessinger, C. Landaverde-Alvarado, M. Cai, A. M. May, C. Slebodnick, D. Zhu, S. D. Senanayake and A. J. Morris, J. Am. Chem. Soc., 2018, 140, 993–1003 CrossRef CAS PubMed.
- T. Matemb Ma Ntep, H. Breitzke, L. Schmolke, C. Schlüsener, B. Moll, S. Millan, N. Tannert, I. El Aita, G. Buntkowsky and C. Janiak, Chem. Mater., 2019, 31, 8629–8638 CrossRef CAS.
- J. H. Carter, X. Han, F. Y. Moreau, I. da Silva, A. Nevin, H. G. W. Godfrey, C. C. Tang, S. Yang and M. Schroder, J. Am. Chem. Soc., 2018, 140, 15564–15567 CrossRef CAS PubMed.
- H. Wang, X. Dong, J. Lin, S. J. Teat, S. Jensen, J. Cure, E. V. Alexandrov, Q. Xia, K. Tan, Q. Wang, D. H. Olson, D. M. Proserpio, Y. J. Chabal, T. Thonhauser, J. Sun, Y. Han and J. Li, Nat. Commun., 2018, 9, 1745 CrossRef PubMed.
- S. Wang, J. S. Lee, M. Wahiduzzaman, J. Park, M. Muschi, C. Martineau-Corcos, A. Tissot, K. H. Cho, J. Marrot, W. Shepard, G. Maurin, J.-S. Chang and C. Serre, Nat. Energy, 2018, 3, 985–993 CrossRef CAS.
- Y. Z. Zhang, T. He, X. J. Kong, X. L. Lv, X. Q. Wu and J. R. Li, ACS Appl. Mater. Interfaces, 2018, 10, 27868–27874 CrossRef CAS PubMed.
- X. Sun, S. Yao, C. Yu, G. Li, C. Liu, Q. Huo and Y. Liu, J. Mater. Chem. A, 2018, 6, 6366–6369 Search PubMed.
- H. Wang, L. Yu, Y. Lin, J. Peng, S. J. Teat, L. J. Williams and J. Li, Inorg. Chem., 2020, 59, 4167–4171 CrossRef CAS PubMed.
- P. T. K. Nguyen, H. T. D. Nguyen, H. N. Nguyen, C. A. Trickett, Q. T. Ton, E. Gutierrez-Puebla, M. A. Monge, K. E. Cordova and F. Gandara, ACS Appl. Mater. Interfaces, 2018, 10, 733–744 CrossRef CAS PubMed.
- L. Zhang, S. Yuan, W. Fan, J. Pang, F. Li, B. Guo, P. Zhang, D. Sun and H. C. Zhou, ACS Appl. Mater. Interfaces, 2019, 11, 22390–22397 CrossRef CAS.
- S. Yuan, P. Zhang, L. Zhang, A. T. Garcia-Esparza, D. Sokaras, J. S. Qin, L. Feng, G. S. Day, W. Chen, H. F. Drake, P. Elumalai, S. T. Madrahimov, D. Sun and H. C. Zhou, J. Am. Chem. Soc., 2018, 140, 10814–10819 CrossRef CAS PubMed.
- Y. Wang, L. Feng, J. Pang, J. Li, N. Huang, G. S. Day, L. Cheng, H. F. Drake, Y. Wang and C. Lollar, Adv. Sci., 2019, 6, 1802059 CrossRef.
- X. Zhu, J. Gu, Y. Wang, B. Li, Y. Li, W. Zhao and J. Shi, Chem. Commun., 2014, 50, 8779–8782 RSC.
- S. Øien-Ødegaard, C. Bazioti, E. A. Redekop, Ø. Prytz, K. P. Lillerud and U. Olsbye, Angew. Chem., Int. Ed., 2020, 59, 21397–21402 CrossRef PubMed.
- Y. Zhang, F. Azambuja and T. N. Parac-Vogt, Coord. Chem. Rev., 2021, 438, 213886 CrossRef CAS.
- G. Kickelbick, D. Holzinger, C. Brick, G. Trimmel and E. Moons, Chem. Mater., 2002, 14, 4382–4389 CrossRef CAS.
- S. O. Baumann, M. Bendova, M. Puchberger and U. Schubert, Eur. J. Inorg. Chem., 2011, 2011, 573–580 CrossRef.
- S. O. Kang, S. Vukovic, R. Custelcean and B. P. Hay, Ind. Eng. Chem. Res., 2012, 51, 6619–6624 CrossRef CAS.
- S. S. Passadis, S. Hadjithoma, A. G. Kalampounias, A. C. Tsipis, S. Sproules, H. N. Miras, A. D. Keramidas and T. A. Kabanos, Dalton Trans., 2019, 48, 5551–5559 RSC.
- C. J. Leggett, B. F. Parker, S. J. Teat, Z. Zhang, P. D. Dau, W. W. Lukens, S. M. Peterson, A. J. P. Cardenas, M. G. Warner, J. K. Gibson, J. Arnold and L. Rao, Chem. Sci., 2016, 7, 2775–2786 RSC.
- D. Sanna, V. Ugone, G. Sciortino, B. F. Parker, Z. Zhang, C. J. Leggett, J. Arnold, L. Rao and E. Garribba, Eur. J. Inorg. Chem., 2018, 1805–1816 CrossRef CAS.
- G. Tian, S. J. Teat, Z. Zhang and L. Rao, Dalton Trans., 2012, 41, 11579–11586 RSC.
- X. Sun, C. Xu, G. Tian and L. Rao, Dalton Trans., 2013, 42, 14621–14627 RSC.
- H. N. Miras, D. Stone, D. L. Long, E. J. L. McInnes, P. Kögerler and L. Cronin, Inorg. Chem., 2011, 50, 8384–8391 CrossRef CAS PubMed.
- M. N. Corella-Ochoa, H. N. Miras, A. Kidd, D.-L. Long and L. Cronin, Chem. Commun., 2011, 47, 8799–8801 RSC.
- J. Yan, D.-L. Long, H. N. Miras and L. Cronin, Inorg. Chem., 2010, 49(4), 1819–1825 CrossRef CAS PubMed.
- E. F. Wilson, H. N. Miras, M. H. Rosnes and L. Cronin, Angew. Chem., Int. Ed., 2011, 50, 3720–3724 CrossRef CAS PubMed.
- H. N. Miras, E. F. Wilson and L. Cronin, Chem. Commun., 2009, 11, 1297–1311 RSC.
- H. N. Miras, D. L. Long, P. Kögerler and L. Cronin, Dalton Trans., 2008, 214–221 RSC.
- H. Zang, A. Surman, D. Long, L. Cronin and H. N. Miras, Chem. Commun., 2016, 52, 9109–9112 RSC.
- H. N. Miras, M. Sorus, J. Hawkett, D. O. Sells, E. J. L. McInnes and L. Cronin, J. Am. Chem. Soc., 2012, 134, 6980–6983 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data for 1 and 2. CCDC 2113958 for 1 and 2113960 for 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03641f |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b,
Haralampos N.
Miras
*b,
Haralampos N.
Miras