
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, molecular structure and fluxional behavior of the elusive [HRu4(CO)12]3− carbonyl anion†
Cristiana
Cesari
 *a,
Marco
Bortoluzzi
b,
Cristina
Femoni
a,
Maria
Carmela Iapalucci
a and
Stefano
Zacchini
a
*a,
Marco
Bortoluzzi
b,
Cristina
Femoni
a,
Maria
Carmela Iapalucci
a and
Stefano
Zacchini
a
aDipartimento di Chimica Industriale “Toso Montanari”, Università di Bologna, Viale Risorgimento 4, 40136 Bologna, Italy. E-mail: cristiana.cesari2@unibo.it
bDipartimento di Scienze Molecolari e Nanosistemi, Ca’ Foscari University of Venice, Via Torino 155, 30175 Mestre (Ve), Italy
First published on 4th January 2022
Abstract
The elusive mono-hydride tri-anion [HRu4(CO)12]3− (4) has been isolated and fully characterized for the first time. Cluster 4 can be obtained by the deprotonation of [H3Ru4(CO)12]− (2) with NaOH in DMSO. A more convenient synthesis is represented by the reaction of [HRu3(CO)11]− (6) with an excess of NaOH in DMSO. The molecular structure of 4 has been determined by single-crystal X-ray diffraction (SC-XRD) as the [NEt4]3[4] salt. It displays a tetrahedral structure of pseudo C3v symmetry with the unique hydride ligand capping a triangular Ru3 face. Variable temperature (VT) 1H and 13C{1H} NMR experiments indicate that 4 is fluxional in solution and reveal an equilibrium between the C3v isomer found in the solid state and a second isomer with Cs symmetry. Protonation–deprotonation reactions inter-converting H4Ru4(CO)12 (1), [H3Ru4(CO)12]− (2), [H2Ru4(CO)12]2− (3), [HRu4(CO)12]3− (4) and the purported [Ru4(CO)12]4− (5) have been monitored by IR and 1H NMR spectroscopy. Whilst attempting the optimization of the synthesis of 4, crystals of [NEt4]2[Ru3(CO)9(CO3)] ([NEt4]2[7]) were obtained. Anion 7 contains an unprecedented CO32− ion bonded to a zero-valent Ru3(CO)9 fragment. Finally, the reaction of 6 as the [N(PPh3)2]+ ([PPN]+) salt with NaOH in DMSO affords [Ru3(CO)9(NPPh3)]− (9) instead of 4. Computational DFT studies have been carried out in order to support experimental evidence and the location of the hydride ligands as well as to shed light on possible isomers.
Introduction
Hydridocarbonylates of the Fe–Ru–Os triad played a fundamental role in the development of the chemistry of metal carbonyl clusters.1–11 The only species structurally characterized for the whole triad are [HM3(CO)11]−,12–14 [HM4(CO)13]−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15–17 and [H2M4(CO)12]2−.10,18,19 In the case of iron, only low nuclearity species comprising 1–5 metal atoms have been structurally characterized, whereas nuclearities up to 10–11 metal atoms have been obtained with Ru and Os (Table 1).20–22 This is mainly due to the fact that Ru–Ru and Os–Os bonds are stronger than Fe–Fe ones, favoring the formation of larger clusters.1,2,23 Moreover, the greater sizes of Ru and Os compared to Fe also seem to play a role, allowing the coordination of more ligands to the same metal cage. In this respect, it is noteworthy that [HRu4(CO)13]− and [HOs4(CO)13]− display a tetrahedral metal cage, whereas [HFe4(CO)13]− adopts a more open butterfly structure in order to accommodate all the ligands.15–17
15–17 and [H2M4(CO)12]2−.10,18,19 In the case of iron, only low nuclearity species comprising 1–5 metal atoms have been structurally characterized, whereas nuclearities up to 10–11 metal atoms have been obtained with Ru and Os (Table 1).20–22 This is mainly due to the fact that Ru–Ru and Os–Os bonds are stronger than Fe–Fe ones, favoring the formation of larger clusters.1,2,23 Moreover, the greater sizes of Ru and Os compared to Fe also seem to play a role, allowing the coordination of more ligands to the same metal cage. In this respect, it is noteworthy that [HRu4(CO)13]− and [HOs4(CO)13]− display a tetrahedral metal cage, whereas [HFe4(CO)13]− adopts a more open butterfly structure in order to accommodate all the ligands.15–17
| Nuclearity | Fe | Ru | Os |
|---|---|---|---|
| 1 | [HFe(CO)4]– | ||
| 2 | [HFe2(CO)8]– | ||
| 3 | [HFe3(CO)11]– | [HRu3(CO)11]– | [HOs3(CO)11]– |
| H2Os3(CO)11 | |||
| H2Os3(CO)10 | |||
| 4 | [HFe4(CO)13]– | [HRu4(CO)13]– | [HOs4(CO)13]– |
| H2Ru4(CO)13 | H2Os4(CO)13 | ||
| [HFe4(CO)12]3– | [HRu4(CO)12]3– (this work) | ||
| [H2Fe4(CO)12]2– | [H2Ru4(CO)12]2– | [H2Os4(CO)12]2– | |
| [H3Ru4(CO)12]– | [H3Os4(CO)12]– | ||
| H4Ru4(CO)12 | H4Os4(CO)12 | ||
| 5 | H2Os5(CO)16 | ||
| [HOs5(CO)15]– | |||
| [HFe5(CO)14]3– | |||
| 6 | H2Os6(CO)19 | ||
| [HRu6(CO)18]– | [HOs6(CO)18]– | ||
| H2Ru6(CO)18 | H2Os6(CO)18 | ||
| H2Ru6(CO)17 | |||
| 7 | H2Os7(CO)22 | ||
| H2Os7(CO)21 | |||
| [HRu7(CO)20]– | |||
| H2Os7(CO)20 | |||
| 8 | [HOs8(CO)22]– | ||
| [H2Ru8(CO)21]2– | |||
| 9 | [HOs9(CO)24]– | ||
| 10 | [H2Ru10(CO)25]2– | ||
| [H4Os10(CO)24]2– | |||
| [H5Os10(CO)24]– | |||
| 11 | [HRu11(CO)27]3– |
It must be remarked that most Ru and Os hydridocarbonylate clusters are neutral or show a low negative charge (charge/nuclearity ≤ 0.5). Conversely, all hydridocarbonylferrates characterized by single crystal X-ray diffraction (SC-XRD) are anionic, often reaching high negative charges (charge/nuclearity = 0.25–1.00). This point is well exemplified by [H4−nM4(CO)12]n− clusters (M = Fe, Ru, and Os; n = 0–3). Iron clusters of this type have been obtained quite recently and only the di-hydride di-anion [H2Fe4(CO)12]2− and mono-hydride tri-anion [HFe4(CO)12]3− are sufficiently stable in order to be isolated and characterized by SC-XRD.9,10 In contrast, Ru and Os congeners have been known for a long time, and neutral tetra-hydrides H4M4(CO)12, tri-hydride mono-anions [H3M4(CO)12]−, and di-hydride di-anions [H2M4(CO)12]2− (M = Ru, Os) have been structurally characterized.11,18,19,24,25 Even though there is spectroscopic evidence of their existence, mono-hydride tri-anions [HM4(CO)12]3− (M = Ru and Os) seem to be rather elusive and they have not been characterized by SC-XRD so far.26,27
Besides historical and fundamental aspects, Ru hydride carbonyl clusters are rather interesting from an application point of view.28 Phosphine derivatives of H4Ru4(CO)12 as well as other Ru–H–CO clusters are active in homogeneous catalysis.29–32 For instance, H4Ru4(CO)12–2x(P–P)x (x = 1 and 2; P–P = chiral diphosphine) species catalyze several asymmetric isomerization, hydroformylation and hydrogenation reactions.33–36 Molecular Ru hydride carbonyl clusters may serve also as models in order to help the location of the hydride ligands within Ru nanoparticles employed for hydrogenation.37,38
In view of this interest for Ru hydridocarbonylates, we have attempted to isolate and fully characterize the elusive mono-hydride tri-anion [HRu4(CO)12]3−. Herein, we report its detailed synthesis as well as its first structural characterization by means of SC-XRD. In addition, the fluxional behavior of this cluster in solution has been investigated by variable temperature (VT) 1H and 13C{1H} NMR experiments. Protonation–deprotonation reactions inter-converting H4Ru4(CO)12 (1), [H3Ru4(CO)12]− (2), [H2Ru4(CO)12]2− (3), [HRu4(CO)12]3− (4) and the purported [Ru4(CO)12]4− (5) were monitored by IR and 1H NMR spectroscopy. Computational DFT studies were carried out in order to support experimental evidence and the location of the hydride ligands as well as to shed light on possible isomers.
Results and discussion
Synthesis, molecular structure and dynamic behavior of [HRu4(CO)12]3− (4)
It was previously reported that 4 is not stable and it decomposes into a mixture of 3 and 5, when obtained by deprotonation of 1.26 This was likely due to the experimental conditions employed, that is, the reaction of 1 with three equivalents of KH in THF, rather than the intrinsic instability of 4. Indeed, it was later reported that 4 could be obtained by reaction of 5 with HBr in THF, even if it was not possible to isolate and structurally characterize it.27Seeking a way to isolate and fully characterize such an elusive species, we investigated the reaction of 2 with an excess of NaOH in a highly polar aprotic solvent such as DMSO. This approach was successful, allowing complete crystallographic and spectroscopic characterization of 4. The same compound could be obtained even more conveniently from the reaction of [HRu3(CO)11]− (6) with an excess of NaOH in DMSO (Scheme 1). Both these syntheses were inspired by our previous findings on the synthesis of isostructural [HFe4(CO)12]3− as well as the highly reduced [Fe6C(CO)15]4−.9,39 Formation of 4 from 2 was just a deprotonation reaction. Conversely, ruthenium was formally reduced from −2/3 to −1 passing from 6 to 4. Electrons required for reduction were produced by oxidation of CO to CO2 promoted by OH− nucleophilic attack at a Ru-coordinated CO ligand. In the presence of an excess of base, CO2 was converted into CO32−. Indeed, we were able to isolate the carbonate-cluster [Ru3(CO)9(CO3)]2− (7) as a side product (see the Experimental section for details). After optimization of the synthetic procedure, we were able to isolate 4 as the [NEt4]+ salt in 77% yields.
 | ||
| Scheme 1 Synthesis of [H4−nRu4(CO)12]n− (n = 0 – 4). | ||
The molecular structure of 4 was determined by SC-XRD (Fig. 1 and Table 2). Moreover, 4 was spectroscopically characterized by IR and VT 1H NMR techniques. In order to gain further insights into its behavior in solution, VT 13C{1H} NMR experiments were performed on a 13CO-enriched sample (ca. 50%) prepared from [HRu3(13CO)11]− (6–13CO).
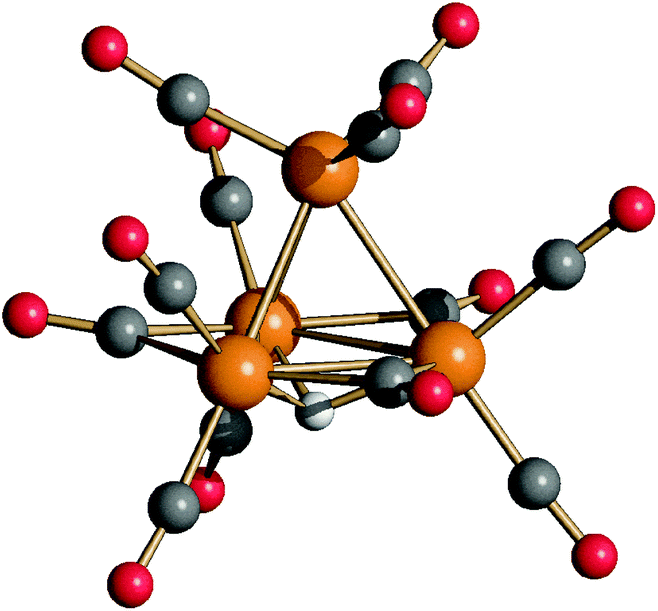 | ||
| Fig. 1 Molecular structure of [HRu4(CO)12]3− (4) (orange Ru; red O; grey C; and white H). | ||
| 4 | 3 | 2 (C2 isomer)b | 2 (C3v isomer)b | |
|---|---|---|---|---|
| a From ref. 11 and 18. b From ref. 11 and 24. | ||||
| Ru(1)–Ru(2) | 2.8032(11) | 2.9771(4) | 2.9032(5) | 2.7733(5) |
| Ru(1)–Ru(3) | 2.8001(11) | 2.9558(4) | 2.7871(5) | 2.7753(5) |
| Ru(1)–Ru(4) | 2.8018(11) | 2.8183(4) | 2.7614(5) | 2.7841(4) |
| Ru(2)–Ru(3) | 2.8106(11) | 2.7735(4) | 2.9423(4) | 2.9380(5) |
| Ru(2)–Ru(4) | 2.8006(11) | 2.7842(4) | 2.7919(4) | 2.9218(5) |
| Ru(3)–Ru(4) | 2.8113(11) | 2.7526(4) | 2.9167(5) | 2.9191(4) |
| H(1)–Ru(1) | — | 1.71(4) | 1.73(4) | — |
| H(1)–Ru(2) | 1.78(4) | 1.86(4) | 1.77(4) | 1.75(4) |
| H(1)–Ru(3) | 1.77(4) | |||
| H(1)–Ru(4) | 1.78(4) | — | — | 1.83(4) |
| H(2)–Ru(2) | — | — | 1.79(4) | 1.74(4) |
| H(2)–Ru(3) | — | 1.87(4) | 1.72(4) | 1.76(4) |
| H(2)–Ru(1) | — | 1.64(4) | — | — |
| H(3)–Ru(3) | — | — | 1.72(4) | 1.72(4) |
| H(3)–Ru(4) | — | — | 1.80(4) | 1.76(4) |
The structure of 4 closely resembles that of [HFe4(CO)12]3−9 and displays an idealized C3v symmetry. It consists of a tetrahedral Ru4 cage and the unique hydride is μ3-coordinated to the triangular basal face. The apical Ru atom is bonded to three terminal carbonyls, whereas each of the three Ru atoms on the basal plane is bonded to two terminal CO ligands. The three remaining carbonyl ligands are in edge bridging positions on the basal triangle. The stereochemistry of the CO ligands of 4 is similar to that of 3,11,18 which contains two edge bridging μ-H hydride ligands on two different Ruapical–Rubasal edges. In contrast, 2 exists in solution as well as in the solid state as two isomers (C2 and C3v) and both contain only terminal carbonyls and edge bridging hydrides.11,24 In agreement with the solid state structure, the IR spectrum of the CH3CN solution of 4 (Fig. S1 in the ESI†) displays three νCO bands in the terminal region [1928(s), 1898(vs) and 1855(m) cm−1] and one in the edge-bridging region [1714(m) cm−1].
The six Ru–Ru bonding contacts of 4 are comprised in a rather narrow range [2.8001(11)–2.8113(11) Å] even if they have different environments. Indeed, three of them [Ru(1)–Ru(2), Ru(1)–Ru(3) and Ru(1)–Ru(4), see Scheme 2 for labeling] are not bridged by any ligand, whereas the three Ru–Ru edges on the basal triangle [Ru(2)–Ru(3), Ru(3)–Ru(4), Ru(2)–Ru(4)] are bridged by three μ-CO ligands and the resulting triangular face is capped by a μ3-H. The shortening of the Ru–Ru distances expected because of the μ-CO ligands is compensated by the elongation due to the face capping hydride, making these three Ru–Ru contacts similar to the non-bridged ones. Indeed, in the case of 3, where the two μ-H and three μ-CO ligands are located on different edges, it is possible to distinguish three sets of Ru–Ru distances.11,18 The two edges bridged by hydride ligands [2.9558(4) and 2.9771(4) Å] are considerably elongated as expected, whereas the three Ru–Ru edges supported by μ-CO ligands [2.7526(4)–2.7842(4) Å] are shortened. The unique Ru–Ru edge not bearing any bridging ligand displays an intermediate value [2.8183(4) Å], very similar to those found in 4. In the case of 2 (C2 and C3v isomers), since all carbonyls are terminally coordinated, there are only two sets of Ru–Ru contacts: those bridged by μ-H ligands [2.9032(5)–2.9423(4) Å, C2 isomer; 2.9191(4)–2.9380(5) Å, C3v isomer] are longer than the unbridged ones [2.7614(5)–2.7919(4) Å, C2 isomer; 2.7735(5)–2.7841(4) Å, C3v isomer].11,24
 | ||
| Scheme 2 Labelling of [H3Ru4(CO)12]− (2) (C2 and C3v isomers), [H2Ru4(CO)12]2− (3), and [HRu4(CO)12]3− (4) (CO omitted for clarity). | ||
The 1H NMR spectrum of 4 recorded at 298 K (Fig. 2 and Fig. S8 in the ESI†) displays broad resonances at −17.65 and −17.95 ppm in CD3CN and DMF-d7 solutions, respectively. The sharp singlets at −15.56 and −19.06 ppm (in DMF-d7 solution) are due to an unknown impurity and 3, respectively. The presence of 3 is due to the fact that 4 is easily protonated (see below for details). Upon lowering the temperature, complete coalescence of the resonance of 4 is observed at 273 K and, then, two resonances appear at lower temperatures. The slow exchange limit is reached at 223 K in DMF-d7 solution, where two singlets at −17.90 and −15.16 ppm are present in a 1.2:1 ratio. The presence of two resonances in the hydride region can be explained by assuming that two exchanging isomers of 4 are present in solution.
 | ||
| Fig. 2 Hydride region of the VT 1H NMR spectra of [NEt4]3[HRu4(CO)12] (4) in DMF-d7. * [H2Ru4(CO)12]2− (3). #, unknown impurity. | ||
Kinetic constants for the exchange process in DMF-d7 solution at different temperatures (Table 3) were obtained by line-shape analysis using the gNMR 5.0.6.0 program for the simulation of VT 1H NMR spectra.40 The Eyring plot yields the following values for the activation parameters: ΔH‡ = +43 ± 6 kJ mol−1 and ΔS‡ = −14 ± 24 J K−1 mol−1. The precision of the entropy term does not allow us to determine if it is zero, positive or negative. In all cases, we could expect a value close to zero. These activation parameters are comparable to those reported for the rapid inter-conversion of the C2 and C3v isomers of 2, that is, ΔH‡ = +46 kJ mol−1 and ΔS‡ = 0.2–8 J K−1 mol−1.41
| T | k |
|---|---|
| 298 K | 6.0 × 104 s−1 |
| 273 K | 3.5 × 103 s−1 |
| 248 K | 8.0 × 103 s−1 |
| 223 K | 1.0 × 102 s−1 |
In order to gain further insight into the structures of the two isomers of 4, VT 13C{1H} NMR experiments were performed on a 13CO-enriched (ca. 50%) sample prepared using 6–13CO (Fig. 3). The 13C{1H} NMR spectrum recorded at 298 K displays, in the carbonyl region, a broad resonance at ca. 226 ppm in agreement with the fluxional behavior observed in the VT 1H NMR experiments. Some sharp resonances are also observed, due to the abovementioned impurities. Moreover, the broad resonance at ca. 220 ppm is attributable to 3. Upon lowering the temperature to 223 K, the broad resonance of 4 is split into several resonances. Four sharp resonances are clearly visible at 268.7, 213.4, 213.0, and 201.7 ppm in a 1:1:1:1 ratio, with the first resonance corresponding to the μ-CO ligands and the other three resonances to terminal carbonyls. These NMR features are in agreement with the C3v symmetry of the major isomer found also in the solid state structure of 4. A similar low temperature 13C{1H} NMR spectrum was previously reported for [HFe4(CO)12]3−.9
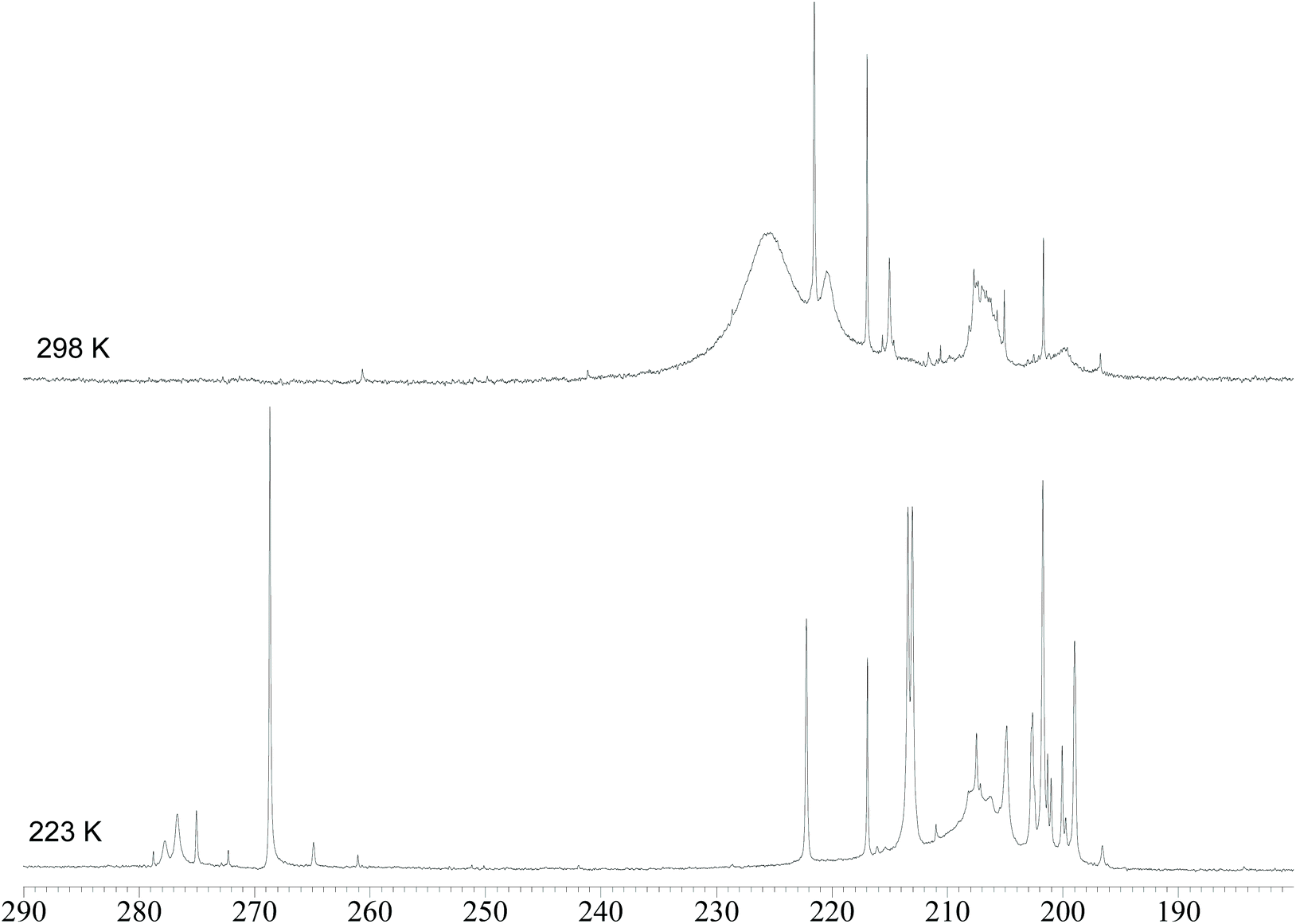 | ||
| Fig. 3 Carbonyl region of the 13C{1H} NMR spectra of [NEt4]3[HRu4(CO)12] (4) in DMF-d7 at 298 K and 223 K. The sample has been enriched with 13CO. | ||
Regarding the second isomer, it must be remarked that, in the 13C{1H} NMR spectrum recorded at 223 K, two further resonances in the μ-CO region are present at 277.8 and 276.7 in a 1:2 ratio. Thus, it can be supposed that the second isomer retains the same stereochemistry of the CO ligands (9 terminal and 3 edge-bridging), whereas the unique hydride is now bridging one Ruapical–Rubasal edge, lowering the symmetry from C3v to Cs. Because of the Cs symmetry, the three μ-CO ligands are not equivalent but split into two sets composed of one and two carbonyls, respectively, as spectroscopically observed. The same should apply also to the terminal CO ligands resulting in six resonances with relative intensities in a ratio of 1:2:1:2:1:2. Indeed, several resonances are present at low temperatures in the 195–220 ppm region but, because of the presence of 3 as well as other impurities, it is not possible to fully assign them.
The location of the hydride in the C3v isomer of 4 was confirmed using DFT calculations (Fig. 4). Moreover, the computational simulations allowed us to optimize the second isomer of 4 with a rough Cs symmetry (R = 0.019), in agreement with the NMR data. The coordination mode of the hydride in the Cs isomer is μ-H, bridging the apical Ru and one of the basal Ru centers. The energies of the two isomers are comparable in the limits of the computational model applied, according to the NMR integration of the hydride resonances (Fig. 2). It is worth noting that in the absence of intermolecular interactions, the Cs isomer is predicted to be slightly more stable than the C3v one.
 | ||
| Fig. 4 DFT-optimized isomers of cluster 4 and relative energy values (orange Ru; red O; grey C; and white H). | ||
The stepwise protonation of 4 with HBF4·Et2O affords the previously reported 3, 2, and 1. The reaction has been monitored by IR and 1H NMR spectroscopy (Fig. 5 and 6; Fig. S1–S5 and S9 in the ESI†). As mentioned above, 4 is a strong base and is readily protonated. Indeed, its 1H NMR spectrum is always contaminated by significant amounts of 3. After the addition of one mole equivalent of acid, 4 completely reacts and the 1H NMR spectrum displays the presence of 3 accompanied by a small amount of 2. The addition of a second mole equivalent of HBF4·Et2O quantitatively converts 3 into 2 with a small amount of 1. The latter becomes the unique species in solution after the addition of a third mole equivalent of acid. Because of the strong basicity of 4, all attempts to deprotonate it and isolate the tetra-anion 5 failed.
 | ||
| Fig. 5 IR spectra in the νCO region of [H4−nRu4(CO)12]n− (n = 0–3) obtained from the stepwise protonation of [HRu4(CO)12]3− (4) with HBF4·Et2O in CH3CN. | ||
 | ||
| Fig. 6 Hydride region of the 1H NMR spectra of [H4−nRu4(CO)12]n− (n = 0–3) obtained from the stepwise protonation of [HRu4(CO)12]3− (4) with HBF4·Et2O in CD3CN at 298 K. | ||
In order to shed light on the different stability of [H4−nRu4(CO)12]n− compared to [H4−nFe4(CO)12]n−, the energy variation associated with the reaction [H4−nRu4(CO)12]n− + [H4−mFe4(CO)12]m− → [H4−mRu4(CO)12]m− + [H4−nFe4(CO)12]n− (n = 1, 2, 3; m = n − 1) was studied. Negative energy variations indicate the higher basicity of the ruthenium cluster with respect to the related iron species. For the sake of comparison, the same isomer was considered for changing the metal centre (see Fig. 7). As reported in Fig. 7, ruthenium clusters were in all cases more basic than the related iron species, and the relative basicity increased with the number of coordinated hydrides.
 | ||
| Fig. 7 Energy variations for the reactions [H4−nRu4(CO)12]n− + [H4−mFe4(CO)12]m− → [H4−mRu4(CO)12]m− + [H4−nFe4(CO)12]n− (m = n − 1) and DFT-optimized structures of the isomers considered (green Fe; orange Ru; red O; grey C; and white H). | ||
As mentioned at the beginning of this section, 7 was obtained as a side product during the synthesis of 4 starting from 6. Its formation is likely due to the partial decomposition of 6 which formally results in a zero-valent unsaturated “Ru3(CO)9” fragment. The latter can bind a CO32− ion which, in turn, is formed from the oxidation of CO to CO2 and subsequent reaction with OH−. This may be viewed as an indirect proof of the fact that 6 is reduced to 4 by oxidation of CO in the presence of OH− ions.
The molecular structure of 7 (Fig. 8) is rather unique as it contains the first example of a CO32− ion bonded to (formally) zero-valent metal atoms. Indeed, the carbonate ion is usually found bonded, also as a multidentate ligand, to metals in a formal positive oxidation state.11,42 In particular, the CO32− ion of 7 is bonded to one Ru atom via one O-atom and to two further Ru atoms through a second (edge bridging) O-atom, whereas the third oxygen is not bonded to any metal atom. Three CO ligands are edge bridging on the Ru3 triangle and, in addition, there are two terminal carbonyls on each Ru atom.
 | ||
| Fig. 8 (a) Molecular structure of [Ru3(CO)9(CO3)]2− (7) (orange Ru; red O; grey C) with a labeling scheme. Main bond distances (Å) and angles (°): Ru(1)–Ru(2) 2.790(3), Ru(1)–Ru(3) 2.800(2), Ru(2)–Ru(3) 2.634(2), Ru(1)–O(1) 2.147(14), Ru(2)–O(2) 2.149(13), Ru(3)–O(12) 2.174(13), C(1)–O(1) 1.34(3), C(1)–O(2) 1.28(3), C(1)–O(3) 1.22(3), Ru(1)–O(1)–C(1) 117.9(14), Ru(2)–O(2)–Ru(3) 75.1(4), O(1)–C(1)–O(2) 118(2), O(1)–C(1)–O(3) 114(2), and O(2)–C(1)–O(3) 128(2). (b) DFT-optimized structure of 7 (orange Ru; red O; and grey C) and selected (3,−1) b.c.p. (green) with density values at b.c.p. (potential energy density values in parentheses). Data in a.u. | ||
The coordination mode of the carbonate ion was confirmed using DFT calculations. The root-mean-square deviation (RMSD) between X-ray and DFT-optimized structures is only 0.019 Å. Three Ru–O (3,−1) bond critical points (b.c.p.) were obtained from the atom-in-molecule (AIM) analysis of the cluster (Fig. 8). The computed Mayer bond order for the Ru–(κ1-O) bond is 0.575, while the bond order for the two Ru–(μ-O) bonds is lower (0.437). In agreement with this result, the electron density (ρ) at b.c.p. is higher and the potential energy density (V) is more negative for the Ru–(κ1-O) interaction with respect to the Ru–(μ-O) ones. AIM data are reported in Fig. 8.
The Hirshfeld partial charges on the Ru atoms are strictly comparable and the average value is 0.105 a.u. The average value for the separated neutral {Ru3(CO)9} fragment is similar, 0.078 a.u., but surprisingly slightly less positive. Charge decomposition analysis (CDA) indicates that CO32− behaves as a donor towards {Ru3(CO)9} by about 0.51 a.u., without back-donation. The increase of the electron density is drained by carbonyl ligands, as confirmed by population analyses. The average CO Hirshfeld charge in 7 is −0.122 a.u., while it is −0.020 a.u. in the related {Ru3(CO)12} fragment. Overall, the computed data do not suggest any change in the formal oxidation state of the Ru atoms upon coordination of CO32−.
Synthesis and molecular structure of [Ru3(CO)9(NPPh3)]− (9)
The reaction of 6 with an excess of NaOH in DMSO under a CO atmosphere affords [Ru2(CO)8]2− (8) rather than 4. Both the compounds contain Ru in the formal oxidation state −1, but 8 has a higher CO/Ru ratio (4) compared to 4 (CO/Ru = 3) in accordance with eqn (1). Compound 8 has been identified by IR spectroscopy and, moreover, its structure has been supported by the SC-XRD pattern of the new [NEt4]2[Ru2(CO)8] salt (Fig. S12 in the ESI†).| [HRu4(CO)12]3− + 4CO ↔ 2[Ru2(CO)8]2− + H+ | (1) |
Interestingly, performing the reaction of 6 with NaOH ion DMSO in the presence of [PPN]+ ions ([PPN]+ = [N(PPh3)2]+) afforded the new [Ru3(CO)9(NPPh3)]− (9) cluster.
Compound 9 has been previously identified by IR and 31P{1H} NMR spectroscopy during the deprotonation of [HRu3(CO)9(NPPh3)] (10) which, in turn, was obtained from the reaction of Ru3(CO)12 with Ph3P = NSiMe3.43 The molecular structure of 9 was determined by SC-XRD as its [NEt4][Ru3(CO)9(NPPh3)] salt (Fig. 9). It consists of a triangular Ru3 cluster with a μ3-NPPh3 ligand, six terminal carbonyls (two per each Ru atom) and three μ-CO ligands on three edges of the Ru3 triangle. In agreement with the solid state structure, its FT-IR spectrum in solution and in the solid state shows νCO bands due to both terminal and edge bridging carbonyls. It must be remarked that two isomers of 10 have been structurally characterized, one displaying the same stereochemistry of the CO ligands as found in 9, whereas the second isomer possesses only terminal carbonyls. It is well known that increasing the anionic charge of a carbonyl cluster favours the bridging mode of the CO ligands.44,45 For instance, 1 and 2 contain only terminal CO ligands, whereas 3 and 4 contain three μ-CO ligands.
 | ||
| Fig. 9 Molecular structure of [Ru3(CO)9(NPPh3)]− (9) (orange Ru; purple P; blue N; red O; grey C; and white H). Main bond distances (Å): Ru–Ru 2.7297(7), 2.7455(7) and 2.7471(7); Ru–N 2.125(5), 2.129(5) and 2.141(5); and N–P 1.597(5). | ||
Cluster 9 is electron precise since it possesses 48 cluster valence electrons (CVE) [3 × 8 (3Ru) + 9 × 2 (9CO) + 1 × 5 (μ3-NPPh3) + 1 (charge)] as expected for a triangular cluster. The μ3-NPPh3 contributes 5 CVE if considered as a neutral ligand and 6 CVE if considered as a mono-anion. A computational analysis of 9 is included in the ESI.†
The 31P{1H} NMR spectrum of 9 in CD2Cl2 at 298 K displays a sharp singlet at δP 57.03 ppm, considerably deshielded compared to the starting [PPN]+ ions (δP 21 ppm).
Conclusions
Synthesis, spectroscopic characterization and structural determination of H4Ru4(CO)12 (1), [H3Ru4(CO)12]− (2) and [H2Ru4(CO)12]2− (3) hydride carbonyl clusters represented a milestone in cluster chemistry.1,11,18,24,25 Spectroscopic evidence that they can be further deprotonated affording the [HRu4(CO)12]3− (4) mono-hydride tri-anion has been reported more than 40 years ago.26,27 Nonetheless, 4 escaped up to now any effort devoted to its isolation and structural characterization. This is likely due to the experimental conditions employed during these attempts, rather than the intrinsic instability of 4. Indeed, by carefully choosing the operational conditions and, in particular, by working in a highly polar aprotic solvent such as DMSO and using a strong base such as NaOH, it has been possible to convert 2 as well as [HRu3(CO)11]− (6) into 4. This allowed its isolation and the determination of its structure by SC-XRD. Similar approaches have been recently adopted for the isolation of [HFe4(CO)12]3− starting from [Fe4(CO)13]2− or [HFe3(CO)11]−,9,10 as well as the synthesis of [Fe6C(CO)15]4− from [Fe6C(CO)16]2−.39 Thus, it seems that the use of strong bases in polar solvents such as DMSO or CH3OH may be a general synthetic pathway towards highly reduced carbonylate clusters.Two isomers of 4 rapidly exchange in solution, whereas a single isomer has been reported for [HFe4(CO)12]3−.9 The latter possesses the same C3v structure with a μ3-H found in the solid state structure of 4. This might be due to the fact that, due to the larger size of Ru compared to Fe, there is more space in a Ru4(CO)12 tetrahedron rather than in Fe4(CO)12 in order to accommodate the hydride ligand on different sites. Indeed, it is well known that both [Ru4(CO)13]2− and [Fe4(CO)13]2− have a tetrahedral structure, which is retained in [HRu4(CO)13]−, whereas [HFe4(CO)13]− shows a more open butterfly structure.15–17 Nonetheless, it must be remarked that 3 and [H2Fe4(CO)12]2− are isostructural.10
Comparison of [H4−nRu4(CO)12]n− (n = 0–3) and [H4−nFe4(CO)12]n− (n = 2–3) indicates that Ru prefers less charged poly-hydride species, whereas more charged anions are favored in the case of Fe. This may be explained by assuming higher basicity for the Ru cluster anions compared to the related Fe species. Indeed, 4 is easily protonated to 3, whereas the protonation of [HFe4(CO)12]3− to [H2Fe4(CO)12]2− requires strictly controlled conditions. Moreover, there is no evidence of less charged Fe poly-hydrides of the type [H3Fe4(CO)12]− and H4Fe4(CO)12.
These findings further corroborate the tendency of Ru toward the formation of poly-hydride clusters. Somehow this is correlated with the involvement of Ru–H complexes, molecular carbonyl clusters and nanoparticles in catalytic processes using hydrogen.28–38
Experimental
General procedures
All reactions and sample manipulations were carried out using standard Schlenk techniques under nitrogen and in dried solvents. All the reagents were commercial products (Aldrich) of the highest purity available and used as received. Analyses of C, H and N were performed using a Thermo Quest Flash EA 1112NC instrument. IR spectra were recorded using a PerkinElmer Spectrum One interferometer in CaF2 cells. 1H, 13C{1H} and 31P{1H} NMR measurements were performed using a Varian Mercury Plus 400 MHz instrument. The proton and carbon chemical shifts were referenced to the non-deuterated aliquot of the solvent. The phosphorus chemical shifts were referenced to external H3PO4 (85% in D2O). Structure drawings were performed using SCHAKAL99.46Synthesis of [NEt4][HRu3(CO)11] (6)47
In a 100 mL autoclave, RuCl3·3H2O (1.57 g, 6 mmol), NaOMe (1.92 g, 35.6 mmol) and [NEt4]Br (0.630 g, 3 mmol) were dissolved in CH3OH (40 mL). The autoclave was pressurized with CO (60 bar) and heated at 120 °C for 14 h. After cooling and venting, the deep red solution containing the already solid product was moved to a 250 mL Schlenk tube and filtered under a nitrogen atmosphere and the residue was washed with methanol (30 mL). Then, the solvent was removed from the filtrate under vacuum and the residue was extracted in CH2Cl2 (40 mL). The solvent was removed from the CH2Cl2 solution under reduced pressure. The residue was washed with water (2 × 30 mL) and toluene (2 × 20 mL) and then extracted with CH2Cl2 (30 mL). The product [NEt4][HRu3(CO)11] was obtained as a red solid with a yield of 75% after the removal of the solvent under reduced pressure. Crystals of [NEt4][HRu3(CO)11] suitable for SC-XRD can be obtained by slow diffusion of n-hexane on the CH2Cl2 solution (yield 1.48 g, 85%).

C19H21NO11Ru3 (742.583): calcd (%): C 30.73, H 2.85, N 1.89; found: C 30.91, H 2.62, N 2.05.
IR (CH2Cl2, 298 K) νCO: 2016(vs), 1987(s), 1952(m) cm−1.
1H NMR (acetone-d6, 298 K) δ (hydride region): −12.51 ppm.
Synthesis of [NEt4]3[HRu4(CO)12] (4) and [NEt4]2[Ru3(CO)9(CO3)] (7)
NaOH (0.800 g, 200 mmol) was added as a solid to a solution of [NEt4][HRu3(CO)11] (0.550 g, 0.741 mmol) in DMSO (10 mL). The resulting mixture was stirred at room temperature for 3 days. The crude product was precipitated by the addition of iPrOH (100 mL) and the solid was recovered by filtration, washed with iPrOH (30 mL), and dried under reduced pressure. The orange solid was further washed with toluene (15 mL), THF (15 mL), and extracted in acetone (15 mL). The acetone solution was layered with n-hexane, affording crystals of [NEt4]2[Ru3(CO)9(CO3)] suitable for SC-XRD (yield 0.175 g, 27%). Then, the residue was extracted in acetonitrile (15 mL) and layered with n-hexane and diisopropyl ether, affording crystals of [NEt4]3[HRu4(CO)12] suitable for SC-XRD (yield 0.349 g, 58%).

Optimized synthesis of [NEt4]3[HRu4(CO)12](4)
[NEt4][HRu3(CO)11] (0.700 g, 0.943 mmol) was stirred at room temperature with 1.50 g of freshly powdered KOH suspended in 10 mL of DMSO for 16 h. The orange solution decanted from KOH powder was added dropwise to a stirred solution of [NEt4]Br (2.00 g) in H2O (15 mL) and iPrOH (150 mL). The crude compound precipitated immediately as an orange powder. The solid was obtained by filtration and vacuum dried. Then, the solid was washed with acetone (10 mL) and the product extracted in CH3CN was identified as [NEt4]3[HRu4(CO)12] by means of NMR and IR spectroscopy (yield 0.616 g, 77%).C36H61N3O12Ru4 (1132.15): calcd (%): C 38.19, H 5.43, N 3.71; found: C 38.31, H 5.07, N 3.49.
Synthesis of [NEt4][Ru3(CO)9(NPPh3)] (9)
NaOH (0.950 g, 23.8 mmol) was added as a solid to a solution of [PPN][HRu3(CO)11] (0.670 g, 0.582 mmol) in DMSO (10 mL) and the resulting mixture was stirred at room temperature overnight. The crude product was filtered and the solution was precipitated by the addition of a saturated solution of [NEt4]Br in H2O. The solid obtained was recovered by filtration, washed with H2O (40 mL), toluene (20 mL) and 2-propanol (20 mL), and then extracted with CH2Cl2 (15 mL). Crystals of [NEt4][Ru3(CO)9(NPPh3)] suitable for SC-XRD were obtained by layering n-pentane on the CH2Cl2 solution (yield 0.352 g, 63%).

C35H35N2O9PRu3 (961.83): calcd (%): C 43.71, H 3.67, N 2.91; found: C 43.56, H 3.38, N 3.11.
IR (CH2Cl2, 298 K) νCO: 2016(w), 1990(s), 1959(vs), 1926(m), 1750(m) cm−1.
IR (Nujol, 298 K) νCO: 2018(w), 1979(ms), 1961(s), 1941(m), 1907(mw), 1890(mw), 1772(m), 1746(s) cm−1.
31P{1H} NMR (CD2Cl2, 298 K) δ: 57.03 ppm.
13C{1H} NMR (CD2Cl2, 298 K) δ (carbonyl region): 221.5 ppm.
X-ray crystallographic study
Crystal data and collection details for [NEt4][HRu3(CO)11], [NEt4][Ru3(CO)9(NPPh3)], [NEt4]2[Ru2(CO)8], [NEt4]3[HRu4(CO)12] and [NEt4]2[Ru3(CO)9(CO3)] are reported in Table 4. The diffraction experiments were carried out on a Bruker APEX II diffractometer equipped with a PHOTON2 detector using Mo–Kα radiation. Data were corrected for Lorentz polarization and absorption effects (empirical absorption correction SADABS).48 Structures were solved by direct methods and refined by full-matrix least-squares based on all data using F2.49 Hydrogen atoms were fixed at calculated positions and refined using a riding model. All non-hydrogen atoms were refined with anisotropic displacement parameters, unless otherwise stated.| [NEt4][HRu3(CO)11] | [NEt4]2[Ru2(CO)8] | [NEt4][Ru3(CO)9(NPPh3)] | [NEt4]3[HRu4(CO)12] | [NEt4]2[Ru3(CO)9(CO3)] | |
|---|---|---|---|---|---|
| Formula | C19H21NO11Ru3 | C24H40N2O8Ru2 | C35H35N2O9PRu3 | C36H61N3O12Ru4 | C26H40N2O12Ru3 |
| f w | 742.58 | 686.72 | 961.83 | 1132.15 | 875.81 |
| T, K | 100(2) | 100(2) | 100(2) | 293(2) | 100(2) |
| λ, Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n | P21/c |
| a, Å | 12.3458(10) | 9.5198(6) | 11.1727(11) | 12.9763(8) | 16.5638(16) |
| b, Å | 11.7217(9) | 13.7870(8) | 11.9062(12) | 19.4012(12) | 10.2807(10) |
| c, Å | 17.4163(15) | 11.8620(7) | 14.3383(14) | 18.0370(11) | 19.8070(19) |
| α, ° | 90 | 90 | 90.095(3) | 90 | 90 |
| β, ° | 97.810(3) | 113.085(2) | 99.075(3) | 97.884(2) | 105.465(3) |
| γ, ° | 90 | 90 | 100.292(3) | 90 | 90 |
| Cell volume, Å3 | 2497.0(4) | 1432.21(15) | 1852.3(3) | 4498.0(5) | 3250.8(5) |
| Z | 4 | 2 | 2 | 4 | 4 |
| D c, g cm−3 | 1.975 | 1.592 | 1.725 | 1.672 | 1.790 |
| μ, mm−1 | 1.847 | 1.100 | 1.305 | 1.375 | 1.436 |
| F(000) | 1448 | 700 | 956 | 2280 | 1752 |
| Crystal size, mm | 0.22×0.18×0.14 | 0.24×0.21×0.19 | 0.18×0.12×0.11 | 0.14×0.12×0.11 | 0.16×0.14×0.10 |
| θ limits, ° | 1.665–26.996 | 2.380–27.994 | 1.439–25.099 | 1.549–26.036 | 2.134–25.074 |
| Index ranges | −15 ≤ h ≤ 15 | −12 ≤ h ≤ 12 | −13 ≤ h ≤ 13 | −15 ≤ h ≤ 15 | −19 ≤ h ≤ 19 |
| −14 ≤ k ≤ 14 | −18 ≤ k ≤ 18 | −14 ≤ k ≤ 14 | −23 ≤ k ≤ 23 | −12 ≤ k ≤ 12 | |
| −22 ≤ l ≤ 22 | −15 ≤ l ≤ 15 | −17 ≤ l ≤ 17 | −22 ≤ l ≤ 22 | −23 ≤ l ≤ 23 | |
| Reflections collected | 30401 | 31227 | 23609 | 56877 | 49201 |
| Independent reflections | 5447 [Rint = 0.0938] | 3453 [Rint = 0.0377] | 6537 [Rint = 0.0604] | 8833 [Rint = 0.0815] | 5706 [Rint = 0.1011] |
| Completeness to θmax | 100.0% | 100.0% | 99.2% | 100.0% | 99.1% |
| Data/restraints/parameters | 5447/0/314 | 3453/0/167 | 6537/414/482 | 8833/825/647 | 5706/252/380 |
| Goodness of fit on F2 | 1.042 | 1.084 | 1.172 | 1.157 | 1.330 |
| R 1 (I > 2σ(I)) | 0.0403 | 0.0145 | 0.0521 | 0.0879 | 0.1398 |
| wR2 (all data) | 0.0837 | 0.0355 | 0.0993 | 0.1983 | 0.3260 |
| Largest diff. peak and hole, e Å−3 | 1.156/−1.005 | 0.292/−0.355 | 1.438/–2.126 | 2.194/−1.663 | 2.880/−2.865 |
Computational details
Geometry optimizations of the clusters were performed in the gas phase using the range-separated hybrid DFT functional ωB97X.50 The basis set used was Ahlrichs'def2 split-valence, with relativistic ECP for Ru.51 The “restricted” approach was used in all the cases. The software used was Gaussian 09.52 The output was used for AIM and Mayer analyses,53 performed using the Multiwfn software, version 3.5.54 The Cartesian coordinates of the DFT-optimized structures are obtained in a separate .xyz file.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Bologna for financial support.References
- C. Cesari, J.-H. Shon, S. Zacchini and L. A. Berben, Chem. Soc. Rev., 2021, 50, 9503–9539 RSC.
- S. Zacchini, Eur. J. Inorg. Chem., 2011, 4125–4145 CrossRef CAS.
- W. Hieber and F. Leutert, Die Naturwissenschaften, 1931, 19, 360–361 CrossRef CAS.
- J. J. Brunet, Chem. Rev., 1990, 90, 1041–1059 CrossRef CAS.
- J. Lewis and P. R. Raithby, J. Organomet. Chem., 1995, 500, 227–237 CrossRef CAS.
- (a) C. R. Eady, B. F. G. Johnson and J. Lewis, J. Chem. Soc., Dalton Trans., 1977, 838–844 RSC; (b) S. A. R. Knox, J. W. Koepke, M. A. Andrews and J. Lewis, J. Am. Chem. Soc., 1975, 97, 3942–3947 CrossRef CAS.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Butterworth-Heinemann, Oxford, 1984 Search PubMed.
- The Chemistry of Metal Cluster Complexes, ed. D. F. Shriver, H. D. Kaesz and R. D. Adams, VCH, New York, 1990 Search PubMed.
- C. Femoni, M. C. Iapalucci, G. Longoni, S. Zacchini and S. Zarra, Inorg. Chem., 2009, 48, 1599–1605 CrossRef CAS PubMed.
- C. Femoni, M. C. Iapalucci, G. Longoni and S. Zacchini, Dalton Trans., 2011, 40, 8685–8694 RSC.
- C. Cesari, M. Bortoluzzi, C. Femoni, M. C. Iapalucci and S. Zacchini, Dalton Trans., 2021, 50, 9610–9622 RSC.
- (a) L. F. Dahl and J. F. Blount, Inorg. Chem., 1965, 4, 1373–1375 CrossRef CAS; (b) E. Isikola, T. A. Pakkanen, T. T. Pakkanen and T. Venalainen, Acta Chem. Scand., 1983, 37, 125–130 CrossRef.
- (a) Y.-C. Liu, W.-Y. Yeh, G.-H. Lee and S.-M. Penn, Organometallics, 2003, 20, 4163–4166 CrossRef; (b) F. Furno, T. Fox, M. Alfonso and H. Berke, Eur. J. Inorg. Chem., 2001, 1559–1565 CrossRef CAS; (c) J. A. Cabeza, M. Damonte, P. Garciá-Álvarez and E. Pérez-Carreño, Chem. Commun., 2013, 49, 2813–2815 RSC.
- J. A. Krause, U. Siriwardane, T. A. Salupo, J. R. Wermer, D. W. Knoeppel and S. G. Shore, J. Organomet. Chem., 1993, 454, 263–271 CrossRef CAS.
- M. Manassero, M. Sansoni and G. Longoni, J. Chem. Soc., Chem. Commun., 1976, 919–920 RSC.
- (a) J. A. Jensen, D. E. Fjare and W. L. Gladfelter, Inorg. Chem., 1983, 22, 1250–1253 CrossRef CAS; (b) S. V. Osintseva, N. A. Shtel'tser, A. S. Peregudov, A. Z. Kreindlin and F. M. Dolgushin, Polyhedron, 2018, 148, 147–160 CrossRef CAS.
- P. A. Dawson, B. F. G. Johnson, J. Lewis, D. A. Kaner and P. R. Raithby, J. Chem. Soc., Chem. Commun., 1980, 961–962 RSC.
- (a) R. Suter, A. A. Bhattacharyya, L.-Y. Hsu, J. A. Krause Bauer and S. G. Shore, Polyhedron, 1998, 17, 2889–2897 CrossRef CAS; (b) C. E. Ellul, J. P. Lowe, M. F. Mahon, P. R. Raithby and M. K. Whittlesey, Dalton Trans., 2018, 47, 4518–4523 RSC.
- B. F. G. Johnson, J. Lewis, P. R. Raithby, G. M. Sheldrick and G. Süss, J. Organomet. Chem., 1978, 162, 179–187 CrossRef CAS.
- P. J. Bailey, M. A. Beswick, B. F. G. Johnson, J. Lewis, M. McPartlin, P. R. Raithby and M. C. Ramirez de Arellano, J. Chem. Soc., Dalton Trans., 1996, 3515–3520 RSC.
- D. Braga, J. Lewis, B. F. G. Johnson, M. McPartlin, W. J. H. Nelson and M. D. Vargas, J. Chem. Soc., Chem. Commun., 1983, 241–243 RSC.
- T. Beringhelli, E. Cairati, C. Dragonetti, S. Galli, E. Lucentini, D. Roberto, A. Sironi and R. Ugo, Inorg. Chim. Acta, 2003, 354, 79–89 CrossRef CAS.
- C. Femoni, M. C. Iapalucci, F. Kaswalder, G. Longoni and S. Zacchini, Coord. Chem. Rev., 2006, 250, 1580–1604 CrossRef CAS.
- (a) M. McPartlin and W. J. H. Nelson, J. Chem. Soc., Dalton Trans., 1986, 1557–1563 RSC; (b) P. F. Jackson, B. F. G. Johnson, J. Lewis, M. McPartlin and W. J. H. Nelson, J. Chem. Soc., Chem. Commun., 1978, 920–921 RSC; (c) C. E. Ellul, M. G. Mahon, O. Saker and M. K. Whittlesey, Angew. Chem., Int. Ed., 2007, 46, 6343–6345 CrossRef CAS PubMed.
- R. D. Wilson, S. M. Wu, R. A. Love and R. Bau, Inorg. Chem., 1978, 17, 1271–1280 CrossRef CAS.
- K. E. Inkrott and S. G. Shore, Inorg. Chem., 1979, 18, 2817–2821 CrossRef CAS.
- A. A. Bhattacharyya, C. C. Nagel and S. G. Shore, Organometallics, 1983, 2, 1187–1193 CrossRef CAS.
- G. Hogarth, S. E. Kabir and E. Nordlander, Dalton Trans., 2010, 39, 6153–5174 RSC.
- M. Zaun, A. Goeppert, R. May, R. Haiges, G. K. S. Prakash and G. A. Olah, ChemSusChem, 2011, 4, 1241–1248 CrossRef PubMed.
- A. F. Abdel-Magied, Y. Theibich, A. K. Singh, A. Rahaman, I. Doverbratt, A. K. Raha, M. Haukka, M. G. Richmond and E. Nordlander, Dalton Trans., 2020, 49, 4244–4256 RSC.
- V. Moberg, R. Duquesne, S. Contaldi, O. Röhrs, J. Nachtigall, L. Damoense, A. T. Hutton, M. Green, M. Monari, D. Santelia, M. Haukka and E. Nordlander, Chem. – Eur. J., 2012, 18, 12458–12478 CrossRef CAS PubMed.
- M. Shieh, Y.-Y. Chu, L.-F. Jang and C.-H. Ho, Inorg. Chem., 2014, 53, 4284–4286 CrossRef CAS PubMed.
- U. Matteoli, M. Bianchi, P. Frediani, F. Piacenti, C. Botteghi and M. Marchetti, J. Organomet. Chem., 1984, 263, 243–246 CrossRef CAS.
- M. Bianchi, G. Menchi, P. Frediani, U. Matteoli and F. Piacenti, J. Organomet. Chem., 1983, 247, 89–94 CrossRef CAS.
- U. Matteoli, V. Beghetto and A. Scrivanti, J. Mol. Catal. A: Chem., 1996, 109, 45–50 CrossRef CAS.
- P. Homanen, R. Persson, M. Haukka, T. A. Pakkanen and E. Nordlander, Organometallics, 2000, 19, 5568–5574 CrossRef CAS.
- (a) I. del Rosal, T. Gutmann, B. Walaszek, I. C. Gerber, B. Chaudret, H.-H. Limbach, G. Buntkowsky and R. Poteau, Phys. Chem. Chem. Phys., 2011, 13, 20199–20207 RSC; (b) T. Gutmann, I. del Rosal, B. Chaudret, R. Poteau, H.-H. Limbach and G. Buntkowsky, ChemPhysChem, 2013, 14, 3026–3033 CrossRef CAS PubMed.
- T. Gutmann, B. Walaszek, X. Yeping, M. Wächtler, I. del Rosal, A. Grünberg, R. Poteau, R. Axet, G. Lavigne, B. Chaudret, H.-H. Limbach and G. Buntkowsky, J. Am. Chem. Soc., 2010, 132, 11759–11767 CrossRef CAS PubMed.
- M. Bortoluzzi, I. Ciabatti, C. Cesari, C. Femoni, M. C. Iapalucci and S. Zacchini, Eur. J. Inorg. Chem., 2017, 3135–3143 CrossRef CAS.
- P. H. M. Budzelaar, gNMR for Windows, NMR Simulation Program (5.0.6.0), IvorySoft, Adept Scientific, Letchworth, U.K, 2006 Search PubMed.
- J. W. Koepke, J. R. Johnson, S. A. R. Knox and H. D. Kaesz, J. Am. Chem. Soc., 1975, 97, 3947–3952 CrossRef CAS.
- L. Maurette, B. Donnadieu and G. Lavigne, Angew. Chem., Int. Ed., 1999, 38, 3707–3710 CrossRef CAS PubMed.
- R. Pattacini, G. Predieri, A. Tiripicchio, C. Mealli and A. D. Phillips, Chem. Commun., 2006, 1527–1529 RSC.
- F. Ragaini, J.-S. Song, D. L. Ramage, G. L. Geoffrey, G. A. P. Yap and A. L. Rheingold, Organometallics, 1995, 14, 387–400 CrossRef CAS.
- C. Cesari, T. Funaioli, B. Berti, C. Femoni, M. C. Iapalucci, F. M. Vivaldi and S. Zacchini, Inorg. Chem., 2021, 60, 16713–16725 CrossRef CAS PubMed.
- E. Keller, SCHAKAL99, University of Freiburg, Germany, 1999 Search PubMed.
- A. Béguin, J. M. Soulié and G. Süss-Fink, Inorg. Synth., 1998, 32, 268–270 Search PubMed.
- G. M. Sheldrick, SADABS, Program for empirical absorption correction, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- (a) Y. Minenkov, Å. Singstad, G. Occhipinti and V. R. Jensen, Dalton Trans., 2012, 41, 5526–5541 RSC; (b) J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC; (c) I. C. Gerber and J. G. Ángyán, Chem. Phys. Lett., 2005, 415, 100–105 CrossRef CAS.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (b) D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian Inc., Wallingford, CT, 2010 Search PubMed.
- (a) F. Jensen, Introduction to Computational Chemistry, Wiley, Chichester, 2nd edn, 2007 Search PubMed; (b) C. J. Cramer, Essentials of Computational Chemistry, Wiley, Chichester, 2nd edn, 2004 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2115041–2115045. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03622j |
| This journal is © The Royal Society of Chemistry 2022 |