
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigations of the reactivity, stability and biological activity of halido (NHC)gold(I) complexes†
Sina Katharina
Goetzfried‡
 a,
Paul
Kapitza‡
a,
Caroline Marie
Gallati
a,
Anna
Nindl
bc,
Monika
Cziferszky
a,
Martin
Hermann
d,
Klaus
Wurst
e,
Brigitte
Kircher
*bc and
Ronald
Gust
*a
a,
Paul
Kapitza‡
a,
Caroline Marie
Gallati
a,
Anna
Nindl
bc,
Monika
Cziferszky
a,
Martin
Hermann
d,
Klaus
Wurst
e,
Brigitte
Kircher
*bc and
Ronald
Gust
*a
aInstitute of Pharmacy, Department of Pharmaceutical Chemistry, University of Innsbruck, Innrain 80-82, 6020 Innsbruck, Austria
bDepartment of Internal Medicine V (Hematology and Oncology), Medical University Innsbruck, Anichstraße 35, 6020 Innsbruck, Austria
cTyrolean Cancer Research Institute, Innrain 66, 6020 Innsbruck, Austria
dDepartment of Anesthesiology and Critical Care Medicine, Medical University Innsbruck, Anichstraße 35, 6020 Innsbruck, Austria
eInstitute for General, Inorganic and Theoretical Chemistry, University of Innsbruck, Innrain 80/82, 6020 Innsbruck, Austria
First published on 8th December 2021
Abstract
The significance of the halido ligand (Cl−, Br−, I−) in halido[3-ethyl-4-phenyl-5-(2-methoxypyridin-5-yl)-1-propyl-1,3-dihydro-2H-imidazol-2-ylidene]gold(I) complexes (2–4) in terms of ligand exchange reactions, including the ligand scrambling to the bis[3-ethyl-4-phenyl-5-(2-methoxypyridin-5-yl)-1-propyl-1,3-dihydro-2H-imidazol-2-ylidene]gold(I) complex (5), was evaluated by HPLC in acetonitrile/water = 50:50 (v/v) mixtures. In the presence of 0.9% NaCl, the bromido (NHC)gold(I) complex 3 was immediately transformed into the chlorido (NHC)gold(I) complex 2. The iodido (NHC)gold(I) complex 4 converted under the same conditions during 0.5 h of incubation by 52.83% to 2 and by 8.77% to 5. This proportion remained nearly constant for 72 h. The halido (NHC)gold(I) complexes also reacted very rapidly with 1 eq. of model nucleophiles, e.g., iodide or selenocysteine (Sec). For instance, Sec transformed 3 in the proportion 73.03% to the (NHC)Au(I)Sec complex during 5 min of incubation. This high reactivity against this amino acid, present in the active site of the thioredoxin reductase (TrxR), correlates with the complete inhibition of the isolated TrxR enzyme at 1 μM. Interestingly, in cellular systems (A2780cis cells), even at a 5-fold higher concentration, no increased ROS levels were detected. The concentration required for ROS generation was about 20 μM. Superficially considered, the antiproliferative and antimetabolic activities of the halido (NHC)Au(I) complexes correlate with the reactivity of the Au(I)–X bond (2 < 3 < 4). However, it is very likely that degradation products formed during the incubation in cell culture medium participated in the biological activity. In particular, the high-cytotoxic [(NHC)2Au(I)]+ complex (5) distorts the results.
Introduction
N-Heterocyclic carbene (NHC) gold(I) complexes have attracted attention as potential anticancer agents.1–5 The “Web of Science” data registered an increase of more than 100% for new publications containing the keywords “gold”, “NHC” and “anticancer” within the last decade. The first (NHC)gold(I) complex was isolated in 1973 by Minghetti et al.6 However, it took more than thirty years until the group of Filipovska discovered the mitochondrial accumulation of bis(1,3-dialkyl-1H-imidazol-2-ylidene)gold(I) complexes in MDA-MB-231 and MDA-MB-468 breast cancer cell lines and the inhibition of thioredoxin reductase (TrxR).7 So far, more than 300 (NHC)gold(I) complexes have been reported and screened against a panel of tumor cells, demonstrating exceptional cytostatic and cytotoxic effects. NHCs have received more attention as ligands for the design of gold complexes, because it is supposed that they form strong and stable bonds to the gold center. Their simple synthesis enables fine-tuning of the complex properties owing to ligand modifications. The changes in hydrophilicity and lipophilicity support the optimization of the complexes for medical purposes.The class of (NHC)gold complexes includes heteroleptic (NHC)Au(I)X (X = e.g., Cl−, Br−, I−, thiol, phosphine) and homoleptic [(NHC)2Au(I)]+ derivatives as well as (NHC)gold(III) species of various structures.1,2,4,5,8–10 The majority of halido (NHC)gold(I) complexes bear either chloride or bromide as a leaving group.11–27 Iodide is rarely used, although (NHC)Au(I)I species have been shown to be highly effective against tumor cells.9,26–28
The mode of action might be multifaceted, but TrxR11,16,24,25,28–33 and glutathione reductase8,24,34 have been discussed as preferred targets. TrxR contains a Gly–Cys–Sec–Gly sequence (reduced form), forming a selenylsulfide bridge (oxidized form) during the reduction of thioredoxin.
In the reduced form, the R-SeH and R-SH groups strongly differ in their acidity. The pKa of the selenol group in selenocysteine (Sec) amounts to 5.2 and documents an ionization grade >99% at physiological pH (7.4). In contrast, the SH group of Cys (pKa = 8.5) preferably exists in its undissociated form. Accordingly, the selenolate anion represents a stronger nucleophile and makes Sec the main target of (NHC)gold(I/III) complexes in TrxR to prevent substrate binding and transformation.35
A variety of cancer cell lines overexpress TrxR to counteract oxidative stress.36–38 Therefore, the inhibition of this NADPH-dependent disulfide reductase affects the intracellular redox balance, leading to an increased content of reactive oxygen species (ROS) and driving the cells into apoptosis.
In previous papers, we reported on the design and in vitro activity of bromido[3-ethyl-4-aryl-5-(2-methoxypyridin-5-yl)-1-propyl-1,3-dihydro-2H-imidazol-2-ylidene]gold(I) complexes and related [(NHC)2Au(I)]+ species.39,40 The latter caused significantly higher antiproliferative effects (IC50 values ≈ 0.5 μM) than their (NHC)Au(I)Br counterparts (IC50 = 3–6 μM). This finding is of particular importance because the (NHC)Au(I)Br complexes undergo ligand rearrangement reactions to the related [(NHC)2Au(I)]+ derivatives in the presence of water. The driving forces are aurophilic interactions and the formation of [(NHC)Au(I)Br]2 dimers.41
In continuation of this study, we synthesized the 4-phenyl-substituted (NHC)Au(I)X analogue with X = Cl− (2), Br− (3) and I− (4) as leaving group and studied their ligand exchange reactions, their reactivity against model nucleophiles (iodide, Sec, Cys, N-acetyl cysteine (NAC)) and their stability under physiological conditions (0.9% NaCl solution). The results are highly relevant and must be considered within the context of the interpretation of in vitro results.
Complexes 2–5 were investigated for antiproliferative and antimetabolic activity in A2780 human ovarian cancer cells (resistant (A2780cis) and sensitive (A2780wt) to Cisplatin), HL 60 acute myeloid leukemia cells and human primary fibroblasts. All of the complexes were further tested for TrxR inhibition and generation of reactive oxygen species (ROS) in A2780cis cells.
Results and discussion
Synthesis and structural characterization
The NHC ligands 1a and 1b were synthesized as already described.39 The iodido (NHC)gold(I) complex 4 was prepared in dichloromethane (DCM)/methanol (MeOH) = 50:50 (v/v) from imidazolium iodide 1a and Ag2O/Me2SAu(I)Cl in the presence of KI (5 eq.) (Scheme 1, method A, yield 34%). The attempt to obtain the analytically pure bromido (NHC)gold(I) complex 3 using an excess of KBr failed.39 The product always contained 10–20% of the iodido analogue 4, formed in the reaction mixture according to Scheme 2(1).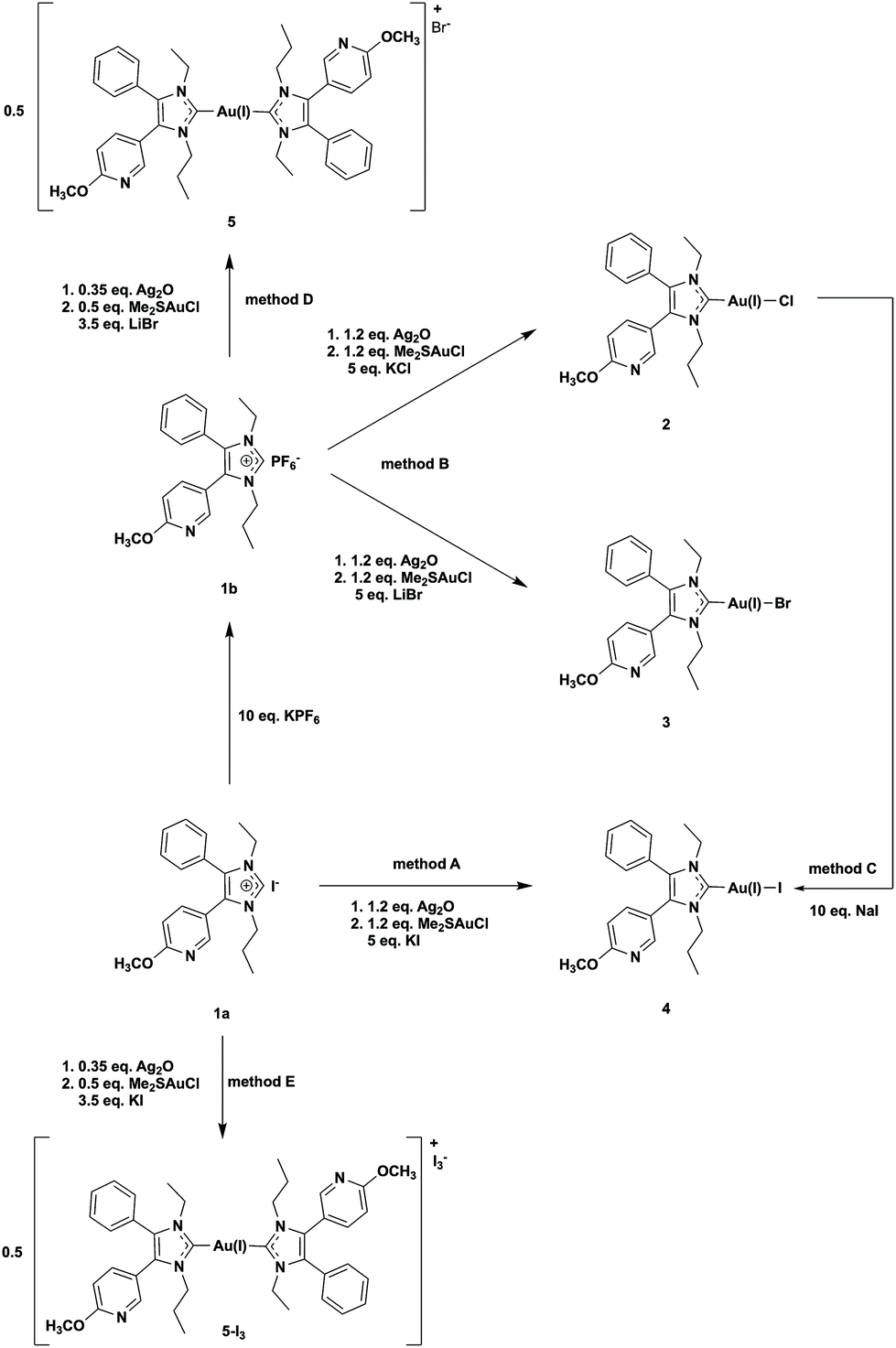 | ||
| Scheme 1 Synthetic routes to obtain complexes 2–5. | ||
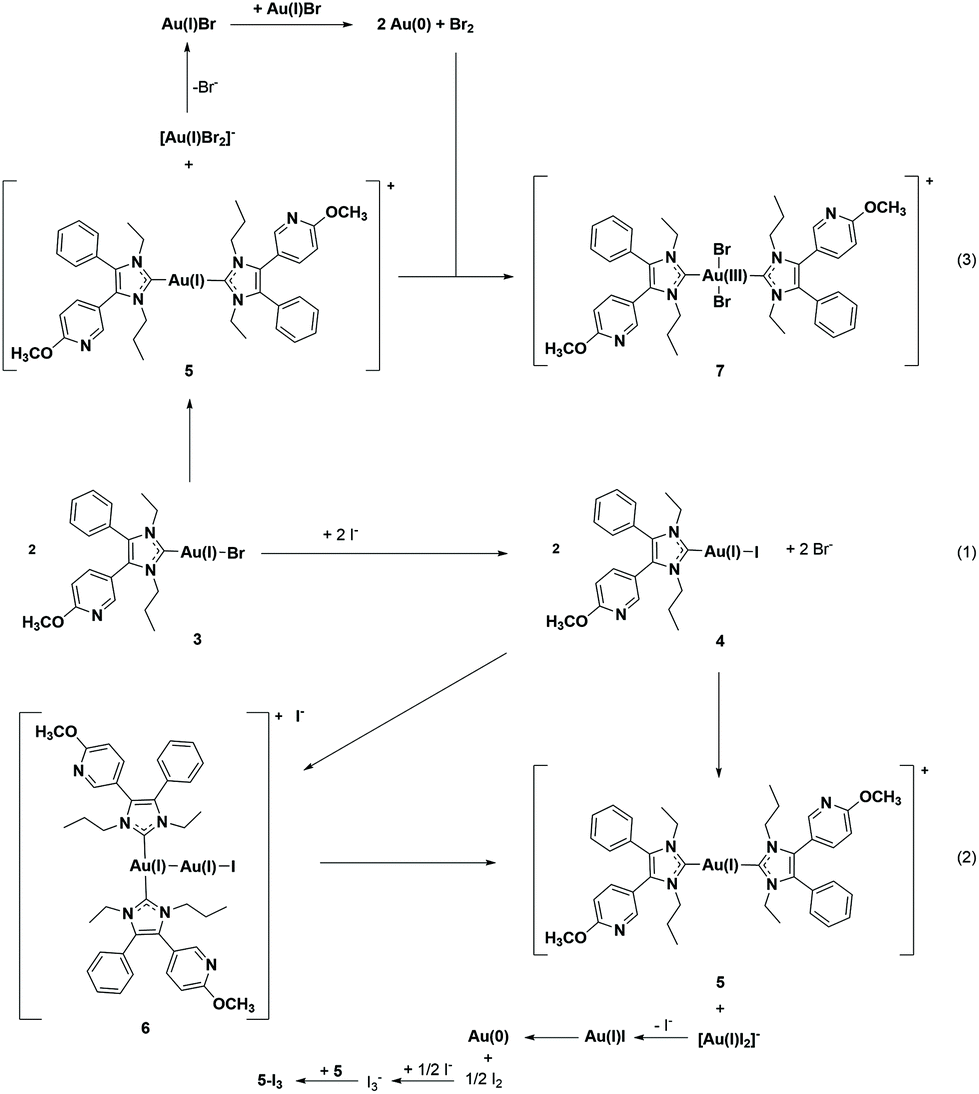 | ||
| Scheme 2 Ligand exchange reactions of halido (NHC)gold(I) complexes. | ||
This finding clearly indicates that iodide is a strong nucleophile for substitution reactions at (NHC)Au(I)X complexes. Therefore, it is necessary to prevent the presence of iodide to avoid contamination with the iodido (NHC)gold(I) complex.
Thus, 1b with the non-coordinative PF6− counter ion was utilized as a precursor. The use of an excess (5 eq.) of KCl or LiBr enabled the isolation of 2 and 3 (Scheme 1, method B).
The high reactivity of iodide against gold(I) complexes also allowed the synthesis of 4 by reacting the chlorido (NHC)gold(I) complex 2 with 10 eq. NaI (Scheme 1, method C). The yield was nearly quantitative (98%) with only marginal amounts of 5.
The [(NHC)2Au(I)]+ species 5 was prepared by the reaction of 1b with 0.35 eq. of Ag2O and 0.5 eq. of Me2SAu(I)Cl in the presence of 3.5 eq. of LiBr (Scheme 1, method D). If 1a and KI were used in this reaction course (Scheme 1, method E), 5-I3 crystallized from a MeOH/water = 50:50 (v/v) mixture with triiodide as a counter ion (Fig. 1, for details see ref. 40). The presence of this anion indicates that iodine was formed during the reaction, which subsequently reacted with iodide to triiodide (Scheme 2(2)).
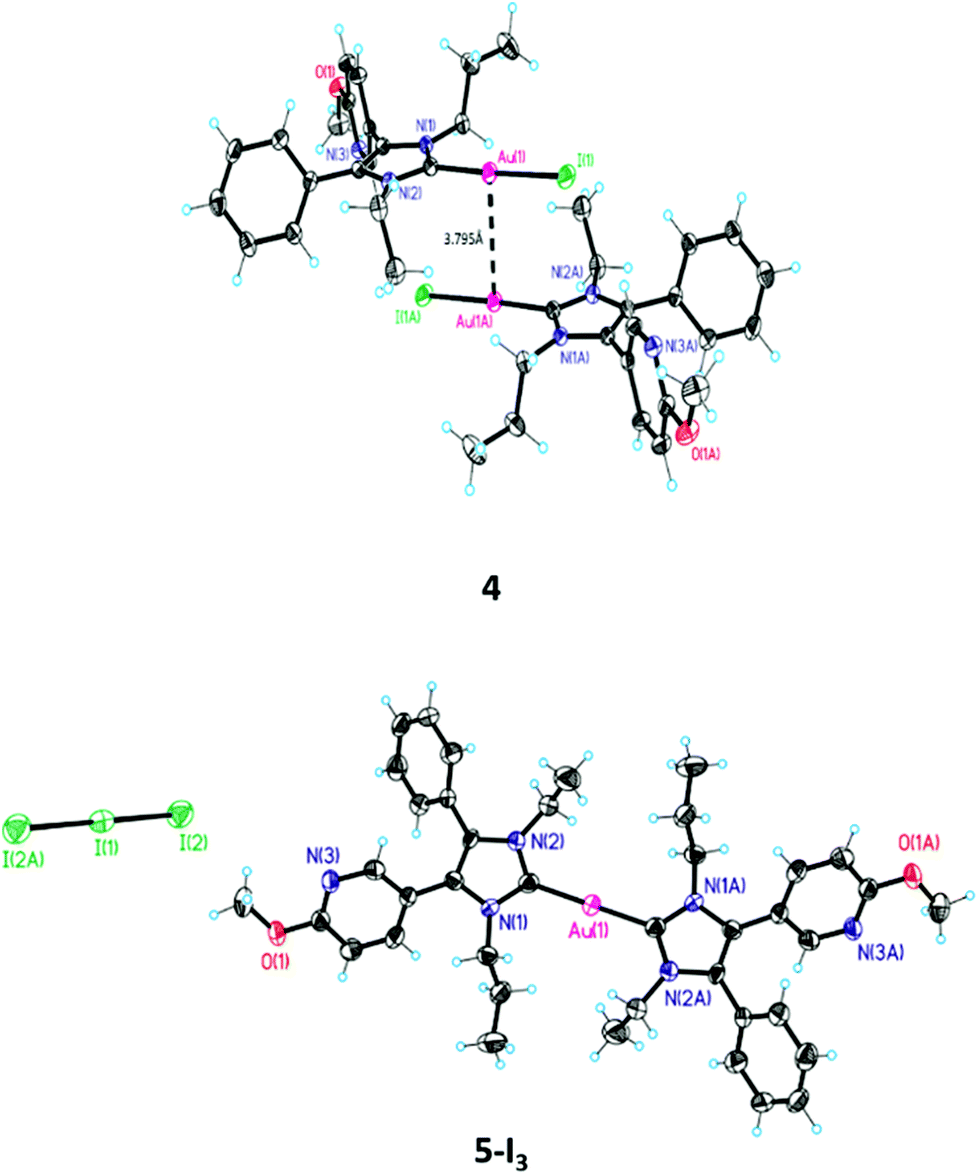 | ||
| Fig. 1 ORTEP of 4 and 5-I3. | ||
Interestingly, although iodine/triiodide was formed during a similar ligand scrambling of 4, the oxidation product [(NHC)2Au(III)I2]+ was never detected during the HPLC analyses.
The complexes were characterized by NMR spectroscopy and high-resolution electrospray ionization mass spectrometry (ESI-MS). Unfortunately, 1H NMR spectra were not meaningful, because the coordinated halide did not influence the resonances of the bound NHC ligand (Fig. S1–S4, ESI†). In contrast, the signal of the C2 carbon in the 13C NMR spectra shifted upon coordination to Au–X from 135 ppm (1a/1b) to 170 (2), 173 (3) and 180 (4) ppm (Fig. S5–S8, ESI†), which is in accordance with the data reported in the literature.25,39
The structure of 4 was further confirmed by crystal structure analysis. It was possible to obtain crystals from a DCM/MeOH mixture (for selected X-ray data see Table 1). The ORTEP of 4 depicted in Fig. 1 displays a nearly linear arrangement (177.49°) of the substituents at the gold(I) center. The NHC–Au(I) distance is 2.02 Å and that of Au(I)–I is 2.55 Å. The related distances for 3 are 2.00 and 2.40 Å.39
| Crystal system | Monoclinic |
| Space group | P21/n (no. 14) |
| Unit cell dimensions | a = 10.2074(2) Å |
| α = 90° | |
| b = 16.3078(4) Å | |
| β = 99.7490(10)° | |
| c = 12.9494(3) Å | |
| γ = 90° | |
| Volume | 2124.43(8) Å3 |
| Z | 4 |
| Density (calculated) | 2.018 mg m−3 |
| Absorption coefficient | 8.391 mm−1 |
| F(000) | 1216 |
| Crystal size | 0.28 × 0.12 × 0.08 mm3 |
| θ range for data collection | 2.38 to 26.00° |
| Index ranges | −12 ≤ h ≤ 12, 19 ≤ k ≤ 20, −15 ≤ l ≤ 15 |
| Reflections collected | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 716 716 |
| Independent reflections | 4168 [R(int) = 0.0429] |
| Absorption correction | None |
| Data/restraints/parameters | 4168/0/235 |
| Goodness-of-fit on F2 | 1.057 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0312, wR2 = 0.0713 |
| R indices (all data) | R 1 = 0.0356, wR2 = 0.0731 |
| Largest difference peak and hole | 2.160 and −1.391 e Å−3 |
Comparison with X-ray structures of other halido[1,3-dihydro-2H-imidazol-2-ylidene]gold(I) complexes42–44 documents only a marginal difference in the NHC–Au(I) distance (NHC–Au(I): 1.94–2.00 Å). The bond length of the Au(I)–X moiety increased with the atomic radius (Au(I)–Cl (∼2.30 Å) < Au(I)–Br (∼2.40 Å) < Au(I)–I (∼2.55 Å)) and complies with the reactivity of the Au(I)–X bond as well as the suitability of the halide as a leaving group (Cl− < Br− < I−).
The identification of 5-I3 by X-ray analysis agrees with our hypothesis on the degradation of (NHC)Au(I)Br complexes in acetonitrile (ACN)/water mixtures. It was postulated that after the ligand scrambling of two molecules giving [(NHC)2Au(I)]+ and [Au(I)Br2]−, the latter undergoes an internal redox reaction to yield elemental gold and Br2. Then Br2 oxidizes [(NHC)2Au(I)]+, 5, to [(NHC)2Au(III)Br2]+, 7 (Scheme 2(3)).41
The presence of triiodide further suggests that during the synthesis of 5, 4 is formed as an intermediate and the final step is not a simple substitution reaction ((NHC)Au(I)I + NHC → [(NHC)Au(I)(NHC)]+ + I−), but a ligand scrambling with the formation of [Au(I)I2]− (Scheme 2(2)) as a side product.
Therefore, the complexes 2–4 were further analyzed by HPLC, using an RP-C18 column and gradient elution of ACN/water (0.1% trifluoroacetic acid (TFA)) from 70:30 to 90:10. The retention times of 2 (tret = 4.81 min), 3 (tret = 5.39 min) and 4 (tret = 6.82 min) document an increasing hydrophobicity dependent on the halide used. The UV–vis spectra obtained during the HPLC runs (Fig. S9–S12, ESI†) showed similar transmissions, with the feature that the metal-to-ligand charge transfer transition is shifted from 254/255 nm in 3 and 4 to 265 nm in 5.
All complexes (concentration 0.250 mM), dissolved in ACN (t = 0 h), were of high purity (>95%, Table S1, ESI†) with only small amounts of the [(NHC)2Au(I)]+ complex 5. Incubation of the solutions for 72 h at room temperature (rt) led to marginal degradation of 2 (0.72%) and 3 (0.96%), but a 15.17% conversion of 4 to 5 (Table S1, ESI†).
Addition of water to the ACN solution (ACN/water = 50:50, (v/v), complex concentration 0.250 mM) only slightly supported the conversion of 2. After 72 h, the proportion of 5 amounted to 9.87%. In contrast, 3 was converted to 5 by 25.12%. The highest transformation to 5 showed 4 with 44.41%. Interestingly, only for 3 a gold(III) species ([(NHC)2Au(III)Br2]+, 7, Scheme 2(3)) was observed as a degradation product (t = 72 h: 10.77%, Fig. 2B).
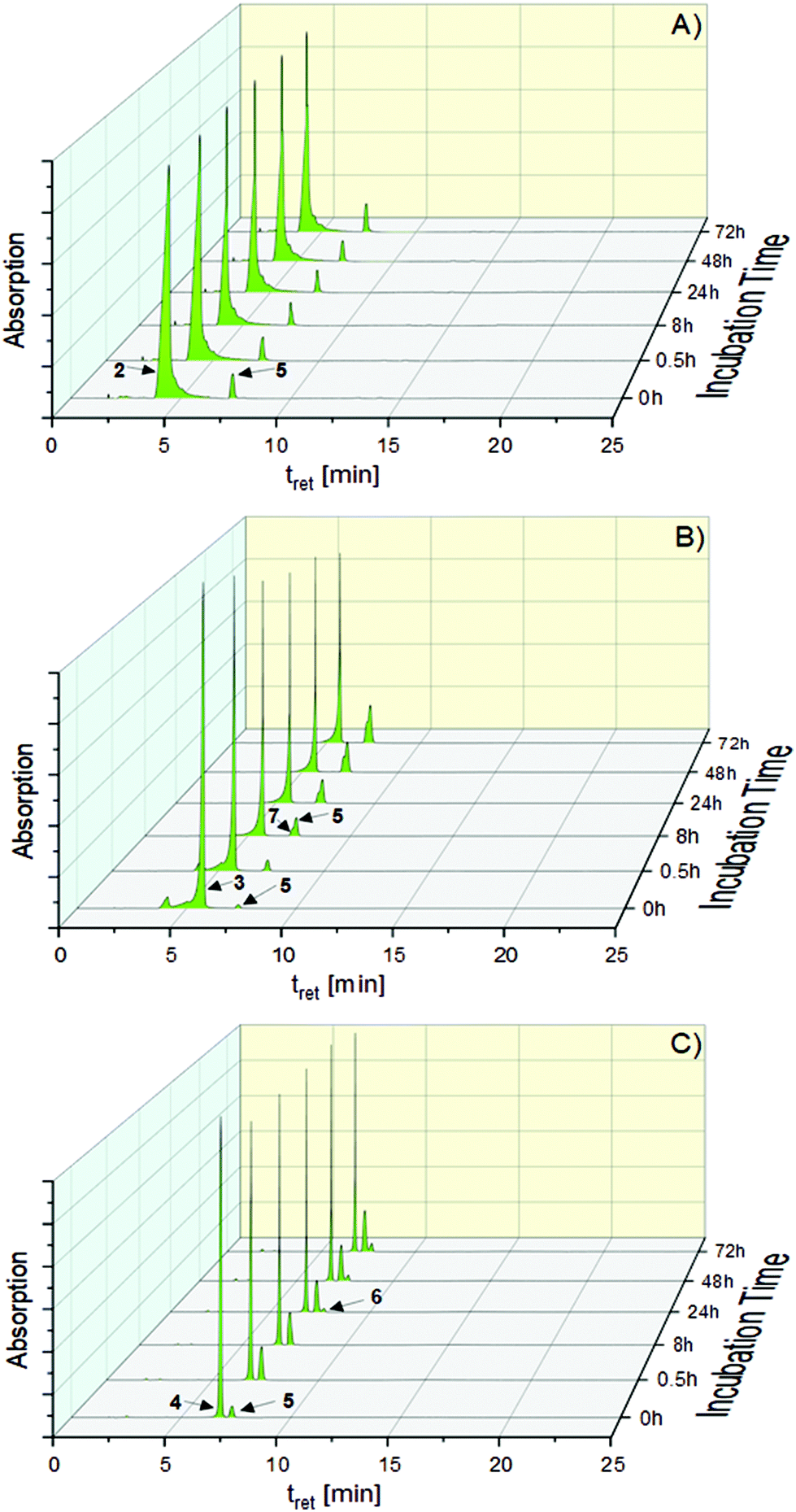 | ||
| Fig. 2 HPLC chromatograms of 2 (A), 3 (B) and 4 (C) measured during 72 h of incubation in ACN/water = 50:50 (v/v) at rt. | ||
A T-shaped intermediate was detected in the cases of 3 and 4 during the HPLC analyses, although in different quantities. While it was formed from 3 only in traces (see also ref. 41 and 45), relatively high amounts were found for 4. The relevant peak of 6 at tret = 7.67 min was observed in the chromatograms for the first time after 12 h (1.92%) and the amount increased to 8.09% during incubation for 72 h (Fig. 2C).
To verify the presence of the intermediate 6, the solution of 4 was additionally investigated using ESI-MS in the positive mode (Fig. S13 and S14, ESI†). The mass spectrum (positive mode) at t = 0 h included as main peaks the cations [(NHC)2Au(I)]+, 5 (m/z = 839.3346), and [(NHC)2Au(I)–Au(I)I]+, 6 (m/z = 1163.2059). During the time of incubation, the distribution shifted in favour of 5 with a higher transformation rate.
These data demonstrate a clear dependence of ligand scrambling on the bound halide ((NHC)Au(I)Cl < (NHC)Au(I)Br < (NHC)Au(I)I). Longer Au–X bonds seem to enhance aurophilic interactions in solution and consequently support the formation of the trigonal transition state at both gold centers followed by conversion to the T-shaped intermediate (see also ref. 41 and 45).
For the interpretation of the biological results, it is necessary to know more about the stability of (NHC)Au(I)X complexes in the first hours of incubation, because it is well known that during this time cellular accumulation is nearly completed.25,28,30,31 Therefore, degradation was followed immediately after dissolution (ACN/water = 50:50 (v/v)) and during the following 8 h incubation period using HPLC (Table S1, ESI†). It is obvious that the first contact with water led to rapid ligand scrambling. After 0.5 h, 5.11% of 2, 4.73% of 3 and 23.99% of 4 were transformed to 5. These portions increased during 8 h to 5.46% (2), 13.01% (3) and 25.60% (4).
The behavior in the presence of 0.9% NaCl is more important. Thus, the respective complex solution in ACN was diluted with 1.8% NaCl in water to achieve a 50:50 (v/v) mixture (complex concentration 0.250 mM).
The chlorido (NHC)gold(I) complex 2 was stable during incubation for 72 h (t = 0 h: 97.68%→97.96% (t = 72 h), Table S1, ESI†). In contrast, 3 reacted immediately after dissolution to 2 (t = 0 h: 99.74%). This proportion remained constant for 72 h (99.07%). Further degradation to the [(NHC)2Au(I)]+ complex 5 did not take place (Fig. 3).
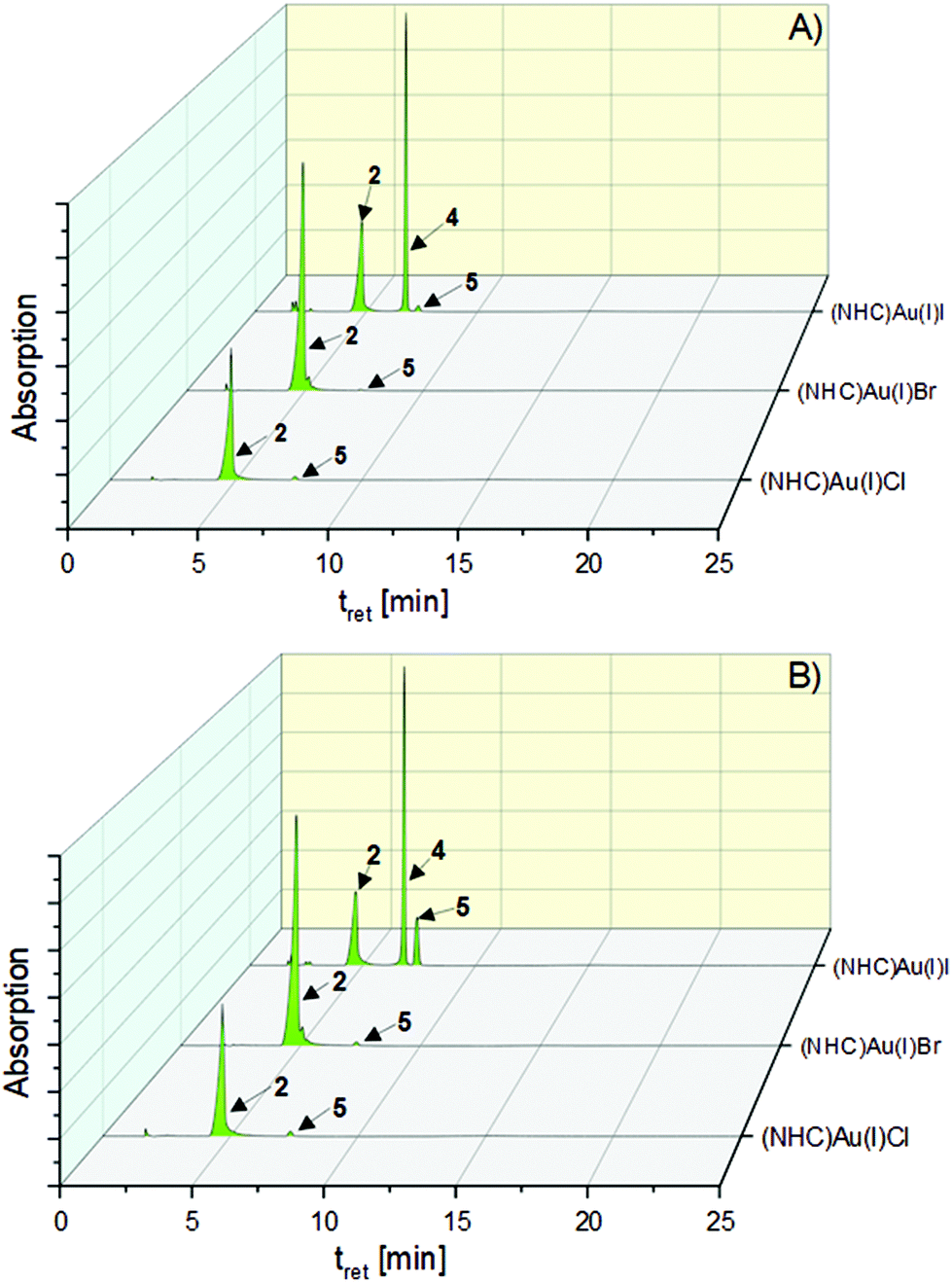 | ||
| Fig. 3 HPLC chromatograms of the halido (NHC)gold(I) complexes 2–4 after 0 h (A) and 72 h (B) of incubation in 0.9% NaCl solution (ACN/water = 50:50 (v/v)) at rt. | ||
The transformation of 4 was more sophisticated. Directly after addition of the aqueous NaCl solution (t = 0 h), 55.36% of 2 and 1.76% of 5 were formed. Within 1 h, the proportion slightly changed to 50.69% (2) and 11.93% (5), which remained nearly constant for 72 h (Fig. 4, 51.35% (5) and 14.07% (4), Table S1, ESI†).
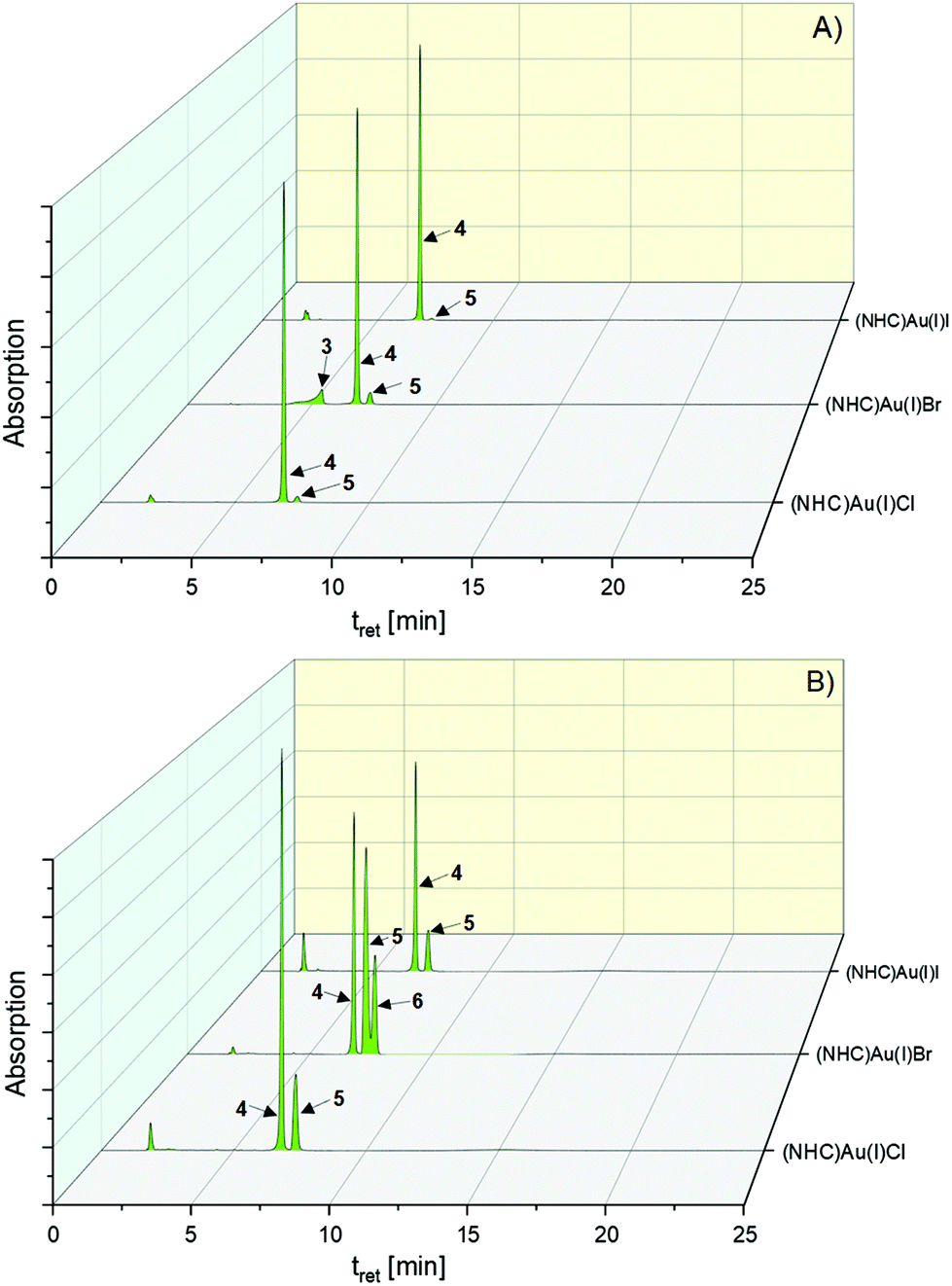 | ||
| Fig. 4 HPLC chromatograms of the halido (NHC)gold(I) complexes 2–4 dissolved in ACN/water = 50:50 (v/v) in the presence of 1 eq. KI at (A) t = 0 h and (B) t = 72 h. | ||
From these findings a question arises about the quality of the halides as a leaving group at the gold(I) center. It is well known from platinum(II) and other complexes that the release of bound halide increases with the atomic radius (Cl− < Br− < I−), more specifically with the bond length to the metal.
The reactivity of metal complexes can be easily investigated using iodide as a model nucleophile. It exchanges the leaving group of platinum(II) complexes, comparable with the nucleobase guanine.46 Therefore, we modified this assay to estimate the reactivity of the halido (NHC)gold(I) complexes towards nucleophiles. The complexes were dissolved in ACN and water containing KI in adequate concentrations was added to achieve a 50:50 (v/v) mixture with 0.250 mM complex concentration.
The exchange of the leaving group in 2 and 3 was very fast in the presence of a 20-fold excess of iodide (Table S1, ESI†). Interestingly, both complexes rapidly reacted at equimolar concentrations with iodide to 4 (Fig. 4 and Table S1, ESI†).
In case of the chlorido (NHC)gold(I) complex 2, only 4 (95.81%) and 5 (4.19%) were detected in the HPLC chromatograms immediately after combination of the solutions (t = 0 h). The degradation in terms of ligand scrambling during 8 h of incubation then followed the same kinetics (→24.20% (5)) as described for 4 dissolved in pure ACN/water (→25.60% (5)).
The behavior of the bromido (NHC)gold(I) complex 3 slightly differed. It initially reacted (t = 0 h) with iodide incompletely to 4 (78.78%) and 5 (5.42%), but reached nearly the same distribution as 2 after 0.5 h (80.20% (4) and 19.80% (5)). During 8 h of incubation, however, formed 4 strongly degraded into 5 (→39.44%) as well as the T-shaped intermediate 6 (→19.95%). The amount of 6 was even distinctly higher than that formed from 4 dissolved in ACN/water. Interestingly, an increased excess of KI (20 eq.) prevented the formation of 6 and 28.31% converted to 5.
The iodido (NHC)gold(I) complex 4 was converted in the proportion of 23.88% to 5 during 8 h of incubation in the presence of 1 eq. KI. The intermediate 6 was not detected in the HPLC chromatograms.
The [(NHC)2Au(I)]+ complex 5 was stable under all of the conditions used. Even incubation with KI did not lead to degradation (Fig. S15, ESI†). The stability of 5 (and related [(NHC)2Au(I)]+ complexes40) in 0.9% NaCl solution points to only marginal degradation under physiological conditions, e.g., in cell culture medium. These findings are consistent with those of other groups that have performed stability tests of [(NHC)2Au(I)]+ complexes in DMSO/DMF and water.47,48
It is worth mentioning that the main turnover was completed after the first few minutes of incubation with only a little further transformation within 24 h.
Biological evaluation
In order to estimate the significance of the bound halide for the biological activity, complexes 2–4 were investigated in A2780 human ovarian cancer cells (resistant and sensitive to Cisplatin; Table 2), as well as in HL 60 acute myeloid leukemia cells (Fig. S16 and S17, ESI†). Additionally, human primary fibroblasts (Fig. 5) were used to evaluate tumor cell selectivity. Cisplatin and/or Auranofin served as positive controls. Furthermore, the potency to inhibit the activity of the isolated TrxR enzyme (Fig. 6) and to induce ROS generation in A2780cis cells was investigated (Fig. 7). Finally, the binding to Sec was studied by HPLC and ESI-MS (Fig. 8). In a reference experiment NAC and Cys were investigated too.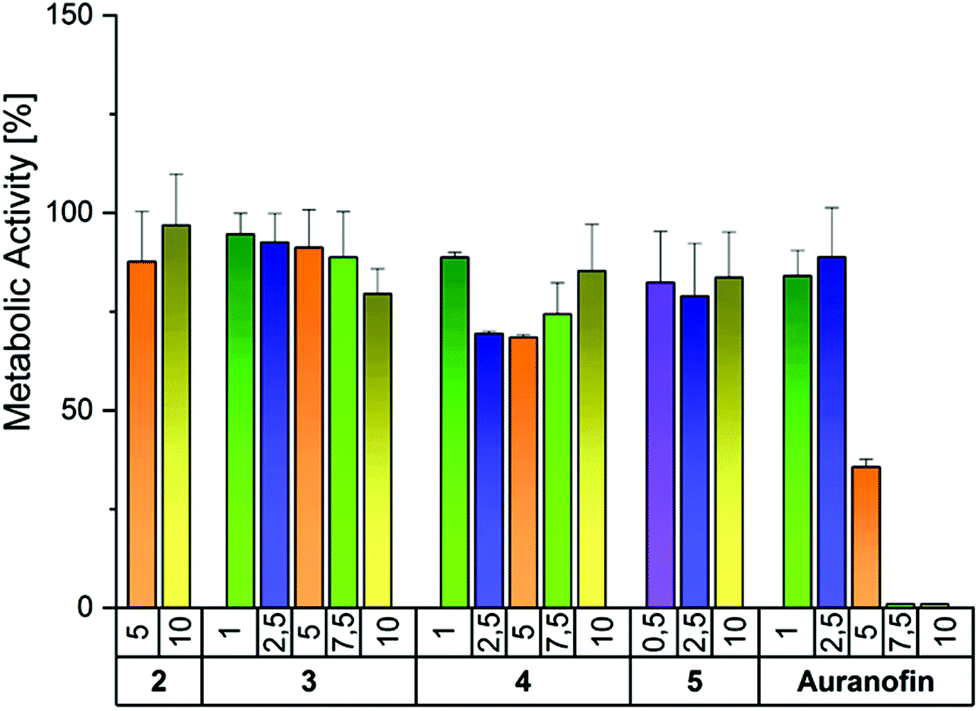 | ||
| Fig. 5 Antimetabolic activity of complexes 2–5 as well as Auranofin in human primary fibroblasts after an incubation time of 72 h (concentration 1–10 μM). Metabolic activity in the absence of the compounds was set to 100%. The mean metabolic activity ± standard error was calculated from three independent experiments. | ||
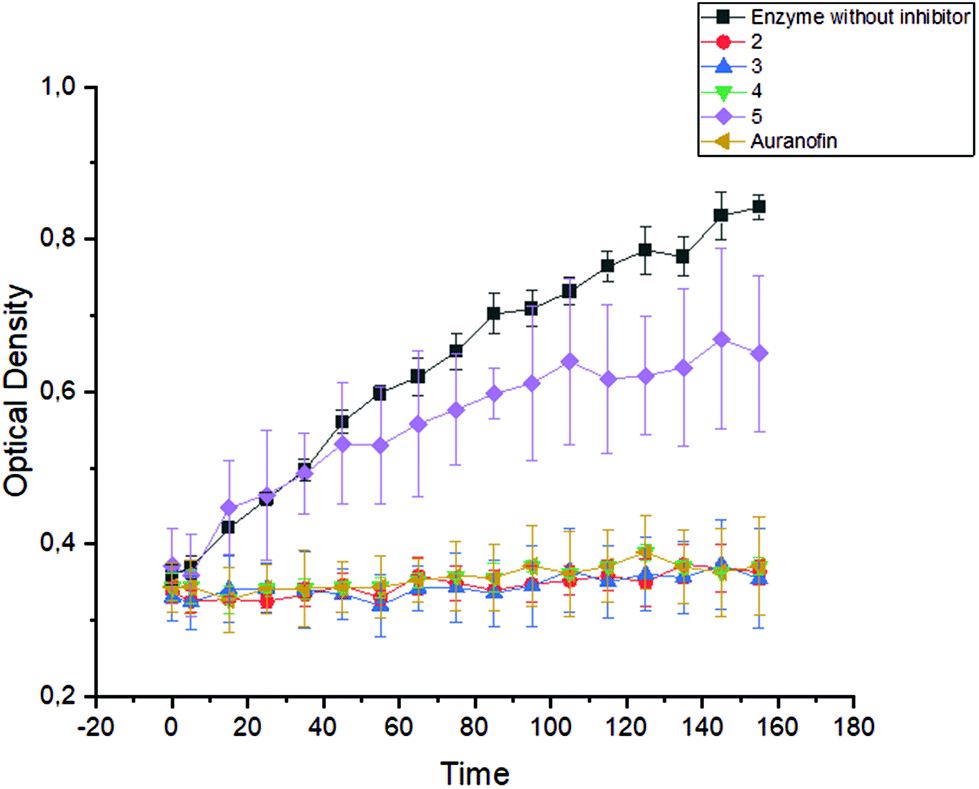 | ||
| Fig. 6 Formation of TNB from DTNB through the isolated TrxR enzyme recorded over a period of 160 min in the presence of 2–5 as well as Auranofin (1 μM each). | ||
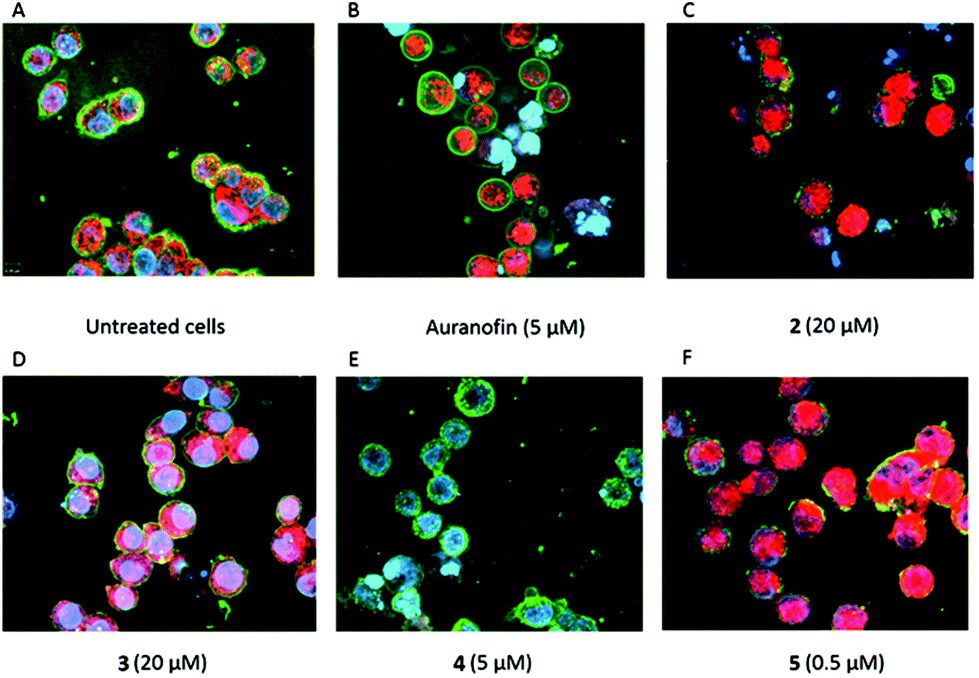 | ||
| Fig. 7 Merged staining of A2780cis cells with Hoechst dye (nuclei), Wheat germ agglutinin conjugate - WGA (cell membrane) and MitoTracker Red (mROS) of (A) untreated cells (panel top), (B) Auranofin (5 μM), (C) complex 2 (20 μM), (D) complex 3 (20 μM), (E) complex 4 (5 μM) and (F) complex 5 (0.5 μM) after 24 h of incubation. | ||
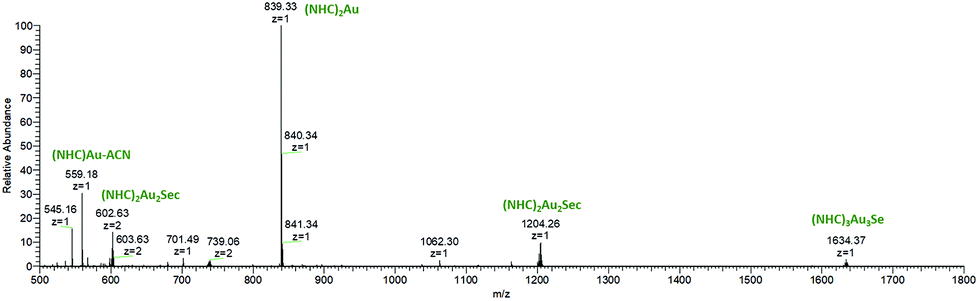 | ||
| Fig. 8 Full ESI-MS spectrum of complex 3 incubated with 1 eq. Sec for 5 min. | ||
| Compound | Proliferation IC50 (μM) | Metabolic activity IC50 (μM) | ||
|---|---|---|---|---|
| A2780wt | A2780cis | A2780wt | A2780cis | |
| 2 | >20 | 10.8 | >20 | 13.8 |
| 3 | 7.4 | 4.0 | 13.6 | 4.0 |
| 4 | 1.3 | 0.6 | 2.0 | 0.8 |
| 5 | 0.3 | 0.2 | 0.4 | 0.2 |
| Auranofin | <0.5 | 2.5 | 1.6 | 3.8 |
| Cisplatin | <1.0 | 4.0 | 4.8 | >10.0 |
It is worth mentioning that the results of 3 and 4 must be treated with caution, because of the significant transformation in the aqueous environment. It is very likely that the degradation products participated in the cellular effects.
From the results depicted in Fig. S18, ESI† the IC50 values were calculated and are listed in Table 2.
The antiproliferative activity of the halido (NHC)gold(I) complexes 2–4 depended on the leaving group, with a slight preference for A2780cis cells (Table 2). The chlorido (NHC)gold(I) complex 2 was nearly inactive in A2780wt cells (IC50 > 20 μM) and inhibited A2780cis cells with IC50 = 10.8 μM. The bromido derivative 3 was marginally more active against A2780cis (IC50 = 4.0 μM) than against A2780wt cells (IC50 = 7.4 μM). The exchange of the bromido leaving group by iodide strongly enhanced the antiproliferative effect. For both cell lines 6-fold lower IC50 values were calculated (A2780wt, 1.3 μM; A2780cis, 0.6 μM). The [(NHC)2Au(I)]+ complex 5 most effectively influenced the cells. It completely stopped the proliferation of A2780cis cells at all tested concentrations (0.25–20 μM). In wild-type cells, a concentration somewhat higher than 0.25 μM was necessary to cause 50% inhibition (IC50 = 0.3 μM).
To test if the antiproliferative activity was accompanied by cytotoxic effects, a modified MTT assay39 was applied to the ovarian cancer cell lines after 72 h of incubation with the compounds (Fig. S18, ESI† and Table 2). Auranofin did not reduce the viability (IC50 = 1.6 μM) of wild-type cells as effectively as their proliferation (IC50 < 0.5 μM), but the antiproliferative (IC50 = 2.5 μM) and antimetabolic activities (IC50 = 3.8 μM) in A2780cis cells were nearly the same. The most active complex 5, diminished the metabolism of A2780 cells as effectively as their proliferation, independently of the resistance status (IC50 = 0.2–0.4 μM).
As already determined in the [3H]-thymidine assay, the metabolism of the cells was reduced by the complexes in the order 2 < 3 < 4 with a slight preference for A2780cis cells. Complex 4 was more active than Auranofin and the activity of 3 equals that of the reference. Only 2 was less active (see Table 2).
At first glance, the bound halide controls the antiproliferative and antimetabolic activity of the complexes. However, the effects can only be ascribed in the case of 2 to a defined compound. The complex was stable in physiological NaCl solution (Fig. 3) and should therefore be taken up into the cells as an unchanged molecule. The same is true for [(NHC)2Au(I)]+ complex 5 and the high activity can unequivocally be assigned to this species.
More difficult is the interpretation of the results obtained with the bromido (NHC)gold(I) complex 3, because it reacted immediately after contact with the 0.9% NaCl solution on the one hand to the (NHC)Au(I)Cl complex 2 and on the other hand to the [(NHC)2Au(I)]+ complex 5. Consequently, the values obtained in the cell culture experiments might be caused by the formed 2 and 5.
The iodido (NHC)gold(I) complex 4 only incompletely reacted in 0.9% NaCl to 2 and 5. Therefore, it is very likely that three derivatives (2, 4 and 5) participate in the cellular effects.
Based on these considerations, we initiated detailed investigations on the behavior of halido (NHC)gold(I) complexes in cell culture medium. In particular, the identification of degradation products that participate in the biological activity is of great interest. This includes reaction with ingredients of the medium, including protein binding, which is likely to influence ligand exchange reactions and availability for cellular uptake.
Tumor selectivity is one of the criteria that has to be fulfilled for successful drug development. Therefore, compounds 2–5 were tested in primary human fibroblasts. Importantly, none of the complexes significantly reduced the metabolic activity even at the highest concentration tested (Fig. 5). In contrast, the reference Auranofin decreased the viability to 35.7% at 5 μM and 0% at 7.5 and 10 μM.
The activity of the enzyme was time-dependently quantified via its ability to convert 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) to 5-thio-2-nitrobenzoic acid (TNB). A sample of enzyme without inhibitor served as a negative control.
The halido (NHC)gold(I) complexes 2–4 reacted very rapidly with TrxR and completely inhibited the enzyme after only 5 min (Fig. 6). This effect was constant for 160 min. Complex 5 showed only low enzyme inhibitory potency, which correlates with its low reactivity towards nucleophiles.
A completely reduced form of Sec can be obtained by treatment of the commercially available material with an excess of either dithiothreitol (DTT) or NaBH4. Although data from the literature documented higher efficiencies for DTT,51 NaBH4 was the reducing agent of choice, because DTT led to the formation of an (NHC)Au(I)-DTT side product during incubation with the (NHC)gold(I) complexes.
Complex 3 was dissolved in ACN (1 mM) for addition to an aqueous solution of Sec (1 eq.), giving a 50:50 (v/v) mixture. The reaction was monitored using HPLC with samples taken after t = 5 min and 1, 2, 4 and 24 h. The products were identified with ESI-MS.
After only 5 min, 3 was quantitatively converted to three compounds with peaks at tret = 2.41, 6.81 and 7.27 min in the chromatogram. The proportions of the species were 73.03, 6.92 and 20.05% (Fig. S19, ESI†), which only marginally varied during 24 h.
The on-line UV–vis spectrum of the compound causing peak 1 taken during the HPLC run exhibited maxima at 203, 225 and 263 nm that were very similar to those of compound 3 (201, 224, and 256 nm). Therefore, this compound was ascribed to the (NHC)Au(I)Sec complex. The retention time of peak 3 (tret = 7.27 min) corresponds to that of the [(NHC)2Au(I)]+ complex 5. The UV–Vis spectrum displayed a strong absorbance band at 275 nm and confirmed this assignment. Peak 2 cannot be unequivocally assigned by UV–Vis spectroscopy (Fig. S20–S22, ESI†).
To complete these assignments, ESI-MS spectra (positive mode) from the solution after 5 min of incubation were measured (Fig. 7). The signal at m/z = 839 corresponds to [(NHC)2Au(I)]+, 5 and represents the dominant species. Additionally, an [(NHC)2Au(I)2Sec] adduct in charge states +1 (m/z = 1204) and +2 (m/z = 602) is visible and was assigned to the complex causing the peak at tret = 6.81 min in the chromatogram of 3. This species should be similarly configurated as the T-shaped intermediate found during ligand scrambling.41
In addition, a low-abundant signal with m/z = 1634 (z = 1) in the mass spectrum corresponds to [(NHC)3Au3Se]. The signal with m/z = 739 (z = 2) could not be assigned to a structure, but the isotopic pattern suggests that this species contains more than one Se atom.
HCD-fragmentation spectra were obtained from all Se-containing species (Fig. S23–S27, ESI†) and showed [(NHC)2Au]+ (m/z = 839) as a fragment in all cases. Complex 5 was investigated using HPLC under the same conditions. Immediately after contact with Sec, 8.24% were transformed to [(NHC)Au(I)Sec]+. This amount only slightly increased to 11.17% during 72 h (Fig. S28, ESI†) and documents the high stability of 5 against nucleophiles. Furthermore, this finding corresponds to the low inhibitory effect of 5 in the TrxR inhibition assay.
In order to demonstrate the difference compared with SH containing amino acids, 3 was incubated with Cys and NAC as well. The complex did not react with NAC and showed only a marginal ligand scrambling to 5 (5.05%) (Fig. S29, ESI†). In contrast, Cys coordinated very rapidly (within 5 min) but incompletely to the complex. The proportion of 70:30 remained constant for the following 24 h. In the HPLC chromatograms only transformation into 5 was observed (6.87% after 24 h) (Fig. S30, ESI†).
Compared to Auranofin, cells treated with 2–4 displayed at 5 μM no/marginal ROS formation (e.g., 4, Fig. 8E). Only at a 4-fold higher concentration (20 μM) increased ROS could be detected in the cells (see, e.g., 2 and 3, Fig. 8C and D). Interestingly, the highest ROS level was identified in cells treated with 5 (concentration 0.5 μM, Fig. 8F), although this compound showed less TrxR inhibitory potency.
Conclusions
Halido[3-ethyl-4-phenyl-5-(2-methoxypyridin-5-yl)-1-propyl-1,3-dihydro-2H-imidazol-2-ylidene]gold(I) complexes (halido = Cl−, Br−, I−) were synthesized and characterized. Of interest was the reactivity of the (NHC)Au(I)–X bond and its relevance to the biological effects. The exchange reaction decreased in the series X = I− < Br− < Cl− < NHC. While the chlorido (NHC)gold(I) complex 2 was relatively stable in aqueous solution, the iodido (NHC)gold(I) derivative 4 reacted within 72 h to about 44.41% to the [(NHC)2Au(I)]+ species 5. In the presence of 0.9% NaCl, 3 was quantitatively transformed to 2, while from 4 both 2 and 5 were formed. The reactivity of the halido (NHC)gold(I) complexes was further tested against iodide as a model nucleophile. Even under equimolar conditions the complexes underwent a nearly quantitative transformation to 4 followed by ligand scrambling to 5. In the case of 3, the T-shaped intermediate 6 was also detected in increased proportions. Generally, the highest stability was observed for 5, which reacted only marginally with nucleophiles. Nevertheless, this complex caused high cytotoxicity and very likely participated in the biological effects of 3 and 4.As the mode of action, TrxR inhibition plays a subordinate role for 2–4, because of a missing correlation of enzyme inhibition, cytotoxicity and the intracellular presence of ROS. In particular, the most cytotoxic complex 5, only marginally inhibited TrxR at 1 μM, but induced high ROS generation and cell growth inhibition at concentrations <0.5 μM.
Experimental section
General material and methods
Chemical reagents and solvents were purchased from commercial suppliers (Sigma-Aldrich, Fluka, Alfa Aesar and Acros) and were used without further purification. Selenocysteine was obtained from Cayman Chemical Company, Ann Arbor, MI, USA. For the analytical thin-layer chromatography, Polygram® SIL G/UV254 (Macherey-Nagel) plastic-backed plates (0.25 mm layer thickness) with fluorescent indicator and Merck TLC Silica gel 60 F254 aluminium backed plates were used. The spots were visualised with UV light (254 nm). For column chromatography silica gel 60 (0.040–0.063 mm) was used. NMR spectra were recorded on a Bruker Avance 4 Neo spectrometer (1H NMR, 400 MHz; 13C NMR, 100 MHz). The centers of the solvent signal and the TMS signal were used as internal standards. Deuterated solvents purchased at Eurisotop were used as solvents. For the HPLC experiments a Shimadzu prominence HPLC with an autosampler (SIL-20A HT), a column oven (CTO-10AS VP), a degasser (DGU-20A), a detector (SPD-M20A) and pumps (LC-20AD) was used; the column was a KNAUER Eurospher 100-5 C18, 250 × 4 mm with a 0.20 μm membrane filter (Carl Roth GmbH + Co. KG, Karlsruhe, Germany). The software used for data processing was LabSolutions. For mass spectra an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) using direct infusion and electrospray ionization was used. MS data analysis was carried out using Xcalibur. Graphics were created with OriginPro 2018G (Origin LabCorporation, Northampton, MA, USA).Synthesis
Method B: After dissolution of the chlorido (NHC)gold(I) complex 2 in dry acetone under an argon atmosphere, 10.0 eq. of sodium iodide were added and the solution was stirred for 20 min. The solvent was removed under reduced pressure. The precipitate was suspended in DCM and purified as described in method A.
Method B: From 49 mg of the chlorido (NHC)gold(I) complex 2. Yellowish solid, 52 mg (98% yield). 1H NMR (CDCl3): δ = 0.85 (t, 3 H, J = 7.4 Hz), 1.31 (t, 3 H, J = 7.2 Hz), 1.76 (qt, 2 H, J = 7.4, 7.4 Hz), 3.93 (s, 3 H), 4.09 (q, 2 H, J = 7.5 Hz), 4.19 (q, 2 H, J = 7.2 Hz) 6.73 (d, 1 H, J = 8.4 Hz), 7.18–7.21 (m, 2 H), 7.37–7.39 (m, 4 H), 8.00 (d, 1 H, J = 2.0 Hz). 13C NMR (CDCl3): δ = 11.1, 16.3, 24.7, 44.1, 50.4, 53.9, 111.3, 116.8, 127.4, 127.9, 129.0, 129.6, 130.4, 131.4, 140.3, 148.6, 164.4, 180.8. ESI-MS m/z: 646.0626 [M + H]+, calculated: 646.0630. Purity calculated using HPLC (peak area): 97.2%.
Crystallography
A Bruker D8 Quest Kappa diffractometer equipped with a Photon 100 detector was used to collect the single-crystal intensity data. Monochromatized MoKα radiation was generated with an Incoatec microfocus X-ray tube (50 kV/1 mA power settings) in combination with a multilayer optic. The supplementary crystallographic data were deposited as CCDC 1923121 (4) ESI.†HPLC and ESI-MS investigations
For all experiments, ACN and water of HPLC grade were used. For experiments in ACN, 0.250 mM solutions of the respective complex were prepared. For experiments in ACN/water, 0.5 mM solutions of the respective complex were prepared in ACN and diluted to a final concentration of 0.250 mM with water.To study the reaction with NaCl/KI, the salt was dissolved at a 2-fold concentration in water, which was then used to prepare an ACN/water mixture (50:50, (v/v)). A solution (0.250 mM in 1.50 mL) of complexes 2–5 in ACN or ACN/water = 50:50 (v/v), also with NaCl (0.9%) or KI (1 eq.), was passed through a 0.20 μm membrane filter and incubated for 72 h at rt. Samples (30 μL each) were taken at appropriate time points (t = 0, 0.5, 1, 1.5, 2, 4, 8, 12, 24, 48 and 72 h) and analyzed with a Shimadzu prominence HPLC system. The mobile phase consisted of ACN and water with 0.1% TFA. To achieve a separation of the compounds a gradient elution from ACN/water = 70:30 to 90:10 was used with a flow rate of 1 mL min−1 at an oven temperature of 35 °C. All solvents were degassed before use. The injection volume was 30 μL and the detection wavelength was set to 254 nm. The data were evaluated and visualized with Origin Pro 2018 (Origin LabCorporation, Northampton, MA, USA).
For the experiments with selenocysteine, 0.046 mg of NaBH4 and 1 eq. of the amino acid were dissolved in water (1.5 mL). The mixture stirred for 1 h at rt until the reduction was completed. Then, 1 eq. of the respective gold(I) complex, dissolved in ACN, was added to the mixture to obtain a concentration of 1 mM. Aliquots of 10 and 80 μL, respectively, were taken to monitor the reaction by means of ESI-MS and HPLC analysis at t = 0, 1, 2, 4 and 24 h. The reaction of 3 (1 mM) with 1 eq. NAC or Cys was analogously performed.
Biology
Abbreviations
| ACN | Acetonitrile |
| cpm | Counts per minute |
| DCM | Dichloromethane |
| DTNB | 5,5′Dithiobis(2-nitrobenzoic acid) |
| DTT | Dithiothreitol |
| eq. | Equivalent |
| ESI | Electrospray ionization |
| HPLC | High-performance liquid chromatography |
| MS | Mass spectrometry |
| MeOH | Methanol |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| mROS | Mitochondrial ROS |
| NHC | N-Heterocyclic carbene |
| NAC | N-Acetyl cysteine |
| ROS | Reactive oxygen species |
| RP | Reversed phase |
| rt | Room temperature |
| TFA | Trifluoroacetic acid |
| TNB | 5-Thio-2-nitrobenzoic acid |
| TrxR | Thioredoxin reductase |
| WGA | Wheat germ agglutinin conjugate |
| wt | Wild-type |
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Austrian Research Promotion Agency FFG (West Austrian BioNMR 858017) is kindly acknowledged.References
- M. Porchia, M. Pellei, M. Marinelli, F. Tisato, F. Del Bello and C. Santini, Eur. J. Med. Chem., 2018, 146, 709–746 CrossRef CAS PubMed.
- I. Ott, Coord. Chem. Rev., 2009, 253, 1670–1681 CrossRef CAS.
- F. M. Vanesa, P. H. Raquel and M. C. Gimeno, Pure Appl. Chem., 2018, 91, 247–269 Search PubMed.
- B. Bertrand and A. Casini, Dalton Trans., 2014, 43, 4209–4219 RSC.
- M. Mora, M. C. Gimeno and R. Visbal, Chem. Soc. Rev., 2019, 48, 447–462 RSC.
- G. Minghetti and F. Bonati, J. Organomet. Chem., 1973, 54, 62–63 CrossRef.
- J. L. Hickey, R. A. Ruhayel, P. J. Barnard, M. V. Baker, S. J. Berners-Price and A. Filipovska, J. Am. Chem. Soc., 2008, 130, 12570–12571 CrossRef CAS PubMed.
- T. Zou, C. T. Lum, C. N. Lok, J. J. Zhang and C. M. Che, Chem. Soc. Rev., 2015, 44, 8786–8801 RSC.
- M. Bian, R. Fan, G. Jiang, Y. Wang, Y. Lu and W. Liu, J. Med. Chem., 2020, 63, 9197–9211 CrossRef CAS PubMed.
- M. Filho, T. Scattolin, P. Dao, N. Tzouras, R. Benhida, M. Saab, K. Van Hecke, P. Lippmann, A. Martin, I. Ott and S. Nolan, New J. Chem., 2021, 45, 9995–10001 RSC.
- J. Arcau, V. Andermark, M. Rodrigues, I. Giannicchi, L. Pérez-Garcia, I. Ott and L. Rodríguez, Eur. J. Inorg. Chem., 2014, 2014, 6117–6125 CrossRef CAS.
- J. Lemke, A. Pinto, P. Niehoff, V. Vasylyeva and N. Metzler-Nolte, Dalton Trans., 2009, 7063–7070 RSC.
- J. Weaver, S. Gaillard, C. Toye, S. Macpherson, S. P. Nolan and A. Riches, Chem. – Eur. J., 2011, 17, 6620–6624 CrossRef CAS PubMed.
- A. M. Al-Majid, S. Yousuf, M. I. Choudhary, F. Nahra and S. P. Nolan, ChemistrySelect, 2016, 1, 76–80 CrossRef CAS.
- S. R. Patrick, A. Collado, S. Meiries, A. M. Z. Slawin and S. P. Nolan, J. Organomet. Chem., 2015, 775, 152–154 CrossRef CAS.
- R. Rubbiani, E. Schuh, A. Meyer, J. Lemke, J. Wimberg, N. Metzler-Nolte, F. Meyer, F. Mohr and I. Ott, MedChemComm, 2013, 4, 942–948 RSC.
- M. Pellei, V. Gandin, M. Marinelli, C. Marzano, M. Yousufuddin, H. V. R. Dias and C. Santini, Inorg. Chem., 2012, 51, 9873–9882 CrossRef CAS PubMed.
- E. Schuh, C. Pflüger, A. Citta, A. Folda, M. P. Rigobello, A. Bindoli, A. Casini and F. Mohr, Eur. J. Med. Chem., 2012, 55, 5518–5528 CrossRef CAS PubMed.
- B. Bertrand, E. Bodio, P. Richard, M. Picquet, P. Le Gendre and A. Casini, J. Organomet. Chem., 2015, 775, 124–129 CrossRef CAS.
- F. Hackenberg, H. Müller-Bunz, R. Smith, W. Streciwilk, X. Zhu and M. Tacke, Organometallics, 2013, 32, 5551–5560 CrossRef CAS.
- B. Bertrand, A. de Almeida, E. P. M. van der Burgt, M. Picquet, A. Citta, A. Folda, M. P. Rigobello, P. Le Gendre, E. Bodio and A. Casini, Eur. J. Inorg. Chem., 2014, 27, 4532–4536 CrossRef.
- C. V. Maftei, E. Fodor, P. G. Jones, M. Freytag, M. H. Franz, G. Kelter, H. H. Fiebig, M. Tamm and I. Neda, Eur. J. Med. Chem., 2015, 101, 431–441 CrossRef CAS PubMed.
- E. Maftei, C. V. Maftei, P. G. Jones, M. Freytag, M. H. Franz, G. Kelter, H. H. Fiebig, M. Tamm and I. Neda, Helv. Chim. Acta, 2016, 99, 469–481 CrossRef CAS.
- R. Rubbiani, I. Kitanovic, H. Alborzinia, S. Can, A. Kitanovic, L. A. Onambele, M. Stefanopoulou, Y. Geldmacher, W. S. Sheldrick, G. Wolber, A. Prokop, S. Wölfl and I. Ott, J. Med. Chem., 2010, 53, 8608–8618 CrossRef CAS PubMed.
- W. Liu, K. Bensdorf, M. Proetto, U. Abram, A. Hagenbach and R. Gust, J. Med. Chem., 2011, 54, 8605–8615 CrossRef CAS PubMed.
- B. Bertrand, L. Stefan, M. Pirrotta, D. Monchaud, E. Bodio, P. Richard, P. Le Gendre, E. Warmerdam, M. H. de Jager, G. M. M. Groothuis, M. Picquet and A. Casini, Inorg. Chem., 2014, 53, 2296–2303 CrossRef CAS PubMed.
- A. Gutiérrez, M. C. Gimeno, I. Marzo and N. Metzler-Nolte, Eur. J. Inorg. Chem., 2014, 2014, 2512–2519 CrossRef.
- W. Liu, K. Bensdorf, M. Proetto, A. Hagenbach, U. Abram and R. Gust, J. Med. Chem., 2012, 55, 3713–3724 CrossRef CAS PubMed.
- B. Bertrand, A. Citta, I. L. Franken, M. Picquet, A. Folda, V. Scalcon, M. P. Rigobello, P. Le Gendre, A. Casini and E. Bodio, J. Biol. Inorg. Chem., 2015, 20, 1005–1020 CrossRef CAS PubMed.
- C. Schmidt, B. Karge, R. Misgeld, A. Prokop, R. Franke, M. Brönstrup and I. Ott, Chem. – Eur. J., 2017, 23, 1869–1880 CrossRef CAS PubMed.
- R. Rubbiani, S. Can, I. Kitanovic, H. Alborzinia, M. Stefanopoulou, M. Kokoschka, S. Mönchgesang, W. S. Sheldrick, S. Wölfl and I. Ott, J. Med. Chem., 2011, 54, 8646–8657 CrossRef CAS PubMed.
- C. Schmidt, L. Albrecht, S. Balasupramaniam, R. Misgeld, B. Karge, M. Brönstrup, A. Prokop, K. Baumann, S. Reichl and I. Ott, Metallomics, 2019, 11, 533–545 CrossRef CAS PubMed.
- D. Curran, O. Dada, M. B. Helge, M. Rothemund, G. Sánchez-Sanz, R. Schobert, X. Zhu and M. Tacke, Molecules, 2018, 23, 2031–2048 CrossRef PubMed.
- S. Nobili, E. Mini, I. Landini, C. Gabbiani, A. Casini and L. Messori, Med. Res. Rev., 2010, 30, 550–580 CrossRef CAS PubMed.
- K. P. Bhabak, B. J. Bhuyan and G. Mugesh, Dalton Trans., 2011, 40, 2099–2111 RSC.
- K. Becker, S. Gromer, R. H. Schirmer and S. Müller, Eur. J. Biochem., 2000, 267, 6118–6125 CrossRef CAS PubMed.
- E. S. J. Arnér and A. Holmgren, Eur. J. Biochem., 2000, 267, 6102–6109 CrossRef PubMed.
- A. Bindoli, M. P. Rigobello, G. Scutari, C. Gabbiani, A. Casini and L. Messori, Coord. Chem. Rev., 2009, 253, 1692–1707 CrossRef CAS.
- C. M. Gallati, S. K. Goetzfried, M. Ausserer, J. Sagasser, M. Plangger, K. Wurst, M. Hermann, D. Baecker, B. Kircher and R. Gust, Dalton Trans., 2020, 49, 5471–5481 RSC.
- C. M. Gallati, S. K. Goetzfried, A. Ortmeier, J. Sagasser, K. Wurst, M. Hermann, B. Kircher and R. Gust, Dalton Trans., 2021, 50, 4270–4279 RSC.
- S. K. Goetzfried, C. M. Gallati, M. Cziferszky, R. A. Talmazan, K. Wurst, K. R. Liedl, M. Podewitz and R. Gust, Inorg. Chem., 2020, 59, 15312–15323 CrossRef CAS PubMed.
- P. de Fre′mont, N. M. Scott, E. D. Stevens and S. P. Nolan, Organometallics, 2005, 24, 2411–2418 CrossRef.
- D. Curran, H. Müller-Bunz, S. I. Bär, R. Schobert, X. Zhu and M. Tacke, Molecules, 2020, 25, 3474 CrossRef CAS PubMed.
- M. Tacke, O. Dada, C. O'Beirne, X. Zhu and H. Müller-Bunz, Acta Crystallogr., Sect. C: Struct. Chem., 2016, 72, 857–860 CrossRef CAS PubMed.
- S. K. Goetzfried, S. M. C. Koenig, C. M. Gallati and R. Gust, Inorg. Chem., 2021, 60, 8546–8553 CrossRef CAS PubMed.
- R. Gust, H. Schönenberger, J. Kritzenberger, K. J. Range, U. Klement and T. Burgemeister, Inorg. Chem., 1993, 32, 5939–5950 CrossRef CAS.
- C. Schmidt, B. Karge, R. Misgeld, A. Prokop, M. Brönstrup and I. Ott, Medchemcomm, 2017, 8, 1681–1689 RSC.
- L. Messori, L. Marchetti, L. Massai, F. Scaletti, A. Guerri, I. Landini, S. Nobili, G. Perrone, E. Mini, P. Leoni, M. Pasquali and C. Gabbiani, Inorg. Chem., 2014, 53, 2396–2403 CrossRef CAS PubMed.
- A. Pratesi, C. Gabbiani, E. Michelucci, M. Ginanneschi, A. M. Papini, R. Rubbiani, I. Ott and L. Messori, J. Inorg. Biochem., 2014, 136, 161–169 CrossRef CAS PubMed.
- Ö. Karaca, V. Scalcon, S. Meier-Menches, R. Bonsignore, J. Brouwer, F. Tonolo, A. Folda, M. Rigobello, F. Kühn and A. Cassini, Inorg. Chem., 2017, 56, 14237–14250 CrossRef PubMed.
- J. N. Burnell, J. A. Karle and A. Shrift, J. Inorg. Biochem., 1980, 12, 343–351 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR of 2–5, ESI-MS spectra of 2 and 5, HPLC chromatograms in ACN/water mixtures, UV–vis spectra, antiproliferative and antimetabolic activity in HL-60 and A2780 wt/cis cells, ESI-MS spectra of complexes 2 and 5 in the presence of Sec. CCDC 1923121. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03528b |
| ‡ These authors contributed equally to the publication. |
| This journal is © The Royal Society of Chemistry 2022 |