
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen-atom and oxygen-atom transfer reactivities of iron(IV)-oxo complexes of quinoline-substituted pentadentate ligands†
Sandip
Munshi
 a,
Arup
Sinha
bc,
Solomon
Yiga
de,
Sridhar
Banerjee
a,
Reena
Singh
b,
Md. Kamal
Hossain
b,
Matti
Haukka
f,
Andrei Felipe
Valiati
g,
Ricardo Dagnoni
Huelsmann
h,
Edmar
Martendal
h,
Rosely
Peralta
g,
Fernando
Xavier
h,
Ola F.
Wendt
*d,
Tapan K.
Paine
*a and
Ebbe
Nordlander
*b
a,
Arup
Sinha
bc,
Solomon
Yiga
de,
Sridhar
Banerjee
a,
Reena
Singh
b,
Md. Kamal
Hossain
b,
Matti
Haukka
f,
Andrei Felipe
Valiati
g,
Ricardo Dagnoni
Huelsmann
h,
Edmar
Martendal
h,
Rosely
Peralta
g,
Fernando
Xavier
h,
Ola F.
Wendt
*d,
Tapan K.
Paine
*a and
Ebbe
Nordlander
*b
aSchool of Chemical Sciences, Indian Association for the Cultivation of Science, Kolkata, India. E-mail: ictkp@iacs.res.in
bChemical Physics, Department of Chemistry, Lund University, Box 124, SE-221 00 Lund, Sweden. E-mail: Ebbe.Nordlander@chemphys.lu.se
cDepartment of Chemistry, School of Advanced Science, Vellore Institute of Technology, Vellore, India
dCenter for Analysis and Synthesis, Department of Chemistry, Lund University, Box 124, SE-221 00 Lund, Sweden. E-mail: Ola.Wendt@chem.lu.se
eDepartment of Chemistry, Makerere University, P. O. Box 7062, Kampala, Uganda
fDepartment of Chemistry, University of Jyväskylä, Box 35, FI-400 14, Jyväskylä, Finland
gDepartment of Chemistry, LABINC, Universidade Federal de Santa Catarina (UFSC), 88040-900 Florianopolis, Santa Catarina, Brazil
hDepartment of Chemistry, Center for Technological Sciences, Universidade do Estado de Santa Catarina (UDESC), 89219-710 Joinville, Santa Catarina, Brazil
First published on 7th January 2022
Abstract
A series of iron(II) complexes with the general formula [FeII(L2-Qn)(L)]n+ (n = 1, L = F−, Cl−; n = 2, L = NCMe, H2O) have been isolated and characterized. The X-ray crystallographic data reveals that metal–ligand bond distances vary with varying ligand field strengths of the sixth ligand. While the complexes with fluoride, chloride and water as axial ligand are high spin, the acetonitrile-coordinated complex is in a mixed spin state. The steric bulk of the quinoline moieties forces the axial ligands to deviate from the Fe–Naxial axis. A higher deviation/tilt is noted for the high spin complexes, while the acetonitrile coordinated complex displays least deviation. This deviation from linearity is slightly less in the analogous low-spin iron(II) complex [FeII(L1-Qn)(NCMe)]2+ of the related asymmetric ligand L1-Qn due to the presence of only one sterically demanding quinoline moiety. The two iron(II)-acetonitrile complexes [FeII(L2-Qn)(NCMe)]2+ and [FeII(L1-Qn)(NCMe)]2+ generate the corresponding iron(IV)-oxo species with higher thermal stability of the species supported by the L1-Qn ligand. The crystallographic and spectroscopic data for [FeIV(O)(L1-Qn)](ClO4)2 bear resemblance to other crystallographically characterized S = 1 iron(IV)-oxo complexes. The hydrogen atom transfer (HAT) and oxygen atom transfer (OAT) reactivities of both the iron(IV)-oxo complexes were investigated, and a Box–Behnken multivariate optimization of the parameters for catalytic oxidation of cyclohexane by [FeII(L2-Qn)(NCMe)]2+ using hydrogen peroxide as the terminal oxidant is presented. An increase in the average Fe–N bond length in [FeII(L1-Qn)(NCMe)]2+ is also manifested in higher HAT and OAT rates relative to the other reported complexes of ligands based on the N4Py framework. The results reported here confirm that the steric influence of the ligand environment is of critical importance for the reactivity of iron(IV)-oxo complexes, but additional electronic factors must influence the reactivity of iron-oxo complexes of N4Py derivatives.
Introduction
High-valent iron-oxo moieties are prevalent in oxygenase enzymes. The presence of a ferryl (FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) moiety in the active form – compound I – of the cytochrome P-450 family of heme oxygenases is well established.1 It has been determined that ferryl units are also formed in reactions catalyzed by non-heme iron oxygenases. Examples include TauD,2 an α-ketoglutarate dependent oxygenase (hydroxylase).3 The non-heme Rieske dioxygenases have been proposed to form active oxidation intermediates containing a perferryl (FeVO) unit.4 Over the last decade, a number of relatively stable bio-inspired non-heme ferryl complexes have been characterized,5 and their reactivities investigated. These complexes have been found to effect a number of different oxidation reactions involving C–H bond activation (hydrogen atom transfer, HAT), e.g. dehydrogenation reactions to form arenes, and oxygen atom transfer (OAT), e.g. sulfoxidation reactions, as well as combinations of HAT and OAT, e.g. aliphatic C–H hydroxylation.6
O) moiety in the active form – compound I – of the cytochrome P-450 family of heme oxygenases is well established.1 It has been determined that ferryl units are also formed in reactions catalyzed by non-heme iron oxygenases. Examples include TauD,2 an α-ketoglutarate dependent oxygenase (hydroxylase).3 The non-heme Rieske dioxygenases have been proposed to form active oxidation intermediates containing a perferryl (FeVO) unit.4 Over the last decade, a number of relatively stable bio-inspired non-heme ferryl complexes have been characterized,5 and their reactivities investigated. These complexes have been found to effect a number of different oxidation reactions involving C–H bond activation (hydrogen atom transfer, HAT), e.g. dehydrogenation reactions to form arenes, and oxygen atom transfer (OAT), e.g. sulfoxidation reactions, as well as combinations of HAT and OAT, e.g. aliphatic C–H hydroxylation.6
Non-heme ligands that have been used to stabilize ferryl complexes are typically tetra- and pentadentate nitrogen-donor ligands. An overwhelming majority of these synthetic ferryl complexes have S = 1 spin state, as opposed to the ferryl moieties in non-heme metalloenzyme active sites, which are found in the high spin state (S = 2). Shaik and coworkers7,8 have proposed that high-spin FeIVO moieties are inherently more reactive, and that hydrogen atom transfer mediated by many low-spin ferryl complexes proceeds via high-spin transition states (two-state reactivity). Studies on synthetic high spin ferryl complexes have not shown enhanced reactivity relative to low-spin complexes, but there is a quest to synthesize stable high spin FeIVO complexes and to investigate their reactivities.9 Ligand design is of central importance in these investigations; it is desirable to use a ligand that exerts a sufficiently weak ligand field to stabilize a high spin ferryl moiety, yet induces sufficient (thermal) stability to enable facile handling of the complex.
Recently, Rasheed et al.,10 Rana et al.11 and Mukherjee et al.12 published iron complexes of the ligand N-[di(2-pyridyl)methyl]-N,N-bis(quinolin-2-ylmethyl)methanamine (L2-Qn)13 (Chart 1). Rasheed et al. synthesized and characterized [FeIV(O)(L2-Qn)]2+ (7, vide infra).10 It was found that this complex was considerably more reactive in hydrogen atom transfer (hydrogen abstraction) reactions than related N5-donor ligands with other nitrogen-containing (-benzimidazolyl, -pyridyl (N4Py)) substituents. Complex 7 is not high spin, but the steric encumbrance exerted by the two quinoline moieties of the pentadentate ligand means that the ligand in the sixth position coordinates in a bent fashion, significantly deviating from the linear axis of an idealized octahedron. This is borne out in the crystal structure of 7,10 where there is a significant tilt of the iron(IV)O unit away from the two quinoline entities, giving an N(amine)–Fe–O angle that deviates from linearity by almost 10°; similar deviations have been observed in the crystal structures of [MnII(OH2)(L2-Qn)]2+,14 [ZnII(NCMe)(L2-Qn)]2+ and [CuII(NO3)(L2-Qn)]+.13 As a consequence of the steric interaction, the Fe–N distances in the equatorial plane are significantly lengthened in comparison to those found for related N4Py-derived ligands.10,15–17 The resultant weakened ligand field is expected to lower the energy gap between the low spin and high spin states and provide enhanced reactivity in agreement with the two state reactivity model proposed by Shaik and coworkers (vide supra).
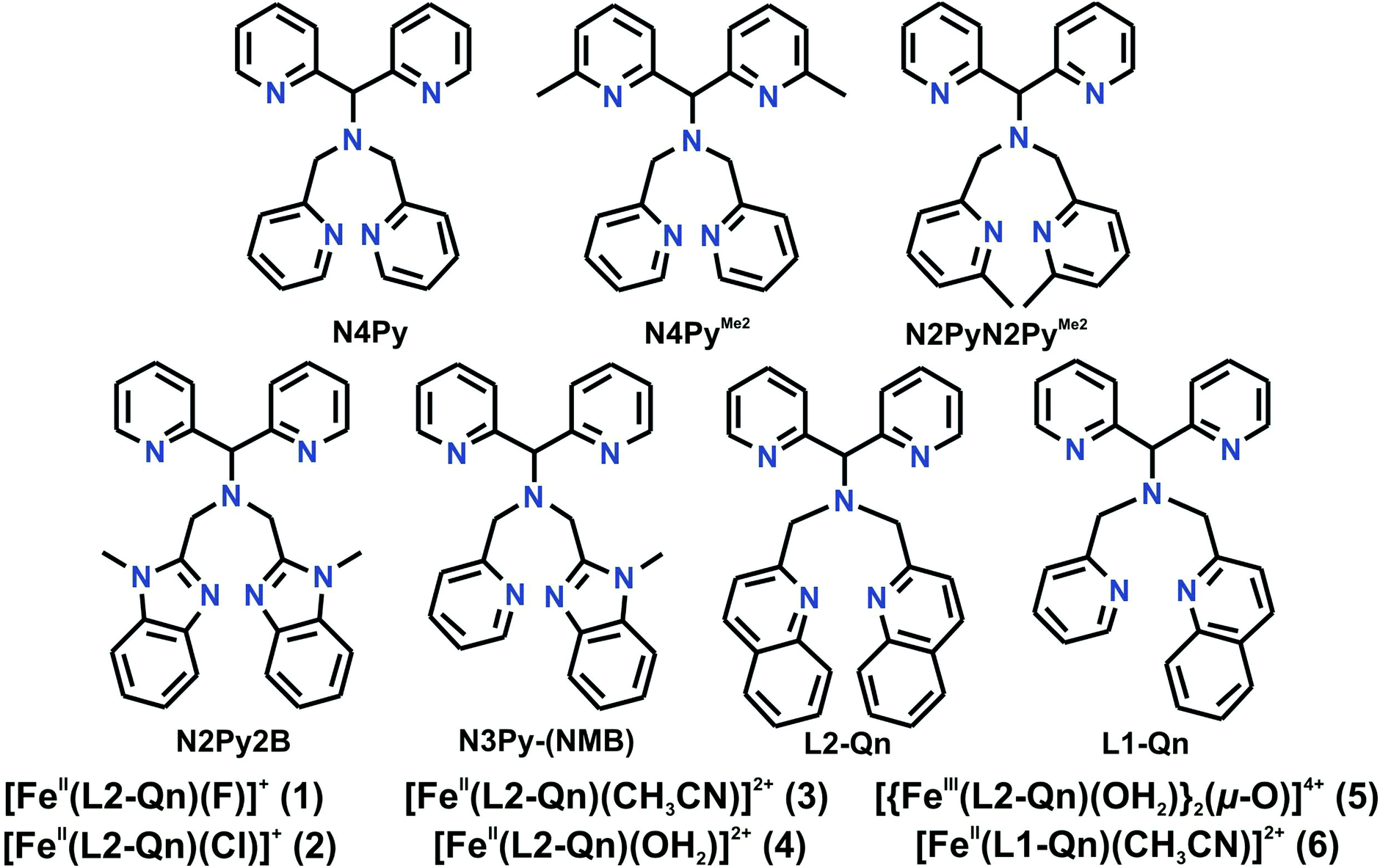 | ||
| Chart 1 Ligands and complexes discussed in this work. | ||
Rana et al.11 also studied the alkane oxidation (hydroxylation) effected by [FeIV(O)(L2-Qn)]2+ and obtained results in agreement with the observations made by Rasheed et al. Computational modeling by Rana et al.11 indicates that the hydroxylation reaction proceeds via an oxygen rebound mechanism, as expected, i.e. initial hydrogen atom abstraction to form an iron(III)-OH intermediate and an alkane radical followed by the rebound of a hydroxyl radical from the intermediate to form a hydroxylated alkane and an iron(II) complex that may be (re)oxidized to form the ferryl complex. Furthermore, these authors demonstrated that the hydrogen abstraction capability of [FeIV(O)(L2-Qn)]2+ can be utilized for halogenation of sp3 C–H bonds by a similar rebound mechanism, with the halide complexes [FeII(X)(L2-Qn)]+ (X = Cl−, Br−) acting as halogen sources. Computational modeling suggests that [FeIV(O)(L2-Qn)]2+ abstracts hydrogen from a suitable alkane substrate to generate [FeIII(OH)(L2-Qn)]2+ and a substrate radical. Exchange of the hydroxide in the latter species with a halide ligand originating from [FeII(X)(L2-Qn)]+ (X = Cl−, Br−) leads to the formation of [FeIII(X)(L2-Qn)]2+ that undergoes a “halogen rebound” with the substrate radical to form the halogenated product.11
Independently, we have studied the reactivity of [FeIV(O)(L2-Qn)]+ and the analogous ferryl complex of the closely related ligand L1-Qn (Chart 1). Here we wish to describe the synthesis and characterization of the complexes [FeII(L2-Qn)(L)]n+ (n = 1, L = F−, Cl−; n = 2, L = NCMe, H2O) and an extended investigation into the HAT and OAT reactivities of [FeIV(O)(L2-Qn)]2+. An optimization of the parameters for catalytic oxidation of cyclohexane by [FeII(NCMe)(L2-Qn)]n+, using hydrogen peroxide as the ultimate oxidant, is also presented. A multivariate methodology using response surfaces for three variables (Box–Behnken design) was used to simultaneously evaluate reaction time, the relative amounts of substrate (cyclohexane), and oxidant (hydrogen peroxide). This strategy was adopted since it was observed in a previous study that these variables strongly interact in such a catalytic system.18 Furthermore, the synthesis of the directly related but asymmetric ligand L1-Qn and its iron(II) complex [FeII(NCMe)(L1-Qn)]2+ is reported. It has also been possible to prepare [FeIV(O)(L1-Qn)]2+ and to determine the crystal structure of [FeIV(O)(L1-Qn)](ClO4)2, and we report a detailed analysis of its reactivity in HAT/OAT reactions. Our results confirm that the steric influence of the ligand environment is of critical importance for the reactivity of iron(IV)O complexes, but additional electronic factors must influence the reactivity of ferryl complexes of N4Py derivatives.
Results and discussion
Ligand syntheses
The pentadentate bisquinoline ligand N-[di(2-pyridyl)methyl]-N,N-bis(quinolin-2-ylmethyl)methanamine (L2-Qn) was synthesized by the method reported by McMorran and co-workers.13 The corresponding monoquinoline ligand 1,1-di(pyridin-2-yl)-N-(pyridin-2-ylmethyl)-N-(quinolin-2-ylmethyl)methanamine (L1-Qn) was prepared by the reaction of N-[di(2-pyridyl)methyl]-N-(2-pyridylmethyl)methylamine19 with 2-chloromethyl quinoline hydrochloride in dry acetonitrile in the presence of K2CO3 and KI (Experimental section).Synthesis and characterization of iron(II) complexes of L2-Qn
Room temperature treatment of Fe(BF4)2 with L2-Qn in acetonitrile yielded a light yellow compound. The X-ray analysis revealed the compound to be [FeII(L2-Qn)(F)](BF4) (1·BF4), instead of the anticipated acetonitrile complex; the fluoride ligand apparently originating from tetrafluoroborate. It is likely that the fluoride ion is formed as the result of hydrolysis of the tetrafluoroborate by adventitious water. It may be noted that in related systems, it has been shown that iron(II) species can abstract fluoride from the BF4− anion to form a fluoro complex.20 The molecular structure of 1 is shown in Fig. 1. A comparison of pertinent Fe–ligand distances for 1 and other complexes reported here is found in Table 1 (vide infra) and relevant crystallographic data for 1 and all other crystal structures reported here are summarized in the ESI (Table S1†). The central iron(II) ion is in a distorted octahedral coordination environment. The data indicate that all the Fe–N distances are in the range 2.219(5)–2.249(5) Å, typical for high-spin iron(II) compounds. As discussed by Rasheed et al.10 (vide supra) the presence of the quinoline moieties in L2-Qn leads to longer Fe–N(quinoline) distances because of the steric bulk of the quinoline moieties and the resultant steric interactions with the axial ligand. Consequently, the equatorial ligand field is decreased and the presence of the axial weak-field fluoride ligand renders complex 1 high spin. The increased ionic radius in the high spin state results in the iron(II) ion being placed 0.536 Å above the ligand equatorial plane.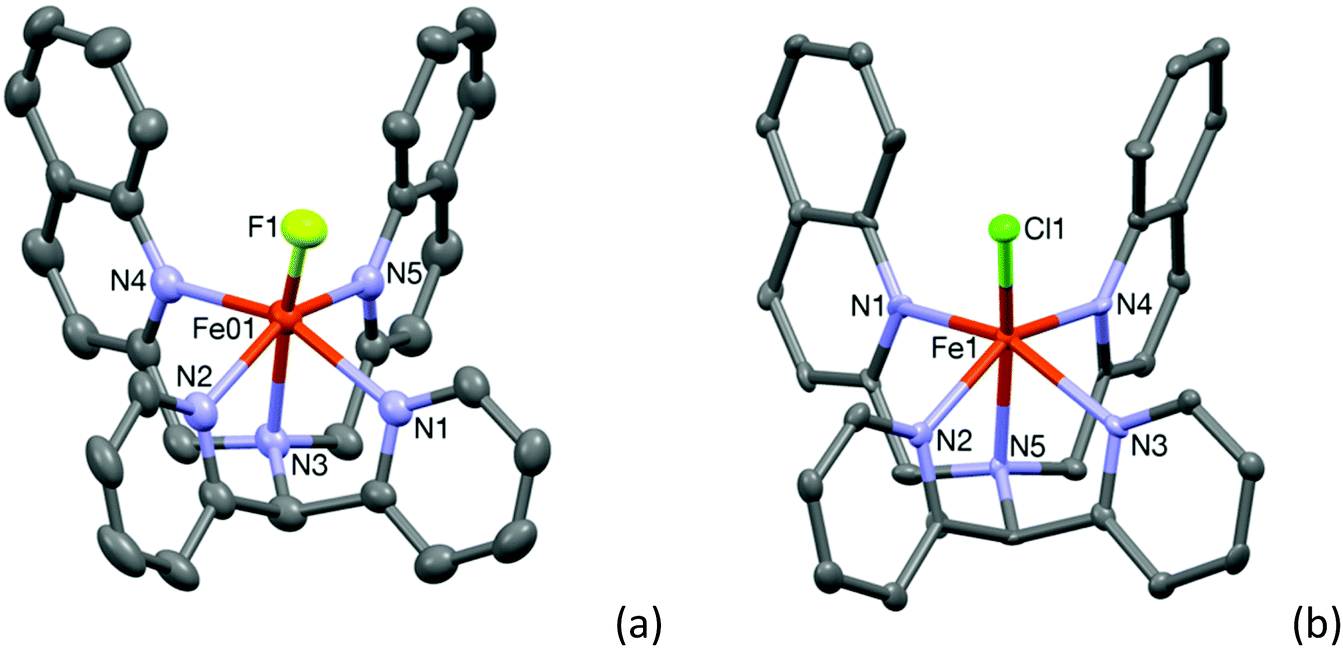 | ||
| Fig. 1 Mercury plots of the molecular structures of the cationic complexes 1 (a) and 2 (b), showing the atom labelling scheme. Thermal ellipsoids are plotted at 30% probability ellipsoids; hydrogen atoms have been omitted for clarity. | ||
| Complex | Fe–Laxial | Fe–Naxial | Fe–NPy (avg.) | Fe–NQn/BzIm/Me2(avg.) |
|---|---|---|---|---|
| 1 | 1.900(2) | 2.248(3) | 2.230 | 2.239 |
| 2 | 2.3010(18) | 2.238(5) | 2.219 | 2.2465 |
| 3 | 1.951(6) | 2.017(5) | 2.0145 | 2.0975 |
| 4 | 2.059(6) | 2.188(6) | 2.191 | 2.200 |
| 6 | 1.937(2) | 1.970(2) | 1.961 | 2.047(18) |
| [FeII(N4Py)(CH3CN)]2+ | 1.915(3) | 1.961(3) | 1.972 | — |
| [FeII(N2Py2B)(CH3CN)]2+ | 1.909(6) | 1.980(6) | 1.953 | 1.977 |
| [FeII(N4PyMe2)(CH3CN)]2+ | 1.889(10) | 1.977(8) | 1.979 | 2.044 |
| [FeII(N2PyN2PyMe2)(CH3CN)]2+ | 1.959(3) | 1.990(6) | 1.993 | 2.081 |
In order to obtain the corresponding acetonitrile complex, L2-Qn was reacted with Fe(ClO4)2 in acetonitrile at room temperature. The reaction mixture was kept in an ethyl acetate bath for two days to yield a dark reddish brown crystalline solid. The ESI mass spectrum of this solid revealed the presence of two complexes. A minor fragment at m/z 557.68 corresponded to the chloride-coordinated monocationic complex [Fe(L2-Qn)(Cl)](ClO4) (2·ClO4), while the major fragment at m/z 281.83 corresponded to the dicationic complex [Fe(L2-Qn)(CH3CN)](ClO4)2 (3·ClO4). Presumably, partial decomposition of the perchlorate ion was the source of the chlorine atom. When the reaction was performed using Fe(ClO4)2·xH2O as a starting material, an oxo-bridged diferric complex, [{FeIII(L2-Qn)(H2O)}2(μ-O)](ClO4)4 (5, cf. Fig. S1, ESI†), was obtained with no trace of 3.
The crystal structure of 2·ClO4 could be determined and the structure of the cation 2 is shown in Fig. 1(b). The crystal structure of [FeII(L2-Qn)(Cl)]Cl (2·Cl), containing cationic 2, has been reported previously (CCDC 1516453), as has that for the bromide analogue of 2 (CCDC 1516421).11 The relatively long Fe–N distances observed for 2 in 2·ClO4, which are in the range 2.214(5)–2.252(6) Å, are consistent with the data reported by Maiti and co-workers11 and are in full agreement with the high-spin electronic configuration in complex 2. In both cases the tilt of the axial chloro ligand is around 19° and the iron atom is 0.556 Å (for complex 2) and 0.542 Å for [FeII(L2-Qn)(Cl)]Cl above the ligand equatorial plane. As discussed above, this is expected due to the steric interactions imposed by the two bulky quinolyl substituents and the presence of the weak-field chloride ligand.
Compound 3·ClO4 was structurally characterized by single crystal X-ray crystallography. The molecular structure of the cation 3 is shown in Fig. 2(a). The structure is similar to that of the “parent” complex [FeII(N4Py)(CH3CN)](ClO4)2 reported by Que and co-workers.21 The Fe–N bond lengths are in the range of 1.951(6)–2.103(5) Å which are slightly longer than those reported for low spin iron(II) centres coordinated to similar pyridine based ligands (Table 1). Again, the presence of the relatively bulky and weakly coordinating quinoline moieties is likely to be the reason for the increase in the metal–ligand bond lengths. Indeed, the iron-pyridine nitrogen atoms are 2.019(5) and 2.010(5) Å apart, while those in the N4Py systems are typically less than 2.0 Å. The iron-quinoline nitrogen atom distances are longer, 2.103(5) and 2.092(5) Å. Mitra et al. reported a similar increase in the Fe–N(pyridine) bond distances upon replacement of a pyridyl moiety by an (N-methyl)benzimidazolyl moiety.16 The iron atom in complex 3 sits 0.295 Å above the ligand equatorial plane. The close proximity of the iron center to the ligand equatorial plane is comparable to that found in the low spin parent complex [FeII(N4Py)(CH3CN)](ClO4)2 where the iron is placed 0.208 Å above the equatorial plane. The relatively shorter Fe–N distances in the equatorial plane of 3, as compared to other high spin complexes in this study, is indicative of a possible mixture of high and low spin states of the iron ion in the solid state, which was corroborated by solution NMR spectroscopy (vide infra), and the bond parameters associated with complex 3 are closer to those expected for a low spin complex of an N4Py-derived ligand.
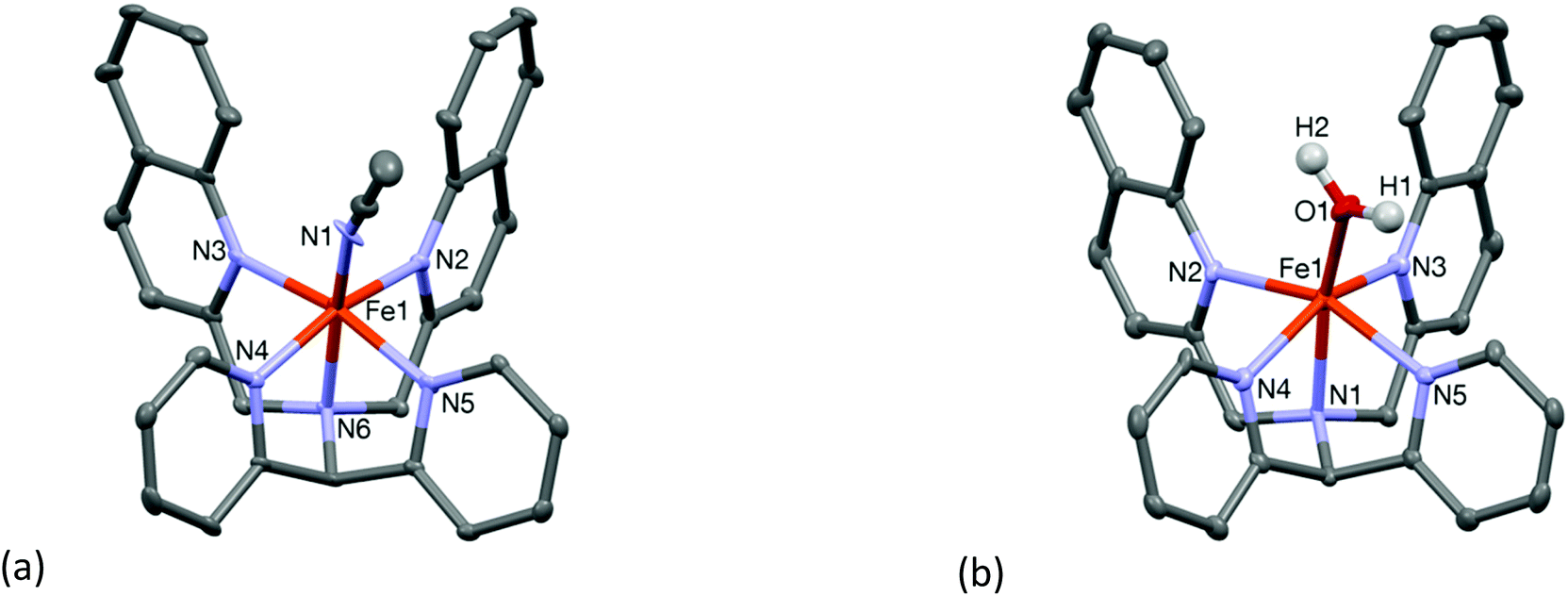 | ||
| Fig. 2 Mercury plots of the molecular structures of the cationic complexes 3 (a) and 4 (b), showing the atom labelling scheme. Thermal ellipsoids are plotted at 30% probability ellipsoids; hydrogen atoms have been omitted for clarity. | ||
The water-coordinated high spin complex [FeII(L2-Qn)(OH2)](OTf)2 (OTf = triflate) (4·OTf) was obtained upon reaction of Fe(OTf)2·2CH3CN with the ligand in dichloromethane and subsequent crystallization from moist acetonitrile. The Fe–N distances are in the range 2.186(6)–2.201(6) Å, while the oxygen atom of the coordinated water molecule is 2.059(6) Å apart from the iron atom (Table 1). The molecular structure of the cationic moiety is shown in Fig. 2(b). As shown in Table 1, the crystallographic data show the changes in metal–ligand bond distances with varying ligand field strengths of the sixth ligand (fluoride, chloride, water and acetonitrile). Similar to the other high spin complexes considered here, the iron ion is dislocated 0.474 Å above the ligand equatorial plane.
In agreement with previous studies (vide supra), the steric effect of the quinoline moieties is reflected in the tilt of the axial ligands from the Fe–Naxial axis. The displacement of the iron ion from the ligand equatorial plane places the Laxial at the vicinity of the H-8 of the quinoline moiety. The magnitude of the tilt increases with increasing distance of the iron atom from the ligand equatorial plane, and vice versa. Accordingly, a higher deviation/tilt (in the range 16.8° to 19.3°) is observed for the high spin complexes 1, 2 and 4, whereas the least deviation (11.5°) is observed for 3.
The spin states of the complexes are further supported by their room temperature 1H NMR spectra. For complexes 1, 2 and 4, the 1H NMR spectra display resonances in the region between 120 ppm and −60 ppm, confirming the high spin state. However, for complex 3 the high spin and low spin forms were found to be in equilibrium, with the high spin forming a minor component in solution at room temperature. Indeed, variable temperature 1H NMR spectroscopy reveals that at −40 °C the low spin state is observed for 3 (Fig. S2, ESI†). The observed spin states are in complete agreement with the ordering of the four ligands in the spectrochemical series, which gives that the halides are all weak-field ligands and that the σ-donor/π-acceptor ligand acetonitrile is a relative strong field ligand: Br− < Cl− < F− < NCMe < pyridine.
Synthesis and characterization of an iron(II) complex of L1-Qn
The iron(II) complex [FeII(L1-Qn)(CH3CN)](ClO4)2 (6) was isolated from the reaction of an equimolar amount of the ligand with Fe(ClO4)2·6H2O in dry acetonitrile at room temperature. The ESI-mass spectrum (positive ion mode in acetonitrile) of complex 6 exhibits ion peaks at m/z 236.50 and 572.02 with the isotope distribution patterns calculated for [FeII(L1-Qn)(CH3CN)]2+ and {[Fe(L1-Qn)](ClO4)}+. In the optical spectrum of 6 in acetonitrile, two charge-transfer bands at 375 nm (ε ∼ 5800 M−1 cm−1) and 460 nm (ε ∼ 5000 M−1 cm−1) are observed (Fig. S3, ESI†). These bands may be attributed to metal-to-ligand charge transfer (MLCT) transitions arising from electron transfer from low-spin iron(II) t2g orbitals to the π* orbitals of the ligand along with ligand based transitions.18 The 1H NMR spectrum of 6 in CD3CN at 298 K indicates the presence of a low spin iron(II) species in solution. However, variable temperature 1H NMR experiments showed that some peaks shifted with variation of temperature (Fig. S4, ESI†). This effect was pronounced in the shifts of the diastereotopic methylene protons of the compound, which were found to display an anti-Curie behavior.22 At 298 K, the 1H NMR spectrum is first order, indicating a weakly coupled AX spin system, consistent with the presence of a paramagnetic iron ion. At 233 K, one set of methylene protons displays a nearly perfect AB quartet typical of a diamagnetic environment. These observations suggest partial population of the high spin state in solution at room temperature, as observed for 3 (vide supra) and [FeII(N2Py2B)(CH3CN)]2+.16 The NMR spectra of these [FeII(N5-donor)(CH3CN)]2+ complexes, in combination with other physical data and reactivity studies, allow us to gauge the relative effective ligand fields of the various related N5-donor ligands (vide infra).The spin state in the solid state was found to be low spin on the basis of its Mössbauer spectrum, which shows isomer shift (δ) and quadrupole splitting (ΔEQ) values of 0.40 mm s−1 and 0.50 mm s−1, respectively (Fig. S5, ESI†). Furthermore, the crystal structure of [FeII(L1-Qn)(CH3CN)](BF4)2 (6·BF4) (Fig. 3) is similar to that of its L2-Qn analogue 3. The complex is chiral but a racemic mixture is formed, and the crystal structure contains two diastereomers related by the inversion center in the monoclinic unit cell; the diastereomer depicted in Fig. 3 displays S and C chiralities23 for the N1 and Fe1 stereogenic centres, respectively. The Fe–N bond distances vary in the range of 2.05–1.96 Å, and are in good agreement with 3 and other reported low-spin iron(II) complexes supported by N4Py-type ligands.16,17 The axial positions of the pseudo-octahedral ligand environment in 6 are occupied by the amine nitrogen (N1) and the nitrogen atom (N6) of the solvent molecule with the N1–Fe1–N6 angle of 171.81(8)°. This deviation from linearity is slightly less than that observed for 3 (vide supra), and consistent with the presence of only one sterically demanding quinoline unit in 6, as opposed to two quinoline entities in 3. A comparison of different Fe–N bond distances in 1–4 and 6 with those of other related complexes is presented in Table 1.
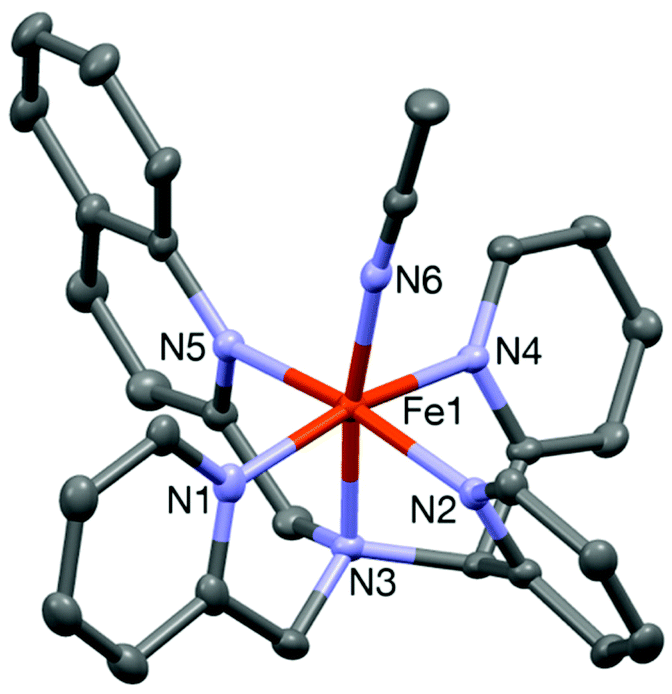 | ||
| Fig. 3 A Mercury plot of the complex cation of 6·(BF4)2 with 30% thermal ellipsoids. All hydrogen atoms have been omitted for clarity. | ||
Generation of [FeIV(O)(L2-Qn)]2+ (7)
Complex 3 was reacted with excess (5 equiv.) solid IBX ester (isopropyl 2-iodoxybenzoate ester) in acetonitrile at room temperature to form the pale green cationic complex [FeIV(O)(L2-Qn)]2+ (7). The physical data for 7 are identical to those determined by Rasheed et al.,10 and Mukherjee et al.12 Further details on the formation and characterization of 7 are found in the ESI.†Generation and characterization of [FeIV(O)(L1-Qn)]2+ (8)
Given that the steric effect of L2-Qn results in the distorted ferryl complex 7 with a resultant weak ligand field and high reactivity, we were interested in assessing whether the asymmetric ligand L1-Qn with only one quinoline donor moiety would exert similar steric influence and thus potentially stabilize an iron(IV)-oxo complex with similar reactivity as that of 7.Upon treatment of 6·(BF4)2 with three equivalents of IBX ester in acetonitrile at 298 K, a pale green intermediate is formed with a corresponding absorption band at 730 nm (Fig. 4) attributable to ligand field transition of the iron(IV) ion. A comparison of different iron(IV)-oxo species of the related pentadentate ligands is presented in Table 2. The ESI-MS spectrum (positive ion mode) of the green species displays ion peaks at m/z 576.3 with the isotope distribution pattern calculated for [Fe(O)(L1-Qn)(BF4)]+. The ion peak shifts two mass units higher to m/z 578.3 upon treatment with H218O with the incorporation of labelled oxygen (40% 18O incorporation) supporting that the intermediate, [Fe(O)(L1-Qn)]2+ (8), can exchange its oxygen atom with water (Fig. 4, inset). The half-life of the intermediate species was found to be 50 h at 298 K, demonstrating the relative thermal stability of the species.
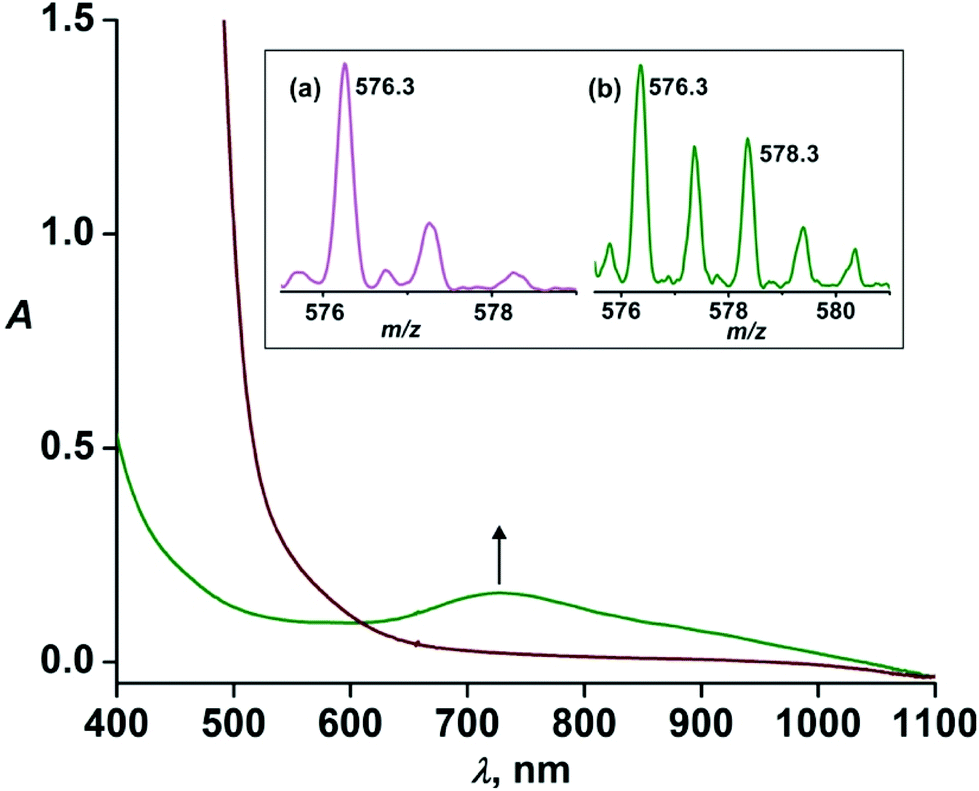 | ||
| Fig. 4 Formation of 8 upon treatment of 6 (0.5 mM) with IBX-ester (1.5 mM) in acetonitrile at 298 K; inset: ESI-mass spectrum (positive ion mode in acetonitrile) of (a) 8 and (b) 8 after treatment with H218O. | ||
| Ligand (L) | λ max, nm (ε, M−1 cm−1) | t 1/2 at 298 K | Mössbauer parameters | Ref. | |
|---|---|---|---|---|---|
| δ (mm s−1) | ΔEQ (mm s−1) | ||||
| L1-Qn | 730 (340) | 50 h | −0.05 | 0.66 | This work |
| N4Py | 695 (400) | 60 h | −0.04 | 0.93 | 26 |
| N3Py-(NMB) | 708 (400) | 40 h | −0.03 | 1.1 | 16 |
| N2Py2B | 725 (380) | 2.5 h | −0.02 | 1.34 | 16 |
| L2-Qn | 770 (380) | 29 min | 0.03 | 0.56 | 10, 11 and this work |
| N4PyMe2 | 740 (220) | 14 min | 0.05 | 0.62 | 27 |
| N2PyN2PyMe2 | 770 (200) | 5.5 min | — | — | 17 and 25 |
Complex 8·(BF4)2 could also be generated by treating an aqueous solution of 6·(BF4)2 with excess ceric ammonium nitrate (CAN). The iron(IV)-oxo complex thus generated was isolated in crystalline form using excess sodium perchlorate (see Experimental section). The resultant green species 8 exhibited the characteristic near-IR absorption at 730 nm (375 M−1 cm−1) in acetonitrile. Considering the molar absorption of the complex, an approximate 91% yield of the complex is generated from 6 in the reaction with IBX-ester in acetonitrile. Complex 8·(BF4)2 in CD3CN was found to exhibit paramagnetically shifted protons in the 1H NMR spectrum, confirming the intermediate spin (S = 1) of the iron(IV) ion (Fig. S7, ESI†). The proton resonances of the pyridine and quinoline rings are assigned based on the spectra of previously reported iron(IV)-oxo complexes.12,16,28
The oxidation state of the iron center of 8 was confirmed by zero-field 57Fe Mössbauer spectroscopy at 293 K. The complex exhibits an isomeric shift (δ) value of −0.05 mm s−1, and a quadrupole splitting (ΔEQ) value of 0.66 mm s−1 (Fig. 5). The Mössbauer parameters are quite similar to those reported for related N-ligated S = 1 iron(IV)-oxo complexes (Table 2).12,19,20
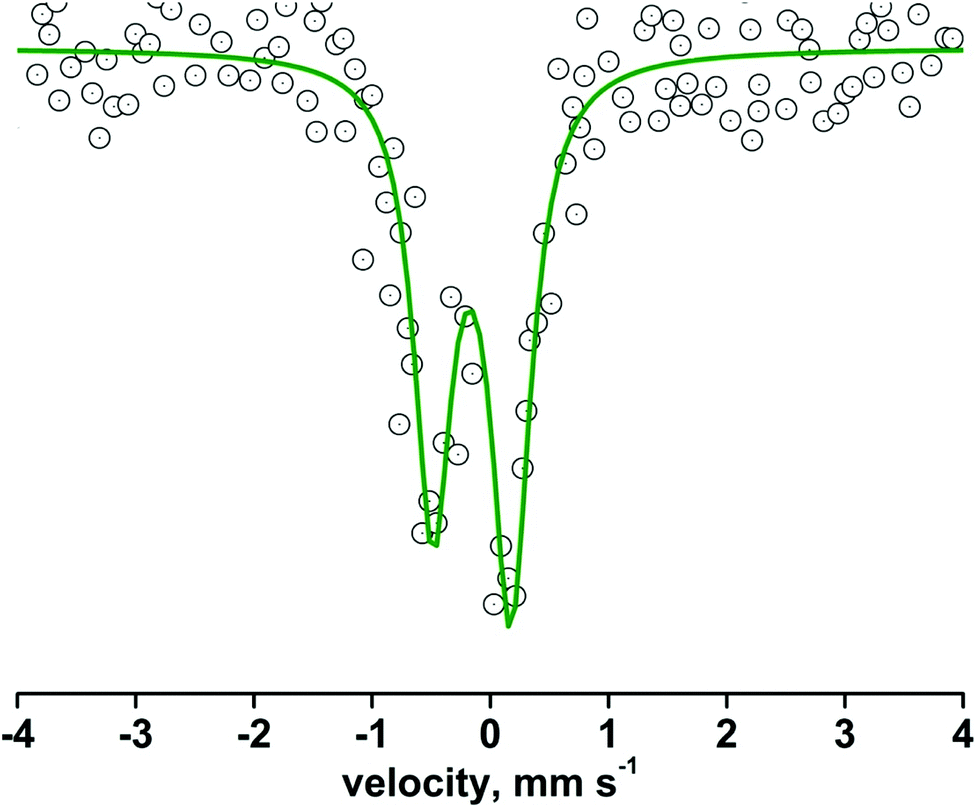 | ||
| Fig. 5 Zero-field Mössbauer spectrum of the solid sample of complex 8 at 293 K. | ||
The complex was further characterized by single crystal X-ray diffraction studies. The molecular structure of the cationic complex 8 is shown in Fig. 6. Like 6, the complex is chiral with stereogenic centers at the N1 nitrogen and iron ion; two diastereomers are found in the unit cell of the complex. The iron center is coordinated by five nitrogen donors from the ligand and the sixth coordination site is occupied by an oxygen atom. The four pyridine nitrogen donors from the pentadentate ligand form the equatorial plane and the axial positions are occupied by the amine nitrogen (N3) and the oxygen atom (O1) with the N3–Fe1–O1 angle of 174.2(2)° (Table 3). The tilt angle (5.8°) lies in between those observed in the iron(IV)-oxo complexes of the N2Py2B and L2-Qn ligand. The Fe1–O1 bond distance of 1.642(5) Å is very close to that reported for other crystallographically characterized S = 1 iron(IV)-oxo complexes.10
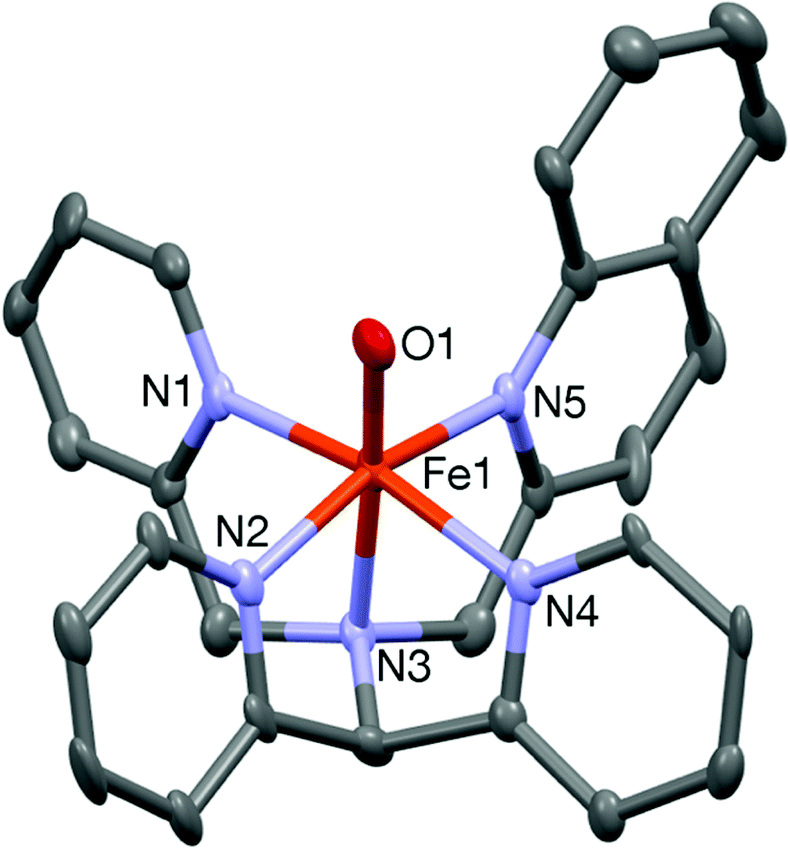 | ||
| Fig. 6 A Mercury plot of the complex cation of 8 with 40% thermal ellipsoids. All hydrogen atoms have been omitted for clarity. | ||
| Fe(1)–O(1) | 1.642(5) | Fe(1)–N(2) | 1.966(5) |
| Fe(1)–N(3) | 2.053(6) | Fe(1)–N(4) | 1.981(5) |
| Fe(1)–N(5) | 2.025(5) | Fe(1)–N(1) | 1.984(6) |
| O(1)–Fe(1)–N(3) | 174.2(2) | N(4)–Fe(1)–N(1) | 164.1(2) |
| N(2)–Fe(1)–N(5) | 163.9(3) |
Reactivity studies on [FeIV(O)(L2-Qn)]2+ (7) – oxidation of thioethers, alkenes and alkanes
Considering the relative instability of [FeIV(O)(L2-Qn)]2+ (7), it was expected that 7 would show a considerably enhanced reactivity relative to other ferryl complexes of N4Py and its derivatives. This has been confirmed by Rasheed et al.,10 who found that both the oxygen atom transfer (OAT) and the hydrogen atom transfer (HAT) reactivities of 7 involve the highest rates of reaction that have thus far been established for a ferryl complex based on the N4Py ligand framework. We have completed a detailed study of the OAT and HAT reactivities of complex 7 with an extended set of thioether, alkene and alkane substrates, as well as catalytic oxidation reactions effected by 3/7. These studies are described in detail in the ESI.†From our reactivity experiments on OAT and HAT reactions it is clear that oxo-complex 7 reacts at faster rates with different hydrocarbon substrates with respect to previously reported complexes having similar ligand environments; a plot of corrected second-order rate constants for HAT vs. bond dissociation energies is found in Fig. 7 (vide infra). It is evident that the quinoline moiety, being a bulkier donor group, exerts steric effects that increase the average Fe–N bond length to more than 2 Å in the parent complex 3. Although the quinoline entity is a relatively strong donor, the weakened equatorial field that is a consequence of the steric effect results in higher HAT and OAT rates relative to other iron complexes containing ligands of the N4Py framework.27
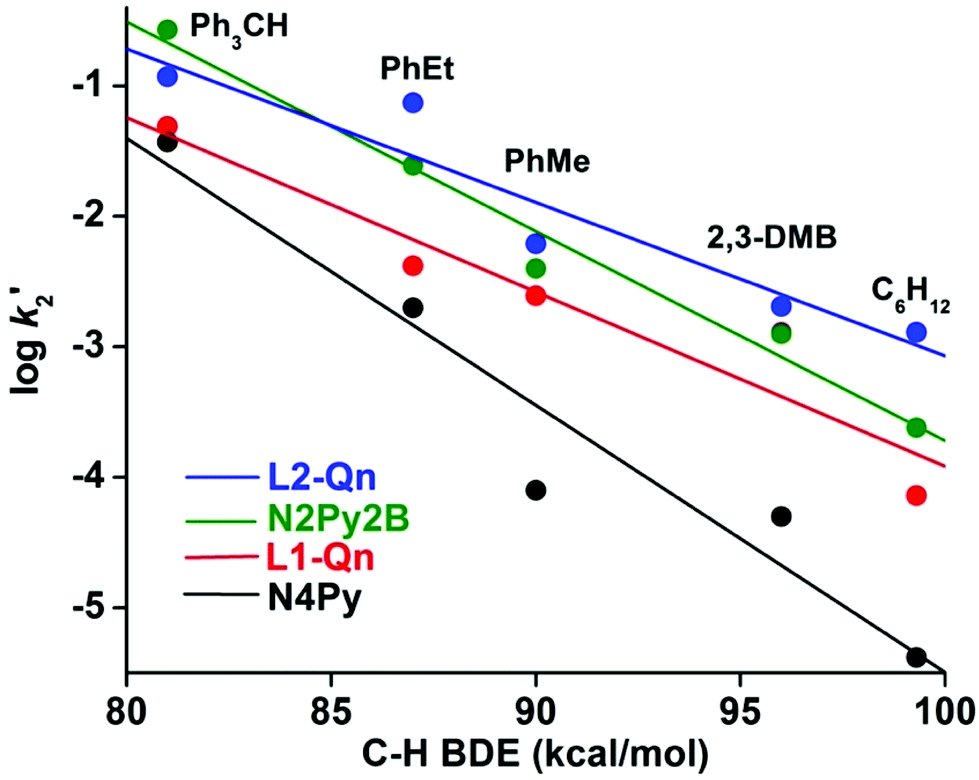 | ||
Fig. 7 Plot of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k′2versus C–H BDEs of different substrates in the oxidation of alkanes by iron(IV)-oxo complexes of different N4Py derivatives. k′2versus C–H BDEs of different substrates in the oxidation of alkanes by iron(IV)-oxo complexes of different N4Py derivatives. | ||
Considering the established high reactivity of 7 and the steric nature of its origin, and the ability of its precursor 3 to function as a (pre)catalyst for alkane and alkene oxidation, we were interested in establishing (i) the optimum conditions for catalytic hydroxylation of hydrocarbons, using 3 as a catalyst and hydrogen peroxide as the terminal oxidant (ii) to what extent the corresponding complexes of the monoquinoline ligand L1-Qn, i.e. [FeII(L1-Qn)(CH3CN)]2+ (6) and [FeIV(O)(L1-Qn)]2+ (8), exhibit reactivities similar to those of 3 and 7, and whether their reactivities can be related to steric factors.
Multivariate analysis of catalytic cyclohexane oxidation using [FeII(L2-Qn)(NCMe)](ClO4)2 (3) as catalyst and H2O2 as oxidant
The optimization of experimental conditions using multivariate statistical approaches such as the Box–Behnken design, a response surface methodology, can be used to study several variables of interest simultaneously.29 This mathematical approach, besides reducing the number of experiments to be performed, generates mathematical models and allows the evaluation of the significance of each factor under study and the interactions between them.30 As it had been demonstrated spectroscopically that complex 3 can generate the FeIVO species 7, and that 3 is catalytically active upon oxidation of organic substrates (vide supra and ESI†), we decided to pursue an optimization of the variables affecting conversion of cyclohexane into cyclohexanol and cyclohexanone. A Box–Behnken experimental design according to Table S5 (ESI†) was performed which generated response surfaces depicted in Fig. S9–S14 (ESI).† Analysis of the response surfaces allowed us to summarize the optimized conditions for the conversion of cyclohexane into cyclohexanol and cyclohexanone, as well as the percent selectivity for the formation of cyclohexanol (Table 4).The amount of catalyst is the most significant difference between the conversions of cyclohexane (CyH) into cyclohexanol (CyOH) or cyclohexanone (CyO) (Fig. S9 and S10, ESI†). This implicates the catalyst as an active species in the initial step to form CyOH as well as in (over)oxidation of CyOH into CyO. Another significant difference can be observed by the strong interaction between the amount of catalyst and peroxide (Fig. S10,† middle and bottom). At lower peroxide concentration, the increase in catalyst concentration does not lead to any significant increase in the amount of CyO formed. On the other hand, at the highest level of peroxide tested, there is a significant increase in CyO formation as the amount of catalyst is increased. This corroborates that the species formed by the interaction between catalyst and peroxide is crucial for the formation of the products, especially CyO. Finally, the results in terms of percent conversion of CyOH, CyO and the percent selectivity for the formation of CyOH can be found in Table 5.
Reactivity studies on [FeIV(O)(L1-Qn)]2+ (8)
| Substrate [BDE (kcal mol−1)] | L1-Qn (k2 × 103) (M−1 s−1) | N4Py (k2 × 103) (M−1 s−1) | N3Py-(NMB) (k2 × 103) (M−1 s−1) | N2Py2B (k2 × 103) (M−1 s−1) | L2-Qn (k2 × 103) (M−1 s−1) | N4PyMe2 (k2 × 103) (M−1 s−1) | N2PyN2PyMe2 (k2 × 103) (M−1 s−1) |
|---|---|---|---|---|---|---|---|
| Ph3CH [81] | 49 | 37 | 31 | 270 | 120 | — | 69 |
| PhEt [87] | 8.4 | 4 | 7.6 | 48 | 147 | 7.6 | 155 |
| PhMe [90] | 7.4 | 0.15 | 1.3 | 12 | 18.5 | 3.5 | 19 |
| 2,3-DMB [96] | 2.6 | 0.12 | 0.32 | 2.5 | 4.1 | 1.8 | — |
| C6H12 [99.3] | 0.86 | 0.05 | 0.3 | 2.9 | 30 | 0.67 | 43.2 |
Complex 8 is efficient in the C–H bond cleavage of aliphatic substrates and shows a linear correlation between logk2′ and the C–H BDE of the substrates (vide infra, k2′ is the second order rate constant corrected for number of “abstractable” hydrogens). The primary kinetic isotope effect (KIE) for the oxidation of toluene and toluene-d8 was found to be ≈19, a value supporting that of a metal-based oxidant (Fig. S16, ESI†). This KIE value is very similar to that of the N4Py system, but higher than the value observed with the iron(IV)-oxo complexes of the N4PyMe2 and N3Py-(NMB) ligands.16,17 Such a large KIE value implies a tunneling mechanism in H atom abstraction, as has been proposed for C–H bond activation by FeIVO species. The linear correlation of k2′ and the observed large kinetic isotope effect indicate that the reactions of 8 with the substrates take place via the transfer of a hydrogen atom in a rate-determining step, as observed for other related Fe(IV)-oxo complexes.31,32
The oxidation of cyclohexane by 8 affords cyclohexanol and cyclohexanone in 12% (TON = 0.37) and 6% (TON = 0.19) yield, respectively, with an alcohol/ketone ratio (A/K) of 1.9. It may be noted that a very poor A/K ratio (0.5) for cyclohexane oxidation was obtained by the [FeIV(O)(N4Py)]2+ complex. Furthermore, 8 was found to oxidize adamantane with a C3/C2 normalized selectivity of 15.5.
The reactivities of 7 and 8 in HAT reactions are compared to those of [FeIV(O)(N4Py)]2+ and [FeIV(O)(N2Py2B)]2+ in Fig. 7. Based on the results discussed above, it may be concluded that the addition of a weak quinoline donor to the N4Py backbone to form L1-Qn is sufficient to increase the reactivity of the resulting iron(IV)-oxo species, as previously reported with the bis-quinoline (L2-Qn) or N-methyl benzimidazole-substituted N4Py ligands (N3Py-(NMB) and N2Py2B), and the reactivity lies in between the N2Py2B and L2-Qn systems.12
Comparative parameters for a number of structurally related ferryl complexes of N4Py and its derivatives are collated in Table 7, in which the bond parameters along with the oxo-tilt angle and the reactivity patterns of the complexes are shown. However, the reactivity patterns cannot be directly correlated with the tilt angle of the Fe–O moiety. The tilt angle of 8 is 5.8°, which lies between the iron(IV)-oxo complexes of the N2Py2B and L2-Qn ligands. However, complex 8 is much less reactive than the iron(IV)-oxo complex of the N2Py2B ligand in cyclohexane oxidation. This finding contradicts the previous report where a linear correlation has been proposed between the tilt angle and the reactivity.10 However, the lengthening of the Fe–O bond in the iron(IV)-oxo complexes does display a correlation with the reactivity (vide infra, Fig. 9).
| Ligand | L1-Qn | N4Py | N2Py2B | L2-Qn |
|---|---|---|---|---|
| Fe–O distance (Å) | 1.642(5) | 1.639(5) | 1.656(4) | 1.677(5) |
| Fe–Namine distance (Å) | 2.053(6) | 2.033(8) | 2.115(6) | 2.084(4) |
| Fe–NPy (avg.) distance (Å) | 1.977 | 1.964(5) | 1.989 | 2.028 |
| Fe–NPy/BzIm/Qn (avg.) distance (Å) | 2.025(5) | 1.964(5) | 1.952 | 2.070 |
| Tilt angle (°) | 5.8 | 0.6 | 3 | 9.57 |
| k 2 for thioanisole oxidation (M−1 s−1) (temp., K) | 0.023 (233) | 0.00024 (233) | 0.033 (243) | 0.31 (243) |
| k 2 for cyclohexane oxidation (M−1 s−1) (298 K) | 0.86 × 10−3 | 0.05 × 10−3 | 2.9 × 10−3 | 30 × 10−3 |
| Ref. | This work | 19 | 10 | 10 |
The reaction of 6 with methyl phenyl sulfide and three equivalents of IBX ester led to the formation of the corresponding sulfoxide in an OAT reaction with a TON of 2.7. No sulfone product was observed. A maximum TON of 33 for sulfoxide product is obtained under catalytic condition with 50 equivalents of IBX-ester. The OAT ability of complex 8 was tested toward the alkenes cyclooctene, 1-octene and cyclohexene (Scheme S2 and Table S6, ESI†). While epoxide is the major, or only, product for cyclooctene and 1-octene, the allylic oxidation is predominant for cyclohexene. The weak allylic C–H bond leads to hydrogen atom abstraction over the OAT pathway. Hammett analyses using different para-substituted thioanisoles yields a ρ value of −1.01 confirming the highly electrophilic nature of 8 (Fig. 8), similar to other iron(IV)-oxo complexes.19,20
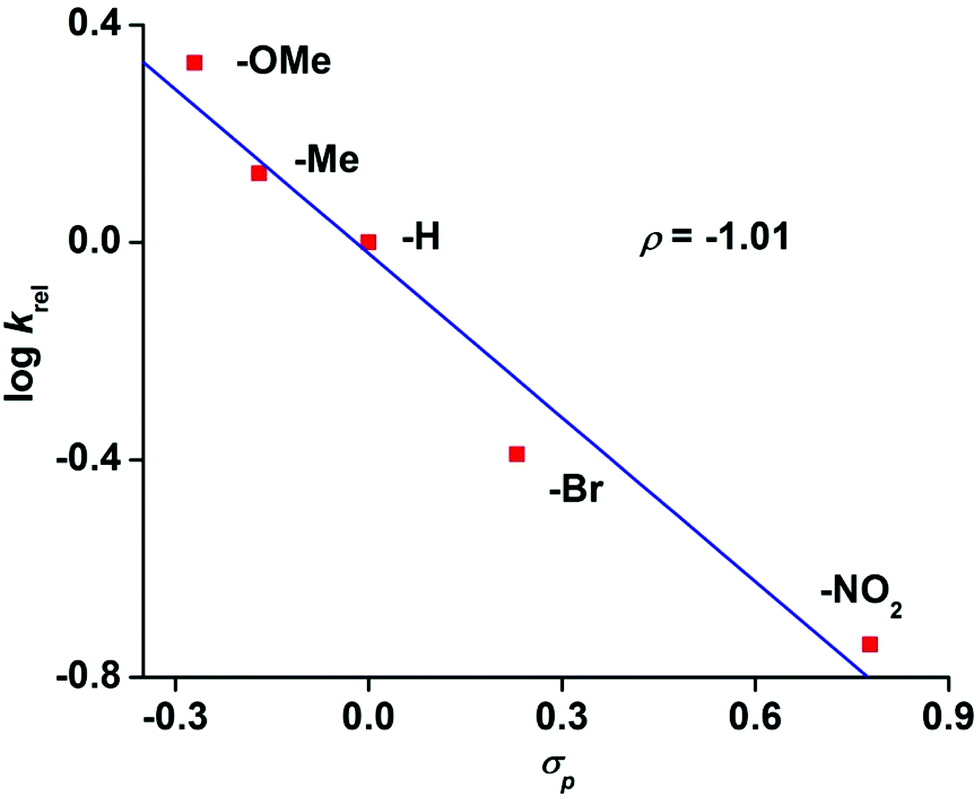 | ||
| Fig. 8 Hammett plot of logkrelvs. σp for the reaction of 8 with p-substituted thioanisoles. | ||
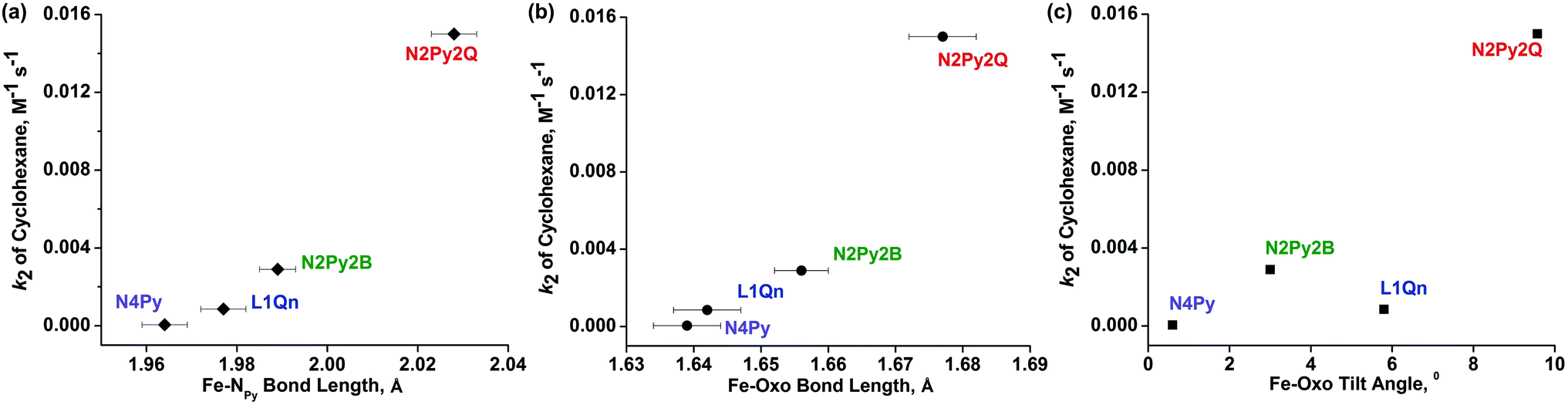 | ||
| Fig. 9 Plots of observed reactivity (k2) for C–H bond activation of cyclohexane vs. (a) Fe-NPy bond length, (b) FeO bond length, and (c) FeO tilt angle for the complexes [FeIV(O)(L)]2+ (L = N4Py, L1-Qn (8), N2Py2B and L2-Qn (7)). | ||
The effective ligand fields exerted by the various pentadentate nitrogen donor ligands discussed here (Chart 1) may be assessed by, inter alia, spin state of [FeII(N5-donor)(CH3CN)]2+ complexes, half-lives of the corresponding [FeIV(O)(N5-donor)]2+ complexes, and the characteristic d–d absorption(s) for these iron(IV) oxo complexes at long wave lengths, the latter of which has been thoroughly analyzed by Solomon, Que and coworkers.33 On the basis of these criteria, we make the following approximate and qualitative ranking of effective (equatorial) ligand field strengths in these complexes, in decreasing order: N4Py > N3Py-(NMB) > L1-Qn >≈ N2Py2B > N4PyMe2 ≈ L2-Qn > N2PyN2PyMe2. The N5-donor ligand field strength affects the reactivity patterns of the iron(IV)-oxo species, but the exact way that the ligand affects that reactivity is difficult to predict (or assess) as the reactivity is clearly related to both steric and electronic factors. The interplay of these factors and their influence on reactivity has not yet been fully elucidated for N4Py-based ligands; for example, it has been shown that a set of such ligands, which includes N4Py, N2Py2B and L2-Qn, influence the relative reactivities of their iron(IV)-oxo and manganese(IV)-oxo systems quite differently.34 While the tilt angle of the iron(IV)O unit is a consequence of the steric influence of the various ligand substituents in the N4Py framework, and thus modulates the ligand field by virtue of affecting Fe–N distances,10,15 it is clearly not the only factor that guides the reactivity of the iron(IV)-oxo species. In Fig. 9, the observed reactivity (k2) for C–H bond activation of cyclohexane (cf.Table 7 and Fig. 7) is related to three steric parameters of four [FeIV(O)(L)]2+ complexes, where L is N4Py, L1-Qn (8), N2Py2B and L2-Qn (7). While the order of relative reactivities (N4Py < L1-Qn < N2Py2B < L2-Qn) correlates with the Fe-NPy and FeO bond lengths (Fig. 9(a) and (b); the longer the bond lengths, the greater the HAT activity), the FeO tilt angle does not, although the complex with the largest FeO tilt angle (complex 7) is clearly the most active.
The propensity for hydrogen atom transfer and oxygen atom transfer reactions should be directly related to the basicity of the iron(IV)O unit, but steric factors may not only influence the ligand field but also affect the ability of substrates to approach this unit. The ligand field strength is expected to affect the energy and the spin state of the transition state for both HAT and OAT reactions, and the ability for the iron(IV) oxo species to undergo two-state reactivity; this is most easily probed by computational methods.7,8,12,16 In two studies, Sastri, de Visser and coworkers (Mukherjee et al.12) have specifically made computational analyses of the HAT and OAT rate enhancements by [FeIV(O)(L2-Qn)]2+ (7), [FeIV(O)(N4PyMe2)]2+ and [FeIV(O)(N2PyN2PyMe2)]2+ relative to the parent complex [FeIV(O)(N4Py)]2+. It is concluded that for 7, the tilt of the FeO moiety enforced by the ligand leads to a significantly lowered spin crossover barrier from a triplet ground state to a quintet transition state (consistent with two-state reactivity) in HAT reactions due to raising of the energies of the π*xz and π*yz orbitals that are Fe–O antibonding orbitals. The tilt also affects the σ*z2 orbital so that this orbital of the complex readily accepts an additional electron in the quintet transition state during HAT.12 These stereoelectronic effects significantly enhance the HAT rates for this complex (via a σ pathway) relative to the N4Py analogue.
We have proposed that the linear nature of the acetonitrile ligand makes the Fe–NNCMe tilt angle in N4Py-derived [FeII(N5-donor)(CH3CN)]2+ complexes an adequate “predictor” of the tilt in the corresponding [FeIV(O)(N5-donor)]2+ complexes.17 This supposition is supported by the computational modeling of [FeIV(O)(N4PyMe2)]2+ and [FeIV(O)(N2PyN2PyMe2)]2+, where the former is found to have a significantly larger tilt angle than the latter (albeit not as large as the tilt angle in [FeII(N4PyMe2)(CH3CN)]2+).27 A conclusion of the comparative experimental and computational study on the latter two iron(IV)-oxo complexes is that the differences in the reactivities of these complexes are caused primarily by steric factors (the positioning of the methyl substituents on the ligand scaffold) rather than electronic factors.27 This is borne out by low reactivity towards bulky substrates, e.g. triphenylmethane which possesses considerable steric bulk but low C–H bond dissociation energy (BDE). Furthermore, Mukherjee et al.27 point out that the reactivities of 7 and [FeIV(O)(N2Py2B)]2+ are primarily governed by electronic rather than steric factors. We note, however, that it is only for the bulky triphenylmethane substrate that the rate of alkane oxidation is higher for [FeIV(O)(N2Py2B)]2+ than for 7 (Fig. 7, vide supra). Given the relatively small BDE of the substrate, we posit that this reflects a tipping point where sterics become more important than electronics; as we have pointed out,16,34 and as is confirmed by the crystal structures of 7 and [FeIV(O)(N2Py2B)]2+,10 the N2Py2B ligand is less sterically hindering than L2-Qn.
Summary and perspectives
A relatively large number of studies on ligands of the N4Py scaffold permit a better understanding of how these ligands affect the reactivities of their M(IV)-oxo (M = Mn, Fe) complexes. As has been shown and discussed, the reactivity of the [Fe(IV)O]2+ entity when coordinated by pentadentate nitrogen-donor ligands of the N4Py scaffold is influenced by both electronic and steric factors. The quinoline-containing ligands featured in this study provide good examples of both influences – sequential introduction of one and two quinolyl moieties weaken the equatorial ligand field and causes a steric interaction with the oxo ligand that further weakens the Fe–N interactions. The afore-mentioned steric interaction is directly related to the number of quinolyl moieties; Rasheed et al.10 have shown that sterics induce a 10° tilt of the FeO moiety in [Fe(O)(L2-Qn)]2+ (7) while a more modest tilt of approximately 6° is observed for [Fe(O)(L1-Qn)]2+ (8). The observed reactivities/rate constants for C–H bond activation for 7 and 8 are in keeping with the relative stabilities indicated by the steric effects (FeO tilt) induced by the two ligands but, as discussed above, this steric/structural criterion is clearly not the only guiding factor in assessing reactivity. It appears that many subtleties of the stereoelectronic effects of this family of pentadentate ligands remain to be fully understood. While reactivities of their metal complexes may be rationalized post factum, it remains difficult to adequately predict reactivity.
Experimental section
Materials
The reagents and solvents were purchased from Sigma-Aldrich and Fisher chemicals and used without any further purification. N-[Di(2-pyridinyl)methy]-N-(2-pyridinylmethyl)methylamine,35 and bis(2-pyridyl)methylamine,36 were prepared according to literature procedures. The ligand N-[di(2-pyridyl)methyl]-N,N-bis(quinolin-2-ylmethyl)methanamine (L2-Qn) was synthesized by a minor modification of the published procedure.13 All manipulations were conducted under an inert gas atmosphere using standard Schlenk, high-vacuum line and glovebox techniques unless otherwise stated. All reagents and solvents used were of commercially available reagent quality. Solvents were purified and dried prior to use. Preparation and handling of air-sensitive materials were carried out under an inert atmosphere by in a glove box. The labeling experiment was carried out with H218O (97 atom%) from Sigma Aldrich.Physical methods
UV/Visible spectra and all kinetic experiments were performed on an Agilent 8453 UV/Vis spectrophotometer equipped with a diode-array detector and a Unisoku cryostat which permits monitoring of the temperature of the experiments from −90 °C to 100 °C. All UV/Vis spectra were measured in a 1 cm quartz cell. NMR spectra were collected on Varian Inova 500 MHz, Bruker Avance 500 MHz, and Bruker DPX-500 Mhz spectrometers in CDCl3 and CD3CN solvents and referenced to the residual signal of the solvent. The mass spectrometry (ESI) was performed with a Bruker HCT ultra mass spectrometer and Waters QTOF Micro YA263 instrument. The high resolution mass spectrum (HRMS) was performed using a Bruker FTICR APEX IV instrument. Fourier transform infrared spectroscopy on KBr pellets was performed on a Shimadzu FT-IR 8400S instrument. Elemental analyses were performed on a Perkin Elmer 2400 series II CHN analyzer. X-band EPR spectra were recorded on a JEOL JES-FA 200 instrument with 100 kHz magnetic modulation, a microwave power of 2.00 mW, and a microwave frequency of 9.1195 GHz. GC-MS measurements were carried out with a PerkinElmer Clarus 600 using an Elite 5 MS (30 m × 0.25 mm × 0.25 μm) column with a maximum temperature of 300 °C. Mössbauer spectra were recorded with a 57Co source in a Rh matrix using an alternating constant acceleration Wissel Mössbauer spectrometer operated in the transmission mode. Isomer shifts are given relative to iron foil at ambient temperature. Simulation of the experimental data was performed with the Igor Pro 8 program.Syntheses
:5) as eluent. Yield: 1.04 g (69%).
Method B: A solution of N-[di(2-pyridinyl)methyl]-N-(2-pyridinylmethyl)amine (1.45 g, 5.3 mmol), 2-chloromethylquinoline hydrochloride (1.24 g, 5.8 mmol), K2CO3 (3.45 g, 25 mmol) and N,N-diisopropylethylamine (1.3 ml, 7.5 mmol) in acetonitrile was heated under reflux for 24 hours. After cooling to room temperature, the solvent was evaporated and the residue was purified by flash column chromatography (neutral alumina) using an ethyl acetate/hexane/triethylamine (10:5:1) mixture as eluent to give L1-Qn (Rf = 0.3, 1.9 g, 86%) as a dark red oil.
1H NMR (400 MHz, CDCl3) δ 8.56 (s, 2H), 8.47 (s, 1H), 8.07 (t, J = 5.6 Hz, 1H), 8.02–7.99 (m, 1H), 7.84 (dd, J = 8.5, 1.5 Hz, 1H), 7.72 (d, J = 7.9 Hz, 3H), 7.69–7.60 (m, 4H), 7.57 (td, J = 7.6, 1.8 Hz, 1H), 7.49–7.43 (m, 1H), 7.13 (s, 2H), 7.06 (s, 1H), 5.39 (s, 1H), 4.16 (s, 2H), 4.02 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 160.7, 159.9, 149.3, 149.1, 147.6, 136.3, 129.3, 129.0, 127.4, 126.0, 124.1, 123.1, 122.1, 121.8, 121.1, 72.0, 58.0, 57.6. ESI-MS:418.2 [M + H]+.
Synthesis of [FeII(L2-Qn)(F)](BF4) (1·BF4)
A solution of Fe(BF4)2·6H2O (233 mg, 0.69 mmol) in CH3CN (3 mL) was added to a solution of L2-Qn (322 mg, 0.69 mmol) in CH3CN (3 mL). The dark red solution was placed in a diethyl ether bath and after 3 days the title compound (400 mg, 0.55 mmol, 80%) was isolated as dark-red crystals. The ESI mass spectrum shows a major fragment at m/z = 542.14 corresponding to the molecular cation. Elemental analysis (C31H25BF5FeN5): calcd; C, 59.17; H, 4.00; N, 11.13, found C 59.23, H 4.12, N 11.21. UV/Vis (in CH3CN): λmax (ε) 375 nm (1000 M−1 cm−1), 483 (1000). HRMS: m/z = 542.1445 ([Fe(L2-Qn)(F)]+) (calcd for [C31H25FeN5F]+, 542.14). Crystals of 1 suitable for X-ray analysis were obtained by vapour diffusion of diethylether into an acetonitrile solution of the compound at 4 °C.Synthesis of [FeII(L2-Qn)(Cl)](ClO4) (2·ClO4)11
Caution: Transition metal perchlorates should be handled with great care and be prepared in small quantities, as metal perchlorates are hazardous and may explode upon heating.The procedure for the synthesis of complex 2 is identical to that for 1. A total of 109 mg (0.23 mmol) of ligand L2-Qn was taken in a vial and dissolved in minimum amount of acetonitrile. To this solution, 58.6 mg (0.23 mmol) of Fe(ClO4)2 in acetonitrile was added under stirring at room temperature under air. A red precipitate appeared within 5 min of stirring. After stirring for about 30 min, the reaction mixture was placed into an ethyl acetate bath and stored overnight. The precipitate was collected by filtration, washed with ethyl acetate, dried under vacuum and obtained as red solid. Yield: 136 mg (77%).
Synthesis of [FeII(L2-Qn)(NCCH3)](ClO4)2 (3·ClO4)
Method A: A total of 100 mg (0.23 mmol) of ligand L2-Qn was taken in a vial and dissolved in a minimum amount of CH3CN. To this solution, 58.6 mg (0.23 mmol) of Fe(ClO4)2 in CH3CN was added under stirring at room temperature under air. A red precipitate appeared within 5 min of stirring. After stirring for about 30 min, the reaction mixture was placed into an ethyl acetate bath and stored overnight. The precipitate was collected by filtration, washed with ethyl acetate, dried under vacuum and obtained as a red solid. Yield: 147 mg (82%). Dark red-colored crystals of 3 suitable for X-ray analysis were obtained by slow evaporation of acetonitrile from its concentrated solution.Method B: A solution of Fe(ClO4)2·6H2O (250 mg, 0.69 mmol) in CH3CN (3 mL) was added to a solution of L2-Qn (322 mg, 0.69 mmol) in CH3CN (3 mL). The dark red solution was placed in a diethyl ether bath and after 3 days the title compound (400 mg, 0.55 mmol, 80%) was isolated as dark-red crystals.
In a similar reaction, the oxo-bridged complex [{FeIII(L2-Qn)(OH2)}2(μ-O)](ClO4)4 (5) was isolated in 25% yield and identified via single crystal X-ray diffraction (cf. ESI†).
Syntheses of [FeII(L1-Qn)(CH3CN)](ClO4)2 (6·ClO4) and [FeII(L1-Qn)(CH3CN)](BF4)2 (6·BF4)
To an acetonitrile solution of the ligand L1-Qn (100 mg, 0.24 mmol) was added of Fe(ClO4)2·6H2O (58.6 mg, 0.23 mmol) in CH3CN under stirring condition at room temperature under air. After stirring the solution for about 1 h, the reaction mixture was placed on an ethyl acetate bath and kept overnight to precipitate a red-brown solid. The solid was collected by filtration, washed with ethyl acetate, dried under vacuum. Yield: 120 mg (73%). IR (KBr) (cm−1): 3435 (br), 3061 (w), 2928 (w), 1603 (s), 1514 (m), 1468 (m), 1443 (s), 1148 (vs), 1090 (vs), 918 (m), 765 (s), 625 (s). ESI-MS (+ve ion mode in CH3CN): m/z = 236.50 for [Fe(L1-Qn)]2+ and 572.02 for {[Fe(L1-Qn)](ClO4)}+. UV-vis, λmax (nm) = 375 (ε ∼ 5800 M−1 cm−1) and 460 nm (ε ∼ 5000 M−1 cm−1). 1H NMR (500 MHz, CD3CN): δ 11.1, 10.8, 10.4, 8.5, 8.3, 8.2, 8.1, 8.0, 7.9, 7.7, 7.5, 6.6, 6.3, 5.3, 4.3, 2.5 ppm.A solution of Fe(BF4)2·6H2O (233 mg, 0.69 mmol) in CH3CN (3 ml) was added to a solution of 1 (290 mg, 0.7 mmol) in CH3CN (3 ml). The dark red solution was placed in a diethyl ether bath and after 3 days dark-red crystals of the title compound (412 mg, 0.6 mmol, 86%) were formed. Elemental Anal. found C 49.79, H 4.09, N 11.50 calcd for C29H26B2F8FeN6; C, 50.63; H, 3.81; N, 12.21.
Hydrogen atom transfer (HAT) reactions
Solutions of the iron(IV) oxo complexes 7/8 (in the concentration ranges 0.5 mM–1.0 mM) were generated in situ by adding excess IBX ester (5 equivalents) into an acetonitrile solution of complex 3/6 (0.5 mM–1.0 mM). On deaeration of the solutions and a temperature equilibration at 25 °C in the UV/Vis cuvette, substrates were added to the stirred solutions. The concentrations of substrates used were ranged from 10 mM to 400 mM and were adjusted to achieve convenient times for the reduction of iron(IV) oxo species. The time course of the decay of the iron(IV) oxo complex was then monitored at 25 °C by the UV/Vis spectrophotometer. Time courses were subjected to pseudo-first order fit and second order rate constants were evaluated from the concentration dependence data.Oxygen atom transfer (OAT) reaction of thioanisole
The iron(IV) oxo solutions were prepared as described before. The solutions were placed in cuvette and the temperature of the UV/Vis instrument was monitored to −40 °C. Then, appropriate amounts of thioanisole substrate were added to the iron(IV) oxo solution and the subsequent decay was monitored. Time courses were subjected to pseudo-first order fit and second order rate constants were evaluated from the concentration dependence data. The products were quantified following the procedure as described before. OAT reactions of other substrates were performed at room temperature.To isolate the organic products, the solutions were passed through a silica column after the end of the reaction, using ethyl acetate as the eluent, in order to remove the metal complex. The ethyl acetate solutions were then analyzed by GC-MS using a known strength of naphthalene solution as the quantification standard.
Isolation of 8
The iron(IV)-oxo species 8 was isolated from the precursor complex 6 using ceric ammonium nitrate (CAN) as an oxidant in aqueous medium following a literature procedure.10 An aqueous solution (2.5 mL) of NaClO4 (350 mg) and CAN (140 mg) was added to a acetonitrile solution (1 mL) of complex 6 (46 mg). The green solution was kept for 4 hours at 5 °C to precipitate of a dark green solid. This solid was filtered, washed with water, dried in high vacuum. Yield: 34 mg (77%).Single crystals suitable for X-ray crystallography were grown by slow evaporation of the acetonitrile solution of the complex and with an aqueous solution of dissolving 20 mg of the solid in 0.2 mL acetonitrile, and adding an aqueous solution (1.8 mL) of NaClO4 (40 mg) at 5 °C.
Isotope labeling experiments
The isotope labeling experiment was carried out by adding 10 μL H218O (97% 18O-enriched) to an acetonitrile solution of 7/8.Reactions with external substrates
Reaction kinetics were performed by adding calculated amounts of substrates (thioanisoles and the alkanes) to 0.5 mM solution of 7/8 in CH3CN under anaerobic condition at 298 K. The rate constants, kobs, were determined by pseudo-first-order fitting of the decay plot of the oxo band (770 nm for 7 and 730 nm for 8). Second-order rate constants k2 were obtained from the slopes of the linear fits of kobsvs. concentration of substrates.Analysis of the products after oxidation of substrates
The iron(II) precursor complex (3 or 6) (0.02 mmol) was dissolved in 5 mL of dry deoxygenated acetonitrile. To the resulting solution, 3 equiv. of IBX-ester was added along with the substrates at 298 K under inert atmosphere. The reaction was allowed to stir without altering the temperature. After the reaction, the resulting solution was passed through silica column (60–120 mesh size) using diethyl ether as eluent. The combined organic phase was then analyzed by GC-MS. For GC analyses, naphthalene was used as an internal standard and the products were identified by comparison of their GC retention times and GC-MS with those of authentic compounds. The oxidation product from thioanisole was identified by 1H NMR spectroscopy and was quantified using 1,3,5-trimethoxybenzene as an internal standard.Control experiments
Control experiments were performed separately for each substrate using an Fe(ClO4)2 and IBX-ester in acetonitrile at 298 K. The small amount of oxidized product, formed in some cases, was considered while calculating the yields.Crystal structure determinations
The crystals of 1 and 6 were immersed in cryo-oil, mounted in a Nylon loop, and measured at a temperature of 100 K. The X-ray diffraction data were collected on Bruker Kappa Apex II and Bruker Kappa Apex II Duo diffractometers using Mo Kα radiation (λ = 0.71073 Å). The APEX237 program package was used for cell refinements and data reductions. The structures were solved by charge flipping technique (SUPERFLIP)38 or direct methods using the SIR201139 program with the Olex240 graphical user interface. A semi-empirical numerical absorption correction based on equivalent reflections (SADABS)41 was applied to all data. Structural refinements were carried out using SHELXL-97.42 The crystal of 1 was diffracting only weakly and therefore atoms N2, C16, C17, C18, C19, and C20 were restrained to have the same Uij components within the standard uncertainty of 0.02. In the structure of 2 one molecule of the acetonitrile of crystallization was disordered over two sites with equal occupancies. Hydrogen atoms were positioned geometrically and were also constrained to ride on their parent atoms, with C–H = 0.95–0.1.00 Å, and Uiso = 1.2 − 1.5Ueq (parent atom).
The X-ray diffraction data for 2, 3, 4 and 8 were collected on a Bruker D8 VENTURE Microfocus diffractometer equipped with a PHOTON II Detector, with Mo Kα radiation, controlled by the APEX3 software package (v2017.3-0). The raw data were integrated and corrected for Lorentz and polarization effects using the Bruker APEX II program suite.43 Absorption corrections were performed by using SADABS. Structure solution and refinement were carried out using the SHELXL program suite44 using the Olex2 interface. Hydrogen atoms at idealized positions were included in the final refinements. The structure was solved by intrinsic methods and subsequent Fourier analyses and refined by the full-matrix least-squares method based on F2 with all observed reflections. The crystallographic details for all structures are summarized in Table S1 (ESI).†
CCDC entries 2113914 (1), 2113532 (2), 2113531 (3), 2113533 (4), 2113535 (5), 2113534 (6), and 2045257 (8)† contain the supplementary crystallographic data for the structures described in this paper.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
TKP acknowledges the Science and Engineering Research Board (SERB), India, for financial support (project: CRG/2019/006393). SM and SB thank the Council of Scientific and Industrial Research (CSIR), India, for research fellowships. The financial support from the Technical Research Center (TRC) at IACS for setting up a Mössbauer spectrometer facility is thankfully acknowledged. AS thanks the Carl Trygger Foundation for a postdoctoral fellowship. MKH thanks the European Commission for an Erasmus Mundus predoctoral fellowship. SY and OFW thank the Swedish Research Council and the SIDA bilateral support program for financial support. FX and EM thank FAPESC for financial support. RA, FX and EN thank CAPES and STINT for a Swedish-Brazilian Joint Collaboration Grant.Notes and references
- (a) J. Rittle and M. Green, Science, 2010, 330, 933–937 CrossRef CAS PubMed; (b) I. Schlichting, J. Berendzen, K. Chu, A. M. Stock, S. A. Maves, D. E. Benson, R. M. Sweet, D. Ringe, G. A. Petsko and S. G. Sligar, Science, 2000, 287, 1615–1622 CrossRef CAS PubMed; (c) I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Chem. Rev., 2005, 105, 2253–2278 CrossRef CAS PubMed; (d) M. Newcomb, R. Zhang, R. E. P. Chandrasena, J. A. Halgrimson, J. H. Horner, T. M. Makris and S. G. Sligar, J. Am. Chem. Soc., 2006, 128(14), 4580–4581 CrossRef CAS PubMed; (e) F. P. Guengerich, ACS Catal., 2018, 8, 10964–10976 CrossRef CAS PubMed; (f) C. M. Krest, E. L. Onderko, T. H. Yosca, J. Livada, J. Rittle and M. T. Green, J. Biol. Chem., 2013, 288, 17074–17081 CrossRef CAS PubMed.
- (a) J. M. Bollinger, J. C. Price, L. M. Hoffart, E. W. Barr and C. Krebs, Eur. J. Inorg. Chem., 2005, 4245–4254 CrossRef CAS; (b) M. Srnec, S. R. Iyer, L. M. K. Dassama, K. Park, S. D. Wong, K. D. Sutherlin, Y. Yoda, Y. Kobayashi, M. Kurokuzu, M. Saito, M. Seto, C. Krebs, J. M. Bollinger and E. I. Solomon, J. Am. Chem. Soc., 2020, 142, 18886–18896 CrossRef CAS PubMed.
- (a) Md. S. Islam, T. M. Leissing, R. Chowdhury, R. J. Hopkinson and C. J. Schofield, Annu. Rev. Biochem., 2018, 87, 585–620 CrossRef CAS PubMed; (b) E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S.-K. Lee, N. Lehnert, F. Neese, A. J. Skulan, Y. S. Yang and J. Zhou, Chem. Rev., 2000, 100, 235 CrossRef CAS PubMed; (c) J. C. Price, E. W. Barr, B. Tirupati, J. M. Bollinger and C. Krebs, Biochemistry, 2003, 42(24), 7497–7508 CrossRef CAS PubMed.
- S. M. Barry and G. L. Challis, ACS Catal., 2013, 3, 2362–2370 CrossRef CAS PubMed.
- (a) A. R. McDonald and L. Que Jr., Coord. Chem. Rev., 2013, 257, 414–428 CrossRef CAS; (b) . M. Puri and L. Que Jr., Acc. Chem. Res., 2015, 48, 2443 CrossRef PubMed; (c) J. Hohenberger, K. Ray and K. Meyer, Nat. Commun., 2012, 3, 720 CrossRef PubMed; (d) J. E. M. N. Klein and L. Que Jr., Biomimetic High–Valent Mononuclear Nonheme Iron–Oxo Chemistry, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, 2016 Search PubMed.
- (a) W. N. Oloo and L. Que Jr., Acc. Chem. Res., 2015, 48, 2612–2621 CrossRef CAS PubMed; (b) W. Nam, Y.-M. Lee and S. Fukuzumi, Acc. Chem. Res., 2018, 51, 2014–2022 CrossRef CAS PubMed; (c) G. Olivo, O. Cussó, M. Borrell and M. Costas, J. Biol. Inorg. Chem., 2017, 22, 425–452 CrossRef CAS PubMed; (d) X. Engelmann, I. Monte-Pérez and K. Ray, Angew. Chem., Int. Ed., 2016, 55, 7632–7649 CrossRef CAS PubMed; (e) A. S. Borovik, Chem. Soc. Rev., 2011, 40, 1870–1874 RSC; (f) M. Milan, M. Bietti and M. Costas, Chem. Commun., 2018, 54, 9559–9570 RSC; (g) W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS PubMed.
- (a) D. Mandal and S. Shaik, J. Am. Chem. Soc., 2016, 138, 2094–2097 CrossRef CAS PubMed; (b) D. Mandal, D. Mallick and S. Shaik, Acc. Chem. Res., 2018, 51, 107–117 CrossRef CAS PubMed.
- S. Shaik, Isr. J. Chem., 2020, 60, 938–956 CrossRef CAS.
- M. Puri and L. Que Jr., Acc. Chem. Res., 2015, 48, 2443–2452 CrossRef CAS PubMed.
- W. Rasheed, A. Draksharapu, S. Banerjee, V. G. Young Jr., R. Fan, Y. Guo, M. Ozerov, J. Nehrkorn, J. Krzyztek, J. Telser and L. Que Jr., Angew. Chem., Int. Ed., 2018, 57, 9387–9391 CrossRef CAS PubMed.
- S. Rana, J. P. Biswas, A. Sen, M. Clemancey, G. Blondin, J.-M. Latour, G. Rajaraman and D. Maiti, Chem. Sci., 2018, 9, 7843–7858 RSC.
- G. Mukherjee, C. W. Z. Lee, S. S. Nag, A. Alili, F. G. C. Reinhard, D. Kumar, C. V. Sastri and S. P. Visser, Dalton Trans., 2018, 47, 14945–14957 RSC.
- W. K. C. Lo, C. J. McAdam, A. G. Blackman, J. D. Crowley and D. A. McMorran, Inorg. Chim. Acta, 2015, 426, 183–194 CrossRef CAS.
- A. A. Massie, A. Sinha, J. D. Parham, E. Nordlander and T. A. Jackson, Inorg. Chem., 2018, 57, 8253–8263 CrossRef CAS PubMed.
- M. Lubben, A. Meetsma, E. C. Wilkinson, B. Feringa and L. Que Jr., Angew. Chem., Int. Ed. Engl., 1995, 34, 1512–1514 CrossRef CAS.
- M. Mitra, H. Nimir, S. Demeshko, S. S. Bhat, S. O. Malinkin, M. Haukka, J. Lloret-Fillol, G. C. Lisensky, F. Meyer, A. A. Shteinman, W. R. Browne, D. A. Hrovat, M. G. Richmond, M. Costas and E. Nordlander, Inorg. Chem., 2015, 54, 7152–7164 CrossRef CAS PubMed.
- R. Singh, G. Ganguly, S. O. Malinkin, S. Demeshko, F. Meyer, E. Nordlander and T. K. Paine, Inorg. Chem., 2019, 58, 1862–1876 CrossRef CAS PubMed.
- A. F. Valiati, R. D. Huelsmann, E. Martendal, A. J. Bortoluzzi, F. R. Xavier and R. A. Peralta, New J. Chem., 2020, 44, 2514–2526 RSC.
- (a) A. G. J. Ligtenbarg, P. Oosting, G. Roelfes, R. M. La Crois, M. Lutz, A. L. Spek, R. Hage and B. L. Feringa, Chem. Commun., 2001, 385–386 RSC; (b) S. Megow, H.-L. Fitschen, F. Tuczek and F. Temps, J. Phys. Chem. Lett., 2019, 10, 6048–6054 CrossRef CAS PubMed; (c) D. Unjaroen, M. Swart and W. R. Browne, Inorg. Chem., 2017, 56, 470–479 CrossRef CAS PubMed; (d) H. Y. Au-Yeung, J. Chan, T. Chantarojsiri and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 15165–15173 CrossRef CAS PubMed; (e) A. Geersing, R. H. de Vries, G. Jansen, M. G. Rots and G. Roelfes, Bioorg. Med. Chem. Lett., 2019, 29, 1922–1927 CrossRef CAS PubMed.
- (a) D. L. Reger, R. P. Watson, J. R. Gardinier, M. D. Smith and P. J. Pellechia, Inorg. Chem., 2006, 45, 10088–10097 CrossRef CAS PubMed; (b) D. L. Reger, E. A. Foley, R. P. Watson, P. J. Pellechia, M. D. Smith, F. Grandjean and G. J. Long, Inorg. Chem., 2009, 48, 10658–10669 CrossRef CAS PubMed; (c) D. L. Reger, A. E. Pascui, M. D. Smith, J. Jezierska and A. Ozarowski, Inorg. Chem., 2012, 51, 11820–11836 CrossRef CAS PubMed; (d) S. Kannan, M. A. Moody, C. L. Barnes and P. B. Duval, Inorg. Chem., 2006, 45, 9206–9212 CrossRef CAS PubMed; (e) B. Butschke, K. L. Fillman, T. Bendikov, L. J. W. Shimon, Y. Diskin-Posner, G. Leitus, S. I. Gorelsky, M. L. Neidig and D. Milstein, Inorg. Chem., 2015, 54(10), 4909–4926 CrossRef CAS PubMed.
- E. J. Klinker, J. Kaizer, W. W. Brennessel, N. L. Woodrum, C. J. Cramer and L. Que Jr., Angew. Chem., Int. Ed., 2005, 44, 3690–3694 CrossRef CAS PubMed.
- I. Morishima, K. Okada, M. Ohashi and T. Yonezawa, J. Chem. Soc. D, 1971, 33–34 RSC.
- The C/A convention for description of chirality can be applied to square pyramidal and octahedral centers. See: Nomenclature of Inorganic Chemistry – IUPAC Recommendations 2005, ed. N. G. Connelly, T. Damhus, R. M. Hartshorn and A. T. Hutton, RSC Publishing, Cambridge, 2005, pp. 201–203; pdf available at https://iupac.org/what-we-do/books/redbook/. We thank Edwin C. Constable (University of Basel, Switzerland), Gerard P. Moss (Queen Mary College, UK) and Andrey Yerin (ACD Labs, Russia) for assistance with the assignment of the chirality of complex Search PubMed.
- G. Roelfes, M. Lubben, K. Chen, R. Y. N. Ho, A. Meetsma, S. Genseberger, R. M. Hermant, R. Hage, S. K. Mandal, V. G. Young Jr., Y. Zang, H. Kooijman, A. L. Spek, L. Que Jr. and B. L. Feringa, Inorg. Chem., 1999, 38, 1929–1936 CrossRef CAS PubMed.
- A. L. Spek , CSD Communication. Private Communication, 2004, CCDC 232796.
- J. Kaizer, E. J. Klinker, N. Y. Oh, J. Rohde, W. J. Song, A. Stubna, J. Kim, E. Münck, W. Nam and L. Que, J. Am. Chem. Soc., 2004, 126, 472–473 CrossRef CAS PubMed.
- G. Mukherjee, A. Alili, P. Barman, D. Kumar, C. V. Sastri and S. P. de Visser, Chem. – Eur. J., 2019, 25, 5086–5098 CrossRef CAS PubMed.
- W. Rasheed, R. Fan, C. S. Abelson, P. O. Peterson, W. Ching, Y. Guo and L. Que Jr., J. Biol. Inorg. Chem., 2019, 24, 533–545 CrossRef CAS PubMed.
- S. L. C. Ferreira, V. A. Lemos, V. S. de Carvalho, E. G. P. da Silva, A. F. S. Queiroz, C. S. A. Felix, D. L. F. da Silva, G. B. Dourado and R. V. Oliveira, Microchem. J., 2018, 140, 176–182 CrossRef CAS.
- L. V. Candioti, M. M. De Zan, M. S. Cámara and H. C. Goicoechea, Talanta, 2014, 124, 123–138 CrossRef PubMed.
- A. Decker, J.-U. Rohde, L. Que and E. I. Solomon, J. Am. Chem. Soc., 2004, 126, 5378–5379 CrossRef CAS PubMed.
- C. V. Sastri, J. Lee, K. Oh, Y. J. Lee, J. Lee, T. A. Jackson, K. Ray, H. Hirao, W. Shin, J. A. Halfen, J. Kim, L. Que Jr., S. Shaik and W. Nam, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19181–19186 CrossRef CAS PubMed.
- A. Decker, J.-U. Rohde, E. J. Klinker, S. D. Wong, L. Que and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 15983–15996 CrossRef CAS PubMed.
- M. C. Denler, A. A. Massie, R. Singh, E. Stewart-Jones, A. Sinha, V. W. Day, E. Nordlander and T. A. Jackson, Dalton Trans., 2019, 48, 5007–5021 RSC.
- (a) M. Goodgame and F. A. Cotton, J. Am. Chem. Soc., 1992, 84, 1543–1548 CrossRef; (b) C. R. Johnson and R. E. Shepherd, Inorg. Chem., 1983, 22, 3506–3513 CrossRef CAS; (c) D. M. L. Goodgame, M. Goodgame and M. J. Weeks, J. Chem. Soc., 1964, 5194–5199 RSC.
- G. Roelfes, M. E. Branum, L. Wang, L. Que Jr. and B. L. Feringa, J. Am. Chem. Soc., 2000, 122, 11517–11518 CrossRef CAS.
- APEX2, in SAINT-Plus and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2008 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, C. Giacovazzo, M. Mallamo, A. Mazzone, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2012, 45, 351–356 CrossRef.
- O. V. Dolomanov, L. J. Borhuis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- Bruker, SADABS, BrukerAXSInc., Madison, Wisconsin, USA, 2001 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- APEX II, 2009 Ed., Bruker Analytical X-ray Systems Inc., Madison, WI, 2009 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2045257 and 2113531–2113535. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03381f |
| This journal is © The Royal Society of Chemistry 2022 |