
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cu(II), Mn(II) and Zn(II) complexes of hydrazones with a quaternary ammonium moiety: synthesis, experimental and theoretical characterization and cytotoxic activity†
Nevena
Stevanović
a,
Matija
Zlatar
 a,
Irena
Novaković
b,
Andrej
Pevec
a,
Dušanka
Radanović
b,
Ivana Z.
Matić
a,
Marija
Đorđić Crnogorac
c,
Tatjana
Stanojković
c,
Miroslava
Vujčić
b,
Maja
Gruden
a,
Dušan
Sladić
a,
Katarina
Anđelković
a,
Iztok
Turel
*d and
Božidar
Čobeljić
*a
a,
Irena
Novaković
b,
Andrej
Pevec
a,
Dušanka
Radanović
b,
Ivana Z.
Matić
a,
Marija
Đorđić Crnogorac
c,
Tatjana
Stanojković
c,
Miroslava
Vujčić
b,
Maja
Gruden
a,
Dušan
Sladić
a,
Katarina
Anđelković
a,
Iztok
Turel
*d and
Božidar
Čobeljić
*a
aUniversity of Belgrade-Faculty of Chemistry, Studentski trg 12–16, 11000 Belgrade, Serbia. E-mail: bozidar@chem.bg.ac.rs
bUniversity of Belgrade-Institute of Chemistry, Technology and Metallurgy, Department of Chemistry, Njegoševa 12, 11000 Belgrade, Serbia
cInstitute of Oncology and Radiology of Serbia, 11000 Belgrade, Serbia
dFaculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, 1000 Ljubljana, Slovenia. E-mail: Iztok.Turel@fkkt.uni-lj.si
First published on 23rd November 2021
Abstract
In this paper, Cu(II), Mn(II) and Zn(II) complexes with N,N,N-trimethyl-2-oxo-2-(2-(1-(thiazol-2-yl)ethylidene)hydrazinyl)ethan-1-aminium chloride (HL1Cl) were synthesized and characterized by single-crystal X-ray diffraction, IR spectroscopy, elemental analysis and DFT calculations. In all three complexes, a ligand (L1) is coordinated in a deprotonated formally neutral zwitterionic form via NNO donor set atoms. Cu(II) and Zn(II) form mononuclear penta-coordinated complexes [CuL1(N3)(CH3OH)]BF4 and [ZnL1(N3)2], respectively, while Mn(II) forms a binuclear [Mn2L12(μ-1,1-N3)2(N3)2]·2CH3OH complex, with unusual distorted trigonal-prismatic geometry around the metal centers. The antimicrobial activity of these complexes was tested against a panel of Gram-negative and Gram-positive bacteria, two yeasts and one fungal strain. The binuclear Mn(II) complex showed antifungal activity of similar intensity to amphotericin B. Based on the results of the brine shrimp test and DPPH radical scavenging activity, the most active Cu(II) and Mn(II) complexes were selected for evaluation of cytotoxic activity against five malignant cancer cell lines (HeLa, A375, MCF7, PC-3 and A549) and one normal cell line HaCaT. Both complexes showed significant activity. It should be pointed out that the activity of the Mn(II) complex against the MCF7 breast cancer cell line is only slightly weaker than that of cisplatin, but with selectivity to the tumor cell line in comparison to normal HaCaT cells, which is non-existent in the case of cisplatin.
Introduction
Metal complexes with hydrazones have been investigated intensively during previous years due to their various pharmaceutical applications as antitumor, antibacterial, antiviral and antifungal agents.1 Of particular interest are hydrazone ligands with the –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NH–C(O)– group, formed by the condensation of aldehydes/ketones with different hydrazides. Girard's T reagent (trimethylaminoacetohydrazide chloride) is attractive due to its ability to form water-soluble hydrazones with various aldehydes/ketones.2 Using aldehydes/ketones with a thiazole ring in their structure for the synthesis of hydrazone ligands, additional coordination atoms are introduced. Combining both properties (additional coordination site due to the thiazole ring and water-solubility due to the positively charged quaternary ammonium moiety) in metal hydrazone complexes can lead to enhanced biological activity.1
N–NH–C(O)– group, formed by the condensation of aldehydes/ketones with different hydrazides. Girard's T reagent (trimethylaminoacetohydrazide chloride) is attractive due to its ability to form water-soluble hydrazones with various aldehydes/ketones.2 Using aldehydes/ketones with a thiazole ring in their structure for the synthesis of hydrazone ligands, additional coordination atoms are introduced. Combining both properties (additional coordination site due to the thiazole ring and water-solubility due to the positively charged quaternary ammonium moiety) in metal hydrazone complexes can lead to enhanced biological activity.1
Copper, manganese and zinc are essential trace elements with many physiological functions. Ions of these elements act as cofactors and as allosteric components for many enzymes.3–5 Cu(II) and Mn(II) complexes have been studied as low molecular weight model compounds that mimic the active site of superoxide dismutase (SOD),6,7 which participate in cell oxidative stress regulation. According to biological evaluations, complexes of Cu(II), Mn(II) and Zn(II) with Schiff base ligands can possess different biological activities, such as effective inhibition against bacteria8–15 and fungi,8,9,11,13,14 as well as cytotoxic activity.8,16–18
Schiff base complexes of Cu(II) and Mn(II), both mononuclear and dinuclear, were previously reported.16,17,19 Most of the biologically active copper and manganese complexes have a mononuclear structure, with the most common being square-planar17,18,20 and square-pyramidal geometry8,16,18,21 around Cu(II) ions and octahedral geometry around Mn(II) ions.13,22–24
In continuation of our previous investigations on the synthesis, characterization and biological activity of complexes with Girard's T reagent-based hydrazones,6,25–29 which showed moderate biological activities, in this paper, three novel Girard's T reagent–based complexes with Cu(II), Mn(II) and Zn(II) ions are described.
Results
General
The reaction of 2-acetylthiazole and Girard's T reagent was performed according to the previously reported method30 which yielded the ligand N,N,N-trimethyl-2-oxo-2-(2-(1-(thiazol-2-yl)ethylidene)hydrazinyl)ethan-1-aminium chloride (HL1Cl) that was used for the synthesis of complexes 1–3 (Scheme 1). The reaction of the ligand HL1Cl with the metal salts Cu(BF4)·6H2O/MnCl2·4H2O/Zn(BF4)·6H2O and NaN3 in the molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:4 in methanol results in the formation of mononuclear Cu(II) complex (1) with the composition [CuL1(N3)(CH3OH)]BF4, binuclear Mn(II) complex 2 with the composition [Mn2L12(μ-1,1-N3)2(N3)2]·2CH3OH, and mononuclear Zn(II) complex (3) with the composition [ZnL1(N3)2] (Scheme 1). The ligand is coordinated in a deprotonated formally neutral zwitterionic form via NNO donor set atoms in all three complexes.
:1:4 in methanol results in the formation of mononuclear Cu(II) complex (1) with the composition [CuL1(N3)(CH3OH)]BF4, binuclear Mn(II) complex 2 with the composition [Mn2L12(μ-1,1-N3)2(N3)2]·2CH3OH, and mononuclear Zn(II) complex (3) with the composition [ZnL1(N3)2] (Scheme 1). The ligand is coordinated in a deprotonated formally neutral zwitterionic form via NNO donor set atoms in all three complexes.
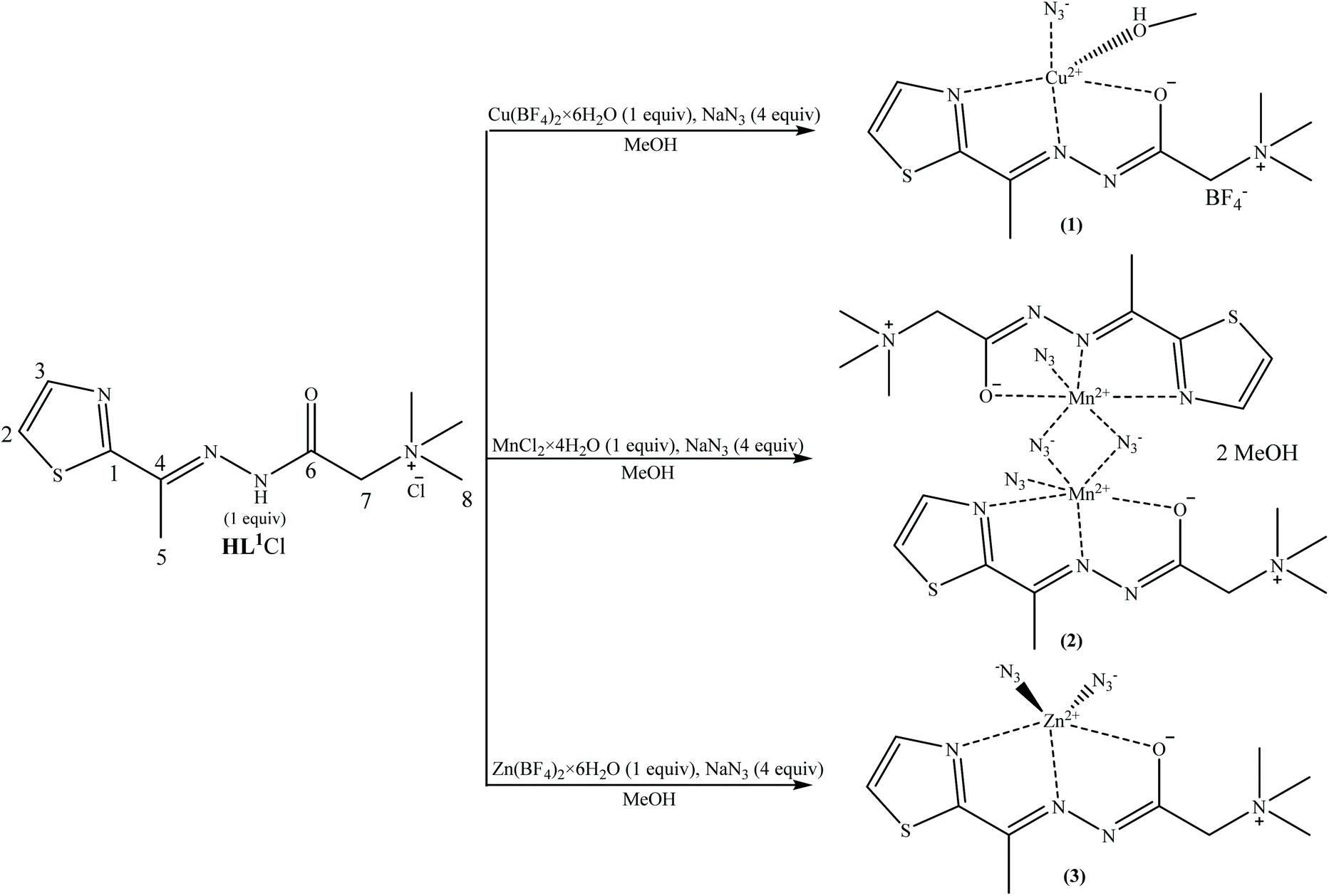 | ||
| Scheme 1 Synthesis of the [CuL1(N3)(CH3OH)]BF4 (1), [Mn2L12(μ-1,1-N3)2(N3)2]·2CH3OH (2) and [ZnL1(N3)2] (3) complexes. | ||
Spectroscopy
O) band at 1701 cm−1, observed in the spectrum of the ligand HL1Cl, new bands at 1698 cm−1, 1688 cm−1 and 1690 cm−1 appeared in the spectra of complexes 1, 2 and 3, respectively, assigned to the ν(–O–CN) vibrations of the deprotonated hydrazide moieties. The coordination of azomethine nitrogen atoms results in the shift of the ν(CN) band from 1612 cm−1 in the spectrum of the ligand HL1Cl to 1604 cm−1, 1595 cm−1, and 1600 cm−1 in the spectra of complexes 1, 2 and 3, respectively.
Crystal structures of [CuL1(N3)(CH3OH)]BF4 (1), [Mn2L12(μ-1,1-N3)2(N3)2]·2CH3OH (2) and [ZnL1(N3)2] (3) complexes
The Cu(II), Mn(II) and Zn(II) ions with L1 form the mononuclear [CuL1(N3)(CH3OH)]BF4 (1) and [ZnL1(N3)2] (3) and binuclear [Mn2L12(μ-1,1-N3)2(N3)2]·2CH3OH (2) complexes in which L1 in a zwitterionic form2 coordinates as a tridentate ligand to M(II) ions through thiazole and imine nitrogen atoms and an enolate oxygen atom. Complexes 1 and 3 crystallize in the monoclinic crystal system with space group No. 14 (P21/n and P21/c cell settings, respectively) and complex 2 in the triclinic crystal system with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (No. 2).
(No. 2).
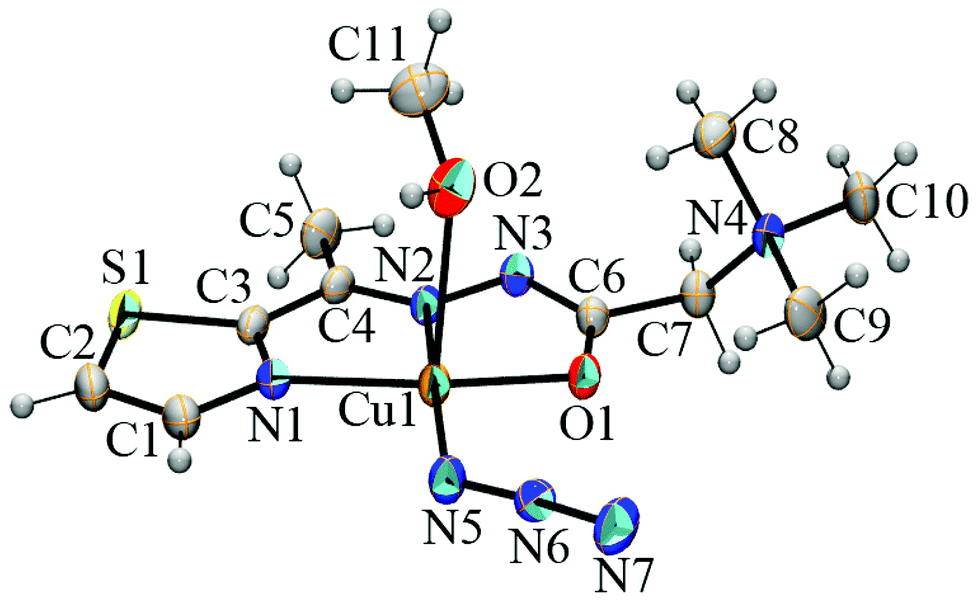 | ||
| Fig. 1 ORTEP representation of the [Cu(L1)(N3)(CH3OH)]+ complex cation in [CuL1(N3)(CH3OH)]BF4. Thermal ellipsoids are drawn at the 30% probability level. | ||
37 with the same ligand, the [Mn2L12(μ-1,1-N3)2(N3)2]·2CH3OH (2) complex shows significantly longer M–NAr and M–Nimine bond distances (Mn–NAr 2.3668(19) Å, Ni–NAr 2.122(2)–2.126(3) Å; Mn–Nimine 2.2500(18) Å, Ni–Nimine 1.997(2)–2.017(3) Å; Mn–Oenolate 2.1879(16) Å and Ni–Oenolate 2.083(2)–2.140(2) Å), indicating the weaker coordination of L1 to Mn(II) ions. Similarly, as in the corresponding Ni(II) complexes,37 in complex 2, the terminal azido ligands are coordinated at trans positions. The N9–N8–Mn1 bond angle of 137.7(2)° shows the bent coordination of the anionic terminals. The central Mn2N2 ring is planar with a Mn–Nazido(end-on)–Mnc bridging angle of 107.37(8)° and an Mn⋯Mnc separation of 3.6031(6) Å. The Mn–Nazido(end-on) bond distances show a discrepancy of 0.0135 Å. In the crystals of 2, binuclear complex units are connected by weak intermolecular C–H⋯Oenolate and C–H⋯Nazide hydrogen bonds38 into chains parallel with [100]. Furthermore, the chains are connected by intermolecular hydrogen bonds that involve solvent methanol molecules serving as hydrogen bond donors (O2–H2A⋯N3) and hydrogen bond acceptors (C7–H7B⋯O2 and C9–H9C⋯O2) into layers parallel with the (0−11) lattice plane (Table S5 and Fig. S2†). The shortest separation of 4.6162(16) Å between the centers of gravity of the 1,3-thiazole rings is observed between the neighboring (0−11) layers.
 | ||
| Fig. 2 ORTEP representation of [Mn2L12(μ-1,1-N3)2(N3)2] in [Mn2L12(μ-1,1-N3)2(N3)2]·2CH3OH. The unlabelled part of the molecule is generated by symmetry operation 1 − x, 1 − y, and 1 − z. Solvent CH3OH molecules have been omitted for clarity. Thermal ellipsoids are drawn at the 30% probability level. | ||
40 (L3 = the condensation product of 2-quinolinecarboxaldehyde and trimethylammoniumacetohydrazide chloride), while the Zn–Oenolate bond is slightly longer—2.230(2) Å vs. 2.146(5)–2.222(2) Å. The azido ligands in 3 are coordinated to the Zn(II) ion in the bent mode, with Zn–N–N angles of 122.5(3) and 122.7(5)°. In the crystals of 3, the complex molecules [ZnL1(N3)2] are self-assembled into supramolecular layers parallel to the (100) lattice plane through weak intermolecular C–H⋯N and C–H⋯O hydrogen bonds38 (Table S6 and Fig. S3†).
 | ||
| Fig. 3 ORTEP representation of the [ZnL1(N3)2] complex. Thermal ellipsoids are drawn at the 30% probability level. | ||
Computational results
DFT calculations were performed to elucidate the structures of the Cu(II), Mn(II), and Zn(II) complexes in DMSO solution. Free energy changes, ΔrG (298 K), of several probable reactions starting from the X-ray determined structures of 1, 2, and 3 were investigated. There is an excellent agreement between the DFT optimized and X-ray structures for all three complexes (Fig. S13†). ΔrG (298 K) was calculated based on the difference in the Gibbs free energy of the products and reactants at the ZORA-M06-2X/TZP-COSMO(DMSO)//ZORA-BP86-D3/TZP-COSMO(DMSO) level of theory. For the Cu(II) complex, we explored the formation of the binuclear complex [Cu2L12(N3)2(CH3OH)2]2+, dissociation of weakly coordinated CH3OH to form square-planar [CuL1(N3)]+, and several potential modes of coordination of DMSO to the Cu(II) center (reactions (1)–(5), Table 1). The results clearly show that the formation of square planar [CuL1(N3)]+ (Fig. 4) from 1 is thermodynamically favored. For the Mn(II) complex, we investigated the dissociation of crystalline CH3OH from 2 and the dissociation of binuclear [Mn2L12(μ-1,1-N3)2(N3)2] to form the mononuclear pentacoordinate complex [MnL1(N3)2] (reactions (6) and (7), Table 1). The subsequent formation of octahedral Mn(II) complexes with a solvent molecule (DMSO) as the sixth ligand is also considered (reactions (8) and (9), Table 1). The results reveal that the thermodynamically most favorable is the formation of the pentacoordinate [MnL1(N3)2] complex (Fig. 5). However, according to the calculated ΔrG of only −1 kcal mol−1 for reaction (7), the binuclear [Mn2L12(μ-1,1-N3)2(N3)2] complex is also expected to be present in the DMSO solution. Two Mn(II) centers in [Mn2L12(μ-1,1-N3)2(N3)2] are weakly ferromagnetically coupled, with the exchange constant J = 6.0 cm−1 calculated by the broken-symmetry DFT approach at the ZORA-M06-2X-COSMO(DMSO)/TZP level of theory. Analogous reactions were considered for the Zn(II) complex, i.e., formation of the binuclear complex [Zn2L12(μ-1,1-N3)2(N3)2] (reaction (10), Table 1) and the formation of six-coordinate complexes (reactions (11) and (12), Table 1). The calculations disclose that the [ZnL1(N3)2] complex (complex 3) is thermodynamically preferred in DMSO solution. | ||
| Fig. 4 Structure of the [CuL1(N3)]+ complex ion optimized at the ZORA-BP86-D3/TZP-COSMO(DMSO) level of theory. | ||
 | ||
| Fig. 5 Structure of the [MnL1(N3)2] complex optimized at the ZORA-BP86-D3/TZP-COSMO(DMSO) level of theory. | ||
| Reaction | ΔrG (298 K) | |
|---|---|---|
| 1 | 2[CuL1(N3)(CH3OH)]+ ⇌ [Cu2L12(N3)2(CH3OH)2]2+ | 8.90 |
| 2 | [CuL1(N3)(CH3OH)]+ ⇌ [CuL1(N3)]+ + CH3OH | −2.90 |
| 3 | [CuL1(N3)(CH3OH)]+ + DMSO ⇌ [CuL1(N3)(CH3OH)(DMSO)]+ | 4.58 |
| 4 | [CuL1(N3)]+ + DMSO ⇌ [CuL1(N3)(DMSO)]+ | 2.96 |
| 5 | [CuL1(N3)]+ + 2DMSO ⇌ [CuL1(N3)(DMSO)2]+ | 7.33 |
| 6 | [Mn2L12(μ-1,1-N3)2(N3)2]·2CH3OH ⇌ [Mn2L12(μ-1,1-N3)2(N3)2] + 2CH3OH | −9.93 |
| 7 | [Mn2L12(μ-1,1-N3)2(N3)2] ⇌ 2 [MnL1(N3)2] | −1.07 |
| 8 | [MnL1(N3)2] + DMSO ⇌ cis-[MnL1(N3)2(DMSO)] | 2.96 |
| 9 | [MnL1(N3)2] + DMSO ⇌ trans-[MnL1(N3)2(DMSO)] | 2.47 |
| 10 | 2 [ZnL1(N3)2] ⇌ [Zn2L12(μ-1,1-N3)2(N3)2] | 10.29 |
| 11 | [ZnL1(N3)2] + DMSO ⇌ cis-[ZnL1(N3)2(DMSO)] | 6.05 |
| 12 | [ZnL1(N3)2] + DMSO ⇌ trans-[ZnL1(N3)2(DMSO)] | 7.85 |
Antimicrobial activity
The antibacterial activity of the synthesized complexes, their precursors HL1Cl, NaN3 and appropriate salts was evaluated against a panel of five Gram-positive and five Gram-negative bacteria. The MIC data are given in Table 2. All three complexes showed antibacterial activity against all tested bacterial strains. For complexes 1 and 2, the precursor compounds either do not have or show low antibacterial activity. The most active complex 2 is also the only binuclear complex in the series. Its activity towards P. aeruginosa is over twice lower than the activity of chloramphenicol, while against P. hauseri, the activity was four times lower than the control compound. Complex 1 displayed the best activity towards E. coli strain and very weak selectivity towards Gram-negative bacteria. The lowest antibacterial activity was obtained for complex 3. In some cases, its activity was lower than that of the parent salt. A comparison of the antimicrobial activity of binuclear azido bridged complexes of Mn(II) (complex 2) and Ni(II) with the same ligand system42 showed that complex 2 has higher antimicrobial activity. Even with this slightly lower activity, the binuclear Ni(II) complex in most cases has higher activity than the mononuclear Cu(II) and Zn(II) complexes. Bearing in mind these two facts, the reason for the higher antibacterial activity of the bimetallic Mn(II) complex can be explained by the existence of two metal centers.| E. coli | P. aeruginosa | P. hauseri | K. pneumoniae | S. enterica | S. aureus | M. flavus | M. luteus | B. spizizenii | C. sporogenes | |
|---|---|---|---|---|---|---|---|---|---|---|
| HL1Cl | — | — | — | — | — | — | — | — | — | — |
| 1 | 1.19 | 2.37 | 4.74 | 2.37 | 2.37 | 4.74 | 4.74 | 4.74 | 4.74 | 4.74 |
| 2 | 1.44 | 1.44 | 1.44 | 1.44 | 1.44 | 2.88 | 1.44 | 1.44 | 1.44 | 2.88 |
| 3 | 2.75 | 2.75 | 5.50 | 2.75 | 2.75 | 2.75 | 2.75 | 2.75 | 5.50 | 5.50 |
| [Ni2L12(μ-1,1-N3)2(N3)2]·4H2O (ref. 42) | 1.56 | 3.12 | 3.12 | 3.12 | 1.56 | 3.12 | 1.56 | 1.56 | 3.12 | 3.12 |
| Cu(BF4)2·6H2O | 7.33 | 7.33 | 7.33 | 7.33 | 7.33 | 7.33 | 7.33 | 7.33 | 7.33 | 7.33 |
| MnCl2·4H2O | 12.64 | 6.32 | 6.32 | 12.64 | 6.32 | 6.32 | 6.32 | 12.64 | 6.32 | 6.32 |
| Zn(BF4)2·6H2O | 0.45 | 7.19 | 7.19 | 7.19 | 3.60 | 3.60 | 7.19 | 1.80 | 3.60 | 3.60 |
| NaN3 | 4.81 | 9.61 | — | — | 9.61 | 19.23 | 19.23 | 38.46 | 38.46 | — |
| Chloramphenicol | 0.19 | 0.77 | 0.39 | 0.19 | 0.10 | 0.05 | 0.10 | 0.05 | 0.05 | 0.77 |
The antifungal activity of the tested compounds is given in Table 3. All tested complexes showed a very good activity towards A. braziliensis and S. cerevisiae and the strongest activity against these strains was displayed by binuclear complex 2. Its activity (MIC 0.09 mM) is comparable to the control compound amphotericin B. All three complexes showed moderate activity against C. albicans.
| A. braziliensis | C. albicans | S. cerevisiae | |
|---|---|---|---|
| HL1Cl | 0.07 | 0.14 | 4.48 |
| 1 | 0.30 | 2.37 | 0.30 |
| 2 | 0.09 | 1.44 | 0.09 |
| 3 | 0.17 | 0.69 | 0.17 |
| Cu(BF4)2·6H2O | 3.67 | 7.33 | 3.67 |
| MnCl2·4H2O | 6.32 | 12.64 | 6.32 |
| Zn(BF4)2·6H2O | 3.60 | 3.60 | 3.60 |
| NaN3 | 0.21 | 0.42 | 1.68 |
| Amphotericin B | 0.04 | 0.02 | 0.01 |
Assessment of toxicity and radical scavenging activity
The obtained toxicity assessment results of the compounds against freshly hatched nauplii Artemia salina as well as their radical scavenging activity are given in Table 4. All synthesized complexes manifested moderate toxicity, with compound 2 exhibiting the highest toxicity. A possible interpretation of this result could be based on its good antibacterial activity. Since the nauplii live in symbiosis with some bacterial strains, it would be reasonable to assume that complex 2 exhibits its toxicity in this way.| LD50 (mM) | DPPH (mM) | |
|---|---|---|
| HL1Cl | 1.143 | 0.489 |
| 1 | 0.567 | 0.094 |
| 2 | 0.315 | 5.934 |
| 3 | 0.869 | 31.680 |
| Cu(BF4)2·6H2O | 0.312 | 29.626 |
| MnCl2·4H2O | 1.406 | — |
| Zn(BF4)2·6H2O | 0.884 | — |
| NaN3 | 0.537 | — |
| K2Cr2O7 | 0.077 ± 0.016 | |
| Ascorbic acid | — | 0.079 ± 0.018 |
Radical scavenging activity was determined by the DPPH test. Complex 1 showed the best activity. This is in line with the structures of the complexes. The central ion of complex 1 is redox-active Cu2+. The radical scavenging activity of complex 1 is comparable to that of ascorbic acid.
Cytotoxic activities of Cu(II) and Mn(II)complexes
The cytotoxic activities of the Cu(II) complex (1), Mn(II) complex (2), Zn(II) complex (3) and their precursor compounds against human cancer cell lines and normal keratinocyte cell line are examined and presented in Table 5. All three complexes showed concentration-dependent cytotoxic effects on the tested cell lines. The Cu(II) complex (1) exerted the highest intensity of cytotoxic activity against melanoma A375, lung carcinoma A549, and prostate adenocarcinoma PC-3 cells with the IC50 values of 18.51 μM, 21.35, and 22.73 μM, respectively. The cytotoxicity of this complex was slightly lower against cervical adenocarcinoma HeLa and breast adenocarcinoma MCF7 cells (the IC50 values are 28.74 μM and 30.45 μM, respectively). The cytotoxic activity of 1 was similar to that of normal keratinocytes HaCaT with an IC50 value of 30.26 μM. The Cu(II) complex (1) demonstrated stronger cytotoxicity against the A375, A549, and PC-3 cancer cell lines compared to its cytotoxicity against normal HaCaT cells. The highest selectivity of the cytotoxic action of 1 was observed against A375 melanoma cells compared to keratinocytes HaCaT (a selectivity coefficient of 1.63). This complex exhibited notably higher cytotoxic activity against all the tested cell lines compared with the cytotoxic activity of its ligand and precursor compounds (Cu(BF4)2·6H2O and NaN3).| HeLa | A375 | A549 | PC-3 | MCF7 | HaCaT | |
|---|---|---|---|---|---|---|
| HL1Cl | ≈200 | 155.65 ± 5.95 | >200 | 197.67 ± 3.29 | 199.23 ± 1.09 | 180.00 ± 4.66 |
| 1 | 28.74 ± 2.93 | 18.51 ± 2.20 | 21.35 ± 0.29 | 22.73 ± 0.52 | 30.45 ± 3.53 | 30.26 ± 4.07 |
| 2 | 40.92 ± 1.30 | 52.07 ± 2.93 | 44.45 ± 2.66 | 50.25 ± 0.36 | 26.66 ± 3.16 | 41.20 ± 1.70 |
| 3 | 289.10 ± 4.11 | 292.63 ± 14.65 | ≈400 | 362.43 ± 2.96 | 375.46 ± 10.87 | 206.92 ± 9.79 |
| NaN3 | >200 | 190.28 ± 13.75 | >200 | >200 | >200 | >200 |
| Cu(BF4)2·6H2O | 123.14 ± 8.04 | 110.92 ± 3.25 | 159.20 ± 4.66 | 96.98 ± 4.75 | 99.05 ± 6.86 | 91.82 ± 1.59 |
| MnCl2·4H2O | 48.68 ± 1.25 | 46.07 ± 5.55 | 153.86 ± 4.33 | 118.94 ± 19.10 | 60.50 ± 3.48 | 79.56 ± 1.21 |
| Zn(BF4)2·6H2O | 171.06 ± 2.85 | 131.85 ± 11.88 | 199.22 ± 1.10 | 192.77 ± 2.63 | 198.78 ± 1.72 | 114.15 ± 8.75 |
| Cisplatin | 4.00 ± 0.47 | 2.46 ± 0.34 | 12.74 ± 1.26 | 12.29 ± 1.60 | 17.82 ± 2.58 | 2.25 ± 0.18 |
The Mn(II) complex (2) exerted the strongest cytotoxic effect on breast adenocarcinoma MCF7 cells with an IC50 value of 26.66 μM. A moderate cytotoxic activity of this complex was observed against the HeLa, A549, A375, and PC-3 cancer cell lines (IC50 values ranging from 40.92 to 52.07 μM). The examined Mn(II) complex (2) exerted moderate cytotoxicity against normal keratinocytes HaCaT with an IC50 value of 41.20 μM. The selectivity of the cytotoxic activity of this complex was shown only against breast cancer MCF7 cells compared with its activity against the normal cell line (selectivity coefficient of 1.55). The cytotoxic activities of its ligand and precursor compounds NaN3 and MnCl2·4H2O were lower than the activity of 2. The only exception of this trend was the slightly lower cytotoxicity of the complex 2 against A375 cells compared with the activity of the salt MnCl2·4H2O against A375 cells.
The Cu(II) complex demonstrated stronger cytotoxic effects against tested cancer cell lines than the Mn(II) complex. However, 2 showed selectivity towards breast adenocarcinoma MCF7 cells in contrast to complex 1. The Zn(II) complex exerted the lowest cytotoxic activity against all the tested cancer cell lines compared to the Cu(II) and Mn(II) complexes including its precursor compounds.
Experimental
Materials and methods
2-Acetylthiazole (99%) was obtained from Acros, Girard's T reagent (99%) from Aldrich, DMSO-d6 from Merck, methanol from Betahem (Belgrade, Serbia), MnCl2·4H2O from Kemika d.d. (Zagreb, Croatia), NaN3 from Riedel-de Haën, and Cu(BF4)2·6H2O and Zn(BF4)2·6H2O from Sigma-Aldrich. IR spectra were recorded using a Nicolet 6700 FT-IR spectrophotometer using the ATR technique in the region 4000–400 cm−1 (vs – very strong, s – strong, m – medium, and w – weak). 1H and 13C NMR spectra were recorded using a Bruker Avance 500 spectrometer (1H at 500 MHz and 13C at 125 MHz) at room temperature using TMS as the internal standard in DMSO-d6. Chemical shifts are expressed in ppm (δ) values and coupling constants (J) in Hz (splitting patterns: s – singlet and d – doublet). Elemental analyses (C, H, and N) were performed by standard micro-methods using an ELEMENTARVario ELIII C.H.N.S.O analyzer.Synthesis
IR (cm−1): 3387 (w), 3128 (w), 3091 (m), 3017 (m), 2955 (s), 1701 (vs), 1612 (w), 1550 (vs), 1486 (s), 1401 (m), 1300 (w), 1201 (s), 1135 (w), 976 (w), 944 (w), 914 (m), 786 (w), 748 (w), 684 (w), 585 (w), 551 (w). (HL1Cl-E). 1H NMR (500 MHz, DMSO-d6), δ (ppm): 2.41 (s, 3H, C5–H), 3.30 (s, 9H, C8–H), 4.60 (s, 2H, C7–H), 7.85 (d, 1H, JC2–H/C3–H = 5.0 Hz, C2–H), 7.93 (d, 1H, JC2–H/C3–H = 5.0 Hz, C3–H), 11.61 (s, 1H, N–H). 13C NMR (125 MHz, DMSO-d6), δ (ppm): 13.90 (C5), 53.65 (C8), 63.01 (C7), 123.33 (C2), 143.94 (C3), 146.98 (C4), 161.23 (C1), 167.04 (C6). (HL1Cl-Z). 1H NMR (500 MHz, DMSO-d6), δ (ppm): 2.53 (s, 3H, C5–H), 3.34 (s, 9H, C8–H), 4.82 (s, 2H, C7–H), 7.85 (d, 1H, JC2–H/C3–H = 5.0 Hz, C2–H), 7.93 (d, 1H, JC2–H/C3–H = 5.0 Hz, C3–H), 11.86 (s, 1H, N–H).13C NMR (125 MHz, DMSO-d6), δ (ppm): 15.05 (C5), 53.89 (C8), 63.76 (C7), 123.65 (C2), 143.97 (C3), 150.80 (C4), 166.78 (C1), 167.34 (C6).
IR (cm−1): 3352 (w), 3317 (w), 3077 (m), 3050 (s), 2970 (m), 2940 (w), 2047 (vs), 1829 (w), 1698 (w), 1604 (w), 1522 (s), 1477 (m), 1444 (m), 1413 (m), 1395 (m), 1325 (m), 1287 (m), 1239. (w), 1159 (w), 1124 (w), 1088 (w), 1053 (m), 1007 (m), 961 (w), 939 (w), 917 (m), 878 (w), 783 (m), 735 (w), 656 (w), 631 (w), 563 (w).
IR (cm−1): 3388 (s), 3086 (m), 3036 (m), 2111 (w), 2042 (vs), 1688 (w), 1643 (w), 1595 (w), 1533 (s), 1481 (s), 1425 (m), 1331 (m), 1273 (m), 1202 (w), 1136 (w), 1115 (w), 1062 (w), 1043 (w), 1004 (w), 928 (w), 907 (w), 879 (w), 768 (w), 723 (w), 640 (w).
IR (cm−1): 3378 (w), 3078 (w), 3054 (w), 3009 (w), 2964 (w), 2057 (vs), 1600 (w), 1540 (s), 1481 (w), 1433 (w), 1407 (w), 1339 (m), 1285 (w), 1203 (w), 1153 (w), 1116 (w), 1079 (w), 1057 (w), 1009 (w), 975 (w), 971 (w), 880 (w), 738 (w), 642 (w). 1H NMR (500 MHz, DMSO-d6), δ (ppm): 2.53 (s, 3H, C5–H), 3.23 (s, 9H, C8–H), 4.13 (s, 2H, C7–H), 7.92 (d, 1H, JC2–H/C3–H = 5.0 Hz, C2–H), 8.04 (d, 1H, JC2–H/C3–H = 5.0 Hz, C3–H). 13C NMR (125 MHz, DMSO-d6), δ (ppm): 15.73 (C5), 53.96 (C8), 67.11 (C7), 124.82 (C2), 143.44 (C3), 147.32 (C4), 165.93 (C1), 171.59 (C6).
X-ray crystallography
Crystal data and the refinement parameters of compounds 1–3 are listed in Table 6. Single crystal X-ray diffraction data were collected at room temperature using an Agilent SuperNova dual-source diffractometer with an Atlas detector equipped with mirror-monochromated Mo–Kα radiation (λ = 0.71073 Å). Data processing was performed with CrysAlis PRO.43 The structures were solved with Olex software44 using SHELXT45 or SHELXS46 and refined by a full-matrix least-squares based on F2 using SHELXL.47 All non-hydrogen atoms were refined anisotropically. All other hydrogen atoms were included in the model at geometrically calculated positions and refined using a riding model. The ORTEP-348 for Windows and MERCURY49 programs were used for graphical presentations. Crystallographic data for complexes 1–3 have been deposited with the Cambridge Crystallographic Data Centre as CCDC 2110386–2110388.†
| 1 | 2 | 3 | |
|---|---|---|---|
| a R = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. c S = {∑[(Fo2 − Fc2)2]/(n/p}1/2 where n is the number of reflections and p is the total number of parameters refined. | |||
| Formula | C11H20BCuF4N7O2S | C22H40Mn2N20O4S2 | C10H16N10OSZn |
| F w (g mol−1) | 464.75 | 822.74 | 389.76 |
| Crystal size (mm) | 0.60 × 0.60 × 0.20 | 0.40 × 0.40 × 0.20 | 0.60 × 0.30 × 0.20 |
| Crystal color | Green | Orange | Yellow |
| Crystal system | Monoclinic | Triclinic | Monoclinic |
| Space group | P21/n |
P |
P21/c |
| a (Å) | 7.0033(3) | 9.6427(5) | 13.0826(10) |
| b (Å) | 10.8941(3) | 10.8396(5) | 10.2506(7) |
| c (Å) | 25.6059(9) | 10.8617(8) | 13.1685(13) |
| α(°) | 90 | 106.971(5) | 90 |
| β (°) | 97.242(4) | 103.497(5) | 111.237(10) |
| γ(°) | 90 | 112.469(5) | 90 |
| V (Å3) | 1938.01(12) | 923.85(10) | 1646.0(3) |
| Z | 4 | 1 | 4 |
| Calcd density (g cm−3) | 1.593 | 1.479 | 1.573 |
| F(000) | 948 | 426 | 800 |
| No. of collected reflns | 17559 |
10077 |
13910 |
| No. of independent reflns | 4425 | 4144 | 3779 |
| R int | 0.0235 | 0.0311 | 0.0315 |
| No. of reflns observed | 3789 | 3319 | 2489 |
| No. parameters | 287 | 232 | 212 |
| R[I > 2σ (I)]a | 0.0388 | 0.0371 | 0.0481 |
| wR2(all data)b | 0.1110 | 0.0982 | 0.1230 |
| Goof, Sc | 1.157 | 1.072 | 1.084 |
| Maximum/minimum residual electron density (e Å−3) | +0.48/−0.34 | +0.28/−0.31 | +0.63/−0.77 |
Computational details
All DFT calculations were done with the ADF program package (version 2017).50–52 Relativistic effects were accounted for by the scalar-relativistic Zeroth-Order Regular Approximation (ZORA).53–55 The all-electron triple-zeta Slater-type orbitals plus one polarization function (TZP) basis set was used for all atoms. All open-shell systems are treated with unrestricted formalism in their high-spin state. The COSMO solvation model,56,57 as implemented in ADF,58 with DMSO as the solvent was used. Geometry optimizations were performed using general gradient functional consisting of Becke's exchange59 and Perdew's correlation60 with Grimme's third-generation dispersion energy correction61 and Becke–Johnson damping,62i.e., BP86-D3. The Cartesian coordinates of all the optimized structures are available in the ESI.† Analytical harmonic frequencies63–65 were calculated at the same level of theory. All normal modes with small frequencies (<50 cm−1) were rescanned numerically66,67 to ascertain that all the optimized structures correspond to the minima on the potential energy surface. Vibrational analysis in the quasi-harmonic approximation as proposed by Truhlar68,69 (frequency cut-off 100 cm−1) was used to evaluate the zero-point effects and the entropic and thermal corrections to the Gibbs free energy at 298 K. Because vibrational analysis is done at a standard state of 1 atm, a conversion to 1 mol dm−3 solution standard state is done. This gives a correction of 1.89 kcal mol−1 to the free energies (at 298 K). This correction is important only for reactions where the number of moles changes. In reactions where DMSO is involved, the free energy correction due to the conversion to the solvent standard state (13.98 mol dm−3) equals 3.46 kcal mol−1 (at 298 K). Entropy correction due to spin-multiplicity (R ln(gs), where gs is the spin-degeneracy of a complex and R is the universal gas constant) was employed. When binuclear [Mn2L12(μ-1,1-N3)2(N3)2] is considered, the low-lying excited states due to the exchange coupling are included in thermochemical analysis. Electronic energies used to calculate the Gibbs free energy were evaluated with the M06-2X70,71meta-hybrid functional, at ZORA-BP86-D3/TZP-COSMO(DMSO) geometries. The LibXC library72 was used for M06-2X calculations. Free energy changes for each considered reaction were corrected for the basis set superposition error by the fragment approach and “ghost atoms” in ADF. The exchange coupling constant J in [Mn2L12(μ-1,1-N3)2(N3)2] was calculated with broken-symmetry DFT formalism73–77 at the ZORA-M06-2X/TZP level of theory at X-ray and ZORA-BP86-D3/TZP-COSMO(DMSO) geometries according to the Yamaguchi approach.78 Broken symmetry solutions are obtained from the high-spin states using the spin–flip method. The calculated exchange coupling constants were used to estimate the relative energies of the low-lying spin-states through diagonalization of the spin Hamiltonian (H = −2JS1S2).
Antimicrobial activity
Antimicrobial activity was tested against a panel of microorganisms including Gram-negative bacteria Escherichia coli (ATCC 25922), Pseudomonas aeruginosa (ATCC 9027), Proteus hauseri (ATCC 13315), Klebsiella pneumoniae (ATCC 10031), and Salmonella enterica subsp. enterica serovar Enteritidis (ATCC 13076), Gram-positive bacteria Staphylococcus aureus (ATCC 6538), Bacillus spizizenii (ATCC 6633), Clostridium sporogenes (ATCC 19404), Micrococcus luteus (ATCC 4698), and Micrococcus luteus (ATCC 10240), yeasts Candida albicans (ATCC 10231) and Saccharomyces cerevisiae (ATCC 9763) and fungal strain Aspergillus brasiliensis (ATCC 16404).Antimicrobial activity was evaluated using the broth microdilution method according to NCCLS [National Committee for Clinical Laboratory Standards, Approval Standard Document M7-A5, Villanova, PA, USA, 2000]. The 96-well plates were prepared by dispensing 100 μl of Mueller–Hinton broth for bacteria and the Sabouraud dextrose broth for yeasts and fungi into each well. A 100 μL aliquot from the stock solution of the tested compounds (concentration 10 mg mL−1 in DMSO) was added into the first row of the plate and double diluted by using a multichannel pipette. The direct colony method was used in the preparation of a suspension of bacteria and yeasts in sterile 0.9% saline, while the process of preparing the suspension of fungal spores included gentle stripping of spores from agar slants with growing aspergilli into sterile 0.9% saline. Suspension turbidity evaluation was conducted by comparison with 0.5 McFarland's standard. 10 μL of diluted bacterial, yeast or spore suspension was added to each well to give a final concentration of 5 × 105 CFU mL−1 for bacteria and 5 × 103 CFU mL−1 for fungi and yeast. Chloramphenicol served as a positive control for bacteria, while amphotericin B served as a positive control for yeasts and fungi.
The inoculated plates were incubated at 37 °C for 24 h for bacteria and at 28 °C for 48 h for the yeasts and fungi. The bacterial growth was visualized by adding 20 μL of 0.5% 2,3,5-triphenyltetrazolium chloride (TTC) aqueous solution.79 Minimum inhibitory concentration (MIC) was defined as the lowest concentration of the compounds that inhibited bacterial growth (red-colored pellet at the bottom of the wells after the addition of TTC).
Brine shrimp assay
About 20 g of commercially purchased lyophilized eggs of Artemia salina was added to 0.5 L of tap water, and air was passed through the suspension by a pump under illumination for 48 h. All tested compounds were dissolved in DMSO and various amounts (0.01–1 mg) were added to 950 μL of artificial seawater with freshly hatched nauplii. After 24 h illumination at room temperature, the number of dead and surviving nauplii were counted and statistically analyzed. LC50 was defined as a concentration of compounds that caused the death of 50% of the nauplii. All samples were done in triplicate.DPPH radical scavenging activity
The 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging activity was determined by the method of Blois.80 Commercially available free radical DPPH was dissolved in methanol at a concentration of 6.58 × 10−5 M, while the tested compounds were dissolved in DMSO. Into a 96-well microplate, 50 μL solutions of the tested compounds at concentrations ranging from 10 to 0.02 mg mL−1 were loaded (50 μL DMSO in the control) and 100 μL of DPPH solution were added. After incubation for 30 min at room temperature in the dark, the absorbance was measured at 517 nm. All the measurements were performed in triplicate and the scavenging activity of the tested derivatives was calculated as| Scavenging activity (%) = 100 × (Acontrol − (Asample − A0))/Acontrol |
The IC50 value was defined as the antioxidant concentration necessary to decrease the amount of the initial DPPH radical by 50% and was calculated from the plotted graph of scavenging activities against the concentrations of the tested compounds. Ascorbic acid was employed as the positive control (concentrations ranging from 50 to 500 μg mL−1).
Determination of cytotoxic activity
The cytotoxic activity of the newly synthesized Cu(II) and Mn(II) complexes and their precursor compounds was examined on five human cancer cell lines: cervical adenocarcinoma HeLa, melanoma A375, lung carcinoma A549, prostate adenocarcinoma PC-3, and breast adenocarcinoma MCF7, as well as against normal human keratinocyte cell line HaCaT. Stock solutions of the compounds were made in DMSO at a concentration of 10 mM, with the exception of the stock solution of Cu(II)complex, which was made at a concentration of 7.5 mM. The human cell lines were grown in a complete nutrient medium RPMI-1640 supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and penicillin–streptomycin solution. HeLa (3000 cells per well), A375 (3000 cells per well), A549 (5000 cells per well), MCF7 (7000 cells per well), PC-3 (5000 cells per well), and HaCaT cells (7000 cells per well) were seeded in 96-well microtiter plates and after 20 h the cells were treated with two complexes and their precursor compounds (five increasing concentrations were tested, ranging from 12.5 μM–200 μM). The nutrient medium was added only to control cells. After 72 h treatment, cell survival was determined by MTT assay according to the method firstly described by Mosmann,81 and modified by Ohno and Abe82 and described in more detail elsewhere.83 Each of the three independent experiments was performed in triplicate. A chemotherapeutic drug, cisplatin, was used as a positive control.Conclusions
Complexes 1–3 have been synthesized and characterized by X-ray crystallographic analysis, elemental analysis and IR spectroscopy. NMR spectroscopy results for the Zn(II) complex showed its stability in solution. The hydrazone ligand is coordinated in a deprotonated form in all three complexes through the thiazole nitrogen, azomethine nitrogen, and carbonyl oxygen atoms. The five-coordination geometry of the Cu(II) ion (mononuclear complex 1) can be described as distorted square-based pyramidal, while in the case of the Zn(II) ion (mononuclear complex 3), the geometry is somewhere in-between square-based pyramidal and trigonal bipyramidal form. The geometry around the Mn(II) ion (binuclear complex 2) is distorted trigonal prism with three donor atoms from the hydrazone ligand, two nitrogen atoms from bridging azide anions, and one nitrogen atom from the terminal azide anion. According to the DFT studies, the Cu(II) complex is the most stable in square-planar [CuL1(N3)]+ geometry in DMSO solution, while in the same solution a mixture of pentacoordinate [MnL1(N3)2] and binuclear Mn(II) complex is predicted.The novel Cu(II) complex showed pronounced cytotoxic effects against tested human cancer cell lines. The complex exerted a higher intensity of cytotoxic activity against A375, A549, and PC-3 cancer cells compared to the intensity of cytotoxicity against normal keratinocytes HaCaT. In addition, the novel Mn(II) complex demonstrated potent cytotoxicity against MCF7 cells. Moderate cytotoxic activity of this complex was observed against other tested cancer cell lines. In general, the Cu(II) complex exhibits more potent cytotoxicity than the Mn(II) complex. However, the activity of complex 2 against the MCF7 breast cancer cell line is very promising as it is only slightly weaker than the activity of cisplatin. However, in contrast to cisplatin, it is selective to the tumor cell line in comparison with the normal cell line (HaCaT).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Ministry of Education, Science and Technological Development of the Republic of Serbia for the financial support (grant numbers: 451-03-9/2021-14/200043, 451-03-9/2021-14/200026 and 451-03-9/2021-14/200168) and the Slovenian Research Agency (P1-0175; funding in 2019–20). The authors thank the EN-FIST Centre of Excellence, Ljubljana, Slovenia, for the use of the SuperNova diffractometer.References
- M. N. Uddin, S. S. Ahmed and S. M. R. Alam, J. Coord. Chem., 2020, 73, 3109–3149 CrossRef CAS.
- M. R. Milenković, B. Čobeljić, K. Anđelković and I. Turel, Eur. J. Inorg. Chem., 2018, 2018, 838–846 CrossRef.
- D. Krajčiová, M. Melník, E. Havránek, A. Forgácsová and P. Mikuš, J. Coord. Chem., 2014, 67, 1493–1519 CrossRef.
- R. C. Balachandran, S. Mukhopadhyay, D. McBride, J. Veevers, F. E. Harrison, M. Aschner, E. N. Haynes and A. B. Bowman, J. Biol. Chem., 2020, 295, 6312–6329 CrossRef CAS PubMed.
- D. S. Auld, BioMetals, 2001, 14, 271–313 CrossRef CAS PubMed.
- M. Stojičkov, S. Sturm, B. Čobeljić, A. Pevec, M. Jevtović, A. Scheitler, D. Radanović, L. Senft, I. Turel, K. Andjelković, M. Miehlich, K. Meyer and I. Ivanović-Burmazović, Eur. J. Inorg. Chem., 2020, 2020, 3347–3358 CrossRef.
- O. Iranzo, Bioorg. Chem., 2011, 39, 73–87 CrossRef CAS PubMed.
- S. Karabasannavar, P. R. Allolli and B. M. Kalshetty, Indian J. Pharm. Educ. Res., 2017, 51, 748–757 CrossRef CAS.
- M. M. E. Shakdofa, F. A. El-Saied, A. J. Rasras and A. N. Al-Hakimi, Appl. Organomet. Chem., 2018, 32, 4376–4393 CrossRef.
- S. M. Emam, S. A. Abouel-Enein and E. M. Abdel-Satar, Appl. Organomet. Chem., 2019, 33, 4847–4872 CrossRef.
- S. M. H. Obaid, J. S. Sultan and A. A. S. Al-Hamdani, Indones. J. Chem., 2020, 20, 1311–1322 CrossRef CAS.
- B. Shaabani, A. A. Khandar, N. Ramazani, M. Fleck, H. Mobaiyen and L. Cunha-Silva, J. Coord. Chem., 2017, 70, 696–708 CrossRef CAS.
- P. Jain, D. Kumar, S. Chandra and N. Misra, Appl. Organomet. Chem., 2020, 34, e5371 CAS.
- L.-W. Xue, X. Fu, G.-Q. Zhao and Q.-B. Li, Acta Chim. Slov., 2021, 68, 17–24 CrossRef CAS.
- L.-H. Wang, X.-Y. Qiu and S.-J. Liu, J. Coord. Chem., 2019, 72, 962–971 CrossRef CAS.
- N. Biswas, S. Saha, S. Khanra, A. Sarkar, D. Mandal, S. Bhattacharjee, A. Chaudhuri, S. Chakraborty and C. R. Choudhury, J. Biomol. Struct. Dyn., 2019, 37, 2801–2822 CrossRef CAS.
- A. Mondal, C. Das, M. Corbella, A. Bauzá, A. Frontera, M. Saha, S. Mondal, K. das Saha and S. K. Chattopadhyay, New J. Chem., 2020, 44, 7319–7328 RSC.
- E. Ramachandran, V. Gandin, R. Bertani, P. Sgarbossa, K. Natarajan, N. S. P. Bhuvanesh, A. Venzo, A. Zoleo, A. Glisenti, A. Dolmella, A. Albinati and C. Marzano, J. Inorg. Biochem., 2018, 182, 18–28 CrossRef CAS PubMed.
- Y.-F. Chen, L. Wei, J.-L. Bai, H. Zhou, Q.-M. Huang, J.-B. Li and Z.-Q. Pan, J. Coord. Chem., 2011, 64, 1153–1164 CrossRef CAS.
- S. Y. Ebrahimipour, M. Mohamadi, J. Castro, N. Mollania, H. A. Rudbari and A. Saccá, J. Coord. Chem., 2015, 68, 632–649 CrossRef CAS.
- A. Th. Chaviara, P. J. Cox, K. H. Repana, A. A. Pantazaki, K. T. Papazisis, A. H. Kortsaris, D. A. Kyriakidis, G. St Nikolov and C. A. Bolos, J. Inorg. Biochem., 2005, 99, 467–476 CrossRef CAS.
- J.-A. Zhang, Y. Li, Y.-Z. Fan, X.-Z. Zou, Y.-J. Liu, L.-J. Zhang and S.-R. Zheng, Inorg. Chem. Commun., 2014, 49, 136–139 CrossRef CAS.
- H. A. EL-Ghamry, K. Sakai, S. Masaoka, K. Y. El-Baradie and R. M. Issa, Chin. J. Chem., 2012, 30, 881–890 CrossRef CAS.
- Y. M. Ahmed, W. H. Mahmoud, M. M. Omar and G. G. Mohamed, J. Inorg. Organomet. Polym. Mater., 2021, 31, 2339–2359 CrossRef CAS.
- B. Čobeljić, I. Turel, A. Pevec, Z. Jagličić, D. Radanović, K. Anđelković and M. R. Milenković, Polyhedron, 2018, 155, 425–432 CrossRef.
- M. Č. Romanović, B. Čobeljić, A. Pevec, I. Turel, S. Grubišić, D. Radanović, K. Anđelković, M. Milenković and M. R. Milenković, J. Coord. Chem., 2017, 70, 3702–3714 CrossRef.
- K. Anđelković, M. R. Milenković, A. Pevec, I. Turel, I. Z. Matić, M. Vujčić, D. Sladić, D. Radanović, G. Brađan, S. Belošević and B. Čobeljić, J. Inorg. Biochem., 2017, 174, 137–149 CrossRef.
- G. Brađan, A. Pevec, I. Turel, I. N. Shcherbakov, M. Milenković, M. Milenković, D. Radanović, B. Čobeljić and K. Anđelković, J. Coord. Chem., 2016, 69, 2754–2765 CrossRef.
- G. Brađan, B. Čobeljić, A. Pevec, I. Turel, M. Milenković, D. Radanović, M. Šumar-Ristović, K. Adaila, M. Milenković and K. Anđelković, J. Coord. Chem., 2016, 69, 801–811 CrossRef.
- T. T. Adejumo, N. V. Tzouras, L. P. Zorba, D. Radanović, A. Pevec, S. Grubišić, D. Mitić, K. K. Anđelković, G. C. Vougioukalakis, B. Čobeljić and I. Turel, Molecules, 2020, 25, 4043–4060 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley-Interscience, New York, 4th edn, 1986 Search PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- T. Keškić, B. Čobeljić, M. Gruden, K. Anđelković, A. Pevec, I. Turel, D. Radanović and M. Zlatar, Cryst. Growth Des., 2019, 19, 4810–4821 CrossRef.
- M. R. Milenković, A. T. Papastavrou, D. Radanović, A. Pevec, Z. Jagličić, M. Zlatar, M. Gruden, G. C. Vougioukalakis, I. Turel, K. Anđelković and B. Čobeljić, Polyhedron, 2019, 165, 22–30 CrossRef.
- T. Keškić, B. Čobeljić, M. Gruden, K. Anđelković, A. Pevec, I. Turel, D. Radanović and M. Zlatar, Cryst. Growth Des., 2019, 19, 4810–4821 CrossRef.
- K. R. Dymock and G. J. Palenik, Inorg. Chem., 1975, 14, 1220–1222 CrossRef CAS.
- T. Keškić, Z. Jagličić, A. Pevec, B. Čobeljić, D. Radanović, M. Gruden, I. Turel, K. Anđelković, I. Brčeski and M. Zlatar, Polyhedron, 2020, 191, 114802–114814 CrossRef.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- B. Čobeljić, A. Pevec, S. Stepanović, M. R. Milenković, I. Turel, M. Gruden, D. Radanović and K. Anđelković, Struct. Chem., 2018, 29, 1797–1806 CrossRef.
- K. Anđelković, A. Pevec, S. Grubišić, I. Turel, B. Čobeljić, M. R. Milenković, T. Keškić and D. Radanović, J. Mol. Struct., 2018, 1162, 63–70 CrossRef.
- M. Č. Romanović, B. Čobeljić, A. Pevec, I. Turel, K. Anđelković, M. Milenković, D. Radanović, S. Belošević and M. R. Milenković, J. Coord. Chem., 2017, 70, 2425–2435 CrossRef.
- N. Stevanović, P. P. Mazzeo, A. Bacchi, I. Z. Matić, M. Đorđić Crnogorac, T. Stanojković, M. Vujčić, I. Novaković, D. Radanović, M. Šumar-Ristović, D. Sladić, B. Čobeljić and K. Anđelković, JBIC, J. Biol. Inorg. Chem. DOI:10.1007/s00775-021-01893-5.
- Agilent, 2014.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- E. J. Baerends, J. Autschbach, D. Bashford, A. Bérces, F. M. Bickelhaupt, C. Bo, P. M. Boerrigter, L. Cavallo, D. P. Chong, L. Deng, R. M. Dickson, D. E. Ellis, M. van Faassen, L. Fan, T. H. Fischer, C. F. Guerra, A. Ghysels, A. Giammona, S. J. A. van Gisbergen, A. W. Götz, J. A. Groeneveld, O. V. Gritsenko, M. Groening, F. E. Harris, P. van den Hoek, C. R. Jacob, H. Jacobsen, L. Jensen, G. van Kessel, F. Kootstra, M. V. Krykunov, E. van Lenthe, D. A. McCormack, A. Michalak, M. Mitoraj, J. Neugebauer, V. P. Nicu, L. Noodleman, V. P. Osinga, S. Patchkovskii, P. H. T. Philipsen, D. Post, C. C. Pye, W. Ravenek, J. I. Rodriguez, P. Ros, P. R. T. Schipper, G. Schreckenbach, M. Seth, J. G. Snijders, M. Sola, M. Swart, D. Swerhone, G. te Velde, P. Vernooijs, L. Versluis, L. Visscher, O. Visser, F. Wang, T. A. Wesolowski, E. M. van Wezenbeek, G. Wiesenekker, S. K. Wolff, T. K. Woo, A. L. Yakovlev and T. Ziegler.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. F. Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- C. van Wüllen and C. van Wüllen, J. Chem. Phys., 1998, 109, 392–399 CrossRef.
- A. Klamt and G. Schoermann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396–408 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- A. Bérces, R. M. Dickson, L. Fan, H. Jacobsen, D. Swerhone, T. Ziegler, A. Brces, R. M. Dickson, L. Fan, H. Jacobsen, D. Swerhone and T. Ziegler, Comput. Phys. Commun., 1997, 100, 247–262 CrossRef.
- H. Jacobsen, A. Bérces, D. P. Swerhone, T. Ziegler, A. Brces, D. P. Swerhone and T. Ziegler, Comput. Phys. Commun., 1997, 100, 263–276 CrossRef CAS.
- S. K. Wolff, Int. J. Quantum Chem., 2005, 104, 645–659 CrossRef CAS.
- L. Fan and T. Ziegler, J. Chem. Phys., 1992, 96, 9005–9012 CrossRef CAS.
- L. Fan and T. Ziegler, J. Am. Chem. Soc., 1992, 114, 10890–10897 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Phys. Chem. Chem. Phys., 2008, 10, 2813–2818 RSC.
- R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2011, 115, 14556–14562 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. A. L. Marques, M. J. T. Oliveira and T. Burnus, Comput. Phys. Commun., 2012, 183, 2272–2281 CrossRef CAS.
- G. Jonkers, C. A. de Lange, L. Noodleman and E. J. Baerends, Mol. Phys., 1982, 46, 609–620 CrossRef CAS.
- L. Noodleman, J. Chem. Phys., 1981, 74, 5737–5743 CrossRef CAS.
- L. Noodleman and E. R. Davidson, Chem. Phys., 1986, 109, 131–143 CrossRef.
- L. Noodleman, J. G. Norman, J. H. Osborne, A. Aizman and D. A. Case, J. Am. Chem. Soc., 1985, 107, 3418–3426 CrossRef CAS.
- F. Neese, Coord. Chem. Rev., 2009, 253, 526–563 CrossRef CAS.
- T. Soda, Y. Kitagawa, T. Onishi, Y. Takano, Y. Shigeta, H. Nagao, Y. Yoshioka and K. Yamaguchi, Chem. Phys. Lett., 2000, 319, 223–230 CrossRef CAS.
- A. Sartoratto, A. L. M. Machado, C. Delarmelina, G. M. Figueira, M. C. T. Duarte and V. L. G. Rehder, Braz. J. Microbiol., 2004, 35, 275–280 CrossRef CAS.
- M. S. Blois, Nature, 1958, 181, 1199–1200 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- M. Ohno and T. Abe, J. Immunol. Methods, 1991, 145, 199–203 CrossRef CAS PubMed.
- I. Z. Matić, I. Aljančić, Ž. Žižak, V. Vajs, M. Jadranin, S. Milosavljević and Z. D. Juranić, BMC Complementary Altern. Med., 2013, 13, 36–48 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2110386–2110388. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03169d |
| This journal is © The Royal Society of Chemistry 2022 |