
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid computational–experimental data-driven design of self-assembling π-conjugated peptides†
Kirill
Shmilovich
a,
Sayak Subhra
Panda
 b,
Anna
Stouffer
b,
John D.
Tovar
b and
Andrew L.
Ferguson
*a
b,
Anna
Stouffer
b,
John D.
Tovar
b and
Andrew L.
Ferguson
*a
aPritzker School of Molecular Engineering, University of Chicago, Chicago, Illinois 60637, USA. E-mail: andrewferguson@uchicago.edu
bDepartment of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, USA
First published on 7th June 2022
Abstract
Biocompatible molecules with electronic functionality provide a promising substrate for biocompatible electronic devices and electronic interfacing with biological systems. Synthetic oligopeptides composed of an aromatic π-core flanked by oligopeptide wings are a class of molecules that can self-assemble in aqueous environments into supramolecular nanoaggregates with emergent optical and electronic activity. We present an integrated computational–experimental pipeline employing all-atom molecular dynamics simulations and experimental UV-visible spectroscopy within an active learning workflow using deep representational learning and multi-objective and multi-fidelity Bayesian optimization to design π-conjugated peptides programmed to self-assemble into elongated pseudo-1D nanoaggregates with a high degree of H-type co-facial stacking of the π-cores. We consider as our design space the 694![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 982 unique π-conjugated peptides comprising a quaterthiophene π-core flanked by symmetric oligopeptide wings up to five amino acids in length. After sampling only 1181 molecules (∼0.17% of the design space) by computation and 28 (∼0.004%) by experiment, we identify and experimentally validate a diversity of previously unknown high-performing molecules and extract interpretable design rules linking peptide sequence to emergent supramolecular structure and properties.
982 unique π-conjugated peptides comprising a quaterthiophene π-core flanked by symmetric oligopeptide wings up to five amino acids in length. After sampling only 1181 molecules (∼0.17% of the design space) by computation and 28 (∼0.004%) by experiment, we identify and experimentally validate a diversity of previously unknown high-performing molecules and extract interpretable design rules linking peptide sequence to emergent supramolecular structure and properties.
1 Introduction
Self-assembling π-conjugated peptides containing a π-core flanked by peptide wings represent a highly tailorable molecular building block for the bottom-up self-assembly of biocompatible supramolecular networks capable of long-range charge transport.1–12 Specific peptide sequences can promote secondary structures within the multi-molecular assemblies akin to beta sheets and guide the quadrupolar association of the π-cores into π-stacked nanostructures with long-range electronic delocalization. Wielding control over the molecular self-assembly of these nanomaterials to tailor the emergent structural, optical, and electronic properties can enable their functional applications as biocompatible, peptide-based field-effect transistors, photoconductors, or solar cells.2,4,7,13 The availability of 20 distinct natural amino acids and various π-cores make these systems extremely tunable and versatile in their optical and electronic properties.14–16 For example, we have previously tuned the steric volume of amino acids directly adjacent to the π-core to engineer tighter or looser packing of the assemblies15,17 and controlled the supramolecular chirality of the nanoaggregates by modulating the length of an alkyl spacer between the peptide wings and the π-core.18 However, most of these materials have been developed by serendipity, intuition, or minor iterative modifications of existing molecules. Systematic data-driven screening approaches present the potential for much deeper and more efficient exploration of sequence space and the discovery of molecules with superior structural and functional properties.The primary goal of the present work is to discover members of the Xn-quaterthiophene-Xn (Xn-4T-Xn) family of π-conjugated peptides capable of self-assembling into pseudo-1D nanoaggregates with in-register stacking of the quaterthiophene π-cores guided by the Xn peptide wings containing n = 1–5 amino acids. Overlap of the π-cores is a structural prerequisite to supramolecular π electron delocalization and the emergence of charge transport functionality. We chose to explore the quaterthiophene π-core due to its demonstrated applications in organic electronic field-effect transistors and photovoltaics19,20 and also our previous measurements of high charge mobilities in oligopeptide–quaterthiophene conjugates.14 The peptide wing containing n = 1–5 amino acids is denoted as Xn, where each X is one of the 20 natural amino acids. We limit the wing length to a maximum of five residues in order to simplify synthetic manipulation. We place two additional design constraints on the peptide wings. First, we require the oligopeptides to be head-to-tail invariant such that they are chemically symmetric about the quaterthiophene core: one peptide wing is symmetric to the other and each molecule possesses two C-termini. This symmetry imposes parity between the left and right sides of the molecular building blocks to promote the formation of linear supramolecular aggregates. Second, we require that the Xn sequence contain at least one acidic residue (i.e., Asp, Glu) and no basic residues (i.e., Arg, His, Lys). The acidic side-chains together with the two carboxyl C-termini allows us to wield pH control of the oligopeptide protonation state such that they possess a formal negative charge of at least (−4)e at high pH and are formally charge neutral at low pH. It has been previously shown through molecular modeling calculations and fluorescence correlation spectroscopy that intermolecular coulombic repulsion and enhanced molecular solvation at high pH disfavors assembly and maintains the system as a mixture of monomers and small oligomers.21 Acidification protonates the ionizable groups to eliminate the coulombic repulsion and serves as a trigger for large-scale supramolecular assembly. Under these two constraints, the Xn-4T-Xn family comprises 694982 unique molecules possessing oligopeptide wings containing between n = 1–5 amino acids. The design challenge is to discover the members of this design space that self-assemble into the most highly ordered linear nanoaggregates.
The large volume of sequence space means that no more than a tiny fraction of molecular candidates can be experimentally explored due to the time and labor costs associated with oligopeptide synthesis and assays. Edisonian trial-and-improvement experimental search is therefore highly inefficient and limited. Chemical intuition can help focus the search, but prior knowledge is restricted to a small number of previously studied candidates and also introduces human bias that can impede the discovery of high-performing, non-intuitive solutions. Computational modeling presents a means to conduct high throughput in silico screening of molecular space. For example, Frederix et al. identified design rules for the assembly of tripeptide sequences by exhaustively simulating all possible amino acid combinations using coarse grained molecular dynamics simulation.22 For more complicated molecules and larger molecular search spaces, exhaustive enumeration becomes intractable and it is profitable to combine computational screening with data-driven modeling and active learning. The essence of this approach is to train on-the-fly sequence–property relationships over all computational screening data collected to date and use these models to guide subsequent rounds of the computational screen within a virtuous feedback loop.23–27 For example, Li et al. used machine learning algorithms such as random forests, gradient boosting, and logistic regression to predict the assembly and formation of hydrogels from possible peptidic precursors.28 Nagasawa et al. employed artificial neural networks and random forests for the discovery of conjugated polymers for organic photovoltaic applications.29 In the context of π-conjugated peptides, we previously combined coarse-grained molecular simulation with deep representational learning and Bayesian optimization to identify molecules predicted to exhibit superior assembly into pseudo-1D linear aggregates30 and we recently synthesized and tested perylene diimide based peptide-π conjugated materials based on quantitative structure property relation models trained over molecular simulation data.17,31
A deficiency of data-driven virtual screening is the weak coupling between computation and experiment. High-throughput virtual screening using computation is used as an initial coarse filtration of the design space that identifies a manageably small number of candidates for synthesis and testing in a subsequent low-throughput experimental screen.32,33 The serial nature of this process means that there is no provision to incorporate experimental feedback into the data-driven search of the design space. This is a lost opportunity since the experimental data can serve as a source of high-quality information to better guide the search and correct for approximations and uncertainties inherent in the computational models. A hybrid data-driven search comprising parallel computational and experimental screens has the potential to offer the best of both worlds – high-throughput approximate computation to achieve broad coverage of the design space and low-throughput experimentation directed towards the most promising candidates. The enabling component of such a procedure is a data-driven model capable of constructing on-the-fly sequence–property relationships from experimental and computational screens that operate asynchronously and in parallel and measure/predict different properties of the molecular system. The trained model is then used within an active learning paradigm to select the most promising molecules for subsequent rounds of computational and experimental screening.
In this work, we develop and deploy a hybrid computational/experimental active learning approach for the data-driven design of Xn-4T-Xn π-conjugated oligopeptides capable of self-assembling into pseudo-1D linear aggregates. We perform high-throughput computational screening using all-atom molecular dynamics simulations that predict the structural morphology of the self-assembled oligopeptide nanoaggregates. We conduct low-throughput experimental oligopeptide synthesis and characterize their assembly using UV-visible spectroscopy. We integrate the computational and experimental screening results to construct on-the-fly sequence–property models that performs asynchronous on-demand selection of the next batch of samples for computational or experimental screening. After sampling only 1181 (∼0.17%) of the 694982 molecules in the design space by computation and only 28 (∼0.004%) by experiment, we discover and experimentally validate a diversity of previously unknown high-performing oligopeptides capable of spontaneously assembling supramolecular aggregates with a high degree of H-type character and extract interpretable design rules linking peptide sequence to emergent supramolecular structure and properties.
2 Methods
In this section, we first present a high-level overview of the hybrid computational/experimental active learning workflow schematically illustrated in Fig. 1. We then present the methodological details of each component. For the interested reader, a more comprehensive explication of the theoretical underpinnings of these techniques, discussion of their numerical implementation, and the codes used to conduct this work are provided in the ESI: Supporting methods.†34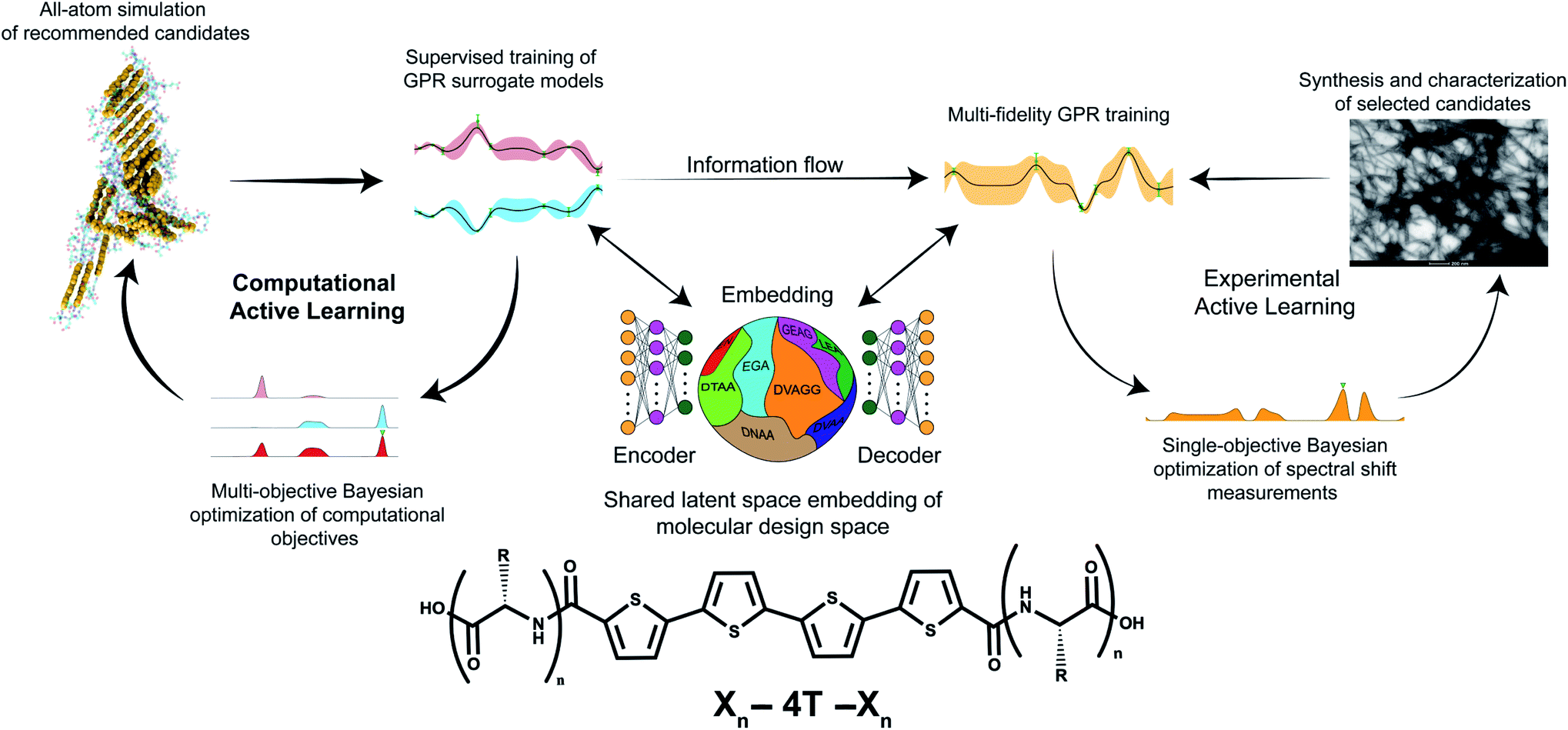 | ||
| Fig. 1 Schematic of the hybrid computational/experimental active learning workflow for the discovery of self-assembling Xn-4T-Xn π-conjugated peptides. We perform separate computational and experimental active learning loops within a shared low-dimensional latent space embedding of the molecular design space learned using regularized autoencoders (RAE). Each active learning loop then consists of three parts. (i) Evaluating the quality of a given molecule k in the Xn-4T-Xn design space by either performing high-throughput all-atom molecular dynamics simulations to measure the average number of contacts per molecule κ(k) and radius of gyration Rg(k) of the self-assembled nanoaggregate or low-throughput experimental synthesis and measurement of the blue-shift λ(k) in the mode of the UV-vis spectrum. (ii) Fitting surrogate models using Gaussian process regression (GPR) to predict the performance of untested candidates given the accumulated simulation and experimental data collected to date. Two separate GPRs are maintained for the two computational objectives GPRκ and GPRRg. We build a multi-fidelity GPR (mfGPR) as our experimental surrogate model GPRλ that also incorporates data from the computational GPRs to improve prediction accuracy beyond what would be possible from the limited experimental data alone. (iii) Employing Bayesian optimization (BO) to interrogate the GPR model and select the next most promising molecular candidates for computation as those lying on the κ − Rg Pareto frontier and for experimentation as those with large values of λ. The molecular renderings in this figure, and throughout the paper, are generated using the Visual Molecular Dynamics (VMD) software.35 | ||
2.1 Overview
We operate a high-throughput computational screening loop to perform all-atom molecular dynamics (MD) simulations to predict the structural morphology of the self-assembled nanoaggregates produced by particular Xn-4T-Xn sequences. The results of the computational screen are used to fit two surrogate sequence–property models relating oligopeptide sequence to the radius of gyration Rg and number of intermolecular contacts κ within the structure. The computational loop seeks to simultaneously maximize κ and Rg to produce pseudo-1D nanoaggregates with in-register π-stacking. We do not a priori know the appropriate relative weights of κ and Rg and so adopt an a posteriori optimization strategy in which we map out the family of Pareto optimal solutions populating the κ − Rg Pareto frontier.36 We construct the sequence–property models over a low-dimensional embedding of the molecular design space extracted using regularized autoencoders37 (RAE) and fit the models using Gaussian process regression38 (GPR). The GPR predictions are passed to multi-dimensional Bayesian optimization39,40 (BO) routines to select the next most promising molecules to simulate for the computational screen.We simultaneously operate a low-throughput experimental screening loop. In this loop, we synthesize Xn-4T-Xn oligopeptides and characterize their assembly by the blue-shift λ in the mode of the UV-visible spectrogram between the unassembled (high-pH) and assembled (low-pH) states as a quantitative measure of the degree of H-type (i.e., co-facial) aggregation. Again we construct a surrogate sequence–property model relating oligopeptide sequence to the spectral shift where we use the same low-dimensional embedding furnished by the RAE. Importantly, the regression model we use to fit this relationship is a multi-fidelity GPR (mfGPR)41 that we train over both the experimental measurements and the computational predictions. Despite measuring different observables of the molecular system – κ and Rgvs. λ – the mfGPR can make use of the voluminous computational predictions to supplement the scarce experimental measurements to furnish a higher accuracy sequence–property model for the spectral shifts than would be possible using the experimental data alone. The computational and experimental screening loops operate asynchronously and in parallel and the data-driven GPR models are continually updated with each new batch of screening data.
The goal of the hybrid computational/experimental screening process is to discover and experimentally validate Xn-4T-Xn molecules with unprecedentedly large values of λ indicative of exceptional in-register π-stacking and H-type character that is a prerequisite for supramolecular electronic delocalization and emergent optical and electronic functionality.
2.2 All-atom molecular dynamics simulations
All-atom molecular dynamics simulations of Xn-4T-Xn molecules were conducted using the GROMACS 2019.2 simulation suite.42 Simulations were initialized in the unassembled state by randomly placing 24 Xn-4T-Xn molecules within a 10 × 10 × 10 nm3 simulation box with three-dimensional periodic boundary conditions and then solvating the system with TIP3P water.43 Peptides were modeled in the electrically neutral state corresponding to low-pH conditions and treated with the AMBER99SB-ILDN forcefield.44 The system was relaxed to T = 300 K and P = 1 bar by steepest descent energy minimization, NVT equilibration45 and NPT46 equilibration. We subsequently conducted 200 ns NPT production runs to observe the spontaneous assembly of supramolecular nanoaggregates. Production runs were of sufficient duration that structural metrics of nanoaggregate formation stabilized over the course of the run (Fig. S1 in the ESI†). Simulation snapshots were extracted and saved every 1 ps for analysis.The fitness of a particular Xn-4T-Xn molecule was defined within our molecular simulations based on its ability to form pseudo-1D linear nanoaggregates with in-register stacking of the π-cores. We quantified this structurally via the average number of contacts per molecule κ and the radius of gyration Rg (ref. 47) of the self-assembled nanoaggregates averaged over the terminal 50 ns of our production runs. An intermolecular contact is defined according to our previously reported “optical distance” dopticali,j that measures the minimum intermolecular distance between any pair of thiophene rings within the π-cores of molecules i and j.30,48–50 We have previously shown that adopting a threshold of dopticali,j < 0.7 nm by which to define an intermolecular contact assures close proximity and in-register π-stacking between at least one pair of thiophene rings.30,49–51 Our computational active learning loop ultimately aims to explore the Pareto frontier of molecules and discover well assembling π-conjugated peptides that possess good thiophene π–π stacking (i.e., large κ) within high-aspect ratio linear nanoaggregates (i.e., large Rg). In general, we found maximization of either objective function alone was insufficient to promote in-register stacked pseudo-1D nanoaggregates: high κ in the absence of high Rg corresponds to globular structures with promiscuous multi-molecular π-stacking, whereas high Rg in the absence of high κ corresponds to weakly associated elongated threads lacking π-core overlaps. As we will show, the hybrid computational/experimental active learning framework learns the appropriate balance between κ and Rg that is most predictive of high-performing experimental candidates with large values of λ.
2.3 Chemical space embedding
Each Xn-4T-Xn molecule in our design space is differentiated by the identity of the peptide wing containing between one and five amino acids. We represent each candidate molecule as a linear amino acid graph where each node is an amino acid and the edges reflect their linear connectivity (Fig. S2 in the ESI†). The molecular design space of 694982 molecules is large, discrete, and high-dimensional. It is possible to perform active learning directly over this space using, for example, kernel models,23,52–57 but superior search efficiencies can be achieved by first projecting the molecular design space into a smooth, continuous, low-dimensional space that is more amenable to the construction of robust regression models and deployment of optimization algorithms.26,30 We learn a bespoke latent space for the Xn-4T-Xn family using a regularized autoencoder37 (RAE) trained on our entire molecular design space (Fig. S3 in the ESI†). An RAE is a deterministic variant of the popular variational autoencoder (VAE) architecture58 that possesses the potential advantage of enabling more expressive flexibility in the data distribution within the low-dimensional embedding by not assuming or enforcing a prior (generally Gaussian) distribution over the latent dimensions. We do not perform a training/validation/testing split since the purpose of our RAE model is to provide a low-dimensional latent space embedding of the complete design space of all 694982 candidate molecules for downstream active learning, not as a predictive tool for out-of-sample inference beyond this design space. As such, we do not use the decoder to generate new candidate molecules and it is therefore not a disadvantage for our applications that the latent distribution is not approximately Gaussian distributed, which is generally desirable for efficient generative sampling. We have previously demonstrated an application of RAEs in the data-driven design of small drug-like cardiolipin-selective molecules.59 This encoder–decoder architecture consists of a message passing neural network60,61 encoder and a decoder performing explicit graph matching to ensure end-to-end invariance of node permutations. The encoder62–64 accepts graphical representations of the Xn peptide sequences the nodes of which are featurized by the 553 single amino acid physiochemical properties contained in the AAindex database and the edges of which are featurized by the 135 pairwise amino acid contact potentials and mutation matrices.65 The bottleneck layer defining the interface between the trained encoder and decoder contains a nonlinear latent embedding of the molecular design space ξ learned by the encoder that preserves relationships between molecular candidates and is sufficiently informative for the decoder to accurately reconstruct the molecular graphs (Fig. S4 in the ESI†). We determine an appropriate dimensionality d = 32 for the latent space using exploratory hyperparameter tuning optimizing the RAE reconstruction loss.66,67 This low-dimensional latent space defines a smooth and continuous representation ξ of the molecular design space over which fit (mf)GPR surrogate models and conduct BO-enabled active learning.
2.4 Computational active learning
The goal of the computational active learning loop is to drive computational discovery of candidate Xn-4T-Xn molecules that simultaneously maximize the average number of contacts per molecule κ and the radius of gyration Rg of the self-assembled aggregates. This amounts to searching for molecules residing upon the κ − Rg Pareto frontier, for which the dual objective functions necessitate a multi-objective optimization strategy. We seeded the active learning screen by conducting MD simulations of 228 initial candidate molecules. To ensure broad initial sampling of the candidate space, we selected 100 molecules as those residing closest to the centroids of a 100-cluster k-means partitioning of the RAE latent space. The remaining 128 molecules were hand-selected to comprise a diversity of peptide wing lengths, residue hydrophobicity, aromaticity, polarity, and presence of heteroatoms. We used the computational predictions of κ and Rg to train two independent Gaussian process regression (GPR) surrogate models38![[small kappa, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e109.gif) = f(ξ) and
= f(ξ) and 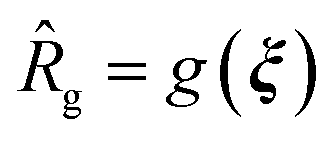 that perform supervised learning of mappings from the latent space coordinates to the two structural observables. The predictions of the two GPRs are passed to a multi-objective BO routine that seeks to simultaneously maximize κ and Rg using the method of random scalarizations.39,40 The trained GPRs for κ and Rg are used to construct two independent BO upper confidence bound (UCB)68 acquisition functions defining the relative desirability of each candidate Xn-4T-Xn molecule in maximizing each of these two objectives. We then collapse these two acquisition functions into a single scalarized acquisition function constructed as a randomly weighted linear sum. The scalarized acquisition function is then used to perform univariate Bayesian optimization. A particular random scalarization corresponds to a particular choice of relative weightings between the two design objectives and defines a vector within the 2D κ − Rg space along which to maximize. Under sufficiently many repeated random scalarizations, the random vectors span the κ − Rg Pareto frontier to discover a family of Pareto optimal solutions. We perform batched selection over different random scalarizations and choices of the UCB hyperparameter balancing the BO exploit–explore tradeoff to propose 25 new candidate molecules per round. MD simulations of these 25 molecules are performed and the three part active learning cycle is iteratively repeated. We assess convergence of the iterative screen by monitoring the set of Pareto optimal points that define the κ − Rg Pareto frontier and terminate sampling once the Pareto frontier ceases to advance with additional rounds of sampling. We conducted 38 rounds of computational screening over which we considered 1181 candidate molecules. A full accounting of the molecules identified in each round of the computational active learning loop is provided in the Data availability statement.34
that perform supervised learning of mappings from the latent space coordinates to the two structural observables. The predictions of the two GPRs are passed to a multi-objective BO routine that seeks to simultaneously maximize κ and Rg using the method of random scalarizations.39,40 The trained GPRs for κ and Rg are used to construct two independent BO upper confidence bound (UCB)68 acquisition functions defining the relative desirability of each candidate Xn-4T-Xn molecule in maximizing each of these two objectives. We then collapse these two acquisition functions into a single scalarized acquisition function constructed as a randomly weighted linear sum. The scalarized acquisition function is then used to perform univariate Bayesian optimization. A particular random scalarization corresponds to a particular choice of relative weightings between the two design objectives and defines a vector within the 2D κ − Rg space along which to maximize. Under sufficiently many repeated random scalarizations, the random vectors span the κ − Rg Pareto frontier to discover a family of Pareto optimal solutions. We perform batched selection over different random scalarizations and choices of the UCB hyperparameter balancing the BO exploit–explore tradeoff to propose 25 new candidate molecules per round. MD simulations of these 25 molecules are performed and the three part active learning cycle is iteratively repeated. We assess convergence of the iterative screen by monitoring the set of Pareto optimal points that define the κ − Rg Pareto frontier and terminate sampling once the Pareto frontier ceases to advance with additional rounds of sampling. We conducted 38 rounds of computational screening over which we considered 1181 candidate molecules. A full accounting of the molecules identified in each round of the computational active learning loop is provided in the Data availability statement.34
2.5 Experimental active learning
The experimental active learning loop aims to maximize the blue-shift λ in the mode of the UV-visible spectrogram between the unassembled (high-pH) and assembled (low-pH) states. The magnitude of this spectral shift λ has been experimentally shown to correlate with co-facial H-type assembly that results in formation of the desired pseudo-1D linear stacks.14,69,70 We seeded the experimental search with a set of 11 molecules hand_selected from the 228 molecules comprising the initial computational round to comprise a diversity of oligopeptide wing lengths and predicted values of κ and Rg. We note that these 11 initial molecules originated from the subset of 128 human-selected molecules from our initial computational round, rather than from the subset of 100 molecules identified via k-means clustering. We trained a GPR model![[small gamma, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e0b2.gif) = h(ξ) to predict the spectral shift λ as a function of latent space coordinates. In this case we have only a single objective function λ but we wish to construct a multi-fidelity surrogate model incorporating both direct experimental measurements of λ and computational predictions of κ and Rg. The rationale is that the computational predictions for κ and Rg should be correlated with and predictive of the experimental measurements of λ. This is expected to be the case since κ and Rg are structural measures of the degree of in-register π-stacking in elongated nanoaggregates that are prerequisites for H-aggregate character manifested in measurements of λ. A multi-fidelity model trained to learn a nonlinear mapping from the low-fidelity computational predictions to high-fidelity experimental measurements can take advantage of the abundant computational data to produce a superior model than that obtained by training over only the sparse experimental data alone. Indeed, by the terminal round of experimental active learning, incorporation of computational screening data within the multi-fidelity paradigm leads to a ∼27% improvement in the predictive accuracy of our surrogate model compared to a single-fidelity model fitted only over the experimental observations (Fig. S5 in the ESI†). This predictive improvement highlights the capabilities of our mfGPR to leverage plentiful low-fidelity data from our computational κ and Rg metrics to improve our predictive performance for our target high-fidelity objective in λ. We observe that the flexibility of the mfGPR framework enables this information transfer even in situations where the low- and high-fidelity observables are measuring different properties, are only weakly correlated or even anti-correlated, and where the degree of correlation may change over the domain.71 The only requirement is that the low-fidelity response surface is informative of the high-fidelity response surface and that this relationship can be extracted from the data within the mfGPR model.
= h(ξ) to predict the spectral shift λ as a function of latent space coordinates. In this case we have only a single objective function λ but we wish to construct a multi-fidelity surrogate model incorporating both direct experimental measurements of λ and computational predictions of κ and Rg. The rationale is that the computational predictions for κ and Rg should be correlated with and predictive of the experimental measurements of λ. This is expected to be the case since κ and Rg are structural measures of the degree of in-register π-stacking in elongated nanoaggregates that are prerequisites for H-aggregate character manifested in measurements of λ. A multi-fidelity model trained to learn a nonlinear mapping from the low-fidelity computational predictions to high-fidelity experimental measurements can take advantage of the abundant computational data to produce a superior model than that obtained by training over only the sparse experimental data alone. Indeed, by the terminal round of experimental active learning, incorporation of computational screening data within the multi-fidelity paradigm leads to a ∼27% improvement in the predictive accuracy of our surrogate model compared to a single-fidelity model fitted only over the experimental observations (Fig. S5 in the ESI†). This predictive improvement highlights the capabilities of our mfGPR to leverage plentiful low-fidelity data from our computational κ and Rg metrics to improve our predictive performance for our target high-fidelity objective in λ. We observe that the flexibility of the mfGPR framework enables this information transfer even in situations where the low- and high-fidelity observables are measuring different properties, are only weakly correlated or even anti-correlated, and where the degree of correlation may change over the domain.71 The only requirement is that the low-fidelity response surface is informative of the high-fidelity response surface and that this relationship can be extracted from the data within the mfGPR model.
We construct multi-fidelity surrogate models fusing the computational (low-fidelity) and experimental (high-fidelity) data using the multi-fidelity Gaussian process regression (mfGPR) formalism.41 The mfGPR model is then passed to a standard BO routine39 employing an expected improvement (EI) acquisition function39,72,73 and the Kringing believer74 batched sampling. We use the BO to propose a batch of molecules for the next round of sampling, which we manually down-select to 8–9 molecules for experimental synthesis and characterization. By incorporating “human-in-the-loop” curation of the selected molecules we hope to balance purely data-driven candidate proposal with chemical intuition and thereby incorporate some degree of prior knowledge and human experience into the search process without, we hope, imposing too much bias on the search. The success of this collaborative human-machine paradigm has been previously demonstrated in the data-driven discovery of molecular organic light emitting diodes.33 The experimental measurements are fed back into the low-throughput experimental active learning loop that is executed asynchronously and in parallel with the high-throughput computational loop. We execute three rounds of experimental active learning over the course of the 38 rounds of computational active learning that are executed at computational rounds 0, 14, and 22. Given the good performance of the candidates studied in the third experimental round together with the relatively modest advances in the Pareto frontier observed over computational rounds 23–38, we elected to terminate our experimental screen after its third round. A full accounting of the molecules identified in each round of the experimental active learning loop is provided in Table 1 and have been made available as detailed in the Data availability statement.34
| Rank | Peptide wing, Xn | Measured spectral shift, λ (nm) | Discovery round | Previously known? |
|---|---|---|---|---|
| 1 | DGG | 55.06 ± 1.00 | 2 | Yes70,86 |
| 2 | DG | 53.42 ± 4.16 | 0 | N |
| 3 | ESA | 50.01 ± 0.53 | 2 | N |
| 4 | EGG | 49.97 ± 1.00 | 0 | Yes14 |
| 5 | ETGG | 45.80 ± 0.62 | 2 | N |
| 6 | DGA | 44.15 ± 1.49 | 2 | N |
| 7 | DDDAA | 42.07 ± 0.46 | 2 | N |
| 8 | DVAA | 41.65 ± 1.15 | 0 | N |
| 9 | DSG | 40.32 ± 1.15 | 1 | N |
| 10 | AEVGA | 40.15 ± 1.12 | 2 | N |
| 11 | DVAG | 35.70 ± 1.49 | 0 | N |
| 12 | DNDN | 29.58 ± 4.76 | 1 | N |
| 13 | DANN | 25.70 ± 0.32 | 2 | N |
| 14 | VEFAG | 21.75 ± 2.07 | 0 | N |
| 15 | VEVEV | 18.43 ± 0.62 | 0 | N |
| 16 | VD | 18.02 ± 2.66 | 0 | N |
| 17 | AAD | 15.98 ± 1.00 | 0 | N |
| 18 | EYIQG | 15.01 ± 7.18 | 1 | N |
| 19 | EV | 14.70 ± 1.20 | 0 | N |
| 20 | DT | 14.70 ± 1.20 | 1 | N |
| 21 | AAED | 13.68 ± 1.46 | 0 | N |
| 22 | SSD | 13.68 ± 1.15 | 1 | N |
| 23 | VEF | 11.99 ± 1.68 | 0 | N |
| 24 | DLAG | 11.49 ± 0.46 | 2 | N |
| 25 | GFGFD | 10.97 ± 1.77 | 1 | N |
| 26 | DGL | 10.25 ± 1.20 | 1 | N |
| 27 | IDSV | 7.70 ± 3.83 | 1 | N |
| 28 | EN | 4.33 ± 1.49 | 1 | N |
2.6 Nonlinear manifold learning of low-dimensional assembly pathways
After completing the hybrid computational/experimental screen we subjected the ensemble of 1181 molecular simulation trajectories of Xn-4T-Xn candidate molecules to nonlinear dimensionality reduction in order to resolve the structural assembly pathways. In doing so, we sought to gain mechanistic understanding of the molecular assembly mechanisms differentiating the top performing molecules identified by our screen. We performed nonlinear dimensionality reduction using diffusion maps manifold learning75,76 to project the configurational coordinate space into a low-dimensional space preserving the leading high-variance collective dynamics of the system.30,48,77–80 Diffusion maps take as an input a pairwise distance matrix measuring the configurational similarity between all 118100 simulation snapshots harvested from the ensemble of 1181 simulation trajectories. We define these pairwise distances using the smooth overlap of atomic positions (SOAP) kernel81–84 between the heavy atoms constituting the 4T π-cores as a distance metric that is naturally invariant to rotations, translations, and permutations of atoms, and which – as a π-core-centric metric – can be applied between oligopeptides with different wing lengths. The influence of the wings is implicitly retained through their impact on the configurations adopted by the π-cores. A density-adaptive variant of diffusion maps85 is then applied to furnish embeddings of the assembly trajectories into a 2D manifold that exposes the assembly pathways and mechanisms followed by the various Xn-4T-Xn molecules. We note that our diffusion maps furnish a low-dimensional embedding of the configurational coordinate space traversed by our MD simulations, and that this embedding is completely independent of the low-dimensional embedding of the molecular design space furnished by the RAE that is employed within our active learning protocol.
2.7 Oligopeptide synthesis
:1 MeOH:water solution with 1% ammonium hydroxide. ESI spectra for each synthesized peptide are provided in the ESI: ESI spectra.†
2.8 UV-visible spectroscopy
UV-vis spectra were obtained using a Varian Cary 50 Bio UV-vis spectrophotometer. Spectroscopic samples were prepared by diluting the peptide solution to the appropriate concentration in Millipore water to achieve an optical density near 0.1, 0.2, and 0.3 in the monomeric/basic solution. The pH was then adjusted by adding 20 μL of 1 M KOH (basic) followed by addition of 40 μL of 1 M HCl (acidic). Approximate concentration of the peptides were 2.25 μM, 4.50 μM, and 6.75 μM for optical density of 0.1, 0.2, and 0.3 respectively.3 Results and discussion
3.1 Hybrid computational/experimental active learning discovers novel high-performing oligopeptides
We report in Fig. 2 the results of our hybrid computational/experiment active learning screen to the molecular design space of 694982 Xn-4T-Xn candidate molecules. We conduct 38 rounds of computational screening to simulate a total of 1181 Xn-4T-Xn molecules interleaved with three rounds of experimental screening in which we synthesize and test a total of 28 molecules. A full accounting of the molecules identified in each round of the computational and experimental active learning loops are provided as detailed in the Data availability statement.34 Round 0 of the computational and experimental screens commence simultaneously, respectively screening 228 and 11 molecules. The two screens then proceed asynchronously and in parallel. The high-throughput computational loop considers 25 candidate molecules per round and iterates more rapidly than the low-throughput experimental screen that considers 8–9 molecules per round. We track the progress of the experimental screen via the measured spectral blue shifts λ upon assembly as a measure of the prevalence of H-type co-facial π-stacking (Fig. 2a). We track the progress of the computational screen via the advancement in the κ − Rg Pareto frontier that we quantify through the mean distance from the origin of all ni molecules cumulatively simulated over the first i rounds (Fig. 2a and b),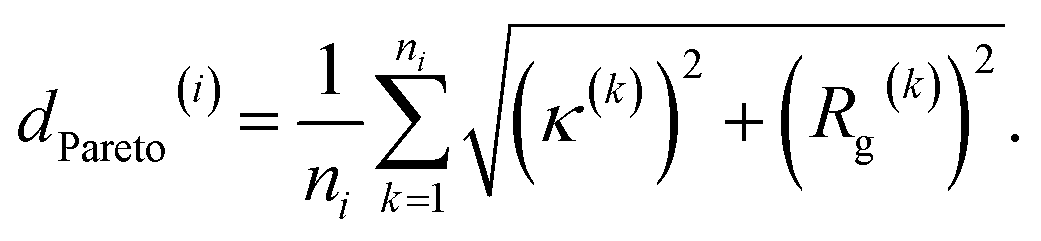 | (1) |
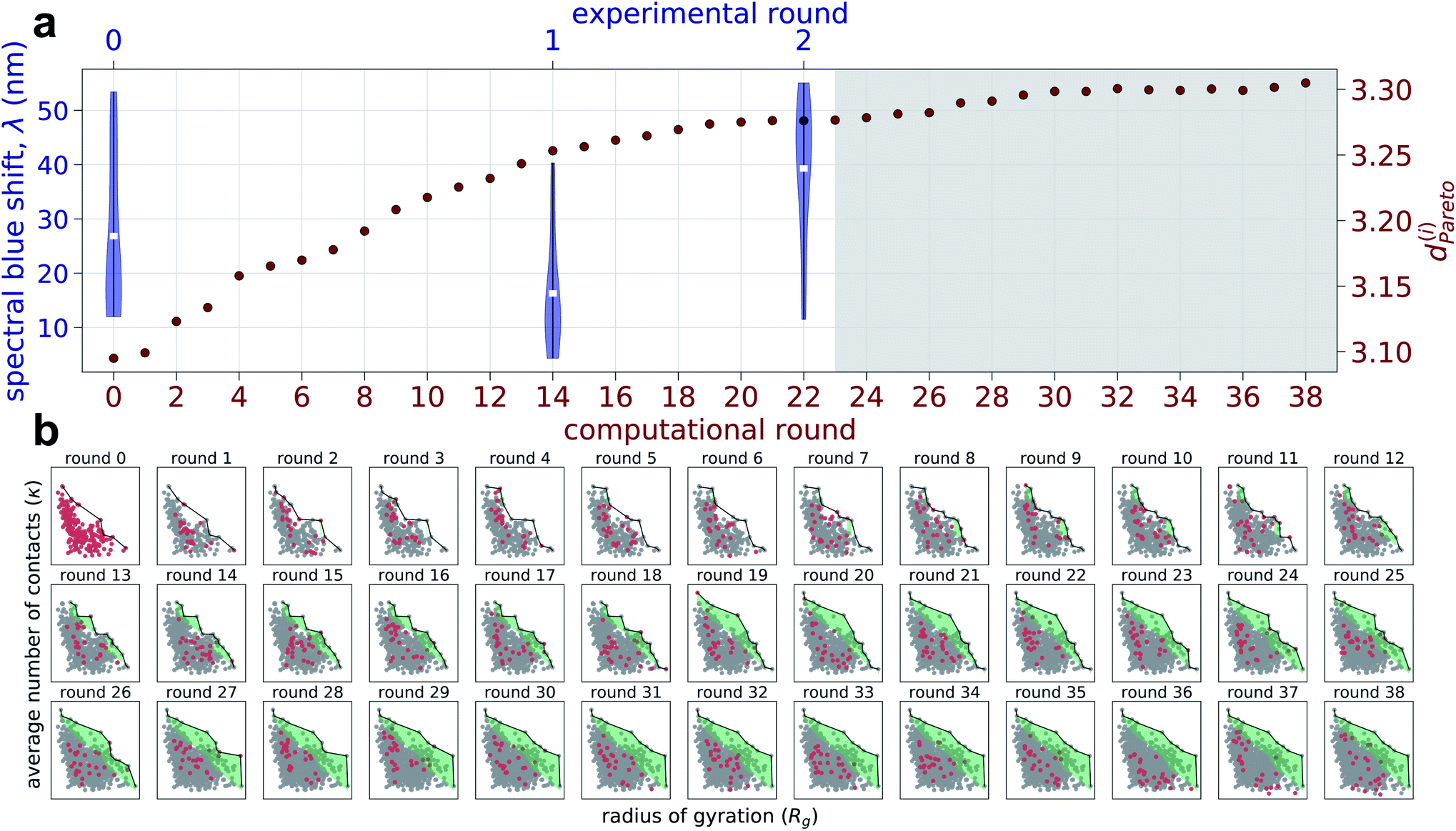 | ||
| Fig. 2 Progress and convergence of the hybrid computational/experimental active learning screen for high-performing Xn-4T-Xn molecules within the design space of 694982 candidates. (a) A total of 38 computational rounds of screening are performed and interleaved with three rounds of experimental screening. The experimental screens are conducted at computational rounds, 0, 14, and 22. Convergence of the computational screen is tracked by dPareto(i), the mean distance from the origin of all molecules cumulatively simulated over the first i rounds within the computational κ − Rg Pareto plot (see panel b), as a measure of the advancement of the optimal frontier (red circles). Convergence of the experimental screen is tracked by the measured spectral blue shift λ quantifying the degree of H-type co-facial π-stacking within the self-assembled nanoaggregates (blue violin plots). Violin plots enclose the range of λ values measured each experimental round and the central white tick denotes the mean λ value for the round. The gray shaded area represents the computational rounds used to verify convergence of the active learning but not used to inform any additional rounds of experimental design. (b) Round-by-round advancement of the computational κ − Rg Pareto frontier over the course of the 38 screening rounds. Within each frame, the points corresponding to Xn-4T-Xn candidates collected within that round are shown in red, those points collected in previous rounds are shown in gray, the Pareto frontier defined by the Pareto optimal points is shown as a black solid line, and the shaded green area indicates the advancement of the Pareto frontier relative to round 0. | ||
We recall that large values of the average number of π-core contacts per molecule κ and radius of gyration of the self-assembled aggregates Rg are anticipated to lead to correlate with the formation of pseudo-1D nanoaggregates with high degrees of H-type character.
Round 1 of the experimental screen is performed upon completing computational round 14, at which time a total of 578 candidate molecules have been computationally assessed. These computational screening data are passed to the experimental surrogate model and Bayesian optimization within the multi-fidelity hybrid computational/experimental active learning framework in order to better inform the design of experimental round 1 than would be possible by analyzing the 11 experimental data points alone. Under our human-in-the-loop selection protocol, we selected nine molecules for round 1 of experimental screening by filtering a 35-molecule list outputted from our BO routine. Down-selection was performed on the basis of high anticipated performance based on our previous experimental and computational work15,30 and maintenance of a diversity of peptide wing compositions and lengths. This human-in-the-loop selection process serves as a simple means to inject prior knowledge into the data-driven search process, which can be particularly valuable in the early stages of the search where the models are trained over small numbers of data points, by directing the search process to regions of molecular design space that are anticipated to be particularly profitable.
Round 2 of the experimental screen is conducted after completing computational round 22, at which point we have simulated 780 candidate molecules. Again, the totality of these computational screening data are used to augment the multi-fidelity experimental surrogate model and used to pick eight candidate molecules for experimental testing down-selected from the 75 top candidates proposed by the BO routine. We inject one addition piece of human intuition into the down-selection process by making a single Tyr to Ala amino acid mutation of one of the predicted sequences – YEVGA to AEVGA – based on prior understanding that aromatic side chains are known to π-stack with the π-cores and therefore liable to disrupt favorable in-register 4T stacking.30 This modification is substantiated by both observing low-ranking candidates possessing bulky aromatic residues in the first two experimental rounds (EYIQG: rank 18/28, VEF: rank 23/28, GFGFD: rank 25/28) along with previous experimental work noting the presence of aromatic residues resulting in reduced UV-vis blue-shifts.15
We continue to conduct an additional 16 rounds of computational screening (rounds 23–38) while experimental round 2 is being completed in anticipation of possibly conducting a fourth experimental round. As illustrated in Fig. 2a, we observe a relatively rapid expansion of the computational exploration of the κ − Rg design space over the course of computational rounds 0–20, which we quantify by dPareto(i) defining the mean distance from the origin of all molecules cumulatively simulated over the first i rounds. This trend, however, plateaus at dPareto ≈ 3.3 by round 29 and exhibits only a 0.7% increase in dPareto relative to round 22. This observation is mirrored by a relatively modest advancement of the Pareto frontier between rounds 23–38 (Fig. 2b). Experimentally, the mean spectral shift λ in experimental round 2 is 46% better than round 0, and the top performing round 2 candidate has a 3% better spectral shift compared to that in round 0. The diminishing returns evinced by the computational dPareto and the successful discovery of a molecule with superior λ impelled us to terminate our search after experimental round 2/computational round 38.
In all, we simulated 1181 molecules comprising ∼0.17% of the 694982 molecules constituting the Xn-4T-Xn design space, corresponding to 236.2 μs of simulation time, and requiring ∼4.97 GPU-years of parallel compute. Experimentally, we synthesized and characterized a total of 28 Xn-4T-Xn molecules over the course of the course of eight months corresponding to exploration of 0.004% of the molecular space.
We present in Fig. 3 an embedding of all 1181 simulated molecules and 28 experimentally tested molecules into the κ − Rg objective function space used to identify high-performing molecules in the computational screening loop. Molecular renderings of the self-assembled nanoaggregates provides qualitative visual conformation that Xn-4T-Xn molecules producing aggregates with large values of both κ and Rg do indeed tend to self-assemble into pseudo-1D structures with good stacking of the 4T π-cores.
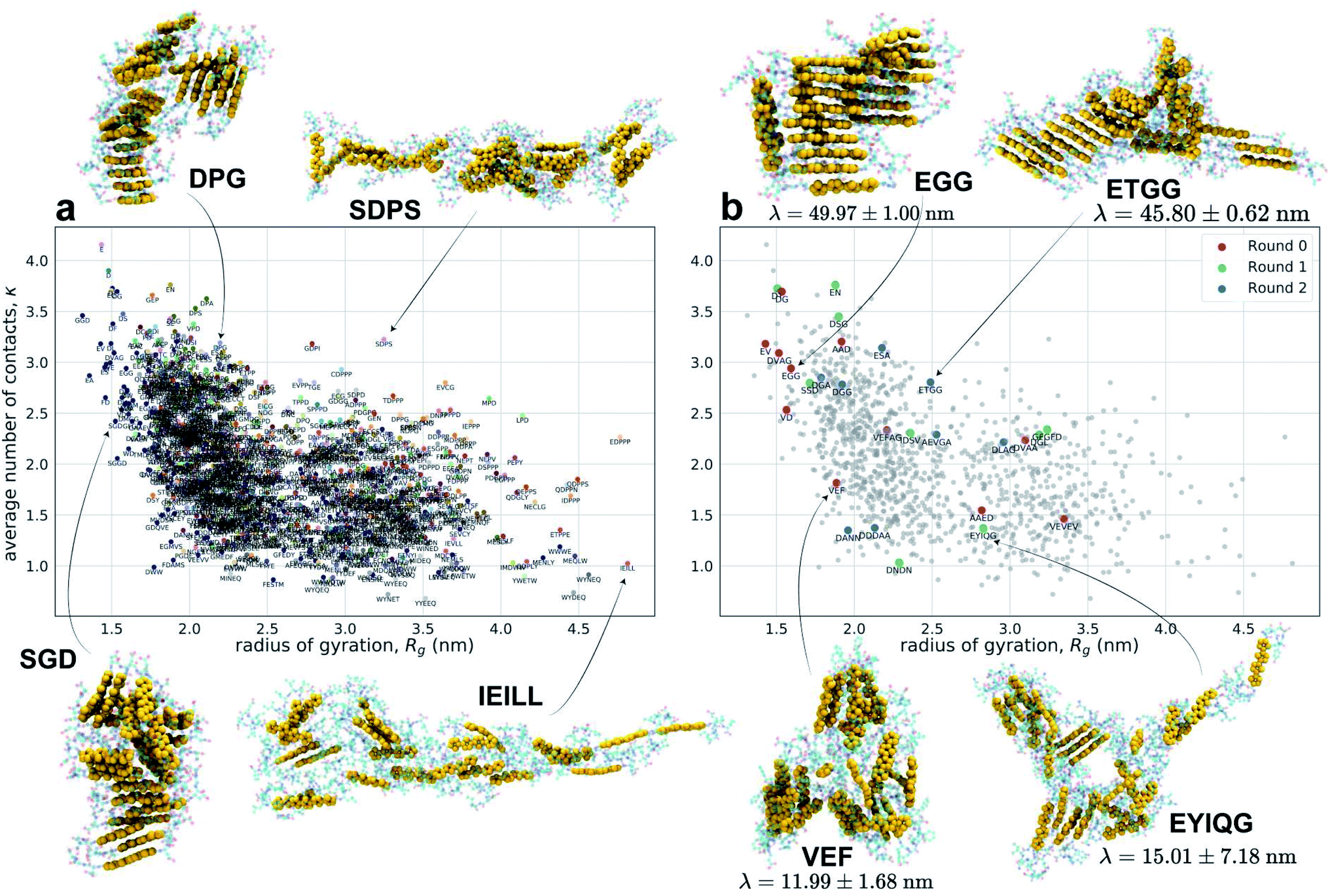 | ||
| Fig. 3 Embedding of the computational and experimental molecules sampled in the active learning screen into the κ − Rg objective function space. (a) Embedding of the 1181 Xn-4T-Xn molecules explored in the computational screen. Points shown in the same color were sampled during the same computational active learning round. The labels associated with each point corresponds to the Xn peptide wing sequence. (b) Highlighting the 28 experimentally tested Xn-4T-Xn molecules (colored points) superposed onto all 1181 computationally simulated points (grey points). The color indicates the round of experimental active learning within which the molecule was tested. Encircling the plot are snapshots from our molecular simulation trajectories showing the terminal self-assembled nanoaggregates. The heavy atoms constituting 4T π-cores are rendered as gold space-filling spheres and the Xn peptide wings as semi-transparent ball-and-stick representations. Molecules producing aggregates with large values of both κ and Rg tend to self-assemble into pseudo-1D structures with good stacking of the 4T π-cores. For the experimentally tested candidates we also report the measured values of the spectral blue shift λ. A full accounting of the computed κ and Rg values and measured λ values for all molecules considered in our screen have been made available as detailed in the Data availability statement.34 | ||
The primary result of our hybrid computational/experimental active learning screen are experimental measurements of 28 Xn-4T-Xn molecules reported in Table 1. The 11 round 0 molecules were selected based on human intuition and used to seed the experimental active learning. The nine round 1 and eight round 2 molecules are the result of our multi-fidelity active learning search. Of these molecules, 26 are completely novel and on par with known high-performing sequences14,70,86 while also possessing greater diversity amino acid sequences previously unknown to correlate with good spectral blue shifts λ. The high values of λ for these molecules are indicative of a high degree of H-type co-facial stacking and the potential for long-range supramolecular electronic delocalization and emergent optoelectronic functionality. Additionally, we were encouraged that our active learning procedure spontaneously discovered DGG-4T-GGD as a previously known high-performing candidate.70,86 A complete list of predicted κ and Rg values from the terminal computational surrogate model and predicted spectral shift measurements λ from the terminal experimental surrogate model for all 694982 molecules within the Xn-4T-Xn design space have been made available as detailed in the Data availability statement.34
3.2 Molecular design rules
Our rank-ordered list of 28 experimentally assayed candidates exposes a number of oligopeptide design precepts, that is relationships between the placement/omission of particular amino acids at specific positions along the oligopeptide wing and the magnitude of the spectral blue shift λ quantifying the degree of H-type co-facial association within the self-assembled nanoaggregates. Despite the relatively small size of the experimental data set, we were able to extract three statistically significant design rules. First, the nine top-ranked molecules within the 28 assayed candidates contain a distal Asp or Glu residue at the C-terminus and a Gly or Ala residue in the position most proximate to the π-core. A statistical analysis using a one-sided Mann–Whitney U test87 reveals a statistically significant (p = 0.0001) increase in the measured blue shifts λ associated with the presence of the (D/E)Xn(A/G) motif. In our prior work on π-conjugated oligopeptides, we typically synthesized the peptide wings with the ionizable residue responsible for actuating pH-triggered assembly located at the C-terminus to locate it as far away as possible from the π-core: our motivation for this design choice was that the hydrophilic and polar nature of these residues which, together with their steric bulk, was anticipated to disrupt good supramolecular assembly of the π-cores.14,15,70,88–91 Interestingly, our active learning screen appears to have also learned this design rule without any explicit human instruction and thus furnished post hoc support for this intuitive choice. Similarly, our recent computational and experimental work30,31,92 is consistent with prior chemical intuition88 that the placement of small non-polar residues adjacent to the π-core should promote good co-facial stacking of the cores. Again, the active learning screen appears to have also learned this design rule within the sequence–property surrogate model and is consistent with a physical rationale that the small steric volume of these amino acids is conducive to in-register π-stacking of the 4T cores. It is more challenging, however, to differentiate between the performance of Gly vs. Ala, with Gly leading to more favorable spectral shifts within a DGX motif – λDGG = (55.05 ± 1.00) nm and λDGA = (44.15 ± 1.49) nm – whereas Ala performs better within a DVAX motif – λDVAG = (35.70 ± 1.49) nm and λDVAA = (41.65 ± 1.15) nm. The absence of a simple modular decomposition of the influence of each amino acid position in the Xn wing reflects the complexity of the self-assembly process and the important role of multi-body interactions, amino acid context, and wing length.Second, consistent with the favorability of core-adjacent Gly and Ala residues, the non-C-terminal amino acids within the Xn peptide sequences of the top-performing molecules tend to also be enriched in small hydrophobic residues such as Ala, Gly or Val (one-sided Mann–Whitney U test, p-value = 0.004). Interestingly, residues containing polar hydroxyl groups such as Ser and Thr are also over-represented within high-performing sequences when Ser or Thr are non-terminal residues and Asp or Glu are C-terminal such as ESA: rank 3/28, ETGG: rank 5/28, and DSG: rank 9/28 (one-sided Mann–Whitney U test, p-value = 0.03). Other polar residues like Asn also perform relatively well in the π-core proximate position when Asp occupies the distal slot (DNDN: rank 12/28; DANN: rank 13/28).
Third, the presence of larger hydrophobic and bulky aromatic residues such as Leu, Ile, Phe, and Tyr at any location are correlated with poorer performing candidates such as DLAG: rank 24/28, DGL: rank 26/28, EYIQG: rank 18/28, and IDSV: rank 27/28, with the poorest-performing candidates enriched in these four amino acids (one-sided Mann–Whitney U test, p-value = 0.002). We have previously shown that favorable interactions between these large hydrophobic residues and the aromatic π-cores can disrupt supramolecular association between the cores.30 This observation is also consistent with our prior observation that oligopeptides possessing oligophenylenevinylene π-cores exhibited larger spectral blue shifts when the peptide wings contained small Gly and Ala residues compared to larger Ile and Val residues.15
Finally, although no Pro containing molecules were sampled in the experimental screen, we note that a number of simulated molecules containing a Pro residue lie at or near the κ − Rg Pareto frontier and were highly ranked in predicted blue shift λ by the terminal mfGPR surrogate model (e.g., DPG: rank 329/694982, AEPP: rank 183/694982, SDPD: rank 10/694982, EAP: rank 13/694982, DDPA: rank 23/694982, GEPG: rank 15/694982). This finding is somewhat surprising because Pro has been largely unexplored in previous experimental and computational π-conjugated peptide studies. Proline, with its unique conformational properties including its conformational rigidity and absence of hydrogen bond donor capacity, appears to be quite favorable in promoting good in-register stacking between the π-cores and the formation of high-aspect ratio nanoaggregates. We suggest that experimental testing and further computational exploration of the molecular mechanisms underpinning these predictions may be a profitable avenue for future investigations.
3.3 Molecular assembly pathways
Having extracted design rules linking amino acid sequence to the degree of H-type stacking within the nanoaggregates, we then sought to analyze our library of molecular simulation trajectories to resolve the molecular self-assembly pathways promoted by the high-performing peptide sequences to gain mechanistic understanding of the link between sequence and the emergent supramolecular structure. We hypothesized that the ensemble of simulation trajectories for 1181 different Xn-4T-Xn molecules collected over the course of our computational screen may admit a low-dimensional clustering within the configurational phase space of assembly pathways, and that the high-performing molecules should follow similar assembly pathways to reach the terminal pseudo-1D nanoaggregates. We report in Fig. 4a and b a 2D embedding into the leading two collective variables ψ2 and ψ3 discovered by diffusion maps. By correlating these collective variables with candidate physical observables, we find ψ2 to be strongly correlated with the instantaneous radius of gyration Rg of the system (ρ(ψ2, Rg) = 0.93) and ψ3 moderately strongly correlated with the instantaneous number of contacts per molecule κ (ρ(ψ3, κ) = 0.69). In addition to providing good physical interpretability of the low-dimensional manifold containing the molecular assembly pathways learned by diffusion maps, the emergence of two collective variables strongly correlated with κ and Rg provides post hoc support for our selection of two observables as the dual objective functions of our computational screen as the leading variables governing the long-time self-assembly dynamics.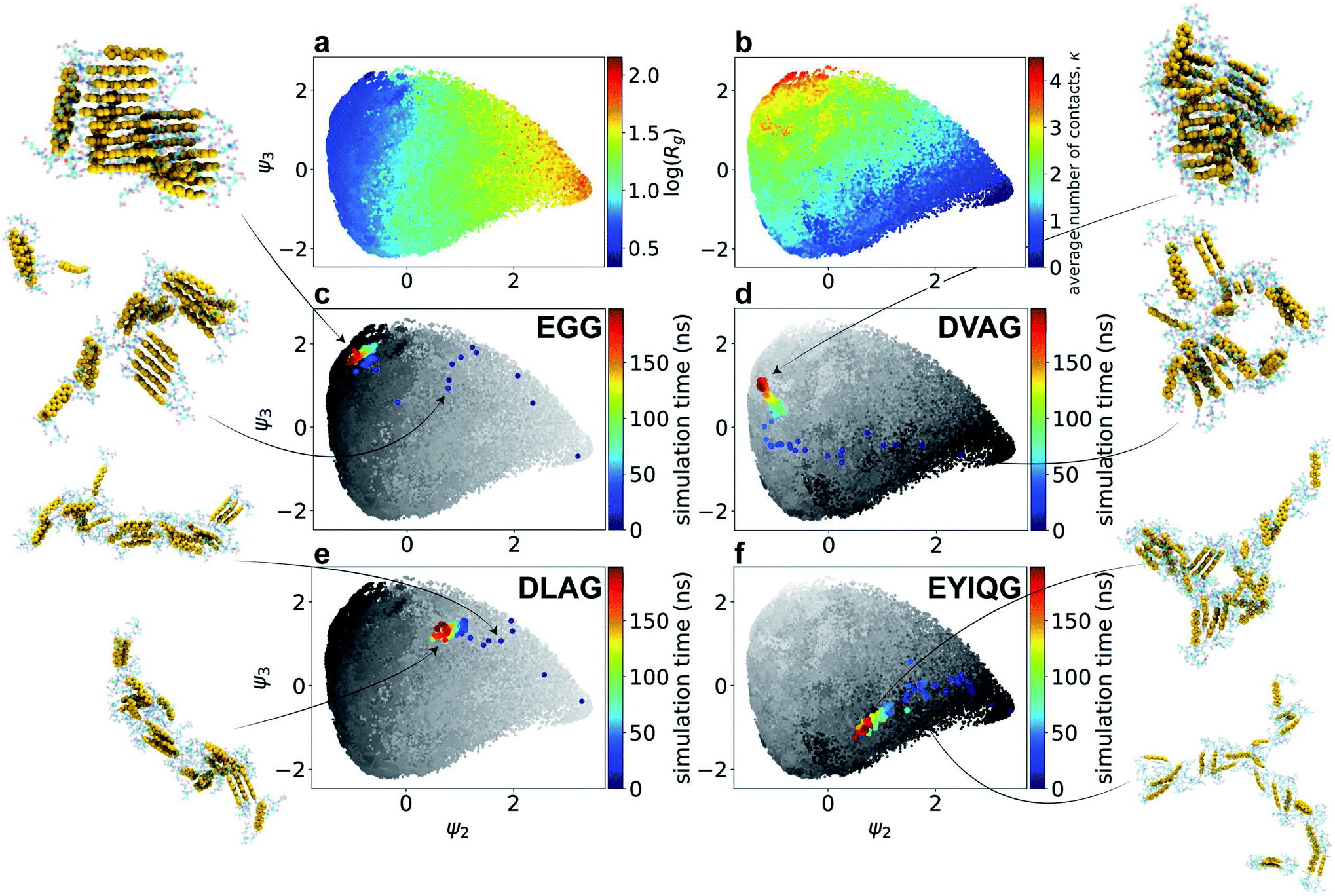 | ||
| Fig. 4 Two-dimensional diffusion map embedding of 118100 simulation snapshots harvested from the molecular simulations of 1181 Xn-4T-Xn molecules collected over the course of the computational active learning screen. Each point represents one simulation snapshot projected into a 2D low-dimensional manifold spanned by the leading collective variables ψ2 and ψ3 learned by diffusion maps. Coloring the points by (a) the log radius of gyration log(Rg) of the self-assembled nanoaggregate and (b) average number of contacts per molecule κ within the aggregate exposes the strong correlation of the two learned collective variables with these two physical observables (ρ(ψ2, Rg) = 0.93, ρ(ψ3, κ) = 0.69). All assembly trajectories commence in a monomeric dispersion contained at the rightmost edge of the manifold. Progression from right-to-left corresponds to a reduction in Rg as the system self-assembles, and progression from bottom-to-top to the formation of more molecular contacts. The progression of the self-assembly pathways over the manifold are shown for molecules (c) EGG-4T-GGE, (d) DVAG-4T-GAVD, (e) DLAG-4T-GALD, and (f) EYIQG-4T-GQIYE, in which points are colored temporally. Grey points in panels c–f represent the embedding of 118100 simulation snapshots shaded in panels c and e by logRg, and panels d and f by κ. Insets show representative molecular renderings throughout the trajectory. | ||
In Fig. 4c–f we illustrate the temporal evolution of the self-assembly pathways for particular Xn-4T-Xn molecules over the ψ2 − ψ3 manifold. Each molecular trajectory begins at the right-most edge of the of the manifold corresponding to the initial monodisperse state. Lateral leftward movement across the manifold corresponds to condensation of the system to smaller Rg values due to the formation of nanoaggregates. Vertical upward movement corresponds to the accumulation of inter-molecular contacts between the π-cores and an elevation in κ. The assembly pathways of the top-performing candidates typically terminate in the upper-left corner of the manifold that contains pseudo-1D nanoaggreates containing κ ≈ 3 intermolecular contacts and Rg ≈ 2 nm corresponding to elongated linear stacks. We observe that Rg necessarily decreases as the system self-assembles from dispersed monomers and that this behavior is not in conflict with the active learning goals of maximizing Rg and κ of the self-assembled nanoaggregates in order to promote high-aspect ratio linear morphologies with good π–π stacking. One prototypical class of assembly pathways for high-performing molecules is exemplified by EGG-4T-GGE (rank 4/28), which traverses the upper edge of the manifold (Fig. 4c). This assembly route corresponds to the rapid formation of small oligomeric stacks, the formation of which is likely promoted by the small size of the peptide wing, that ultimately associate into an elongated aggregate with good in-register and global stacking. DVAG-4T-GAVD (rank 11/28) is another high-performing candidate that is prototypical of a different assembly route followed by high-performing molecules (Fig. 4d). This pathway commences with an initial rapid hydrophobic aggregation of the system corresponding to a rapid leftward lateral motion over the manifold. The absence of any early upward vertical motion is indicative of no initial substantive increase in κ due to the larger peptide wings seemingly preventing good π-core stacking. This initial collapse is, however, then followed by a more gradual structural ripening as the cores do achieve good stacking and we observe late upward motion over the manifold corresponding to an increase in κ.
Trajectories that terminate within the bulk of the manifold and far from the upper-left corner typically fail to form nanoaggregates containing globally connected pseudo-1D stacks. DLAG-4T-GALD (rank 24/28) is emblematic of a poor-performing molecule that initially builds a reasonable number of intermolecular contacts, but then fails to further condense into an in-register stacked nanoaggregate (Fig. 4e). Differing only in a V to L mutation relative to the high-performing DVAG-4T-GAVD, the presence of the bulkier hydrophobic Leu residue appears to preclude structural ripening into the desired elongated stack. Finally, molecules rich in large hydrophobic side chains such as EYIQG-4T-GQIYE (rank 18/28) tend to exhibit moderate leftward motion over the manifold corresponding to hydrophobic collapse but accompanied with unfavorable downward motion indicative of the formation of very few intermolecular π-core contacts (Fig. 4f). This behavior can be attributed to the bulky aromatic hydrophobes that stack against the π-cores and prevent the formation of core–core contacts.
Whereas Fig. 4 provided anecdotal insights into the self-assembly trajectories traced out by particular representative Xn-4T-Xn molecules, in Fig. 5 we present the entire distribution of trajectory end points for all molecules considered in our active learning screen. In Fig. 5a and b we illustrate the end points of the 1181 molecules sampled in our computational screen colored by the Rg and κ values of the terminal nanoaggregates and in Fig. 5c the 28-molecule subset of these candidates that were experimentally tested colored by the measured spectral blue shift λ. Focusing on the 28 experimental molecules, we observe a clustering of 17 molecules in the upper left region of the manifold that we bound by a purple box. As anticipated by the understanding exposed by the diffusion map, these molecules tend to be high-performers comprising nine of the top 11 experimentally-tested molecules with spectral blue shifts λ ≥ 35 nm. Further, the molecules within the box possess a mean spectral shift of λ = 31 nm compared to the mean value for those outside the box with λ = 21 nm (one-sided Mann–Whitney U test, p-value = 0.07). The correlation between (Rg, κ) and λ within the diffusion map embedding provides a strong post hoc substantiation for the use of the former measures as a computational proxy for the latter within the active learning screen, and demonstrates the power and value of the high-throughput computational screen in focusing and guiding the low-throughput experimentation.
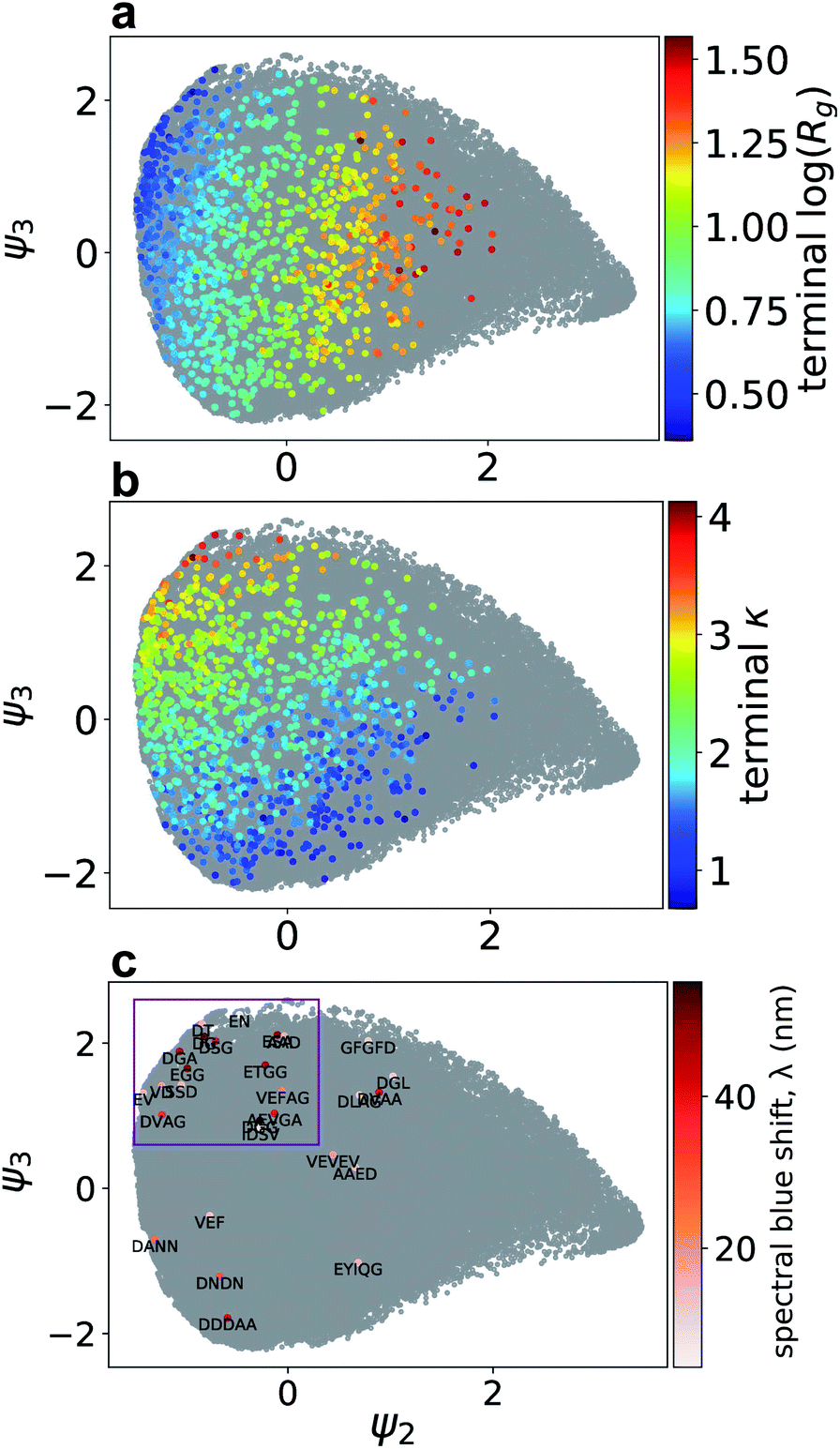 | ||
| Fig. 5 Terminal locations of the Xn-4T-Xn self-assembly trajectories over the 2D diffusion map manifold. End points of the 1181 molecules considered in our computational screen colored by the (a) log of the radius of gyration log(Rg) and (b) average number of contacts per molecule κ computed over the terminal 50 ns of the trajectory. The 118100 simulation snapshots used to construct the diffusion map embedding are shown in grey. (c) Terminal locations of the 28 experimentally tested molecules colored by their measured spectral blue shift λ and annotated with the sequence of the peptide wing. The purple box bounds a cluster of high-performing experimental candidates residing in the upper-left region of the manifold possessing high values of λ. | ||
4 Conclusions
In this work, we have reported an integrated computational/experimental iterative design strategy to discover synthetic π-conjugated oligopeptides within the Xn-4T-Xn family with the capacity to self-assemble into highly-ordered linear aggregates with in-register stacking of the π-cores. These supramolecular assemblies are desirable as biocompatible nanoaggregates possessing emergent optoelectronic properties and potential applications as peptide-based field-effect transistors, photoconductors, or solar cells. The Xn-4T-Xn design space consisting of symmetric oligopeptide wings containing between one and five amino acids comprises 694982 candidate molecules, making its exhaustive exploration impracticable by either simulation or experiment. By fusing computational and experimental data streams within an integrated computational–experimental active learning framework, we perform a data-driven efficient traversal the space of Xn-4T-Xn peptides that minimizes computational and experimental burden required to discover and validate new high-preforming candidates. Our platform employs a combination of all-atom molecular dynamics simulations, deep representational learning, single- and multi-fidelity Gaussian process regression, and single- and multi-objective Bayesian optimization. A computational active learning loop serves as a high-throughput and cheaply available experimental proxy used to refine a surrogate model that predicts the experimental performance of untested candidates. Using this platform, we discovered a diversity of high-performing new molecules experimentally validated to form pseudo-1D linear nanoaggregates after sampling only 1181 molecules (∼0.17% of the design space) by computation and 28 (∼0.004%) by experiment. Subsequent interrogation of our experimental screening data exposed molecular design rules linking sequence to the emergent structure and function of the self-assembled nanoaggregates. Analysis of the computational screening results revealed two prototypical assembly mechanisms and pathways shared by the high-performing molecules: (i) hierarchical assembly of small in-register supramolecular oligomers that undergo further assembly into a single linear aggregate with ordered π-stacking and (ii) rapid hydrophobic collapse followed by slow structural ripening and the emergence of in-register ordering of the π-cores.
This work exposes new understanding of how variation in oligopeptide sequence in π-conjugated peptides impacts assembly behavior using an integrated experimental-computational active learning platform. Our findings corroborate prior physico-chemical understanding and chemical intuition of π-conjugated peptide assembly, but also reveals new design rules and understanding of molecular assembly mechanisms. Our hybrid computational/experimental active learning platform demonstrates the power of tightly integrated collaboration between theory and experiment, and this paradigm is transferable to other generic molecular design and discovery applications.
Data availability
Data providing a full accounting of all molecules simulated and experimentally tested throughout the active learning process with associated measurements for the average number of contacts κ, radius of gyration Rg and spectral blue shift λ, and terminal GPR and mfGPR predicted κ, Rg and λ; neural network weights and training codes; GPR training codes; RAE embeddings; active learning workflow.34Author contributions
KS performed the simulations, data analysis and developed the active learning workflow methodology. SSP and AS performed the peptide synthesis and characterization. KS, SSP and ALF designed the research and wrote the manuscript. ALF and JDT supervised the research.Conflicts of interest
A. L. F. is a co-founder and consultant of Evozyne, Inc. and a co-author of US Patent Application 16/887,710, US Provisional Patent Applications 62/853,919, 62/900,420, and 63/314,898 and International Patent Applications PCT/US2020/035206 and PCT/US2020/050466.Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. DMR-1841807 and DMR-1728947, and a National Science Foundation Graduate Research Fellowship to K. S. under Grant No. DGE-1746045. This work was completed in part with resources provided by the University of Chicago Research Computing Center. We gratefully acknowledge computing time on the University of Chicago high-performance GPU-based cyberinfrastructure supported by the National Science Foundation under Grant No. DMR-1828629.Notes and references
- S. S. Panda, H. E. Katz and J. D. Tovar, Chem. Soc. Rev., 2018, 47, 3640–3658 RSC.
- R. J. Kumar, J. M. MacDonald, T. B. Singh, L. J. Waddington and A. B. Holmes, J. Am. Chem. Soc., 2011, 133, 8564–8573 CrossRef CAS PubMed.
- H. A. M. Ardoña and J. D. Tovar, Bioconjugate Chem., 2015, 26, 2290–2302 CrossRef PubMed.
- M. A. Khalily, G. Bakan, B. Kucukoz, A. E. Topal, A. Karatay, H. G. Yaglioglu, A. Dana and M. O. Guler, ACS Nano, 2017, 11, 6881–6892 CrossRef CAS PubMed.
- D. A. Stone, A. S. Tayi, J. E. Goldberger, L. C. Palmer and S. I. Stupp, Chem. Commun., 2011, 47, 5702–5704 RSC.
- H. Shao, J. Seifert, N. C. Romano, M. Gao, J. J. Helmus, C. P. Jaroniec, D. A. Modarelli and J. R. Parquette, Angew. Chem., 2010, 122, 7854–7857 CrossRef.
- W.-W. Tsai, I. D. Tevis, A. S. Tayi, H. Cui and S. I. Stupp, J. Phys. Chem. B, 2010, 114, 14778–14786 CrossRef CAS PubMed.
- T. Kitamura, S. Nakaso, N. Mizoshita, Y. Tochigi, T. Shimomura, M. Moriyama, K. Ito and T. Kato, J. Am. Chem. Soc., 2005, 127, 14769–14775 CrossRef CAS PubMed.
- R. S. Moghaddam, E. R. Draper, C. Wilson, H. Heidari and D. J. Adams, RSC Adv., 2018, 8, 34121–34125 RSC.
- G. L. Eakins, R. Pandey, J. P. Wojciechowski, H. Y. Zheng, J. E. Webb, C. Valéry, P. Thordarson, N. O. Plank, J. A. Gerrard and J. M. Hodgkiss, Adv. Funct. Mater., 2015, 25, 5640–5649 CrossRef CAS.
- M. Pandeeswar, H. Khare, S. Ramakumar and T. Govindaraju, Chem. Commun., 2015, 51, 8315–8318 RSC.
- J. López-Andarias, M. J. Rodriguez, C. Atienza, J. L. López, T. Mikie, S. Casado, S. Seki, J. L. Carrascosa and N. Martín, J. Am. Chem. Soc., 2015, 137, 893–897 CrossRef PubMed.
- T. Lee, S. S. Panda, J. D. Tovar and H. E. Katz, ACS Nano, 2020, 14, 1846–1855 CrossRef CAS PubMed.
- K. Besar, H. A. M. Ardona, J. D. Tovar and H. E. Katz, ACS Nano, 2015, 9, 12401–12409 CrossRef CAS PubMed.
- B. D. Wall, A. E. Zacca, A. M. Sanders, W. L. Wilson, A. L. Ferguson and J. D. Tovar, Langmuir, 2014, 30, 5946–5956 CrossRef CAS PubMed.
- B. D. Wall, S. R. Diegelmann, S. Zhang, T. J. Dawidczyk, W. L. Wilson, H. E. Katz, H.-Q. Mao and J. D. Tovar, Adv. Mater., 2011, 23, 5009–5014 CrossRef CAS PubMed.
- S. S. Panda, K. Shmilovich, N. S. Herringer, N. Marin, A. L. Ferguson and J. D. Tovar, Langmuir, 2021, 37(28), 8594–8606 CrossRef CAS PubMed.
- S. S. Panda, K. Shmilovich, A. L. Ferguson and J. D. Tovar, Langmuir, 2019, 35, 14060–14073 CrossRef CAS PubMed.
- G. Horowitz, Adv. Mater., 1998, 10, 365–377 CrossRef CAS.
- A. Mishra, C.-Q. Ma and P. Bauerle, Chem. Rev., 2009, 109, 1141–1276 CrossRef CAS PubMed.
- L. R. Valverde, B. A. Thurston, A. L. Ferguson and W. L. Wilson, Langmuir, 2018, 34, 7346–7354 CrossRef CAS PubMed.
- P. W. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30 CrossRef CAS PubMed.
- C. Kim, A. Chandrasekaran, A. Jha and R. Ramprasad, MRS Commun., 2019, 9, 860–866 CrossRef CAS.
- J. Ling, M. Hutchinson, E. Antono, S. Paradiso and B. Meredig, Integrating Materials and Manufacturing Innovation, 2017, 6, 207–217 CrossRef.
- R. Barrett and A. D. White, J. Chem. Inf. Model., 2021, 61, 95–105 CrossRef CAS PubMed.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed.
- A. van Teijlingen and T. Tuttle, J. Chem. Theory Comput., 2021, 17, 3221–3232 CrossRef CAS PubMed.
- F. Li, J. Han, T. Cao, W. Lam, B. Fan, W. Tang, S. Chen, K. L. Fok and L. Li, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11259–11264 CrossRef CAS PubMed.
- S. Nagasawa, E. Al-Naamani and A. Saeki, J. Phys. Chem. Lett., 2018, 9, 2639–2646 CrossRef CAS PubMed.
- K. Shmilovich, R. A. Mansbach, H. Sidky, O. E. Dunne, S. S. Panda, J. D. Tovar and A. L. Ferguson, J. Phys. Chem. B, 2020, 124, 3873–3891 CrossRef CAS PubMed.
- B. A. Thurston and A. L. Ferguson, Mol. Simul., 2018, 44, 930–945 CrossRef CAS.
- P. V. Balachandran, B. Kowalski, A. Sehirlioglu and T. Lookman, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- R. Gómez-Bombarelli, J. Aguilera-Iparraguirre, T. D. Hirzel, D. Duvenaud, D. Maclaurin, M. A. Blood-Forsythe, H. S. Chae, M. Einzinger, D.-G. Ha and T. Wu, et al. , Nat. Mater., 2016, 15, 1120–1127 CrossRef PubMed.
- K. Shmilovich, S. S. Panda, A. Stouffer, J. D. Tovar and A. L. Ferguson, Supporting data for: “Hybrid computational–experimental data-driven design of self-assembling π-conjugated peptides”, 2021, DOI:10.5281/zenodo.5048397.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- V. Pareto, Cours d'Économie Politique, Librairie Droz, 1964, vol. 1 Search PubMed.
- P. Ghosh, M. S. Sajjadi, A. Vergari, M. Black and B. Schölkopf, 2019, arXiv preprint arXiv:1903.12436.
- C. E. Rasmussen and C. K. I. Williams, Gaussian Processes for Machine Learning (Adaptive Computation and Machine Learning), The MIT Press, 2005 Search PubMed.
- E. Brochu, V. M. Cora and N. De Freitas, 2010, arXiv preprint arXiv:1012.2599.
- B. Paria, K. Kandasamy and B. Póczos, Proceedings of The 35th Uncertainty in Artificial Intelligence Conference, 2020, pp. 766–776 Search PubMed.
- P. Perdikaris, M. Raissi, A. Damianou, N. D. Lawrence and G. E. Karniadakis, Proc. R. Soc. A, 2017, 473, 20160751 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1, 19–25 CrossRef.
- P. Mark and L. Nilsson, J. Phys. Chem. A, 2001, 105, 9954–9960 CrossRef CAS.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins: Struct., Funct., Bioinf., 2010, 78, 1950–1958 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- D. N. Theodorou and U. W. Suter, Macromolecules, 1985, 18, 1206–1214 CrossRef CAS.
- J. Wang and A. L. Ferguson, J. Phys. Chem. B, 2016, 120, 8016–8035 CrossRef CAS PubMed.
- R. A. Mansbach and A. L. Ferguson, J. Phys. Chem. B, 2017, 121, 1684–1706 CrossRef CAS PubMed.
- R. A. Mansbach and A. L. Ferguson, Org. Biomol. Chem., 2017, 15, 5484–5502 RSC.
- R. A. Mansbach and A. L. Ferguson, J. Phys. Chem. B, 2018, 122, 10219–10236 CrossRef CAS PubMed.
- M. K. Warmuth, J. Liao, G. Rätsch, M. Mathieson, S. Putta and C. Lemmen, J. Chem. Inf. Comput. Sci., 2003, 43, 667–673 CrossRef CAS PubMed.
- D. A. Pertusi, M. E. Moura, J. G. Jeffryes, S. Prabhu, B. W. Biggs and K. E. Tyo, Metab. Eng., 2017, 44, 171–181 CrossRef CAS PubMed.
- R. Varela, W. P. Walters, B. B. Goldman and A. N. Jain, J. Med. Chem., 2012, 55, 8926–8942 CrossRef CAS PubMed.
- V. Khanna and S. Ranganathan, BMC Bioinf., 2011, 1–12 Search PubMed.
- D. Reker, P. Schneider and G. Schneider, Chem. Sci., 2016, 7, 3919–3927 RSC.
- A. W. Naik, J. D. Kangas, C. J. Langmead and R. F. Murphy, PLoS One, 2013, 8, e83996 CrossRef PubMed.
- D. P. Kingma and M. Welling, 2013, arXiv preprint arXiv:1312.6114.
- B. Mohr, K. Shmilovich, I. S. Kleinwächter, D. Schneider, A. L. Ferguson and T. Bereau, Chem. Sci., 2022, 13, 4498–4511 RSC.
- J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals and G. E. Dahl, International Conference on Machine Learning, 2017, pp. 1263–1272 Search PubMed.
- P. W. Battaglia, J. B. Hamrick, V. Bapst, A. Sanchez-Gonzalez, V. Zambaldi, M. Malinowski, A. Tacchetti, D. Raposo, A. Santoro and R. Faulkneret al., 2018, arXiv preprint arXiv:1806.01261.
- J. Chung, C. Gulcehre, K. Cho and Y. Bengio, 2014, arXiv preprint arXiv:1412.3555.
- Y. Li, D. Tarlow, M. Brockschmidt and R. Zemel, 2015, arXiv preprint arXiv:1511.05493.
- O. Vinyals, S. Bengio and M. Kudlur, 2015, arXiv preprint arXiv:1511.06391.
- S. Kawashima, P. Pokarowski, M. Pokarowska, A. Kolinski, T. Katayama and M. Kanehisa, Nucleic Acids Res., 2007, 36, D202–D205 CrossRef PubMed.
- D. P. Kingma and J. Ba, 2014, arXiv preprint arXiv:1412.6980.
- A. Paszke, S. Gross, F. Massa, A. Lerer, J. Bradbury, G. Chanan, T. Killeen, Z. Lin, N. Gimelshein and L. Antiga, et al. , Adv. Neural Inf. Process. Syst., 2019, 32, 8026–8037 Search PubMed.
- P. Auer, J. Mach. Learn. Res., 2002, 3, 397–422 Search PubMed.
- M. Kasha, H. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS.
- H. A. M. Ardoña, K. Besar, M. Togninalli, H. E. Katz and J. D. Tovar, J. Mater. Chem. C, 2015, 3, 6505–6514 RSC.
- A. L. Ferguson and K. A. Brown, Annu. Rev. Chem. Biomol. Eng., 2022, 13, 1–20 Search PubMed.
- D. J. Lizotte, PhD thesis, University of Alberta, 2008.
- J. Močkus, Optimization Techniques IFIP Technical Conference, 1975, pp. 400–404 Search PubMed.
- D. Ginsbourger, R. Le Riche and L. Carraro, 2008, HAL preprint hal-00260579.
- R. R. Coifman and S. Lafon, Appl. Comput. Harmon. Anal., 2006, 21, 5–30 CrossRef.
- B. Nadler, S. Lafon, R. R. Coifman and I. G. Kevrekidis, 2005, arXiv preprint math/0506090.
- A. W. Long and A. L. Ferguson, Mol. Syst. Des. Eng., 2018, 3, 49–65 RSC.
- J. Wang and A. L. Ferguson, Mol. Simul., 2018, 44, 1090–1107 CrossRef CAS.
- Y. Ma and A. L. Ferguson, Soft Matter, 2019, 15, 8808–8826 RSC.
- A. L. Ferguson, A. Z. Panagiotopoulos, P. G. Debenedetti and I. G. Kevrekidis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13597–13602 CrossRef CAS PubMed.
- A. P. Bartók, R. Kondor and G. Csányi, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 184115 CrossRef.
- L. Himanen, M. O. Jäger, E. V. Morooka, F. F. Canova, Y. S. Ranawat, D. Z. Gao, P. Rinke and A. S. Foster, Comput. Phys. Commun., 2020, 247, 106949 CrossRef CAS.
- S. De, A. P. Bartók, G. Csányi and M. Ceriotti, Phys. Chem. Chem. Phys., 2016, 18, 13754–13769 RSC.
- M. O. Jäger, E. V. Morooka, F. F. Canova, L. Himanen and A. S. Foster, npj Comput. Mater., 2018, 4, 1–8 CrossRef.
- J. Wang, M. A. Gayatri and A. L. Ferguson, J. Phys. Chem. B, 2017, 121, 4923–4944 CrossRef CAS PubMed.
- B. A. Thurston, E. P. Shapera, J. D. Tovar, A. Schleife and A. L. Ferguson, Langmuir, 2019, 35, 15221–15231 CrossRef CAS PubMed.
- H. B. Mann and D. R. Whitney, Ann. Math. Stat., 1947, 50–60 CrossRef.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- A. M. Sanders, T. J. Dawidczyk, H. E. Katz and J. D. Tovar, ACS Macro Lett., 2012, 1, 1326–1329 CrossRef CAS PubMed.
- G. S. Vadehra, B. D. Wall, S. R. Diegelmann and J. D. Tovar, Chem. Commun., 2010, 46, 3947–3949 RSC.
- S. S. Panda and J. D. Tovar, Organic Materials, 2021 Search PubMed.
- S. S. Panda, K. Shmilovich, A. L. Ferguson and J. D. Tovar, Langmuir, 2020, 36, 6782–6792 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d1dd00047k |
| This journal is © The Royal Society of Chemistry 2022 |