
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemo- and regio-selective amidation of indoles with isocyanates using borane Lewis acids†
Ayan
Dasgupta‡
 a,
Michael G.
Guerzoni‡
a,
Nusaybah
Alotaibi
a,
Yara
van Ingen
a,
Kaveh
Farshadfar
bd,
Emma
Richards
*a,
Alireza
Ariafard
*c and
Rebecca L.
Melen
*a
a,
Michael G.
Guerzoni‡
a,
Nusaybah
Alotaibi
a,
Yara
van Ingen
a,
Kaveh
Farshadfar
bd,
Emma
Richards
*a,
Alireza
Ariafard
*c and
Rebecca L.
Melen
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, Cymru/Wales, UK. E-mail: RichardsE10@cardiff.ac.uk; MelenR@cardiff.ac.uk
bDepartment of Chemistry, Islamic Azad University, Central Tehran Branch, Poonak, Tehran, 1469669191, Iran
cSchool of Physical Sciences, University of Tasmania, Private Bag 75, Hobart, Tasmania 7001, Australia. E-mail: alireza.ariafard@utas.edu.au
dResearch Group of Computational Chemistry, Department of Chemistry and Materials Science, Aalto University, FI-00076 Aalto, Finland
First published on 25th August 2022
Abstract
The efficacy of boron-based catalysts has drawn considerable attention from the scientific community due to their relatively low toxicities and high selectivities. Formation of a new carbon–carbon or carbon–nitrogen bond to generate an amide/urea functionality using mild, catalytic reaction protocols has always been an important challenge, as functionalised amides and urea derivatives are important scaffolds in medicinal chemistry. Herein we report a facile and mild catalytic reaction protocol towards the amidation of N-methyl indoles/pyrroles (17 examples, yields up to 58%) using B(C6F5)3 (30 mol%). Moreover, our investigation revealed that although catalytic amounts of B(C6F5)3 (10 mol%) are efficient towards the N-carboxamidation of unprotected indoles, catalytic BCl3 (5 mol%) is capable of producing near quantitative yields of the N-carboxamidation products (21 examples, yields up to 95%). In contrast with previous literature reports, the reaction between 2-(alkynyl)anilines and aryl isocyanates using catalytic BCl3 (5 mol%) afforded N–H inserted products (9 examples, yields up to 71%) chemo-selectively as opposed to the intramolecular hydroamination product. Comprehensive DFT studies have been undertaken to understand the mechanistic details of the N–H functionalisation of indoles.
The activation of small molecules using main group element based catalysts has increased in popularity over the last two decades and opens up new avenues for bond forming reactions that may offer alternative methods towards the synthesis of fine chemicals.1 Whilst precious transition metal-based catalysts have been largely used, their toxicities and limited abundance cause problems in developing active molecules for applications in food-, agro-, and medicinal-chemistry.2 The partially filled d-orbitals present in transition metal species can synergistically accept and donate electrons to and from small molecules, resulting in the activation of both polar and inert bonds.3 Boranes on the other hand only act as an acceptor whereby the presence of an empty p-orbital renders them Lewis acidic and thus able to activate polar, Lewis basic substrates.4 The ability to finetune the activity, selectivity, and stability of the borane catalyst can be achieved by altering the ligands attached to the central boron atom. In particular, the use of sterically demanding and electron withdrawing perfluoroaryl ligands (e.g., C6F5) has been particularly attractive giving highly active and selective catalysts that can be employed in a range of organic reactions.5
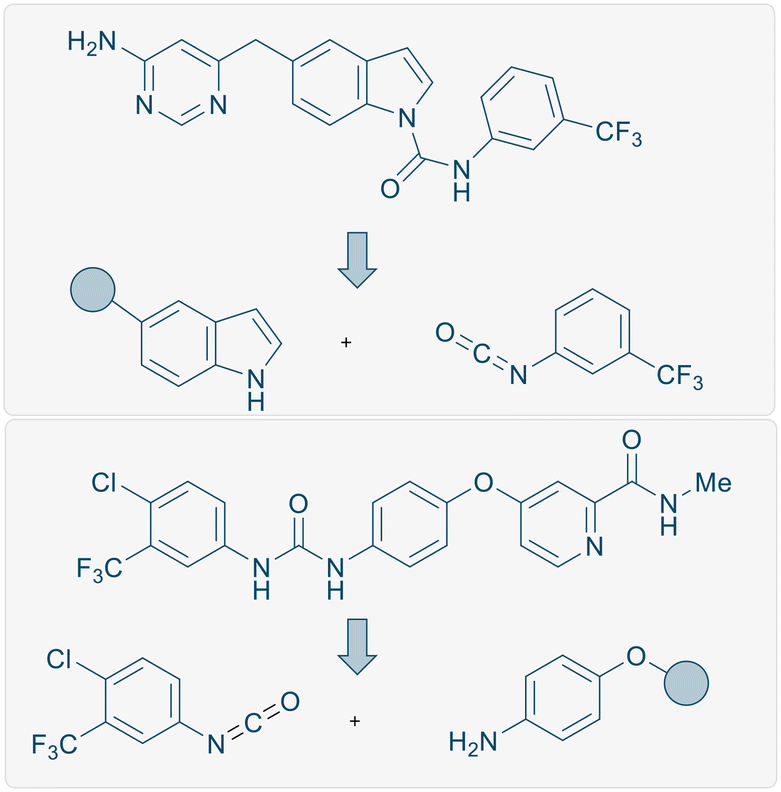
Methods to functionalise nitrogen-containing heterocycles6 are an important and ongoing area of research due to their notable biological and medicinally relevant activities (Scheme 1).7 The introduction of an amide group at the C3 position of an indole can be carried out through amidation reactions using isocyanate derivatives. Extensive studies have been carried out to establish facile synthetic routes to form such functionalised moieties using transition metals, including Pd, Cu, Zn, and Rh.8 In addition, in 2016, the carbamoylation of 1-methylindole using stoichiometric Me2AlCl was investigated by Hattori and co-workers.9
 | ||
| Scheme 1 Medicinally important functionalised urea derivatives. | ||
The N1 functionalisation of unprotected indoles with isocyanates to generate urea moieties has also been extensively studied. A range of transition metal catalysts including Pd,10 Cu,11 Ru,12 and Rh (ref. 13) have been used for N-carboxamidation of unprotected indoles through the formation of a new N–C bond. CsOH·H2O has also been employed as an alternative to the transition metal catalysts.14 In 2017, Camp and co-workers reported the functionalisation of ureas obtained from the reaction between isocyanates and secondary amines using cyrene as a solvent.15 In both cases, either the isocyanate scope was limited, or the reaction was only performed with more nucleophilic amines. Strong non-nucleophilic bases, such as NaH, have also been employed in stoichiometric amounts (or a large excess) for the reaction between unprotected indoles and aryl/alkyl isocyanates to afford urea derivates.16
Another route often pursued for the in situ generation of indole scaffolds relies on the employment of 2-alkynyl anilines which, upon intramolecular cyclisation, can be further functionalised at the C3 position with various electrophiles. For example, stoichiometric n-BuLi in combination with ZnCl2 afforded 3-zinciobenzoheteroles which could be further transformed into C3 Cu complexes and quenched with various electrophiles to afford C3 substituted indole derivatives.17 In 2017, Paradies and co-workers demonstrated catalytic B(C6F5)3 can be employed for the synthesis of indole derivatives starting from 2-alkynyl anilines. Such intramolecular hydroamination of internal alkynes proceeds through 5-and 6-endo-dig cyclisation with proto-deborylation.18
In this study, we chose to investigate the regio-and chemo-selective functionalisation of both protected and unprotected indoles with isocyanates using catalytic amounts of borane Lewis acids. We also investigated the reactivities of 2-alkynyl anilines derivatives towards isocyanates using catalytic BCl3 (Scheme 2).
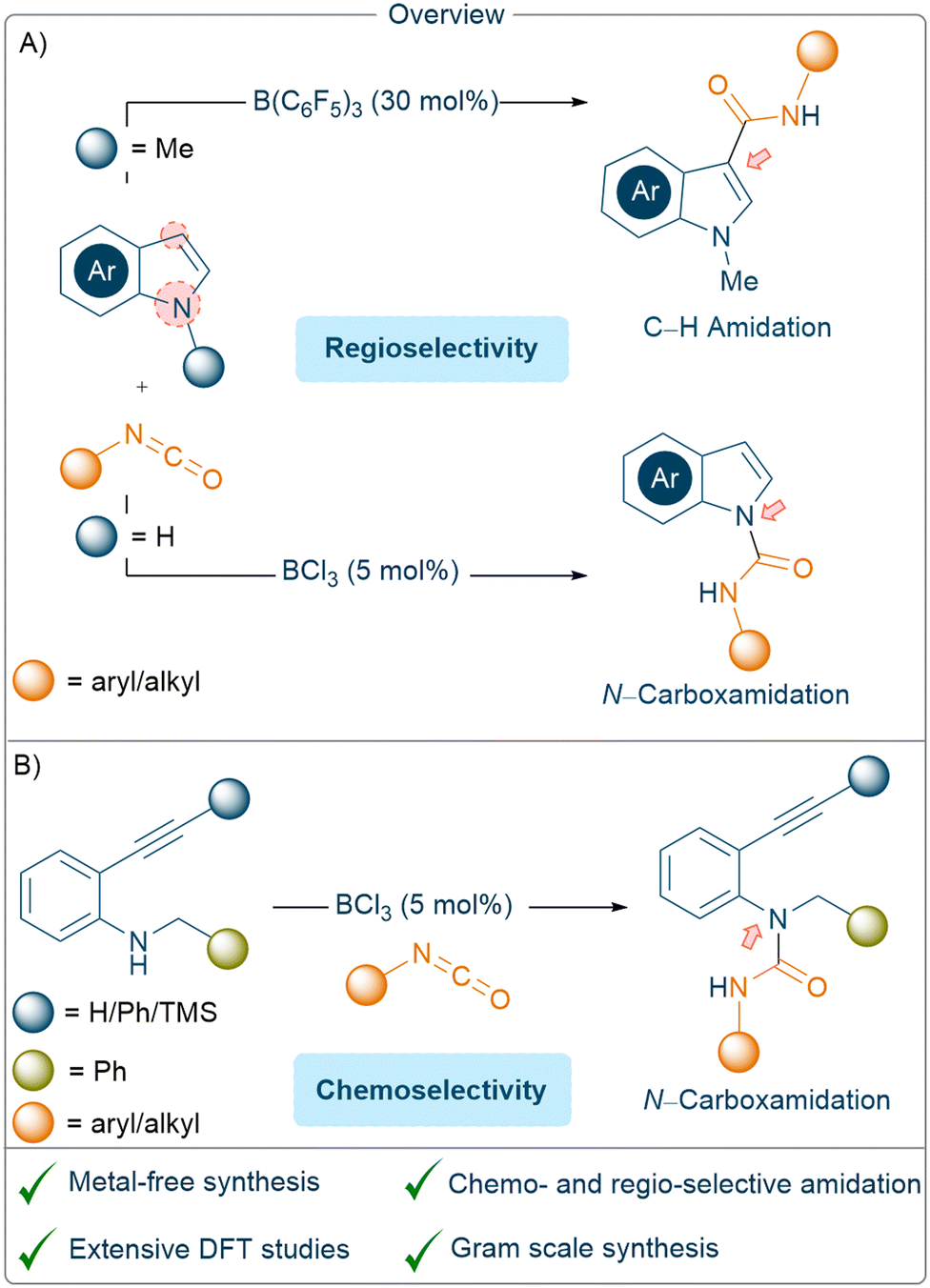 | ||
| Scheme 2 An overview of indole amidation reactions: (A); an intramolecular reaction vs. N–H insertion of 2-alkynyl anilines, with isocyanates (B). | ||
Initially, the borane mediated reaction between 1-methylindole and phenyl isocyanate was examined under various reaction conditions (see ESI† for the optimisation, Table S1). After extensive screening of the borane, solvent, temperature, loading, and time, we found that the optimum conditions for the reaction were 30 mol% B(C6F5)3, trifluorotoluene (TFT) solvent, and 80 °C (Fig. 1, top). Importantly, we could crystallise the product 1b from the reaction and, when employing stoichiometric B(C6F5)3, the reaction led to the formation of triaryl borane adduct 1b·B(C6F5)3. Both 1b and 1b·B(C6F5)3 could be characterised by single crystal X-ray diffraction (Fig. 1, bottom). Using our optimised reaction conditions, various indole derivatives including 1-methylindole, 1,2-dimethylindole, 5-bromo-1-methylindole, and 1-methyl-2-phenylindole were reacted with aryl isocyanates to afford the corresponding C3 amide substituted products (1a–1j) in moderate to good yields (up to 58%, Scheme 3). Interestingly, the reaction between 5-bromo-1-methylindole and 4-chlorophenyl isocyanate did not afford the expected C3 functionalised product.
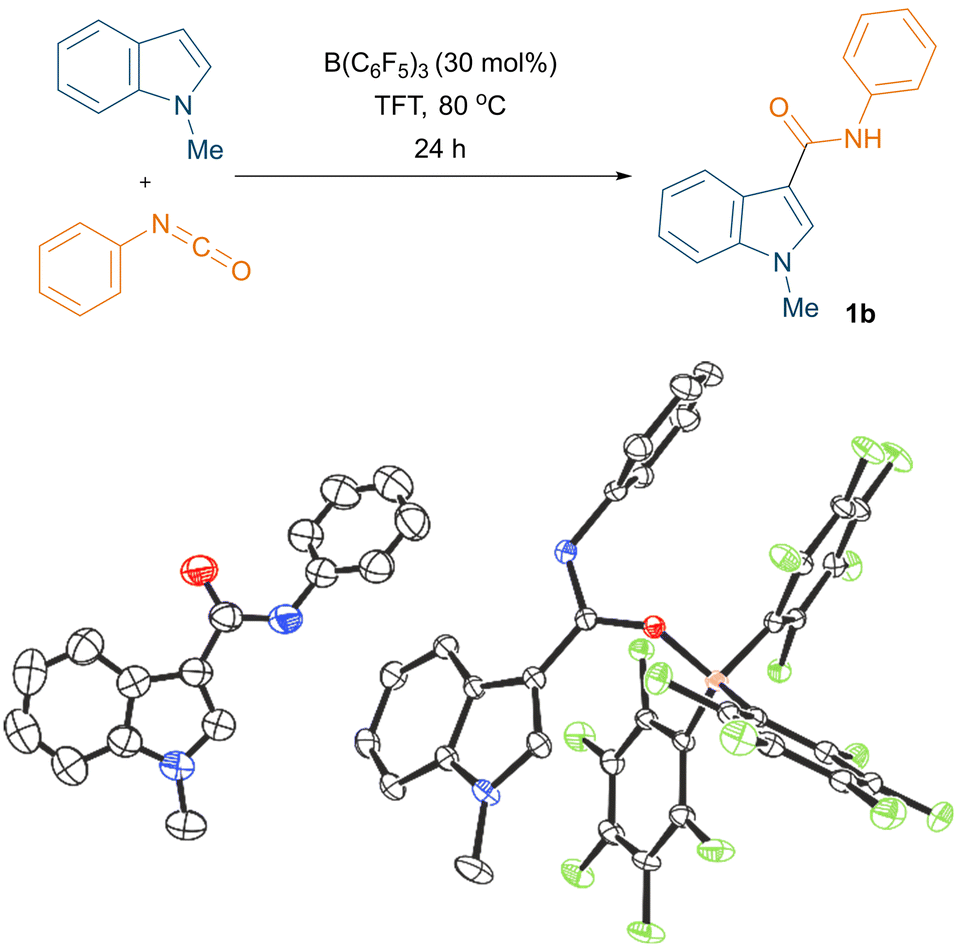 | ||
| Fig. 1 Reaction of 1-methylindole and phenyl isocyanate (top). Crystal structures of 1b (bottom left) and 1b·B(C6F5)3 (bottom right), thermal ellipsoids shown at 50%. H atoms omitted for clarity. Carbon: black; oxygen: red; nitrogen: blue; fluorine: green; boron: pink. | ||
 | ||
| Scheme 3 C3 amidation of indoles/pyrroles using aryl isocyanates mediated by B(C6F5)3. All the reactions were carried out on a 0.1 mmol scale; yields reported are isolated yields. | ||
Instead, slow evaporation from a CH2Cl2 solution of two isolated fractions afforded single crystals suitable for X-ray diffraction measurements, which revealed the formation of boron adduct 2 (Fig. S126†) and a C6F5-transfer product 3 (see ESI,† Fig. S127) in 39% and 41% yield respectively (Scheme 4, top). We infer that these isolated products, showing catalyst termination, could explain the relatively low yields obtained for other substrates, as well as the high loading of B(C6F5)3 needed for the reaction. This insightful result led us to hypothesise possible further functionalisation of the amidic nitrogen with another molecule of isocyanate. Treatment of compound 1b with 4-chlorophenyl isocyanate under the optimised conditions pleasingly afforded the N-carboxamidated product 4, albeit in a low yield of 21% (Scheme 4, bottom). Slow evaporation of the solvent from a concentrated ethyl acetate solution of 4 afforded single crystals suitable for X-ray diffraction measurements which confirmed the structure of the product (Fig. S128†).
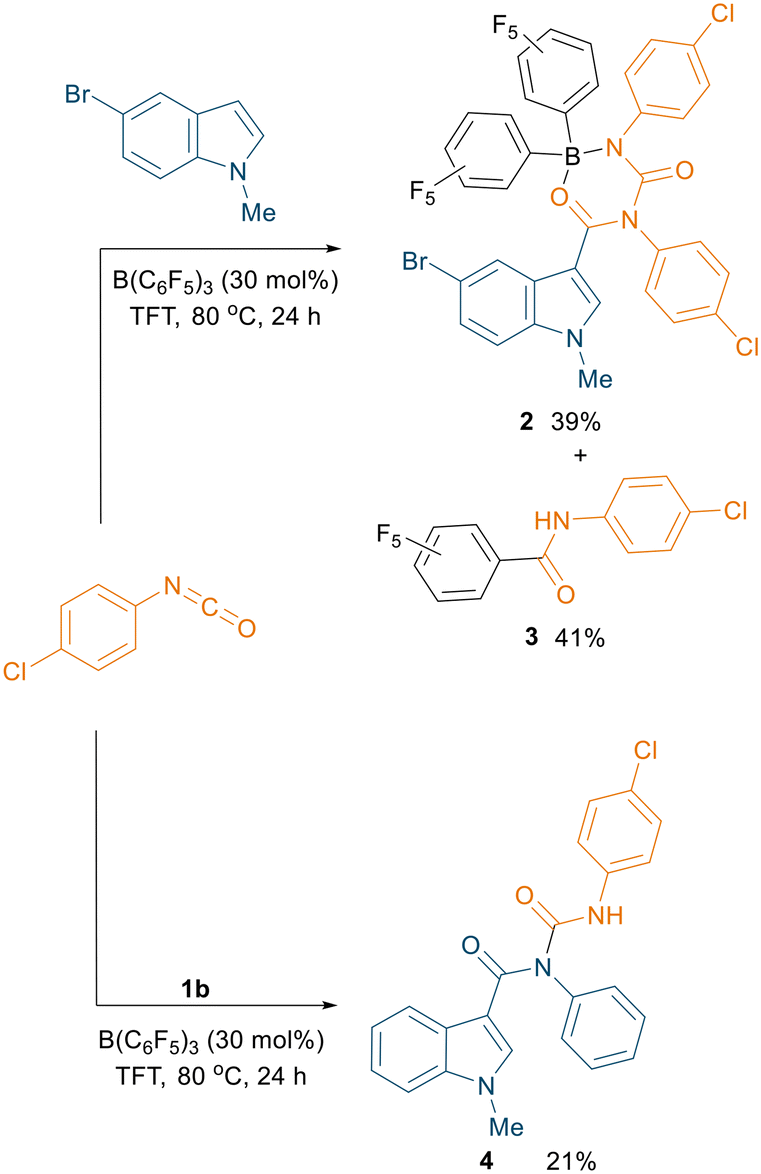 | ||
| Scheme 4 Reactivity observed when using 5-bromo-1-methylindole with 4-chlorophenyl isocyanate to afford 2 and 3 (top). Amidation of 1b using 30 mol% B(C6F5)3 to afford 4 (bottom). All the reactions were carried out on a 0.1 mmol scale; yields reported are isolated yields. | ||
In addition to indoles, we also investigated the reactivity of aryl isocyanates towards pyrrole substrates. Using the same reaction conditions, 1,2,5-trimethyl-pyrrole was employed for the reaction with various isocyanates to give the corresponding C3 amidation products 1k–1n in moderate yields (up to 52%, Scheme 3).
During the investigation of the substrate scope, we also examined the reactivities of 2,5-substituted pyrroles with aryl isocyanates. Using our optimised reaction conditions (30 mol% B(C6F5)3, TFT, 80 °C), a mixture of two regio-isomeric products was obtained. Although the C2 amide functionalised products (1o–1p, see ESI† Scheme S1) were formed as the major products (yields up to 50%), in each case the C3 functionalised products (1o′–1p′, see ESI† Scheme S1) were isolated as minor isomers (yields up to 20%). Due to poor conversion, when 4-methoxy phenyl isocyanate was used, only the C2 functionalised product (1q) was isolated in 17% yield.
Following the C3 amidation of N-protected indoles and pyrroles, we turned our attention to the reactivities of unprotected indoles towards isocyanates. Employing the same reaction conditions used for the C3 C–H amidation of N-protected indoles, a reaction between 1H-indole and phenyl isocyanate was investigated. In contrast to our above results, NMR (1H and 13C) spectroscopic data clearly indicated the formation of N-carboxamidated product 5a in 17% yield (see ESI,† Table S2). Given the presence of unreacted 1H-indole starting material in the reaction mixture, we optimised the conditions to improve the yield of 5a (see ESI,† Table S2). Optimisation of the borane catalyst and conditions revealed that BCl3 (5 mol%) was an efficient catalyst for the N-carboxamidation of 1H-indole with phenyl isocyanate at 60 °C in 1,2-C2H4Cl2 to afford 5a in near quantitative yield (93%). The superior reactivity and catalytic property of BCl3 over B(C6F5)3 is attributed to its higher Lewis acidity. From both computational and experimental metrics of Lewis acidity, it can be determined with fair confidence that BCl3 is more Lewis acidic than B(C6F5)3 by Gutmann–Beckett, Childs, and fluoride ion affinity (FIA) measurements.19,20,21 This can be explained by the increased accessibility of the boron atom in BCl3 upon coordination with a suitable Lewis base, rendering lower strain upon pyramidalisation of the boron centre. Additionally, the global electrophilicity index (GEI) addresses intrinsic Lewis acidity, without dependence on a base, again showing a higher Lewis acidity for BCl3 at 2.077 eV than B(C6F5)3 at 1.408 eV.22
Using the optimised reaction conditions of 5 mol% BCl3 in 1,2-C2H4Cl2 at 60 °C, a substrate scope was explored to show the robustness of this methodology (Scheme 5). 1H-indole, 5-chloro-1H-indole, 3-methyl-1H-indole, and 5-methoxy-1H-indole were combined with various aryl isocyanates (phenyl isocyanate, p-tolyl isocyanate, p-chlorophenyl isocyanate, and p-methoxyphenyl isocyanate) afford the corresponding urea derivatives in excellent yields (5a–5k; yields up to 98%). We further examined the reactivities of carbazole and 1H-benzo[d]imidazole, towards phenyl isocyanate. Satisfyingly, excellent yields of the corresponding urea derivatives were again obtained (5l–5m, yields up to 97%). Product 5n was obtained from the reaction between indolin-2-one and phenyl isocyanate in lower yields (48%), presumably due to the strong interaction between the carbonyl functionality and the highly Lewis acidic borane.
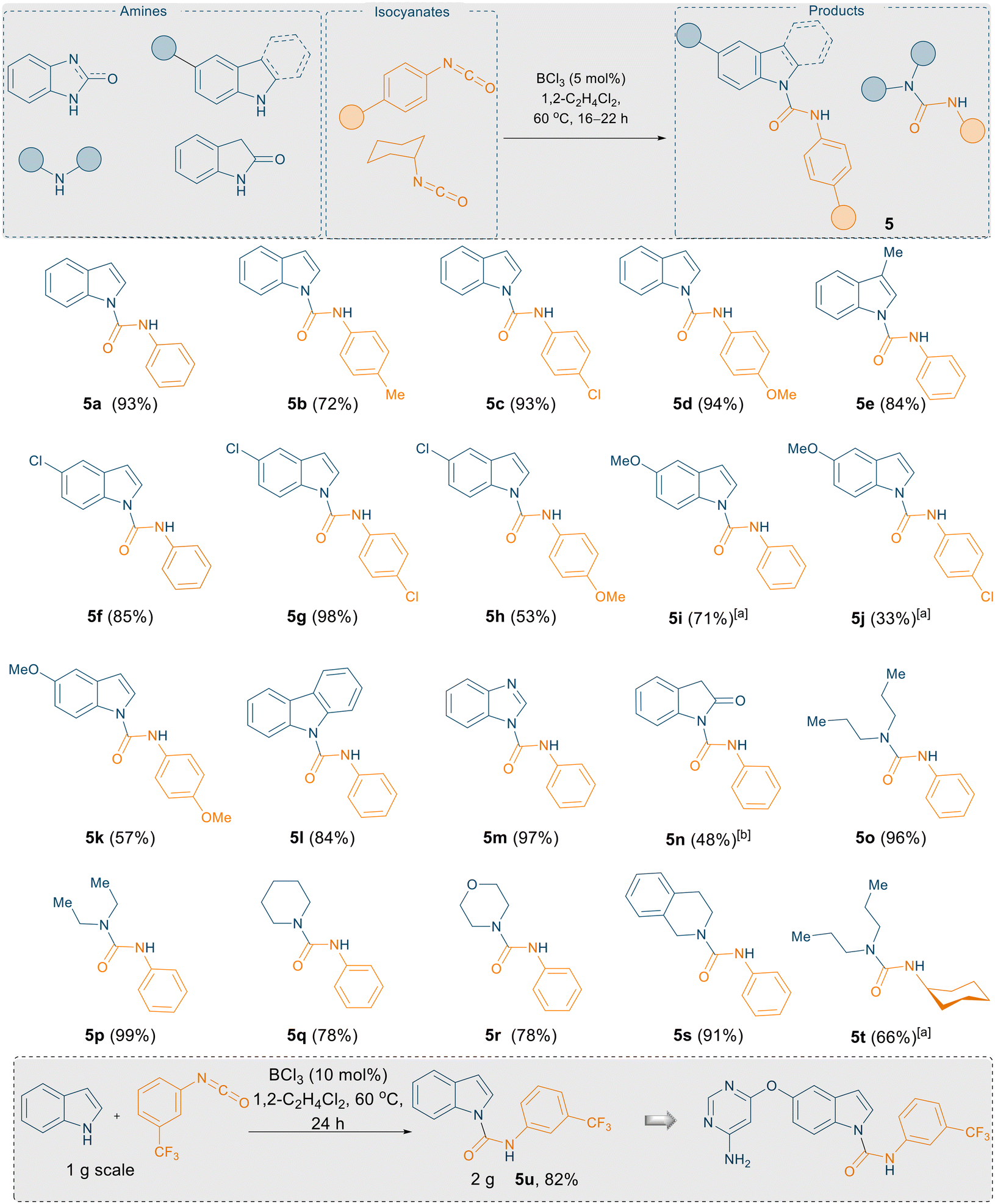 | ||
| Scheme 5 BCl3 catalysed N-carboxamidation of 1H-indoles using isocyanates. All the reactions were carried out on a 0.1 mmol scale; yields reported are isolated yields, [a] 10 mol% BCl3 was used, [b] 48 h. | ||
After this successful outcome, we extended the substrate scope to non-aromatic aliphatic secondary amines such as dipropylamine and diethylamine treated with phenyl/cyclohexyl isocyanate. All corresponding urea derivatives (5o–5t) were obtained in near quantitative yields (up to 99%). It is noteworthy that in 2017, Bousfield and Camp reported the synthesis of 5o–5s using bio-alternative solvent cyrene without the aid of any catalyst, albeit in lower yields.15 For example, the synthesis of 5o and 5q are reported in 44% and 61% yields whereas using our optimised catalytic reaction conditions, 5o and 5q can be obtained in 98% and 79% yield respectively.
Finally, to demonstrate the utility of this methodology, we employed our procedure towards the synthesis of a core structure of an anti-vascular endothelial growth factor therapeutic compound 5u (Scheme 5, insert) which is the core structure of a bioactive molecule (Scheme 1, top).16 A key synthetic step of this oral VEGFR-2 inhibitor was reported using a large excess of NaH (2 equiv.) to couple the isocyanate and indole motifs, or by using 1 mol% copper iodide as a catalyst.16 Using our slightly modified reaction protocol, the N–H inserted product 5u could be generated in 82% yield from the reaction of 1H-indole with 3-(trifluoromethyl)phenyl isocyanate on gram scale (Scheme 5, insert).
As evidenced from recent literature, 2-alkynyl anilines can undergo an intramolecular cyclisation to afford 2-substituted indoles using catalytic amounts of B(C6F5)3 (5 mol%).18 We were curious whether an intramolecular hydroamination concept could be applied to generate the indole in situ and subsequently substitute the nucleophilic C3 position with an isocyanate to generate the corresponding C3 amides in a one pot reaction. With this target in mind, 2-alkynyl anilines bearing TMS, phenyl, and unsubstituted alkynes were prepared according to literature procedures and employed for the reaction with aryl isocyanates (Scheme 6). Initially, B(C6F5)3 was used (20 mol%) as a catalyst and the reaction was carried out in TFT at 80 °C. A new product was formed and crystallised via slow evaporation from CH2Cl2. Single crystal X-ray diffraction data revealed the formation of N-carboxamidation product 6a instead of the expected cyclised-C3-functionalised product (Fig. S132†), which was isolated in 21% yield.
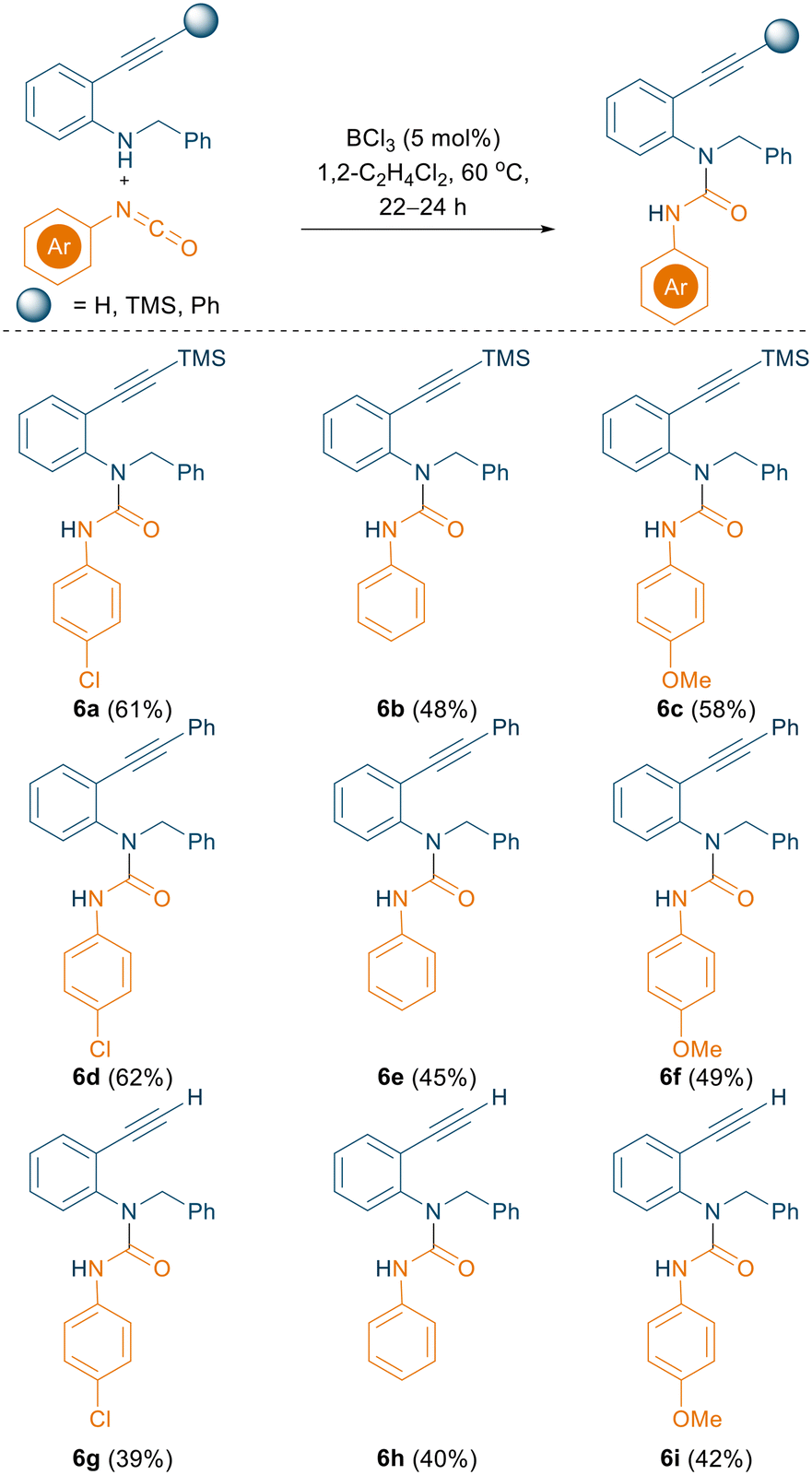 | ||
| Scheme 6 BCl3 catalysed N-carboxamidation of 2-alkynyl anilines using aryl isocyanates. All the reactions were carried out on a 0.1 mmol scale; yields reported are isolated yields. | ||
The yield of 6a was subsequently improved from 21% to 61% by using the optimised reaction conditions for the N-carboxamidation of 1H-indoles (5 mol% BCl3, 1,2-C2H4Cl2, 60 °C). 2-Alkynyl anilines in combination with various aryl isocyanates selectively afforded the corresponding N-carboxamidated products (6a–6i, Scheme 6) in good yields (up to 62%).
The complete chemo-selectivity observed for unprotected indoles motivated us to establish the most plausible reaction pathway for the N-carboxamidation. We undertook thorough DFT calculations at the SMD/M06-2X/def2-TZVP//SMD/M06-2X/6-31G(d) level of theory in CH2Cl2.
According to the literature, the Lewis acid-catalysed reaction between indole and isocyanate was proposed to take place via two conventional mechanisms depicted in Fig. S134A† (cycles A and B). In both cycles the reaction begins with coordination of the acid catalyst to the isocyanate electrophile, thereby activating it towards the nucleophilic attack of the indole. It has been proposed that when N-protected indoles are used, the catalytic reaction occurs via cycle A, whereas unprotected indoles favour cycle B.23 To determine the validity of this proposal, we calculated the energy profiles of cycles A and B for the reaction between the unprotected indole and phenyl isocyanate, the results of which are shown in Fig. S134A (left).† In a recent study, we revealed that coordination of BCl3 to the nitrogen atom of isocyanate activates it more effectively than coordination to its oxygen atom.24 As a result, we only considered this pathway in this work when assuming activation of the isocyanate by BCl3. To begin our computational investigations, we evaluated the stability of various adducts formed by the interaction of BCl3 with indole and isocyanate. Adduct 7 in which the BCl3 is bonded to the nitrogen atom of the indole is found to be more stable than the other possible adducts (Fig. S134A,† insert). As a result, adduct 7 is set as the reference structure in all energy profiles calculated in this study.
The calculations for cycle A indicate that nucleophilic attack of the C3 atom of the indole on the BCl3-activated isocyanate results in the formation of 15. Once formed, a proton transfer from the C3 atom to the nitrogen atom bonded to BCl3 yields product 17 while simultaneously regenerating the catalyst BCl3. As illustrated in Fig. S134† (Cycle A), we used a second indole as a proton transfer agent and determined that this reaction occurs with an overall activation free energy of 23.7 kcal mol−1.
The calculations for cycle B consider the case where the indole acts as a nucleophile attacking the activated isocyanate via its nitrogen atom, yielding intermediate 18 (Fig. S134A,† right). The resultant intermediate is then subjected to a proton transfer process, affording the experimentally observed product 5a (Fig. S134B,† bottom). According to literature, cycle B should be more favourable than cycle A for unprotected indoles.23 Contrary to this belief, we found that deprotonation in this pathway via transition structure TS9 requires an activation free energy of 29.7 kcal mol−1, which is higher than the free energies calculated for all transition structures of cycle A (Fig. S134A†). The unfavourable character of this pathway is mainly due to the much lower stability of intermediate 18 compared to its counterpart on cycle A (intermediate 15). As a result, the formation of product 5avia cycle B is unlikely, and it should be formed via a pathway different to those described in previous reports.
After conducting extensive research into an alternative pathway, we identified another cycle (Fig. 2), which accurately accounts for the formation of product 5avia a lower-energy pathway (Fig. 2). Interestingly, the key species in the proposed catalytic cycle is an imine → BCl3 adduct (10·BCl3) formed by an easy proton transfer process, as described below. As shown in Fig. 2A, the deprotonation of adduct 7 through TS7–9 by another molecule of indole generates iminium ion 8 and the BCl3-stabilised indolyl anion 9. Subsequent re-protonation of 9 at the C3 position by 8 yields adduct 10·BCl3 through TS9–10. Our calculations indicate that the formation of adduct 10·BCl3 is exergonic with ΔGrxn = −11.0 kcal mol−1 and occurs by surmounting an overall activation free energy of 13.5 kcal mol−1. This result implies that in the presence of BCl3, the indole should initially form adduct 10·BCl3, which is the catalyst resting state. It is this species, which reacts with the isocyanate via transition state TS10–12 to form intermediate 10. The relatively high activation free energy of 32 kcal mol−1 for the reaction to proceed through catalytic cycle shown in Fig. 2 may be explained by the overestimation of the entropy effect for the bimolecular transformation 10·BCl3 + 11 → TS10–12.25 The resultant intermediate 12 then undergoes a series of proton transfer processes to form intermediate 13, with an activation free energy of 9.9 kcal mol−1 (Fig. S2†). The formation of the product proceeds through 5a·BCl3 and regenerates the catalyst. Adduct 10′ is formed immediately before transition structure TS10–12. The high basicity of imine 10 results in the corresponding adducts being generated with relative energies of 20.9 (11.3) kcal mol−1. This adduct is highly reactive toward C–N bond formation, yielding 12 by overcoming an activation barrier as low as 0.8 kcal mol−1via transition structure TS10–12. The calculations reveal that although adduct 10′ has lower potential energy than separated structures 10 and 11, it has higher free energy due to the entropy effect. This explains why TS10–12 and the separated structures 10 and 11 exhibit the same energy trend. The key transition with a relative free energy of 21.7 kcal mol−1 is lower in energy than the key transition structures on cycles A and B (see ESI,† Fig. S134), implying that product 5a is most likely formed via this unprecedented mechanism (Fig. 2B). To support our theoretical calculations, we independently synthesised the 10·BCl3 adduct according to literature procedures26 and tested it as a catalyst for the reaction. Using the optimised reaction conditions and 5 mol% 10·BCl3, the reaction between 1H-indole and phenyl isocyanate showed evidence for formation of 5a (yield: 75%) as shown by crude 1H NMR spectroscopy (see ESI,† Fig. S121). A question could be raised that the spontaneous dissociation of 10·BCl3 into BCl3 in solution could potentially catalyse the reaction to afford 5a. But based on our both experimental and as well DFT calculation data these possibilities can be ruled out. When BCl3 is released from 10·BCl3, the imine 10 is formed, which has a very high nucleophilic character due to its high basicity. This statement is supported by the fact that species 10 can trap BCl3-activated species 11 with an activation barrier as low as 8.5 kcal mol−1 (Fig. 2A). This barrier is lower than the other activation barriers calculated for other chemical steps discussed in this study. This implies that the formation of 5a is feasible using 10·BCl3 as a catalyst. To further validate this claim, two sets of stoichiometric reactions were carried out between 10·BCl3/phenyl isocyanate and 1-H indole/phenyl isocyanate/BCl3. Both reactions afforded the 5a in a poor yield (approx. 10% calculated from the 1H NMR spectra of the crude reaction mixture). This can be further justified as the formation of 5a·BCl3 is calculated to be an endergonic process with ΔGrxn = +0.3 kcal mol−1. Combining the DFT studies and experimental results, it's clear that the catalytic reaction most likely proceeds via the mechanism proposed in Fig. 2B.
 | ||
| Fig. 2 DFT calculated reaction pathway at the SMD/M06-2X/def2-TZVP//SMD/M06-2X/6-31G(d) level of theory in CH2Cl2 for the N-carboxamidation of 1H-indole using phenyl isocyanate and BCl3 as a catalyst (A). Free energies (potential energies) are given in kcal mol−1. DFT proposed catalytic cycle for the N-carboxamidation of 1H-indole with phenyl isocyanate and catalytic BCl3 (B). Crystal structure of 10·BCl3 (insert, top right). Thermal ellipsoids shown at 50% probability. H atoms omitted for clarity. Carbon: black; nitrogen: blue; chlorine: green; boron: pink. | ||
Conclusions
In conclusion, a facile metal-free synthetic route has been developed for the amidation of indoles. When using B(C6F5)3,N-protected indoles undergo C3 C–H amidation, whereas when using BCl3, unprotected indoles regioselectively formed N-carboxamidation products in near quantitative yields. A broad substrate scope has been demonstrated to show the wide applicability of the reported methodology. Although 2-alkynyl anilines have been reported to undergo intramolecular hydroamination reactions to afford indole derivatives when using catalytic B(C6F5)3, we observed the chemo-selective N-carboxamidation. A detailed mechanistic study revealed the role of the borane catalyst in the initial activation of the indole through imine → BCl3 adduct formation. This reported methodology furthers the understanding of borane catalysts for metal-free amidation reactions of nitrogen containing heterocyclic compounds.Experimental
General procedures
Data availability
ESI includes detailed experimental procedures, NMR spectra, DFT data, and X-ray data. Crystallographic data for 1b, 1a·B(C6F5)3, 1b·B(C6F5)3, 2, 3, 4, 5a, 5i, 5t, 6a, and 10·BCl3 have been deposited in the Cambridge Crystallographic Data Centre (CCDC) under accession numbers CCDC: 2125082, 2125086, 2125087, 2125085, 2163367, 2143129, 2125083, 2163369, 2167662, 2163272 and 2163368. Information about the data that underpins the results presented in this article, including how to access them, can be found in the Cardiff University data catalogue at https://doi.org/10.17035/d.2022.0217686022.Author contributions
A. D. G. designed and optimised the synthetic method, developed the substrate scope, drafted the manuscript, and directed the project. M. G. G. and N. A. have carried out synthetic work including the reaction optimisations and substrate scope. Y. v. I. undertook X-ray measurements and solved all the crystal structures. A. A. and K. F. carried out the DFT calculations. R. L. M. and E. R. directed the project and wrote the manuscript. All authors analysed the data and proofread the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
A. D. G., M. G. G., E. R., and R. L. M. would like to acknowledge the Leverhulme Trust for funding (RPG-2020-016). N. A. acknowledges support from the Saudi Ministry of Education and the King Faisal University, Saudi Arabia. A. A. and K. F. thank the Australian Research Council (ARC) for project funding (DP180100904) and the Australian National Computational Infrastructure and the University of Tasmania for the generous allocation of computing time. R. L. M. would like to thank the EPSRC for a research Fellowship (EP/R026912/1). R. L. M. would like to thank Universities Wales for Global Wales International Research Mobility funding (UNIW/RMF-CU/08).Notes and references
- (a) L. Li, Z. Wu, H. Zhu, G. H. Robinson, Y. Xie and H. F. Schaefer, J. Am. Chem. Soc., 2020, 142, 6244–6250 CrossRef CAS PubMed; (b) R. D. Dewhurst, M. A. Légaré and H. Braunschweig, Commun. Chem., 2020, 3, 8–11 CrossRef; (c) M. A. Légaré, M. Rang, G. Bélanger-Chabot, J. I. Schweizer, I. Krummenacher, R. Bertermann, M. Arrowsmith, M. C. Holthausen and H. Braunschweig, Science, 2019, 363, 1329–1332 CrossRef PubMed; (d) P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed; (e) G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- (a) E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef; (b) K. S. Egorova and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 12150–12162 CrossRef CAS.
- J. Takaya, Chem. Sci., 2021, 12, 1964–1981 RSC.
- For selected review see: J. L. Carden, A. Dasgupta and R. L. Melen, Chem. Soc. Rev., 2020, 49, 1706–1725 RSC.
- For a selected review see: (a) A. Dasgupta, E. Richards and R. L. Melen, ACS Catal., 2022, 12, 442–452 CrossRef CAS; also see (b) X. Tao, C. G. Daniliuc, R. Knitsch, M. R. Hansen, H. Eckert, M. Lübbesmeyer, A. Studer, G. Kehr and G. Erker, Chem. Sci., 2018, 9, 8011–8018 RSC; (c) G.-Q. Chen, G. Kehr, C. G. Daniliuc, M. Bursch, S. Grimme and G. Erker, Chem. – Eur. J., 2017, 23, 4723–4729 CrossRef CAS PubMed; (d) I. Chatterjee and M. Oestreich, Angew. Chem., Int. Ed., 2015, 54, 1965–1968 CrossRef CAS PubMed; (e) T. Stahl, H. F. T. Klare and M. Oestreich, ACS Catal., 2013, 3, 1578–1587 CrossRef CAS; (f) S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2008, 47, 5997–6000 CrossRef CAS PubMed.
- For selected reviews see: (a) D. Maiti, R. Das and S. Sen, J. Org. Chem., 2021, 86, 2522–2533 CrossRef CAS PubMed; (b) K. Urbina, D. Tresp, K. Sipps and M. Szostak, Adv. Synth. Catal., 2021, 363, 2723–2739 CrossRef CAS; (c) J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618–5627 CrossRef CAS; (d) G. Broggini, E. M. Beccalli, A. Fasana and S. Gazzola, Beilstein J. Org. Chem., 2012, 8, 1730–1746 CrossRef CAS.
- For selected reviews on N-heterocycles see: (a) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed; (b) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS; (c) C.-V. T. Vo and J. W. Bode, J. Org. Chem., 2014, 79, 2809–2815 CrossRef CAS; For selected reviews on indoles see: (d) K. Urbina, D. Tresp, K. Sipps and M. Szostak, Adv. Synth. Catal., 2021, 363, 2723–2739 CrossRef CAS; (e) S. Dadashpour and S. Emami, Eur. J. Med. Chem., 2018, 150, 9–29 CrossRef CAS PubMed; (f) P. T. Singh and M. O. Singh, Mini-Rev. Med. Chem., 2018, 18, 9–25 CrossRef PubMed.
- For C3 amidation of indoles using Pd catalyst see: (a) J. Peng, L. Liu, Z. Hu, J. Huang and Q. Zhu, Chem. Commun., 2012, 48, 3772–3774 RSC; (b) Z. Hu, D. Liang, J. Zhao, J. Huang and Q. Zhu, Chem. Commun., 2012, 48, 7371–7373 RSC; Using Cu catalysts see: (c) K. Hirano, T. Satoh and M. Miura, Org. Lett., 2011, 13, 2395–2397 CrossRef CAS; (d) N. Matsuda, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2012, 77, 617–625 CrossRef CAS PubMed; Using Rh catalysts see: (e) Z. Hu, X. Tong and G. Liu, Org. Lett., 2016, 18, 2058–2061 CrossRef CAS PubMed; Using Zn catalysts see: (f) A. Pews-Davtyan and M. Beller, Org. Biomol. Chem., 2011, 9, 6331–6334 RSC; Also see: (g) G. Qiu, Q. Ding and J. Wu, Chem. Soc. Rev., 2013, 42, 5257–5269 RSC; (h) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873–2920 CrossRef CAS.
- K. Nemoto, S. Tanaka, M. Konno, S. Onozawa, M. Chiba, Y. Tanaka, Y. Sasaki, R. Okubo and T. Hattori, Tetrahedron, 2016, 72, 734–745 CrossRef CAS.
- S. Ye, Q. Ding, Z. Wang, H. Zhou and J. Wu, Org. Biomol. Chem., 2008, 6, 4406–4412 RSC.
- J. Chen, L. Hu, H. Wang, L. Liu and B. Yuan, Eur. J. Org. Chem., 2019, 2019, 3949–3954 CrossRef CAS.
- N. Pannilawithana and C. S. Yi, ACS Catal., 2020, 10, 5852–5861 CrossRef CAS.
- T. Jeong, S. Han, N. K. Mishra, S. Sharma, S. Y. Lee, J. S. Oh, J. H. Kwak, Y. H. Jung and I. S. Kim, J. Org. Chem., 2015, 80, 7243–7250 CrossRef CAS.
- W. Yan, W. Shan, Z. Hailing, C. Rui and H. Shuhua, Chin. J. Org. Chem., 2019, 39, 3567–3573 CrossRef.
- L. Mistry, K. Mapesa, T. W. Bousfield and J. E. Camp, Green Chem., 2017, 19, 2123–2128 RSC.
- E. L. Meredith, N. Mainolfi, S. Poor, Y. Qiu, K. Miranda, J. Powers, D. Liu, F. Ma, C. Solovay, C. Rao, L. Johnson, N. Ji, G. Artman, L. Hardegger, S. Hanks, S. Shen, A. Woolfenden, E. Fassbender, J. M. Sivak, Y. Zhang, D. Long, R. Cepeda, F. Liu, V. P. Hosagrahara, W. Lee, P. Tarsa, K. Anderson, J. Elliott and B. Jaffee, J. Med. Chem., 2015, 58, 9273–9285 CrossRef CAS.
- M. Nakamura, L. Ilies, S. Otsubo and E. Nakamura, Org. Lett., 2006, 8, 2803–2805 CrossRef CAS PubMed.
- S. Tussing, M. Ohland, G. Wicker, U. Flörke and J. Paradies, Dalton Trans., 2017, 46, 1539–1545 RSC.
- (a) P. Erdmann and L. Greb, Angew. Chem., Int. Ed., 2022 DOI:10.1002/anie.202114550; (b) R. J. Mayer, N. Hampel and A. R. Ofial, Chem. – Eur. J., 2021, 27, 4070–4080 CrossRef CAS; (c) I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270–271, 75–88 CrossRef CAS.
- (a) I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270–271, 75–88 CrossRef CAS; (b) R. F. Childs, D. L. Mulholland and A. Nixon, Can. J. Chem., 1982, 60, 809–812 CrossRef CAS.
- P. Erdmann, J. Leitner, J. Schwarz and L. Greb, ChemPhysChem, 2020, 21, 987–994 CrossRef CAS PubMed.
- (a) A. R. Jupp, T. C. Johnstone and D. W. Stephan, Inorg. Chem., 2018, 57, 14764–14771 CrossRef CAS PubMed; (b) A. R. Jupp, T. C. Johnstone and D. W. Stephan, Dalton Trans., 2018, 47, 7029–7035 RSC.
- B. Yuan, J. Wan, X. Guo, Y. Gong, F. Zhang, Q. Li, G. Wang, J. Chen and R. He, New J. Chem., 2020, 44, 9878–9887 RSC.
- A. Dasgupta, Y. van Ingen, M. Guerzoni, K. Farshadfar, J. M. Rawson, E. Richards, A. Ariafard and R. L. Melen, Chem. – Eur. J., 2022 DOI:10.1002/chem.202201422.
- The relatively high activation free energy of 32 kcal mol−1 for the reaction to proceed through catalytic cycle shown in Fig. 2 may be explained by the overestimation of the entropy effect for the bimolecular transformation 10·BCl3 +11 → TS10–12.
- S. Guidotti, I. Camurati, F. Focante, L. Angellini, G. Moscardi, L. Resconi, R. Leardini, D. Nanni, P. Mercandelli, A. Sironi, T. Beringhelli and D. Maggioni, J. Org. Chem., 2003, 68, 5445–5465 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy01441f |
| ‡ Both the authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |