
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTris(2,4,6-trimethoxyphenyl)phosphine – a Lewis base able to compete with phosphazene bases in catalysing oxa-Michael reactions†
Susanne M.
Fischer
 ab,
Petra
Kaschnitz
b and
Christian
Slugovc
*ab
ab,
Petra
Kaschnitz
b and
Christian
Slugovc
*ab
aChristian Doppler Laboratory for Organocatalysis in Polymerization, Stremayrgasse 9, 8010 Graz, Austria
bInstitute for Chemistry and Technology of Materials, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria. E-mail: slugovc@tugraz.at
First published on 1st September 2022
Abstract
The performance of the strong Lewis base tris(2,4,6-trimethoxyphenyl)phosphine (TTMPP) in catalysing oxa-Michael reactions is assessed and compared with other electron-rich tertiary arylphosphines and, as the benchmark, with the Brønsted base 1-tert-butyl-2,2,4,4,4-pentakis-(dimethylamino)-2λ5,4λ5-catenadi-(phosphazen) (P2-tBu). A matrix of five varyingly strong Michael acceptors and four varyingly acidic alcohols is used to evaluate the activity of the catalysts. The study demonstrated that TTMPP shows a significant superiority over other arylphosphine based Lewis bases and a similar activity to P2-tBu under highly concentrated, quasi solvent free conditions. Furthermore, the performance of TTMPP and P2-tBu is compared in the oxa-Michael polymerisation reactions of 2-hydroxyethyl acrylate (HEA) and of 1,4-butanediol diacrylate (BDDA) with diols under solvent free conditions. In the case of HEA, TTMPP is preferred over P2-tBu because the latter gave a not fully soluble polymeric product. TTMPP is the first Lewis base capable of catalysing the oxa-Michael polymerisation of diacrylates and diols, albeit P2-tBu catalysis results in higher molar masses in this polymerisation reaction. Furthermore, the performance of the catalysts under diluted conditions was assessed and the activity of TTMPP is distinctly more concentration dependent than the activity of P2-tBu. The use of the polar protic solvent t-butanol mitigates the negative impact of dilution exerted by nonpolar aprotic and polar aprotic solvents such as toluene or dimethylformamide. Finally, data are shown confirming TTMPP to have limited but still acceptable stability to oxygen for practical work in air.
Introduction
Lewis base catalysis1 with tertiary phosphines, often referred to as nucleophilic phosphine catalysis, relies on the conjugate addition of a sufficiently electron rich tertiary phosphine to an electron deficient multiple bond forming an energetically disfavoured zwitterionic species. This zwitterion can then be trapped with nucleophiles, electrophiles or a combination of both, resulting in manifold applications of this methodology in preparative chemistry.2 A special case of a reaction with an electrophile is the proton transfer of an acidic proton from another reagent, e.g. an alcohol, to the zwitterion. This reaction results in the formation of an ion pair consisting of the corresponding phosphonium cation and the corresponding base of the acidic reagent as the newly formed anion, e.g. an alkoxide (Fig. 1). In other words, the Lewis-basic phosphine is creating a zwitterionic Brønsted base, which in turn is protonated by another more acidic reagent.3 This mode of action is exploited in Michael addition chemistries where pronucleophiles, i.e. C–H acidic compounds like malonates or thiols, have to be activated upon deprotonation. Since the first demonstration in 1973,4 phosphine catalysed Michael reactions of carbon, sulphur and oxygen donors have been frequently employed in all areas of organic chemistry,2 including polymer synthesis.5 Among Michael donor reagents, oxygen based Michael donors are less frequently used, which has been rationalized by a lack of reactivity and selectivity of these reagents compared to e.g. carbon or sulphur based Michael donors.6 However, within the last decades more and more oxa-Michael reactions were reported6 and oxa-Michael polymerisations, also named hydrogen-transfer polymerisations,7 were conducted. Typically, oxa-Michael reactions are Brønsted base catalysed8,9 and the most potent catalysts for oxa-Michael polymerisations are phosphazene bases.10 Phosphazene bases are extremely strong, uncharged Brønsted bases that contain a phosphorous atom bonded to four nitrogen atoms and were disclosed by Schwesinger.11 Some of them, like 1-tert-butyl-2,2,4,4,4-pentakis-(dimethylamino)-2λ5,4λ5-catenadi-(phosphazen) (P2-tBu, Fig. 1), are stable in an ambient atmosphere making their use particularly attractive. The toxicity and the relatively high prices of phosphazene bases can be regarded as their downsides. Furthermore, their high basicity might be problematic when base labile substrates need to be used. In such cases, a Lewis base catalyst might be the better choice. Commonly used Lewis base catalysts are tertiary phosphines,3,12 amines like 4-dimethylaminopyridine13 and N-hetereocyclic carbenes.14 Highly reactive nucleophiles such as trialkylphosphines or NHCs often suffer from poor air and moisture stability or require a strong base to form from their precursors. Therefore, the quest for an air and moisture insensitive Lewis base catalyst with high activity is ongoing. Recently, we have reported a step towards this direction by showing that the electron rich tris(4-methoxyphenyl)phosphine (TMPP, Fig. 1) is more active in converting poor and intermediate Michael acceptors than triphenylphosphine (TPP).15 Regrettably, the improvement in activity was not that pronounced that TMPP could rival the performance of phosphazene bases.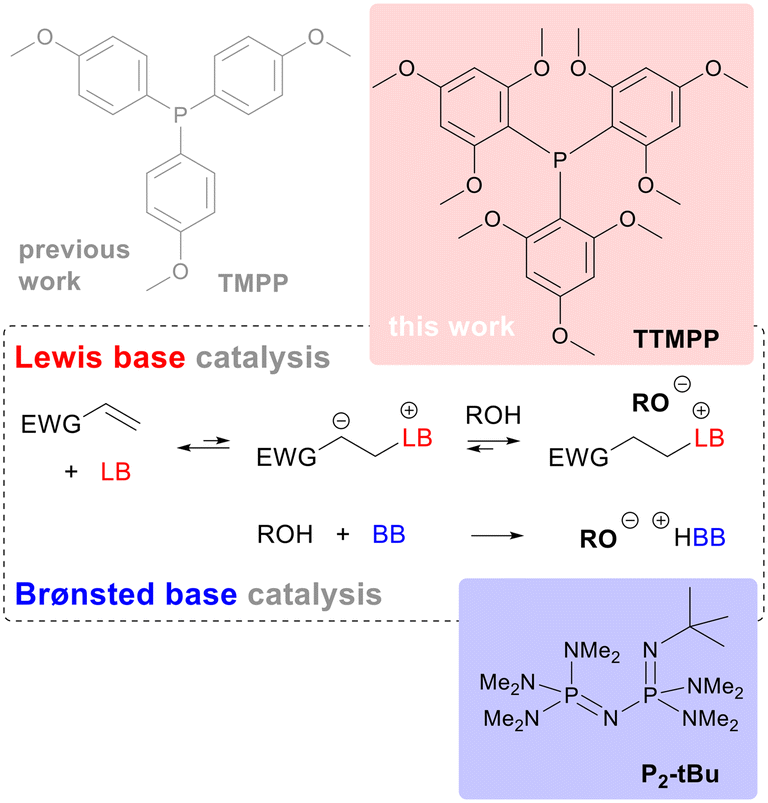 | ||
| Fig. 1 Generation of alkoxides in Lewis base or in Brønsted base catalysis showing the Lewis and Brønsted bases used in this study. | ||
Herein, we wish to report the activity of tris(2,4,6-trimethoxyphenyl)phosphine (TTMPP, Fig. 1) as a Lewis base catalyst in oxa-Michael reactions and compare its performance with that of the phosphazene base P2-tBu. Moreover, we investigate the oxidation stability of TTMPP.
TTMPP was first disclosed in 1984 by Wada and Higashizaki and has been described as a very strong nucleophile.16 It acts as a Lewis base catalyst in various reactions such as chemo- and stereoselective deacetylations, ring opening of aziridines, and Henry and sila-Morita–Baylis–Hillman reactions.17 In polymerisation reactions, the group transfer polymerisation and, in another work, the Lewis-pair catalysed polymerisation of alkyl methacrylates were demonstrated using TTMPP.18 Epoxy-phenol resins were cured with TTMPP as the catalyst19 and the reaction of TTMPP with epoxides has been described.20 Another important application of TTMPP is as a reagent for the preparation of alkali-stable phosphonium cation bearing hydroxide exchange membranes.21 Recently, also ionic liquids have been prepared from TTMPP.22 Considering that TTMPP is commercially available from major specialty chemical suppliers, the relatively small number of reports on the use of this compound as a Lewis base catalyst is surprising.
Results and discussion
The catalytic activity of TTMPP was evaluated by reacting varyingly strong Michael acceptors, namely divinyl sulfone (1), acrylonitrile (2), acrylamide (3), t-butyl acrylate (4) and N,N-dimethylacrylamide (5), with alcohols of different acidity. All reactions were carried out with 2.0 equiv. alcohol (3.0 equiv. for 1) and 1 mol% catalyst (with respect to the Michael acceptor) at 25 °C. The conversion of double bonds after 24 h was determined by 1H NMR spectroscopy. Results for TTMPP are compared with those for TPP, TMPP and the phosphazene base P2-tBu and are displayed in Fig. 2.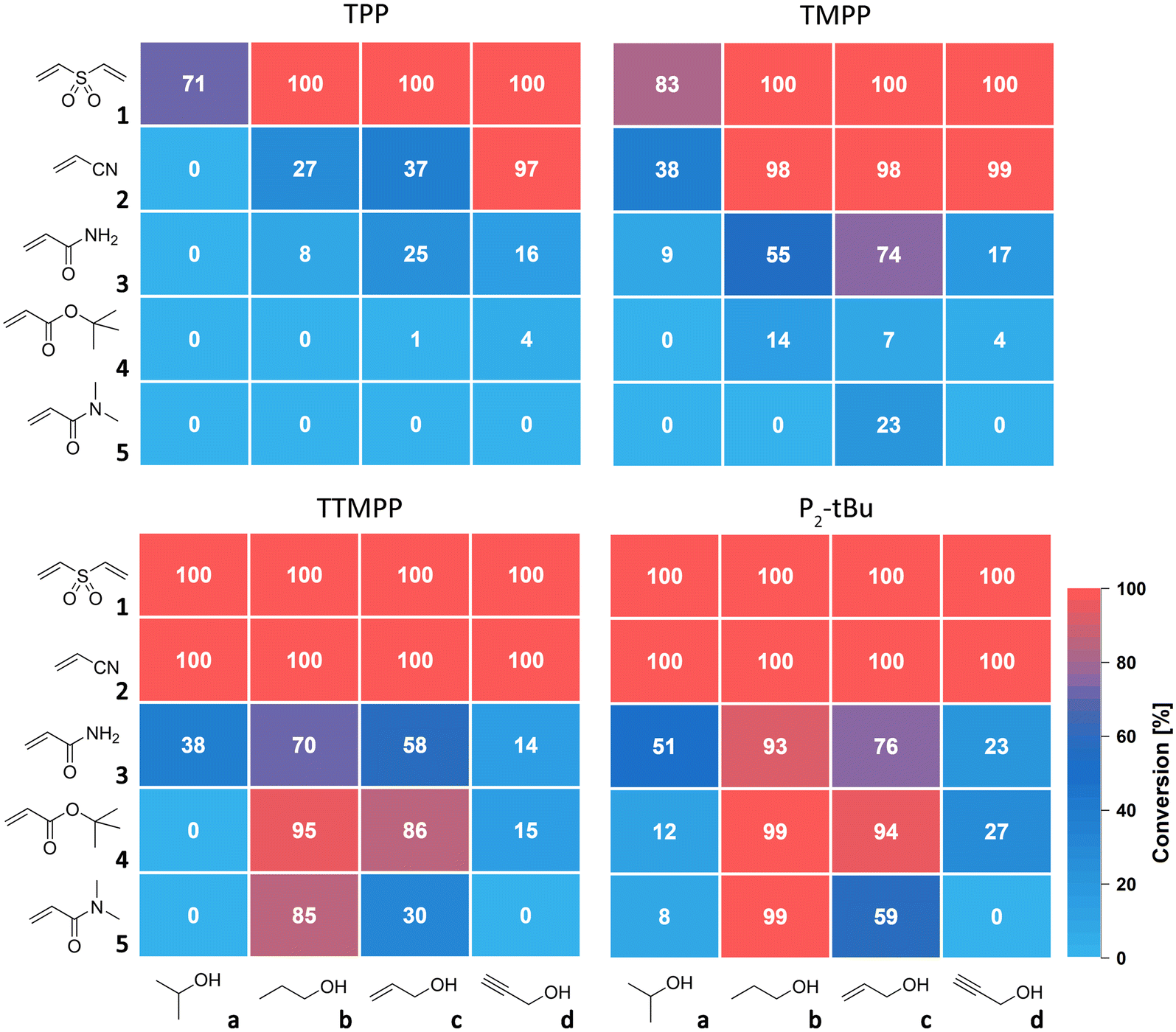 | ||
| Fig. 2 Heatmap visualization of the double bond conversion obtained in the benchmarking of different catalysts in the oxa-Michael addition of alcohols with Michael acceptors; reaction conditions: 1.0 equiv. Michael acceptor, 2.0 equiv. alcohol (3 equiv. when reacted with 1), 0.01 equiv. catalyst, solvent-free, 25 °C, 24 h. | ||
Strong Michael acceptors like divinyl sulfone (E = −18.36, for phenyl vinyl sulfone23) and acrylonitrile (E = −19.05 (ref. 23)) are fully converted by the Lewis base TTMPP and the Brønsted base P2-tBu after 24 h. In contrast, less electron rich TMPP gives (almost) full conversion with primary alcohols (b–d), but not with i-propanol (a). Least active TPP fully converts the most reactive Michael acceptor 1 using primary alcohols.15 Interestingly, acrylonitrile and i-propanol did not react within 24 h using TPP as the catalyst. Weaker Michael acceptors 3, 4 and 5 are not or only marginally converted with TPP as the catalyst. With TMPP, the situation improves, in particular for acrylamide (no E value available) but with t-butyl acrylate (E = −20.22 (ref. 23)) and N,N-dimethylacrylamide (E = −23.54 (ref. 23)) no or very low conversions with all alcohols under investigation are obtained. In contrast, TTMPP and P2-tBu with propanol and allyl alcohol give satisfactory conversions of all weak Michael acceptors, but not with i-propanol and propargyl alcohol. For t-butyl acrylate, transesterification was observed as a side reaction (see ESI†). Importantly, TTMPP and P2-tBu perform similarly in all investigated combinations and the activity of TTMPP was found to be only slightly lower than that of P2-tBu. These findings indicate that alkoxide generation by Lewis base catalysis with TTMPP under the selected conditions is similarly efficient to using the strong Brønsted base and not dependent on the acidity of the alcohol. This is in contrast to reactions of strong acceptors like 1 and 2 catalysed with the weaker Lewis bases TPP and TMPP. In these cases, the acidity of the alcohol was found to be decisive for the speed of the reaction. As particularly evident from the reaction of 2 catalysed with TPP, the least acidic alcohol a gave the lowest conversion and the most acidic alcohol d the highest conversion after 24 h (Fig. 2).15 This correlation between the alcohol's acidity and the obtained conversion can be rationalized by a more efficient trapping of the intermediately formed zwitterion by more acidic alcohols and thus a faster formation of alkoxide. When strong Michael acceptors react, all alkoxides are nucleophilic enough to form the desired Michael adducts. However, when going to weaker Michael acceptors like 3–5, the nucleophilicity of the alkoxides becomes the critical factor for the reaction to proceed. The poor conversions of 4 and 5 with alcohols a and d can be explained by the relatively poor nucleophilicity of the corresponding alkoxides. Moreover, the obtained conversions correlate with the thermodynamic stabilities of the products (see ESI,† Tables S1 and S2). n-Propoxide is the most potent oxa-Michael donor studied here. Its high nucleophilicity is rationalized by a low steric congestion and the lack of stabilization of the negative charge through electronic effects like in c or d.24
Solvents and dilution
Although the solvent-free conditions used so far are desirable, it is sometimes unavoidable to use solvents, for example, when the donor and acceptor are not soluble in each other. Therefore, the performance of TTMPP and P2-tBu (0.01 equiv.) in solution was studied by monitoring the conversion of differently concentrated solutions of acrylonitrile (1 equiv., [2] = 2.0, 1.0, 0.50 or 0.25 mol L−1) in benzene-d6 upon reaction with n-propanol (1.5 fold excess) by 1H NMR spectroscopy (Fig. 3, left). At a relatively high concentration of 2 mol L−1, the use of TTMPP results in a fast reaction (half-life period τ½ = 1 min 20 s). However, upon increasing the dilution, a pronounced deceleration of the reaction is observed. This deceleration is particularly striking when compared to the P2-tBu catalysed variants at the same dilution. At a concentration of 1 mol L−1, τ½ increased to 6 min 30 s when TTMPP is used while the P2-tBu catalysed reaction is distinctly faster (τ½ < 1 min).25 Further halving the concentration of 2 to 0.5 mol L−1 resulted in a τ½ value of 162 min for the TTMPP catalysed reaction. For P2-tBu, the Michael addition is still fast (τ½ = 1 min 20 s). In other words, all differently diluted Brønsted base catalysed reactions reached conversions >95% after 15 min reaction time ([2] = 1–0.25 mol L−1). In sharp contrast, the speed of Lewis base catalysed reactions is strongly concentration dependent. Only the 2 M reaction reaches a conversion >90% within 15 min and more diluted reactions exhibit conversions of 66% ([2] = 1 mol L−1), 12% ([2] = 0.5 mol L−1) and 0.5% ([2] = 0.25 mol L−1), at that time. These observations suggest a strong concentration dependence of the speed of alkoxide formation through protonation of the intermediate zwitterion by the alcohol (Fig. 1).26 This hypothesis is based on the results obtained in the study of different solvents at the same dilution. The same reaction was carried out in either toluene (apolar aprotic), N,N-dimethylformamide (polar aprotic) or t-butanol (polar protic) in a dilution of 1.0 and 0.5 mol L−1 (Fig. 3, right). After 15 min, a significantly higher conversion of 2 could be observed when the reaction was carried out in t-butanol (66.2% at 0.5 M) instead of toluene (19.8% at 0.5 M) or N,N-dimethylformamide (17.7% at 0.5 mol L−1). Only the desired product 2b formed in all cases and no evidence for a conjugate addition of t-butanol under these conditions could be retrieved. The superiority of t-butanol as the solvent can be rationalized by its hydrogen bonding donor ability, which activates the Michael acceptor, stabilizes anionic intermediates and promotes the hydrogen transfer to trap the zwitterionic intermediate (Fig. 1).27 Accordingly, the speed of alkoxide formation in Lewis base catalysis increases with increasing nucleophilicity of the Lewis base, increasing electrophilicity of the Michael acceptor, increasing acidity of the alcohol and decreasing dilution of the reaction mixture. The negative effect of dilution can be mitigated to some extent by using polar protic solvents (of low nucleophilicity and lower acidity than the alcohol intended for carrying out the addition reaction).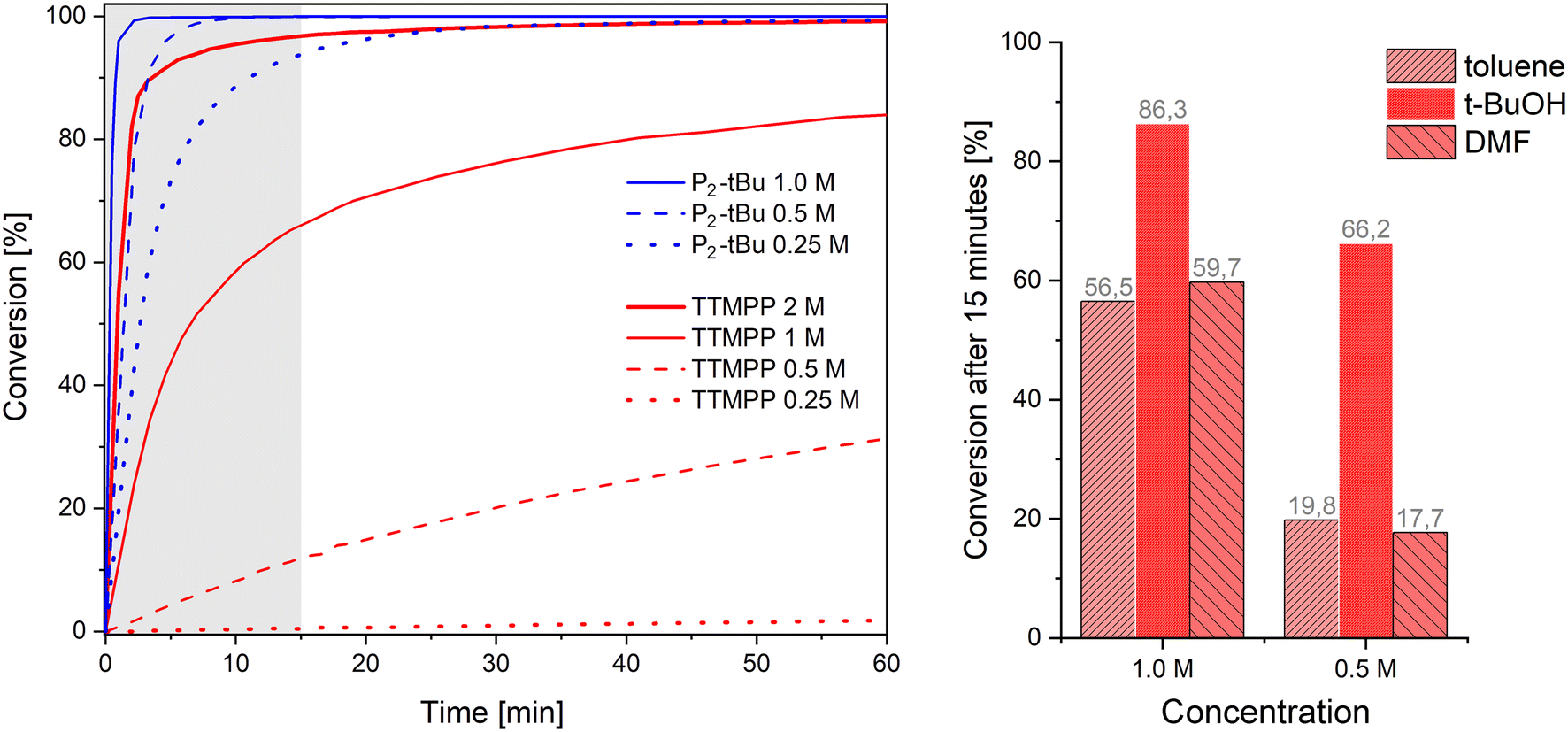 | ||
| Fig. 3 Left: conversion over time of the reaction of acrylonitrile (1 equiv.) with n-propanol (1.5 equiv.) in the presence of 1 mol% catalyst at various concentrations (solvent: benzene-d6) and room temperature; right: bar graph showing the conversion of the same reaction at 15 min vs. the concentration; toluene, t-butanol and dimethylformamide were used as the solvents. | ||
Nucleophile or base?
The above described results strongly suggest that TTMPP acts as a Lewis base catalyst and P2-tBu as a Brønsted base catalyst. Also theoretically, TTMPP should not be able to deprotonate the most acidic alcohol in this study, propargyl alcohol (pKa 14.1), because the pKa value of its conjugated acid is calculated to be 4.9.28 Nevertheless, to retrieve further evidence, the reaction of acrylonitrile with 2.0 equiv. allyl alcohol and 5 mol% catalyst (TPP, TTMPP and P2-tBu) was monitored with an IR thermal imaging camera. In Fig. 4, the temperature profiles are displayed. The respective catalyst was added to the alcohol and in the case of TPP and TTMPP, no increase of the temperature of the reaction mixture was noted. In contrast, the addition of P2-tBu to the alcohol resulted in a significant temperature rise due to the acid base reaction between c and the phosphazene base. Finally, upon addition of the Michael acceptor, a temperature surge was detected for the TTMPP and P2-tBu catalysed runs, indicating the rapid oxa-Michael addition reaction, whereas no temperature increase was observed in the case of TPP. The reaction was also studied by 31P-NMR spectroscopy revealing that the signal for TTMPP (at −65.8 ppm relative to 85% H3PO4 in DMSO-d6) is not changed when allyl alcohol was added. This disagrees with the formation of a phosphonium species since protonation of TTMPP causes a deshielding of the phosphorous atom (ESI,† Fig. S19). Only upon addition of acrylonitrile a new peak at 5.1 ppm appeared in the 31P spectrum, which increased in intensity after 24 h. This signal is tentatively attributed to the β-phosphonium species forming after conjugate addition to acrylonitrile. For comparison, chemical shifts of zwitterions of TTMPP and methacrylates were reported to be located at 2.4 and 2.9 ppm (in toluene-d8)18b and the chloromethyl phosphonium salt of TTMPP has a 31P NMR shift of 8.5 ppm (in DMSO-d6, ESI† Fig. S22). Moreover, the formation of the corresponding phosphine oxide (7.10 ppm) was observed. Accordingly, solid evidence for TTMPP being a Lewis base catalyst in these reactions is available.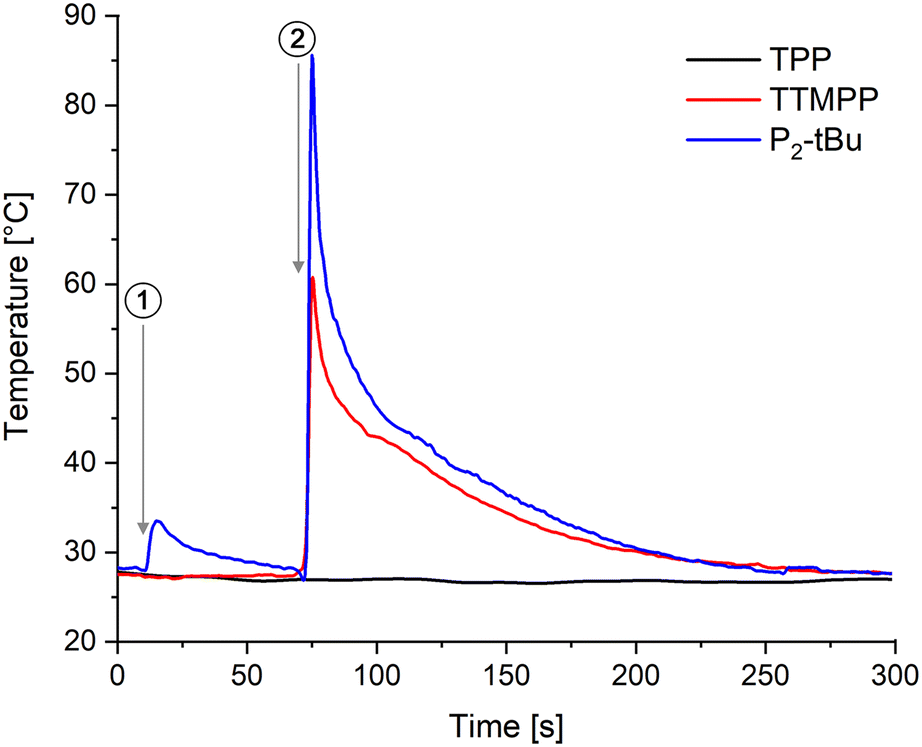 | ||
| Fig. 4 Temperature profiles of the reaction of allyl alcohol with acrylonitrile and TTP, TTMPP or P2-tBu as the catalysts. Reaction conditions: 2 equiv. allyl alcohol, 0.05 equiv. of catalyst was added at point in time 1; afterwards, at point in time 2, 1 equiv. acrylonitrile was added. | ||
Oxa-Michael polymerisation
The performance of TTMPP (0.05 equiv.) was further evaluated in the oxa-Michael polymerisation of 2-hydroxyethyl acrylate (HEA). The reaction progress was checked after 1 h and 24 h by sampling an aliquot and analyzing it by 1H NMR spectroscopy and size exclusion chromatography (SEC). The double bond conversions reached a high value of 95% already after 1 h, which further increased to 97–98% after 24 h. Using TTMPP or P2-tBu catalysis resulted in the same double bond conversions, which were higher than that obtained with TPP (1 h: 48%; 24 h: 75%) or TMPP (1 h: 80%; 24 h: 90%).15 Number average molecular masses (Mn) and dispersities (Đ) were obtained by subjecting the reaction mixture to an SEC machine after 24 h reaction time and Mn and Đ values of 1280 ± 100 g mol−1 and 2.1 ± 0.1 were determined for the TTMPP catalysed reactions (ESI,† Fig. S27). The use of phosphazene base resulted in lower Mn values of 770 ± 50 g mol−1 and in higher Đ values of 2.7 ± 0.1. However, the reaction mixture in these cases is not fully soluble in THF since a small residue was noticed in the syringe filter used during sample injection. Accordingly, the results from SEC have to be considered with precaution. Performing the polymerisation of HEA at higher temperatures resulted in partly insoluble polymers for both catalysts. For comparison, the HEA polymerisation with TMPP at room temperature resulted in similar molecular masses (Mn = 1160 g mol−1, Đ = 1.8) to TTMPP.15Next, the oxa-Michael polymerisation of 1,4-butanediol diacrylate (BDDA) and butane-1,4-diol was investigated. The performance of TTMPP and P2-tBu was compared with that of other commonly used organocatalysts, namely the triarylphosphines TPP and TMPP and amine based nucleophiles such as 1-azabicyclo[2.2.2]octane (ABCO), 4-dimethylaminopyridine (DMAP), 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU), and 1,1,3,3-tetramethylguanidine (TMG). All reactions were performed with 5 mol% catalyst loading and at room temperature without any solvent. Results are displayed in Fig. 5. Double bond conversions higher than 85% could only be observed in the polymerisations catalysed by TTMPP (86 ± 1%) and by P2-tBu (93 ± 1%) after 1 h. Other catalysts clearly fall behind with TMPP and TMG giving the best results as evident from double bond conversions of about 25 ± 2% after 1 h. This situation does not significantly change after 24 h, as double bond conversions increase only slightly in the case of TTMPP (88 ± 1%) or stay unaltered as in the case of P2-tBu. The other catalysts somewhat improved the double bond conversion over time, but in all cases no values higher than 50% were obtained. After 24 h reaction time, SEC was performed for all reaction mixtures and only TTMPP and P2-tBu catalysed reactions resulted in the formation of macromolecules. Lewis base catalysis gave poly2 at 1400 ± 150 g mol−1 and relatively small Đ values of 1.6 ± 0.1 while P2-tBu catalysis produced somewhat higher molar masses (Mn = 2500 ± 150 g mol−1, Đ = 2.0 ± 0.1). Switching to other diols, namely more acidic (Z)-butene-1,4-diol and butyne-1,4-diol, yielded the corresponding polymers poly3 and poly4. NMR spectroscopy investigations revealed significant amounts of repeating units derived from transesterification reactions and the oxa-Michael addition of released butane-1,4-diol which has already been observed before for alkyl acrylates (cf. ESI† for details).13 SEC performed after 24 h revealed Mn values of 1150 ± 50 g mol−1 (Đ = 1.6) for poly3 and 1350 ± 50 g mol−1 (Đ = 1.7) for poly4 when TTMPP catalysis is used. In the case of P2-tBu catalysis, somewhat higher values were obtained for poly3 (1400 ± 100 g mol−1, Đ = 1.8) and poly4 (1650 ± 250 g mol, Đ = 2.0). Accordingly, it can be said that TTMPP is the first Lewis base capable of catalysing the oxa-Michael polymerisation of diacrylates and diols.
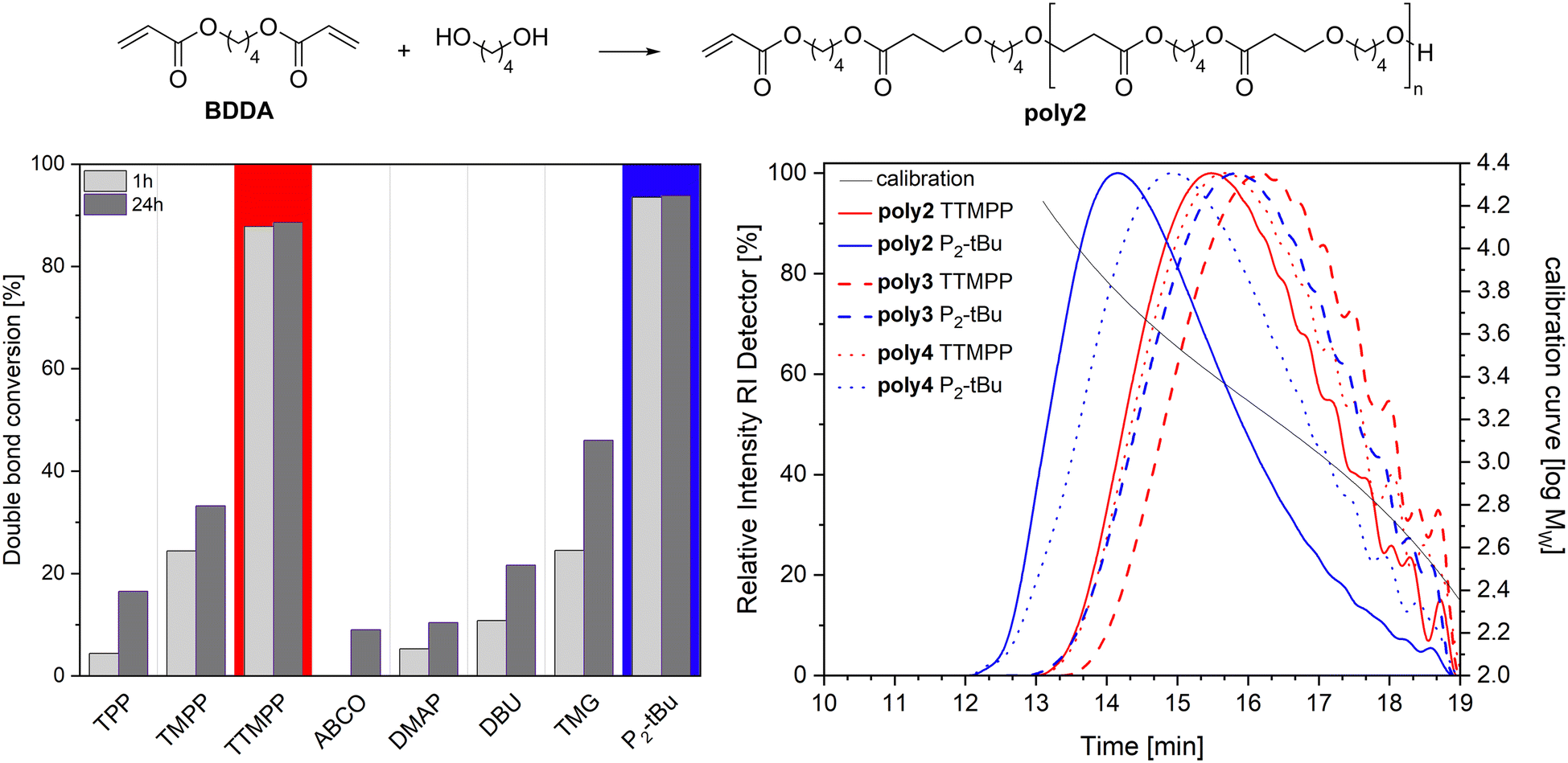 | ||
| Fig. 5 Left: double bond conversion of 1,4-butanediol diacrylate (1 equiv.) in the reaction with butane-1,4-diol (1 equiv.) in the presence of a catalyst (0.05 equiv.) after 1 h and 24 h; polymerisation performed at 25 °C; no solvent used. Right: size exclusion chromatograms of poly2 (alcohol = butane-1,4-diol), poly3 (alcohol = butene-1,4-diol), and poly4 (alcohol = butyne-1,4-diol) obtained after 24 h without any workup of the polymerisation reaction. | ||
Oxidation stability of TTMPP
Finally, the oxidation stability of TTMPP was tested. For this purpose, re-crystallized TTMPP was exposed to air for 5 d at room temperature under different conditions. The samples were either stored in the dark, under daylight or were irradiated with a 250 W tungsten-halogen lamp. The samples were dissolved in benzene-d6, DMSO-d6, chloroform-d and acetone-d6 or were stored as solids. The amount of phosphine oxide was then determined via31P-NMR spectroscopy (Fig. 6). First, the results obtained from the samples stored in the dark are discussed. In contrast to our expectations, TTMPP is relatively stable as a solid and in solution when stored in the dark. As a solid and dissolved in benzene, no phosphine oxide can be detected after 5 d. In acetone-d6, 11% phosphine oxide forms, indicating that the amount of phosphine oxide is dependent on the oxygen solubility of the solvent.29 When the samples are stored under light, oxidation stability is significantly decreased. The sample stored in acetone-d6 under daylight was determined to be fully oxidized after 5 d. In DMSO, the relative amount of phosphine oxide is with 70% slightly lower. Finally, turning to the results obtained in chloroform-d. After 5 d in the dark, 16% phosphine oxide and 13% of another species (characterized by a 31P-NMR shift of 24.9 ppm) formed. Considering the high nucleophilicity of TTMPP, a reaction with chloroform is conceivable, as TTMPP is known to react with dichloromethane to give the chloromethyl phosphonium salt within 15 min at room temperature (see ESI† for the full characterization of this species).16b Moreover, another side reaction, namely the formation of the corresponding phosphinate, could potentially occur in the presence of singlet oxygen.30 To obtain some evidence for such phosphinate species, a solution containing the singlet oxygen sensitizer Pd(II) meso-tetra(4-fluorophenyl)tetrabenzoporphyrin (Pd4F) and TTMPP in benzene-d6 was irradiated with a 250 W metal-halogen lamp for 20 min. In 31P NMR spectroscopy, a new signal with a chemical shift of 22.05 ppm was observed (approx. 5% with respect to TTMPP) and mass spectrometry revealed a newly formed compound with a mass of 565.5 g mol−1 (ESI,† S34). Both findings point to the formation of the phosphinate species, particularly because the irradiation of a solution of TTMPP without Pd4F results in the formation of 14% phosphine oxide, but no phosphinate species.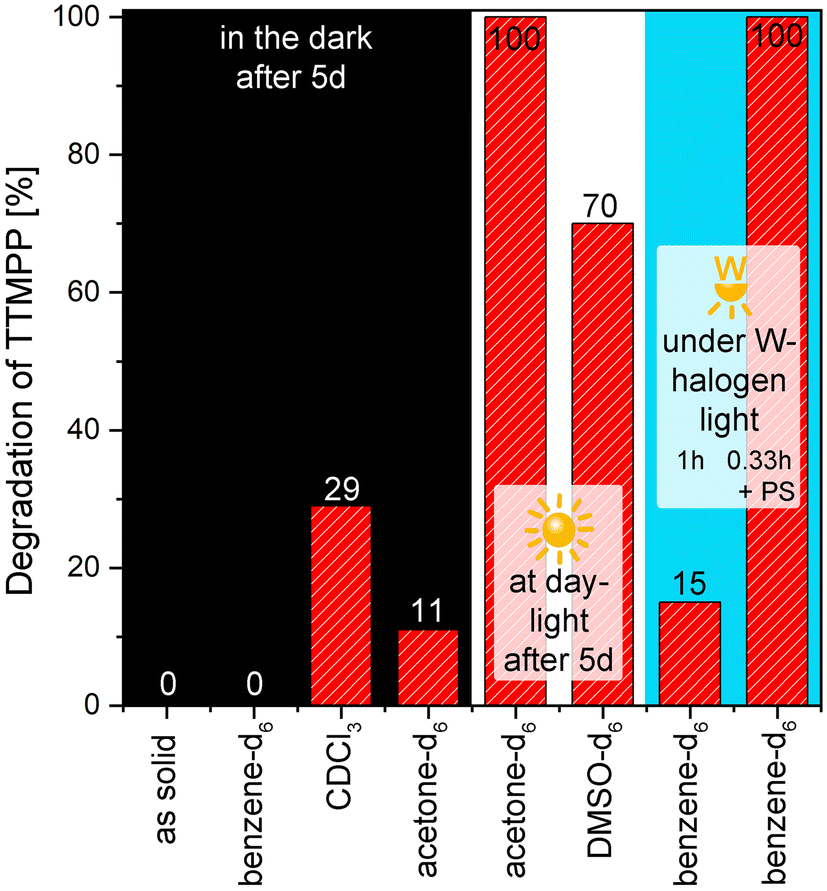 | ||
| Fig. 6 Degradation of TTMPP in the presence of air as investigated by 31P-NMR spectroscopy at room temperature after storage of TTMPP as a solid or dissolved in the solvent (x-axis) for 5 d in the dark (black background), for 5 d under daylight (white background) and upon illumination with a tungsten-halogen lamp (blue background) for 1 h and for 20 min in the presence of a photosensitizer (PS). | ||
Overall, TTMPP is not particularly sensitive towards oxidation and it can be used as a catalyst without the unconditional need to exclude oxygen. However, aliphatic halogenated solvents, in particular dichloromethane, should be avoided.
Conclusion
The activity of the Lewis base TTMPP in catalysing oxa-Michael reactions was evaluated and benchmarked against the strong Brønsted base P2-tBu. Under solvent free or highly concentrated conditions, TTMPP is only slightly less active than P2-tBu but considerably more active than other Lewis bases investigated so far. Upon dilution of the reaction mixture, the favourable activity of TTMPP is quickly lost and P2-tBu becomes clearly superior under these conditions. This phenomenon can be understood by the underlying mechanism of the Lewis base catalysed oxa-Michael reaction, which involves the formation of an energetically disfavoured intermediate zwitterion. This zwitterion either decomposes again or is trapped upon protonation by an alcohol, thus generating an alkoxide stabilized by the corresponding phosphonium cation. Under concentrated conditions, the alkoxide generation is, thanks to the high nucleophilicity of TTMPP, fast and not the rate determining step of the overall oxa-Michel reaction, while under diluted conditions the trapping of the zwitterion, i.e. alkoxide generation, becomes the rate determining step. Considering that TTMPP is fairly air-stable and exclusion of atmospheric oxygen is not mandatory for typical reactions carried out with this catalyst, this commercially available Lewis base is a powerful alternative to previously established Lewis base catalysts.Funding
Funding from the Christian Doppler Research Association (Austrian Federal Ministry for Digital and Economic Affairs and the National Foundation for Research, Technology and Development) is gratefully acknowledged.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Karin Bartl for performing SEC measurements, A. Daniel Boese for providing access to HPC resources, Sergey Borisov and Matthias Schwar for providing Pd(II) meso-tetra(4-fluorophenyl)tetrabenzoporphyrin and help with photooxidation experiments and Karin Ratzenböck for useful discussions.References
- S. E. Denmark and G. L. Beutner, Lewis Base Catalysis in Organic Synthesis, Angew. Chem., Int. Ed., 2008, 47, 1560–1638, DOI:10.1002/anie.200604943.
- (a) C. Xie, A. J. Smaligo, X.-R. Song and O. Kwon, Phosphorus-Based Catalysis, ACS Cent. Sci., 2021, 7, 536–558, DOI:10.1021/acscentsci.0c01493; (b) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Phosphine Organocatalysis, Chem. Rev., 2018, 118, 10049–10293, DOI:10.1021/acs.chemrev.8b00081; (c) H. Ni, W.-L. Chan and Y. Lu, Phosphine-Catalyzed Asymmetric Organic Reactions, Chem. Rev., 2018, 118, 9344–9411, DOI:10.1021/acs.chemrev.8b00261.
- I. C. Stewart, R. G. Bergman and F. D. Toste, Phosphine-Catalyzed Hydration and Hydroalkoxylation of Activated Olefins: Use of a Strong Nucleophile to Generate a Strong Base, J. Am. Chem. Soc., 2003, 125, 8696–8697, DOI:10.1021/ja035232n.
- D. A. White and M. M. Baizer, Catalysis of the Michael reaction by tertiary phosphines, Tetrahedron Lett., 1973, 14, 3597–3600, DOI:10.1016/S0040-4039(01)86980-X.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Michael addition reactions in macromolecular design for emerging technologies, Prog. Polym. Sci., 2006, 31, 487–531, DOI:10.1016/j.progpolymsci.2006.03.001.
- (a) C. F. Nising and S. Bräse, The oxa-Michael reaction: from recent developments to applications in natural product synthesis, Chem. Soc. Rev., 2008, 37, 1218–1228, 10.1039/B718357G; (b) C. F. Nising and S. Bräse, Recent developments in the field of oxa-Michael reactions, Chem. Soc. Rev., 2012, 41, 988–999, 10.1039/C1CS15167C; (c) Y. Wang and D.-M. Du, Recent advances in organocatalytic asymmetric oxa-Michael addition triggered cascade reactions, Org. Chem. Front., 2020, 7, 3266–3283, 10.1039/D0QO00631A.
- T. Saegusa, S. Kobayashi and Y. Kimura, Hydrogen-Transfer Polymerization of Hydroxyalkyl Acrylates, Macromolecules, 1975, 8(6), 950–952, DOI:10.1021/ma60048a051.
- (a) S. Thiyagarajan, V. Krishnakumar and C. Gunanathan, KOt Bu-Catalyzed Michael Addition Reactions Under Mild and Solvent-Free Conditions, Chem. – Asian J., 2020, 15, 518–523, DOI:10.1002/asia.201901647; (b) D. Csókás, A. X. Y. Ho, R. O. Ramabhadran and R. W. Bates, How an early or late transition state impacts the stereoselectivity of tetrahydropyran formation by intramolecular oxa-Michael addition, Org. Biomol. Chem., 2019, 17, 6293–6304, 10.1039/C9OB00750D.
- (a) H. Si, K. Wang, B. Song, A. Qin and B. Z. Tang, Organobase-catalyzed hydroxyl–yne click polymerization, Polym. Chem., 2020, 11, 2568–2575, 10.1039/D0PY00095G; (b) M. L. Tran-Do, N. Eid, C. Totée, O. Gimello and B. Améduri, Does the oxa-Michael reaction of 2-trifluoromethacrylic acid lead to fluorinated polyesters?, Polym. Chem., 2021, 12, 4508–4523, 10.1039/D1PY00685A.
- (a) H. Yang, Y. Zuo, J. Zhang, Y. Song, W. Huang, X. Xue, Q. Jiang, A. Sun and B. Jiang, Phosphazene-catalysed oxa-Michael addition click polymerization, Polym. Chem., 2018, 9, 4685–4800, 10.1039/C8PY01089G; (b) Q. Jiang, Y. L. Zhang, Y. Du, M. Tang, L. Jiang, W. Huang, H. Yang, X. Xue and B. Jiang, Preparation of hyperbranched polymers by oxa-Michael addition polymerization, Polym. Chem., 2020, 11, 1298–1306, 10.1039/C9PY01686D; (c) Q. Jiang, Y. Du, Y. Zhang, L. Zhao, L. Jiang, W. Huang, H. Yang, X. Xue and B. Jiang, pH and thermo responsive aliphatic tertiary amine chromophore hyperbranched poly(amino ether ester)s from oxa-Michael addition polymerization, J. Polym. Sci., 2020, 58, 2718–2727, DOI:10.1002/pol.20200432; (d) Q. Jiang, L. Zhao, Y. Du, W. Huang, X. Xue, H. Yang, L. Jiang, Q. Jiang and B. Jiang, Synthesis of thermoresponsive nonconjugated fluorescent branched poly(ether amide)s via oxa-Michael addition polymerization, Polym. Chem., 2022, 13, 631–639, 10.1039/D1PY01437D.
- R. Schwesinger and H. Schlemper, Peralkylated Polyaminophosphazenes – Extremely Strong, Neutral Nitrogen Bases, Angew. Chem., Int. Ed. Engl., 1987, 26, 1167–1169, DOI:10.1002/anie.198711671.
- S. Strasser and C. Slugovc, Nucleophile-mediated oxa-Michael addition reactions of divinyl sulfone - a thiol-free option for step-growth polymerisations, Catal. Sci. Technol., 2015, 5, 5091–5094, 10.1039/c5cy01527h.
- (a) K. Ratzenböck, D. Pahovnik and C. Slugovc, Step-growth polymerisation of alkyl acrylates via concomitant oxa-Michael and transesterification reactions, Polym. Chem., 2020, 11, 7476–7480, 10.1039/D0PY01271H; (b) K. Ratzenböck, M. M. U. Din, S. M. Fischer, E. Žagar, D. Pahovnik, A. D. Boese, D. Rettenwander and C. Slugovc, Water as a monomer: synthesis of an aliphatic polyethersulfone from divinyl sulfone and water, Chem. Sci., 2022, 13, 6920–6928, 10.1039/D2SC02124B; (c) S. Strasser, C. Wappl and C. Slugovc, Solvent-free macrocyclisation by nucleophile-mediated oxa-Michael addition polymerisation of divinyl sulfone and alcohols, Polym. Chem., 2017, 8, 1797–1804, 10.1039/C7PY00152E; (d) D. Edinger, H. Weber, E. Žagar, D. Pahovnik and C. Slugovc, Melt-polymerization of acrylamide initiated by nucleophiles: a route towards highly branched and amorphous polyamide 3, ACS Appl. Polym. Mater., 2021, 3, 2018–2026, DOI:10.1021/acsapm.1c00084.
- (a) E. M. Phillips, M. Riedrich and K. A. Scheidt, N-Heterocyclic Carbene-Catalyzed Conjugate Additions of Alcohols, J. Am. Chem. Soc., 2010, 132, 13179–13181, DOI:10.1021/ja1061196; (b) S. Matsuoka, S. Namera and M. Suzuki, Oxa-Michael addition polymerization of acrylates catalyzed by N-heterocyclic carbenes, Polym. Chem., 2015, 6, 294–301, 10.1039/C4PY01184H; (c) W. N. Ottou, D. Bourichon, J. Vignolle, A.-L. Wirotius, F. Robert, Y. Landais, J.-M. Sotiropoulos, K. Miqueu and D. Taton, From the N-Heterocyclic Carbene-Catalyzed Conjugate Addition of Alcohols to the Controlled Polymerization of (Meth)acrylates, Chem. – Eur. J., 2015, 21, 9447–9453, DOI:10.1002/chem.201500594.
- S. M. Fischer, S. Renner, A. D. Boese and C. Slugovc, Electron-rich triarylphosphines as nucleophilic catalysts for oxa-Michael reactions, Beilstein J. Org. Chem., 2021, 17, 1689–1697, DOI:10.3762/bjoc.17.117.
- (a) M. Wada and S. Higashizaki, A highly basic triphenylphosphine, [2,4,6-(MeO)3C6H2]3P, J. Chem. Soc., Chem. Commun., 1984, 482–483, 10.1039/C39840000482; (b) K. R. Dunbar and S. C. Haefner, Synthesis and properties of tris(2,4,6-trimethoxyphenyl)phosphine and tris(2,4,6-trimethoxyphenyl)phosphine oxide, Polyhedron, 1994, 13, 727–736, DOI:10.1016/S0277-5387(00)81676-9.
- (a) K. Yoshimoto, H. Kawabata, N. Nakamichi and M. Hayashi, Tris(2,4,6-trimethoxyphenyl)phosphine (TTMPP): A Novel Catalyst for Selective Deacetylation, Chem. Lett., 2001, 30, 934–935, DOI:10.1246/cl.2001.934; (b) S. Matsukawa and K. Tsukamoto, TTMPP: An efficient organocatalyst in the ring-opening of aziridines with silylated nucleophiles, Org. Biomol. Chem., 2009, 7, 3792–3796, 10.1039/B908322G; (c) A. Trofimov and V. Gevorgyan, Sila-Morita−Baylis−Hillman Reaction of Arylvinyl Ketones: Overcoming the Dimerization Problem, Org. Lett., 2009, 11, 253–255, DOI:10.1021/ol8026522; (d) J. A. Weeden and J. D. Chisholm, Phosphine-catalyzed nitroaldol reactions, Tetrahedron Lett., 2006, 47, 9313–9316, DOI:10.1016/j.tetlet.2006.10.107; (e) S. Chuprakov, D. A. Malyshev, A. Trofimov and V. Gevorgyan, Sila Morita−Baylis−Hillman Reaction of Cyclopropenes, J. Am. Chem. Soc., 2007, 129, 14868–14869, DOI:10.1021/ja077437s; (f) H. Kawabata and M. Hayashi, Lewis base-catalyzed transformation of α,β-unsaturated aldehydes to saturated carboxylic acids, esters, and amides, Tetrahedron Lett., 2002, 43, 5645–5647, DOI:10.1016/S0040-4039(02)01133-4; (g) N. Hirata and M. Hayashi, Nitroaldol reaction catalyzed by tris(2,4,6-trimethoxyphenyl)phosphine (TTMPP), Synth. Commun., 2007, 37, 1653–1657, DOI:10.1080/00397910701263833; (h) S. Matsukawa and I. Sekine, TTMPP-catalyzed addition of alkynes using trimethylsilylacetylenes, Synth. Commun., 2009, 39, 1718–1721, DOI:10.1080/00397910802585878; (i) S. Matsukawa and E. Kitazaki, Catalytic cyanomethylation of carbonyl compounds and imines with highly basic phosphine, Tetrahedron Lett., 2008, 49, 2982–2984, DOI:10.1016/j.tetlet.2008.02.155.
- (a) M. Fevre, J. Vignolle, V. Heroguez and D. Taton, Tris(2,4,6-trimethoxyphenyl)phosphine (TTMPP) as Potent Organocatalyst for Group Transfer Polymerization of Alkyl (Meth)acrylates, Macromolecules, 2012, 45, 7711–7718, DOI:10.1021/ma301412z; (b) W. N. Ottou, E. Conde-Mendizabal, A. Pascual, A.-L. Wirotius, D. Bourichon, J. Vignolle, F. Robert, Y. Landais, J.-M. Sotiropoulos, K. Miqueu and D. Taton, Organic Lewis Pairs Based on Phosphine and Electrophilic Silane for the Direct and Controlled Polymerization of Methyl Methacrylate: Experimental and Theoretical Investigations, Macromolecules, 2017, 50, 762–774, DOI:10.1021/acs.macromol.6b02205.
- C. S. Tyberg, P. Shih, K. N. E. Verghese, A. C. Loos, J. J. Lesko and J. S. Riffle, Latent nucleophilic initiators for melt processing phenolic–epoxy matrix composites, Polymer, 2000, 41, 9033–9048, DOI:10.1016/S0032-3861(00)00298-6.
- M. Wada and A. Tsuboi, Reactions of tris(2,6-dimethoxyphenyl)phosphine with epoxides, J. Chem. Soc., Perkin Trans. 1, 1987, 151–154, 10.1039/P19870000151.
- S. Gu, R. Cai, T. Luo, Z. Chen, M. Sun, Y. Liu, G. He and Y. Yan, A soluble and highly conductive ionomer for high-performance hydroxide exchange membrane fuel cells, Angew. Chem., Int. Ed., 2009, 48, 6499–6502, DOI:10.1002/anie.200806299.
- J. Chen, M. Li, M. Li, X. Lin and T. Qiu, Self-Solidifying Quaternary Phosphonium-Containing Ionic Liquids as Efficient and Reusable Catalysts for Biodiesel Production, ACS Sustainable Chem. Eng., 2020, 8, 6956–6963, DOI:10.1021/acssuschemeng.9b07432.
- D. S. Allgäuer, H. Jangra, H. Asahara, Z. Li, Q. Chen, H. Zipse, A. R. Ofial and H. Mayr, Quantification and Theoretical Analysis of the Electrophilicities of Michael Acceptors, J. Am. Chem. Soc., 2017, 139, 13318–13329, DOI:10.1021/jacs.7b05106.
- (a) X. Chen and J. I. Brauman, Hydrogen Bonding Lowers Intrinsic Nucleophilicity of Solvated Nucleophiles, J. Am. Chem. Soc., 2008, 130, 15038–15046, DOI:10.1021/ja802814a; (b) T. B. Phan and H. Mayr, Comparison of the nucleophilicities of alcohols and alkoxides, Can. J. Chem., 2005, 83, 1554–1560, DOI:10.1139/v05-170.
- The P2-tBu catalysed reaction at [2] = 2 M is too fast to be measured by this technique.
- S. Huang, K. Kim, G. M. Musgrave, M. Sharp, J. Sinha, J. W. Stansbury, C. B. Musgrave and C. N. Bowman, Determining Michael acceptor reactivity from kinetic, mechanistic, and computational analysis for the base-catalyzed thiol-Michael reaction, Polym. Chem., 2021, 12, 36193628, 10.1039/D1PY00363A.
- (a) S. Huang, K. Kim, G. M. Musgrave, M. Sharp, J. Sinha, J. W. Stansbury, C. B. Musgrave and C. N. Bowman, Determining Michael acceptor reactivity from kinetic, mechanistic, and computational analysis for the base-catalyzed thiol-Michael reaction, Polym. Chem., 2021, 12, 36193628, 10.1039/D1PY00363A; (b) A. Fedotova, E. Kondrashov, J. Legros, J. Maddaluno and A. Y. Rulev, Solvent effects in the aza-Michael addition of anilines, C. R. Chim., 2018, 21, 639–643, DOI:10.1016/j.crci.2018.03.006; (c) K. De, J. Legros, B. Crousse and D. Bonnet-Delpon, Solvent-Promoted and -Controlled Aza-Michael Reaction with Aromatic Amines, J. Org. Chem., 2009, 74, 6260–6265, DOI:10.1021/jo9012699; (d) A. Genest, S. Binauld, E. Pouget, F. Ganachaud, E. Fleury and D. Portinha, Going beyond the barriers of aza-Michael reactions: controlling the selectivity of acrylates towards primary amino-PDMS, Polym. Chem., 2017, 8, 624–630, 10.1039/C6PY01802E.
- Calculated using the pKa prediction platform available at https://pka.luo-group.com see: Q. Yang, Y. Li, J.-D. Yang, Y. Liu, L. Zhang, S. Luo and J.-P. Cheng, Holistic Prediction of the pKa in Diverse Solvents Based on a Machine-Learning Approach, Angew. Chem., Int. Ed., 2020, 59, 19282–19291, DOI:10.1002/anie.202008528.
- T. Sato, Y. Hamada, M. Sumikawa, S. Araki and H. Yamamoto, Solubility of Oxygen in Organic Solvents and Calculation of the Hansen Solubility Parameters of Oxygen, Ind. Eng. Chem. Res., 2014, 53, 19331–19337, DOI:10.1021/ie502386t.
- D. Zhang, B. Ye, D. G. Ho, R. Gao and M. Selke, Chemistry of singlet oxygen with arylphosphines, Tetrahedron, 2006, 62, 10729–10733, DOI:10.1016/j.tet.2006.07.112.
Footnote |
| † Electronic supplementary information (ESI) available: Containing experimental details, characterization of oxa-Michael adducts, polymers and TTMPP derived compounds and computational results. See DOI: https://doi.org/10.1039/d2cy01335e |
| This journal is © The Royal Society of Chemistry 2022 |