
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of SO2 poisoning on undoped and doped Mn-based catalysts for selective catalytic reduction of NO†
Javier
Ruiz-Martínez
 *a,
Lieven E.
Gevers
a,
Linga R.
Enakonda
a,
Ameen
Shahid
a and
Fei
Wen
b
*a,
Lieven E.
Gevers
a,
Linga R.
Enakonda
a,
Ameen
Shahid
a and
Fei
Wen
b
aKing Abdullah University of Science and Technology, KAUST Catalysis Center, Catalysis Nanomaterials and Spectroscopy (CNS), Thuwal 23955, Saudi Arabia. E-mail: javier.ruizmartinez@kaust.edu.sa
bUmicore AG & Co. KG, Rodenbachen Chaussee 4, 63457 Hanau-Wolfgang, Germany
First published on 29th September 2022
Abstract
In this work, the poisoning effect of SO2 was investigated in binary MnTi and ternary MnCeTi mixed oxides for the NH3-SCR reaction under conditions relevant for mobile applications. For the binary MnTi sample, catalytic activity increases up to 250 °C, and then drops due to the oxidation of ammonia to NOx. The addition of Ce decreases the catalytic activity at 150 °C but widens the optimal operational temperature and reaches high conversion at 350 °C. Upon performing activity test with 100 ppm of SO2 in the gas stream, catalytic activity drastically decreases in all catalyst samples. The shape of the deactivation curve and SO2 concentrations at the outlet of the reactor suggest a strong adsorption and poisoning of SO2 on all the catalysts. Although samples containing large amounts of Ce display a better SO2 tolerance, this is insufficient to be considered for practical applications. Deactivated samples were investigated by a wide range of characterization tools. N2 physisorption measurements reveal a drop in the surface area that could partially explain catalyst deactivation. TGA reveals the absence of (NH4)2SO4 on the deactivated samples and suggests the formation of Mn and Ce sulfates on the catalyst surface. XPS results confirm the formation of MnSO4 and also show a decrease in the Mn and Ce oxidation states. Analysis of the redox function by catalytic NO oxidation and H2-TPR experiments shows a strong loss of redox function upon SO2 deactivation, which could explain the decrease of NH3-SCR catalytic activity. Upon unraveling the effect and cause of deactivation, a doping study was performed. As in the binary MnTi and ternary MnCeTi, catalytic activity strongly decreases upon the introduction of SO2 in the gas stream. None of the dopants investigated was able to suppress SO2 deactivation, which suggest that other dopants or strategies should be pursued to commercialize Mn-based catalysts for low-temperature applications.
1. Introduction
Selective catalytic reduction (SCR) with ammonia is one of the most promising technologies to abate nitric oxides (NOx, which includes NO and NO2) under lean-burning conditions encountered in mobile lean-burn engines and stationary applications.1–3 Currently, there is a search for more efficient SCR catalysts motivated by the ever-increasing demand of stringent global legislation for improving air quality.4 V2O5–WO3/TiO2 catalysts provide high NOx removal efficiency (over 90%) at high gas-hourly space velocities (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–90000 h−1) and between 250 and 400 °C.5–8 Unfortunately, the emission of toxic V oxides during operation has restricted such catalysts for mobile applications. Cu-CHA zeolites (Cu-SSZ-13 and Cu-SAPO-34) have been revolutionary in SCR applications due to their high activity and broader operational temperature (200–450 °C).9–11 Still, great advances in engine technology have brought more efficient combustion engines and consequently lower exhaust temperatures, which makes NOx more refractory to abate, even for Cu-zeolites. Due to this situation, low-temperature (below 200 °C) catalysts are required. In this field, Mn-based catalysts have exhibited optimal NH3-SCR activity due to their strong redox functionality. Typically, Mn-oxide catalysts contain other metal oxides that serve as supports and modulate their catalytic properties. Although Mn-oxide catalysts have shown their superior performance at low-temperature NH3-SCR, they are not commercialized in automotive applications. One of the main reasons is their strong intolerance to SO2,12–15 which is due to the combustion of sulfur contaminants in fuels and lubricants. Although the SO2 concentration could be as low as a few ppm, those are sufficient to effectively poison Mn-oxide catalyst's active sites.
000–90000 h−1) and between 250 and 400 °C.5–8 Unfortunately, the emission of toxic V oxides during operation has restricted such catalysts for mobile applications. Cu-CHA zeolites (Cu-SSZ-13 and Cu-SAPO-34) have been revolutionary in SCR applications due to their high activity and broader operational temperature (200–450 °C).9–11 Still, great advances in engine technology have brought more efficient combustion engines and consequently lower exhaust temperatures, which makes NOx more refractory to abate, even for Cu-zeolites. Due to this situation, low-temperature (below 200 °C) catalysts are required. In this field, Mn-based catalysts have exhibited optimal NH3-SCR activity due to their strong redox functionality. Typically, Mn-oxide catalysts contain other metal oxides that serve as supports and modulate their catalytic properties. Although Mn-oxide catalysts have shown their superior performance at low-temperature NH3-SCR, they are not commercialized in automotive applications. One of the main reasons is their strong intolerance to SO2,12–15 which is due to the combustion of sulfur contaminants in fuels and lubricants. Although the SO2 concentration could be as low as a few ppm, those are sufficient to effectively poison Mn-oxide catalyst's active sites.
Several studies have discussed the detrimental effect of SO2 on Mn-based catalysts and the deactivation effect has been explained by two main reasons. The first one is due to the reaction of SO2 and NH3 and H2O to form (NH4)2SO4 and NH4HSO4, which deposit on the catalyst surface and have a high decomposition temperature (230 and 350 °C), respectively.16,17 The second one is related to the formation of metal sulfates that might modify or deactivate the active sites. Bliek et al. investigated the SO2 deactivation of MnOx/Al2O3 catalysts by IR spectroscopy and found that the formation of MnSO4 is the main source of deactivation.18 There are reports describing the regeneration of SO2-poisoned catalysts can be efficient by water washing.19 Unfortunately, this approach is unfeasible for in situ catalyst regeneration in a real mobile application. To solve this deactivation process, catalyst formulations were modified with Fe,20–23 Co,24,25 Ni,26,27 W28 and Sm29,30 to improve their tolerance to SO2 and H2O. For example, Wang et al. shown that Fe and Co co-doping reduces the adsorption of SO2 on Mn–Ce/TiO2 catalysts.23 Sm-doped mixed oxide catalysts were shown to exhibit considerably enhanced catalytic activity and SO2 tolerance. Sun et al.29 found that the electron transfer from Sm2+ to Mn4+ restrained the electron transfer from SO2 to Mn4+, thereby suppressing the formation of SO3 and sulfate species. Meng et al.31 found that a SmOx–MnOx catalyst had an ideal SO2 tolerance at low temperature, and they suggested that the incorporation of Sm could induce the formation of bulk-like sulfates on the Sm sites and weaken the influence of SO2 on the Mn sites. However, the origin of the effect of Sm doping into the catalysts on the SO2 tolerance is still unclear, and an in-depth study is still needed.
In this work, we aimed to understand the poisoning effect of SO2 on a MnTi binary mixed oxide. Then, the role of Ce in catalyst deactivation was investigated in MnCeTi tertiary mixed oxides with different amounts of Ce. For this purpose, NH3-SCR reactions were performed by adding an amount of SO2 equivalent to the total amount of S that a catalyst will be exposed in the exhaust of a real internal combustion engine after 200000 km. The activity recovery was assessed by a regeneration step under high temperature and NH3-SCR conditions. These conditions were applied to simulate a theoretical regeneration in a real mobile application. After that, the catalysts were promoted by a wide range of metal dopants to evaluate the effect on poisoning and on catalyst regeneration.
2. Experimental
A series of individual, binary and ternary materials with different molar concentrations were prepared. Titanium(IV) sulfate solution (Ti(SO4)2, Pfaltz & Bauer., 30% in H2SO4), cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O, Sigma-Aldrich, 99.999% trace metals basis), manganese(II) nitrate hydrate (Mn(NO3)2·xH2O, Sigma-Aldrich, 99.999% trace metals basis), and ammonium hydroxide (NH4OH, Alfa Aesar, ACS grade, 28.0–30.0%) were used as received, without further purification. The binary MnTi and ternary MnCeTi were prepared by a controlled co-precipitation method described in our previous publication.32 The method was designed to precipitate all metals at the same pH level to obtain a homogeneously well-mixed metal oxide system.For the doping of the ternary MnCeTi catalysts, precipitation and impregnation methods were explored. In the precipitation method, a ternary MnCeTi was first suspended in an aqueous solution (50 mL). Then, 50 ml aqueous solution with the dopants were added slowly to the MnCeTi suspension, then the suspension was stirred for 30 min. After that, an aqueous ammonium hydroxide solution was added dropwise to the suspension under continuous stirring until a pH of 10.5 was reached. This was performed to precipitate all metal precursors on the catalyst surface. Afterwards, the suspension was stirred for another 1 h and then filtered. The solid product was washed with water 5–6 times, then dried in a hot air-oven overnight. For the impregnation method, 3 g of ternary MnCeTi sample were suspended in 50 ml of water. After 30 min of stirring, 50 ml of an aqueous solution of the dopant precursor were slowly added to the catalyst suspension. After stirring for 24 h, the suspension was evaporated and the solid was dried overnight. All catalysts were calcined at 500 °C for 6 h.
For the TG/DTA-MS analysis, 10–20 mg of sample was used. The samples were first kept at 30 °C for 30 min to stabilize the mass-loss signal. Then, the sample was heated with a ramp of 10 °C min−1 to 950 °C at 10 ml min−1 of air. The analysis of the gases was performed by mass spectrometry by following the signal from H2O (m/e = 18), NO (m/e = 30), N2O (m/e = 44) and SO2 (m/e = 48).
X-ray diffraction patterns were obtained using a Bruker D8 Advance A25 diffractometer in the Bragg–Brentano geometry with a Cu Kα,β radiation source operated at 40 kV and 40 mA. β radiation was filtered out with a Ni plate. The diffractograms were measured with a step size of 0.05° in the 2θ range of 10–80°.
Nitrogen adsorption and desorption isotherms of the samples were measured at 77 K using a Micromeritics ASAP-2420 surface area and porosity analyzer instrument. The samples were previously evacuated at 300 °C for 3 hours. Specific surface areas and pore size distribution were calculated according to the multi-point Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods, respectively. From the adsorption data, total pore volumes were estimated at P/P0 = 0.99. The elemental compositions (Mn, Ce, Ti) of the samples were determined using an inductively coupled plasma spectrometer (Model 8900, Agilent Technologies). The samples were dissolved in HF and HCl.
H2-TPR experiments were performed on an Autochem 2950 instrument equipped with a thermal conductivity detector and a dry ice/isopropanol cold trap (−78 °C) to remove H2O generated during the reduction. All catalysts (100 mg) were pretreated in a U-shaped quartz tubular microreactor in a flow of Ar at 250 °C for 2 h to yield a clean surface, and then cooled down to 40 °C temperature. Then, the temperature was raised from 40 to 1000 °C at a rate of 10 °C min−1 under a flow of 10 vol% H2 (90 vol% Ar).
Temperature-programmed desorption of ammonia (NH3-TPD) was used to determine the acidity of the fresh samples and samples deactivated by SO2. The experiments were performed in a fixed bed quartz tube reactor. Prior to the measurement, the samples were first pretreated at 500 °C under a N2 flow. The reactor was cooled down at 100 °C and the samples were saturated with 1050 ppm NH3 for 30 min. The samples were flushed with N2 for 30 min at room temperature, and then the temperature was increased to 500 °C at a rate of 10 K min−1. The outlet gas composition (NH3, NO, NO2, N2O) was monitored by using a MultiGas™ 2030 FTIR continuous gas analyzer.
For the XPS study, a Kratos Axis Ultra X-ray photoelectron spectrometer equipped with a monochromatic Al Kα source was used to determine the surface composition and chemical states of the samples. The C 1s signal for adventitious carbon (284.8 eV) was used to calibrate the sample energy. The chemical states of manganese and cerium in the catalysts were determined by peak modeling in CasaXPS software. To model the Mn 2p3/2 peaks of the catalysts, pure MnO, Mn2O3, and MnO2 samples were used as references. Manganese(IV) oxide (99.997% – metals basis) was acquired from Alfa Aesar (Fisher US), manganese(III) oxide (99.9% – trace metals basis) was acquired from Sigma Aldrich, and manganese(II) oxide (99.99% – trace metal basis) was acquired from Acros Organics (VWR). The fitting parameter data (FWHM and peak positions) obtained from the peak modeling of the standard samples were used for the calculation of the chemical state of manganese in our catalysts. More details about the fitting procedure and measurements can be obtained in our previous publication.32
The catalytic activity measurements of the catalysts in the NH3-SCR reaction were carried out in a fixed bed quartz tube reactor loaded with 0.5 ml of sample (PID Eng&Tech). Before loading, the catalysts were pressed into pellets, crushed and sieved to obtain a fraction between 500 and 710 μm. The application-relevant catalyst particle size, space velocity and gas composition were applied. The inlet NOx composition contained no NO2 to avoid higher conversions coming from the “fast SCR” mechanism when this gas is present. The total flow rate was maintained at 1000 ml min−1, and the reaction condition corresponds to a GHSV of 120000 hr−1. The flow rate of gases was controlled using Bronkhorst mass flow controllers. A controlled evaporation and mixing (CEM) system from Bronkhorst was used for evaporation to achieve the target steam content in the gas feed before entering the reactor. The inlet gas stream contained 450 ppm NO, 500 ppm NH3, 5% O2, 5% H2O and N2 balance. A MultiGas™ 2030 FTIR continuous gas analyzer was used to analyze the inlet and outlet gas compositions (NO, NO2, NH3, N2O). The catalytic tests were performed in the temperature range of 150–500 °C (with an interval of 50 °C) at ambient pressure. NO conversion and N2O selectivities were calculated under steady-state conditions. The SCR activity (NO conversion) and N2 selectivity are calculated as follows:
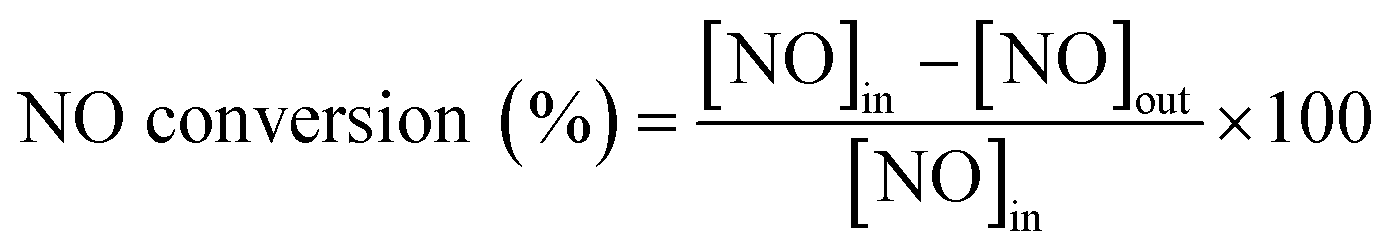 | (1) |
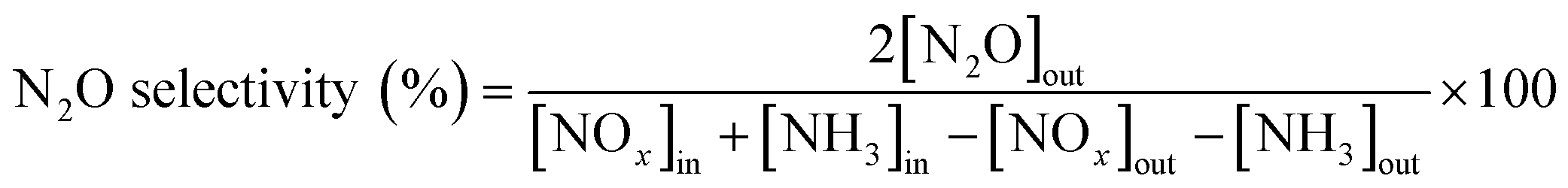 | (2) |
The NO oxidation reaction was performed in the same fashion as the NH3-SCR. In this case, 250 ppm of NO and 5% O2 were fed to the reactor and balanced with N2. In the experiments under wet conditions, 5% of vapor H2O was included in the inlet gas stream.
3. Results
Activity measurements
NOx conversion of the MnTi binary and MnCeTi ternary mixed oxides as a function of the reaction temperature is presented in Fig. 1. The data at 150 °C were determined to be free of external and internal mass transport limitations (more details in the ESI†). In general, conversion increases until reaching a maximum, and then decreases with the reaction temperature. NH3 conversion (ESI† Fig. S1) follows the same trend as NOx conversion, but monotonically increases when the conversion drops. This mismatch in conversions is related to the undesired combustion of ammonia, which leads to the formation of additional NOx in the gas stream. The binary MnTi catalyst is more active at low temperatures but lacks N2 selectivity, showing a high concentration of N2O at the outlet (see Fig. 1b). The addition of Ce has several effects on catalytic performance. First, low-temperature activity decreases suggesting that Mn species are less active due to the interaction with Ce, as we previously reported.32 Second, the addition of Ce widens the operational window, as was also reported previously,3 and finally promotes N2 selectivity.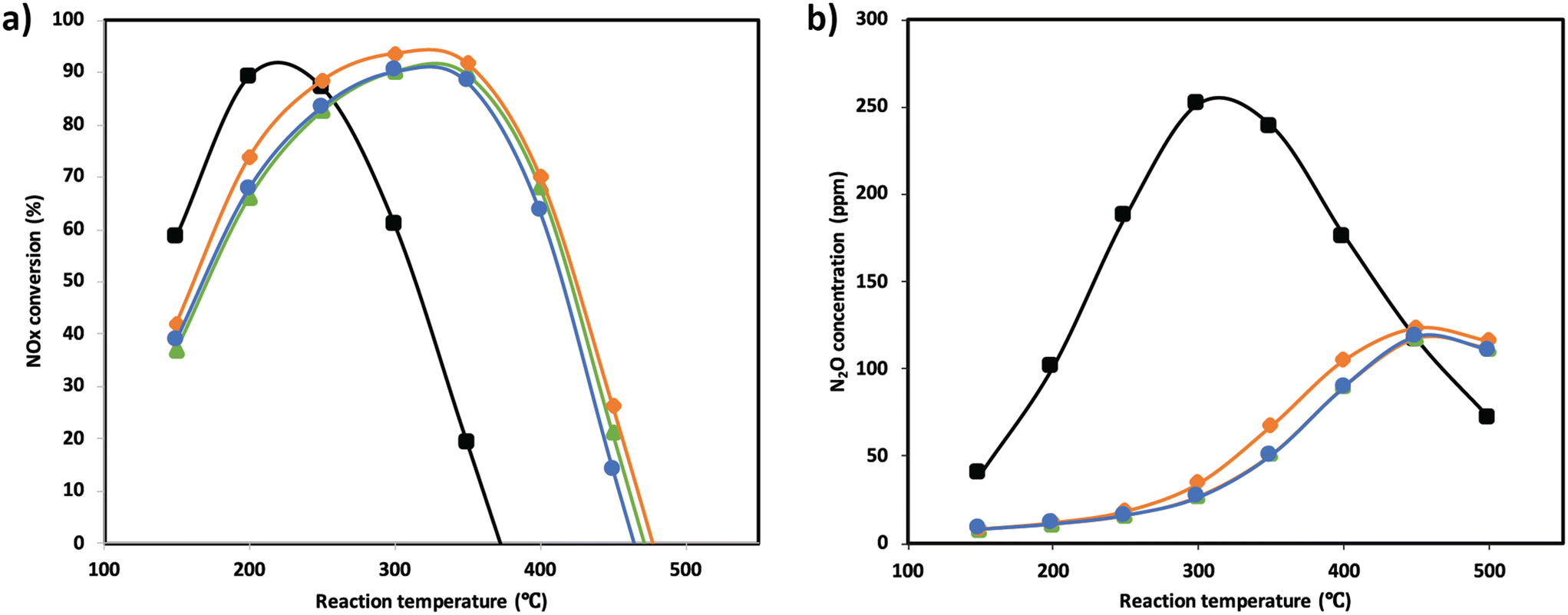 | ||
| Fig. 1 NOx conversion (a) and N2O concentration (b) as a function of the reaction temperature for Mn0.35Ce0.00Ti0.65 (black), Mn0.37Ce0.04Ti0.59 (orange), Mn0.33Ce0.07Ti0.60 (green), and Mn0.30Ce0.19Ti0.51 (blue). | ||
In order to understand the effect of SO2 poisoning, experiments were performed where NOx conversion was measured before and during SO2 poisoning under NH3-SCR conditions. Fig. 2 shows the NH3-SCR conversion at 250 °C before and during SO2 deactivation. When 100 ppm SO2 is introduced in the stream, the conversion of all samples drastically decreased from ≈90% to ≈20% in 4 hours. Interestingly, the sample with the largest amount of Ce (Mn0.30Ce0.19Ti0.51) displays a better sulfur tolerance. The S-type shape of the deactivation curves suggest that SO2 is adsorbing strongly on the catalyst surface and deactivating the catalyst bed from the inlet to the outlet. Deactivation rates were consistently compared using second-order deactivation kinetics by applying the equation da/dt = −kD·a2 where a is the normalized NOx activity. Deactivation rate constants, in Fig. 2a, are more pronounced for the binary MnTi catalyst and the samples with a small amount of Ce (Mn0.37Ce0.04Ti0.59). Then, deactivation constants drop when the amount of Ce increases, suggesting a lessening of deactivation with increasing Ce content. After removal of SO2 from the feed gas, NOx conversion is marginally recovered indicating that SO2 deactivation is mostly irreversible.
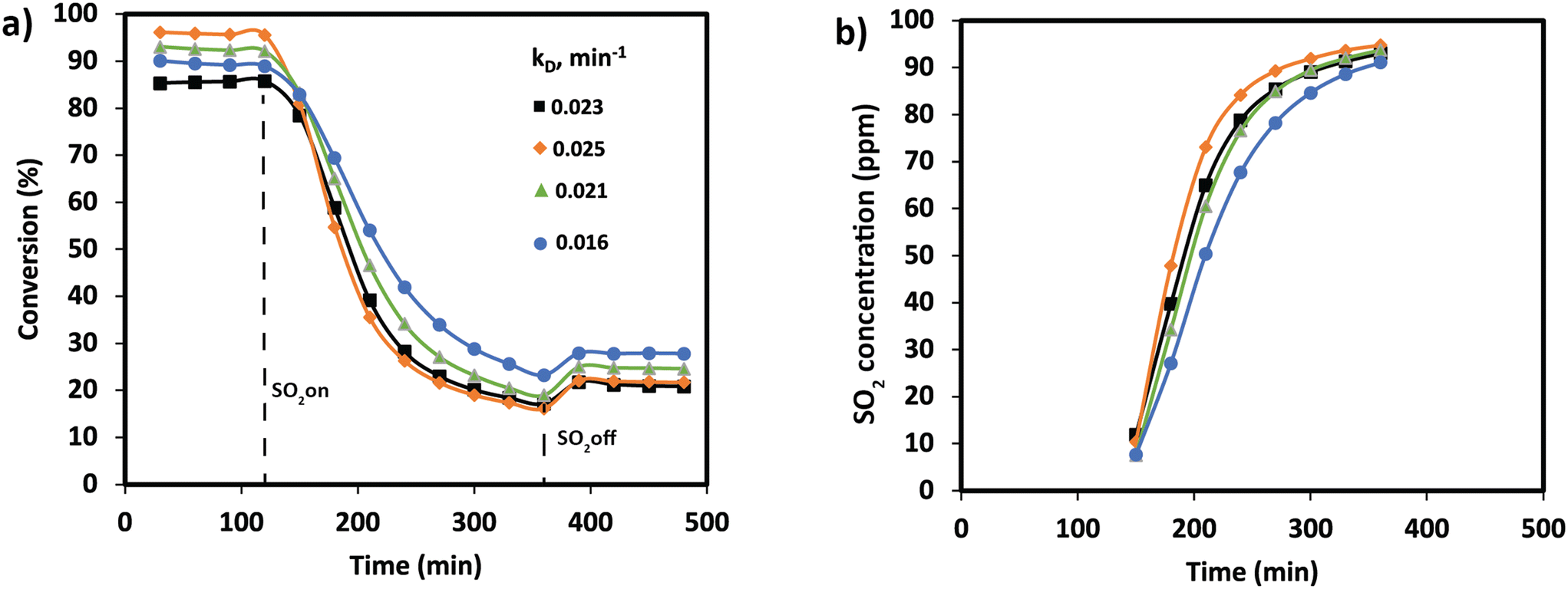 | ||
| Fig. 2 a) Conversion plots and deactivation constants and b) SO2 concentration at the outlet during the deactivation experiments performed at 250 °C for Mn0.35Ce0.00Ti0.65 (black), Mn0.37Ce0.04Ti0.59 (orange), Mn0.33Ce0.07Ti0.60 (green), and Mn0.30Ce0.19Ti0.51 (blue). | ||
SO2 uptakes were calculated by monitoring the SO2 concentration at the reactor outlet in Fig. 2b. Clearly, the drop in NOx conversion is strongly related to the SO2 uptake. The results show that all catalysts are adsorbing all SO2 at the first stage of the experiment. Then, the SO2 concentration is monotonically increasing until almost reaching the inlet concentration. SO2 uptakes, in Table 1, show an increase in the uptake for MnCeTi ternary catalysts with higher Ce loading. To rule out the effect of the surface area on the SO2 uptake, a surface normalization was applied. After such normalization, the results show a nearly constant SO2 adsorption in the MnCeTi ternary systems and therefore an independency from the amount of Ce. In the case of the binary MnTi, the SO2 uptake normalized by the surface area shows a twofold increase with respect to the Ce-containing samples. Our previous study showed that the surface of the binary MnTi samples is enriched with Mn,32 which would suggest that sulfur prefers Mn to either Ti or Ce.
| Sample | SO2 uptake (μmol g−1) | SO2 uptake (μmol m−2) |
|---|---|---|
| Mn0.35Ce0.00Ti0.65 | 139 | 1.2 |
| Mn0.37Ce0.04Ti0.59 | 119 | 0.6 |
| Mn0.33Ce0.07Ti0.60 | 165 | 0.7 |
| Mn0.30Ce0.19Ti0.51 | 182 | 0.7 |
In order to understand if there are sulfates formed and their chemistry, thermogravimetric analysis (TGA) was performed with the samples before and after SO2 deactivation. The chemistry of the sulfate species was inferred by comparing the results with the TGA analysis results of pure (NH4)2SO4, TiOSO4, MnSO4, and Ce(SO4)2. These results are included in the ESI† Fig. S2. The MnTi binary sample, in Fig. 3, shows a continuous decrease in mass loss after the normal NH3-SCR reaction, which could be related to the removal of species adsorbed after the reaction. In the NH3-SCR reaction with SO2, there is a lack of mass loss at around 400 °C for the SO2 deactivated sample, which indicates that (NH4)2SO4 is not present in the deactivated material. In contrast, there is a remarkable increase in the weight loss at around 780 °C, which is a temperature slightly lower than the decomposition temperature of MnSO4 (see ESI†). Combined mass spectrometry measurements (in ESI† Fig. S3a) show an evolution of SO2 coming out of the sample, suggesting the decomposition of a metal sulfate. We hypothesized that the weight loss is due to the decomposition of MnSO4 and the fact that the decomposition temperature is slightly lower than that of pure bulk sulfates could be due to the fact that the sulfates might be finely dispersed on the catalyst surface. The existence of TiOSO4 is discarded due to the lack of weight loss and SO2 evolution at the decomposition temperature of such species.
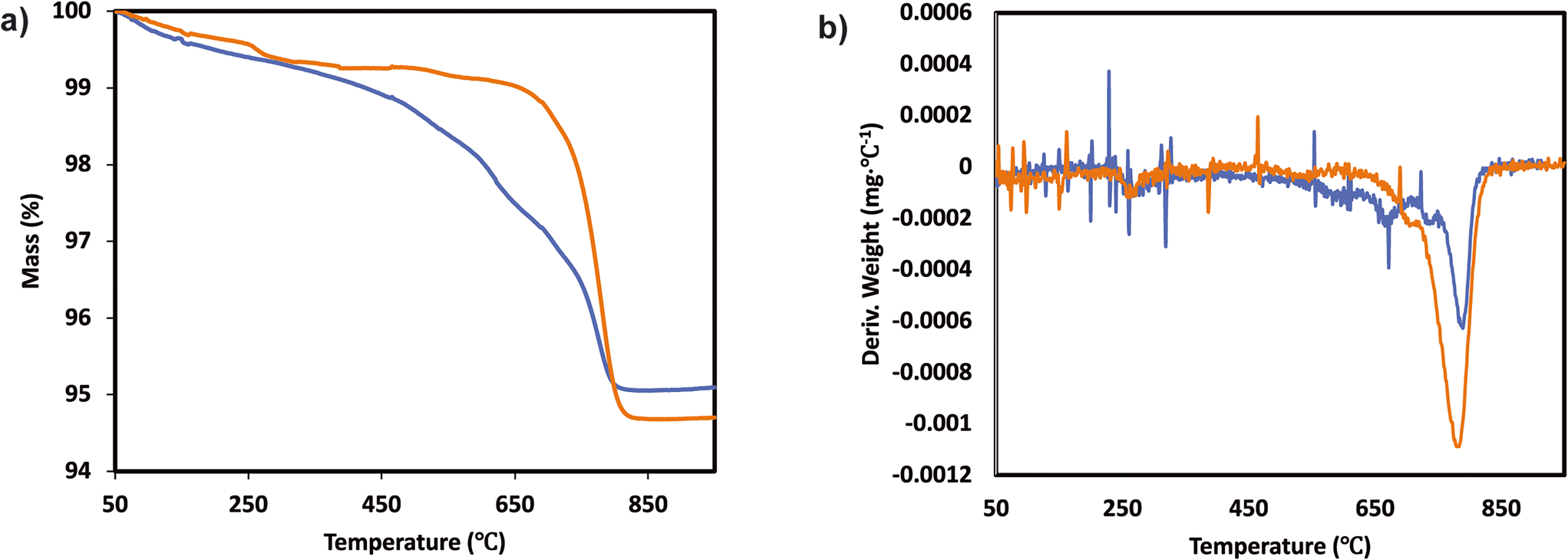 | ||
| Fig. 3 a) TGA analysis and b) derivative thermogravimetric curves of the Mn0.35Ce0.00Ti0.65 sample after normal NH3-SCR reaction (blue) and NH3-SCR reaction with SO2 (yellow). | ||
The TGA analysis of the ternary Mn0.37Ce0.04Ti0.59 and Mn0.30Ce0.19Ti0.51 samples is shown in Fig. 4 and 5. The samples without deactivation show a slight mass loss around 800 °C with an SO2 evolution, suggesting that there are some sulfates formed during the catalyst synthesis due the use of TiOSO4 as a titanium precursor. All samples have also mass loss around 650 °C, which could be related to the decomposition of TiOSO4. However, this cannot be confirmed as there is no evolution of SO3 in the mass spectrometer. Measuring SO3 is quite challenging due to its highly reactive nature. Upon SO2 deactivation, there are several decomposition events at around 800 °C, which could be ascribed to the formation of both Ce and Mn sulfates. The existence of sulfates is corroborated by the evolution of SO2 during the TGA experiments.
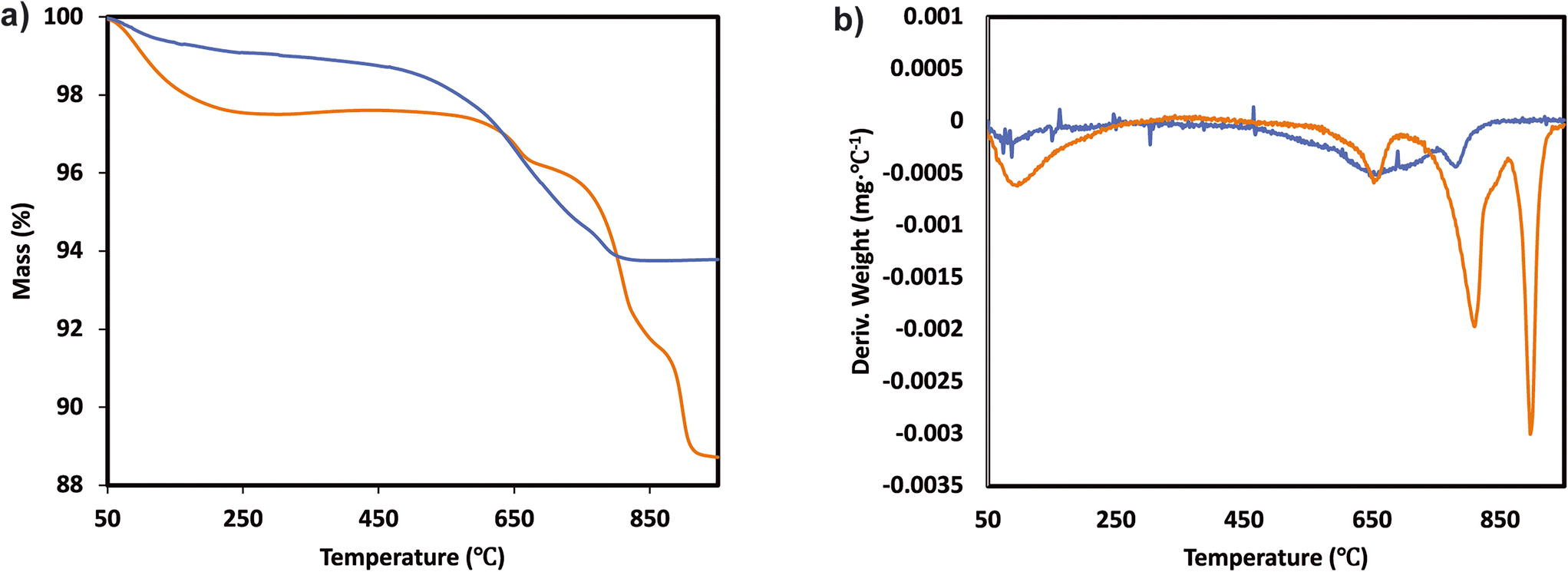 | ||
| Fig. 4 a) TGA analysis and b) derivative thermogravimetric curves of the Mn0.37Ce0.04Ti0.59 ternary system after normal NH3-SCR reaction (blue) and NH3-SCR reaction with SO2 (yellow). | ||
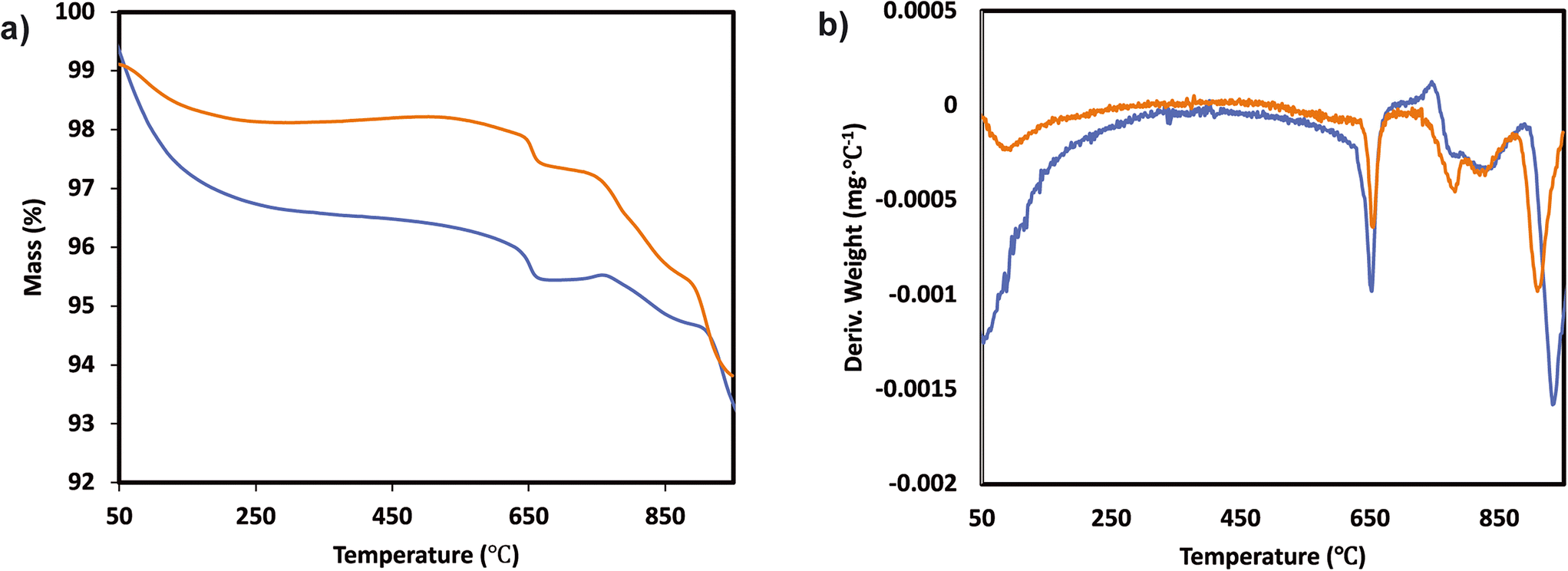 | ||
| Fig. 5 a) TGA analysis and b) derivative thermogravimetric curves of the Mn0.30Ce0.19Ti0.51 ternary system after normal NH3-SCR reaction (blue) and NH3-SCR reaction with SO2 (yellow). | ||
To understand the structural changes occurring upon SO2 deactivation, the crystalline structure of the samples before and after deactivation was investigated by X-ray diffraction (XRD). The results, in Fig. 6, show that the MnCe binary oxide show reflections of rutile TiO2, with no appearance of MnOx phases. In the case of the MnCeTi ternary systems, the diffraction patterns lack any reflection indicating the amorphous nature of the samples. After SO2 deactivation, the diffraction patterns resemble the fresh counterparts. The lack of additional crystalline phases indicates that the metal sulfates have an amorphous nature, most likely due to the formation of the sulfates solely on the catalyst surface.
 | ||
| Fig. 6 X-ray diffraction patterns of the Mn0.35Ce0.00Ti0.65 (black), Mn0.37Ce0.04Ti0.59 (orange), Mn0.33Ce0.07Ti0.60 (green), and Mn0.30Ce0.19Ti0.51 (blue) samples: a) fresh and b) after SO2 deactivation. | ||
To further investigate the location of the sulfur species on the catalysts, the bulk and surface composition of sulfur was investigated by ICP and XPS, respectively. Table 2 shows the sulfur composition in mol%. Clearly, all the samples show a sulfur enrichment on the catalyst surface compared to the total bulk composition, in line with the hypothesis that sulfur species are created more preferentially on the catalyst surface.
| Sample | Surface composition (mol%) | Bulk composition (mol%) |
|---|---|---|
| Mn0.35Ce0.00Ti0.65 | 6.4 | 3 |
| Mn0.37Ce0.04Ti0.59 | 6.9 | 5.3 |
| Mn0.30Ce0.19Ti0.51 | 7.8 | 3.3 |
Experiments to understand the acidity of the samples upon SO2 deactivation were performed by temperature-programmed desorption of ammonia (NH3-TPD). The temperature-programmed evolution of the binary Mn0.35Ce0.00Ti0.65 sample is plotted in ESI† Fig. S4. The amount of NH3 desorbed is more than double after SO2 deactivation (see ESI† Table S1), which reflect the formation of new acid sites. This insight has also been reported previously and indicates the acidic nature of the new sulfate species formed on the surface.33–35 Additional information about the reactivity of oxygen species on the catalyst could be extracted from the evolution of N2O during NH3-TPD. Less N2O is formed at higher temperature after SO2 deactivation, which points to the loss of active oxygen during deactivation.
To gain more insight into the effect of SO2, the chemistry of Mn and Ce was investigated by XPS. Oxidation states of Mn were rigorously fitted from a set of Gaussian–Lorentzian components per oxidation state, due to the multiplet splitting between the unpaired electrons in Mn 2+, 3+ and 4+. The set of components of the discrete oxidation states were obtained from measurements of pure MnSO4, MnO, Mn2O3 and MnO2 oxides and the results were compared with previously reported measurements.36 The Mn 2p spectra of the reference samples and catalysts (fresh and SO2 deactivated) are plotted in ESI† Fig. S5 and S6. Table 3 shows the surface composition and chemistry of Mn and Ce on selected samples. After SO2 deactivation, Mn surface composition decreases whereas Ce remains nearly constant. The Mn (2p3/2) spectra show an overall change in the composition of the samples to more reduced Mn species. Although Mn2O3 is dominant in the fresh samples, MnO is the most predominant species upon SO2 deactivation. Additionally, a substantial amount of MnSO4 is formed. Overall, around 60% of Mn4+ and Mn3+ gets reduced to Mn2+. Ce (3d) spectra show an increase in the concentration of Ce3+ upon deactivation (see also ESI† Fig. S7), which could be explained by the oxidation of SO2 to SO4−2 and the reduction of Ce4+ to Ce3+ with subsequent formation of sulfates.37
| Sample | Surface composition (at%) | Manganese | Cerium | |||||
|---|---|---|---|---|---|---|---|---|
| Mn | Ce | MnO2 | Mn2O3 | MnO | MnSO4 | Ce(IV) | Ce(III) | |
| Mn0.35Ce0.00Ti0.65 | 15.2 | 0.0 | 17.9 | 66.1 | 16.0 | 0.0 | 0.0 | 0.0 |
| Mn0.35Ce0.00Ti0.65 SO2 | 12.3 | 0.0 | 1.1 | 33.4 | 56.0 | 9.5 | 0.0 | 0.0 |
| Mn0.37Ce0.04Ti0.59 | 23.4 | 2.4 | 12.3 | 75.8 | 11.9 | 0.0 | 70.7 | 29.3 |
| Mn0.37Ce0.04Ti0.59 SO2 | 18.2 | 2.7 | 19.6 | 18.8 | 53.6 | 8.0 | 36.3 | 63.7 |
| Mn0.30Ce0.19Ti0.51 | 14.5 | 6.3 | 20.6 | 27.3 | 42.3 | 9.7 | 66.2 | 33.8 |
| Mn0.30Ce0.19Ti0.51 SO2 | 10.5 | 6.0 | 18.9 | 17.2 | 42.4 | 21.3 | 38.1 | 61.9 |
The N2 physisorption results, in Table 4, show a surface area of the MnTi binary sample of 117 m2 g−1. After the addition of Ce, there is an increase in the surface area and total pore volume, mostly related to the formation of an amorphous mixed-oxide phase. The samples after SO2 deactivation show a drop in the specific surface area. In the case of the binary MnTi, 23% of the specific surface area is lost after deactivation. For the samples with Ce, the loss in the specific surface area increases with the amount of Ce (28, 29 and 40% for Mn0.33Ce0.07Ti0.60, Mn0.37Ce0.04Ti0.59 and Mn0.30Ce0.19Ti0.51, respectively). Other authors have also observed a decrease in the surface area upon SO2 deactivation on Mn-based catalysts,18,38,39 mostly due to the formation of surface sulphate species. Indeed, this loss in the surface area has a direct impact on catalyst deactivation. However, in our study, it can only explain catalyst deactivation to a limited extent. Therefore, this is not the main deactivation mechanism.
| Sample | BET surface area (m2 g−1) | Total pore volume (cm3 g−1) | Pore size (nm) |
|---|---|---|---|
| Mn0.35Ce0.00Ti0.65 | 117 | 0.45 | 14.0 |
| Mn0.37Ce0.04Ti0.59 | 200 | 0.53 | 9.5 |
| Mn0.33Ce0.07Ti0.60 | 222 | 0.63 | 10.0 |
| Mn0.30Ce0.19Ti0.51 | 243 | 0.86 | 12.5 |
| Mn0.35Ce0.00Ti0.65 SO2 | 90 | 0.36 | 20.4 |
| Mn0.37Ce0.04Ti0.59 SO2 | 142 | 0.51 | 13.1 |
| Mn0.33Ce0.07Ti0.60 SO2 | 159 | 0.49 | 11.6 |
| Mn0.30Ce0.19Ti0.51 SO2 | 147 | 0.44 | 11.2 |
To gain more insight into the effect of SO2 poisoning on catalytic performance, NO oxidation to NO2 of the Mn0.37Ce0.04Ti0.59 ternary sample was performed before and after SO2 deactivation. This is a model reaction to investigate the behavior of the catalyst redox function, which is strongly related to its NH3-SCR performance at low temperatures. Additionally, NO oxidation plays a key role in the fast SCR reaction, which is believed to be ten times faster than the standard reaction of NO with NH3.40,41 As the NH3-SCR reaction is performed under the presence of H2O, the experiments were performed under dry and wet conditions. The results, in Fig. 7, show a clear drop in NO oxidation upon SO2 deactivation in the range of temperatures investigated, suggesting that the redox function is drastically affected upon deactivation as it was also observed during the evolution of N2O in the NH3-TPD experiments. Several authors have also observed similar effects on CeO2–MnOx42 and MnOx/TiO2 (ref. 43) catalysts and postulate that the reason is the formation of Mn sulfates.
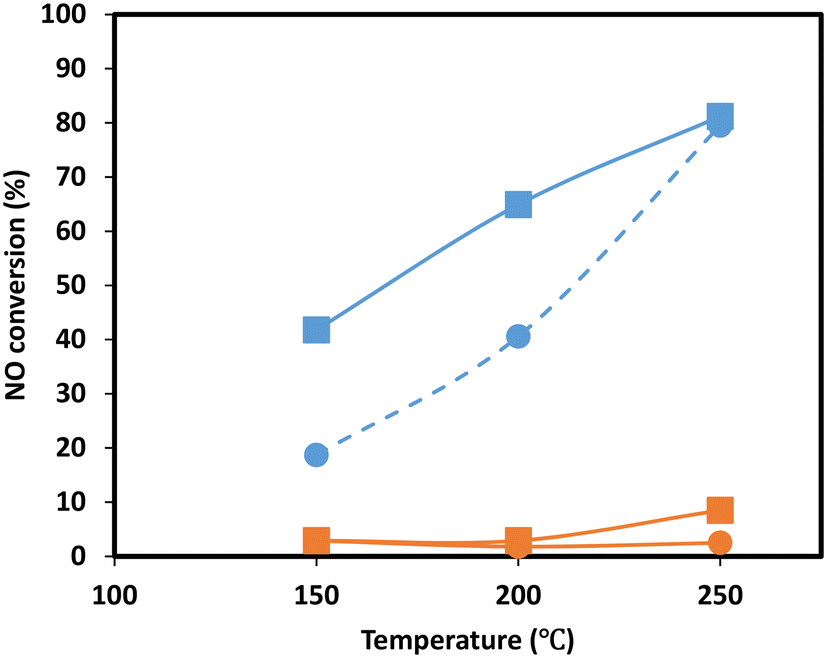 | ||
| Fig. 7 NO oxidation experiments of Mn0.37Ce0.04Ti0.59 under dry (square) and wet (circle) conditions for the fresh sample (blue) and sample upon SO2 deactivation (orange). | ||
Since the redox function seems to be strongly affected after SO2 deactivation, additional insights were obtained by temperature-programmed reduction (TPR) experiments with H2. The H2-TPR results of the samples (fresh and after SO2 deactivation) are shown in Fig. 8. Overall, we ascribe the reduction of the H2 consumption peak at 200–450 °C to the reduction of Mn4+ and Mn3+ to Mn2+.44,45 Upon SO2 deactivation, there is a decrease in the H2 consumed at those temperatures, most likely due to the overall reduction in the Mn oxidation state, as suggested by the XPS results. Additionally, H2 consumption is shifted to higher temperatures, which could be rationalized as a consequence of the interaction of sulfate species with Mn. The strong H2 consumption at 550–600 °C is related to the reduction of Mn and Ce sulfates.33,34,46
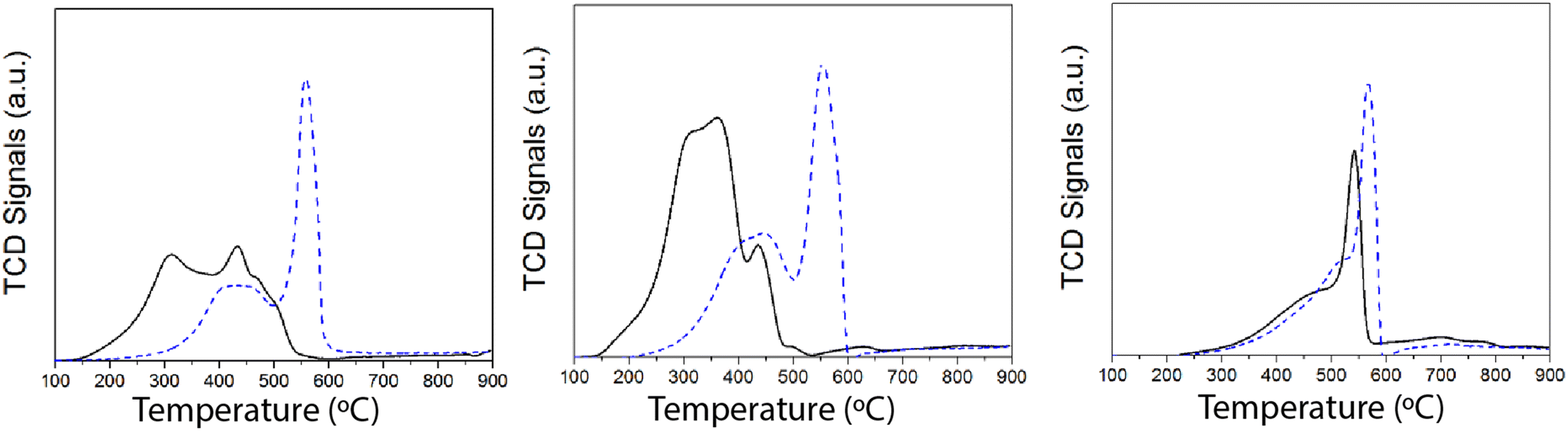 | ||
| Fig. 8 H2-TPR results of S15, S24 and S28: fresh (solid line) and SO2 deactivated (dashed line). | ||
Overall, our results shows that catalyst deactivation is mainly due to the formation of metal sulfates, most likely Mn and Ce sulfates. These metal sulfates are decreasing the oxidation states of Mn and Ce, which have a strong impact on the redox function of such metals. Additionally, SO2 deactivation decreases the catalysts' total specific surface area, but this is not the main deactivation mechanism.
In order to improve the catalytic properties of the samples upon deactivation, a thorough doping study was performed. Catalytic performances of the selected samples (fresh and upon SO2) and NH3-SCR regeneration at 500 °C for 30 min are shown in Table 5. On the selected metal-doped catalysts, relatively high NH3-SCR activities were obtained with the samples doped with Sm, V, La and Eu. Upon catalytic reaction with 100 ppm of SO2, the NH3-SCR activity was drastically decreased for both the undoped and doped samples. Therefore, the dopant seems to be ineffective in avoiding catalyst deactivation under the investigated conditions.
| Sample | Conversion before deactivation (%) | Conversion after SO2 deactivation (%) | Conversion after SO2 regeneration (%) | ||
|---|---|---|---|---|---|
| 150 °C | 250 °C | 250 °C | 150 °C | 250 °C | |
| Mn0.37Ce0.04Ti0.59 | 47 | 95 | 48 | 4 | 28 |
| 1% Sm/Mn0.37Ce0.04Ti0.59 (ppt) | 53 | 98 | 35 | 2 | 24 |
| 1% V/Mn0.37Ce0.04Ti0.59 (ppt) | 57 | 98 | 38 | 6 | 58 |
| 1% La/Mn0.37Ce0.04Ti0.59 (ppt) | 68 | 98 | 47 | 6 | 34 |
| 1% Mo/Mn0.37Ce0.04Ti0.59 (imp) | 45 | 96 | 28 | 4 | 52 |
| 1 wt% Eu/Mn0.37Ce0.04Ti0.59 (imp) | 72 | 95 | 25 | 3 | 34 |
To recover the catalyst activity, the samples were treated at 500 °C in 20% oxygen, as these conditions could be applied in a real exhaust of a combustion engine. The results show that in some samples, such conditions are even more detrimental, decreasing the catalyst activity upon SO2 uptake. As exceptions, samples doped with V and Mo display an almost twofold increase in catalytic activity at 250 °C. As described in the literature, V and Mo are acidic metals and could inhibit the adsorption of SO2 to some extent.47 Unfortunately, activity at 150 °C is still poor, indicating that the addition of such metals is not sufficient to avoid the formation of Mn and Ce sulfates and therefore the loss of redox function. We hypothesized that the high activities at 250 °C are due to V and Mo themselves, which are active metals for NH3-SCR and are not that drastically affected by SO2 deactivation. However, such metals are not sufficiently active at low temperatures. Our results are contradicting other previously reported data, for example summarized elsewhere,3 where dopants improve sulfur tolerance. While reviewing those studies, one can see that most of them were performed at lower space velocities and/or in the absence of steam, which is not representative of a real application.
4. Conclusions
In this work, the poisoning effect of SO2 was investigated in binary MnTi and ternary MnCeTi mixed oxides for the NH3-SCR reaction under conditions relevant for mobile applications. Upon performing activity test with 100 ppm of SO2 in the gas stream, the catalytic activity drastically decreases in all catalyst samples. The shape of the deactivation curve and SO2 concentrations out of the reactor suggest a strong adsorption and poisoning of SO2 on all the catalysts. Although samples containing large amounts of Ce display a better SO2 tolerance, this is insufficient to be considered for realistic practical applications. Spent samples were investigated by a wide range of characterization tools. N2 physisorption measurements reveal a drop in the surface area that could partially explain catalyst deactivation. TGA reveals the absence of (NH4)2SO4 on the deactivated samples and suggests the formation of Mn and Ce sulfates on the catalyst surface. XPS results confirm the formation of MnSO4 and also show a decrease in the Mn and Ce oxidation states. Analysis of the redox function by catalytic NO oxidation and H2-TPR experiments shows a strong loss of redox function upon SO2 deactivation, which could explain the decrease in NH3-SCR catalytic activity. Upon unraveling the effect and cause of deactivation, the catalysts were modified by doping with a wide range of metals. As in the binary MnTi and ternary MnCeTi, catalytic activity strongly decreases upon the introduction of SO2 in the gas stream. None of the dopants investigated was able to suppress SO2 deactivation nor regenerate catalyst activity at low temperatures, which suggest that other dopants or strategies should be pursued to commercialize Mn-based catalysts for low-temperature applications.Conflicts of interest
The authors declare no competing interest.Acknowledgements
The authors thank the financial support from Umicore N.V. (award number RGC/3/3291-01). The research was supported by the resources and facilities provided by the King Abdullah University of Science and Technology. The authors also acknowledge the KAUST Imaging and Characterization Core Lab. Lutz Ruwisch (Umicore) is kindly acknowledged for fruitful discussion.References
- J. L. Sorrels, Selective Catalytic Reduction. U.S. Enviromental Protection Agency. https://www.epa.gov/economic-and-cost-analysis-air-pollution-regulations/chapter-2-selective-catalytic-reduction.
- R. Zhang, N. Liu, Z. Lei and B. Chen, Selective Transformation of Various Nitrogen-Containing Exhaust Gases toward N2 over Zeolite Catalysts, Chem. Rev., 2016, 116(6), 3658–3721 CrossRef CAS PubMed.
- L. Han, S. Cai, M. Gao, J. Y. Hasegawa, P. Wang, J. Zhang, L. Shi and D. Zhang, Selective Catalytic Reduction of NOx with NH3 by Using Novel Catalysts: State of the Art and Future Prospects, Chem. Rev., 2019, 119(19), 10916–10976 CrossRef CAS PubMed.
- T. V. Johnson, Diesel emission control in review, SAE Int. J. Fuels Lubr., 2009, 1(1), 68–81 CrossRef CAS.
- J. Chen and R. Yang, Role of WO3 in mixed V2O5-WO3/TiO2catalysts for selective catalytic reduction of nitric oxide with ammonia, Appl. Catal., A, 1992, 80(1), 135–148 CrossRef CAS.
- T. Xu, X. Wu, Y. Gao, Q. Lin, J. Hu and D. Weng, Comparative study on sulfur poisoning of V2O5-Sb2O3/TiO2 and V2O5-WO3/TiO2 monolithic catalysts for low-temperature NH3-SCR, Catal. Commun., 2017, 93, 33–36 CrossRef CAS.
- X. Tian, Y. Xiao, P. Zhou, W. Zhang and X. Luo, Investigation on performance of V2O5–WO3–TiO2–cordierite catalyst modified with Cu, Mn and Ce for urea-SCR of NO, Mater. Res. Innovations, 2014, 18(sup2), 202–206 Search PubMed.
- Z. Lian, Y. Li, W. Shan and H. He, Recent Progress on Improving Low-Temperature Activity of Vanadia-Based Catalysts for the Selective Catalytic Reduction of NOx with Ammonia, Catalysts, 2020, 10(12), 1421 CrossRef CAS.
- D. W. Fickel and R. F. Lobo, Copper coordination in Cu-SSZ-13 and Cu-SSZ-16 investigated by variable-temperature XRD, J. Phys. Chem. C, 2010, 114(3), 1633–1640 CrossRef CAS.
- J. Girard, G. Cavataio, R. Snow and C. Lambert, Combined Fe-Cu SCR systems with optimized ammonia to NOx ratio for diesel NOx control, SAE Int. J. Fuels Lubr., 2009, 1(1), 603–610 CrossRef CAS.
- J. H. Kwak, R. G. Tonkyn, D. H. Kim, J. Szanyi and C. H. Peden, Excellent activity and selectivity of Cu-SSZ-13 in the selective catalytic reduction of NOx with NH3, J. Catal., 2010, 275(2), 187–190 CrossRef CAS.
- B. Jiang, Y. Liu and Z. Wu, Low-temperature selective catalytic reduction of NO on MnOx/TiO2 prepared by different methods, J. Hazard. Mater., 2009, 162(2), 1249–1254 CrossRef CAS PubMed.
- S. Deng, T. Meng, B. Xu, F. Gao, Y. Ding, L. Yu and Y. Fan, Advanced MnOx/TiO2 Catalyst with Preferentially Exposed Anatase {001} Facet for Low-Temperature SCR of NO, ACS Catal., 2016, 6(9), 5807–5815 CrossRef CAS.
- E. Park, M. Kim, H. Jung, S. Chin and J. Jurng, Effect of Sulfur on Mn/Ti Catalysts Prepared Using Chemical Vapor Condensation (CVC) for Low-Temperature NO Reduction, ACS Catal., 2013, 3(7), 1518–1525 CrossRef CAS.
- S. Yu, Y. Lu, Y. Cao, J. Wang, B. Sun, F. Gao, C. Tang and L. Dong, Composite catalytic systems: A strategy for developing the low temperature NH3-SCR catalysts with satisfactory SO2 and H2O tolerance, Catal. Today, 2019, 327, 235–245 CrossRef CAS.
- I. S. Nam, J. W. Eldridge and J. R. Kittrell, Deactivation of a vanadia-alumina catalyst for nitric oxide reduction by ammonia, Ind. Eng. Chem. Prod. Res. Dev., 1986, 25(2), 192–197 CrossRef CAS.
- Q. Wang, J. Zhou, J. Zhang, H. Zhu, Y. Feng and J. Jin, Effect of ceria doping on catalytic activity and SO2 resistance of MnOx/TiO2 catalysts for selective catalytic reduction of NO with NH3 at low temperature, Aerosol Air Qual. Res., 2020, 20, 477–488 CAS.
- W. Sjoerd Kijlstra, M. Biervliet, E. K. Poels and A. Bliek, Deactivation by SO2 of MnOx/Al2O3 catalysts used for the selective catalytic reduction of NO with NH3 at low temperatures, Appl. Catal., B, 1998, 16(4), 327–337 CrossRef CAS.
- Z. Sheng, Y. Hu, J. Xue, X. Wang and W. Liao, SO2 poisoning and regeneration of Mn-Ce/TiO2 catalyst for low temperature NOx reduction with NH3, J. Rare Earths, 2012, 30(7), 676–682 CrossRef CAS.
- S. Wu, X. Yao, L. Zhang, Y. Cao, W. Zou, L. Li, K. Ma, C. Tang, F. Gao and L. Dong, Improved low temperature NH3-SCR performance of FeMnTiOx mixed oxide with CTAB-assisted synthesis, Chem. Commun., 2015, 51(16), 3470–3473 RSC.
- F. Liu, H. He, Y. Ding and C. Zhang, Effect of manganese substitution on the structure and activity of iron titanate catalyst for the selective catalytic reduction of NO with NH3, Appl. Catal., B, 2009, 93(1), 194–204 CrossRef CAS.
- B. Q. Jiang, Z. B. Wu, Y. Liu, S. C. Lee and W. K. Ho, DRIFT Study of the SO2 Effect on Low-Temperature SCR Reaction over Fe−Mn/TiO2, J. Phys. Chem. C, 2010, 114(11), 4961–4965 CrossRef CAS.
- F. Wang, B. Shen, S. Zhu and Z. Wang, Promotion of Fe and Co doped Mn-Ce/TiO2 catalysts for low temperature NH3-SCR with SO2 tolerance, Fuel, 2019, 249, 54–60 CrossRef CAS.
- H. Hu, S. Cai, H. Li, L. Huang, L. Shi and D. Zhang, Mechanistic Aspects of deNOx Processing over TiO2 Supported Co–Mn Oxide Catalysts: Structure–Activity Relationships and In Situ DRIFTs Analysis, ACS Catal., 2015, 5(10), 6069–6077 CrossRef CAS.
- L. Huang, X. Hu, S. Yuan, H. Li, T. Yan, L. Shi and D. Zhang, Photocatalytic preparation of nanostructured MnO2-(Co3O4)/TiO2 hybrids: The formation mechanism and catalytic application in SCR deNOx reaction, Appl. Catal., B, 2017, 203, 778–788 CrossRef CAS.
- J. Liu, X. Li, R. Li, Q. Zhao, J. Ke, H. N. Xiao, L. Wang, S. Liu, M. O. TadÈ and S. Wang, Facile synthesis of tube-shaped Mn-Ni-Ti solid solution and preferable Langmuir-Hinshelwood mechanism for selective catalytic reduction of NO x by NH 3, Appl. Catal., A, 2018, 549, 289–301 CrossRef CAS.
- L. Chen, R. Li, Z. Li, F. Yuan, X. Niu and Y. Zhu, Effect of Ni doping in NixMn1−xTi10 (x = 0.1–0.5) on activity and SO2 resistance for NH3-SCR of NO studied with in situ DRIFTS, Catal. Sci. Technol., 2017, 7(15), 3243–3257 RSC.
- X. Wang, X. Li, Q. Zhao, W. Sun, M. Tade and S. Liu, Improved activity of W-modified MnOx–TiO2 catalysts for the selective catalytic reduction of NO with NH3, Chem. Eng. J., 2016, 288, 216–222 CrossRef CAS.
- C. Sun, H. Liu, W. Chen, D. Chen, S. Yu, A. Liu, L. Dong and S. Feng, Insights into the Sm/Zr co-doping effects on N2 selectivity and SO2 resistance of a MnOx-TiO2 catalyst for the NH3-SCR reaction, Chem. Eng. J., 2018, 347, 27–40 CrossRef CAS.
- B. Wang, M. Wang, L. Han, Y. Hou, W. Bao, C. Zhang, G. Feng, L. Chang, Z. Huang and J. Wang, Improved Activity and SO2 Resistance by Sm-Modulated Redox of MnCeSmTiOx Mesoporous Amorphous Oxides for Low-Temperature NH3-SCR of NO, ACS Catal., 2020, 10(16), 9034–9045 CrossRef CAS.
- D. Meng, W. Zhan, Y. Guo, Y. Guo, L. Wang and G. Lu, A Highly Effective Catalyst of Sm-MnOx for the NH3-SCR of NOx at Low Temperature: Promotional Role of Sm and Its Catalytic Performance, ACS Catal., 2015, 5(10), 5973–5983 CrossRef CAS.
- L. E. Gevers, L. R. Enakonda, A. Shahid, S. Ould-Chikh, C. I. Q. Silva, P. P. Paalanen, A. Aguilar-Tapia, J.-L. Hazemann, M. N. Hedhili, F. Wen and J. Ruiz-Martínez, Unraveling the structure and role of Mn and Ce for NOx reduction in application-relevant catalysts, Nat. Commun., 2022, 13(1), 2960 CrossRef CAS PubMed.
- L. Zhang, L. Li, Y. Cao, X. Yao, C. Ge, F. Gao, Y. Deng, C. Tang and L. Dong, Getting insight into the influence of SO2 on TiO2/CeO2 for the selective catalytic reduction of NO by NH3, Appl. Catal., B, 2015, 165, 589–598 CrossRef CAS.
- M. Waqif, P. Bazin, O. Saur, J. C. Lavalley, G. Blanchard and O. Touret, Study of ceria sulfation, Appl. Catal., B, 1997, 11(2), 193–205 CrossRef CAS.
- J. Li, C. Zhang, Q. Li, T. Gao, S. Yu, P. Tan, Q. Fang and G. Chen, Promoting mechanism of SO2 resistance performance by anatase TiO2 {001} facets on Mn-Ce/TiO2 catalysts during NH3-SCR reaction, Chem. Eng. Sci., 2022, 251, 117438 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni, Appl. Surf. Sci., 2011, 257(7), 2717–2730 CrossRef CAS.
- R. M. Ferrizz, R. J. Gorte and J. M. Vohs, TPD and XPS Investigation of the Interaction of SO2 with Model Ceria Catalysts, Catal. Lett., 2002, 82(1), 123–129 CrossRef CAS.
- B. Jiang, B. Deng, Z. Zhang, Z. Wu, X. Tang, S. Yao and H. Lu, Effect of Zr Addition on the Low-Temperature SCR Activity and SO2 Tolerance of Fe–Mn/Ti Catalysts, J. Phys. Chem. C, 2014, 118(27), 14866–14875 CrossRef CAS.
- J. Liu, R.-T. Guo, M.-Y. Li, P. Sun, S.-M. Liu, W.-G. Pan, S.-W. Liu and X. Sun, Enhancement of the SO2 resistance of Mn/TiO2 SCR catalyst by Eu modification: A mechanism study, Fuel, 2018, 223, 385–393 CrossRef CAS.
- M. Koebel, M. Elsener and G. Madia, Reaction Pathways in the Selective Catalytic Reduction Process with NO and NO2 at Low Temperatures, Ind. Eng. Chem. Res., 2001, 40(1), 52–59 CrossRef CAS.
- G. Madia, M. Koebel, M. Elsener and A. Wokaun, The Effect of an Oxidation Precatalyst on the NOx Reduction by Ammonia SCR, Ind. Eng. Chem. Res., 2002, 41(15), 3512–3517 CrossRef CAS.
- F. Lin, Y. He, Z. Wang, Q. Ma, R. Whiddon, Y. Zhu and J. Liu, Catalytic oxidation of NO by O2 over CeO2–MnOx: SO2 poisoning mechanism, RSC Adv., 2016, 6(37), 31422–31430 RSC.
- N. Tang, Y. Liu, H. Wang and Z. Wu, Mechanism Study of NO Catalytic Oxidation over MnOx/TiO2 Catalysts, J. Phys. Chem. C, 2011, 115(16), 8214–8220 CrossRef CAS.
- F. Arena, T. Torre, C. Raimondo and A. Parmaliana, Structure and redox properties of bulk and supported manganese oxide catalysts, Phys. Chem. Chem. Phys., 2001, 3(10), 1911–1917 RSC.
- X. Wu, Q. Liang, D. Weng, J. Fan and R. Ran, Synthesis of CeO2–MnOx mixed oxides and catalytic performance under oxygen-rich condition, Catal. Today, 2007, 126(3), 430–435 CrossRef CAS.
- S. Yang, Y. Guo, H. Chang, L. Ma, Y. Peng, Z. Qu, N. Yan, C. Wang and J. Li, Novel effect of SO2 on the SCR reaction over CeO2: Mechanism and significance, Appl. Catal., B, 2013, 136-137, 19–28 CrossRef CAS.
- D. W. Kwon, K. H. Park and S. C. Hong, Enhancement of SCR activity and SO2 resistance on VOx/TiO2 catalyst by addition of molybdenum, Chem. Eng. J., 2016, 284, 315–324 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy01151d |
| This journal is © The Royal Society of Chemistry 2022 |