
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synchrotron PXRD deconvolutes nickel particle and support changes in Ni/ZrO2 methanation catalysts†
Mariam L.
Schulte‡
 ab,
Sebastian
Weber‡
ab,
Linda
Klag
a,
Jan-Dierk
Grunwaldt
ab and
Thomas L.
Sheppard
*ab
ab,
Sebastian
Weber‡
ab,
Linda
Klag
a,
Jan-Dierk
Grunwaldt
ab and
Thomas L.
Sheppard
*ab
aInstitute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstr. 20, 76131 Karlsruhe, Germany. E-mail: thomas.sheppard@kit.edu; Tel: +49 72160847989
bInstitute of Catalysis Research and Technology, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 13th July 2022
Abstract
Understanding catalyst deactivation is important for future knowledge-based design of catalysts with improved stability. Deactivation by thermal aging is particularly relevant for exothermic reactions, here demonstrated with CO2 methanation using nickel-based catalysts. A series of five Ni/ZrO2 catalysts is studied which differ by calcination temperature of the ZrO2 support prior to Ni deposition, leading to different textural properties of the support. Artificial thermal aging of the Ni/ZrO2 series is then performed and monitored by operando synchrotron-based powder X-ray diffraction (SPXRD). This reveals the dependence of catalyst stability on the support properties and shows that different deactivation mechanisms take place. Catalyst deactivation is mainly correlated either to changes of the support or to changes in nickel crystallite size, depending on the calcination temperature of the support before nickel deposition. By preparing a targeted series of supports with different textural properties, it is possible to deconvolute these effects. Operando SPXRD is shown as a powerful tool to follow both textural and structural changes during thermal catalyst deactivation, which is mostly only performed by post mortem ex situ analysis.
1 Introduction
Exothermic heterogeneous catalytic processes are challenging in regard to safety aspects and catalyst stability. Reliable heat management is necessary in order to maintain safe operation under process conditions. Thermal catalyst deactivation can be classified in three effects: (i) loss of active sites due to metal particle agglomeration; (ii) collapse of support structure leading to a decrease in surface area or porosity changes; (iii) formation of undesired alloy or mixed metal oxide phases, by reaction of the active phase and catalyst support.1,2 Distinguishing between these three effects for a given chemical process is difficult, since all effects can occur simultaneously and in addition to other types of deactivation, e.g., poisoning or fouling. Detailed investigation of temperature-induced catalyst deactivation is needed in order to unravel the impact of each degradation process.A suitable exothermic model reaction for studying catalyst deactivation is CO2 hydrogenation to CH4, also known as CO2 methanation. This process involves significant heat generation and therefore forms distinct temperature gradients (hot-spots),3 which have been simulated in fixed-bed reactors4 and observed experimentally using spatially-resolved measurements.5 A wide range of catalysts have been investigated for CO2 methanation and the reviews of Aziz et al.6 and Frontera et al.7 provide a comprehensive overview on catalyst development. Ni-based catalysts are most frequently studied, due to relatively low cost of the metal and high activity and selectivity in CO and CO2 methanation.7
Possible deactivation pathways for CO2 methanation catalysts have also been reviewed.8–10 These mainly include fouling/coking, poisoning, (redox) reaction of the active metal sites, and thermal degradation. Although coking is discussed as a deactivation phenomena for CO2 methanation, it is more relevant for CO or mixed CO/CO2 methanation where the Boudouard reaction favors coke formation from CO-containing feed.8–11 Poisoning is mainly relevant for applications of CO2 methanation associated with coal or biomass based CO2 feeds, and can be attributed to sulfur species present in the feed.8,10,12 Changes in oxidation state of active metallic Ni sites are discussed in several studies especially in terms of fluctuating feeds such as H2 dropout scenarios, which can lead to irreversible or partially irreversible Ni oxidation to NiO.13–17 However, the above deactivation pathways can generally be reversed by reactivation, compared to thermal degradation,18 which is a permanent feature due to the exothermic nature of the reaction.
As an example, long-term tests were reported for co-precipitated Ni/Al2O3 catalysts at reaction temperatures of around 380–400 °C by Abelló et al.19 and Koschany et al.,20 where a slow decline in activity was observed over several hundreds of hours of operation, eventually leading to constant conversion after 320 h. This was traced back to gradual sintering of Ni particles, which Beierlein et al.18 studied in more detail during rapid ageing tests by exposing the catalysts to hydrothermal conditions at 600 to 700 °C. They observed increasing Ni particle sizes and additional changes in the support structure on thermal aging.18 In addition, potential deactivation by an oxidized or restructured Ni surface was discussed.18 Similar findings were reported by Mebrahtu et al.21 showing the influence of nickel hydroxide-like species formed under hydrothermal conditions, resulting in enhanced formation of mixed oxides or sintering. Ewald et al.22 attributed the long term deactivation of co-precipitated Ni/Al2O3 catalysts to Ni particle sintering, textural changes, i.e., loss of surface area and reduction of medium basic sites, which is associated with changes of the CO2 adsorption capacity. In addition, they report the potential incorporation of Ni into γ-Al2O3 or the beginning of mixed oxide phase formation such as NiAl2O4.22
Both the support and the metal nanoparticles can change upon (hydro)thermal ageing at the same time. Hence, the challenge is to deconvolute the many different deactivation pathways which may occur, e.g., sintering, oxidation, composite formation. Post mortem or ex situ analysis can hardly reveal the stage at which deactivation happens. This requires in situ or operando measurements sensitive to the aforementioned sample changes.23–25 It should be noted that there are mainly two different mechanisms proposed for CO2 methanation,7,8,10 where aside from Ni metal as the active species, the support might also be involved in the reaction by adsorption of CO2 as part of an associative mechanism. The behavior of both the active metal and the support may therefore be important. In the current study, we present a systematic examination of deactivation effects of Ni/ZrO2 methanation catalysts by operando synchrotron-based powder X-ray diffraction (SPXRD) during artificial thermal aging. The aim is to deconvolute contributions of support and Ni nanoparticle changes to catalyst deactivation. A designed model series of different Ni/ZrO2 catalysts was prepared using the same ZrO2 support, which was calcined at various temperatures prior to Ni deposition. Thus, distinct thermal stability and textural properties of the supports can be achieved, while similar Ni particles are initially present on each support, as illustrated in Fig. 1. A Ni/ZrO2 system with the monoclinic ZrO2 polymorph was chosen as a typically employed combination for methanation catalysts6,7 and because ZrO2 exhibits a more defined crystalline structure compared to more commonly applied γ-Al2O3 supports. Furthermore, monoclinic ZrO2 is typically a non-reducible support and mixed oxide formation with Ni is less likely compared to γ-Al2O3. ZrO2 is therefore an ideal model support system to study thermally induced catalyst changes, as the expected changes are mainly related to textural properties. Operando SPXRD measurements were used to reveal structural and textural changes of the support and Ni nanoparticles based on crystallite size analysis at different deactivation temperatures during CO2 methanation. In this way, the distinct contributions of active metal or support changes on catalyst deactivation can be deconvoluted. This allows to derive structure–deactivation relationships for the Ni/ZrO2 catalyst series. Therefore, this study presents an approach to distinguish changes occurring in different components of the solid catalyst during artificial thermal deactivation cycles, using a single method, i.e., in situ/operando SPXRD. The catalysts used in this study may be considered as model systems for the purpose of monitoring deactivation, without optimization regarding their catalytic performance.
 | ||
| Fig. 1 Illustration of the initially prepared Ni/ZrO2 catalysts applied in artificial thermal aging experiments. Before Ni deposition the ZrO2 support was calcined at different temperatures to alter the support textural properties (surface area, pore volume) and to vary the thermal stability of the support. Ni blue, ZrO2 grey, the grey color gradient illustrates changing textural properties of the support. | ||
2 Experimental
2.1 Catalyst synthesis
Ni/ZrO2 catalysts were prepared using the homogeneous deposition–precipitation method adapted from Mutz et al.26 As support material commercial ZrO2 pellets (Saint-Gobain) were crushed and thermally aged at different temperatures (indicated in abbreviation) for 12 h (10 °C min−1) resulting in samples Z350, Z500, Z650, Z800 and Z1000. The calcined powders were finely granulated to a mesh size ≤100 μm and 1.6 g of each powder was suspended in an aqueous solution (230 mL) of 2.002 g Ni(NO3)2·6H2O (Merck 99%) with 2.906 g of urea (7 eq., Carl Roth, crystalline, 99.6%), which corresponds to an target Ni loading of 20 wt%. The suspension was stirred for 1 h at room temperature (pH = 4) and afterwards heated to 90 °C. The mixture was heated for 18 h under reflux, cooled to room temperature, then stirred for another 1 h (pH = 8). The solid was filtered off, washed three times with 100 mL of deionized water and dried overnight at 90 °C. Subsequently, the catalysts were calcined at 500 °C for 4 h (10 °C min−1).2.2 Laboratory catalyst characterization
Catalyst composition was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) with a Perkin Elmer Optima 8000 instrument equipped a Scott/crossflow sample injection system.Specific surface area (ABET) and pore volume (Vp) were obtained from N2 physisorption using a BELSORP-mini II system (Microtrac) with the method of Brunauer, Emmett and Teller (BET).27 Prior to the measurement the samples were degassed at 300 °C for 2 h under vacuum.
Mercury intrusion porosimetry (MIP) analysis was conducted using a ThermoScientific PASCAL 140 (lower pressures) and a ThermoScientific PASCAL 440 (higher pressures). The pore width (wP) distribution was calculated using the Washburn equation (eqn (1)).28 The used contact angle (θ) was 140° with a surface tension (σ) of 0.48 N m−1. Here pL and pG are the pressure of liquid and gas, respectively, and dp is the pore diameter.
 | (1) |
Transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM) images were obtained for three spent catalysts after catalytic testing on a Tecnai F20 (Philips) electron microscope with 200 kV acceleration. A copper grid was used as specimen holder and element distribution was studied energy dispersive X-ray analysis (EDX) using an EDAX s-UTW detector.
2.3 Catalyst activity testing
The catalytic performance of the catalysts was studied in a continuous flow setup with a stainless steel tubular fixed-bed reactor (length = 410 mm, inner diameter di = 7 mm, outer diameter do = 9.5 mm) (Fig. S14, ESI†), described in previous work.26 The catalyst bed (length = 30 to 35 mm) contained 300 mg of catalyst (sieve fraction 300–450 μm) diluted in 1.15 g of SiC (210 μm, Carborundum, VWR Chemicals). The spare void of the reactor was densely packed with SiC (500 μm) and the different layers were separated with quartz glass wool to ensure a defined gas flow. Thermocouples (K-type) were placed in front and behind the catalyst bed for automated temperature control, using the average temperature of the front and end positions. Gases were dosed using mass flow controllers (Bronkhorst) calibrated using a gas flow meter (Bios DryCal Definer 220, MesaLabs). Gaseous product analysis was performed by gas chromatography (GC) using an Agilent 490 micro gas chromatograph (μ-GC) (channel 1: 10 m PoraPLOT Q, 0.25 mm diameter, carrier gas He; channel 2: 10 m mole sieve column with 5 Å, 0.25 mm diameter, carrier gas Ar). Prior to each reaction, the catalysts were activated in the reactor in a 50% H2/N2 mixture (both purity 99.9999% from Air Liquide) (600 mL min−1) at 500 °C for 2 h. After cooling to reaction temperature, the gas feed was changed to 10% CO2/40% H2/N2 (CO2 purity 99.995% from Air Liquide) resulting in a weight hourly space velocity (WHSV) of 60 L gcat−1 h−1. The catalytic performance was tested at three different temperatures (250 °C, 350 °C and 400 °C) and a hold time of 1 h each to obtain stable catalytic activity. Conversion of CO2 (XCO2) was calculated similarly to earlier measurements by Mutz et al. using N2 as internal standard (see ESI†).262.4 Operando synchrotron powder X-ray diffraction (SPXRD) experiments
SPXRD measurements were performed at the X04SA-MS beamline of the Swiss Light Source (Paul Scherrer Institute, Villigen, Switzerland).29 An X-ray energy of 30.5 keV (λ = 0.406739 Å) was used to ensure high transmission of the samples, which was required due to the strongly absorbing ZrO2 support. 2D diffraction patterns were acquired with a Pilatus 6M detector, which were azimuthally integrated using the Dioptas software.30Operando SPXRD studies were carried out using a quartz capillary (WJM-Glas Müller GmbH) microreactor setup of 1.0 mm diameter.31 About 4 mg of the undiluted calcined catalyst (sieve fraction 100–200 μm) resulting in a bed length of ≈4 mm was used for each experiment. The reactor was heated using a hot-air blower (LE MINI SENSOR KIT, Leister Technologies), gases were dosed using mass flow controllers (Bronkhorst) and reactor outlet gases were analyzed by on-line mass spectrometry (MS) with an OMNI-Star GSD 320 (Pfeiffer Vacuum). Before the experiments, the temperature inside an empty capillary was calibrated using a type-K-thermocouple (for calibration data see Fig. S16, ESI†). It should be noted that an online temperature control was used during the whole experiment, but the recording of the temperature values failed due to an error in the control software. A LaB6 NIST 640b standard was measured inside of a 1.0 mm quartz capillary as a calibrant for 2θ scale and to retrieve an instrumental profile function.
The initial calcined catalyst was first activated in a total flow of 10 mL min−1 of 25% H2/He at 500 °C for 2 h with a heating rate of 10 °C min−1. Afterwards, it was cooled to 400 °C and switched to reaction conditions of 5% CO2/20% H2/He to determine the initial activity of the catalyst sample at reaction step R1 for 30 min. Subsequently, the catalysts underwent three artificial thermal deactivation cycles with 5 °C min−1 ramps and a holding time of 15 min at temperatures of 500 °C, 575 °C and 625 °C, still under reaction conditions. In between each cycle, changes in catalyst activity were studied at 400 °C labelled as reaction steps R2, R3 and R4. Afterwards, the sample was cooled to room temperature with 10 °C min−1. Diffraction patterns were measured with 9 s acquisition time during activation and final cool-down as well as 3 s during reaction conditions and artificial thermal deactivation cycles.
The on-line MS data was used to estimate the conversion of CO2 (XCO2) and H2 (XH2) during operando SPXRD. Xi for the respective m/z ratio and thus component i is calculated according to eqn (2):
 | (2) |
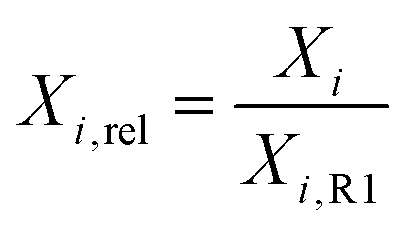 | (3) |
2.5 Analysis of SPXRD data
Rietveld refinements32,33 of the integrated operando SPXRD data were performed using Topas (v.6, Bruker AXS).34 The measured LaB6 NIST 640b standard was used to derive an instrumental profile function, described by a pseudo-Voigt Thompson–Cox Hastings peak shape. A micro-structure analysis was performed using the double-Voigt approach by Balzar as implemented in Topas to describe crystallite size (Lvol) and strain (η).35 The monoclinic ZrO2 phase was described by the structure reported by Smith et al.,36 NiO phase by a structure from Rodic et al.,37 and the Ni phase with a structure by Rouquette et al.38 Sequential Rietveld refinements were performed by adapting templates available from the Topas wiki website.39 More details on the Rietveld refinements are provided in the ESI.†An average Lvol,avg of the catalyst is defined as following:
| Lvol,avg = ∑Lvol,i·ωi | (4) |
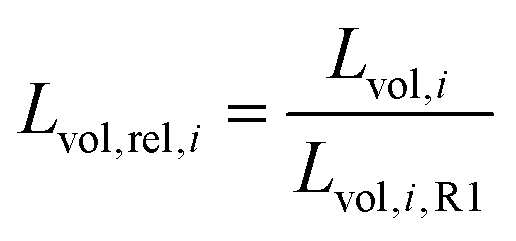 | (5) |
 | (6) |
 | (7) |
 | (8) |
3 Results and discussion
3.1 Synthesis of catalysts with varying support texture
A series of five ZrO2 supports was synthesized with different calcination temperatures ranging from 350 °C to 1000 °C (supports Z350 to Z1000, Table S1, ESI†). Following Ni deposition, the resulting Ni/ZrO2 catalysts were named accordingly as NZ350 to NZ1000 (Table 1). The aim was to prepare a catalyst series with distinct textural properties of the support (e.g., ABET, VP) dependent only on one parameter – variation in calcination temperature (Fig. 1). In addition, the supported Ni particles were intended to exhibit a similar initial size distribution across all catalysts.| Catalysts | ω Ni/wt% | A BET/m2 g−1 | V p/cm3 g−1 | L vol,ZrO2/nm | L vol,NiO/nm | L vol,Ni/nm | D/% | TOF/s−1 |
|---|---|---|---|---|---|---|---|---|
| NZ350 | 14.1 | 66 | 0.23 | 10.4 | 10.7 | 34 | 11.0 | 2.3 × 10−2 |
| NZ500 | 13.2 | 53 | 0.14 | 13.6 | 9.6 | 32 | 11.2 | 1.8 × 10−2 |
| NZ650 | 14.7 | 33 | 0.070 | 23 | 10.3 | 26 | 12.9 | 1.2 × 10−2 |
| NZ800 | 15.2 | 26 | 0.055 | 45 | 8.1 | 31 | 11.5 | 7.7 × 10−3 |
| NZ1000 | 14.7 | 18 | 0.056 | 95 | 7.9 | 29 | 11.8 | 2.6 × 10−3 |
The synthesized catalysts were first characterized by various methods to study the structural and textural properties as summarized in Fig. 2 and Table 1. The support textural properties were studied by N2 physisorption to gain information about the surface area (ABET) and the pore volume (Vp) (Table S1, ESI†). A steady decrease in ABET from 93 m2 g−1 for the support calcined at 350 °C to 5 m2 g−1 for the support calcined at 1000 °C was observed (Fig. 2a, light grey). The Vp changed analogously from 0.23 cm3 g−1 to 0.009 cm3 g−1 with increasing calcination temperature (Fig. 2d, light grey). The as-prepared Ni/ZrO2 catalysts exhibited similar tendencies as the pure supports. A decrease in ABET from 66 m2 g−1 to 18 m2 g−1 (Fig. 2a, dark grey) and in Vp from 0.23 cm3 g−1 to 0.056 cm3 g−1 (Fig. 2d, dark grey) were observed with increasing calcination temperature of the initial support (Table 1). An increased density and therefore decrease in volume of the ZrO2 powders with higher support calcination temperatures was visually apparent following synthesis (Fig. 2i), which correlates with the N2 physisorption results. In general, the ABET values increased following Ni deposition, excluding for the NZ350 catalyst. The relatively large decrease in ABET between Z350 (93 m2 g−1) and NZ350 (66 m2 g−1) can be explained by the subsequent calcination of all catalysts at 500 °C after Ni deposition. For all catalysts except NZ350, 500 °C was an equal or lower temperature treatment than during the initial support calcination step prior to Ni deposition, and would therefore not be expected to induce significant changes in the support. Notably for the NZ350 catalyst ABET (66 m2 g−1) and Vp (0.23 cm3 g−1) remained higher than for NZ500 (52 m2 g−1, 0.14 cm3 g−1). At higher support calcination temperatures (650–1000 °C) the catalysts exhibited larger ABET and Vp than the pure support, which can be explained by additional contribution of the nickel to the surface area and pore volume. The ABET of the catalysts is comparable to other Ni/ZrO2 catalysts in literature. Everett et al. reported 66 m2 g−1 (calcined at 500 °C) for a 5 wt% Ni/ZrO2 catalyst with commercial m-ZrO2 from Saint-Gobain.40 Martinez et al. presented surface areas of 53 and 19 m2 g−1 for 20 wt% Ni/ZrO2 catalysts (calcined at 500 °C) with mixed ZrO2 phases (m-ZrO2 and t-ZrO2).41 Pandey et al. reported Ni and Ni–Fe supported on m-ZrO2 (calcined at 500 °C) with surface areas between 34 and 23 m2 g−1.42
 | ||
| Fig. 2 (a) ABET obtained by N2 physisorption of the pure calcined supports (light grey) and as-prepared calcined catalysts (dark grey). (b and c) Pore width (wP) distribution obtained by MIP for as-prepared calcined NZ500 and NZ1000. (d) Vp obtained by N2 physisorption of the pure calcined supports (light grey) and as-prepared calcined catalysts (dark grey). (e and f) TEM images of the catalysts NZ500 and NZ1000 after CO2 methanation. (g) ZrO2 crystallite sizes (Lvol,ZrO2) of the pure calcined supports (light grey) and the as-prepared calcined catalysts (dark grey). (h) Crystallite size Lvol for NiO of the as-prepared catalysts (Ni/ZrO2-ap) and Ni of the spent catalysts after CO2 methanation (Ni/ZrO2-ar). (i) An equal mass of the as-prepared catalysts NZ500, NZ650, NZ800 and NZ1000 showing volume loss and colour change. | ||
The pore structure of the as-prepared catalysts was further studied by mercury intrusion porosimetry (MIP) (Fig. S1–S5†). The wP distribution for NZ500 and NZ1000 are depicted in Fig. 2b and c. A shift to larger mean wP values (from 17 nm to 43 nm) was observed with rising calcination temperature. In addition, TEM analysis showed growth and agglomeration of the support particles during the aging process, leading to the formation of intraparticle voids (Fig. 2e and f). For the NZ1000 catalyst, the support structure mainly consists of large agglomerated particles with no visible internal porosity. Compared to NZ1000, the NZ500 catalyst has smaller particles, which seem to be packed more densely. Hence, an increase of intraparticle voids with rising support calcination temperature can be observed combined with particle growth, explaining the wP changes from MIP. This further substantiates the N2 physisorption results which showed a strong decrease of surface area, and the visually apparent increase in density of supports calcined at higher temperature.
The trend of increasing support particle sizes can further be confirmed with laboratory powder X-ray diffraction (PXRD). The PXRD for the calcined supports, as-prepared and spent catalysts are shown in Fig. S6–S9, ESI.† For the support almost all reflections can be assigned to m-ZrO2 with small contributions of t-ZrO2 for the supports Z25 and Z350 (Fig. S6 and S7, ESI†). The t-ZrO2 phase is not detected in NZ350, which might be related to the calcination after Ni deposition. By using Rietveld refinement, the crystallite sizes (Lvol) of ZrO2 for the pure support and the catalysts were determined (Tables S2–S7, ESI†). The zirconia crystallite size (Lvol,ZrO2) increased for the pure support together with temperature from ≈10 nm (Z350) to ≈116 nm (Z1000) (Fig. 2g, light grey, Table S2, ESI†). For the catalysts (dark grey) a similar trend with increasing Lvol,ZrO2 from ≈10 nm (NZ350) to ≈95 nm (NZ1000) was observed (Fig. 2g, Table 1). The difference in Lvol,ZrO2 for Z1000 (116 nm) and NZ1000 (95 nm) could be due to support changes during the synthesis and/or calcination after Ni deposition. In summary, the textural properties ABET and VP decreased, whereas Lvol,ZrO2 increased with rising support calcination temperature. Therefore, the temperature can be used as a single parameter to adjust the textural properties of the initial supports prior to Ni deposition.
Despite the textural differences of the supports, it was aimed to have similar Ni loading and particle size across the catalyst series. Elemental analysis showed that all catalysts have a comparable Ni loading of ≈13–15 wt% (Table 1). The crystallite sizes of NiO (as-prepared catalysts) and Ni (spent catalysts) were investigated using PXRD. A comparable Lvol,NiO for all as-prepared catalysts of ≈10 nm was determined (Fig. 2h, light blue). After the reaction, Lvol,Ni was ≈32 nm for all catalysts except NZ650 (26 nm) (Fig. 2h, dark blue). The crystallite growth from 10 to 32 nm could be due to agglomeration during reduction and/or reaction. The size of Ni crystallites is comparable to other Ni/ZrO2 catalysts reported in literature. Pandey et al.42 reported Lvol,Ni of 58 nm for a 10 wt% Ni/ZrO2 catalyst and Ren et al.43 34 nm for a 30 wt% Ni/ZrO2 catalyst.
For a 17 wt% Ni/Al2O3 catalyst, which was synthesized with the same method used in this study, Lvol,Ni of 5.7 nm was reported.17,26 However the different support material (Al2O3) limits comparability to this study, while tuning the Ni particle size towards lower values was not the focus of the present work. A common technique to obtain particle size distributions is TEM/STEM. However, the obtained contrast between ZrO2 and Ni was too low to distinguish these from each other, which did not allow for reasonable particle size analysis using TEM/STEM. Therefore, EDX mapping was applied for specific areas of selected catalysts (Fig. S10–S13, ESI†). Both large (≥100 nm) and small Ni particles (10–30 nm) were found for all catalysts by EDX. With the large Ni particles, the number of obtained images was not sufficient to retrieve statistically meaningful particle size distributions. Therefore, solely PXRD is used to quantitatively estimate the particle/crystallite size based on structural refinement. In conclusion, a catalyst series (NZ350–1000) with similar Ni centers was obtained based on ZrO2 supports with varying textural properties. This series allows to further investigate the activity and stability of the catalysts depending on the support properties.
3.2 Catalyst testing results
Catalytic activity for CO2 methanation was monitored at three reaction temperatures (Tr: 250 °C, 350 °C, 400 °C) (Fig. 3a). At 250 °C between 1 to 8% XCO2 was reached, which is far below the equilibrium conversion of 97% and therefore suitable for probing catalyst stability.44 A trend of decreasing XCO2 with increasing calcination temperature of the support could be observed during reaction at 250 °C. At higher temperatures (350 °C, 400 °C) no specific trend was observed, except for the strong decrease in activity for NZ1000. At 350 °C conversions between 73% and 78% were obtained for NZ350 to NZ800 and 33% for NZ1000. At 400 °C the NZ1000 catalyst showed an increase in activity with 58% XCO2, whereas the rest of the series achieved similar XCO2 as for 350 °C (≈78%), close to but significantly below equilibrium conversion (Fig. 3a, black stars). For better comparison of the activity data, the conversion was normalized to the Ni amount of the tested catalyst (Fig. 3b). With the normalization a similar trend to the one observed at 250 °C can be found also at higher temperatures for the catalysts NZ800 and NZ1000, which exhibited decreased activity compared to NZ350, NZ500 and NZ650. | ||
| Fig. 3 Catalytic activity of the Ni/ZrO2 series in CO2 methanation at 250 °C, 350 °C and 400 °C. (a) CO2 conversion (XCO2) with equilibrium conversion (black stars). (b) XCO2 normalized to the Ni amount in the reactor. | ||
All catalysts exhibited 100% selectivity towards CH4 at 250 °C (Fig. S15, ESI†). With increase of Tr, the formation of CO slightly increases. CO formation is probably due to the reverse water gas shift (RWGS) reaction, which is endothermic and therefore more favored at higher temperatures.44 The catalysts showed almost no differences in CH4 selectivity depending on the support calcination temperature except for NZ1000. At 350 °C CH4 selectivity decreased from 99% (NZ350) to 86% (NZ1000), while at 400 °C a decrease from 97% (NZ350) to 84% (NZ1000) was found. The decline in SCH4 is accompanied with a rise in SCO from 1% (NZ350) to 14% (NZ1000) at 350 °C and from 3% (NZ350) to 16% (NZ1000) at 400 °C. A similar phenomena was observed by Beierlein et al. by applying rapid aging tests on Ni/Al2O3 catalysts.18 They could observe an increase in SCO for samples aged at higher temperatures. Zhao et al.45 used different combustion methods to synthesize 15 wt% Ni/ZrO2 catalysts and tested them in CO2 methanation. They obtained 1–15% XCO2 at 250 °C, 40–80% at 350 °C and 60–82% at 400 °C. The selectivity towards CH4 was 100% at 250 °C and at higher temperatures a slight decrease (93%) for some catalysts was observed.
For better comparison with literature the turnover frequency (TOF) was calculated (see details in ESI†) for Tr of 250 °C with XCO2 far below the thermodynamic equilibrium (Table 1). The dispersion (D) was calculated using the Lvol,Ni obtained from Rietveld refinements of the spent samples. For all catalysts a similar D ranging from 11 to 12% was determined (Table 1). The calculated TOF values ranged between 2.3 × 10−2 s−1 for NZ350 and 2.6 × 10−3 s−1 for NZ1000 (Table 1). Tan et al.46 reported a comparable TOF of 3.8 × 10−3 s−1 at 200 °C for a 12 wt% Ni/ZrO2 catalyst with similar XCO2 as at 250 °C reported here. The value is similar to NZ1000 at 250 °C with 2.6 × 10−3 s−1, which showed the lowest activity in this series compared to NZ350 with a nearly one order of magnitude higher TOF of 2.3 × 10−2 s−1. Jia et al.47 obtained a TOF value of 4.0 × 10−2 s−1 for a 10 wt% Ni/ZrO2 catalyst at 235 °C which is similar to NZ350. At 200 °C the group of Liu et al.48 found a TOF of 4.9 × 10−3 s−1 with a similar XCO2 as for 250 °C in this study. In conclusion, the activity of the catalysts can be influenced by the support calcination temperature, while showing comparable performance to literature values. Notably, the loading, dispersion, and crystallite size of nickel was similar for all catalysts. A decrease in XCO2 and TOF with increased aging temperature can be observed, especially at Tr of 250 °C. Following activity tests, the stability of the catalyst during thermal aging experiments was further investigated to distinguish between deactivation caused by changes of the support or Ni particles.
3.3 Operando SPXRD experiments
Operando SPXRD studies were carried out for the Ni/ZrO2 catalyst series to investigate various potential deactivation mechanisms. Distinctions in catalyst thermal stability were expected due to the different initial calcination temperatures of the ZrO2 supports prior to Ni deposition, and the observed differences in catalytic performance depending on the support properties. The experimental setup and applied temperature profile are shown in Fig. 4a and b, respectively. The acquired MS data for the samples are shown in Fig. S17, S19, S21, S23 and S25, ESI.† All catalysts were first activated in 25% H2/He at 500 °C for 2 h with the SPXRD results shown in Fig. 4d. During activation the NiO reflexes completely vanish and Ni reflexes appear in the same temperature region (≈200 to 250 °C) for all five samples, indicating complete reduction to Ni. | ||
| Fig. 4 (a) Scheme of the experimental setup used at the beamline. (b) Overview of the experimental conditions during operando SPXRD experiments with activation in 25% H2/He (blue), reaction conditions 5% CO2/20% H2/He (green) and thermal deactivation cycles under reaction conditions (red) with each reaction condition step labelled as R1–4. (c) CO2 conversion normalized by the Ni mass inside the reactor for operando SPXRD at reaction step R1. (d) 2D overview plots of the diffraction patterns showing the Ni (∘) formation from NiO (*) during activation of the five samples as well as (e) under reaction conditions and thermal deactivation cycles, showing no crystalline phase changes. | ||
The activation was followed by switching to reaction conditions 5% CO2/20% H2/He at 400 °C for 30 min, which is labelled as the first reaction step R1. R1 is used to determine the initial activity of the catalysts based on the determined XCO2 and XH2 (Fig. S18, S20, S22, S24 and S26, ESI†). The XCO2/Ni is shown in Fig. 4c. The activity differences between the samples show comparable trends to those found during laboratory testing (Fig. 3). R1 was followed by artificial thermal deactivation cycles (Fig. 4b), with temperatures of 500 °C, 575 °C and 625 °C. After each cycle the activity of the catalysts was measured again at 400 °C for 15 min, labelled as reaction steps R2, R3 and R4, respectively. This allows to determine catalyst deactivation depending on three different applied temperatures. During thermal deactivation cycles no changes of the crystalline phase composition were found as shown by the SPXRD in Fig. 4e. Minor changes of the reflex positions (Fig. 4e), can be attributed to temperature induced lattice expansion or contraction, which matches the temperature program applied to the capillary.
3.4 Crystallite size changes during operando SPXRD
In general, textural properties such as surface area are strongly connected to catalyst performance. In particular adsorption of reactants or products takes place on the surface, but the available surface area is also linked to stabilization of metal nanoparticles. The specific surface area (SSA) can be calculated from particle sizes as shown in eqn (7), where for crystalline compounds the particle size can be approximated by the crystallite size. However, this relation is only valid for spherical particles. In the present case, two distinct crystalline phases are present, which limits the definition of an overall particle size based on the crystallite sizes as required for eqn (7). Additionally, exact knowledge about ρ is hardly possible during operando experiments due to textural changes of the sample, which might lead to density changes. Therefore, in a first step an average crystallite size (Lvol,avg) is calculated for the catalysts in the initial calcined state based on eqn (4) taking into account Lvol,ZrO2 and Lvol,NiO. Details on the respective refinements are provided in the ESI.† This Lvol,avg is then correlated to ABET (Fig. 5a), which is available for the catalysts in the initial state. This correlation is fitted by eqn (8), which is similar to eqn (7) without the requirement of knowing the exact value of ρ and approximating it via a fitting parameter. The fitting results are shown in Fig. 5a with the theoretical curve for pure monoclinic ZrO2 (eqn (7), ρ = 5.86 g cm−3 (ref. 36)) showing a similar trend.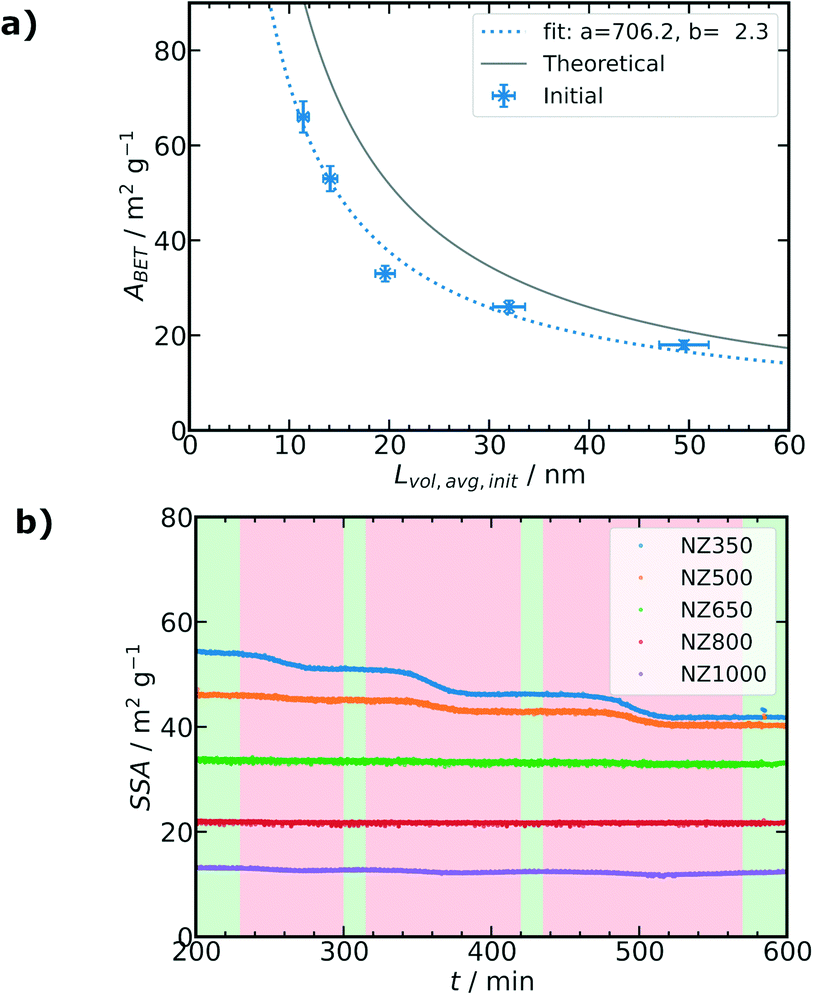 | ||
| Fig. 5 (a) Fitted correlation (blue dotted line) of the measured ABET to Lvol,avg (blue crosses, mass fraction weighted average of Lvol,ZrO2 and Lvol,NiO) initially at the start of the operando SPXRD experiments based on eqn (8) with fitting variables a and b, error bars estimated as 5% standard deviation. Theoretical relation of ABET and Lvol for spherical particles based on eqn (7) (grey line). (b) Changes of SSA during reaction conditions and thermal deactivation cycles of the operando SPXRD experiments as calculated from Lvol,avg (mass fraction weighted average of Lvol,ZrO2 and Lvol,Ni) and the fit values from (a) according eqn (8) (shaded areas as Fig. 4b). | ||
Based on the fitted values it is possible to calculate the SSA of the catalysts during the operando SPXRD experiments using Lvol,avg of the ZrO2 and Ni phases present after activation. However, it should be noted that the values are an approximation as the correlation was obtained for the calcined state and not the activated state. Nevertheless, as all catalysts show a very similar phase composition and no crystalline phase changes occur during reaction conditions and thermal deactivation cycles, SSA can be approximated in this way to unravel textural changes of the catalysts. The same correlation was also studied for the pure supports as shown in Fig. S39, ESI,† resulting in similar fit values supporting the correlation approach. The SSA starting from R1 evolves differently for the catalysts as shown in Fig. 5b. Similarly as for ABET analysis (Fig. 2a), it can be observed that the initial SSA of the catalysts (at t = 200 min) decreased with increasing calcination temperature of the ZrO2 support before Ni deposition. The calculated initial SSA for all catalysts is lower compared to the measured ABET values of the initial calcined samples (Table 1), which can be attributed to the increased crystallite sizes of Ni compared to NiO by a factor of ≈3 (for details see Tables S8–S13 ESI†). For NZ350 a pronounced step-wise decrease of the SSA can be observed, with each step related to one of the thermal deactivation cycles. NZ500 exhibits a decrease only significantly with the second and third thermal deactivation step, while NZ650, NZ800 and NZ1000 show no significant reduction of SSA following the thermal deactivation cycles. Besides the study of the overall textural changes based on the calculated SSA, Rietveld refinement together with microstructural analysis allows to investigate crystallite size changes of each crystalline phase individually. Fig. 6a and b show the evolution of the selected (111) reflex for ZrO2 and Ni phases during operando SPXRD, respectively. The different peak shapes of the ZrO2 (111) reflection (Fig. 6a) illustrate the different ZrO2 crystallite sizes depending on the calcination temperature of the support prior to Ni deposition. A representative pattern during each of the reaction steps R1 to R4 is highlighted. For NZ350 and NZ500 significant changes of the reflex depending on the reaction step can be observed, which are more pronounced for NZ350. This indicates crystallite growth of the ZrO2 phase during thermal deactivation cycles. For NZ650, NZ800 and NZ1000 no significant variation of the reflex can be directly observed. In the case of the Ni (111) reflex (Fig. 6b), all samples show significant changes depending on the reaction step. The variations become more pronounced with higher calcination temperature of the ZrO2 support. Additionally to the qualitative description of the peak shape, a microstructural refinement as part of Rietveld refinement of the SPXRD provides quantitative information about crystallite size changes during the thermal deactivation cycles. Fig. 6c–e show the evolution of Lvol,ZrO2, Lvol,Ni and the mass fraction weighted average Lvol,avg of both, respectively. Complementary to the absolute values, Fig. 6f–h show the relative changes compared to the initial reaction step R1, respectively, indicating sequential changes during the subsequent thermal aging cycles.
 | ||
| Fig. 6 (a) Changes of the ZrO2 (111) reflex during thermal deactivation cycles highlighted by R1, R2, R3 and R4 steps, (b) analogous changes of the Ni (111) reflex. Evolution of different crystallite sizes obtained from sequential Rietveld refinements for the different catalysts during operando SPXRD experiments: (c) Lvol,ZrO2, (d) Lvol,Ni and (e) Lvol,avg (mass fraction weighted average of Lvol,ZrO2 and Lvol,Ni). Relative changes of Lvol normalized to the initial reaction step R1 according eqn (5): (f) ZrO2 phase, (g) Ni phase and (h) average crystallite size of the catalysts. Shaded areas as Fig. 4b. | ||
L vol,ZrO2 depends on the calcination temperature of the support prior to Ni deposition and ranges from about 9 nm to 70 nm in the initial reaction step R1. During thermal deactivation cycles a step-wise increase of Lvol,ZrO2 can be observed for each temperature step for NZ350. Although all catalysts were initially activated for 2 h at 500 °C, for NZ350 a crystallite growth can be observed already at the first thermal deactivation temperature of 500 °C. For NZ500 a significant increase of the crystallite size is only detected with the second and third thermal deactivation steps. The catalysts NZ650, NZ800 and NZ1000 show no significant increase of Lvol,ZrO2 during those cycles. The reversible increase of Lvol,ZrO2 of NZ1000 during the thermal deactivation cycles (Fig. 6c and f) might be explained by larger crystalline domains due to lattice expansion or reversible ordering/disordering, but the differences are small compared to the refinement error and could also be caused by refinement artifacts. The evolution of Lvol,Ni is contrary to the ZrO2 phase. The initial Lvol,Ni at reaction step R1 range from about 25 to 35 nm for the different catalysts (Fig. 6d). For NZ350, NZ500 and NZ650, the Lvol,Ni only slightly increased with thermal deactivation after the second reaction step R2 (Fig. 6d and g). This is different for the NZ800 and NZ1000 samples where Lvol,Ni already increased with the first thermal deactivation step, while the behavior is most pronounced for NZ1000. The changes can be also observed for the Lvol,avg in Fig. 6e and h. As Lvol,avg represents the mass fraction weighted average for ZrO2 and Ni it mainly reflects changes of the ZrO2 phase. Therefore, here the NZ350 and NZ500 samples show the most pronounced changes. On the other hand, it can be observed that the strong changes of Lvol,Ni result in a significant increase of the Lvol,avg for NZ1000. The strong Ni crystallite size increase in NZ1000 might be explained by the support reaching such a small SSA that Ni particle growth is strongly favored. In contrast, NZ650 and NZ800 were less impacted as a whole by the thermal deactivation procedure.
In summary, the crystallite size changes during thermal deactivation cycles could be quantified and show for NZ350 and NZ500 mainly changes of Lvol,ZrO2, while for NZ800 and NZ1000 especially changes of Lvol,Ni were observed.
3.5 Correlation of crystallite size and thermal deactivation
The conversion of CO2 and H2 obtained from MS analysis were used to determine the activity of the catalysts during operando SPXRD. The absolute values for XCO2 and XH2 at the different reaction steps R1–4 are shown in Fig. S27 and S28, ESI,† respectively. The initial conversions for the catalysts at R1 are in the range of 57 to 67%. Therefore, it can be reasonably assumed that the reaction is performed far from thermodynamic equilibrium conditions or full conversion (≈80%, Fig. 3a), which is a prerequisite to detect possible catalyst deactivation. The changes of XCO2 and XH2 in comparison to the initial activity at R1 (ΔX) are shown in Fig. 7a and d, respectively. All studied catalysts show a step-wise deactivation during the thermal deactivation cycles. Therefore, the deactivation can be clearly related to the temperature applied during each of the cycles. Less pronounced deactivation occurs for the NZ650 and NZ800 catalysts, which suffer a loss of ≈1% in XCO2 and XH2 after thermal deactivation cycles. The other three catalysts lost more than 2% in XCO2 and XH2, while there are differences in the extent of activity loss between XCO2 and XH2. | ||
| Fig. 7 (a) Changes of the XCO2 for each reaction step as difference to the initial activity at R1. (b) Relative changes ΔLvol,rel,ZrO2 of the ZrO2 crystallite size according eqn (6) depending on the reaction step referenced to R1. (c) Relative CO2 conversion XCO2,rel (eqn (3)) divided by the relative crystallite size Lvol,rel (eqn (5)) for Ni and ZrO2 phases. (d) Changes of the XH2 for each reaction step as difference to the initial activity at R1. (e) Relative changes ΔLvol,rel,Ni of the Ni crystallite size according eqn (6) depending on the reaction step with R1 as reference. (f) Relative H2 conversion XH2,rel (eqn (3)) divided by the relative crystallite size Lvol,rel (eqn (5)) for Ni and ZrO2 phases. | ||
Besides the activity differences, the changes of Lvol,rel,ZrO2 and Lvol,rel,Ni in reference to the initial reaction step R1 are shown in Fig. 7b and e, respectively. The Lvol,rel,ZrO2 of NZ350 is increased by about 40% and for NZ500 by about 17% compared to the initial values at R1, while the other three catalysts show only minor changes of the Lvol,rel,ZrO2 after thermal deactivation cycles of less than 3%. This is different for Lvol,rel,Ni, where NZ1000 shows an increase of almost 100% followed by the NZ800 sample of ≈20%. The other three samples show increases of ≈10% or below.
To estimate the influence of the different Lvol changes on catalyst deactivation, the relative conversions (XCO2,rel, XH2,rel, eqn (3)) are correlated with the relative crystallite sizes (Lvol,rel,ieqn (5)) with respect to the initial reaction step R1 in Fig. 7c and f, respectively. This correlation analysis allows to retrieve information regarding which of the two phases might have a stronger influence on catalyst deactivation. For both evaluated conversions, the strongest observed correlations are for the changes of Lvol,Ni of the NZ1000 catalyst, followed by the correlations of Lvol,ZrO2 for NZ350. In general, it can be observed that there is a greater correlation between Lvol,Ni and the conversions for NZ1000, NZ800 and NZ650. This is different for NZ350 and NZ500, where the correlation of Lvol,ZrO2 with the conversions is larger than Lvol,Ni.
3.6 Overall discussion of the thermal deactivation
The five different studied catalysts essentially differ by the calcination temperature and consequently the surface area and pore volume of the ZrO2 support prior to Ni deposition, while the following calcination and activation steps are identical for all samples. In addition, the initial properties of the active metal Ni particles in terms of loading, dispersion, and crystallite size were similar for all catalysts following synthesis and calcination. As observed in the operando SPXRD the activity trends of the catalysts after activation at R1 are similar compared to laboratory tests, showing generally decreasing activity with higher calcination temperature of the support, especially for NZ1000. The activated catalysts at R1 are mainly distinguished by their Lvol,ZrO2, while the Lvol,Ni is in a comparable range for all catalysts with minor differences. The Lvol,Ni is relatively large at around 30 nm. Therefore, it can be reasonably assumed that no major differences in the reaction mechanism depending on the Ni particle size as shown by Vogt et al.15 are present, since the type of surface sites present are similar and deactivation as reported by Mutz et al.14 should be negligible. The resulting activity should mainly depend on the available active surface area of the catalysts.Typically, the active surface area and the specific surface area of catalysts are determined via gas sorption methods in post mortem analysis.49,50 However, applying these methods under operando conditions is hardly possible. Contrary to this, PXRD with microstructural refinement as shown here allows to determine textural changes of the ZrO2 support and Ni nanoparticles as long as there are no complex phase changes occurring during thermal deactivation. In the present case, the five catalysts show different crystallite size changes and deactivation behavior depending on the calcination temperature of the support. The most pronounced catalyst deactivation was observed for NZ350, NZ500 and NZ1000 catalysts (Fig. 7a and d), while for those three samples also the strongest crystallite size changes were found. For NZ350 and NZ500, mainly an increase of Lvol,ZrO2 and thus sintering of the support was detected, while for NZ1000 the Lvol,Ni after thermal aging was much larger and thus Ni particle sintering occured. Based on the correlation analysis shown in Fig. 7c and f the major reason for catalyst deactivation might be divided into: (i) dominant changes of the ZrO2 support, (ii) dominant changes of the Ni phase and (iii) an intermediate region with both effects as schematically illustrated in Fig. 8.
 | ||
| Fig. 8 Summary of the different proposed deactivation mechanism found by operando SPXRD. NZ350 and NZ500 show predominant ZrO2 sintering and NZ650, NZ800 and NZ1000 dominant Ni sintering and an expected intermediate region as origin of deactivation. Ni blue, ZrO2 grey. The grey color gradient illustrates changing textural properties of the support associated to a decrease in surface area and pore volume due to sintering. | ||
Despite that all catalysts were activated at 500 °C for 2 h, NZ350, NZ500 and NZ1000 show a slight deactivation after only 15 min at 500 °C under reaction conditions. The deactivation of NZ350 and NZ500 can be mainly related to textural changes of the ZrO2 support. For NZ650, NZ800 and NZ1000 sintering of Ni particles appears to be the main reason for deactivation, with NZ650 and NZ800 showing highest stability among the tested samples. The stability of NZ650 and NZ800 might be related to the calcination temperature of the supports during catalyst preparation, which were higher than the subsequent deactivation temperatures. The deactivation due to Ni sintering can be explained by a decrease in the active metal surface area. Literature studies have discussed changes of Ni nanoparticles, the support material and a complex interplay of the effects as reasons for deactivation.18–22 However, deconvolution of these effects was not previously possible due to entirely post mortem analysis and thus the missing link with activity data provided by the operando analysis shown here. Sintering of the ZrO2 support as shown in the present work might influence the catalyst stability in different ways. For Ni/ZrO2 the associative mechanism for CO2 methanation would require CO2 adsorbates on the ZrO2 support to take part in the reaction.7,8,10 Therefore the amount of CO2 adsorbed on the support is lower with decreasing support surface area leading to lower catalytic activity. Furthermore, it is possible that the textural changes of the support can result in blocking of active Ni surface sites.
Additionally, SPXRD did not reveal formation of bulk NiO species and the quantitative phase analysis from Rietveld refinement matches the values obtained from elemental analysis. Thus, a fully reduced state of Ni throughout the experiment was assumed. The formation of surface NiO cannot be fully excluded by the applied methods, however due to the relatively large and similar Ni particle sizes in the catalyst series, similar redox properties are assumed for all catalysts. Possible deactivation due to formation of surface oxide species, if this occurs, should therefore be similar for all five catalysts due to identical reaction conditions. The significant quantity of H2 in the feed makes formation of NiO species unlikely in any case, also according to previous studies using operando XAS.13,14,16,17 In these previous studies, NiO was only significantly present during H2-dropout tests. However, to unravel the different potential effects in more detail, further kinetic and spectroscopic studies of the active and deactivated catalysts would be required. In the present study, the focus was on presenting SPXRD as a single powerful and relatively routinely available method to differentiate thermal deactivation effects sensitive towards textural and crystallographic changes under operando conditions, with the possibility to distinguish support and metal nanoparticle changes.
4 Conclusions
In the present study, a bottom-up designed series of five different Ni/ZrO2 catalysts was prepared via an adapted homogeneous deposition precipitation method. The catalysts are based on a commercial ZrO2 support material, which was calcined at different temperatures prior to Ni deposition in order to change the textural properties, i.e., specific surface area and pore volume of the support systematically. The catalysts were characterized by conventional laboratory methods confirming a decrease in surface area and pore volume with increasing support calcination temperature. Laboratory catalytic tests for CO2 methanation showed comparable activity towards literature reported samples with TOF ranging from 2.3 × 10−2 s−1 to 2.6 × 10−3 s−1 at 250 °C. The catalysts exhibit similar Ni crystallite size (≈30 nm), dispersion and Ni loading. As the five catalysts essentially only differ by a single parameter, the support calcination temperature, the series is well suited for thermal deactivation studies.During operando SPXRD studies with artificial thermal deactivation cycles, the catalysts showed different extent of deactivation with NZ350, NZ500 and NZ1000 showing the strongest activity loss and NZ650 and NZ800 the least. No crystalline phase changes were observed during thermal deactivation, but rather crystallite size changes of the ZrO2 support and Ni nanoparticles. These changes were dependent on the ZrO2 calcination temperature along with the related textural properties. The five catalysts can be distinguished into samples that mainly deactivate due to changes of the support (NZ350 and NZ500), or changes of the Ni crystallite size (NZ650, NZ800 and NZ1000). The activity loss associated with Ni sintering can be explained by a decrease of the specific metal surface area. For the samples with dominant support sintering, either loss in CO2 adsorption sites due to decreased support surface area or blocking of accessible metal surface area can be an explanation for the observed deactivation.
The presented approach of using operando SPXRD to study catalyst deactivation due to textural changes of the active site (metal nanoparticles) or the support is a valuable tool to understand temperature induced catalyst deactivation. In general, it can be emphasized to not only focus on changes of the metal nanoparticles, but also to consider the role of the support. This approach can be used in general to differentiate influences on catalyst stability under reaction conditions, which are not directly accessible with established methods including gas sorption or electron microscopy. The possibility to follow catalysts changes while they occur is especially important for potential dynamic process operation related to fluctuating renewable energy supply.51 There, (ir)reversible changes of the catalyst structure are difficult to follow by ex situ characterization, but are important to reveal for a fundamental understanding of catalyst behavior.51 In future, promising developments of 3D spatially-resolved techniques, e.g., operando XRD or PDF tomography,52,53 may provide additional information about the location of the textural changes with respect to the reactor position (e.g., due to hot-spots) or within specific catalyst particles. Additionally, recent progress in hard X-ray nanotomography methods allows to investigate textural sample changes in situ approaching routinely sub 50 nm resolution, which can be sufficient to reveal the processes observed here.54
Author contributions
Conceptualization, SW; data curation, MS, SW, TS; formal analysis, MS, SW; funding acquisition, TS; investigation, MS, SW, LK, TS; methodology, MS, SW; project administration, SW, JDG, TS; resources, JDG, TS; supervision, SW, JDG, TS; validation, MS, SW, JDG, TS; visualization, MS, SW; writing–original draft preparation, MS, SW; writing–review and editing, all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Gefördert durch die Deutsche Forschungsgemeinschaft (DFG)-406914011, funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)-406914011. Dr. Marc-André Serrer is acknowledged for support of the laboratory catalyst testing experiments. We thank Dr. Eduardo Barbosa and Dr. Sumeet Kale (Karlsruhe Institute of Technology) for performing N2 sorption experiments. Thanks to Heike Rudzik, Ken Luca Abel and Prof. Roger Gläser (Universität Leipzig) for providing ICP-OES analysis. We acknowledge Maximilian Franz and Prof. Dirk Enke (Universität Leipzig) for carrying out MIP experiments. We thank Dr. Florian Maurer for assistance in the graphical preparation of the schemes. This work was partly carried out with the support of the Karlsruhe Nano Micro Facility (KNMF), a Helmholtz Research Infrastructure at Karlsruhe Institute of Technology (KIT), which provided access to TEM instruments via proposal 2019-022-026980. The authors thank Xiaohui Huang, Dr. Reihaneh Pashminehazar and Prof. Christian Kübel (Karlsruhe Institute of Technology) for support with the TEM experiments. The authors acknowledge the Paul Scherrer Institut, Villigen, Switzerland for provision of synchrotron radiation beamtime at beamline X04SA-Materials Science of the Swiss Light Source and thank Dr. Nicola Casati, Michael Lange (Paul Scherrer Institut) and Shweta Sharma (Karlsruhe Institute of Technology) for support during the beamtime. Open access funding enabled by the Read & Publish agreement between Karlsruhe Institute of Technology and the Royal Society of Chemistry.Notes and references
- C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17–60 CrossRef CAS.
- M. D. Argyle and C. H. Bartholomew, Catalysts, 2015, 5, 145–269 CrossRef CAS.
- J. Bremer and K. Sundmacher, React. Chem. Eng., 2019, 4, 1019–1037 RSC.
- J. Bremer, K. H. G. Rätze and K. Sundmacher, AIChE J., 2017, 63, 23–31 CrossRef CAS.
- M. Serrer, M. Stehle, M. L. Schulte, H. Besser, W. Pfleging, E. Saraci and J.-D. Grunwaldt, ChemCatChem, 2021, 13, 3010–3020 CrossRef CAS.
- M. A. Aziz, A. A. Jalil, S. Triwahyono and A. Ahmad, Green Chem., 2015, 17, 2647–2663 RSC.
- P. Frontera, A. Macario, M. Ferraro and P. Antonucci, Catalysts, 2017, 7, 59 CrossRef.
- S. Rönsch, J. Schneider, S. Matthischke, M. Schlüter, M. Götz, J. Lefebvre, P. Prabhakaran and S. Bajohr, Fuel, 2016, 166, 276–296 CrossRef.
- M. Younas, L. Loong Kong, M. J. Bashir, H. Nadeem, A. Shehzad and S. Sethupathi, Energy Fuels, 2016, 30, 8815–8831 CrossRef CAS.
- B. Miao, S. S. K. Ma, X. Wang, H. Su and S. H. Chan, Catal. Sci. Technol., 2016, 6, 4048–4058 RSC.
- B. Mutz, P. Sprenger, W. Wang, D. Wang, W. Kleist and J.-D. Grunwaldt, Appl. Catal., A, 2018, 556, 160–171 CrossRef CAS.
- R. P. Struis, T. J. Schildhauer, I. Czekaj, M. Janousch, S. M. Biollaz and C. Ludwig, Appl. Catal., A, 2009, 362, 121–128 CrossRef CAS.
- B. Mutz, H. W. Carvalho, S. Mangold, W. Kleist and J.-D. Grunwaldt, J. Catal., 2015, 327, 48–53 CrossRef CAS.
- B. Mutz, A. M. Gänzler, M. Nachtegaal, O. Müller, R. Frahm, W. Kleist and J.-D. Grunwaldt, Catalysts, 2017, 7, 279 CrossRef.
- C. Vogt, E. Groeneveld, G. Kamsma, M. Nachtegaal, L. Lu, C. J. Kiely, P. H. Berben, F. Meirer and B. M. Weckhuysen, Nat. Catal., 2018, 1, 127–134 CrossRef CAS.
- M. A. Serrer, K. F. Kalz, E. Saraci, H. Lichtenberg and J.-D. Grunwaldt, ChemCatChem, 2019, 11, 5018–5021 CrossRef CAS.
- M.-A. Serrer, A. Gaur, J. Jelic, S. Weber, C. A. Fritsch, A. H. Clark, E. Saraci, F. Studt and J.-D. Grunwaldt, Catal. Sci. Technol., 2020, 10, 7542–7554 RSC.
- D. Beierlein, D. Häussermann, Y. Traa and E. Klemm, Catal. Lett., 2021, 1, 3 Search PubMed.
- S. Abelló, C. Berrueco and D. Montané, Fuel, 2013, 113, 598–609 CrossRef.
- F. Koschany, D. Schlereth and O. Hinrichsen, Appl. Catal., B, 2016, 181, 504–516 CrossRef CAS.
- C. Mebrahtu, S. Perathoner, G. Giorgianni, S. Chen, G. Centi, F. Krebs, R. Palkovits and S. Abate, Catal. Sci. Technol., 2019, 9, 4023–4035 RSC.
- S. Ewald, M. Kolbeck, T. Kratky, M. Wolf and O. Hinrichsen, Appl. Catal., A, 2019, 570, 376–386 CrossRef CAS.
- M. A. Bañares, Catal. Today, 2005, 100, 71–77 CrossRef.
- R. Portela, S. Perez-Ferreras, A. Serrano-Lotina and M. A. Bañares, Front. Chem. Sci. Eng., 2018, 12, 509–536 CrossRef.
- I. L. Buurmans and B. M. Weckhuysen, Nat. Chem., 2012, 4, 873–886 CrossRef CAS PubMed.
- B. Mutz, M. Belimov, W. Wang, P. Sprenger, M. A. Serrer, D. Wang, P. Pfeifer, W. Kleist and J.-D. Grunwaldt, ACS Catal., 2017, 7, 6802–6814 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. W. Washburn, Phys. Rev., 1921, 17, 273–283 CrossRef.
- P. R. Willmott, D. Meister, S. J. Leake, M. Lange, A. Bergamaschi, M. Böge, M. Calvi, C. Cancellieri, N. Casati, A. Cervellino, Q. Chen, C. David, U. Flechsig, F. Gozzo, B. Henrich, S. Jäggi-Spielmann, B. Jakob, I. Kalichava, P. Karvinen, J. Krempasky, A. Lüdeke, R. Lüscher, S. Maag, C. Quitmann, M. L. Reinle-Schmitt, T. Schmidt, B. Schmitt, A. Streun, I. Vartiainen, M. Vitins, X. Wang and R. Wullschleger, J. Synchrotron Radiat., 2013, 20, 667–682 CrossRef CAS PubMed.
- C. Prescher and V. B. Prakapenka, High Pressure Res., 2015, 35, 223–230 CrossRef CAS.
- J.-D. Grunwaldt, M. Caravati, S. Hannemann and A. Baiker, Phys. Chem. Chem. Phys., 2004, 6, 3037–3047 RSC.
- H. M. Rietveld, Acta Crystallogr., 1967, 22, 151–152 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- DIFFRAC.SUITE TOPAS – XRD Software Bruker, 2020, https://www.bruker.com/de/products/x-ray-diffraction-and-elemental-analysis/x-ray-diffraction/xrd-software/topas.html Search PubMed.
- D. Balzar, Int. Union Crystallogr. Monogr. Crystallogr., 1999, 10, 94–126 CAS.
- D. K. Smith and W. Newkirk, Acta Crystallogr., 1965, 18, 983–991 CrossRef CAS.
- D. Rodic, V. Spasojevic, V. Kusigerski, R. Tellgren and H. Rundlof, Phys. Status Solidi B, 2000, 218, 527–536 CrossRef CAS.
- J. Rouquette, J. Haines, G. Fraysse, A. Al-Zein, V. Bornand, M. Pintard, P. Papet, S. Hull and F. Gorelli, Inorg. Chem., 2008, 47, 9898–9904 CrossRef CAS PubMed.
- https://topas.dur.ac.uk/topaswiki/doku.php?id=topas, accessed on 18/10/2021.
- O. E. Everett, P. C. Zonetti, O. C. Alves, R. R. de Avillez and L. G. Appel, Int. J. Hydrogen Energy, 2020, 45, 6352–6359 CrossRef CAS.
- J. Martínez, E. Hernández, S. Alfaro, R. López Medina, G. Valverde Aguilar, E. Albiter and M. Valenzuela, Catalysts, 2018, 9, 24 CrossRef.
- D. Pandey and G. Deo, J. Ind. Eng. Chem., 2016, 33, 99–107 CrossRef CAS.
- J. Ren, X. Qin, J. Z. Yang, Z. F. Qin, H. L. Guo, J. Y. Lin and Z. Li, Fuel Process. Technol., 2015, 137, 204–211 CrossRef CAS.
- J. Gao, Y. Wang, Y. Ping, D. Hu, G. Xu, F. Gu and F. Su, RSC Adv., 2012, 2, 2358–2368 RSC.
- K. Zhao, W. Wang and Z. Li, J. CO2 Util., 2016, 16, 236–244 CrossRef CAS.
- J. Tan, J. Wang, Z. Zhang, Z. Ma, L. Wang and Y. Liu, Appl. Surf. Sci., 2019, 481, 1538–1548 CrossRef CAS.
- X. Jia, X. Zhang, N. Rui, X. Hu and C. J. Liu, Appl. Catal., B, 2019, 244, 159–169 CrossRef CAS.
- Z. Liu, X. Ding, R. Zhu, Y. Li, Y. Wang, W. Sun, D. Wang, L. Wu and L. Zheng, ChemistrySelect, 2022, 7, e202103774 CAS.
- M. Králik, Chem. Pap., 2014, 68, 1625–1638 Search PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- K. F. Kalz, R. Kraehnert, M. Dvoyashkin, R. Dittmeyer, R. Gläser, U. Krewer, K. Reuter and J.-D. Grunwaldt, ChemCatChem, 2017, 9, 17–29 CrossRef CAS PubMed.
- A. Vamvakeros, S. D. Jacques, M. Di Michiel, D. Matras, V. Middelkoop, I. Z. Ismagilov, E. V. Matus, V. V. Kuznetsov, J. Drnec, P. Senecal and A. M. Beale, Nat. Commun., 2018, 9, 1–11 CrossRef PubMed.
- S. D. Jacques, M. Di Michiel, S. A. Kimber, X. Yang, R. J. Cernik, A. M. Beale and S. J. Billinge, Nat. Commun., 2013, 4, 1–7 Search PubMed.
- S. Weber, A. Diaz, M. Holler, A. Schropp, M. Lyubomirskiy, K. L. Abel, M. Kahnt, A. Jeromin, S. Kulkarni, T. F. Keller, R. Gläser and T. L. Sheppard, Adv. Sci., 2022, 9, 2105432 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy00972b |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |