
Selective production of formate over a CuO electrocatalyst by electrochemical and photoelectrochemical biomass valorisation†
Ping-Chang
Chuang
and
Yi-Hsuan
Lai
 *
*
Department of Materials Science and Engineering, National Cheng Kung University, No.1, University Road, Tainan City 701, Taiwan. E-mail: yhlai@gs.ncku.edu.tw
First published on 21st July 2022
Abstract
Electrochemical and photoelectrochemical valorisation of biomass is an attractive technology to provide value-added chemicals such as formate in a sustainable way. Monoclinic CuO acting as an efficient non-precious electrocatalyst for formate production from glucose and cellulose is reported. Notably, a high faradaic efficiency of 94.1 ± 1.5% and a turnover frequency of 240.2 ± 16.3 h−1 for formate production at an applied potential of 1.5 V vs. reversible hydrogen electrode (RHE) are achieved using monoclinic CuO as the electrocatalyst for glucose oxidation. The mechanism of formate generation from electrocatalytic oxidation of glucose is elucidated by product analyses and electron paramagnetic resonance (EPR) study. The results suggest that one glucose is oxidised to one formate and one pentose mediated by the generation of OH radicals during the electrolysis. Electrocatalytic formate production using purified α-cellulose and “real world” biomass wastes was also investigated. In particular, a moderate faradaic efficiency of 41.4 ± 9.7% was observed using rice straw as the substrate. Furthermore, monoclinic CuO acts as an efficient cocatalyst on a hematite photoanode, showing a faradaic efficiency of 97.3 ± 2.8% for formate production through photoelectrochemical glucose valorisation.
Introduction
Conversion of biomass waste to value-added chemicals has been envisioned as a promising strategy for generating sustainable resources and enabling carbon-neutral cycles.1 Due to the abundance and availability, glucose and cellulose are the most widely used substrates for biomass valorisation. Diverse value-added chemical feedstocks such as formic acid,1a gluconic acid, and glucaric acid2 are available from the reforming of glucose and cellulose. Among the commodities, formic acid has driven lots of attention, not only because it is highly in demand in the chemical, pharmaceutical, rubber, agriculture, and textile industries, but also because it is a promising hydrogen energy carrier due to its high volumetric capacity (53 g H2 L−1) and high chemical stability under ambient conditions.1a,3Traditional methods for producing formic acid from biomass include pyrolysis,4 wet oxidation,5 catalytic oxidation,6 and acid hydrolysis.7 Although high yields of formic acid can be produced using these conventional methods, several inherent disadvantages are also present. For example, a high temperature (450–650 °C) is generally required in pyrolysis to obtain high yields of formic acid.4 On the other hand, additional oxidants and high-pressure O2 (1–5 Mpa) are typically needed for wet and catalytic oxidation processes, respectively.5,8 In contrast, electrochemical oxidation is an emerging methodology in biomass valorisation. Electrochemical oxidation eliminates the need for hazard oxidants and is generally operated under ambient or mild conditions. The product selectivity is possibly tuned by the applied potentials.2,9 In addition, hydrogen and value-added chemicals can be co-produced through hydrogen evolution and biomass oxidation at the cathode and anode, respectively, in an electrochemical system.2,10 Last but not least, since biomass oxidation is less energy demanding than water oxidation, the efficiency of converting solar light into storable chemical energy in a photoelectrochemical (PEC) biomass cell is expected to be higher than that of a PEC water splitting cell.11
Several noble metals and alloys, such as Au,9,12 Pt,9 and PdAu,13 have been investigated as efficient electrocatalysts for converting glucose to gluconic acid and glucaric acid by the oxidation aldehyde group on C1 and the hydroxymethyl group on C6, respectively (Table S1†). However, it is essential to explore other selective and efficient electrocatalysts made of inexpensive and earth-abundant elements if (photo)electrochemical biomass valorisation is to be a viable technology. For glucose valorisation, rare examples of earth-abundant electrocatalysts, including NiFeOx,2 Cu,9 and CuBi2O4,14 have been recently reported. It is found that only Cu-based materials can effectively catalyse C–C bond breaking, resulting in the formation of formate. However, mixtures of low molar mass carboxylic acids, gluconic acid, and glucaric acid are co-present in the products.9 The low selectivity of formate might result from the complex surface states on Cu. In particular, it is known that Cu2O, CuO, and Cu(OH)2 are co-present on the surface of Cu under anodic potentials higher than 1.0 V in alkaline solutions.15
On the other hand, CuO is relatively stable compared to Cu under anodic conditions.15a CuO has also been widely used as an electrochemical sensor for glucose detection.16 The limited reports on CuO acting as a selective electrocatalyst for biomass valorisation include glycerol oxidation to dihydroxyacetone.17 However, to the best of our knowledge, there is no report on a pure phase of CuO acting as a highly selective electrocatalyst for formate production from glucose, along with detailed product analyses and mechanism discussion.
In this work, we synthesised a monoclinic CuO thin film on a fluorine-dope tin oxide (FTO) electrode. CuO shows high selectivity in terms of faradaic efficiency for formate production (FEformate) from glucose in alkaline solutions. Formate also can be electro-catalytically generated at CuO by using cellulose, a homopolymer of glucose, as the substrate with a moderate FEformate. We also demonstrate that formate production is also applicable to the electrooxidation of crude sources of cellulose, including tissue paper and rice straw. Last but not least, for the first time, CuO was decorated onto a hematite (α-Fe2O3) photoanode for selective PEC glucose valorisation. This composite photoelectrode made of only earth-abundant materials performs photoelectrochemical valorisation with a FEformate of approximately one unit.
Results and discussion
A CuO precursor electrode can be firstly prepared by spin coating a copper nitrate-containing sol–gel precursor followed by evaporating the solvent on a hot plate at 100 °C in air for 10 min. Monoclinic CuO was obtained by further heat-treating the precursor layer on a hot plate at 450 °C for one hour (see Experimental for details). The above process was repeated three times, and the resulting electrode is denoted as m-CuO. The low-magnification planar SEM image indicates that m-CuO was homogeneously covered on the FTO substrate (Fig. S1a†). The high magnitude planar SEM image and side-view image show that m-CuO has a nanoparticulate morphology and a film thickness of approximately 60 nm, respectively (Fig. 1a and S1b†). Grazing incident X-ray diffraction (GI-XRD), transmission electron microscopy (TEM) and X-ray photoelectron spectroscopy (XPS) were performed to confirm the composition of m-CuO. Except for the peaks arising from the FTO substrate, two peaks at 2 theta of 35.50 and 38.70 are only observed in GI-XRD, corresponding to the (11![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and (111) planes of the monoclinic phase of CuO (JCPDS 48-1548). The TEM images support that m-CuO is composed of agglomerates of nanoparticles (Fig. 1c and S2a†). The selected area electron diffraction (SAED) result corresponding to a low magnification TEM image indicates the polycrystalline feature of m-CuO, while the elemental composition mapping images and energy dispersive spectrum (EDS) show the uniform dispersion of Cu and O with a ratio close to 1
) and (111) planes of the monoclinic phase of CuO (JCPDS 48-1548). The TEM images support that m-CuO is composed of agglomerates of nanoparticles (Fig. 1c and S2a†). The selected area electron diffraction (SAED) result corresponding to a low magnification TEM image indicates the polycrystalline feature of m-CuO, while the elemental composition mapping images and energy dispersive spectrum (EDS) show the uniform dispersion of Cu and O with a ratio close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S2†). The lattice fringes of 3.36 Å and 2.49 Å in the high-resolution TEM (HR-TEM) image are consistent with the d-spacing of the (010) and (11) crystal faces of monoclinic CuO, respectively (Fig. 1d). The corresponding narrow beam electron diffraction (NBED) pattern of Fig. 1d suggests that m-CuO is well crystallised. Only signals belonging to Cu and O are observed in the XPS survey spectrum (Fig. S3†), suggesting that there are no other impurities in m-CuO. In the Cu 2p core region, the signals centred at 933.3 eV (2p3/2) and 953.1 eV (2p1/2) and the corresponding shake-up satellites are attributed to the presence of CuO on m-CuO (Fig. 1e).18 Additionally, in the O 1s region, peaks centred at 529.9 eV and 531.1 eV belong to the binding energy of lattice O and defect O in m-CuO, respectively (Fig. 1f).18b
:1 (Fig. S2†). The lattice fringes of 3.36 Å and 2.49 Å in the high-resolution TEM (HR-TEM) image are consistent with the d-spacing of the (010) and (11) crystal faces of monoclinic CuO, respectively (Fig. 1d). The corresponding narrow beam electron diffraction (NBED) pattern of Fig. 1d suggests that m-CuO is well crystallised. Only signals belonging to Cu and O are observed in the XPS survey spectrum (Fig. S3†), suggesting that there are no other impurities in m-CuO. In the Cu 2p core region, the signals centred at 933.3 eV (2p3/2) and 953.1 eV (2p1/2) and the corresponding shake-up satellites are attributed to the presence of CuO on m-CuO (Fig. 1e).18 Additionally, in the O 1s region, peaks centred at 529.9 eV and 531.1 eV belong to the binding energy of lattice O and defect O in m-CuO, respectively (Fig. 1f).18b
 | ||
| Fig. 1 (a) SEM planar view image, (b) GI-XRD, (c) bright-field TEM image, (d) HR-TEM image, (e) XPS Cu 2p core level spectrum and (f) XPS O 1s core level spectrum of m-CuO. The inset in (a) shows the photograph image of m-CuO. F in (b) indicates that the signals are contributed by the FTO substrate. The inset in (d) shows the corresponding NBED pattern of Fig. 1d. | ||
The electrooxidation performance of m-CuO was first evaluated in 0.1 M NaOH with various concentrations of glucose. Without the addition of glucose, no noticeable electrocatalytic current is observed until the potential is more positive than 1.7 V vs. reversible hydrogen electrode (RHE), which is assigned to water oxidation reaction (Fig. S4†). In contrast, in the presence of glucose, oxidative currents increase dramatically with an onset potential of approximately 1.2 V vs. RHE in the cyclic voltammograms (CVs). The oxidative currents increase with an increment of glucose concentration and reach the saturated catalytic current at a glucose concentration of 20 mM. Further enhancement in the glucose concentration does not result in further enhancement in the catalytic current, and this might be attributed to the limited active sites on m-CuO for glucose oxidation.
The prepared m-CuO was subsequently subjected to the 1 h controlled potential electrolysis (CPE) in a NaOH (0.1 M) containing 20 mM glucose at potentials of 1.3 V, 1.4 V, 1.5 V, and 1.6 V vs. RHE (Fig. 2b). The generation rate and electrocatalytic index, including the turnover frequency of formate (TOFformate) and FEformate, are shown in Fig. 2c. High-performance liquid chromatography (HPLC) analyses were performed for formate quantification (see Experimental for details). We are aware that a trace amount of formate was possibly generated by a self-degradation of glucose during the prolonged electrolysis in an alkaline solution (Table S2†). For accurate quantification of FEformate and TOFformate, the amount of formate produced by the chemical reaction was excluded from the total amount of formate (see Table S2† and Experimental for details). The results indicate that formate can be generated by electrocatalytic oxidation using m-CuO at potentials higher than 1.3 V vs. RHE with a formate production rate (Rformate) rate of 2.00 ± 0.49 μmol h−1 cm−2. Rformate dramatically increased to 154.73 ± 10.53 μmol h−1 cm−2 and 175.62 ± 5.97 μmol h−1 cm−2 at 1.5 V and 1.6 V vs. RHE, respectively. The TOFformate value of m-CuO is estimated by dividing Rformate (μmol h−1 cm−2) by the amount of the catalyst (μmol cm−2) and the duration of CPE (h). Cu was found to be 0.64 μmol cm−2 by inductively coupled plasma (ICP) measurements (see Experimental for details). The TOFformate of m-CuO is highly dependent on the applied potential, ranging from approximately 3.1 to 272.6 h−1 between 1.3 and 1.6 V vs. RHE (Fig. 2c). On the other hand, m-CuO shows a similarly high FEformate (>80%) in the potential window between 1.3 and 1.6 V vs. RHE and reaches the highest FEformate value of 94.1 ± 1.5% at 1.5 V. The slight decrease of FEformate at an applied potential >1.5 V is possibly attributed to water oxidation to oxygen or the further electrochemical oxidation of formate.
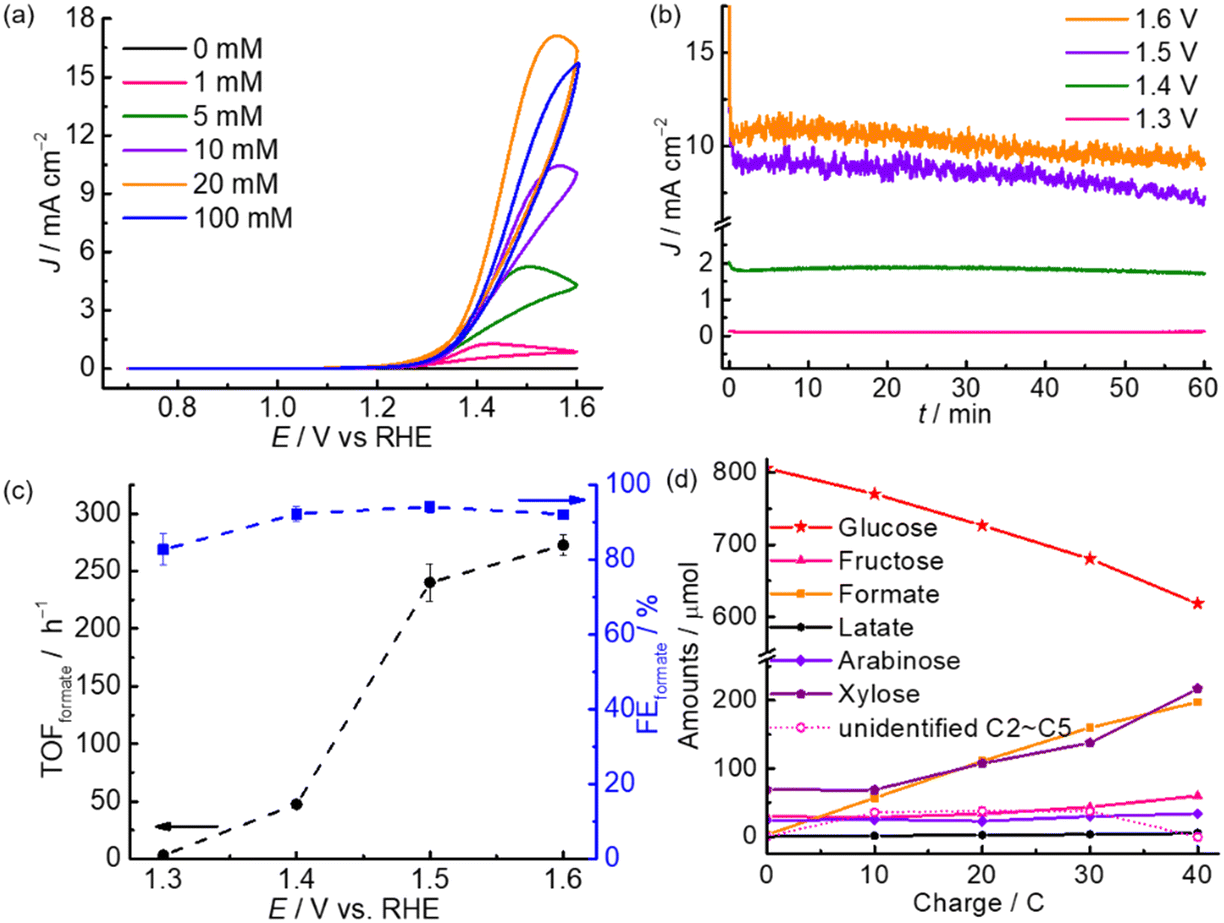 | ||
| Fig. 2 (a) CVs of m-CuO recorded at a scan rate of 50 mV s−1 in the NaOH solution (0.1 M) containing glucose of various concentrations. (b) CPE measurements of m-CuO at various potentials (vs. RHE). (c) TOFformate and FEformate at m-CuO from 1 h CPE at various potentials. (d) Amounts of glucose and products (solid lines) and unidentified C2–C5 products (dash line) as a function of charge passed of m-CuO at 1.5 V vs. RHE. The amounts of glucose, fructose, formate, lactate, arabinose and xylose were quantified by HPLC analyses. The amount of unidentified C2–C5 was derived from the difference between the amount of glucose consumption and the generation of fructose, xylose, and arabinose. For (b)–(d), electrolysis was carried out in NaOH solution (0.1 M) containing 20 mM glucose. | ||
To monitor the dynamic performance of m-CuO toward electrocatalytic glucose valorisation, HPLC analyses were performed at different charge quantities(Fig. 2d and S5†). The corresponding FEformate is summarised in Fig. S6,† and the results indicate that formate is selectively generated with over 90% FEformate in the entire electrolysis. Except for formate, significant amounts of fructose and pentose (xylose and arabinose), as well as a trace amount of lactate, were found (Fig. 2d). It is known that fructose can be generated from the isomerisation of glucose in an alkaline solution.19 Other typical glucose oxidation products, such as glucaric acid and gluconic acid, were not observed (Fig. S5†). It is worth noting that the amount of formate and pentose continuously increased with the passed charge, while glucose was constantly consumed. After the charge of 40 C was passed, approximately 194 μmol of formate and 158 μmol of pentose (xylose and arabinose) were generated, while approximately 188 μmol of glucose was consumed. However, only 158 μmol of glucose was consumed by electrolysis since approximately 30 μmol of glucose was isomerised to fructose. The molar ratio of formate and pentose generation to glucose consumption by electrolysis is 1.2:1:1 (formate:pentose:glucose), suggesting that one glucose might be electro-oxidised to one formate and one pentose. The slight deviation from the 1:1 ratio of formate generation and glucose consumption possibly resulted from that extra amount of formate produced by the chemical and electrochemical degradation of C2–C5 during the electrolysis.
For comparison, electrocatalytic glucose oxidation has also been performed with Cu foil and pre-oxidised Cu foil. Pre-oxidised Cu foil was prepared by electro-oxidising Cu foil in an alkaline solution. An oxidation peak of Cu foil is observed at approximately 1.03 V in the CV (Fig. S7†). Therefore, Cu foil was subjected to oxidising at 1.03 V in 0.5 M KOH solution for 1 h to obtain the pre-oxidised Cu foil. The XPS core-level spectrum of Cu 2p confirms that the surface of pre-oxidised Cu foil is composed of a mixture of Cu, CuO, and Cu(OH)2 (Fig. S8a†), although the main composition is still Cu. Furthermore, the XPS core-level spectrum of O 1s also supports the existence of CuO and Cu(OH)2 on Cu foil after the pre-oxidised treatment (Fig. S8b†). The catalysts showed a decreased electrocatalytic activity toward formate production from electro-oxidising glucose in the sequence of m-CuO, pre-oxidised Cu foil, and Cu foil (Table S3†) in terms of Rformate. In particular, Cu foil has a Rformate of 76.03 μmol cm−2 h−1, approximately only half of that generated using m-CuO. Although the pre-oxidised Cu foil has a comparable Rformate with m-CuO, it has a lower FEformate of 82.0% compared to m-CuO (94.1%). Since the sequence (Cu foil < pre-oxidised Cu foil < m-CuO) of electroactivity toward the formate production from glucose is consistent with the sequence of contents of CuO, CuO is most likely the active material toward C–C breaking.
There is a controversy about the mechanism of glucose oxidation by CuO. It has been previously accepted that Cu3+ plays the dominant role in glucose oxidation, in which Cu3+ is firstly formed by the oxidation of Cu2+ under anodic potential followed by directly oxidising glucose.20 However, Cu3+ species are only formed at a highly anodic potential where water oxidation occurs.21 The CV results indicate that m-CuO catalyse water oxidation along with the formation of Cu3+ at approximately 1.7 V vs. RHE in 0.1 M NaOH, while a reduction peak assigned to the reduction of Cu3+ is observed approximately at 1.65 V vs. RHE (Fig. S4†). However, m-CuO can perform electrochemical oxidation of glucose to formate at potentials of 1.3 to 1.6 V vs. RHE, below the potential of Cu3+ formation. The results suggest that other mechanisms mediated the reaction at m-CuO from 1.3 to 1.6 V vs. RHE.
Hydroxyl (OH) radical-mediated glucose oxidation has been recently proposed.16,21b,22 CuO is a p-type semiconductor,23 and the p-type feature of m-CuO can be confirmed by the negative slope in the Mott–Schottky analysis (Fig. S9†). For a p-type material, vacancies accumulate in the interface region when the applied potential is more positive than its flat band potential (VFB).24 It is believed that when the applied potential is more positive than the VFB of CuO, the absorbed OH ions can be oxidised by the accumulation of vacancies in the interface region of CuO followed by the generation of OH radicals.21b The OH radicals, acting as strong oxidants, subsequently react with glucose. In order to elucidate the mechanism of m-CuO electrocatalytic glucose oxidation, EPR analyses were performed by using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as the trapping agent of OH radicals. The VFB of m-CuO is approximately 1.15 V vs. RHE, as determined by Mott–Schottky analyses (Fig. S9†). The EPR spectrum shows CuO in 1 M NaOH solution after 10 min electrolysis at 1.5 V (Fig. 3a), revealing a well-defined four-line pattern with an intensity ratio of 1:2:2:1 and hyperfine splitting of αN = αH = 15 G, corresponding to the characteristic of a DMPO–OH adduct,25 which confirms the presence of OH radicals.
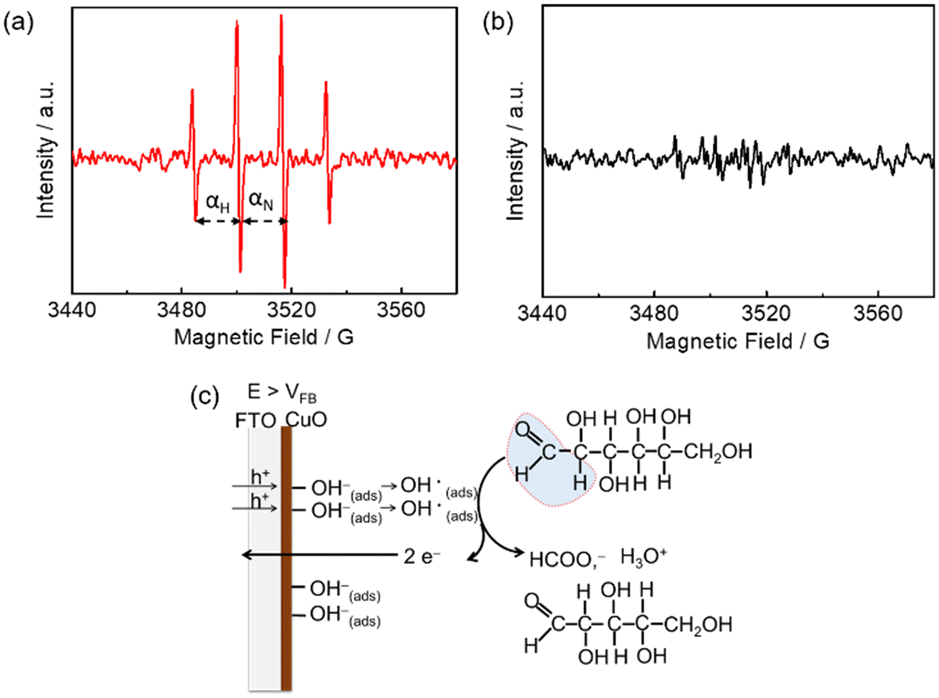 | ||
| Fig. 3 EPR spectra of m-CuO after 10 min electrolysis at 1.5 V vs. RHE in (a) 1 M NaOH solution and (b) 1 M NaOH solution containing 20 mM glucose. (c) The proposed mechanism of electrochemical formate production at m-CuO in the potential regions where the potentials are more positive than the VFB of m-CuO. | ||
It should be noted that, however, prior to electrolysis, the signals for the DMPO–OH adduct were present (Fig. S10†). However, no significant difference in the intensity of signals between the background signal and electrolysis at the OCP or 1.2 V vs. RHE, indicating that potentials before or near VFB of m-CuO do not result in the formation of further OH radical by electrolysis. However, m-CuO exhibited significantly higher intensity at potentials of 1.5 V vs. RHE, indicating that electrolysis at a potential more positive than the VFB of m-CuO generated more OH radicals. The OH radicals were subsequently consumed by glucose, which was supported by the signals of the DMPO–OH adduct completely disappeared in the presence of glucose (Fig. 3b).
The mechanism of formate production from glucose oxidation at m-CuO is therefore proposed based on the electrochemical study, product analyses, and the EPR spectra. At the potentials between 1.3–1.6 V vs. RHE, where the potentials are higher than the VFB of m-CuO, OH radicals are formed by the absorbed OH ions with the accumulated vacancies at the m-CuO surface. Subsequently, two OH radicals react with one glucose to generate one formate and one pentose (Fig. 3c).
Since m-CuO acts as a selective electrocatalyst for formate production from glucose, we were interested in determining if formate can be efficiently electrochemically produced at m-CuO by using α-cellulose, a homopolymer of glucose, which is also the most abundant form of forest biomass as the substrate. However, α-cellulose has very low solubility in solutions. Therefore, a pretreatment process involving stringing the α-cellulose in a strongly alkaline solution was applied before the electrochemical study (see Experimental for details). Trace amounts of glucose of approximately 0.038 mM were found in the pretreated α-cellulose solution. An onset oxidation potential at 1.2 V vs. RHE and an oxidation wave centred at 1.58 V vs. RHE followed by water oxidation at 1.7 V vs. RHE are observed in the presence of cellulose (Fig. 4a). On the other hand, no noticeable oxidation current is observed without α-cellulose at potentials of 1.2–1.7 V vs. RHE. However, a reduction wave centred at 1.65 V vs. RHE can be observed in both solutions with and without cellulose. The reduction wave can be attributed to the reduction of Cu3+ to Cu2+, whereas the oxidation wave of Cu2+ to Cu3+ overlapped with water oxidation.26 The electrochemical results suggest that cellulose and its derivatives can be oxidised at potentials more positive than 1.2 V vs. RHE. In addition, the CV curves of m-CuO negatively shifted with the increase of the pH value with a rate of 0.059 × pH, suggesting that hydroxide ions followed by the formation of OH radicals participate in α-cellulose oxidation at potentials before water oxidation. The electrochemical features are similar to those of glucose oxidation at m-CuO, and presumably, glucose oxidation contributed to at least a partial oxidation current.27
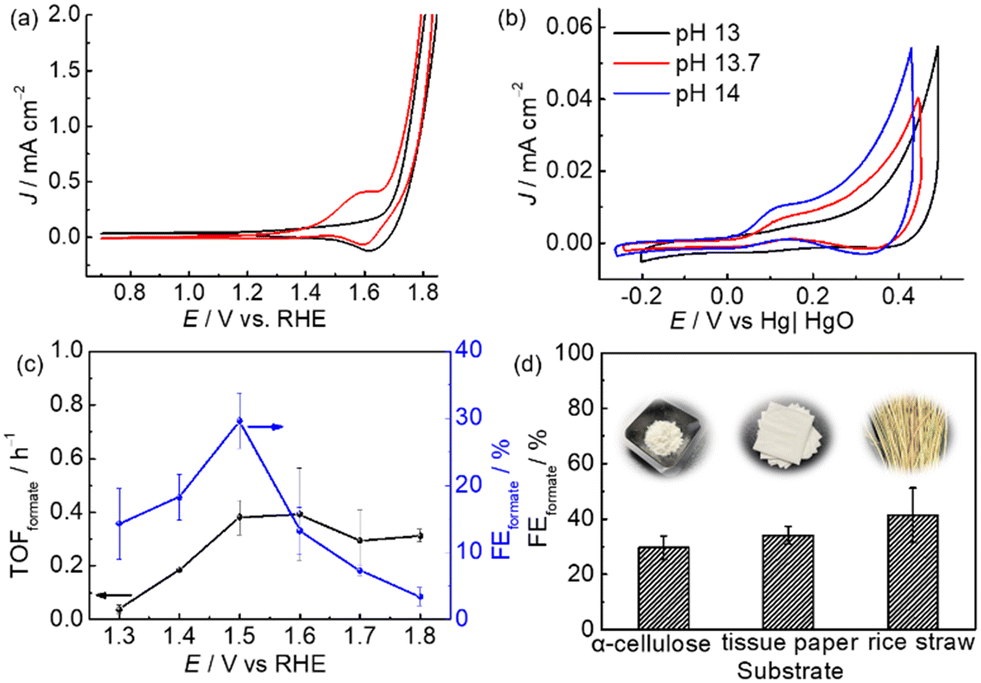 | ||
| Fig. 4 CVs of m-CuO recorded at a scan rate of 50 mV s−1(a) in the NaOH solution (black line) and NaOH solution containing α-cellulose (red line) at pH 13 and (b) in NaOH solution containing cellulose at different pH values. (c) TOFformate and FEformate at m-CuO from CPE at the various potentials. NaOH solution containing α-cellulose (pH 13) was used as the electrolyte. (d) FEformate from 2.5 h CPE at 1.5 V vs. RHE using m-CuO as the electrocatalyst in purified and raw cellulose samples. | ||
Subsequently, experiments of m-CuO electrocatalysing α-cellulose oxidation at various potentials were performed. m-CuO shows moderate catalytic activity towards reforming α-cellulose to formate, and the FEformate increase from 14.3 ± 5.3% to 29.7 ± 4.2% at 1.3 V vs. RHE and 1.5 V vs. RHE, respectively. Although m-CuO generated a higher current at 1.6 V than 1.5 V (Fig. 4c and S11†), the TOFformate value was similar to that at 1.5 V (0.39 h−1vs. 0.38 h−1). Additionally, the FEformate value dramatically decreases at potentials higher than 1.5 V vs. RHE, presumably due to the side reaction of water oxidation starting at 1.6 V vs. RHE. Replacement of purified α-cellulose with raw “real-world” biomass waste results in even high FEformate by using m-CuO as the electrocatalyst. FEformate values of 34.1 ± 3.3% and 41.4 ± 9.7% were quantified by using tissue paper and rice straw as the substrate, respectively, from 2.5 h CPE at 1.5 V vs. RHE. In particular, rice straw has over 2.5-fold Rformate compared to purified α-cellulose from 2.5 h CPE at 1.5 V (Table S4†).
m-CuO was subsequently coated on a hematite (α-Fe2O3) electrode to demonstrate that m-CuO can be used as a cocatalyst on a photoanode to perform selective photoelectrochemical glucose valorisation. Hematite is robust in alkaline solutions. It has been immensely investigated as a photoanode for solar-driven water oxidation due to its narrow bandgap and proper electronic band structure, capable of using visible light to drive water oxidation. However, hematite shows low selectivity toward organic compounds oxidation such as glycerol.28
Nanorod structure hematite (nanoFe2O3) was converted by heating the substrate of β-FeOOH at 800 °C for 20 min. β-FeOOH was synthesised by a chemical bath deposition method (see Experimental for details and Fig. S12†). To prepare m-CuO modified nanoFe2O3 (nanoFe2O3|m-CuO), one layer of the copper precursor was spun onto nanoFe2O3, followed by the same heat treatment process used for preparing m-CuO (Fig. 5a). The cherry red colour of nanoFe2O3 became wine red colour after m-CuO modification (insets of Fig. 5b and c). GI-XRD confirms that the nanoFe2O3 is consistent with the phase of α-Fe2O3 (JCPDS No. 33-0664). Additional peaks at 2 theta of 32.50 and 38.80 are observed in nanoFe2O3|m-CuO, which belong to the (110) and (111) planes of the monoclinic phase of CuO (JCPDS 48-1548), respectively. The SEM images indicate that nanoFe2O3 has a rod-like structure with a rod diameter and length of approximately 60 nm and 0.4 μm, respectively, similar to its FeOOH template (Fig. 5b, b′ and S12†). On the other hand, nanoFe2O3|m-CuO remains to be the rod structure of nanoFe2O3, although its rod diameter increases to 90–200 nm (Fig. 5c, c′).
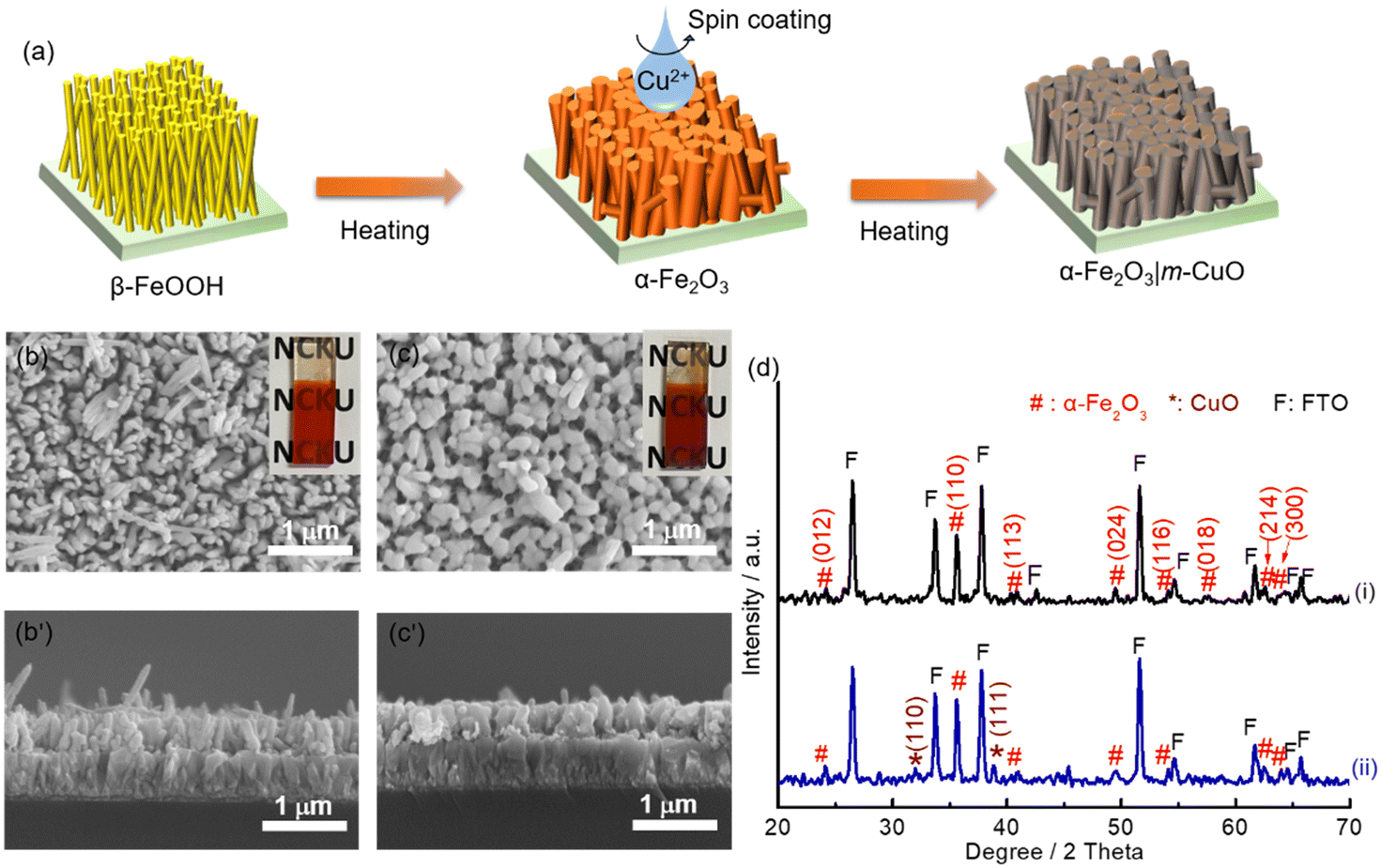 | ||
| Fig. 5 (a) Schematic representation of the preparation process of nanoFe2O3 and nanoFe2O3|m-CuO. SEM images of (b, b′) nanoFe2O3 and (c, c′) nanoFe2O3|m-CuO. The insets in (b) and (c) are the photograph images of nanoFe2O3 and nanoFe2O3|m-CuO, respectively. (d) XRD patterns of (i) nanoFe2O3 and (ii) nanoFe2O3|m-CuO. | ||
The electronic structure of nanoFe2O3 and m-CuO can be estimated from their UV-vis spectra and Mott–Schottky analyses. The direct bandgap of m-CuO and nanoFe2O3 is 1.7 eV and 2.15 eV, respectively, consistent with the reported values (Fig. S13 and S14†).23a,29 The positive slope in the Mott–Schottky analysis plot of nanoFe2O3 confirms its n-type feature, and its flat band edge is estimated at 0.35 V vs. RHE from the extrapolated intercepts. For an n-type semiconductor, its conduction band is close to its flat band edge. Therefore, the valence band edge of nanoFe2O3 can be estimated from its bandgap and conduction band edge and is approximately 2.5 V vs. RHE (Fig. 6a). The water and glucose oxidation potentials are 1.23 V vs. RHE and 0 V, respectively.30 Under solar light illumination, the holes of nanoFe2O3 can provide sufficient driving force for both water oxidation and glucose oxidation. However, the sluggish water oxidation kinetics of nanoFe2O3 resulted in its onset potential of 0.7 V vs. RHE, significantly deviating from its flat band edge of 0.35 V vs. RHE (Fig. 6c(i)). In the presence of glucose, the onset potential negatively shifts to 0.5 V vs. RHE, and the photocurrents increased more than two times at 1.23 V vs. RHE, which indicates that the glucose oxidation kinetics is more facile than water oxidation. Nevertheless, since nanoFe2O3 catalyses both water and glucose oxidation under light illumination, it shows only a moderate FEFormate of approximately 60.8 ± 1.5% toward glucose oxidation at 1.0 V vs. RHE (Fig. 6f).
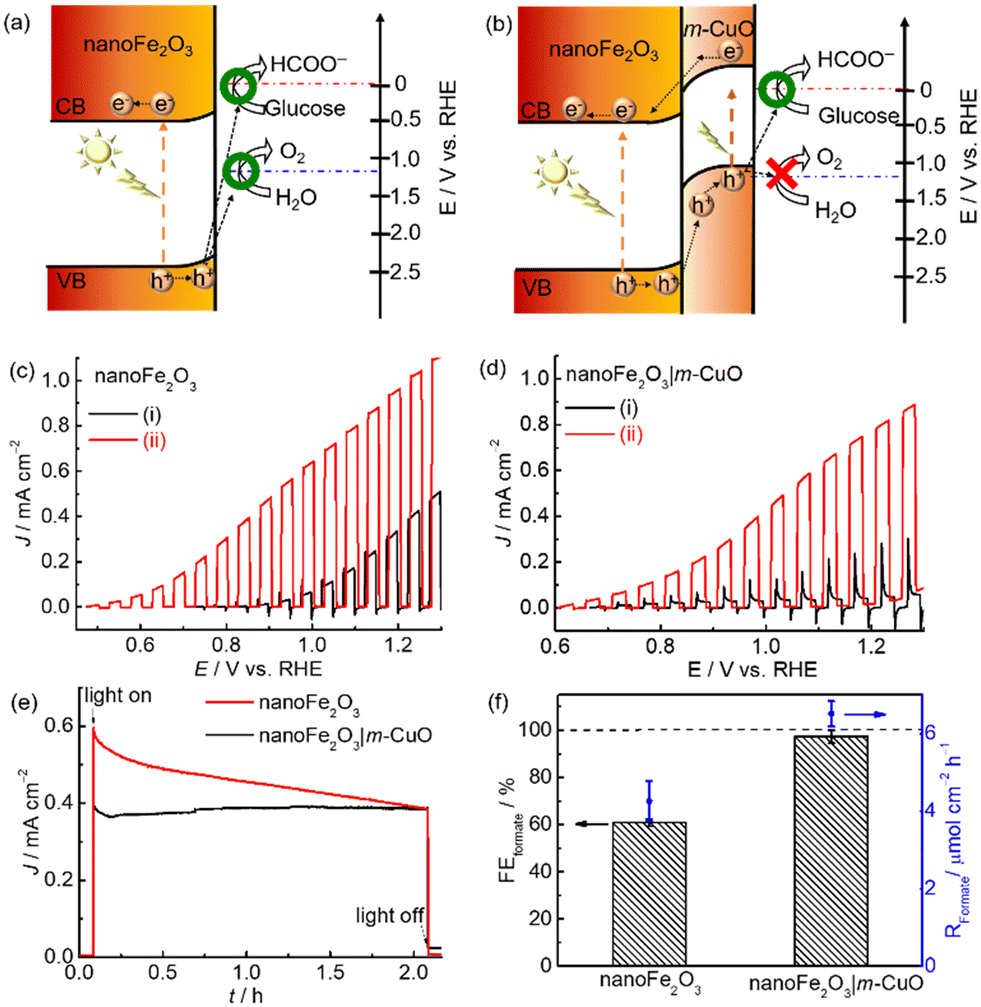 | ||
| Fig. 6 Schematic representation of the electronic band structures of (a) nanoFe2O3 and (b) nanoFe2O3|m-CuO. Current density (J)-potential (E) curves with a scan rate of 5 mV s−1 of (c) nanoFe2O3 and (d) nanoFe2O3|m-CuO recorded in (i) 0.1 M NaOH solution and (ii) 0.1 M NaOH solution containing 20 mM glucose under chopped solar light irradiation (AM 1.5G, 100 mW cm−2). (e) CPE measurements and (f) FEformate and Rformate of nanoFe2O3 and nanoFe2O3|m-CuO at 1.0 V vs. RHE in 0.1 M NaOH solution containing 20 mM glucose under solar light irradiation (AM 1.5G, 100 mW cm−2). | ||
The unsatisfactory selectivity of formate production from glucose oxidation on nanoFe2O3 is possibly alleviated by passivating it with m-CuO. Firstly, m-CuO can serve as an efficient electrocatalyst on nanoFe2O3. Secondly, the oxidative force of the light-induced holes is possibly tuned by the n–p heterojunction of nanoFe2O3 and m-CuO (Fig. 6b). The valence band edge of m-CuO is located near its flat band potential of 1.15 eV. Therefore, the light-induced holes of the heterostructure of nanoFe2O3|m-CuO are only able to oxidise glucose. This hypothesis can be evident from the current density-potential curves under chopped light illumination. The nanoFe2O3 photoelectrochemical water oxidation was almost eliminated by passivating m-CuO (Fig. 6d(i)). In addition, the photocurrent of nanoFe2O3|m-CuO in NaOH solution containing glucose is 15 times higher than that without glucose at potentials 1.23 V vs. RHE. NanoFe2O3|m-CuO exhibited stable photocurrent and a high FEformate of 97.3 ± 2.8% in NaOH solution containing 20 mM glucose at 1.0 V vs. RHE (Fig. 6e and f). This serves as a rare example of using only earth-abundant materials for the highly selective photoelectrochemical valorisation of organic compounds to formate (Table S5†).
Conclusions
A non-precious electrocatalyst, m-CuO, for the selective production of formate from the electrochemical and photoelectrochemical glucose oxidation was explored and investigated. m-CuO effectively catalyses C–C bond breaking of glucose, resulting in the formation of formate and pentose, while the EPR study suggests that the electro-generated OH radicals mediate the C–C bond breaking at the potentials of 1.3–1.6 V vs. RHE. α-cellulose, tissue paper and rice straw can also be used as the substrate in the electrocatalytic valorisation over m-CuO with moderate FEformate. In addition, the photoelectrochemical water oxidation on nanoFe2O3 can be almost entirely suppressed by modifying with m-CuO, resulting in a significant enhancement of FEformate from 60.8 ± 1.5% to 97.3 ± 2.8%.Experimental
Preparation of electrodes
m-CuO electrode was prepared on fluorine-doped tin oxide (FTO) coated glass (Pilkington TEC Glass™ 7) by spin coating a copper nitrate-containing sol–gel precursor followed by heating. FTO was cleaned in an NH4OH (Sigma Aldrich, 30%)–H2O2 (J.T. Baker, 30%)–H2O solution with a volume ratio of 1:1:5 at 70 °C for 40 min and dried at room temperature under N2 prior to use. To prepare the copper nitrate-containing sol–gel, Cu(NO3)2·3 H2O (2 mmol, Showa, 98%) and citric acid (2 mmol, Showa, 99%) were dissolved in ethanol (10 mL, Echo Chemical Co., Ltd., >99.5%) and stirred for 15 min. Subsequently, 0.25 mL ethylene glycol (Sigma Aldrich, 99.8%) was added to the above mixture, and the resulting solution was further stirred overnight. The well-dispersed copper nitrate-containing sol–gel solution was spin-coated onto the FTO substrates at 3000 rpm for 60 s, followed by heating on a hot plate at 100 °C for 10 min and then at 450 °C for 1 h in air. The spin coating and heating procedures were repeated 3 times to obtain m-CuO.
NanoFe2O3 was synthesised by a chemical bath deposition method followed by a heat-treatment process. Briefly, FTO glass was immersed into a bottle containing 0.15 M FeCl3·H2O (Sigma Aldrich, 99%) and 1 M NaNO3 (J.T. Baker, 99%) aqueous solution. The bottle was then sealed and heated at 100 °C for 6 h. The resultant bright yellow β-FeOOH nanorod electrode was subsequently converted to nanoFe2O3 in a preheated tube furnace by annealing at 800 °C for 20 min. NanoFe2O3|m-CuO was synthesised by spin-coating one layer of the CuO precursor, and the heat treatments were the same as that for the preparation of the m-CuO electrode.
Material characterisation
The morphologies of the electrodes were characterised by SEM (HITACHI SU-5000). TEM images, SAED patterns, and NBED patterns were recorded using a TEM (JEOL JEM-2010F CS STEM), whereas the elemental composition mapping and EDS analyses were performed by energy-dispersive X-ray spectroscopy (Oxford Aztec Ultimax). The sample for TEM analysis was prepared by scraping m-CuO from the FTO substrate followed by dispersing the m-CuO particles in ethanol solution. The resulting solution was dropped on carbon-coated Ni grids (Ted Pella, Inc.) for analysis. GI-XRD analyses were performed by Bruker D8 Discover. XPS spectra were measured using a Thermo Scientific Theta Probe X-ray spectrometer with an Al anode as the X-ray source. The binding energies were corrected by referencing the C 1s peak to 284.6 eV. ICP was used to quantify the amount of copper in m-CuO by using a Horiba Jobin Yvon J. Y. 2000-2 ICP optical emission spectrometer, and it was found to be 0.64 ± 0.07 μmol cm−2. EPR was performed at room temperature using a Bruker EMXplus-10/12/P/L SYSTEM with the X-band operating at a frequency of 9.84 GHz and a microwave power of 20 mW. DMPO was used as the trapping agent of the OH radicals. The UV-vis absorption spectra were recorded using a UV-vis spectrophotometer (Agilent Cary 60) equipped with a diffuse reflectance accessory. The Kubelka–Munk function (F(R)) was used to convert reflectance into Tauc plots.31Electrochemical and PEC characterisation
Electrochemical and photoelectrochemical measurements were recorded with a potentiostat (CHI 7273E) with an electrochemical cell with two compartments separated by an anion exchange membrane (AEM-025, Johnson Matthey, UK). Hg|HgO (1 M NaOH) and Pt foil were used as the reference and counter electrodes, respectively. 0.5 cm2 and 1.0 cm2 geometric areas of the working electrodes were defined using polyester tape (3M, 1350FY-1) for CV and CPE, respectively. The precise geometric areas were quantified after every experiment. Prior to the electrochemical experiments, the potential of the reference electrode was calibrated by a commercial saturated calomel electrode (ALS Co., Ltd, RE-2BP). Unless otherwise noted, all the potentials are converted to RHE by using the equation E (V vs. RHE) = E (V vs. Hg|HgO, 1 M NaOH) + 0.118 + 0.059 × pH. The cell's internal resistance was measured before performing CV or CPE, and the iR compensation was done with 90% compensation. For CV measurements, all the data shown here are from the 3rd cycle of the scans. All the electrolytes were purged with N2 gas for 30 min before measurement and continuously purged during the whole experiment. For reforming glucose, the D-(+)-glucose (Sigma Aldrich) was added just before the measurements to avoid glucose degradation in the 0.1 M NaOH solution. For α-cellulose reforming experiments, 1 g of α-cellulose (Sigma Aldrich) was added to 20 ml of 10 M NaOH solution and stirred for 24 h, followed by dilution to a total volume of 100 ml by H2SO4 solution with a final pH of 13 and removing the undissolved α-cellulose by filtration. The same procedures were used for reforming paper tissue (Kirkland Signature) and rice straw (Taiken 11) by replacing α-cellulose with paper tissue or rice straw. For the PEC study, a solar light simulator (SAN-ElECTRIC, XES-40S2-CE, AAA class) equipped with an air mass 1.5 global (AM 1.5G) filter was used, and the light intensity was calibrated using a monocrystalline silicon solar cell to 1 sun (100 mW cm−2).Product analysis
All samples for reforming glucose were analysed by Shimadzu Nexera-i LC2040C 3D Puls (PDA model) HPLC equipped with a refractive index detector (RID-20A). A photodiode array (PDA) detector set at 207 nm was used to detect and quantify xylose, fructose, formate and other organic acids. A refractive index detector (RID) detector thermostated at 40 °C was used to detect and quantify glucose and arabinose. The eluent was 2 mM sulfuric acid with a flow rate of 0.6 ml min−1 at 60 °C. In each analysis, the sample (1 ml) was neutralised with 0.5 ml of 0.1 M H2SO4. Subsequently, the 50 μl neutralised samples were injected directly into a Shodex SH1821 column. The identification and quantification of the products were determined from the calibration curves of the corresponding standard solutions. We are also aware that trace amounts of formate might be generated by the self-degradation of glucose in an alkaline solution rather than by the electrochemical reaction. In order to determine the amount of formate generated by the self-degradation, control experiments were performed by keeping the electrolyte at the same duration as the electrolysis, followed by the quantification of formate. For PEC experiments, the control experiments for determining the amount of formate generated by the self-degradation were performed by the same procedures as those for electrolysis, except that the samples were additionally illuminated with simulated solar light (100 mW cm−2) during the experiment. The amount of formate resulting from self-degradation was excluded from the total amount of formate quantified after the electrolysis to calculate FEformate and TOFformate.The FEformate and TOFformate are calculated using the following equations (eqn (1) and (2)):
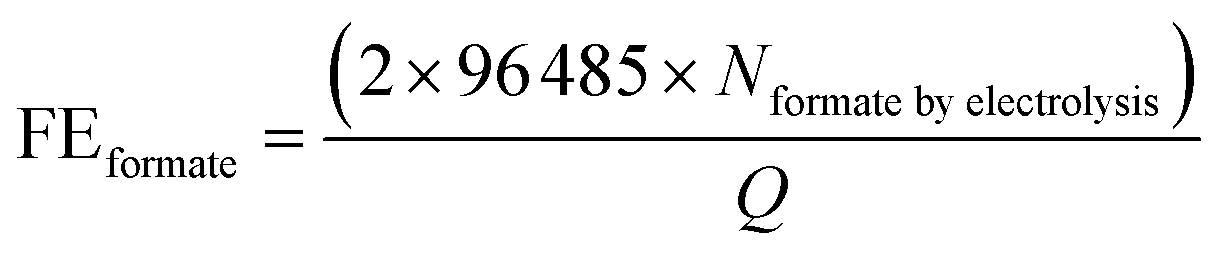 | (1) |
 | (2) |
For reforming α-cellulose, tissue paper and rice straw, formate was detected by 883 Basic IC plus ion chromatography (Metrohm) equipped with a Metrosep organic acids guard (4.6 × 50 mm) and a Metrosep organic acids column (7.8 × 250 mm) rather than HPLC to avoid the interference from carbohydrate signals.
Author contributions
P.-C. Chuang: investigation, methodology, validation and writing – original draft. Y.-H. Lai: conceptualisation, supervision, funding acquisition, and writing – original draft and review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Ministry of Science and Technology, Taiwan (MOST 110-2636-E-006-023 and MOST 111-2636-E-006-017) is gratefully acknowledged. This research was supported in part by Higher Education Sprout Project, Ministry of Education to the Headquarters of University Advancement at National Cheng Kung University (NCKU). We appreciate the fruitful suggestions from Prof. Chia-Yu Lin. The authors also gratefully acknowledge the use of EM000700 and EM000800 of MOST 110-2731-M-006-001 belonging to the Core Facility Center of National Cheng Kung University and the Electron Paramagnetic Resonance spectrometer of Joint Center for High Valued Instruments at National Sun Yat-sen University.References
- (a) F. Shen, R. L. Smith Jr, J. Li, H. Guo, X. Zhang and X. Qi, Green Chem., 2021, 23, 1536–1561 RSC; (b) D. A. Bulushev and J. R. H. Ross, ChemSusChem, 2018, 11, 821–836 CrossRef CAS PubMed; (c) Z. Huang, N. Luo, C. Zhang and F. Wang, Nat. Rev. Chem., 2022, 6, 197–214 CrossRef; (d) M. J. Hülsey, H. Yang and N. Yan, ACS Sustainable Chem. Eng., 2018, 6, 5694–5707 CrossRef; (e) Y. Holade, N. Tuleushova, S. Tingry, K. Servat, T. W. Napporn, H. Guesmi, D. Cornu and K. B. Kokoh, Catal. Sci. Technol., 2020, 10, 3071–3112 RSC; (f) G. C. de Assis, I. M. A. Silva, T. G. dos Santos, T. V. dos Santos, M. R. Meneghetti and S. M. P. Meneghetti, Catal. Sci. Technol., 2021, 11, 2354–2360 RSC; (g) D. K. Lee, S. R. Kubota, A. N. Janes, M. T. Bender, J. Woo, J. R. Schmidt and K.-S. Choi, ChemSusChem, 2021, 14, 4563–4572 CrossRef CAS PubMed.
- W.-J. Liu, Z. Xu, D. Zhao, X.-Q. Pan, H.-C. Li, X. Hu, Z.-Y. Fan, W.-K. Wang, G.-H. Zhao, S. Jin, G. W. Huber and H.-Q. Yu, Nat. Commun., 2020, 11, 265 CrossRef CAS PubMed.
- (a) J. Eppinger and K.-W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS; (b) A. K. Singh, S. Singh and A. Kumar, Catal. Sci. Technol., 2016, 6, 12–40 RSC; (c) J. Hietala, A. Vuori, P. Johnsson, I. Pollari, W. Reutemann and H. Kieczka, in Ullmann's Encyclopedia of Industrial Chemistry, 2016, pp. 1–22 Search PubMed; (d) J. Li, Y. Kuang, Y. Meng, X. Tian, W.-H. Hung, X. Zhang, A. Li, M. Xu, W. Zhou, C.-S. Ku, C.-Y. Chiang, G. Zhu, J. Guo, X. Sun and H. Dai, J. Am. Chem. Soc., 2020, 142, 7276–7282 CrossRef CAS PubMed; (e) S. A. Al-Tamreh, M. H. Ibrahim, M. H. El-Naas, J. Vaes, D. Pant, A. Benamor and A. Amhamed, ChemElectroChem, 2021, 8, 3207–3220 CrossRef CAS.
- R. Vinu and L. J. Broadbelt, Energy Environ. Sci., 2012, 5, 9808–9826 RSC.
- (a) C. Wang, X. Chen, M. Qi, J. Wu, G. Gözaydın, N. Yan, H. Zhong and F. Jin, Green Chem., 2019, 21, 6089–6096 RSC; (b) F. Jin and H. Enomoto, Energy Environ. Sci., 2011, 4, 382–397 RSC.
- D. Voß, H. Pickel and J. Albert, ACS Sustainable Chem. Eng., 2019, 7, 9754–9762 CrossRef.
- M. Sert, A. Arslanoğlu and L. Ballice, Renewable Energy, 2018, 118, 993–1000 CrossRef CAS.
- W. Wang, M. Niu, Y. Hou, W. Wu, Z. Liu, Q. Liu, S. Ren and K. N. Marsh, Green Chem., 2014, 16, 2614–2618 RSC.
- G. Moggia, T. Kenis, N. Daems and T. Breugelmans, ChemElectroChem, 2020, 7, 86–95 CrossRef CAS.
- H. Luo, J. Barrio, N. Sunny, A. Li, L. Steier, N. Shah, I. E. L. Stephens and M.-M. Titirici, Adv. Energy Mater., 2021, 11, 2101180 CrossRef CAS.
- (a) H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed; (b) G. Bharath, K. Rambabu, A. Hai, N. Ponpandian, J. E. Schmidt, D. D. Dionysiou, M. Abu Haija and F. Banat, Appl. Catal., B, 2021, 298, 120520 CrossRef.
- Y. Holade, K. Servat, T. W. Napporn, C. Morais, J.-M. Berjeaud and K. B. Kokoh, ChemSusChem, 2016, 9, 252–263 CrossRef CAS PubMed.
- T. Rafaïdeen, S. Baranton and C. Coutanceau, Appl. Catal., B, 2019, 243, 641–656 CrossRef.
- C.-Y. Lin, S.-Y. Lin, M.-C. Tsai and C.-H. Wu, Sustainable Energy Fuels, 2020, 4, 625–632 RSC.
- (a) M. Pourbaix, Lectures on Electrochemical Corrosion, Plenum Press, New York, 1973 CrossRef; (b) L. Ostervold, S. I. Perez Bakovic, J. Hestekin and L. F. Greenlee, RSC Adv., 2021, 11, 31208–31218 RSC.
- (a) N. J. Cory, E. Visser, J. Chamier, J. Sackey, F. Cummings and M. Chowdhury, Appl. Surf. Sci., 2022, 576, 151822 CrossRef CAS; (b) A. Scandurra, M. Censabella, S. Boscarino, G. G. Condorelli, M. G. Grimaldi and F. Ruffino, Nanotechnology, 2022, 33, 045501 CrossRef CAS PubMed.
- C. Liu, M. Hirohara, T. Maekawa, R. Chang, T. Hayashi and C.-Y. Chiang, Appl. Catal., B, 2020, 265, 118543 CrossRef CAS.
- (a) Y. J. Jang, A. E. Lindberg, M. A. Lumley and K.-S. Choi, ACS Energy Lett., 2020, 5, 1834–1839 CrossRef CAS; (b) M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334 CrossRef CAS.
- (a) C. Kooyman, K. Vellenga and H. G. J. De Wilt, Carbohydr. Res., 1977, 54, 33–44 CrossRef CAS; (b) G. De Wit, A. P. G. Kieboom and H. van Bekkum, Carbohydr. Res., 1979, 74, 157–175 CrossRef CAS.
- J. M. Marioli and T. Kuwana, Electrochim. Acta, 1992, 37, 1187–1197 CrossRef CAS.
- (a) Y. Deng, A. D. Handoko, Y. Du, S. Xi and B. S. Yeo, ACS Catal., 2016, 6, 2473–2481 CrossRef CAS; (b) J. T. C. Barragan, S. Kogikoski, E. T. S. G. da Silva and L. T. Kubota, Anal. Chem., 2018, 90, 3357–3365 CrossRef CAS PubMed.
- M. Sajadpour, H. Siampour, S. Abbasian, M. Amiri, R. Rameshan, C. Rameshan, A. Hajian, H. Bagheri and A. Moshaii, J. Electrochem. Soc., 2019, 166, B1116–B1125 CrossRef CAS.
- (a) R. Siavash Moakhar, S. M. Hosseini-Hosseinabad, S. Masudy-Panah, A. Seza, M. Jalali, H. Fallah-Arani, F. Dabir, S. Gholipour, Y. Abdi, M. Bagheri-Hariri, N. Riahi-Noori, Y.-F. Lim, A. Hagfeldt and M. Saliba, Adv. Mater., 2021, 33, 2007285 CrossRef PubMed; (b) Z. Wang, L. Zhang, T. U. Schülli, Y. Bai, S. A. Monny, A. Du and L. Wang, Angew. Chem., Int. Ed., 2019, 58, 17604–17609 CrossRef CAS PubMed.
- A. W. Bott, Curr. Sep., 1998, 17, 87–92 CAS.
- J. K. Kim and I. S. Metcalfe, Chemosphere, 2007, 69, 689–696 CrossRef CAS PubMed.
- B. Miller, J. Electrochem. Soc., 1969, 116, 1675 CrossRef CAS.
- V.-C. Nguyen, D. B. Nimbalkar, L. D. Nam, Y.-L. Lee and H. Teng, ACS Catal., 2021, 11, 4955–4967 CrossRef CAS.
- N. Perini, C. Hessel, J. L. Bott-Neto, C. T. G. V. M. T. Pires, P. S. Fernandez and E. Sitta, J. Solid State Electrochem., 2021, 25, 1101–1110 CrossRef CAS.
- A. G. Tamirat, J. Rick, A. A. Dubale, W. N. Su and B. J. Hwang, Nanoscale Horiz., 2016, 1, 243–267 RSC.
- M. F. Kuehnel and E. Reisner, Angew. Chem., Int. Ed., 2018, 57, 3290–3296 CrossRef CAS PubMed.
- D. G. Barton, M. Shtein, R. D. Wilson, S. L. Soled and E. Iglesia, J. Phys. Chem. B, 1999, 103, 630–640 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy00950a |
| This journal is © The Royal Society of Chemistry 2022 |