
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Primary amines from lignocellulose by direct amination of alcohol intermediates, catalyzed by RANEY® Ni†
Xianyuan
Wu
 a,
Mario
De Bruyn
b and
Katalin
Barta
*ab
a,
Mario
De Bruyn
b and
Katalin
Barta
*ab
aStratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG, The Netherlands
bInstitute for Chemistry, University of Graz, Heinrichstrasse 28/II, 8010, Graz, Austria. E-mail: katalin.barta@uni-graz.at
First published on 19th August 2022
Abstract
Primary amines are crucially important building blocks for the synthesis of a wide range of industrially relevant products. Our comprehensive catalytic strategy presented here allows diverse primary amines from lignocellulosic biomass to be sourced in a straightforward manner and with minimal purification effort. The core of the methodology is the efficient RANEY® Ni-catalyzed hydrogen-borrowing amination (with ammonia) of the alcohol intermediates, namely alkyl-phenol derivatives as well as aliphatic alcohols, obtained through the two-stage LignoFlex process. Hereby the first stage entails the copper-doped porous metal oxide (Cu20PMO) catalyzed reductive catalytic fractionation (RCF) of pine lignocellulose into a crude bio-oil, rich in dihydroconiferyl alcohol (1G), which could be converted into dihydroconiferyl amine (1G amine) in high selectivity using ammonia gas, by applying our selective amination protocol. Notably also, the crude RCF-oil directly afforded 1G amine in a high 4.6 wt% isolated yield (based on lignin content). Finally it was also shown that the here developed Ni-catalysed heterogeneous catalytic procedure was equally capable of transforming a range of aliphatic linear/cyclic primary/secondary alcohols – available from the second stage of the LignoFlex procedure – into their respective primary amines.
1. Introduction
Primary amines hold vast importance for the manufacturing of a wide range of industrially relevant compounds, among which are polymers and pharmaceuticals.1–5 Their efficient and sustainable synthesis from widely available alcohols that may be obtained from renewable lignocellulosic biomass and this preferentially by direct coupling with ammonia as an abundant nitrogen source, and utilizing heterogeneous catalysts, is a highly desired objective.6–9An attractive, highly atom-economic method for the direct conversion of alcohols into their respective amines is via the “hydrogen borrowing” strategy. Typically such procedure comprises three consecutive stages,10–12 namely a) the dehydrogenation of a alcohol to its corresponding carbonyl compound, b) imine formation by reaction of the carbonyl intermediate with the amine reaction partner, accompanied with the release of water as only side-product, and c) reduction of the imine to the desired amine product, using the hydrogen equivalents ‘borrowed’ in the first step. The dehydrogenation and hydrogenation steps are mediated by a suitable transition metal catalyst. In fact, the challenge of finding such suitable catalytic systems to accomplish efficient ‘hydrogen-borrowing’ transformations in high selectivity, especially when using ammonia, has been met by a number of research groups.5–12 Exemplary in this matter are the use of Ni/SiO2–Al2O3,13 Ni/θ-Al2O3 (or Ni/γ-Al2O3),14 Ni/CaSiO3 (ref. 15) NiAl hydrotalcite,16 PdCo/CeO2,17 and Ru–MgO/TiO2 (ref. 18) as heterogeneous catalytic agents. However, while these hydrogen-borrowing catalysts tend to display great activity/selectivity for the amination of aliphatic alcohols, they are markedly less performing in the amination of lignin-derived substituted primary alcohols also comprising an phenol moiety, prone to preferential coordination, especially with supported metal catalysts comprising basic supports.19 In parallel, notable cases of the direct reductive amination of phenols to cyclohexylamines have been reported by the De Vos' group.20,21
Reductive catalytic fractionation (RCF) has recently emerged as a powerful tool22–25 to the depolymerization of lignin into lignin-oil that contains aromatic monomers in high selectivity. However, due to the high purification cost and the tedious downstream processing, commercializing of these monomer-rich lignin oils has proven very challenging26,27 and targeting higher value products offers to be a beneficial mitigation strategy.28–30
We have previously reported on the flexible use of Cu20PMO28,31,32 in a unique two stage catalytic lignocellulose disassembly process. In the first step, a reductive catalytic fractionation is performed at 140–180 °C, to deliver lignin-oil particularly rich (up to 90% selectivity) in dihydroconyferyl alcohol (1G).28 In the second step, the solid residues mainly consisting of cellulose(s) and unreacted lignin, were converted to a mixture of aliphatic alcohols in supercritical methanol, whereby the reducing equivalents for these processes originated from the solvent upon reforming.33,34 Strikingly, both these catalytic steps have delivered aliphatic alcohols, which are ideally suited for the development of sustainable amination protocols by the borrowing hydrogen strategy. As a central and representative example, we attempted the direct conversion of dihydroconiferyl alcohol (1G) to dihydroconiferyl amine (1G amine), an interesting building block for the making of polymeric materials35 as well as the synthesis of a range of pharmaceuticals (e.g. cuspareine,36 capsazepine,37 galipeine36) (Fig. 1A). This is a particularly challenging transformation as a) the obtained primary amine is prone to overalkylation, that way creating a secondary, or tertiary amine and b) dehydrogenated 1G (= the corresponding aldehyde product of the first step of the hydrogen borrowing procedure) is susceptible to decarbonylation, that way forming undesired 4-ethyl guaiacol.28
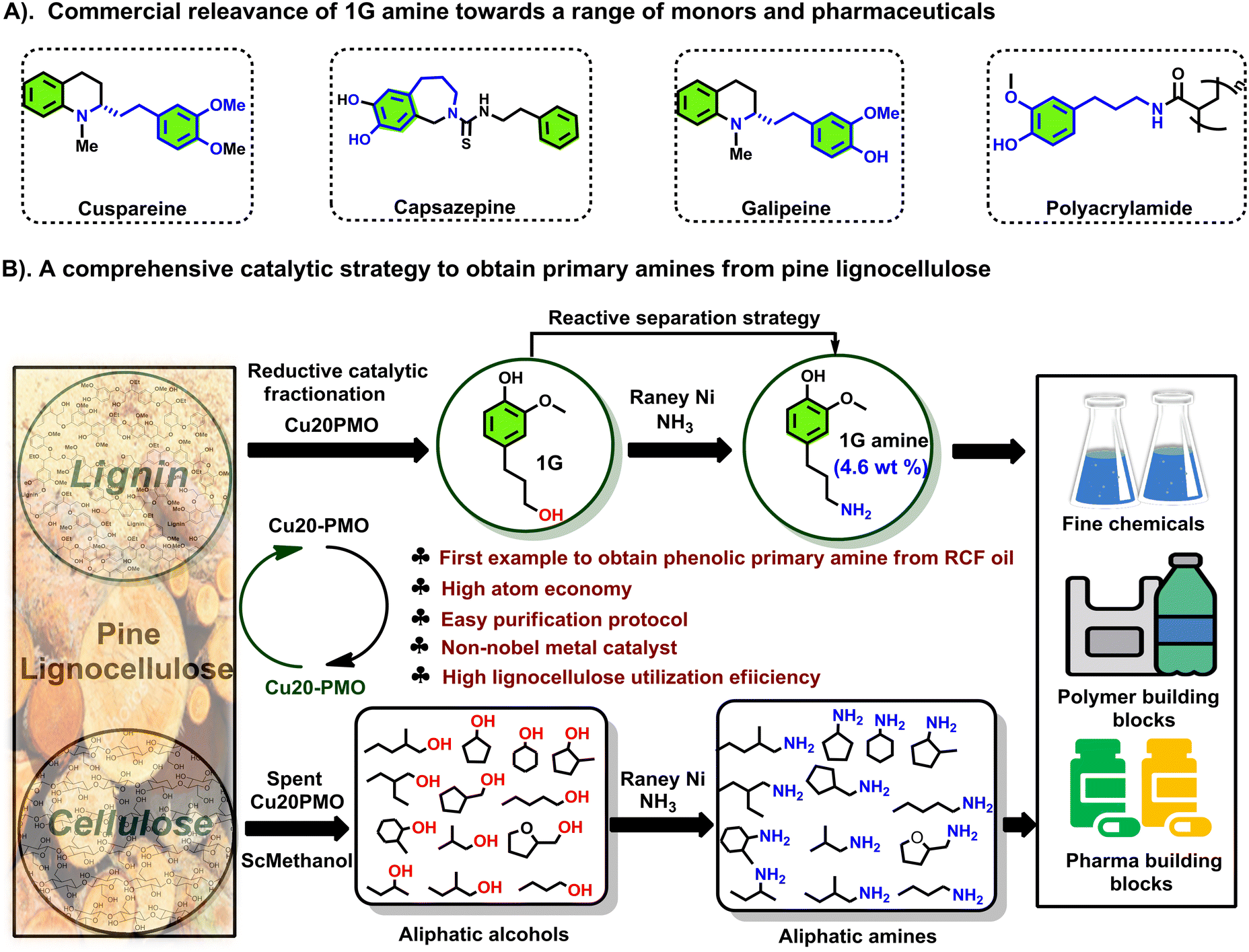 | ||
| Fig. 1 Strategy for the formation of primary amines from both cellulose- and lignin-derived aromatic/aliphatic alcohols, and this with specific attention to real life applications. | ||
Notably, the here developed heterogeneous catalytic procedure using RANEY® Ni as catalyst delivered excellent product selectivities. This was further validated in the amination of two other lignin-derived phenolic primary alcohols, namely dihydrosinapyl alcohol (1S) and 4-(3-hydroxypropyl)phenol (1H) as well as a range of aliphatic alcohols (primary and secondary), available from the second step of the LignoFlex procedure.28 In summary, this work presents a catalytic strategy for the depolymerization and upgrading of pine lignocellulose to primary amines (Fig. 1B).
2. Experimental
2.1 Catalyst preparation
The Cu20PMO catalyst was prepared according to our previously reported procedure.28 In a typical procedure, a solution containing AlCl3·6H2O (12.07 g, 0.05 mol), Cu(NO3)2·2.5H2O (6.98 g, 0.03 mol) and MgCl2·6H2O (24.4 g, 0.12 mol) in deionized water (200 mL) was dropwise added to a solution containing Na2CO3 (5.30 g, 0.05 mol) in water (300 mL) at 60 °C under vigorous stirring. The pH value was always kept between 9 and 10 by addition of small portions of a 1 M solution of NaOH. The mixture was vigorously stirred at 60 °C for 72 h. After cooling to room temperature, the light blue solid was filtered and resuspended in a 2 M solution of Na2CO3 (300 mL) and stirred overnight at 40 °C. The catalyst precursor was filtered and washed with deionized water until chloride free. After drying the solid for 6 h at 100 °C followed by the calcination at 460 °C for 24 h in air, 9.5 g of Cu20PMO was obtained.2.2 Characterizations
Gas Chromatography (GC) was used for products identification as well as determination of conversion and selectivity values.Products identification was performed by GC-MS (5975C MSD) equipped with an HP-5MS column, and helium as carrier gas. The temperature program started at 50 °C for 5 min, heated by 10 °C min−1 to 325 °C and held for 5 min. Conversion and products selectivity were determined by GC-FID (Shimadzu Agilent 8890 GC) equipped with an HP-5MS column using nitrogen as carrier gas. Nuclear Magnetic Resonance (NMR) spectroscopy: 1H, and 13C NMR spectra were recorded on a Bruker Avance III 300 MHz (300 and 75 MHz, respectively) and 2D NMR spectra were recorded on a Bruker Avance III 700 MHz with Cryoplatform and a 5 mm Triple-Resonance cryoprobe (700 and 175 MHz, respectively). 1H,13C NMR and 2D NMR spectra were recorded at RT. Chemical shift values are reported in ppm with the solvent resonance as the internal standard (CDCl3: 7.26 for 1H, 77.0 for 13C; CD3OD: 3.31 for 1H, 49.0 for 13C; DMSO-d6: 2.50 for 1H, 39.5 for 13C). Data are reported as follows: chemical shifts, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br. = broad, m = multiplet), coupling constants (Hz), and integration.
2.3 Method
3. Results and discussion
3.1 Catalytic amination of lignin-derived 1G to 1G amine
Our first goal was to accomplish the highly selective amination of the 4-(3-propanol) phenol derivatives, products of reductive catalytic fractionation (RCF) of lignocellulose, directly with ammonia gas. While this transformation appears straightforward, achieving high selectivity towards the primary amine is very challenging, due to a series of side reactions that may take place under the operating conditions, as summarized on Fig. 2A. An ideal catalytic system should favour alcohol dehydrogenation and facilitate imine-to amine reduction, both crucial steps in the hydrogen borrowing sequence. With this goal in mind, we have evaluated a range of commercially available heterogeneous catalysts towards the direct amination of 1G to 1G amine. Here, t-amyl alcohol was selected because it has been proved effective as a solvent towards the production of primary amines.19,38 Surprisingly, very low 1G conversions (typically under 10%) were observed with a range of catalysts at 150 °C, notably Pd/C, Pt/C, Rh/C, Ru/Al2O3, Ni/SiO2–Al2O3, Ni/SiO2, indicating low efficiency of alcohol dehydrogenation with these catalysts, under these conditions. Notably, in these runs, rather the corresponding 1G nitrile was seen as main product in no less than 90% selectivity (Table S1,† entries 1–6), which could be attributed to higher dehydrogenation activity than hydrogenation of 1G imine over these catalysts in the absence of H2 pressure. Best selectivity towards 1G nitrile (up to 99%) was observed with group VIII containing heterogeneous catalysts – notably Pd/C, Pt/C, Rh/C and Ru/Al2O3 (Table S1,† entries 1–4).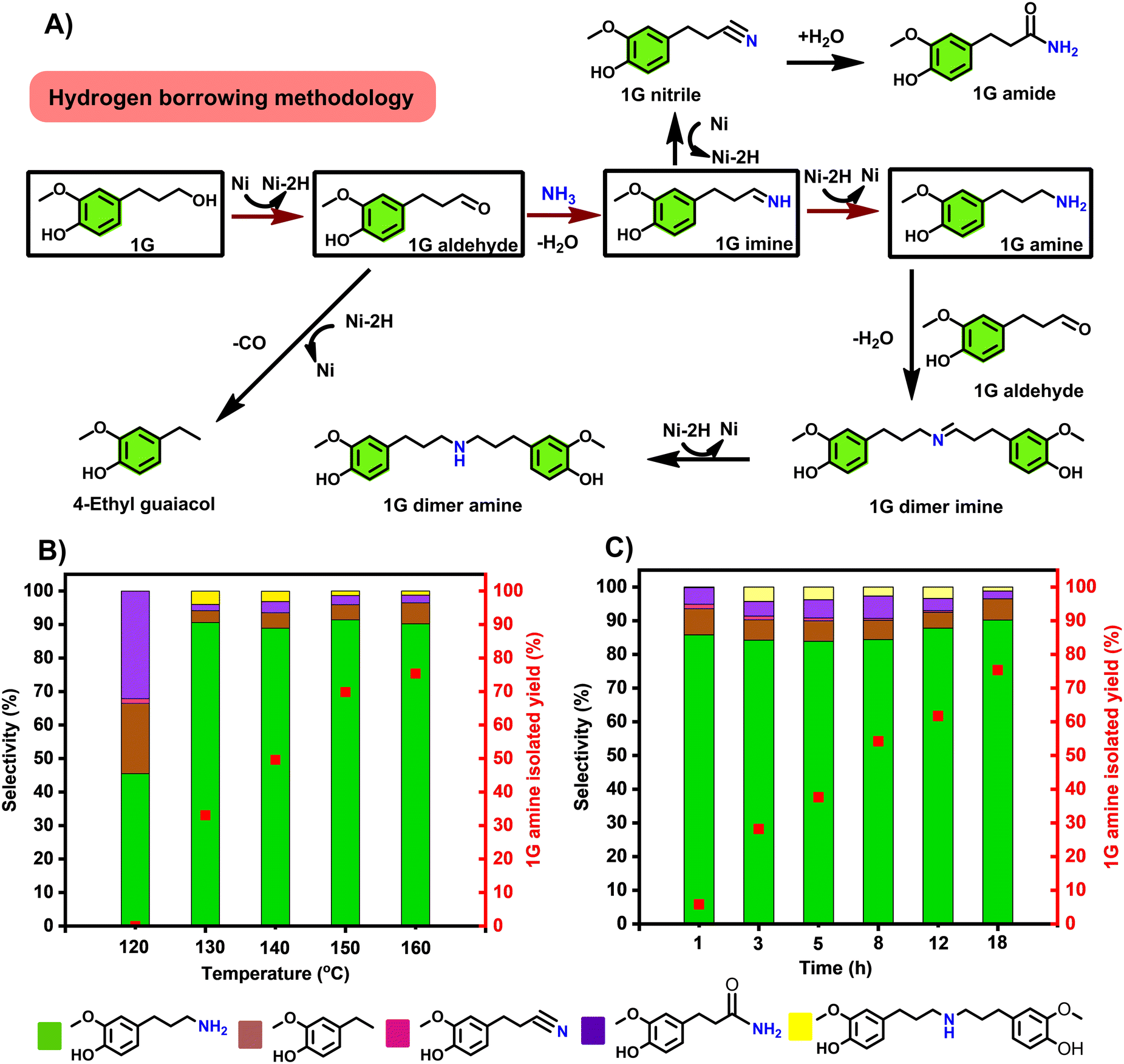 | ||
| Fig. 2 A) A proposed reaction network detailing desired main reactions towards the 1G amine and typical expected side-reactions, in relation to the experimentally observed major and minor products; B) influence of the reaction temperature on the reaction selectivity and the 1G amine isolated yield. Reaction time: 18 h; C) influence of the reaction time on reaction selectivity and the 1G amine isolated yield. Reaction temperature: 160 °C; general reaction conditions: 0.5 mmol 1G, 100 mg RANEY® Ni, 2.5 mL t-amyl alcohol, 120–160 °C, 1–18 h, 7 bar NH3. Selectivity and conversion were determined by GC-FID based on the peak area (see calculation methods in supplementary information). 1G amine was isolated as its HCl salt. For numerical values see Tables S2 and S3.† | ||
The formation of 1G nitrile likely involved 1G imine dehydrogenation which could also undergo subsequent hydration to form 1G amide – the other side product.39 With the base metal catalysts Ni/SiO2–Al2O3 and Ni/SiO2, the 4-ethylguaiacol was the dominant side product (Table S1,† entries 5 and 6). The latter compound derives from the decarbonylation of 1G aldehyde.28 The inactivity of supported metal catalyst was possibly attributed to preferred adsorption of phenol moiety of 1G on supports such as Al2O3 and SiO2, leaving aliphatic –OH moiety uncoordinated.40
Interestingly, in sharp contrast to all the other catalysts tested, the application of RANEY® Ni gave rise to the formation of 1G amine in 91.4% selectivity at a conversion level of 87.6% (Table S1, entry 7, Fig. S1†). From this reaction mixture 1G amine could be obtained as its HCl salt in up to 70% isolated yield. Overalkylation of 1G amine to 1G dimer amine was only detected in very small amounts (Table S1,† entry 7). The high catalytic reactivity was mainly ascribed to the fact that RANEY® Ni is highly efficient in a range of hydrogen transfer transformations, including amination by hydrogen-borrowing reactions.41 It has to be noticed that using atmospheric pressure of NH3 shifted towards the production of 4-ethyl guaiacol, with a selectivity of >99%. (Table S4†) Therefore this catalyst system was selected for detailed optimization over a wide temperature range (120–160 °C), the results having been summarized in Fig. 2B. A high 1G amine selectivity was obtained in all these runs. As though the lower temperature range represented incomplete conversions (Tables S2 and S3†), the isolated 1G amine yield was found to increase with the applied reaction temperature, reaching a maximum of 75.8% at 160 °C (Fig. 2B). While the 1G amine selectivity was found independent of the applied reaction time, the isolated 1G amine yield increased with the reaction time (Fig. 2C). In addition, catalyst loading was also investigated (Table S5†). It was found the optimal catalyst loading is 100 mg, which gives the best 1G conversion of 94.6% and isolated yield (75.8%) to 1G amine. While using decreased catalyst loading only lower the 1G conversion, proceeding 25.8 and 66.2%, respectively. Upscaling the reaction using 500 mg 1G led to slightly decreased catalytic reactivity, with a conversion of 86.6% and 80.5% selectivity (Table S6†).
In a further advancement we studied the synthetic possibilities of 1G amine towards the formation of important pharmaceutical and polymeric building blocks (see Fig. 3 for an overview) and proposed suitable future applications, inspired by recent literatures. First, demethylation of 1G amine to dopa 1G amine (Fig. 3-1B) was achieved in 92% isolated yield. The latter compound is an essential building block to the medicine capsazepine37 (see also Fig. 1A). Next, 1G amine was further reacted with vanillyl acid and ferulic acid which can be in principle sourced from lignin, that way providing for a range of polymerizable bio-based bisguaiacols42,43 (Fig. 3-2B and 3B). The respective isolated yields of 2B and 3B are 94.8% and 83.0%. Furthermore, the functionalization of 1G amine with methyl bromoacetate gave a potential polyamide building block 4B,44 while the reaction of 1G amine with epichlorohydrin yields a multiepoxy containing monomer 5B obtained in 34.8% isolated yield (Fig. 3-5B), which is highly suitable for the creation of epoxy resins. Alternatively, functionalization of 1G amine with acrylic acid yields a suitable monomer (6B) for (controlled) radical and cationic polymerization.35 The isolated yield of 6B was 85.2%.
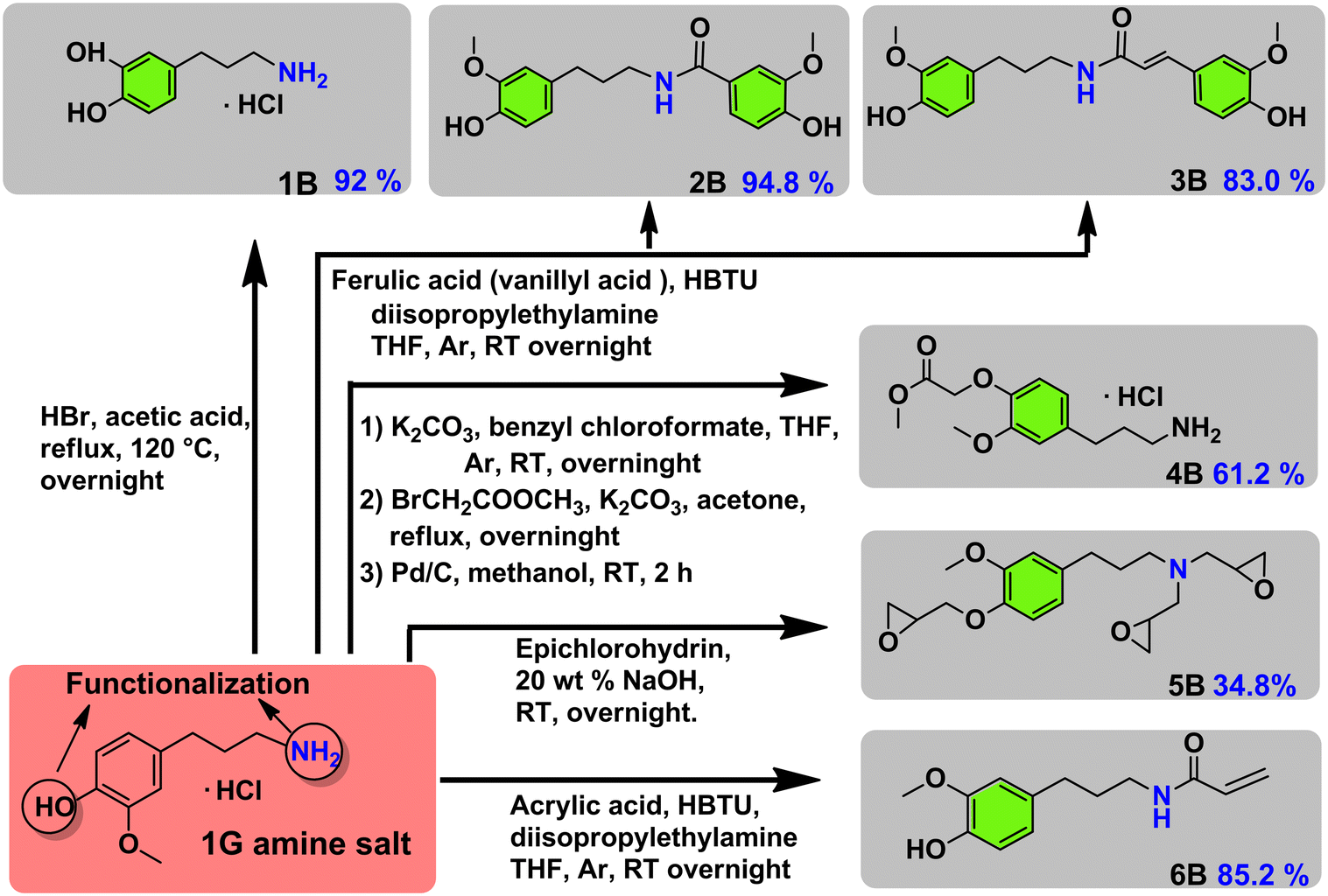 | ||
| Fig. 3 Potential usage of 1G amine as a synthetic building block for a range of pharmaceutical and polymeric building blocks. | ||
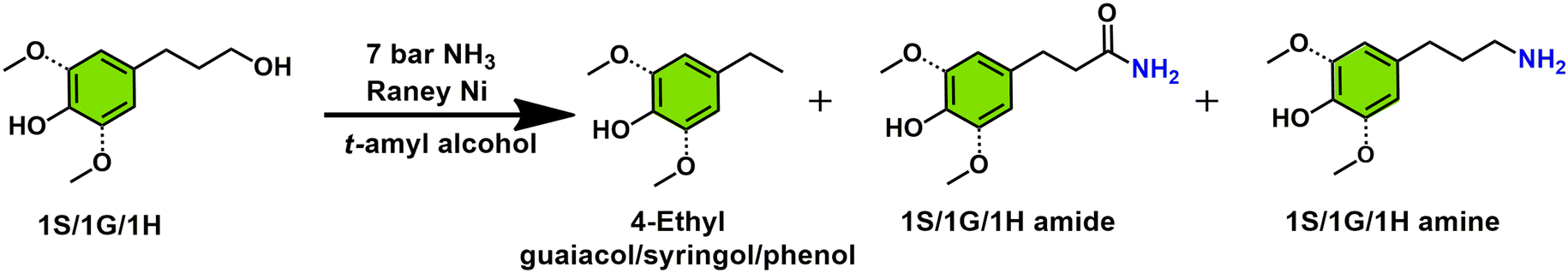
Next to 1G amine formation, the usage of RANEY® Ni as a hydrogen borrowing amination catalyst was also tested, under optimal reaction conditions, in the amination of two other lignin-derived platform molecules that may originate from the RCF of hardwood, 1S and 1H. This leads respectively to the formation of dihydrosinapyl amine (1SA) and 4-(3-aminopropyl)phenol (1HA), two structural motives omnipresent in pharmaceuticals (e.g. trimethoprim).45 In comparing Table 1, it can be seen that the presence an additional methoxy group vis-à-vis1G, as in compound 1S, leads to a marked drop in conversion, going from 94.6% for 1G to 66.4% for 1S. Importantly though, the selectivity for 1S amine is maintained at 91.2%, slightly higher than the one observed for 1G amine (90.2%). Conversely, with compound 1H as substrate near quantitative conversion is obtained (99.5%) at a similarly high selectivity (89.4%) as observed with the formation of 1G amine. With 1S the conversion level could be further increased from 66.4% (seen at 160 °C) to 89.2% by using a somewhat higher reaction temperature of 180 °C, with only a slight decrease in 1S amine selectivity (89.4%). Isolated yields of the HCl salts of 1S amine and 1H amine were 64.5% and 53.4% respectively.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Substrate | T (°C) | Conv. (%) | Sel. (%) | GC yield (%) | Isolated yield (%) | ||
| 4-Ethyl/guaiacol syringol | 1S/1G/1H amide | 1S/1G/1H amine | |||||
| a Reaction conditions: 0.5 mmol substrate, 100 mg RANEY® Ni, 2.5 mL t-amyl alcohol, 18 h, 7 bar NH3. Selectivity, conversion and yield were determined by GC-FID based on the peak area (see calculation methods in ESI†). | |||||||
| 1H | 160 | 99.5 | 4.1 | 6.4 | 89.4 | 90.0 | 53.4 |
| 1G | 160 | 94.6 | 5.0 | 2.5 | 90.2 | 85.3 | 75.8 |
| 1S | 160 | 66.4 | 3.2 | 1.8 | 91.2 | 60.6 | 32.4 |
| 1S | 180 | 89.2 | 3.5 | 2.2 | 89.4 | 79.7 | 64.5 |
3.2 Catalytic upgrading of native-lignin into 1G amine
Next, we attempted to apply our RANEY® Ni-based amination protocol developed on model compounds, directly to the crude depolymerized lignin oil obtained by RCF of pine lignocellulose over previously developed procedure using 2 g pine lignocellulose and Cu20PMO as catalyst at reaction conditions (2 g pine, 0.4 g Cu20PMO, 20 mL methanol, 40 bar H2, 180 °C, 18 h). When applying the here developed amination procedure directly to crude aromatic bio-oil (see Fig. 4A), consisting mainly of monomers (1G, 46 mg, 2G, 4.1 mg, 3G, 0.6 mg) but also a range of other compounds typical for such bio-oil such as dimers, oligomers and sugars,28,46 no amination could be observed. This is tentatively attributed to the presence of oligomers/polysaccharides and a small amount of organic acids.46,47 To alleviate these problems the crude aromatic bio-oil was extracted with ethyl acetate (EtOAc), followed by a treatment with small amount of NaHCO3/brine, that way yielding a purified bio-oil (EtOAc extracts) rich in 1G and lesser amounts of 2G and 3G, as characterized by GC-FID (Fig. 4B). Gratifyingly in applying the amination protocol to such EtOAc extracts, 1G amination was found to proceed cleanly (Fig. 4B). Moreover, inherent purification of 1G amine was found possible by precipitation as its HCl salt, characterized by 1H NMR and 13C NMR (see Fig. 4C), giving a 4.6 wt% isolated yield on lignin basis. | ||
| Fig. 4 Methodology to obtain 1G amine from pine lignocellulose. A) Established protocol for the 1G amine formation from pine lignocellulose; step 1: 2 g pine lignocellulose, 400 mg Cu20PMO, 20 mL methanol, 180 °C, 18 h; step 2: 200 mg RANEY® Ni, 2.5 ml t-amyl alcohol, 7 bar NH3, 160 °C, 18 h; B) GC-FID traces of the EtOAc extracts of the original crude RCF bio-oil and the crude aminated bio-oil C) 1H NMR and 13C NMR spectra of the 1G amine salt isolated from the crude aminated lignin oil using HCl. | ||
3.3 Direct amination aliphatic alcohol intermediates originating from cellulose residues
As the second step of the LignoFlex procedure converts the remaining polysaccharides over Cu20PMO in supercritical methanol, into a mixture consisting of predominantly aliphatic primary/secondary linear/cyclic alcohols,28 our heterogeneous catalytic amination methodology was also evaluated on individual model compounds representing the main compounds of these lignocellulose-derived product mixtures. As can be inferred from Fig. 5, most depicted amination reactions gave GC determined yields larger than 80%, and isolated yields between 40–70%. It can further be seen that steric hindrance adjacent to the alcohol moiety tends to decrease the obtained yield (1aversus2a, 5aversus6a, 7aversus13a). Also, the substitution of a cyclopentane ring for a tetrahydrofuran one markedly influenced the obtained amine yield, reaching 93.1% and 60.8%, respectively. No distinct difference in reactivity between primary and secondary aliphatic alcohols could be discerned.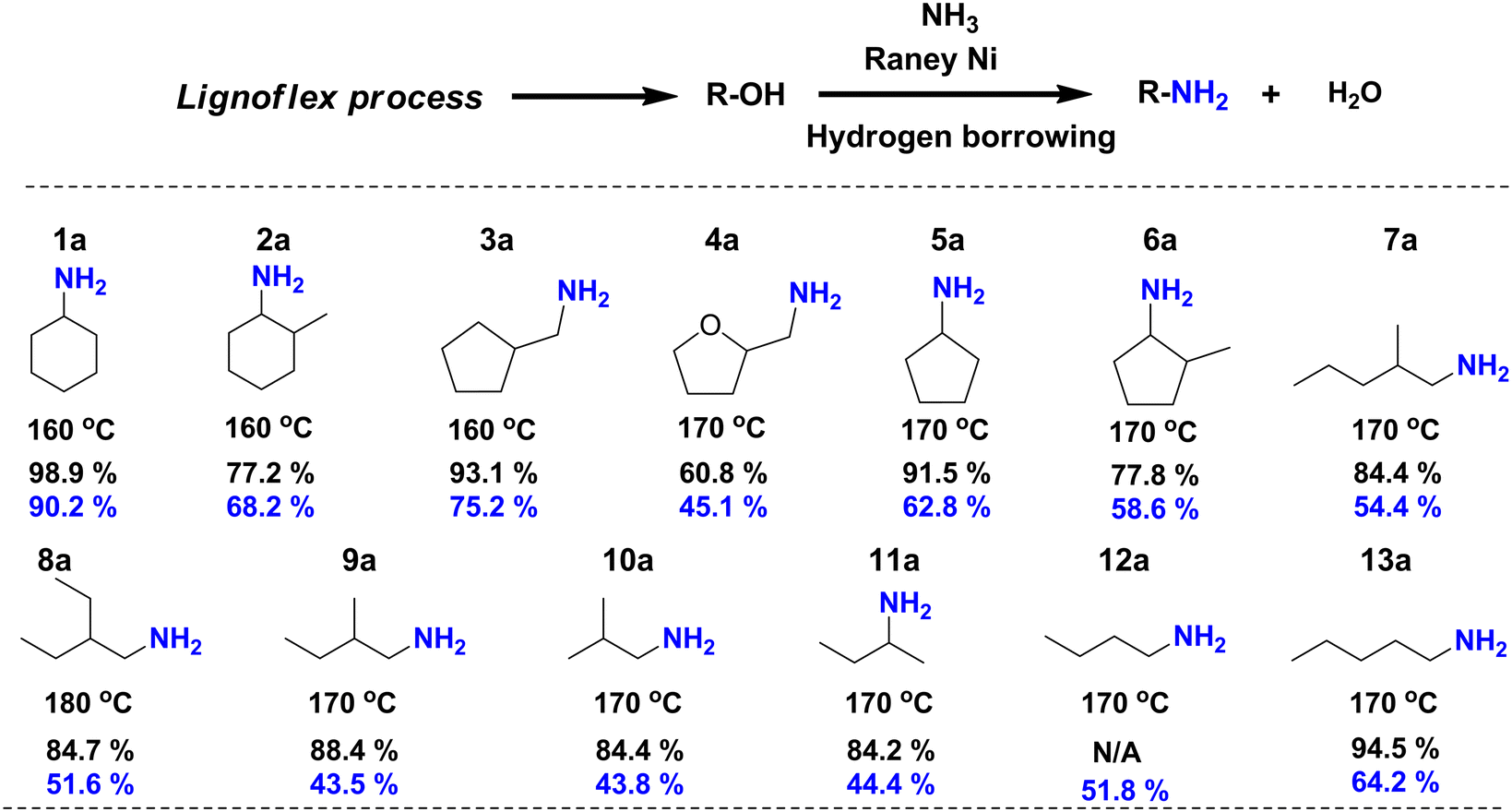 | ||
| Fig. 5 Catalytic direct amination of polysaccharide-derived aliphatic primary/secondary linear/cyclic alcohols to their corresponding primary/secondary amines. All aliphatic alcohols present here were separately treated by catalytic amination procedure to corresponding primary amines. Reaction conditions: 0.5 mmol substrates, 100 mg RANEY® Ni catalyst, 2.5 mL t-amyl alcohol, 7 bar NH3, 160–180 °C, 18 h. Yield: the black numbers refer to the yield determined by GC-FID based on the peak area (see calculation methods in ESI†) while the blue numbers relate to isolated yields. For compound 12a, the GC-FID peak is overlapped with solvent peak. | ||
We note, that a gap between isolated yield and GC yield is observed especially for linear/branched short chain aliphatic amine salts (e.g.7a–13a) compared to a lower than 20% gap for amine products bearing cyclic aliphatic or aromatic moieties. We attribute this to the more challenging or incomplete isolation of the respective amine salts, rather than selectivity reasons.
The rationale of using single model alcohol compounds over artificial/real mixtures of alcohols lies in the fact that future alcohol producing biorefineries – such as those based on the Lignoflex procedure – would be able to separate pure product streams by distillation of such alcohol mixtures, before converting them to amines. This is rationalized by the fact that 1) mixtures of amines have lower boiling points as well as a narrower spread of the boiling points than mixtures of alcohols and 2) in converting alcohol mixtures to their corresponding amines one often creates undesired azeotropes. In these respects it is also noteworthy that industrial amine separation revolves around multi-distillation trains, extensive extraction steps and recycling of various streams, which all comes at high energy and materials costs.48 Nonetheless, the direct application of our method to alcohol mixtures to create mixtures of amines for certain specific applications (e.g. surfactants, lubricants) may also be an industrially relevant scenario.
4. In conclusion
We have developed a RANEY® Ni-based amination methodology capable of transforming lignin-derived phenolic primary alcohols with differing aromatic substitution patterns into their corresponding primary amines in high selectivities (∼90%) at high conversions (>90%) while keeping the phenol moiety intact. Moreover, this was achieved using an atom efficient hydrogen borrowing approach, the only side product being water. Furthermore, provided a first extraction step, RANEY® Ni was found capable to aminate a bio-oil rich in 1G (as obtainable through the LignoFlex procedure) yielding the corresponding amino-alkyl phenol (1G amine) in 4.6% isolated yield on lignin basis. To the best of our knowledge the capability of aminating phenolic primary alcohols obtained from real RCF oil directly using ammonia, with a heterogeneous catalyst, and following an atom economic hydrogen borrowing methodology, has not yet been reported on. Furthermore, by providing relevant synthetic pathways, we have concisely shown that such phenolic primary amines can serve as suitable pharmaceutical and polymeric building blocks. Moreover, applying the developed amination methodology to a range of model aliphatic primary/secondary linear/cyclic alcohols, such as those obtained from the LignoFlex process, we demonstrated the applicability of the same method on converting cellulose-derived platform chemicals into primary amines in high overall yield.Overall, the capability to derive a broad range of amines from different biomass constituents: lignin as well as (hemi)cellulose, using a non-noble heterogeneous catalyst, an atom economic hydrogen borrowing methodology, and involving minimal purification efforts (minimal extraction and facile isolation), holds potential to broadening the scope of sustainable biorefineries producing a range of value-added amines.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. B. is grateful for financial support from the European Research Council, ERC Starting Grant 2015 (CatASus) 638076 and ERC proof of Concept Grant 2019 (PURE) 875649. This work is part of the research programme Talent Scheme (Vidi) with project number 723.015.005 (K. B.), which is partly financed by The Netherlands Oganization for Scientific Research (NWO). X. W. is grateful for financial support from the China Scholarship Council (grant number 201808330391).References
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- V. Froidevaux, C. Negrell, S. Caillol, J. P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University, Cambridge, 2004 Search PubMed.
- D. T. Smith, M. D. Delost, H. Qureshi and J. T. Njarðarson, Top 200 Pharmaceutical Products by Retail Sales in 2016, https://njardarson.lab.arizona.edu/sites/njardarson.lab.arizona.edu/files/2016Top200PharmaceuticalRetailSalesPosterLowResV3_0.pdf Search PubMed.
- B. M. Stadler, C. Wulf, T. Werner, S. Tin and J. G. de Vries, ACS Catal., 2019, 9, 8012–8067 CrossRef CAS.
- X. Chen, S. Song, H. Li, G. K. Gözaydın and N. Yan, Acc. Chem. Res., 2021, 54, 1711–1722 CrossRef CAS PubMed.
- M. Pelckmans, T. Renders, S. Van de Vyver and B. Sels, Green Chem., 2017, 19, 5303–5331 RSC.
- K. Murugesan, T. Senthamarai, V. G. Chandrashekhar, K. Natte, P. C. Kamer, M. Beller and R. V. Jagadeesh, Chem. Soc. Rev., 2020, 49, 6273–6328 RSC.
- S. S. Wong, R. Y. Shu, J. G. Zhang, H. C. Liu and N. Yan, Chem. Soc. Rev., 2020, 49, 5510–5560 RSC.
- A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed.
- S. Koller, P. Klein, K. Reinhardt, L. Ochmann, A. Seitz, C. Jandl, A. Pothig and L. Hintermann, Helv. Chim. Acta, 2021, 104, e202100175 CrossRef.
- K. I. Shimizu, Catal. Sci. Technol., 2015, 5, 1412–1427 RSC.
- L. A. Y. K. Leung, K. Hellgardt and K. K. Hii, ACS Sustainable Chem. Eng., 2018, 6, 5479–5484 CrossRef.
- K. I. Shimizu, K. Kon, W. Onodera, H. Yamazaki and J. N. Kondo, ACS Catal., 2013, 3, 112–117 CrossRef CAS.
- K. I. Shimizu, S. Kanno, K. Kon, S. M. A. H. Siddiki, H. Tanaka and Y. Sakata, Catal. Today, 2014, 232, 134–138 CrossRef CAS.
- K. Zhou, R. H. Xie, M. T. Xiao, D. R. Guo, Z. D. Cai, S. M. Kang, Y. J. Xu and J. J. Wei, ChemCatChem, 2021, 13, 2074–2085 CrossRef CAS.
- T. Tong, W. J. Guo, X. H. Liu, Y. Guo, C. W. Pao, J. L. Chen, Y. F. Hu and Y. Q. Wang, J. Catal., 2019, 378, 392–401 CrossRef CAS.
- Y. Kita, M. Kuwabara, S. Yamadera, K. Kamata and M. Hara, Chem. Sci., 2020, 11, 9884–9890 RSC.
- Y. Z. Liu, A. Afanasenko, S. Elangovan, Z. H. Sun and K. Barta, ACS Sustainable Chem. Eng., 2019, 7, 11267–11274 CrossRef CAS PubMed.
- P. Tomkins, C. Valgaeren, K. Adriaensen, T. Cuypers and D. E. De Vos, ChemCatChem, 2018, 10, 3689–3693 CrossRef CAS.
- T. Cuypers, T. Morias, S. Windels, C. Marquez, C. Van Goethem, I. Vankelecom and D. E. De Vos, Green Chem., 2020, 22, 1884–1893 RSC.
- Y. M. Questell-Santiago, M. V. Galkin, K. Barta and J. S. Luterbacher, Nat. Rev. Chem., 2020, 4, 311–330 CrossRef CAS.
- T. Renders, G. Van den Bossche, T. Vangeel, K. Van Aelst and B. Sels, Curr. Opin. Biotechnol., 2019, 56, 193–201 CrossRef CAS PubMed.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2016, 9, 1544–1558 CrossRef CAS PubMed.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Roman-Leshkov, J. S. M. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- E. Cooreman, T. Vangeel, K. Van Aelst, J. Van Aelst, J. Lauwaert, J. W. Thybaut, S. Van den Bosch and B. F. Sels, Ind. Eng. Chem. Res., 2020, 59, 17035–17045 CrossRef CAS.
- M. Tschulkow, T. Compernolle, S. Van den Bosch, J. Van Aelst, I. Storms, M. Van Dael, G. Van den Bossche, B. Sels and S. Van Passel, J. Cleaner Prod., 2020, 266, 122022 CrossRef CAS.
- Z. H. Sun, G. Bottari, A. Afanasenko, M. C. A. Stuart, P. J. Deuss, B. Fridrich and K. Barta, Nat. Catal., 2018, 1, 82–92 CrossRef CAS.
- Z. H. Sun and K. Barta, Chem. Commun., 2018, 54, 7725–7745 RSC.
- Z. H. Sun, J. L. Cheng, D. S. Wang, T. Q. Yuan, G. Y. Song and K. Barta, ChemSusChem, 2020, 13, 5199–5212 CrossRef CAS PubMed.
- K. Barta, T. D. Matson, M. L. Fettig, S. L. Scott, A. V. Iretskii and P. C. Ford, Green Chem., 2010, 12, 1640–1647 RSC.
- T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2011, 133, 14090–14097 CrossRef CAS PubMed.
- K. Barta and P. C. Ford, Acc. Chem. Res., 2014, 47, 1503–1512 CrossRef CAS PubMed.
- M. De Bruyn, Z. H. Sun and K. Barta, Adv. Inorg. Chem., 2021, 77, 59–98 CrossRef.
- H. F. Liu, B. Lepoittevin, C. Roddier, V. Guerineau, L. Bech, J. M. Herry, M. N. Bellon-Fontaine and P. Roger, Polymer, 2011, 52, 1908–1916 CrossRef CAS.
- S. G. Davies, A. M. Fletcher, I. T. Houlsby, P. M. Roberts, J. E. Thomson and D. Zimmer, J. Nat. Prod., 2018, 81, 2731–2742 CrossRef CAS PubMed.
- https://en.wikipedia.org/wiki/Capsazepine .
- X. Y. Wu, M. V. Galkin and K. Barta, Chem Catal., 2021, 1, 1466–1479 CrossRef.
- K. Yamaguchi, H. Kobayashi, Y. Wang, T. Oishi, Y. Ogasawara and N. Mizuno, Catal. Sci. Technol., 2013, 3, 318–327 RSC.
- A. Popov, E. Kondratieva, J. P. Gilson, L. Mariey, A. Travert and F. Mauge, Catal. Today, 2011, 172, 132–135 CrossRef CAS.
- Z. H. Sun, Z. H. Zhang, T. Q. Yuan, X. H. Ren and Z. M. Rong, ACS Catal., 2021, 11, 10508–10536 CrossRef CAS.
- A. W. Bassett, C. M. Breyta, A. E. Honnig, J. H. Reilly, K. R. Sweet, J. J. La Scala and J. F. Stanzione, Eur. Polym. J., 2019, 111, 95–103 CrossRef CAS.
- K. H. Nicastro, C. J. Kloxin and T. H. Epps, ACS Sustainable Chem. Eng., 2018, 6, 14812–14819 CrossRef CAS.
- F. Poulhes, D. Mouysset, G. Gil, M. P. Bertrand and S. Gastaldi, Polymer, 2013, 54, 3467–3471 CrossRef CAS.
- https://en.wikipedia.org/wiki/Trimethoprim .
- S. Elangovan, A. Afanasenko, J. Haupenthal, Z. H. Sun, Y. Z. Liu, A. K. H. Hirsch and K. Barta, ACS Cent. Sci., 2019, 5, 1707–1716 CrossRef CAS PubMed.
- X. Y. Wu, M. V. Galkin, T. Stern, Z. H. Sun and K. Barta, Nat. Commun., 2022, 13, 3376 CrossRef CAS PubMed.
- W. Featherstone, Amine separation process, US Pat., US4407703A, 1983 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy00864e |
| This journal is © The Royal Society of Chemistry 2022 |