
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Covalent triazine-based frameworks with cobalt-loading for visible light-driven photocatalytic water oxidation†
Hongmei
Chen
a,
Adrian M.
Gardner
b,
Guoan
Lin
c,
Wei
Zhao
a,
Mounib
Bahri
 d,
Nigel D.
Browning
e,
Reiner Sebastian
Sprick
ae,
Xiaobo
Li
*fa,
Xiaoxiang
Xu
*c and
Andrew I.
Cooper
*a
d,
Nigel D.
Browning
e,
Reiner Sebastian
Sprick
ae,
Xiaobo
Li
*fa,
Xiaoxiang
Xu
*c and
Andrew I.
Cooper
*a
aDepartment of Chemistry and Materials Innovation Factory, University of Liverpool, Liverpool L7 3NY, UK. E-mail: aicooper@liverpool.ac.uk
bStephenson Institute for Renewable Energy, University of Liverpool, Liverpool L69 7ZF, UK
cClinical and Central Lab, Putuo People's Hospital and Shanghai Key Lab of Chemical, Assessment and Sustainability, School of Chemical Science and Engineering, Tongji University, Shanghai 200060, China. E-mail: xxxu@tongji.edu.cn
dAlbert Crewe Centre for Electron Microscopy, University of Liverpool, Liverpool L69 3GL, UK
eDepartment of Pure and Applied Chemistry, University of Strathclyde, Thomas Graham Building, 295 Cathedral Street, Glasgow G1 1XL, UK
fKey Laboratory of the Ministry of Education for Advanced Catalysis Materials, Zhejiang Key Laboratory for Reactive Chemistry on Solid Surfaces, Institute of Physical Chemistry, Zhejiang Normal University, Jinhua 321004, China. E-mail: xiaobo.li@zjnu.edu.cn
First published on 27th July 2022
Abstract
Conjugated polymers have received significant attention as photocatalysts. However, photocatalytic oxygen evolution has only been reported for a few polymers so far. Here, we present a bipyridine based covalent triazine-based framework containing metal coordination sites (Bpy-CTF). The material is highly active for sacrificial photocatalytic oxygen evolution with a rate of 322 μmol g−1 h−1 under visible light illumination (≥420 nm) after post-synthetic cobalt coordination. An analogous photocatalyst containing biphenyl was found to be less active as it is not able to coordinate cobalt. Transient absorption spectroscopy studies showed that the cobalt coordinated in the bipyridine units of Bpy-CTF promotes charge separation and transfer, thus increasing water oxidation activity. The study demonstrates the growing potential of polymer photocatalysts for oxygen evolution by structural engineering and post-synthetic metalation.
1. Introduction
Solar-driven photocatalytic water splitting is an attractive method for producing green hydrogen.1–4 To effectively split water into hydrogen and oxygen, the hydrogen evolution half reaction must be balanced by an oxygen evolution reaction. Water oxidation is typically the more challenging reaction,5 involving O–O bonds formation that requires a four-electron transfer, which is usually associated with a high overpotential and sluggish kinetics.Due to their structural diversity and tunable photophysical properties, organic semiconductor photocatalysts have received interest for photocatalytic water oxidation reaction.6–10 Carbon nitride was the first reported organic photocatalyst for oxygen production in 2009.11 In the last decade, the photocatalysis efficiency of carbon nitride has been promoted by structure engineering,12 such as constructing element defects and element doping, cocatalyst loading, and hybrid semiconductor systems.
Beyond carbon nitrides,13,14 some covalent triazine-based frameworks (CTFs)12,15–17 have shown activity for photocatalytic water oxidation reactions. Wang et al. synthesised CTP-2 using a superacid-catalysed polycondensation method and this framework showed a sacrificial oxygen evolution reaction (OER) activity of 50 μmol g−1 h−1 under visible light irradiation (>420 nm).18 Tang et al. synthesised CTF-1-100W using a microwave method, which gave a sacrificial OER rate of 140 μmol g−1 h−1 under visible light illumination (>420 nm), with an apparent quantum yield (AQY) of 3.8% at 420 nm.16 Tang et al. prepared CTF-0-I using an ionothermal trimerization method, which gave an AQY of 5.2% at 420 nm for photocatalytic water oxidation.15 Recently, a mild condensation strategy to produce CTFs from aldehydes and amidines has been reported;19 these CTFs possessed high surface areas and showed high activities for the sacrificial photocatalytic hydrogen evolution half-reaction. This mild synthesis also allows for facile scale-up to synthesis at the multigram level. However, there have been no reports of CTFs synthesised using this mild aldehyde condensation strategy for application in photocatalytic water oxidation.
The enhancement of active site design in photocatalysts has attracted much attention since the successes in carbon nitride-based photocatalysts.12,20 Incorporating bipyridine units into a polymer photocatalyst has been shown to improve the photocatalytic water oxidation activity. A bipyridine-based COF (Bp-COF) structure containing bipyridine units that were used to coordinate cobalt as a cocatalyst for oxygen evolution was reported.21 Bipyridine units have also been used to stabilize metal atoms in linear polymers,22 COFs,23–25 and MOFs26 for hydrogen evolution and CO2 reduction. Here, we introduced the bipyridine units into the CTF-1 structure via the mild aldehyde condensation reaction. The resulting Bpy-CTF material possesses high surface areas, which contributes to its improved activity for photocatalytic water oxidation under visible light illumination. The oxygen evolution rates were significantly enhanced after coordinating cobalt onto the bipyridine units of Bpy-CTF due to the improved charge separation and the presence of catalytic active sites for sacrificial water oxidation.
2. Experimental section
2.1 Materials
2,2′-Bipyridyl-5,5′-dialdehyde was obtained from ABCR. Benzene-1,4-dicarbonitrile, 4,4′-biphenyl-dicarboxaldehyde, and cobalt(II) nitrate hexahydrate were purchased from TCI Europe. Lithium bis(trimethylsilyl)azanide (1.0 mol L−1 in THF), caesium carbonate (Cs2CO3), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich. Hydrochloric acid, ethanol, methanol, and tetrahydrofuran (THF) were from Fisher Scientific. All chemicals were used as received.2.2 Catalysts synthesis
Bpy-CTF and B-CTF were synthesized by using a modified previous reported condensation method.27 In a typical synthesis, terephthalamidine dihydrochloride (117.5 mg, 0.5 mmol), caesium carbonate (245 mg, 0.75 mmol), and dimethyl sulfoxide (10 mL) were stirred at 100 °C for 30 min under an N2 atmosphere. 2,2′-Bipyridyl-5,5′-dialdehyde (53 mg, 0.25 mmol) was dissolved in dimethyl sulfoxide (20 mL) by sonication and then added to the previously formed mixture at 90 μL min−1 by a syringe pump. The mixture was kept stirring at 100 °C for 24 hours, and then heated at 180 °C for 72 hours, equipped with a condenser and a CaCO3 drying tube. The reaction mixture was cooled to room temperature and quenched with water. The precipitate was filtered and washed with water, ethanol, and acetone, before Soxhlet extraction with methanol and tetrahydrofuran. The final product Bpy-CTF (103 mg, yield: 83%) was dried in a vacuum oven at 80 °C overnight. B-CTF (97 mg, yield: 78%) was synthesized by the same procedure by using 4,4′-biphenyl-dicarboxaldehyde (52.5 mg, 0.25 mmol).Bpy-CTF (20 mg) was dispersed in tetrahydrofuran (20 mL) by sonication, and then predefined quantity of a Co(NO3)2·6H2O tetrahydrofuran solution (10 mg mL−1) was added. The suspension was stirred at 65 °C for 2 hours and then filtered. The resulting solid was washed with ethanol and dried in a vacuum oven at 80 °C overnight. The same method was used for cobalt loading onto B-CTF. The resulting products were named Bpy-CTF–Co-x and B-CTF–Co-x (x refers to the cobalt loading amount).
2.3 Catalysts characterizations
1H solution nuclear magnetic resonance (NMR) spectra were measured using a Bruker Avance 400 NMR spectrometer at 400 MHz. Samples were dissolved in DMSO solution. 13C cross polarization magic angle spinning nuclear magnetic resonance (13C CP/MAS NMR) measurements were carried out at 100.63 MHz using a Bruker Avance III HD spectrometer and 4 mm (rotor o.d.) probe. Spectra were acquired at a spin rate of 10 kHz. Crosspolarisation (CP) spectra were recorded with TOSS spinning sideband suppression, 1 ms contact time and with a recycle delay of 1 s. Carbon spectral referencing is relative to neat tetramethylsilane, carried out by setting the high frequency signal from an external sample of adamantane to 38.5 ppm. Fourier-transform infrared spectroscopy (FT-IR) were recorded on a Bruker alpha spectrometer by attenuated total reflectance (ATR). X-ray photoelectron spectroscopy (XPS) analysis was performed using a Kratos Axis SUPRA XPS fitted with a monochromated Al Kα X-ray source (1486.7 eV), a spherical sector analyser and 3 multichannel resistive plate, 128 channel delay line detectors. Data was analysed using CasaXPS v2.3.20PR1.0 and the spectra were calibrated with C 1s peak at 284.8 eV. Element analysis (EA) was performed on a Thermo EA1112 Flash CHNS–O analyzer using standard microanalytical procedures. Powder X-ray diffraction (PXRD) data were collected on a Panalytical Empyrean diffractometer in transmission mode. Powder samples were positioned on a Mylar film. A high throughput screening XYZ stage, X-ray focusing mirror, and PIXcel detector were equipped, and Cu Kα (λ = 1.541 Å) was used as light source. Thermogravimetric analysis (TGA) was carried out on EXSTAR6000 ranging from room temperature to 800 °C (10 °C min−1) under a 10 ml min−1 nitrogen flow. The nitrogen adsorption isotherms of synthesized samples (77.3 K) were measured by a Micromeritics ASAP 2020 volumetric adsorption analyser. The powder samples were degassed at 393 K for 12 h under vacuum (10–5 bar) before analysis. The BET surface areas were calculated in the relative pressure range (P/P0) from 0.05 to 0.12. Pore size distributions of samples were obtained by fitting the adsorption data with the nonlocal density functional theory (NL-DFT) model. Water vapor isotherms were collected at 293 K on an IGA gravimetric adsorption apparatus (Hiden Isochema, Warrington, UK), equipped with an anti-condensation system and an ultrahigh vacuum system. Transmittance of sample dispersions were measured on a Formulaction S.A.S. Turbiscan AGS system. An 880 nm NIR diode was used as light source, and a detector was fixed at 180° compared to the position of light source. For each measurement, 5 mg sample powders was dispersed in 20 ml water by sonication, and transmittance result is a number average from 4 mm to 30 mm height of sample transmittance. Static light scattering measurements (SLS) were carried out on a Malvern Mastersizer 3000 Particle Sizer, at room temperature. Samples were dispersed in aqueous solutions by sonication. UV-visible light absorption spectra of the solid state samples were obtained using a Cary 5000 UV-vis-NIR spectrometer in reflectance mode. Steady-state photoluminescence spectra of samples (2 mg catalyst suspended in 3 mL ethanol) were recorded on an Edinburgh Instruments LS980-D2S2-STM spectrometer. Time correlated single photon counting (TCSPC) measurements were carried out on an Edinburgh Instruments LS980-D2S2-STM spectrometer, with picosecond pulsed LED excitation sources and a R928 detector, with a stop count of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. An EPL-405 diode (λ = 405 nm, instrument response 100 ps, fwhm) for emission detection was used. The instrument response (IRF) was performed using colloidal silica (LUDOX HS-40, Sigma-Aldrich). FAST software was used to fit decay times considering lifetime estimates. 2 mg sample was dispersed in 3 mL ethanol by sonication for measurement. Scanning electron microscopy (SEM) images were captured using a Hitachi S-4800 cold field emission scanning electron microscope (FE-SEM), at a working voltage of 3 kV and working distance of 8 mm. The upper and lower secondary electron detectors were used as a combination. Transmission electron microscopy (TEM) images and element analysis were recorded on a JEOL 2100FCs microscopy, equipped with an Energy-Dispersive Spectroscopy (EDS) detector, at an accelerating voltage of 200 kV. Inductively coupled plasma optical emission spectrometry (ICP-OES) measurements were conducted on an ICP-OES Agilent 5110. Samples were digested in concentrated nitric acid (67–69%, trace metal analysis grade) by microwave, and then diluted using distilled water.
000. An EPL-405 diode (λ = 405 nm, instrument response 100 ps, fwhm) for emission detection was used. The instrument response (IRF) was performed using colloidal silica (LUDOX HS-40, Sigma-Aldrich). FAST software was used to fit decay times considering lifetime estimates. 2 mg sample was dispersed in 3 mL ethanol by sonication for measurement. Scanning electron microscopy (SEM) images were captured using a Hitachi S-4800 cold field emission scanning electron microscope (FE-SEM), at a working voltage of 3 kV and working distance of 8 mm. The upper and lower secondary electron detectors were used as a combination. Transmission electron microscopy (TEM) images and element analysis were recorded on a JEOL 2100FCs microscopy, equipped with an Energy-Dispersive Spectroscopy (EDS) detector, at an accelerating voltage of 200 kV. Inductively coupled plasma optical emission spectrometry (ICP-OES) measurements were conducted on an ICP-OES Agilent 5110. Samples were digested in concentrated nitric acid (67–69%, trace metal analysis grade) by microwave, and then diluted using distilled water.
2.4 Transient absorption (TA) measurements
The apparatus employed to obtain transient absorption, TA, has been described previously.24 Briefly, ∼1 W from a Ytterbium laser system (PHAROS Short-Pulse 10 W, PH1-SP-10W, Light Conversion) with an output wavelength of 1028 nm, a repetition rate of 10 kHz, and pulse duration of ∼180 fs is used to drive an optical parametric amplifier, OPA (ORPHEUS, Light Conversion) in tandem with a second harmonic generation module (LYRA, Light Conversion) in order to generate radiation centred at 365 nm with a bandwidth (FWHM) of 3 nm. This 365 nm output was used as the pump source for subsequent TA measurements, which employed a commercial TA spectrometer (HARPIA, Light Conversion). The probe light was a 500–1000 nm or 400–500 nm white light supercontinuum generated by focusing 1030 nm or 515 nm radiation, respectively, onto a sapphire window. The pump and probe beams were focused to 600 μm spots at the sample. The pump laser beam was chopped, resulting in an effective pumping repetition rate of 5 kHz. The power of the chopped beam incident on the sample was 1.0 mW. The samples were suspensions of the CTF (2.0 mg) of interest in H2O (20 ml), purged with Ar, sonicated for 10 minutes and held within a quartz cuvette with a 10 mm path length. The suspension was agitated using a magnetic stirrer to maintain a constant suspension of CTFs and to prevent over exposure of sample to the laser beams. An uncoated 2.5 cm focal length N-BK7 lens (LA1252, Thorlabs) was placed after the sample to collect and collimate the probe light following transmission through the very scattering suspensions. A 400 nm long pass filter (FEL0400, Thorlabs) was placed after the sample to reduce the level of scattered pump light detected at 2λ. The probe light was spectrally dispersed by a spectrograph (Kymera 193i, Andor), employing a grating of 150 lines per mm, blazed at 800 nm (500–1000 nm probe) or 350 nm (400–500 nm probe) and detected using an NMOS detector (S3901, Hamamatsu). Data was analysed using Carpetview software (Light Conversion).2.5 Photocatalytic oxygen evolution experiments
Photocatalytic oxygen production of the samples was measured in a top-irradiation-type reactor, connected to a closed gas circulation and evacuation system (Perfect Light, Labsolar-IIIAG). The temperature of the reactor was kept constant at 8 °C by a water jacket. An online gas chromatograph (TECHCOMP, GC7900), equipped with a TCD detector (5 Å molecular sieve column and Ar carrier) was used to analyse the gas components in the reactor. In each measurement, 10 mg of the sample and 0.2 g La2O3 were sonicated in 100 mL 0.01 M aqueous silver nitrate solution to obtain a uniform suspension. The reactor was sealed and evacuated several times before light irradiation using a 300 W Xe light source (Perfect Light, PLX-SXE300). A UV cut-off filter (λ ≥ 420 nm) was used to obtain visible light irradiation.Quantum efficiency was determined by using monochromatic light illumination that was produced by adding suitable bandpass filters (Perfect Light) in front of the output of the 300 W Xe light source. A quantum meter was used to analyse the photon flux (Apogee MP-300). The apparent quantum efficiencies (AQY) were calculated by the following equation:

3. Results and discussion
3.1 Catalysts characterizations
The CTFs, named Bpy-CTF and B-CTF, were synthesized by condensation reaction of aldehyde and amidine monomers using dimethyl sulfoxide (DMSO) as solvent and caesium carbonate (Cs2CO3) as a base (Fig. 1a, details in Experimental section). The structures of Bpy-CTF and B-CTF were characterized by Fourier-transform infrared spectroscopy (FT-IR), 13C cross polarization magic angle spinning nuclear magnetic resonance (13C CP/MAS NMR), and X-ray photoelectron spectrometry (XPS). The successful formation of the triazine units was demonstrated by the characteristic vibrations at 1500 and 1343 cm−1 in the FT-IR spectra of both Bpy-CTF and B-CTF (Fig. 1b), which are in agreement with those reported in the literature.19 The chemical shifts at 170.2 ppm and 170.3 ppm in the 13C CP/MAS NMR spectra of Bpy-CTF and B-CTF is assigned to the triazine carbon signal, which also proved the existence of triazine rings in the structural framework (Fig. 1c). In the 13C CP/MAS NMR spectra of Bpy-CTF, the chemical shifts as 157.8 ppm can be assigned to the carbon signal in pyridine rings.28 | ||
| Fig. 1 (a) Molecular structures of the monomers and the framework structures of the corresponding CTFs. (b) FT-IR spectra, (c) solid-state 13C CP/MAS NMR spectra of Bpy-CTF and B-CTF. | ||
XPS spectra show the elements and bonding types in the structures of Bpy-CTF and B-CTF. The low-resolution XPS survey spectra show that Bpy-CTF and B-CTF are consists of carbon, nitrogen, and oxygen (Fig. S1a†). The high-resolution XPS spectra of N 1s shows that Bpy-CTF and B-CTF mainly consists of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NC (399.0 eV), but also has defect sites as C–N–C (400.0 eV) (Fig. S1b†).29 The high-resolution C 1s spectra of Bpy-CTF and B-CTF, Fig. 2c, can be deconvoluted into three peaks. The peak located at 284.8 eV is assigned to the adventitious carbon used for calibration and carbon in the aromatic ring (Fig. S1c†). Considering the CTFs structures, the peak at 287.1 eV can be attributed to the carbon of NC–N. From literature values,30 the 285.6 eV shoulder can be assigned to carbon in –C–OH groups. The deconvoluted high-resolution O 1s spectra showed peaks at 531.2 eV and 533.1 eV, which can be assigned to C–OH, and CO (Fig. S1d†).31 Consistent with the presence of these groups, the peaks at 1673 cm−1 and 1041 cm−1 in the FT-IR spectra of Bpy-CTF and B-CTF can be assigned to –CO and –C–O, respectively (Fig. 1b). Combing XPS with FT-IR analysis, the residual functional groups in both CTFs could be –CO and –C–OH groups, which might be formed by the hydrolysis of the unreacted end groups in the edges or defects in the framework, is in agreement with previously reported CTFs structures.31
NC (399.0 eV), but also has defect sites as C–N–C (400.0 eV) (Fig. S1b†).29 The high-resolution C 1s spectra of Bpy-CTF and B-CTF, Fig. 2c, can be deconvoluted into three peaks. The peak located at 284.8 eV is assigned to the adventitious carbon used for calibration and carbon in the aromatic ring (Fig. S1c†). Considering the CTFs structures, the peak at 287.1 eV can be attributed to the carbon of NC–N. From literature values,30 the 285.6 eV shoulder can be assigned to carbon in –C–OH groups. The deconvoluted high-resolution O 1s spectra showed peaks at 531.2 eV and 533.1 eV, which can be assigned to C–OH, and CO (Fig. S1d†).31 Consistent with the presence of these groups, the peaks at 1673 cm−1 and 1041 cm−1 in the FT-IR spectra of Bpy-CTF and B-CTF can be assigned to –CO and –C–O, respectively (Fig. 1b). Combing XPS with FT-IR analysis, the residual functional groups in both CTFs could be –CO and –C–OH groups, which might be formed by the hydrolysis of the unreacted end groups in the edges or defects in the framework, is in agreement with previously reported CTFs structures.31
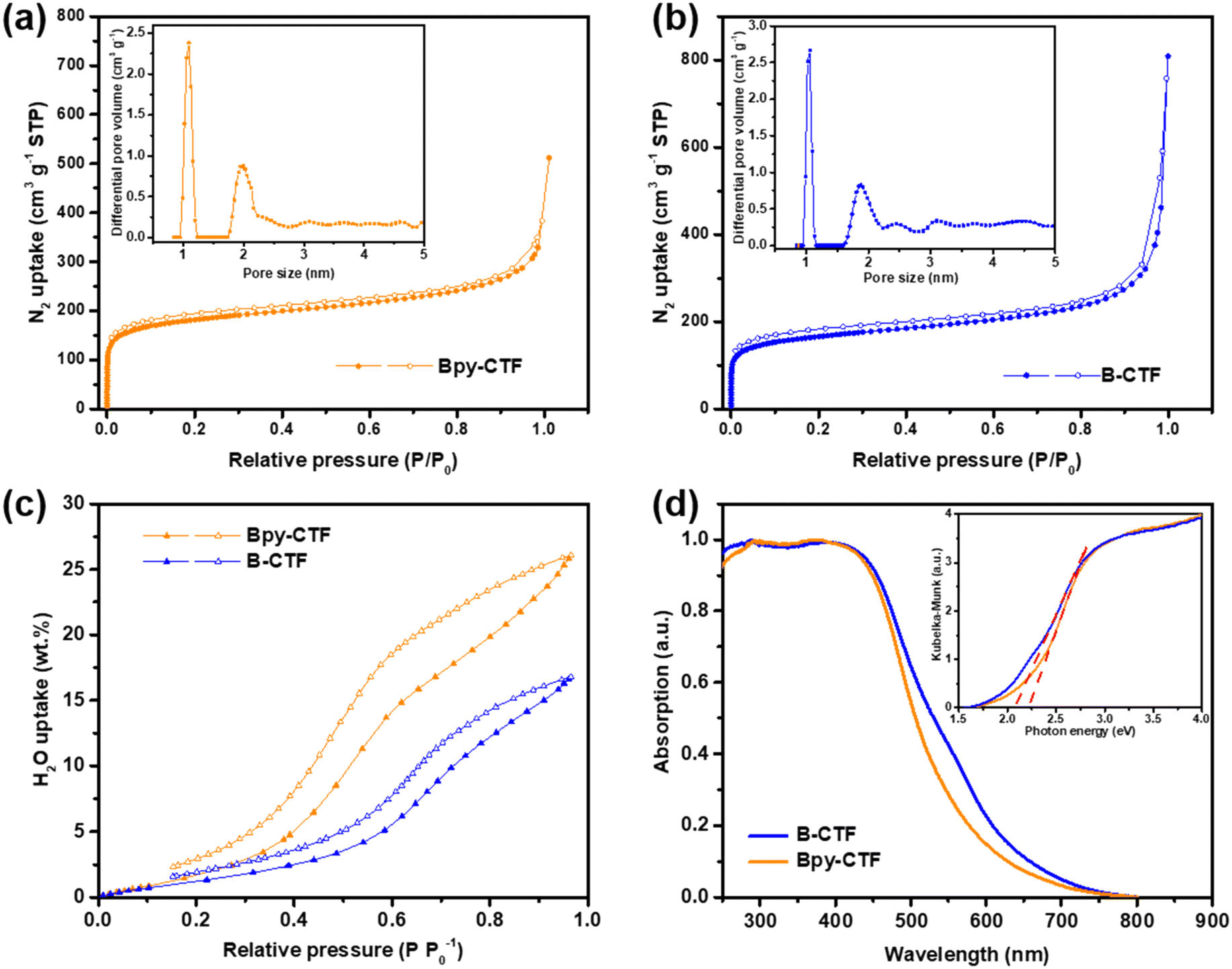 | ||
| Fig. 2 Nitrogen adsorption (filled symbol) and desorption isotherms (open symbol) of (a) Bpy-CTF and (b) B-CTF measured at 77 K (insets: pore size distributions). (c) Water adsorption (filled symbol) and desorption (open symbol) isotherms of Bpy-CTF and B-CTF measured at 293 K. (d) UV/visible light absorption spectra of Bpy-CTF and B-CTF in the solid state (insets: Kubelka–Munk transformation of diffuse reflectance data). | ||
The structural ordering of the synthesized CTFs was investigated using powder X-ray diffraction (PXRD). The PXRD patterns of Bpy-CTF and B-CTF showed a broad peak at around 6°, which suggests a very little long-range order (Fig. S2†). The thermal stability of Bpy-CTF and B-CTF were studied by thermal gravimetric analysis (TGA), both CTFs show high thermal stability and are stable up to 600 °C without significant loss of mass under a nitrogen atmosphere (Fig. S3†).
The porosity of Bpy-CTF and B-CTF was analysed by nitrogen gas sorption isotherm measurements at 77 K. The nitrogen isotherm curves demonstrated a microporous structure for both CTFs, indicated by a steep rise at a low relative pressure (Fig. 2a and b). Another steep rise is shown at high relative pressure, which represents macropores between the highly aggregated particles. Brunauer–Emmett–Teller (BET) and nonlocal density functional theory (NL-DFT) methods were used to calculate the surface areas and pore size distributions of CTFs. The surface areas of Bpy-CTF and B-CTF were calculated to be 675 and 612 m2 g−1, respectively. The pore size distributions of Bpy-CTF and B-CTF was shown in the insets of Fig. 2a and b. The water adsorption properties of Bpy-CTF and B-CTF were also investigated at 293 K. The two CTFs have different water uptake capabilities, which are related to their pore wettability: Bpy-CTF (25.98 wt% at 1.0 bar and 293 K) has a higher water uptake than B-CTF (16.69 wt% at 1.0 bar and 293 K) (Fig. 2c). The bipyridine units in the Bpy-CTF structure promote water-framework interactions and act as a strong adsorption site for water molecules.32 B-CTF lacks these favourable adsorption sites.
Contact angle measurements show that B-CTF has a contact angle at 68.2° with pure water, which is relatively low compared to most organic polymers (60–110°) (Fig. S4a†). Bpy-CTF swelled when exposed to a water drop on the disc surface, consistent with the findings of our water isotherm measurements and demonstrating that Bpy-CTF is much more hydrophilic than B-CTF (Fig. S4b†). This is important because wettability of photocatalysts is critical for water oxidation. Light obscuration measurements were further conducted to study the dispersibility of the two CTFs. The transmittance values indicate that a material disperses well (low transmittance values) or settles/creams quickly (high transmittance values). The CTF samples were dispersed in pure water giving low transmittance values of 1.95% and 1.38% for Bpy-CTF and B-CTF as expected from these hydrophilic materials. Particle size is another factor for photocatalytic efficiency as smaller particles offer more interface with water and the scavenger, thus allowing for more reactive sites to be accessed.33 To probe this, we carried out static light scattering measurements of the two CTFs after dispersion in water. The particle size distribution of B-CTF fell mostly in range 0.5–1 μm, whereas the distribution was somewhat broader for Bpy-CTF (0.5–2 μm; Fig. S5†).
UV-vis light absorption spectra of Bpy-CTF and B-CTF were measured for the powders in reflection mode. These spectra showed that the absorption onset for Bpy-CTF and B-CTF were at 575 and 610 nm, respectively (Fig. 2d). Compared to Bpy-CTF, the absorption onsets for B-CTF were red shifted by 35 nm. The band gaps of Bpy-CTF and B-CTF were 2.21 and 2.07 eV, as determined by Kubelka–Munk function based on their UV-vis absorption spectra (insets of Fig. 2d). Both CTFs have, in principle, a sufficient thermodynamic driving force for photocatalytic oxygen evolution based on DFT calculations (Fig. S6†).29
3.2 The status of cobalt
Owing to their broad visible light absorption, combined with their hydrophilic nature, both Bpy-CTF and B-CTF were interesting candidates for photocatalytic water oxidation. Cobalt was introduced into the frameworks because it is a widely-used co-catalyst for water oxidation.6,34,35 Both Bpy-CTF and B-CTF were loaded with cobalt by reflux in tetrahydrofuran containing Co(NO3)2·6H2O, obtaining Bpy-CTF–Co-x and B-CTF–Co-x (x refers to the cobalt loading amount). The successful introduction of Co(NO3)2 in Bpy-CTF–Co-3 and B-CTF–Co-3 was demonstrated by XPS. The high-resolution XPS N 1s spectra of Bpy-CTF–Co-3 showed a new peak located at 406.6 eV, which is assigned to N atoms in the –NO3 group (Fig. S7a†).21 Another new peak at 400.4 eV corresponds to the N atom of pyridine unit coordinated with Co(II).21 The Co 2p1/2 and Co 2p3/2 peaks of Bpy-CTF–Co-3 at 796.8 and 780.9 eV further confirmed the existence of Co(II) (Fig. S7b†). By comparison, no Co signal could be detected for as made Bpy-CTF (Fig. S7c†). The N 1s spectra of B-CTF–Co-3 has no difference from that of B-CTF (Fig. S7d†), demonstrating that there is no nitrogen coordination site for Co within this structure. Despite this, the Co 2p1/2 and Co 2p3/2 peak at 796.8 and 780.9 eV is observed in B-CTF–Co-3 indicative that cobalt was physiosorbed on the surface of B-CTF (Fig. S7e†). The ICP results further confirm that the successful loading of Co onto Bpy-CTF and B-CTF (Table S3†). The morphology and distribution of cobalt on the CTFs surface was captured by scanning electron microscopy (SEM) and Transmission electron microscopy (TEM). SEM images show that Bpy-CTF–Co-1 and B-CTF–Co-1 have aggregated microplate-like structures (Fig. S8a–d†). TEM images further demonstrated the layered morphology of Bpy-CTF–Co-1 and B-CTF–Co-1, and no cobalt nanoparticles or clusters were found (Fig. S7a–d†). TEM element mapping for Bpy-CTF–Co-1 showed a uniform distribution of Co over the samples (Fig. S10†).3.3 Photocatalytic oxygen evolution
Photocatalytic oxygen evolution activities of the synthesized photocatalysts were evaluated in a sacrificial system under visible light irradiation (λ ≥ 420 nm). Control experiments were performed first in the absence of either photocatalysts or light; no oxygen was detectable in these measurements. By contrast, oxygen was observed upon light illumination of the photocatalysts in the sacrificial solutions. The temporal oxygen evolution profile of catalysts under continuous light irradiation is shown in Fig. 3a. Over a 5-hour illumination period, Bpy-CTF produced almost twice as much oxygen as B-CTF. After cobalt loading, Bpy-CTF–Co-3 and B-CTF–Co-3 showed a significantly enhanced photocatalytic oxygen evolution activity compared with Bpy-CTF and B-CTF. | ||
| Fig. 3 (a) Photocatalytic oxygen evolution of Bpy-CTF, Bpy-CTF–Co-3, B-CTF and B-CTF–Co-3 under visible light illumination (λ ≥ 420 nm); a typical measurement: 10 mg photocatalysts, 0.01 M AgNO3 aqueous solution as a sacrificial reagent, and 0.2 g La2O3 as a pH buffer. (b) Photocatalytic oxygen evolution of Bpy-CTF loaded with different amounts of Co. (c) The apparent quantum efficiency (AQE) of photocatalytic water oxidation for Bpy-CTF–Co-3 under different irradiation wavelengths. | ||
The photocatalytic oxygen evolution performance of Bpy-CTF was further studied by varying the cobalt content, and the amount of oxygen evolved under continuous light irradiation is shown in Fig. 3b. It was found that 3 wt% Co loading was optimum for Bpy-CTF (Bpy-CTF–Co-3), providing a significant photoactivity improvement over the catalyst where no Co is present. Further increasing the amount of Co (5 wt%) resulted in a negative effect on oxygen evolution. This could be due to the blocking of light and photogenerated hole transferring by excess Co. However, without co-catalyst loading, Bpy-CTF had a much lower activity for water oxidation. The oxygen evolution rate of Bpy-CTF was enhanced significantly even with a low amount of Co loading (0.5 wt%, Bpy-CTF–Co-0.5). This sacrificial photocatalytic oxygen evolution performance is comparable to the state-of-the-art polymer photocatalysts reported in the literature under comparable conditions (Table S5†).
The wavelength dependence of photocatalytic oxygen evolution was studied by measuring apparent quantum efficiencies (AQE) under monochromatic light. As shown in Fig. 3c, AQE of Bpy-CTF–Co-3 is consistent with the optical absorption of the photocatalyst, which is greatest at 420 nm (0.56%) and decreases with a longer wavelength, consistent with decreasing light absorption at these wavelengths, with photocatalytic activity observed to wavelengths ∼600 nm. FT-IR spectra of Bpy-CTF–Co-3 collected after oxygen evolution show no changes that suggested the photocatalyst decomposed (Fig. S11†). The decrease in activity observed over time is likely a result of the formation of silver coating produced from reduction of the Ag+ sacrificial electron acceptor required for the water oxidation half reaction, prohibiting the light absorption by the photocatalyst.32
To investigate the charge recombination during light illumination, photoluminescence (PL) emission spectra was collected by 405 nm laser excitation. The photoluminescence emission intensity of Bpy-CTF–Co-3 is quenched in comparison with that of Bpy-CTF (Fig. 4a), indicating that the emissive relaxation of the initially formed exciton is significantly lowered in the former, implying a non-radiative relaxation pathway is opened, likely charge migration, leading to the inhibition of charge recombination, in line with literature reports.16,22,36 This observation rationalises the higher photocatalytic oxygen evolution activity of Bpy-CTF with cobalt coordination. In addition, the time-correlated single-photon counting (TCSPC) measurements were carried out to estimate the excited-state emissive lifetimes for these materials (Fig. 4b). The average weighted lifetime of Bpy-CTF and Bpy-CTF–Co-3 was estimated to be τavg = 1.99 and 1.31 ns, respectively (Table S4†).
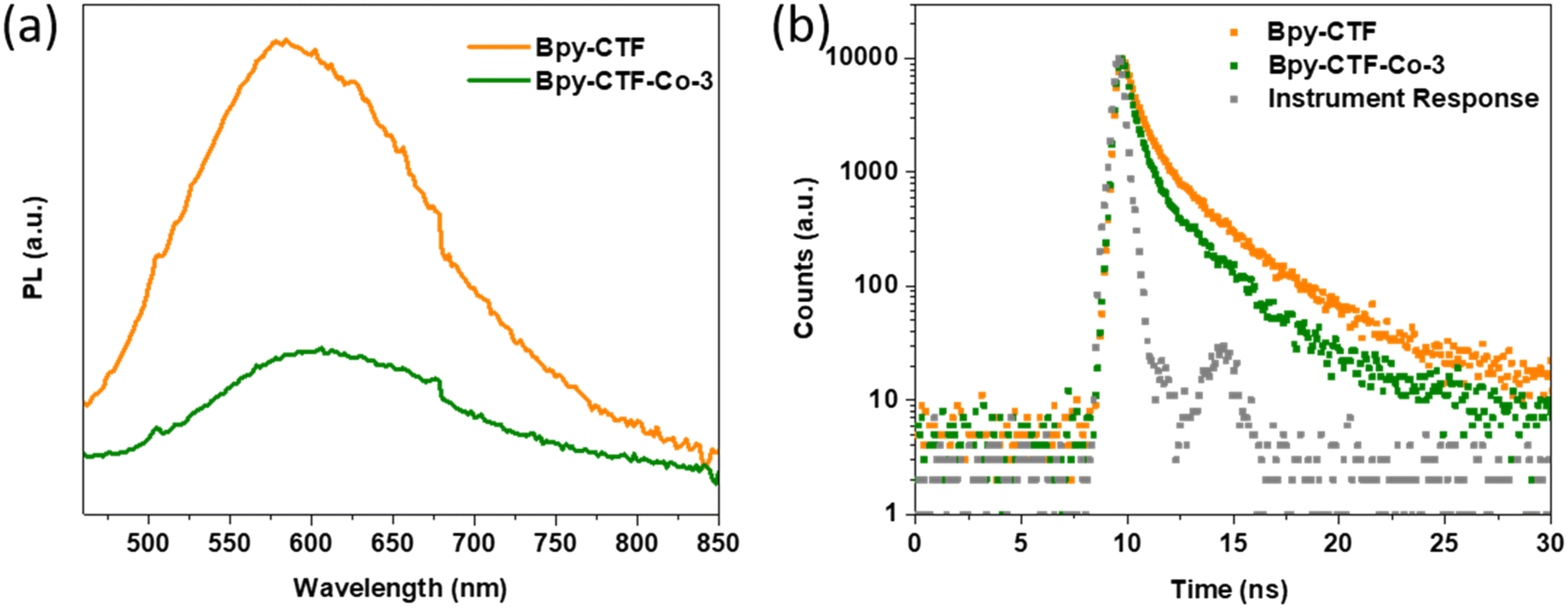 | ||
| Fig. 4 (a) PL spectra of Bpy-CTF and Bpy-CTF–Co-3 (λex = 405 nm); (b) TCSPC experiment of Bpy-CTF and Bpy-CTF–Co-3. The samples were excited with a λexc = 405 nm laser and emission was observed at λem = 600 nm. | ||
3.4 The role of Co coordination from transient absorption (TA) spectroscopy
To further explore the potential of increased charge separation of Bpy-CTF upon the incorporation of Co, as hypothesised by the PL and TCSPC experiments, we performed ultrafast transient absorption (TA) spectroscopic measurements on both Bpy-CTF and Bpy-CTF–Co-1 in pure H2O, shown in Fig. 5. A pump wavelength of 365 nm was employed for all TA experiments, coincidental with the minimum transmission observed for suspensions of Bpy-CTF in pure H2O (Fig. S12†). At ∼1 ps, the TA spectra of Bpy-CTF and Bpy-CTF–Co-1 have a similar appearance (Fig. 5a and d): a narrow negative signal with a minimum at ∼425 nm, and a broad Photo-Induced Absorption (PIA) with peaks at ∼550 and 875 nm. This structure is remarkably similar to that observed in TA spectra at early pump–probe delays recently reported for other CTFs.37,38 For both CTFs, the intensity of these bands decreases with increasing pump–probe delay. At longer wavelengths, the broad PIA decays at a similar rate for both CTFs (Fig. 5i). Although the ∼550 nm band initially decays at similar rates for both CTFs, at delays >5 ps, this feature begins to decay more quickly for Bpy-CTF–Co-1 (Fig. 5g). The kinetics are more complex in the 600–800 nm wavelength range (Fig. 5e and h), where a broad PIA centred at ∼675 nm grows between 5–50 ps for Bpy-CTF–Co-1, which is seen to decay at delays >100 ps. Careful inspection of the 50 ps spectrum of Bpy-CTF highlights that a weak band is also observed at ∼650 nm. While the PIA and negative bands lose intensity, little change in the shape of the TA spectrum of Bpy-CTF occurs between 50 ps–1 ns, however for Bpy-CTF–Co-1, the broad band between ∼550–700 has lost considerable intensity over this timescale, changing the shape of the spectra. The shape of the spectra continue to evolve, and by ∼3 ns, the TA spectrum is broadly similar for both CTFs: a weak positive signal at ∼920 nm with a tail to shorter wavelengths is observed. A narrow negative band at ∼440 nm is observed for Bpy-CTF, along with a shoulder between 500–550 nm showing similar structure to that at ∼20 ps, however, for Bpy-CTF–Co-1, a flat, negative, baseline is observed between ∼450–600 nm. | ||
| Fig. 5 TA spectra (365 nm pump), normalised at the global maximum ΔA, at key pump–probe wavelengths as indicated (in ps) for (a) and (b) Bpy-CTF and (d) and (e) Bpy-CTF–Co-1 in pure H2O. DADS from compartments 2-5 for (c) Bpy-CTF and (f) Bpy-CTF–Co-1 from a 6-compartment parallel fit; decay for each compartment are indicated. The grey area at ∼500 nm and ∼900 nm indicates where spectra obtained in different spectral ranges (400–500 nm, 500–900 nm and 900–1000 nm) have been spliced and between 710–755 nm where spectra obtained in a single spectral range (500–900 nm) are contaminated by detected pump laser scatter (2λ). Kinetic traces of Bpy-CTF (black) and Bpy-CTF–Co-1 (red) at probe wavelengths (g) 550 nm, (h) 760 nm and (i) 950 nm. | ||
In order gain further insight of the complex dynamics observed for these CFTs, Global Lifetime Analysis (GLA) has been performed for the TA data shown in Fig. 5. Based on the description of the TA spectra, above, a five compartment parallel model was initially chosen to describe: (i) the fast initial decay of the excitonic structure, (ii) the formation of the broad PIA between ∼600–800 nm, (iii) the decay of this feature, (iv) the slower evolution of the spectrum between 1–3, ns and (v) the decay of the long-lived (>3 ns) population, however for both CTFs, an additional compartment which decays with the instrument response (∼300 fs) was required to result in a satisfactory fit of the data hence a 6-compartment parallel scheme was employed. Decay Associated Difference Spectra (DADS) for compartment 2-5, which result from this analysis are shown in Fig. 5c and f for Bpy-CTF and Bpy-CTF–Co-1, respectively, those for compartments 0 and 1 are omitted for clarity and are shown in (Fig. S13†). DADS are wavelength dependent for each lifetime component with a positive feature describing decay of a positive TA feature, or growth of a negative TA feature, while a negative feature describes the decay of a negative TA feature, or growth of a positive TA feature. Compartment 0 and 1, which decays in ∼0.1 and ∼2 ps, for both CTFs, describes decay of the initially formed exciton, (Fig. S13c and f†). The DADS of compartment 2 for Bpy-CTF shows continued decay of the narrow negative TA band and the broad PIA, except between 600–750 nm, where little change is observed. For Bpy-CTF–Co-1, the DADS of compartment 2 has similar structure to that of Bpy-CTF, however, a negative signal is observed between ∼600–750 nm describing the growth of the PIA for this CTF between 5–50 ps, discussed above. Such behaviour is consistent with the formation of a species with charge separated character from the initially formed exciton for both CTFs. Interestingly, this species is produced in similar times for both CTFs (17 and 18 ps for Bpy-CTF–Co-1 and Bpy-CTF, respectively), however the yield of this species is significantly greater for Bpy-CTF–Co-1 than Bpy-CTF. Consistent with this assignment, a recent study has assigned a long-lived band at ∼750 nm in the TA spectra of related CTFs materials to an electron polaron.37 The steady state PL spectrum (Fig. 4a) largely overlaps with the broad negative going bands observed in the DADS of compartment 2 of both CTFs; deconvoluting the change in stimulated emission (SE) of the initially formed exciton in this wavelength range from the PIA of the charge-separated species is unfortunately not possible. The DADS of compartment 3 (τ ∼ 300 ps) for Bpy-CTF–Co-1 has a similar, but inverted, structure to compartment 2 between 550–900 nm, indicating that this compartment largely describes decay of the species produced in compartment 2, along with a change in shape of the negative band at wavelengths <500 nm Notably, although the PIA at wavelengths >700 nm, is observed to decay in the DADS of compartment 3 of Bpy-CTF–Co-1, the shape does not reflect that of compartment 2; instead, the DADS of compartment 4 (τ ∼ 900 ps) shows the decay of the 700–850 nm feature formed in compartment 2, contemporaneously as the narrow negative band at wavelengths <500 nm disappears. Owing to this, both compartment 3 and 4 for Bpy-CFT-Co-1 are assigned to relaxation of the charge-separated species produced in compartment 2. For Bpy-CTF, the DADS of compartment 3 shows decay of the broad PIA, and changing structure of the narrow negative band <500 nm, and can be assigned to continued excitonic relaxation. The DADS of compartment 4 (τ ∼ 1 ns) of Bpy-CTF shows a similar, but inverted shape to compartment 2, hence is assigned to relaxation of the charge-separated state. The final compartments describe the slowest decay processes, hence provide a description of the relaxation of the longest-lived transient species observed, and have structure reflecting that described above for the TA spectra at 3 ns. 5-Compartment parallel GLA for both Bpy-CTFs in which the ∼1 ns decay is excluded shows poor agreement between the 3 ns spectrum and the longest lived DADS, (Fig. S14c and f†) and highlights the decay of the charge separated state observed in compartment 4 of the 6-compartment GLA of both Bpy-CTFs (Fig. 5c and f).
To probe whether h+ transfer to Co provides a possible route to charge separation, TA experiments were performed for both Bpy-CTF and Bpy-CTF–Co-1 in a 0.01 M AgNO3 aqueous solution, with Ag+ expected to act as a sacrificial electron acceptor (SEA), as in the photocatalytic experiments reported herein. TA spectra, and DADS obtained from a subsequent GLA (6-compartment parallel scheme) of this data are shown in Fig. S15.† The 1 ps spectra (Fig. S15a and c†) are remarkably similar to those obtained in pure water. By 5 ps, significant loss in intensity of the narrow ∼425 nm negative band and loss of PIA structure has occurred for both CTFs, with the PIA between ∼650–750 nm having greater intensity than at 1 ps. By 3 ns, the TA spectra of both CTFs consists of a broad negative baseline onto which the residual excitonic ∼500 nm band is superimposed. The DADS of compartment 1 (Fig. S15b and d†) have similar structure to those of compartment 2 in pure H2O (Fig. 5c and f), assigned to the formation of a charge-separated species. Should this feature be assigned to a fully separated h+ polaron in AgNO3, it would be expected that a band at 650–800 nm would also be observable immediately following its formation (as in Bpy-CTF–Co-1 in pure H2O) and at long pump–probe delays. However, owing to the relatively unstructured spectra observed in the 650–800 nm wavelength range for pump–probe delays >5 ps for both CTFs in AgNO3, we assign the decay of compartment 1 to quenching of the initially formed exciton through rapid (∼2 ps for both CTFs) e− transfer to Ag+. No new bands are observed to evolve contemporaneously with quenching, hence we conclude no spectral signatures of the polymer h+ polaron are observed in the 400–900 nm wavelength range. Owing to this observation, the broad 600–800 nm PIA band produced from decay of compartment 2 in Bpy-CTF–Co-1 in pure H2O can be assigned to the spectral signature of an e− polaron, consistent with recent literature assignments of TA bands of CTFs.37 As deposited, the dominant oxidation state of Co is +2 in Bpy-CTF–Co-1. Reported spectroelectrochemical measurements has shown a large increase in absorption is expected across the visible (550–900 nm) spectral region upon oxidation of CoII to CoIII.39 The DADS of the longest-lived compartment of both CTFs are compared in (Fig. S15e†); although the ∼500 nm feature is slightly narrower in Bpy-CTF–Co-1, the overall shapes of the two DADS are remarkably similar, hence we conclude h+ transfer to Co does not occur on a timescale <3 ns.
Returning to the TA spectra of the CTFs in pure H2O, the considerable loss in intensity as observed in the DADS of compartment 3 and 4, of the broad 600–800 nm band a for Bpy-CTF–Co-1, assigned to an e− polaron, cannot be assigned to continued charge-separation by h+ transfer to Co. No new bands are observed contemporaneously with the decay of this feature. We propose that the degree of charge-separation is limited (a polaron pair, for example), and charge-recombination provides a significant, and undesired, loss mechanism of this species. Interestingly, as noted above, in Bpy-CTF–Co-1, the charge-separated species is observed to decay with two lifetimes (τ ∼ 300 and 900 ps), indicating that species with a range of separation are likely produced for this CTF.
TA experiments were performed for both B-CTF and B-CTF–Co-1 in pure H2O. TA spectra are shown in Fig. S16† along with DADS which resulted from 5-compartment GLA of the TA data. A 6-component GLA of B-CTF converged to give a second compartment with a lifetime within the IRF, while a significant improvement to the fit was not obtained for B-CTF–Co-1 (Fig. S17c and f†). At 0.5 ps, the TA spectra of both B-CTFs are similar, and significant oscillating structure is observed between 400 and 650 nm, which largely decays by ∼5 ps, (Fig. S16a and d†) by which time, the spectral shape resembles that obtained for the Bpy-CTFs; we assign this early timescale evolution to changing SE structure for the B-CTFs. The DADS of compartment 2 of the B-CTFs are remarkably similar to those obtained for the corresponding Bpy-CTF. Growth of a new PIA band is observed for B-CTF–Co-1 at ∼650 nm, whereas little change in PIA is observed for B-CTF around ∼600 nm, contemporaneously as the PIA bands at ∼500 and ∼900 nm and negative band at 435 nm decay (Fig. S16c and f†). Through comparison with the Bpy-CTFs, we assign this to the spectral signature of a e− polaron with partial charged-separated character. The DADS of compartment 3 shows a very broad positive baseline between 500–900 nm for both B-CTFs, however, for B-CTF–Co-1, superimposed on this is a narrow, negative going band at ∼620 nm. This is clearly reflected by the broader positive going band which is superimposed on the negative baseline of compartment 4, with the breadth resulting from partial overlap with a weak band observed at ∼610 nm in the 10 ps TA spectrum (Fig. S16e†). These observations suggest that the partially charge-separated state formed in compartment 2 undergoes relaxation in compartment 3 forming a state with a narrower spectral signature at longer timescales. Interestingly, although the yield of the initially formed charge-separated state appears to be lower for B-CTF–Co-1 than Bpy-CTF–Co-1, its lifetime is longer. Unlike B-CTF–Co-1, a clear band at ∼620 nm is not observed in the DADS of compartment 4 of B-CTF, consistent with the much lower yield of the initially formed charge-separated state for B-CTF.
The initial yield of charge-separated species is significantly greater in Bpy-CTF–Co-1and B-CTF–Co-1 than Bpy-CTF and B-CTF, despite no evidence of h+ to Co being observed. We hypothesise that the presence of Co ions aids charge-separation through induction of polarisation within the CTFs as achieved by structural modification of related CTFs.37 The results of the TA experiments highlight a potential facile route of enhancing charge separation in CTF photocatalysts for complete water splitting by the incorporation of charged metal oxygen evolution catalysts in either a Z-scheme, or by co-loading with a common hydrogen evolution catalyst (e.g. Pt or Pd). The long (>3 ns) h+ transfer to Co results in significant loss in population of the initially formed charge-separated state, which we propose occurs through charge-recombination. Incorporating oxygen evolution catalysts which allow rapid h+ transfer from the CTF will be an important step required to achieve high activities without the use of sacrificial reagents.
4. Conclusions
In summary, Bpy-CFT is a promising photocatalyst for sacrificial oxygen evolution. Bpy-CTF shows efficient photocatalytic water oxidation activity under visible light illumination. The oxygen evolution rate of Bpy-CTF was significantly enhanced after incorporation of a cobalt co-catalyst. The highest oxygen evolution rate achieved by Bpy-CTF–Co-3 was 322 μmol g−1 h−1 (≥420 nm), which is comparable to the state-of-the-art for sacrificial photocatalytic oxygen evolution with polymer photocatalysts. The efficient oxygen evolution performance of Bpy-CTF–Co-3 is attributed to its wettability, porosity, large surface area, and efficient charge separation. We have demonstrated that incorporating Co results in an increased yield of charge-separated species in the absence of sacrificial reagents. When strongly coordinating Co is incorporated through the introduction of binding sites within the CTF structure when bipyridine linkers are employed, the yield of charge-separated species is further enhanced. This work expands the diversity of organic photocatalysts for water oxidation, especially under visible light irradiation, which is the highest intensity spectral range of sunlight reaching Earth's surface. In the future, such materials could form part of a Z-scheme for overall water splitting consisting of coupled organic proton reduction and water oxidation subunits.Author contributions
H. Chen synthesized, characterized the photocatalysts and analysed the photocatalysis results. A. Gardner performed TAS measurements and analysed the TAS data. G. Lin performed the photocatalytic oxygen evolution measurements. W. Zhao synthesized the terephthalamidine dihydrochloride monomer and provided useful suggestions in the synthesis of CTFs. M. Bahri and N. Browning captured TEM images. A. Cooper, X. Xu, and X. Li directed the project. The manuscript was prepared by H. Chen, X. Li, A. Gardner, R. Sprick, X. Xu, and A. Cooper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr. S. Guan at Cardiff University for the XPS measurement, Dr. H. Ai and Dr. H. Yang at University of Liverpool for the help with SEM images. The authors thank Dr X. Wang at Huazhong University of Science and Technology for useful discussions. The authors acknowledge funding from the Engineering and Physical Sciences Research Council (EPSRC) (EP/N004884/1). H. Chen thanks the China Scholarship Council for a PhD studentship. Prof. Xu thanks the National Natural Science Foundation of China (Grant No. 51972233, 52172225), Natural Science Foundation of Shanghai (Grant No. 19ZR1459200) for financial support. Transient absorption spectroscopic measurements were carried out at the University of Liverpool ECR Laser Laboratory supported by UKRI-EPSRC grant EP/S017623/1 and the University of Liverpool. The X-ray photoelectron (XPS) data collection was performed at the EPSRC National Facility for XPS (“HarwellXPS”), operated by Cardiff University and UCL, under Contract No. PR16195.References
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC
.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS
- C. Zhao, Z. Chen, R. Shi, X. Yang and T. Zhang, Adv. Mater., 2020, 32, 1–52 Search PubMed
- L. Wang, P. Cai, Z. Liu, Z. Xie and Y. Fang, J. Colloid Interface Sci., 2022, 607, 203–209 CrossRef CAS PubMed
- D. Kong, Y. Zheng, M. Kobielusz, Y. Wang, Z. Bai, W. Macyk, X. Wang and J. Tang, Mater. Today, 2018, 21, 897–924 CrossRef CAS
- Y. Fang, Y. Hou, X. Fu and X. Wang, Chem. Rev., 2022, 122, 4204–4256 CrossRef CAS PubMed
- Z. Zhang, X. Chen, H. Zhang, W. Liu, W. Zhu and Y. Zhu, Adv. Mater., 2020, 1907746, 1–6 Search PubMed
- J. Wang, W. Shi, D. Liu, Z. J. Zhang, Y. Zhu and D. Wang, Appl. Catal., B, 2017, 202, 289–297 CrossRef CAS
- K. C. Ranjeesh, L. George, V. C. Wakchaure, Goudappagouda, R. N. Devi and S. S. Babu, Chem. Commun., 2019, 55, 1627–1630 RSC
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed
- Y. Li, X. Li, H. Zhang, J. Fan and Q. Xiang, J. Mater. Sci. Technol., 2020, 56, 69–88 CrossRef
- L. Lin, Z. Lin, J. Zhang, X. Cai, W. Lin, Z. Yu and X. Wang, Nat. Catal., 2020, 3, 649–655 CrossRef CAS
- Z. Luo, M. Zhou and X. Wang, Appl. Catal., B, 2018, 238, 664–671 CrossRef CAS
- D. Kong, X. Han, J. Xie, Q. Ruan, C. D. Windle, S. Gadipelli, K. Shen, Z. Bai, Z. Guo and J. Tang, ACS Catal., 2019, 9, 7697–7707 CrossRef CAS PubMed
- J. Xie, S. A. Shevlin, Q. Ruan, S. J. A. Moniz, Y. Liu, X. Liu, Y. Li, C. C. Lau, Z. X. Guo and J. Tang, Energy Environ. Sci., 2018, 11, 1617–1624 RSC
- J. Bi, W. Fang, L. Li, J. Wang, S. Liang, Y. He, M. Liu and L. Wu, Macromol. Rapid Commun., 2015, 36, 1799–1805 CrossRef CAS PubMed
- Z. A. Lan, Y. Fang, Y. Zhang and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 470–474 CrossRef CAS PubMed
- K. Wang, L. M. Yang, X. Wang, L. Guo, G. Cheng, C. Zhang, S. Jin, B. Tan and A. Cooper, Angew. Chem., Int. Ed., 2017, 56, 14149–14153 CrossRef CAS PubMed
- Y. Wang, P. Li, J. Wang, Z. Liu, Y. Wang, Y. Lu, Y. Liu, L. Duan, W. Li, S. Sarina, H. Zhu and J. Liu, Catal. Sci. Technol., 2021, 11, 4429–4438 RSC
- J. Chen, X. Tao, C. Li, Y. Ma, L. Tao, D. Zheng, J. Zhu, H. Li, R. Li and Q. Yang, Appl. Catal., B, 2020, 262, 1–8 Search PubMed
- L. Li, R. G. Hadt, S. Yao, W. Y. Lo, Z. Cai, Q. Wu, B. Pandit, L. X. Chen and L. Yu, Chem. Mater., 2016, 28, 5394–5399 CrossRef CAS
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS PubMed
- Z. Fu, X. Wang, A. M. Gardner, X. Wang, S. Y. Chong, G. Neri, A. J. Cowan, L. Liu, X. Li, A. Vogel, R. Clowes, M. Bilton, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 543–550 RSC
- X. Wang, Z. Fu, L. Zheng, C. Zhao, X. Wang, S. Y. Chong, F. McBride, R. Raval, M. Bilton, L. Liu, X. Wu, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Mater., 2020, 32, 9107–9114 CrossRef CAS
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang, X. Meng, J. Zhang and J. Ye, Angew. Chem., 2016, 128, 14522–14526 CrossRef
- M. Liu, K. Jiang, X. Ding, S. Wang, C. Zhang, J. Liu, Z. Zhan, G. Cheng, B. Li, H. Chen, S. Jin and B. Tan, Adv. Mater., 2019, 31, 1807865 CrossRef PubMed
- M. Liu, Q. Huang, S. Wang, Z. Li, B. Li, S. Jin and B. Tan, Angew. Chem., Int. Ed., 2018, 57, 11968–11972 CrossRef CAS PubMed
- L. Guo, X. Wang, Z. Zhan, Y. Zhao, L. Chen, T. Liu, B. Tan and S. Jin, Chem. Mater., 2021, 33, 1994–2003 CrossRef CAS
- D. H. Wang, Y. Hu, J. J. Zhao, L. L. Zeng, X. M. Tao and W. Chen, J. Mater. Chem. A, 2014, 2, 17415–17420 RSC
- S. Zhang, G. Cheng, L. Guo, N. Wang, B. Tan and S. Jin, Angew. Chem., Int. Ed., 2020, 59, 6007–6014 CrossRef CAS PubMed
- R. S. Sprick, Z. Chen, A. J. Cowan, Y. Bai, C. M. Aitchison, Y. Fang, M. A. Zwijnenburg, A. I. Cooper and X. Wang, Angew. Chem., 2020, 132, 18854–18859 CrossRef
- C. M. Aitchison and R. S. Sprick, Nanoscale, 2021, 13, 634–646 RSC
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC
- L. Yang, H. Chen, Y. Xu, R. Qian, Q. Chen and Y. Fang, Chem. Eng. Sci., 2022, 251, 117435 CrossRef CAS
- J. X. Cui, Y. M. Fu, B. Meng, J. Zhou, Z. Y. Zhou, S. M. Liu and Z. M. Su, J. Mater. Chem. A, 2022, 13418–13427 RSC
- C. Wu, Z. Teng, C. Yang, F. Chen, H. Bin Yang, L. Wang, H. Xu, B. Liu, G. Zheng and Q. Han, Adv. Mater., 2022, 2110266, 1–9 Search PubMed
- Y. Zou, S. Abednatanzi, P. Gohari Derakhshandeh, S. Mazzanti, C. M. Schüßlbauer, D. Cruz, P. Van Der Voort, J. W. Shi, M. Antonietti, D. M. Guldi and A. Savateev, Nat. Commun., 2022, 13, 1–13 Search PubMed
- Y. Ma, A. Kafizas, S. R. Pendlebury, F. Le Formal and J. R. Durrant, Adv. Funct. Mater., 2016, 26, 4951–4960 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy00773h |
| This journal is © The Royal Society of Chemistry 2022 |