
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel thymidine phosphorylase to synthesize (halogenated) anticancer and antiviral nucleoside drugs in continuous flow†
Ana I.
Benítez-Mateos
 ,
Calvin
Klein
,
David
Roura Padrosa
and
Francesca
Paradisi
*
,
Calvin
Klein
,
David
Roura Padrosa
and
Francesca
Paradisi
*
Department of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: francesca.paradisi@unibe.ch
First published on 8th August 2022
Abstract
Four pharmaceutically relevant nucleoside analogues (5-fluoro-2′-deoxyuridine, 5-chloro-2′-deoxyuridine, 5-bromo-2′-deoxyuridine, and 5-iodo-2′-deoxyuridine) have been synthesized by using a novel thymidine phosphorylase from the halotolerant H. elongata (HeTP). Following enzyme immobilization on microbeads, the biocatalyst was implemented as a packed-bed reactor for the continuous production of halogenated nucleosides, achieving up to 90% conversion at the 10 mM scale with 30 min residence time. Taking the synthesis of floxuridine (5-fluoro-2′-deoxyuridine) as a study case, we obtained the highest space–time yield (5.5 g L−1 h−1) reported to date. In addition, bioinformatic tools such as MD analysis and CapiPy have contributed to shine light on the catalytic performance of HeTP as well as its immobilization, respectively.
Introduction
Viral infections and cancer disorders are among the most concerning diseases of our society. Nucleoside analogues are classified as an important class of anticancer and antiviral drugs, with many examples already on the market.1 Specifically, halogenated derivatives have been proven to be very effective therapeutics due to their enhanced solubility and bioavailability.2 For instance, floxuridine (5-fluoro-2′-deoxyuridine) is effective against different types of cancer and it is also an active substance used in the manufacturing of vaccines against SARS-related coronaviruses.3–5Since the 1950s, nucleoside analogues have been mainly synthesized by chemical strategies which generally require multi-step pathways, hazardous and extreme conditions, and large waste production.6 More recently, enzymatic synthesis has evolved as an attractive alternative to overcome those issues.7 Apart from an exquisite substrate selectivity, enzymes are biocompatible and biodegradable catalysts that can work at mild physiological conditions. Although enzymes still suffer from poor substrate solubility and low substrate loading, discovery of more robust enzymes together with state-of-the-art technologies (i.e. enzyme immobilization, flow chemistry) bear a high potential towards more sustainable synthesis of nucleoside drugs.7–9
Thymidine phosphorylases (TP) (EC 2.4.2.4) are part of the family II of nucleoside phosphorylases and catalyze the cleavage of the glycosidic bond of thymidine in presence of phosphate. TP are highly selective for 2′-deoxy pyrimidine nucleosides while they accept a broad variety of modifications at the C5 position of the nucleobase.10 These catalytic properties make TP ideal biocatalysts for the two-step production of halogenated thymidine analogues in just one pot (Scheme 1).
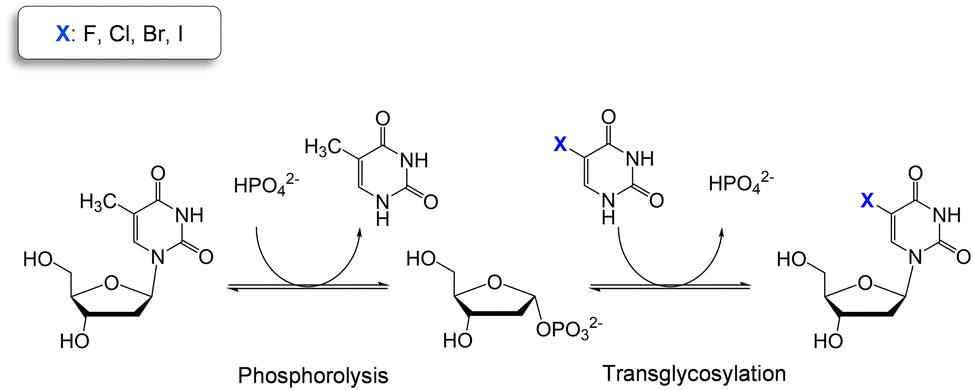 | ||
| Scheme 1 Two-step one-pot enzymatic reaction for the synthesis of halogenated pyrimidine nucleosides using thymidine as sugar donor. | ||
Although the biocatalytic synthesis of nucleoside drugs has been thoroughly studied during the last decades with some industrial examples,11,12 new approaches are still desired to develop scalable enzymatic syntheses.7 While the biocatalyst's performance can be improved by protein engineering, enzyme discovery, or enzyme immobilization,13–15 flow chemistry is emerging as a very powerful tool to intensify the process reaction, minimize the waste and bring biocatalysis closer to industrial scale.16
Here we present a highly stable thymidine phosphorylase from Halomonas elongata (HeTP) as a robust and efficient candidate to synthesize 5-halogenated-2′-deoxyuridines. HeTP has been characterized in terms of activity and stability against temperature, pH, and presence of organic co-solvent which are often needed during the synthesis of nucleoside analogues. The HeTP biocatalyst was further improved by covalent immobilization on methacrylate microbeads which then allowed its integration into a packed-bed reactor (PBR) for the intensification of the process to synthesize nucleoside drugs under flow conditions. Flow biocatalysis technology as well as in silico analyses have been adopted in this work to boost the biocatalytic potential of HeTP. Finally, we have investigated the substrate scope of HeTP towards other pyrimidine nucleoside drugs providing new insights into the active pocket of HeTP.
Results and discussion
Characterization of TP from the halotolerant H. elongata (HeTP)
The efficiency and scale-up of synthetic processes are generally linked to high temperatures and presence of co-solvents to which enzymes are not naturally adapted. These conditions are certainly needed to enhance the poor water-solubility of nucleoside analogues. For this reason, we screened the genome of the halotolerant organism Halomonas elongata which we have extensively studied in our lab as a source of robust enzymes, with an inherited tolerance towards co-solvents.17–22 HeTP was expressed in E. coli as a soluble enzyme. After optimization of the protein expression (Fig. S1†), HeTP harbouring a (6×)His-tag in the C-terminal was purified by IMAC obtaining 25–35 mg of protein per L of culture media (Fig. S2†).The activity of HeTP towards the phosphorolysis of thymidine reached 112.8 U mg−1 at 70 °C in 0.5 M phosphate buffer at pH 7.0 (Fig. S3†). Interestingly, HeTP presented an increasing activity up to 0.5 M of phosphate, and still retained activity even at 0.9 M phosphate, which may be due to its origin from a halotolerant microorganism (Fig. S3B†). To simplify the reaction set up, the temperature for the standard activity assay was set at 30 °C where HeTP maintains in any case 80% of the initial activity after 6 h (Fig. S4†). Moreover, the enzyme can be stored at 4 °C or room temperature for 2 weeks while retaining most of the activity (Fig. S5†). In terms of pH, HeTP was stable between pH 6.0 and 9.0 for 3 h (Fig. S6†). In addition, HeTP retained 80% of the phosphorolysis activity when 10% of DMSO or MeOH was added to the reaction media (Fig. S7A†). Interestingly, HeTP activity was mostly retained (at least 75%) after incubation in various organic co-solvents such 20% dimethyl sulfoxide (DMSO), 20% methanol (MeOH), 10% ethanol (EtOH), 20% dimethyl formamide (DMF), 10% acetonitrile (MeCN) (Fig. S7B†). While the stability with 20% DMF and 20% MeCN is comparable to the E. coli TP, the stability with 20% EtOH is clearly superior to other pyrimidine nucleoside phosphorylases which suffered total loss of activity.13,23 More comparative analysis could not be conducted due to the lack of previous results for other TPs.
HeTP activity towards different nucleosides
HeTP phosphorolysis towards thymidine, the natural substrate, reached the equilibrium with 62% conversion at 5 mM scale in 2 h (Fig. S8†). The enzyme presented a KM towards thymidine of 320 μM with a Vmax of 73 U mg−1 (Fig. S9A†). For phosphate, a KM ∼360 μM was estimated (Fig. S9B†). These values are similar to the ones reported for the E. coli and human thymidine phosphorylases, while KM towards thymidine is significantly lower than the TP from M. hyorhinis and T. thermophilus (473 μM and ∼2400 μM, respectively).15,24–26The substrate scope of HeTP towards other natural nucleosides was then probed. As an intrinsic characteristic of the thymidine phosphorylase family, HeTP did not accept purine nucleoside analogues (Table S1†),10 and it converted only the pyrimidine nucleoside analogues containing a 2′-deoxyribose and the O4 in the nucleobase ring (thymidine or deoxyuridine) (Table S2†). Noteworthy, HeTP did not show activity towards 4′-amino nucleobases which are building blocks for other pharmaceutically relevant nucleosides.27 In an attempt to shift the activity towards this class of molecules, homology modelling and site-directed mutagenesis to favor the binding of the amino group was carried out (Fig. S10†). Arg171 was identified as a hotspot as it acts as one of the electrostatic stabilizers during the catalysis. Arg171 was replaced by Gln171 and Glu171 which could potentially stabilize the 4-amino moiety, as predicted through docking, MD (molecular dynamics), and amino acid conservation analysis (Fig. S11–S13†). As expected, the substitution R171E led to a loss of activity towards the natural substrate (thymidine),28 while R171Q showed 10% of activity towards thymidine compared to the wild-type (WT) HeTP. However, none of the variants showed activity towards any of the 4-amino substituted substrates tested despite trying different reaction conditions (Fig. S14, see ESI† for more details).
Biosynthesis of halogenated nucleoside analogues
WT HeTP was applied to the biosynthesis of four 5-substituted nucleoside analogues (5-fluoro-2′-deoxyuridine, 5-chloro-2′-deoxyuridine, 5-bromo-2′-deoxyuridine, and 5-iodo-2′-deoxyuridine) (Scheme 1). Using thymidine as the sugar donor, HeTP was employed to catalyze the one-pot single-enzyme phosphorolysis and transglycosylation of the halogenated nucleoside drugs (Fig. 1). In this first screening, before any optimization of the reaction conditions was carried out, up to 50% of conversion with the four halogenated drugs was achieved in 24 h. Similar conversions were previously achieved for the nucleoside analogues containing F and Br by using 5-fold the amount TP from E. coli, but adding half of the sugar donor (thymidine).13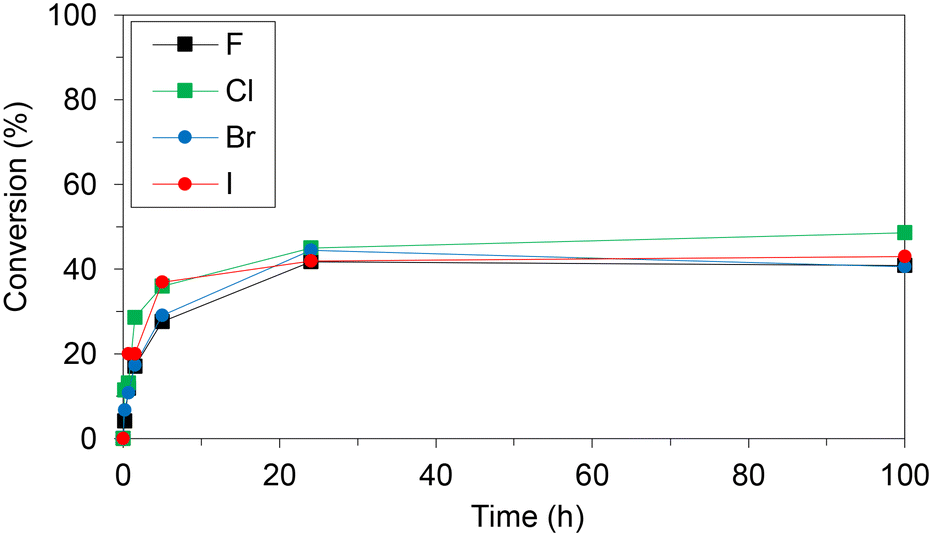 | ||
| Fig. 1 Monitoring the transglycosylation conversions to produce halogenated nucleoside analogues using free HeTP. The reaction mixture contained 10 mM of the halogenated nucleobase and 4 eq. of thymidine in 20 mM phosphate buffer at pH 7.5. The reactions were triggered with 2 units of free HeTP and incubated at 37 °C and shaking. | ||
Immobilization of HeTP on commercial resins
To improve the efficiency and sustainability of the biocatalytic process, HeTP was immobilized on microparticles that allow the reuse of the biocatalyst as well as its integration into a continuous reactor. We began testing methacrylate (EP403/S and EP400/SS) and agarose microbeads functionalized either with epoxy and cobalt chelates (Ep/Co2+) or glyoxyl (Gx) groups which are commercial supports and enable the covalent irreversible immobilization of HeTP as observed by SDS-PAGE (Fig. S15†).29 Although the immobilization yielded more than 85%, the recovered enzymatic activity was low (12–15%) compared to the free enzyme (Table 1). Hence, we decided to explore other supports for enzyme immobilization such as glass microparticles (EziG) and silica microbeads (Si2200). HeTP activity after irreversible immobilization on Si2200 decreased even more (5%). However, the HeTP immobilized on EziG yielded up to 3-fold higher recovered activity (38%) as the enzyme was reversibly bound to the support by metal coordination affinity which maintains the flexibility of the enzyme (Table 1).| Support code | Reactive group | Enzyme-support binding | Matrix material | Immobilization yield (%) | Recovered activity (%) | Expressed activity (U g−1) |
|---|---|---|---|---|---|---|
| Ep/Co2+-EP403/S | Epoxy/Co2+ |
 Irreversible Irreversible |
Methacrylate | 100 | 19 | 11.9 |
| Ep/Co2+-AG | Epoxy/Co2+ | Agarose | 98 | 19 | 12.2 | |
| Ep/Co2+-Si2200 | Epoxy/Co2+ | Silica | 65 | 6 | 3.2 | |
| Gx-EP403/S | Aldehyde |
 Irreversible Irreversible |
Methacrylate | 97 | 23 | 20.5 |
| Gx-EP400/SS | Aldehyde | Methacrylate | 85 | 13 | 10.9 | |
| Gx-AG | Aldehyde | Agarose | 87 | 18 | 11.2 | |
| EziG1 | Fe3+ |
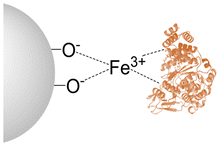 Reversible Reversible |
Hydrophilic glass | 97 | 38 | 35.9 |
| EziG2 | Fe3+ | Hydrophobic polymer | 99 | 27 | 25.6 | |
| EziG3 | Fe3+ | Semi-hydrophilic polymer | 98 | 26 | 25.0 |
Analysis of HeTP using CapiPy,30 revealed that the C-terminal His-tag is completely exposed and on the backside of the catalytic cleft, hence explaining the high recovered activity when immobilizing only through the (6×)-HisTag (Table 1 and Fig. S16A†). On the other hand, HeTP has 14 solvent-accessible lysines per monomer scattered throughout the surface. Therefore, an irreversible and covalent multipoint attachment could reduce the necessary domain movement for the catalytic activity of HeTP and create a distortion of the protein structure leading to lower recovered activity. Moreover, in the open conformation of the enzyme, multiple lysines in the active site are also accessible and could abolish activity when directly involved in the immobilization (Fig. S16B†).
The reusability of the different immobilized HeTP preparations was then tested. We found that the immobilized HeTP on Gx-EP403/S and Ep/Co-EP403/S retained more than 90% of the initial activity after 10 reaction cycles in batch, while the immobilized HeTP on EziG lost 30–50% of activity after only 5 reaction cycles (Fig. S17†). Therefore, Gx-EP403/S was selected as support for the subsequent studies, considering the recovered activity of the enzyme, the low cost of the support, and the operational stability of the biocatalyst in batch. The protein loading on Gx-EP403/S was also optimized, with 3 mg g−1 of HeTP resulting in the best preparation (Fig. S18†).
Enzyme immobilization often enhances the enzyme stability under harsher conditions. Thus, the temperature and DMSO stability of the irreversibly immobilized HeTP was tested. After 24 h of incubation at 30–45 °C, the immobilized HeTP retained more than 85% of the initial activity, while the free enzyme lost up to 50% (Fig. S19†). The stability of HeTP in presence of 10–20% DMSO was also increased after enzyme immobilization as HeTP maintained full initial activity after 3 h (Fig. S20†). Some of these results are comparable to the solvent stability of the immobilized TP from E. coli, but only DMF and MeCN were used in that work.13
Optimization of the synthesis of halogenated nucleoside analogues with immobilized HeTP
The Gx-EP403/S immobilized HeTP was then applied for the synthesis of floxuridine (5-fluoro-2′-deoxyuridine) as a model reaction due to its relevance as antiviral, antibacterial and anticancer drug. The sugar donor concentration as well as the phosphate buffer were optimized for the transglycosylation. 10 mM phosphate buffer and 10 eq. of sugar donor were selected as the best conditions to reach the maximum conversion (85%) while using the least of reagents (Fig. S21†). Further attempts to improve the conversion by increasing the nucleobase concentration, the amount of biocatalyst, and the temperature were not successful, in agreement with previous studies on nucleoside phosphorolysis (Table S3†).31 In contrast, the conversion was maintained when the nucleobase concentration was increased up to 20 mM with 10 eq. sugar donor (Table S3†), as it could be expected from the law of mass action considering relative concentrations.32 To the best of our knowledge, this is the highest substrate concentration used for the enzymatic synthesis of floxuridine reported to date.With the optimized conditions, we also performed the biosynthesis of the other halogenated nucleoside analogues in batch mode. The transglycosylation conversion reached 70–80% for the Br, I, and Cl nucleoside analogues (Table 2).
| Halogen in nucleobase | 2 h | 24 h | 120 h | |||
|---|---|---|---|---|---|---|
| %P | %T | %P | %T | %P | %T | |
| F | 5 | 33 | 17 | 84 | 18 | 85 |
| Cl | 4 | 28 | 14 | 76 | 15 | 79 |
| Br | 4 | 20 | 10 | 67 | 10 | 70 |
| I | 6 | 35 | 12 | 80 | 12 | 83 |
Flow biosynthesis of halogenated nucleoside drugs: process intensification
To increase the productivity of the biocatalytic system, the immobilized HeTP was implemented into a packed-bed reactor (PBR) for the continuous biosynthesis of halogenated nucleoside drugs. To do this, the nucleobase and the sugar donor were mixed with a T-tube before entering the PBR. HPLC pumps were used to modulate the flow-rate with residence times (R.T.) from 15 to 45 min, reaching the equilibrium at 30 min of R.T. for the F, Br, and Cl nucleoside drugs (Table 3). To the best of our knowledge, this is the first time that a thymidine phosphorylase is implemented under flow conditions for the synthesis of halogenated nucleoside drugs. In fact, to date, only one study describes the biocatalysed flow synthesis of the F and I analogues but with a different enzyme (2′-deoxyribosyl transferase) immobilized on epoxy silica.33|
|
||||||
|---|---|---|---|---|---|---|
| Halogen in nucleobase | 15 min | 30 min | 45 min | |||
| %P | %T | %P | %T | %P | %T | |
| F | 9 | 53 | 15 | 82 | 15 | 87 |
| Cl | 6 | 55 | 14 | 85 | 15 | 86 |
| Br | 9 | 63 | 14 | 84 | 15 | 88 |
| I | 7 | 55 | 11 | 74 | 13 | 83 |
The scale of the flow biosynthesis of floxuridine was increased to 50 mM of the nucleobase while the sugar donor was maintained at 100 mM. Unsurprisingly, the conversion decreased from 87% to 17% while the amount of the final product (8 mM) was maintained (Table S4†). As the nucleobase concentrations used herein are unusually high, a level of inhibition by the nucleobase may be observed, resulting in a slower reaction rate.15,34 At 20 mM nucleobase with 10 eq. of sugar donor, which is the maximum achievable in terms of water solubility with this set up, 85% conversion was again achieved but in this case the amount of product was 2-fold higher at 16.8 mM (Table S5†). This system provided the highest space–time yield reported to date for the synthesis of floxuridine (Table 4).13,33,35,36
| Nucleobase (mM) | Conversion (%) | STY (g L−1 h−1) | Catalyst productivity (mgproduct mgenzyme−1 h−1) |
|---|---|---|---|
| 10 | 87 ± 1 | 2.9 | 0.95 |
| 20 | 84 ± 2 | 5.5 | 1.84 |
In terms of the biocatalyst stability in flow, after 20 column volumes (10 h), more than 60% conversion was maintained (Fig. S22†). These results show a significant improvement over the previously reported example in which just 35% conversion was retained after 10 h.33 Besides, we stored the PBR at 4 °C for 2 months while not in use, preserving more than 90% of the enzymatic activity.
Conclusions
We have expanded the toolbox of robust thymidine phosphorylases with the halotolerant HeTP. This is the first time that a thymidine phosphorylase has been implemented in a flow reactor. Moreover, we have pioneered the biosynthesis of 5-chloro-2′-deoxyuridine by using isolated enzymes.35This work also showcases the potential of two key technologies in biocatalysis: enzyme immobilization and flow chemistry. The immobilized HeTP has enhanced stability against high temperatures, co-solvents and long-term storage compared with its free counterpart. Enzyme immobilization also enabled the reuse of the biocatalyst for 10 cycles retaining the full enzymatic activity, which means a decrease in waste and process costs. Finally, continuous flow technology speeds up biosynthetic processes, while offering an automatized and controlled handling of the system. This has allowed the process intensification of floxuridine reaching the highest STY and productivity results reported so far. Hence, our work is timely in progressing the biosynthetic strategies of such important molecules. Specifically with the relevance of floxuridine as anticancer and building block in SARS-related vaccines.
Experimental section
Materials
All chemical reagents were acquired from Sigma Aldrich (Gillingham, UK), Thermo Fisher (Loughborough, UK), Acros Organics (Reinach, Switzerland), or Fluorochem (Hadfield, UK) and used without further purification. Plain agarose (6BCL) was purchased from Agarose Bead Technologies (Madrid, Spain). The methacrylate supports were purchased from Resindion S.R.L (Milan, Italy). The EziG™ (glass microparticles) were kindly donated by EnginZyme.HeTP expression and purification
The gene deoA codifying for the HeTP (A.N. Q7CP66) was cloned into the pET28b plasmid (NcoI/XhoI) and transformed into E. coli BL21(DE3) star competent cells by heat-shock at 42 °C. Then, 1 L flasks containing 300 mL of autoinduction medium (ZYM) with 50 μg mL−1 kanamycin were inoculated with a colony and incubated at 37 °C for 20 h. Protein expression in LB and TB media were also tested (Fig. S1†). The resulting cells were harvested by centrifugation at 4500 rpm and resuspended in 50 mM phosphate buffer, 300 mM NaCl and 30 mM imidazole at pH 7.0. The suspension was placed on ice and sonicated at 40% amplitude for 8 min, with pulses of 5 s ON, 10 s OFF. After centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300g for 45 min, the supernatant was filtered with a 0.45 μm filter and HeTP was purified from the supernatant by a Ni-NTA column in the AKTA-pure. HeTP was eluted in 50 mM phosphate buffer, 300 mM NaCl and 300 mM imidazole at pH 8.0. The purified enzyme was dialyzed twice in 100 mM phosphate buffer at pH 7.0. The protein expression and purification were analyzed by SDS-PAGE.
300g for 45 min, the supernatant was filtered with a 0.45 μm filter and HeTP was purified from the supernatant by a Ni-NTA column in the AKTA-pure. HeTP was eluted in 50 mM phosphate buffer, 300 mM NaCl and 300 mM imidazole at pH 8.0. The purified enzyme was dialyzed twice in 100 mM phosphate buffer at pH 7.0. The protein expression and purification were analyzed by SDS-PAGE.
Determination of protein concentration
The protein concentration was estimated by measuring the absorbance at 280 nm in the EPOCH2 (nanodrop Tek3 plate) and using the predicted extinction coefficient 14105 M × cm−1 and the molecular weight 47736 Daltons obtained from https://web.expasy.org/protparam. Low protein concentrations (<1 mg mL−1) were determined by Bradford assay by measuring the absorbance at 595 nm of a solution containing 5 μL of protein sample with 250 μL of Bradford reagent. A standard curve was done with BSA.
Activity assay
A modified photometric assay previously reported was used to assess the activity of the soluble HeTP. Briefly, 990 μL of 1 mM thymidine and 0.5 M potassium phosphate at pH 7.0 were added to a 1 mL-cuvette and the reaction was triggered with 10 μL of HeTP (0.25 mg mL−1). The absorbance at 290 nm was recorded for 2 min at 30 °C. Alternatively, the activity was monitored in a 96-well plate by mixing 300 μL of substrate solution and 5 μL of HeTP (d = 0.75 cm). In the time frame of linear absorbance decrease, regression was performed to calculate the slope of the absorbance decrease per minute. One unit is defined as the amount of enzyme that will convert 1 μmol of thymidine to thymine per minute. The specific enzyme activity (U mg−1) was calculated as reported by Krenitsky et al.,37 based on the difference in millimolar extinction coefficients (ε) between thymidine and thymine which under the assay conditions equals 1. Specific activity was then calculated with eqn (1):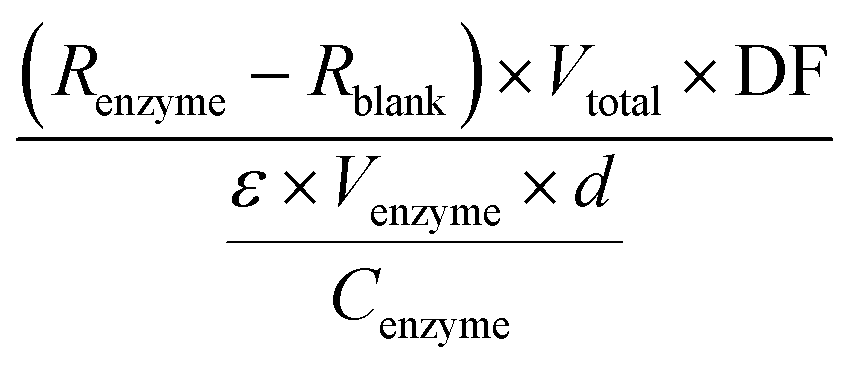 | (1) |
R enzyme = slope of the reaction regression (min−1)
R blank = slope of the blank regression (min−1)
DF = dilution factor of enzyme
ε = difference in millimolar extinction coefficient between thymidine and thymine (M−1 cm−1)
V enzyme = volume of enzyme added (mL)
V total = total volume of the reaction (mL)
C enzyme = concentration of the added enzyme solution (mg mL−1)
d = pathlength (cm).
Biotransformations in batch mode
Batch reactions with soluble enzyme (2 units) were incubated at 37 °C and under shaking for 24 h in 1 mL of reaction mix containing the sugar donor, the nucleobase, and phosphate buffer at pH 7.0 at the specified concentrations. Batch reactions with immobilized enzyme were triggered adding 50 mg of 3 mg g−1 immobilized HeTP, unless otherwise specified. The reactions were monitored by HPLC.The conversion (%) was calculated from the HPLC analyses of the reaction mixtures following the eqn (2):
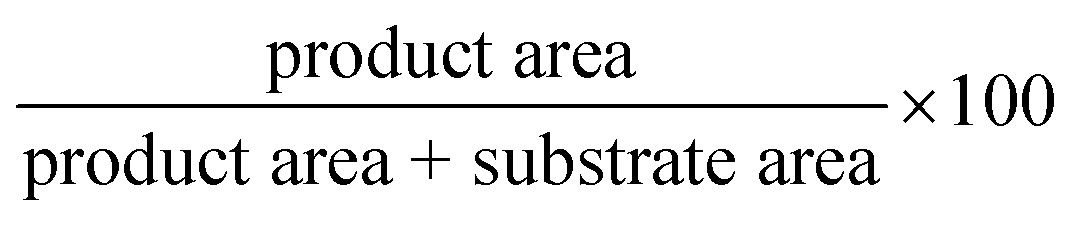 | (2) |
Activation of supports and enzyme immobilization
Stability tests
HeTP was incubated in 1 mL of buffer. At different time points, 100 μL of enzyme solution/suspension were withdrawn to measure the activity as described above.Flow reactions
Flow biotransformations were performed using a R2S/R4 Vapourtec flow reactor equipped with a V3 pump and an Omnifit glass column (6.6 mm i.d. × 100 mm length) filled with the immobilized enzyme (2 g with 3 mg g−1 of protein loading) as a packed-bed reactor (PBR) of 2.4 mL. A first equilibration step was performed by running 10 mM phosphate buffer pH 7.0 at 0.4 mL min−1 for 10 min. Then, the solutions of substrates at different concentrations were mixed in a T-tube and pumped through the PBR containing the immobilized biocatalyst. The flow-rate was adapted depending on the desired residence time for each reaction (R.T. 15 min corresponds to 0.160 mL min−1; R.T. 30 min corresponds to 0.080 mL min−1; R.T. 45 min corresponds to 0.053 mL min−1) unless otherwise specified. Samples were collected after each column volume and analyzed by HPLC.HPLC analysis
The samples were prepared by mixing 100 μL of the reaction medium with 200 μL of MeCN and 200 μL of 0.2% HCl in H2O (or 400 μL of MeOH). The samples were filtered with a 0.45 μm PTFE filter prior submission to HPLC Dionex UltiMate 3000 (Thermo Fisher, Loughborough, UK) equipped with a C18 column 3.5 μm, 2.1 × 100 mm (Waters, Elstree, UK). 2 μL of sample were injected. For the phosphorolysis analysis, a gradient method 5:95 to 95:5 (H2O:MeCN containing 0.1% TFA) over 4 min with a flow rate of 0.8 mL min−1 at 45 °C was employed. Thymidine (4 min) and thymine (3.1 min) were detected by UV at 265 nm. For the analysis of both phosphorolysis and transglycosylation, a gradient method 1:55 to 55:1 (H2O:MeOH) over 8 min was used. 5′-Azacytosine (1.8 min), 5′-aza-2′-deoxycitidine (2.9 min), were detected by UV at 250 nm. Thymidine (5.1 min), thymine (4.1 min), 5-fluorouracil (2.6 min), and 5-fluoro-2′-deoxyuridine (4.4 min) were detected by UV at 265 nm. 5-Chlororouracil (3.9 min), 5-chloro-2′-deoxyuridine (5.2 min), 5-bromorouracil (4.3 min), 5-bromo-2′-deoxyuridine (5.5 min), 5-iodouracil (5.1 min), and 5-iodo-2′-deoxyuridine (6 min), cytosine (2 min), 2′-deoxycitidine (3.76 min) were detected by UV at 280 nm.
Modelling and docking analysis
HeTP was modelled using SWISS-MODEL via the Expasy web server. Visualization of the active site pocket was performed by using the CASTp web server. The resulting PDB file was then loaded into Chimera alongside the original HeTP model and colored for visualization. The model was used for the docking studies, performed with Autodock Vina.40 The substrates were manually build using the Build function in UCSF Chimera from their SMILES code.41 The results were analyzed and visualized in UCSF Chimera and PyMOL.42MD analysis
To perform the molecular dynamics studies, the Enlighten2 plugin for PyMOL was used.43 Simulations were performed with the monomeric structure of HeTP and with mobility only on the residues at 12 Å surrounding the docked substrate. After minimization of the system, the MD simulation was performed for 1 ns. Results were analyzed using the built-in tools of UCSF Chimera.Author contributions
F. P. supervised and guided the project. A. I. B. M. and F. P. conceptualized the idea and secured the funding. A. I. B. M. conducted most of the experimental part and wrote the first draft of the manuscript. C. K. carried out the first characterization of the HeTP. D. R. P. performed MD and CapiPy analysis. All the authors discussed the results and agreed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the SNSF (200021_192274, F.P) and the University of Bern “Seal of Excellence Fund” Postdoctoral Fellowship (SELF19-03 BIORPHANDRUG, A.I.B.M).References
- L. P. Jordheim, D. Durantel, F. Zoulim and C. Dumontet, Nat. Rev. Drug Discovery, 2013, 12, 447–464 CrossRef CAS PubMed.
- D. Benedetto Tiz, L. Bagnoli, O. Rosati, F. Marini, L. Sancineto and C. Santi, Molecules, 2022, 27, 1643 CrossRef CAS PubMed.
- W. S. Yeo, R. Arya, K. K. Kim, H. Jeong, K. H. Cho and T. Bae, Sci. Rep., 2018, 8, 1–10 CAS.
- C. M. Galmarini, Electron. J. Oncol., 2002, 3, 22–32 Search PubMed.
- HS/CN8 classification reference list for dataset ‘EU trade since 2015 of COVID-19 medical supplies’, https://ec.europa.eu/eurostat/documents/6842948/11003521/Corona+related+products+by+categories.pdf, (accessed 11 April 2022).
- M. J. Lapponi, C. W. Rivero, M. A. Zinni, C. N. Britos and J. A. Trelles, J. Mol. Catal. B: Enzym., 2016, 133, 218–233 CrossRef CAS.
- S. C. Cosgrove and G. J. Miller, Expert Opin. Drug Discovery, 2022, 17, 355–364 CrossRef CAS PubMed.
- F. Kaspar, M. R. L. Stone, P. Neubauer and A. Kurreck, Green Chem., 2021, 23, 37–50 RSC.
- Y. Ni, D. Holtmann and F. Hollmann, ChemCatChem, 2014, 6, 930–943 CrossRef CAS.
- S. Kamel, H. Yehia, P. Neubauer and A. Wagner, in Enzymatic and Chemical Synthesis of Nucleic Acid Derivatives, ed. J. Fernández-Lucas and M.-J. Camarasa Rius, First, 2019, pp. 1–28 Search PubMed.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS PubMed.
- J. A. McIntosh, T. Benkovics, S. M. Silverman, M. A. Huffman, J. Kong, P. E. Maligres, T. Itoh, H. Yang, D. Verma, W. Pan, H. I. Ho, J. Vroom, A. M. Knight, J. A. Hurtak, A. Klapars, A. Fryszkowska, W. J. Morris, N. A. Strotman, G. S. Murphy, K. M. Maloney and P. S. Fier, ACS Cent. Sci., 2021, 7, 1980–1985 CrossRef CAS.
- I. Serra, T. Bavaro, D. A. Cecchini, S. Daly, A. M. Albertini, M. Terreni and D. Ubiali, J. Mol. Catal. B: Enzym., 2013, 95, 16–22 CrossRef CAS.
- S. Rocchietti, D. Ubiali, M. Terreni, A. M. Albertini, R. Fernández-Lafuente, J. M. Guisán and M. Pregnolato, Biomacromolecules, 2004, 5, 2195–2200 CrossRef CAS PubMed.
- F. Kaspar, P. Neubauer and A. Kurreck, ChemBioChem, 2021, 22, 1385–1390 CrossRef CAS PubMed.
- A. I. Benítez-Mateos, M. L. Contente, D. Roura Padrosa and F. Paradisi, React. Chem. Eng., 2021, 6, 599–611 RSC.
- D. Roura Padrosa, A. I. Benítez-Mateos, L. Calvey and F. Paradisi, Green Chem., 2020, 22, 5310–5316 RSC.
- L. Cerioli, M. Planchestainer, J. Cassidy, D. Tessaro and F. Paradisi, J. Mol. Catal. B: Enzym., 2015, 120, 141–150 CrossRef CAS.
- D. Roura Padrosa, Z. Nissar, F. Paradisi and D. M. Reactors, Catalysts, 2021, 11, 520 CrossRef.
- D. Roura Padrosa, V. De Vitis, M. L. Contente, F. Molinari and F. Paradisi, Catalysts, 2019, 9, 232 CrossRef.
- A. I. Benítez-Mateos and F. Paradisi, ChemSusChem, 2022, 15, 1–9 CrossRef.
- M. L. Contente, N. Fiore, P. Cannazza, D. Roura Padrosa, F. Molinari, L. Gourlay and F. Paradisi, ChemCatChem, 2020, 12, 5679–5685 CrossRef CAS.
- N. Hori, M. Watanabe and Y. Mikami, Biocatalysis, 1991, 4, 297–304 CrossRef CAS.
- N. G. Panova, C. S. Alexeev, A. S. Kuzmichov, E. V. Shcheveleva, S. A. Gavryushov, K. M. Polyakov, A. M. Kritzyn, S. N. Mikhailov, R. S. Esipov and A. I. Miroshnikov, Biochemistry, 2007, 72, 21–28 CAS.
- J. Vande Voorde, F. Gago, K. Vrancken, S. Liekens and J. Balzarini, Biochem. J., 2012, 445, 113–123 CrossRef CAS.
- T. Oh and M. H. El Kouni, PLoS One, 2018, 13, 1–18 Search PubMed.
- L. Zhong, Y. Li, L. Xiong, W. Wang, M. Wu, T. Yuan, W. Yang, C. Tian, Z. Miao, T. Wang and S. Yang, Signal Transduction Targeted Ther., 2021, 6, 201 CrossRef PubMed.
- E. Mitsiki, A. C. Papageorgiou, S. Iyer, N. Thiyagarajan, S. H. Prior, D. Sleep, C. Finnis and K. R. Acharya, Biochem. Biophys. Res. Commun., 2009, 386, 666–670 CrossRef CAS PubMed.
- A. I. Benítez-Mateos and M. L. Contente, Catalysts, 2021, 11, 814 CrossRef.
- D. Roura Padrosa, V. Marchini and F. Paradisi, Bioinformatics, 2021, 37, 2761–2762 CrossRef PubMed.
- F. Kaspar, R. T. Giessmann, P. Neubauer, A. Wagner and M. Gimpel, Adv. Synth. Catal., 2020, 362, 867–876 CrossRef CAS.
- C. S. Alexeev, I. V. Kulikova, S. Gavryushov, V. I. Tararov and S. N. Mikhailov, Adv. Synth. Catal., 2018, 360, 3090–3096 CrossRef CAS.
- F. Rinaldi, J. Fernández-Lucas, D. de la Fuente, C. Zheng, T. Bavaro, B. Peters, G. Massolini, F. Annunziata, P. Conti, I. de la Mata, M. Terreni and E. Calleri, Bioresour. Technol., 2020, 307, 123258 CrossRef CAS.
- F. Kaspar, F. Brandt, S. Westarp, L. Eilert, S. Kemper, A. Kurreck, P. Neubauer, C. R. Jacob and A. Schallmey, ChemRxiv, 2022, preprint, DOI:10.26434/chemrxiv-2022-xjvpw.
- C. W. Rivero, C. N. Britos, M. E. Lozano, J. V. Sinisterra and J. A. Trelles, FEMS Microbiol. Lett., 2012, 331, 31–36 CrossRef CAS.
- M. B. Méndez, C. W. Rivero, F. López-Gallego, J. M. Guisán and J. A. Trelles, J. Biotechnol., 2018, 270, 39–43 CrossRef.
- T. Krenitsky and S. Bushby, US Pat., 4178212, Burroughs Wellcome Co., 1979 Search PubMed.
- C. Mateo, G. Fernández-Lorente, E. Cortés, J. L. Garcia, R. Fernández-Lafuente and J. M. Guisan, Biotechnol. Bioeng., 2001, 76, 269–276 CrossRef CAS.
- J. M. Guisán, Enzyme Microb. Technol., 1988, 10, 375–382 CrossRef.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS.
- W. L. Delano, CCP4 Newsl, Protein Crystallogr.
- K. Zinovjev and M. W. Van Der Kamp, Bioinformatics, 2020, 36, 5104–5106 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy00751g |
| This journal is © The Royal Society of Chemistry 2022 |