
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic performance of mixed MxCo3−xO4 (M = Cr, Fe, Mn, Ni, Cu, Zn) spinels obtained by combustion synthesis for preferential carbon monoxide oxidation (CO-PROX): insights into the factors controlling catalyst selectivity and activity†
Camillo
Hudy
*a,
Olga
Długosz
b,
Joanna
Gryboś
a,
Filip
Zasada
a,
Aneta
Krasowska
a,
Janusz
Janas
a and
Zbigniew
Sojka
 *a
*a
aFaculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Krakow, Poland. E-mail: hudy@mail.ch.uj.edu.pl; sojka@chemia.uj.edu.pl
bFaculty of Engineering and Chemical Technology, Cracow University of Technology, 31-155 Krakow, Poland
First published on 5th April 2022
Abstract
A series of mixed cobalt spinel catalysts (MxCo3−xO4 (M = Cr, Fe, Mn, Ni, Cu, Zn)) was synthesized and tested in the CO-PROX reaction and in sole CO oxidation and H2 oxidation as references. The catalysts were thoroughly characterized by XRF, XRD, XPS, Raman, and IR spectroscopy, by TEM/STEM/EDX and with SEM techniques. Depending on the u parameter value (oxygen centricity between the tetra- and octahedral cations), two types of mixed spinel catalyst with distinctly different catalytic behavior in the CO-PROX reaction were distinguished. The A-type spinels (Co, Ni, Cu) with u ∼ 0.2625 exhibited overall higher activity in CO and H2 oxidation and lower selectivity in the high-temperature range (T > 180 °C), whereas B-type spinels (Cr, Fe, Mn) with u < 0.2625 were less active yet more selective in the high-temperature range. The work function of the spinel catalysts was found to be a useful concise parameter in accounting for their CO-PROX performance, supporting the proposed categorization of the mixed spinels. Two heuristic descriptors, EO2p + kΔ|<χM> − χO| and ΔEM–O = (EM3d − EO2p), based on the position of the oxygen 2p (EO2p) and the metal 3d (EM3d) band centers and on the average metal <χM> and oxygen (χO) electronegativity difference, were proposed for the rationalization of the mixed spinel performance in terms of their intrinsic electronic properties. It was established that the activity depends on the volcano-type fashion of the ΔEM–O values, whereas the selectivity is correlated with the EO2p + kΔ|<χM> − χO| parameter in a monotonous fashion. The unique behavior of the Zn–Co spinel results from the lifting of the O2p center above the 3d band center of the redox Co3+ cations, in contrast to the other mixed spinel catalysts.
1. Introduction
Preferential carbon monoxide oxidation in hydrogen-rich streams (CO-PROX) is a promising and economically appealing technology for the purification of H2 down to levels below 100 ppm,1–3 which is required for practical applications in polymer electrolyte membrane fuel cells (PEMFC) or in ammonia synthesis.4–6 The development of highly active, selective and stable CO-PROX catalysts and a mechanistic understanding of the reaction course that controls their activity and selectivity are intensively explored research topics in catalytic chemistry.7–10Supported PGM (platinum group metals) systems, such as Pd/zeolites,11 Pt/Al2O3,12,13 Au/Al2O3,14 Au/Ce1−xZrxO2,15 and Ru/Al2O3,16 have high costs and appear to be not selective enough in the presence of H2O and CO2.17 Methanation of CO/CO2, which leads to the undesirable consumption of hydrogen, is another problem observed for noble metal-containing catalysts, exemplified by Au/Co3O4.3 Although cheaper supported Ag-containing systems exhibit high selectivity for CO at low temperature, their overall behavior in the PROX reaction is not satisfactory.18
Catalytic systems based on transition metal oxides, in bulk or supported forms, provide an alternative class of materials with promising total CO oxidation19–23 and CO-PROX activities.8,24 CuOx/CeO2 catalysts are some of the most widely investigated systems, which profit from the unique oxygen release and storage properties of the ceria support.25,26 Recently, high activity and stability have been reported for CoVOx catalysts, where divalent cobalt cations in cobalt oxide are stabilized by the incorporation of vanadium.27 Another important class of CO-PROX catalysts includes oxides of perovskite (ABO3)6,28,29 and spinel (AB2O4)30–32 structures, where the choice of appropriate A and B cations may be used for tuning their catalytic performance. The replacement of the A or B cations in spinels, such as Co3O4 or Mn3O4, by alien ions can change the valence state of the parent cations. This significantly influences the formation of the surface oxygen adspecies and oxygen vacancies, the catalytic chemistry of which is of prime importance for the oxidation reactions33 and for the CO-PROX performance in particular. Among spinel materials, several mixed and supported catalysts, such as CuFe2O4,34 NiFe2O4 (ref. 35) or CuMn2O4/CeO2,36 along with various cobalt spinel-based systems, including the bare Co3O4,37 also ZnxCo3−xO4,38 Co3O4–CeO2,39 Co3O4–CuO,40 Co3O4/ZrO2 and Co3O4/CeO2–ZrO2,41 ZnCo3O4/Al2O3,42 have been reported to exhibit promising CO-PROX performance.
In line with other catalytic oxidation reactions,33,43,44 the mechanism of the CO-PROX reaction is often discussed in terms of the involvement of the suprafacial (Langmuir–Hinshelwood) and/or lattice oxygen species (Mars–van Krevelen) in the CO and H2 oxidation events, which appear to be sensitive to the catalyst structure, faceting of the crystallites and the reaction conditions.37,45–47 Despite the progress made, some problems related to the intrinsic structural and mechanistic issues controlling the CO vs. H2 selectivity, as well as the H2O and/or CO2 susceptibility of the spinel catalyst surface to the active site blocking, remain to be addressed and understood in more detail. In the case of mixed spinels, the influence of the cation distribution between tetrahedral and octahedral sites, and segregation of oxide phases of the dopant ions on the catalytic performance are important issues to be resolved. The replacement of the cobalt cations by alien divalent or trivalent transition metal ions (M) in the parent spinel ((Co2+)A[Co23+]BO4) matrix may lead to the inhomogeneous distribution of the cations, giving rise to the formation of a partially inverted spinel structure, where either the foreign and parent cobalt ions can occupy the octahedral and tetrahedral sites. The resultant distribution of the divalent dopants in the mixed cobaltite spinels can be written as (M2+x−λCo2+1−xCo3+λ)A[M2+λCo3+2−λ]BO4, and for the trivalent dopants as (M3+x−λCo2+1−x)A[Mλ3+Co3+2−λCox2+]BO4, where 0 < x ≤ 1, and λ stands for the degree of inversion of the spinel structure. Since the inversion is driven by the configurational entropy increase, its extent depends strongly on the thermal conditions at which the samples were obtained, and therefore, should be determined specifically for the catalysts prepared by the applied synthesis method.
Owing to its simplicity, easy scale-up, and application versatility, combustion synthesis is often used for the preparation of oxide catalysts.47,48 This method is based on the redox reaction between metal precursor salts acting as oxidants (usually nitrates) and organic fuel, often possessing multiple functional groups that behave as ligands. The fuel may act as an ion complexant, fostering the homogeneous mixing of the cations in solution, thereby preventing the undesired selective precipitation of different metals when mixed oxide catalysts are prepared.49,50 Upon heating the sample, self-propagating oxidation initiates the actual combustion process.51 Combustion synthesis has been often used for the successful preparation of Co3O4-based catalysts for applications in various redox reactions.52,53 The resultant mixed oxide catalysts usually exhibit rather good crystallinity, small grain size and good dispersion.54 However, the rapid progression of the highly exothermic reaction may lead to the formation of structural defects and/or phase segregation.55,56
Herein, we examine the catalytic performance in the CO-PROX reaction for a series of mixed cobalt spinels containing alien cations with various numbers of 3dn electrons (Cr, Fe, Mn, Ni, Cu, Zn). Encouraged by the previous literature,57 the catalysts were obtained by combustion synthesis and were thoroughly examined to ascertain their morphology, structure, oxidation states of the cations and their distribution between the tetrahedral and octahedral sites. The observed changes in the activity and selectivity were rationalized in terms of the catalyst work function, and the spinel band structure modifications induced by doping.
2. Experimental
2.1. Materials
A series of mixed spinel oxide catalysts, MxCo3−xO4 (where M = Cr, Mn, Fe, Ni, Cu, and Zn), was synthesized via the combustion method, using nitrate precursors M(NO3)2·nH2O (≥99.0%, Sigma Aldrich) and citric acid (C6H8O7·H2O, ≥99.0%, Sigma Aldrich) as the reagents:In the standard protocol, a mixture of M(NO3)2·nH2O and Co(NO3)2·6H2O with the molar ratio M/Co = 0.5 was dissolved in 15 ml of distilled water, followed by the addition of 3.34 g of citric acid. The as-prepared gel was preheated at 400 °C for 15 minutes, then the obtained powder was ground and calcined at 600 °C for 4 hours, and cooled to room temperature.

The synthesized mixed spinel samples are labelled hereafter as Cr–Co, Mn–Ce, Fe–Co, Ni–Co, Cu–Co, Co–Co and Zn–Co.
2.2. Methods
The chemical composition of the prepared samples was determined using an Energy-Dispersive XRF spectrometer (Thermo Scientific, ARL QUANT'X). X-rays of 4–50 kV (1 kV step) with the beam size of φ = 1 mm, generated by the Rh anode, were used. The XRF spectra were obtained using the 3.5 mm Si(Li) drifted crystal detector with Peltier cooling. Calibration of the apparatus was done by measuring a series of metallic standards, and the quantitative analysis was performed using the UniQuant software.X-ray photoelectron spectroscopy was used to examine the chemical states of the spinel constituents. The spectra were recorded on a Prevac XPS instrument with the monochromatized aluminum X-ray source. The binding energies were adjusted to the reference C 1s peak at 284.8 eV. The peak deconvolution was performed with the Casa XPS software,58 using Gaussian–Lorentzian or Pearson VII line shapes, and nonlinear Shirley-type background subtraction.
X-ray diffractograms were recorded in the 2θ range of 10–70° with steps of 0.02° (3 s per step), using a Bruker D8-advance diffractometer with CuKα radiation (λ = 1.540598 Å). The obtained XRD diffractograms were thoroughly analyzed by the Rietveld method, using the Match! Software with FullProf and the pseudo-Voigt line shape function. The size of the crystallites and the lattice strains were assessed based on the Williamson–Hall method.
Laser Raman spectra were recorded using a Renishaw InVia spectrometer with an excitation wavelength of 785 nm. The spectra were collected in the range of 100–900 cm−1 with the resolution of 1 cm−1, and nine scans were accumulated to ensure a satisfactory signal-to-noise ratio.
The IR spectra were recorded using a Nicolet 6700 spectrometer in the transmission mode. The spectra were collected in the range of 550–4000 cm−1 upon accumulation of 64 scans using an MCT detector cooled with liquid N2.
Scanning electron microscopic (SEM) imaging of the samples was carried out on a Tescan-Vega 3 instrument, equipped with the LaB6 emitter at the 30 kV acceleration voltage.
The work function (ϕ) measurements were conducted by dynamic condenser method with a KP6500 probe (McAllister Technical Services), under vacuum at 10−7 mbar. The standard procedure includes the standardization of the pelletized samples (100 mg, φ = 11 mm) at 350 °C for 30 minutes, followed by measurements of the contact potential difference (CPD) at 150 °C until the signal stabilization. The work function values were calculated as ϕsample = ϕref − CPD, where ϕref = 4.2 eV is the work function of the reference stainless steel vibrating electrode.
Analysis of the morphology and elemental composition of the samples was performed by means of transmission electron microscopy techniques, using an FEI Tecnai Osiris microscope equipped with an X-FEG Schottky field emitter (200 kV), and a high angle annular dark-field (HAADF) detector. Mapping of the constituent elements was performed with energy-dispersive X-ray spectroscopy (EDX), using a Super-EDX windowless detector system with the 4-sector silicon drift detector (SDD), and the Bruker Esprit software. Prior to the microscopic analyses, the samples were deposited on a lacey carbon film placed on a copper or golden grid (Agar Scientific, London, UK, 300 mesh).
The multipoint BET specific surface areas of the investigated samples were determined by N2 physisorption at 77 K using a Quantachrome Autosorb Station. Before the measurements, the samples were pretreated at 400 °C in helium.
Temperature programmed reduction (TPR) experiments with 5% H2 diluted in argon were performed using a Quantachrome Chembet Pulsar instrument equipped with the TCD detector. For typical measurements, 12 mg of sample was placed in a U-shaped quartz reactor and then heated up to 800 °C with the rate of 8 °C min−1 under the 37 ml min−1 gas flow.
The catalytic tests were carried out in a gradient-less quartz reactor (ϕ = 16 mm, catalyst layer height ∼2 mm), described and evaluated elsewhere.59 The Peclet (Pe ≈ 0.005) and Damkohler (Da ≈ 0.1) numbers indicate that a practically complete mixing regime was achieved for all the measurements. A QMS spectrometer (hidden) was used for the detection of the diagnostic lines with m/z = 2(H2), 4(He), 18(H2O), 28(CO), 32(O2), 40(Ar), 44(CO2). The mass of the catalysts was 200 mg, and the temperature was varied from 25 °C to 300 °C, with a heating rate of 10 °C min−1. Prior to each PROX reaction, the catalysts were standardized by heating to 600 °C in the 5% O2 gas stream. The gas feed in the CO oxidation (2% CO in He), H2 oxidation (5% H2 in He) and PROX reaction (2% CO, 2% O2, 30% H2 and He balance) was supplied by mass flow controllers (Bronkhorst) with the total flow rate of 100 ml min−1. The O2 excess with regard to CO was set to λ = 2. Carbon monoxide conversion (XCO) and the selectivity of the PROX process towards CO2 formation were calculated as described elsewhere.60 The surface-normalized specific activities (apparent conversion rates, ri) were calculated according to the following:46
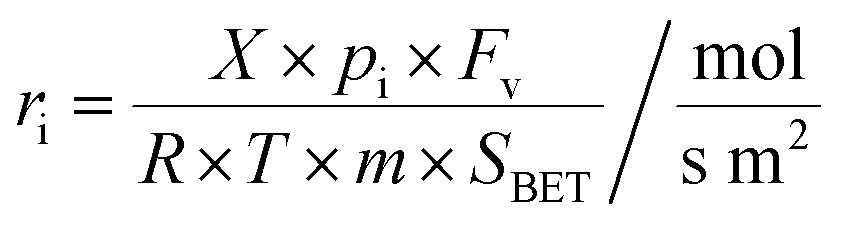
3. Results and discussion
3.1. Characterization of the mixed spinel catalysts
The phase composition of the synthesized catalyst was examined by the XRD method. The obtained diffractograms are shown in Fig. 1a, along with variations of the lattice parameter and oxygen u parameter upon cobalt spinel doping (Fig. 1b, Table 1). The latter describes the position of the O atoms in the spinel unit cell and by influencing the Madelung energy, it plays an important role in cation distribution between the octahedral and tetrahedral sites,61,62 redox behavior of spinels,63 as well as the modification of their surface properties.64 | ||
| Fig. 1 XRD patterns of the synthetized spinel catalysts (a), along with the variation of the lattice parameter (b). The colors of the XRD profiles correspond to the colors of the chemical symbols of the alien cations in (b). The pink circles indicate a minor ZnO phase, the red asterisk CuO, the cyan triangles NiO, and the blue squares indicate a secondary Fe–Co mixed spinel phase (see Table 1 for more information). | ||
| Catalyst | Phase structure | Composition/% | Spinel lattice parameter/Å | Oxygen u parameter |
|---|---|---|---|---|
| Co–Cr | (Cr0.22Co0.78)[Co1.15Cr0.85]O4 | 100.0 | 8.335 | 0.261 |
| Co–Mn | (Mn0.11Co0.89)[Co1.03Mn0.97]O4 | 100.0 | 8.254 | 0.248 |
| Co–Fe | (Fe0.34Co0.66)[Co1.61Fe0.39]O4 | 79.9 | 8.144 | 0.257 |
| (Fe0.44Co0.56)[Co1.03Fe0.97]O4 | 20.1 | 8.359 | ||
| Co3O4 | Co3O4 | 100.0 | 8.080 | 0.263 |
| Co–Ni | (Ni0.32Co0.68)(Co1.39Ni0.61)O4 | 93.5 | 8.096 | 0.264 |
| NiO | 6.5 | |||
| Co–Cu | (Cu0.30Co0.70)[Cu0.42Co1.58]O4 | 82.3 | 8.089 | 0.264 |
| CuO | 17.7 | |||
| Co–Zn | (Zn0.83Co0.17)Co2O4 | 95.9 | 8.097 | 0.264 |
| Zn0.85Co0.15O | 4.1 |
The observed dominant peaks 19.02° (111), 31.26° (220), 36.84° (311), 38.54° (222), 44.80° (400), 55.64° (422), 59.34° (511) and 65.22° (440) were indexed within the Fd3m space group, confirming the cubic spinel structure (JCPDS card #43-1003). Upon introduction of the alien cations into the spinel matrix the diffraction peaks shifted accordingly, indicating their successful incorporation.65 The observed line broadening may, in turn, be caused either by the lattice stress due to the ionic radii mismatch, or the grain size effect (Table S1†).66,67
The sole spinel phase was observed in the case of the Mn–Co, Cr–Co and Fe–Co samples, whereas for Zn–Co, Co–Ni and Co–Cu, a small amount of segregated Zn0.85Co0.15O (∼4%) (marked by circles in Fig. 1), NiO (6.4%, triangles) and a higher amount of CuO (17.7%, asterisks) were stated. The crystallite size of the synthesized mixed spinels was calculated using the Williamson–Hall method, and the results are collated in Table S1.† For the spinels doped with Fe, Mn and Cr, the average diameters of the nanocrystals (20.8–27.4 nm) were distinctly smaller than those for Co–Co (49.6 nm) and Ni–Co (46.6 nm), whereas for Cu–Co and Zn–Co they tend to be slightly larger.
As revealed by the SEM imaging (Fig. S1†), the rounded spinel grains were of uniform size and were agglomerated, forming rather compact structures in the cases of Cr–Co, Ni–Co and Zn–Co, while for the Mn–Co, Co–Co and Fe–Co samples the agglomerates were distinctly more loosely interconnected. Generally, the grain size decreases upon doping from 220–300 nm for Co–Co, Cu–Co and Zn–Co, to 160–200 nm in the case of Cr–Co and Ni–Co, becoming the smallest (140–160 nm) for the Mn, Fe-containing spinels. More detailed insight into the shape, size and composition of the nanometric spinel crystallites was obtained by subsequent S/TEM measurements, and the results are presented in Fig. 2. As can be inferred from the TEM and HAADF/STEM images (Fig. 2a1–g1 and a3–g3), overall, the spinel nanocrystals exhibited subhedral shapes. The highest tendency for agglomeration was observed for cobalt spinel nanocrystals doped with chromium, iron and copper (Fig. 2b1, d1 and f1, respectively). The Co–Mn nanocrystals have a strong tendency for oriented attachment, forming branched chains (Fig. 2c1). The associated SAED patterns, presented in Fig. 2a2–g2, confirmed high crystallinity and the spinel structure of the samples. The results of the elemental analysis (EDX maps) are presented in Fig. 2. Bare cobalt spinel nanocrystals (Fig. 2a4 and a5) preserve the stoichiometric Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O ratio (0.74) well. Chromium (Fig. 2b4 and b5), manganese (Fig. 2c4 and c5) and iron (Fig. 2d4 and d5)-doped nanocrystals exhibited a small variation in the (M,Co):O ratio, yet preserving the single spinel structure. In the case of the Ni–Co, Cu–Co and Zn–Co samples, apart from the mixed spinel phase, segregated NiO (Fig. 2e4 and e5), CuO (Fig. 2f4 and f5) and ZnO (Fig. 2g4 and g5) phases were also observed in accordance with the XRD results.
:O ratio (0.74) well. Chromium (Fig. 2b4 and b5), manganese (Fig. 2c4 and c5) and iron (Fig. 2d4 and d5)-doped nanocrystals exhibited a small variation in the (M,Co):O ratio, yet preserving the single spinel structure. In the case of the Ni–Co, Cu–Co and Zn–Co samples, apart from the mixed spinel phase, segregated NiO (Fig. 2e4 and e5), CuO (Fig. 2f4 and f5) and ZnO (Fig. 2g4 and g5) phases were also observed in accordance with the XRD results.
 | ||
| Fig. 2 Microscopic analysis of the morphology and elemental composition of the bare and doped cobalt spinel nanocrystals. TEM images (a1–g1), SAED patterns (a2–g2), HAADF STEM images (a3–g3), and EDX maps (a4–g5). Color coding: blue: cobalt cations, green: selected dopants, red: oxygen. | ||
The observed segregation of ZnO, CuO and NiO minor phases and their origins, have been discussed previously.68
 | ||
| Fig. 3 Raman (a) and infrared (b) spectra of the synthetized spinel catalysts. | ||
The RS spectrum of the pristine Co3O4 sample shows the presence of five characteristic bands at 191 cm−1, 516 cm−1 and 617 cm−1 (attributed to the F2g vibrational modes), 477 cm−1 (Eg mode), along with the most intense peak at 688 cm−1 associated with the A1g vibrations in the cubic spinel structure.69 However, a simple association of the observed bands with the vibrations of the tetrahedral or octahedral cations is not straightforward.70 This is caused by the fact that the RS modes are associated mainly with the movement of the oxygen atoms.71 Thus, the oxygen vibrations in the spinel tetrahedra and octahedra are coupled, and only in favorable cases may they be conditionally treated individually.70,71 The strongest peak due to the A1g mode has been attributed to the breathing vibration of the A–O4 tetrahedron,72 which is valid only when the A–O bonds are much stronger than B–O.73 The A1g vibration consists of the stretching of the A–O bond and the concomitant deformation of the three B–O bonds. It has been proposed that the Ag1 vibration is mainly determined by the nature of the highest-valence cation and its bond distance to the oxygen atoms, irrespective of whether it is located at the tetrahedral or octahedral sites.74,75 The F2g(1) and F2g(2) peaks are sensitive to the cations occupying the tetrahedron.76 The Eg mode arises from an asymmetric bending motion of the oxygen atoms connected to the A and B cations.
The Raman spectrum of Cr–Co reveals characteristic spinel modes with slight differences in the Raman shifts (Fig. 3a). An additional broad band (marked by an asterisk) at the 548 cm−1 was also observed. As the Cr ions exhibit a strong preference to the occupation of the octahedral sites, the observed new band can be attributed to the shifted F2g vibration mode, arising from the partial substitution of Cr3+ for Co3+ in the octahedral sites.77,78 The preferential location of Cr in the B-sites is also reflected in the broadening of the Eg and F2g bands. Such location was additionally confirmed by the XPS spectra and Rietveld analysis of the XRD patterns (see below). The RS spectrum of the Mn–Co sample is distinctly different from that of Co3O4, and shows only 3 much broader peaks at 177 cm−1, 488 cm−1 and at 648 cm−1 (Fig. 3a), in accordance with previous findings.79 The observed significant broadening (F2g (ref. 1)) and coalescence of the spinel bands, F2g (ref. 2) with Eg and F2g (ref. 3) with A1g, are associated with a random distribution of the Mn and Co cations in the octahedral sublattice of the spinel structure,80–82 and with the lattice strain induced by the Mn3+ cations (Table S1†), which are Jahn–Teller active. The apparent decrease in their frequencies with respect to Co3O4 resulted from the expansion of the cell volume due to the replacement of the low-spin Co3+ cations with a smaller radius (0.65 Å) by the high-spin Mn3+ cations of the significantly higher radius (0.785 Å). The obtained RS spectra for Fe–Co, Ni–Co and Cu–Co do not differ much from that of the bare Co3O4 sample, in terms of the number of lines and their shape. Larger deviations in the peak positions and broadening were observed for Fe–Co than for the Ni–Co and Cu–Co spinels. In contrast, the Zn–Co catalyst exhibited significant changes in both the frequencies and intensities of the resolved bands (Fig. 3a), with the peak at 486 cm−1 becoming the strongest, and a set of additional peaks at 151 cm−1, 206 cm−1 and 710 cm−1, which can be attributed to ZnO and ZnxCo3−xO4, respectively,83 in accordance with the TEM/EDX results (Fig. 2) The significant displacement of the A1g peak confirmed the localization of Zn in the A-sites.
Among the oscillation modes characterizing the spinel crystal structure, the F1u vibrations are IR active and involve the direct movement of the metal A and B atoms, in contrast to the RS modes. They appear as two strong peaks, clearly observed in the IR spectra of the examined samples (Fig. 3b). In the case of Co3O4, they are located at 579 cm−1 (v1) and 667 cm−1 (v2), usually assigned to the Co3+ vibrations in the octahedral and the Co2+ vibrations in the tetrahedral sites, respectively.84 The changes in the position, broadening and/or doubling of those bands observed in all doped samples confirmed the successful incorporation of the hetero-ions into the spinel matrix. The most significant broadening of the v1 and v2 bands was observed for the samples with the hetero-cations that prefer to occupy the octahedral sites (Cr, Mn, Fe), which also shifted towards lower frequencies. Such behavior can plausibly be related to the deformation of the tetrahedral (squeezing) and octahedral (elongation) sites, caused by the displacement of the common oxygen atom from the B into the A atom, as revealed by the oxygen u parameter (Table 1). Noteworthily, a distinct peak around 668 cm−1 observed in the v2 bands resulted from the oscillations of the intact CoO4 groups, present in the doped cobalt spinels. In the case of the Zn–Co sample with the zinc cations occupying the tetrahedral sites, all IR bands were displaced to higher frequencies, despite Zn being heavier than Co. This observation implies the enhancement of the Zn–O–Co bond strengths.
The bulk chemical composition and the surface elemental composition of the catalysts were examined by XRF and XPS, respectively. The calculated bulk and surface M/Co ratios are listed in Table S2,† together with the resultant chemical formula of synthesized catalysts. For determination of the oxidation states of the cations, narrow-scan XPS spectra of the corresponding diagnostic 2p regions were obtained (Fig. S2†). In the case of the Mn–Co sample, the Mn 3s region was additionally measured. The photoelectron Co 2p spectrum of the pristine Co3O4 exhibited a Co 2p3/2 peak at 779.9 eV and a Co2p1/2 peak shifted by 15.2 eV.85,86 Deconvolution of the 2p spectrum (Fig. S2a2†) revealed the presence of the 2p3/2 and 2p1/2 component signals of Co3+ located at 779.84 eV and 794.77 eV, and the corresponding two Co2+ signals at 781.51 eV and 796.29 eV, along with three satellites, S1 at 783.12 eV and S2 at 788.83 eV (associated with the octahedral Co3+), and S3 at 804.04 eV (associated with the tetrahedral Co2+).87
In the case of the Cr–Co sample, deconvolution of the Cr 2p3/2 band revealed the presence of three peaks (Fig. S2b†). Those located at 575.2 eV and 576.4 eV can be assigned to Cr3+ in the octahedral coordination, probably reflecting the presence of the Cr–Cr and Cr–Co motifs, whereas the signal at 579.1 eV corresponds to the minor tetra-coordinated Cr3+ species.88 The calculated Co3+/Co2+ = 0.96 was in agreement with the lowering of the Co3+ content due to the incorporation of the Cr3+ cations. For the Mn–Co sample, apart from the Co 2p and Mn 2p regions, the Mn 3s band was also analyzed (Fig. S2c†). The Mn 2p region was deconvoluted considering the complications resulting from the pronounced spin multiplet splitting,89 and the possible simultaneous presence of the Mn2+, Mn3+ and Mn4+ cations (Fig. S2c2,3†). For this purpose, inspired by previous XPS analysis of the Mn spectrum of manganese oxides,89 we applied a comprehensive peak fitting model for each oxidation state of Mn, taking into account the multiplet splitting. The resultant effective peak profile, approximated by the Pearson VII line shape, was used for fitting the Mn 2p spectra of the examined Mn–Co sample. The performed analysis assigned the dominant peak at 642.6 eV to the Mn3+ cations in the octahedral positions, and the weaker lines to Mn2+ (640.5 eV) and Mn4+ (643.5).90 The appearance of Mn4+ and Mn3+ in the mixed cobalt manganese spinels was previously observed by XANES measurements and explained by the redox reaction between the Mn3+ and Co3+cations,91 or by the partial oxidation of manganese cations in the near-surface layers.92 The observed speciation of manganese is consistent with its average oxidation state (AOS) estimated from the empirical relationship, AOS = 8.956–1.126 × ΔEs, where ΔEs is the splitting of the Mn 3s peak shown in the inset of Fig. S2c3.93 The observed peak separation of 5.37 eV corresponds to AOS = 2.9, in accordance with the Mn 2p deconvolution results. Thus, the majority of the manganese is located in the octahedral sites as Mn3+; the existence of a small amount of Mn2+ in the tetrahedral sites and Mn4+ in the octahedral sites was also disclosed. For the Fe–Co sample, the lower Co3+/Co2 ratio again implies the incorporation of the Fe3+ ions into the spinel host. However, the unambiguous location of the iron cations in the particular oxidation state is not straightforward as the Co 2p and Fe 2p regions overlap with the bands resulting from the Auger transitions.94 Taking these obstacles into account, the Fe 2p region was cautiously deconvoluted as shown in Fig. S2d2,3.† The 2p3/2 lines at 709.9 eV and 711.7 eV were assigned to the coexistence of the Fe2+ and Fe3+ cations, respectively,95,96 whereas the peak at 718.3 eV is due to the satellite of Fe2+. Unfortunately, the position of the Fe3+ 2p line is virtually insensitive to cation coordination. Our previous Mossbauer investigations of Co3−xFexO4 spinels revealed that for x < 1, the Fe cations tend to occupy the tetrahedral positions, and then the octahedral ones.97 Thus, for the iron content around x ∼ 1, both positions are expected to be occupied by the iron cations, which was then substantiated by the Rietveld refinement (see below).
The Ni 2p region (Fig. S2e2,3†) was deconvoluted into 3 peaks located at 854.04 eV (assigned to octahedral Ni2+ in the spinel and the segregated NiO phases), 855.6 eV (Ni3+ in octahedral sites), 856.30 eV (tetrahedral Ni3+), and the shake-up satellite peak at 861.14 eV.98–100 The asymmetric Cu 2p peak is accompanied by an intense shake-up satellite at the high binding energy side (Fig. S2f2,3†), which is diagnostic for the Cu2+cations;101 its apparent, yet discernible doubling implies the presence of Cu2+ in two different chemical environments, as previously observed for CuCo2O4.102 The deconvolution of the 2p3/2 line confirmed the existence of two overlapping signals at 932.6 eV and 933.9 eV due to the Cu2+ species. The narrower peak at higher binding energy can be attributed to Cu2+ accommodated in the tetrahedral sites,103 whereas the broader peak at the lower binding energy is attributed to Cu2+ located in the octahedral sites of the CuCo2O4 spinel, in accordance with previous literature.104 The satellite-main peak energy gap, ΔCu ≈ 9.5 eV, and the peak ratio of the Cu 2p3/2 line to its satellite, Isat/Imain ≈ 0.41, are close to those found for copper oxide105,106 so the contribution of the Cu2+ located in the spinel B-sites is masked by the contribution of the octahedral Cu2+ in the segregated CuO. This point was further addressed by the Rietveld phase analysis of this sample. The XPS spectrum of the Zn–Co sample was dominated by the 2p3/2 peak at 1021.13 eV (Fig. S2g2,3†), attributed to the Zn2+ cations in the A-sites and/or in the partly segregated ZnO,107 where Zn also occupies the tetrahedral positions. A significant increase in the Co3+/Co2+ ratio in the corresponding Co 2p3/2 spectrum is consistent with the replacement of the Co2+ cations by Zn2+ in the A-sites.
The results of the XPS spectral analysis concerning the location and the oxidation state of the hetero-cations were next used to corroborate a more detailed analysis of the XRD results. Rietveld refinement of the XRD patterns (Fig. S3†) allowed the precise determination of the phase composition of the synthesized samples and substantiated the cation distribution between the A and B-sites. The experimental patterns are shown as orange dots, whereas the calculated ones are black solid lines. The individual R factors, along with the cell parameters and volumes of the elemental cell are summarized in Table S3,† and the phase compositions of the examined catalysts are shown in Table 1, together with the calculated values of the lattice and oxygen u parameters. The position of oxygen atoms in the spinel cell, defined by the u parameter, varied from 0.248 for the Mn–Co sample to 0.264 in the case of the Ni, Cu and Zn-doped catalysts, whereas for the pristine Co3O4, u = 0.263. Significant changes (especially pronounced for the Mn–Co sample) indicate a noteworthy deformation of the spinel structure (centricity of the oxygen atoms that are shared by the constituting octahedra and tetrahedra).
Based on the spectroscopic data and Rietveld refinement, the cationic distribution and the level of substitution x in MxCo3−xO4 were determined. In the particular case of Mn–Co with the lattice constant aMn–Co = 8.254 Å, application of the Vegard law, a = 8.080 + 0.194x,108 leads to x = 0, 90, in good agreement with the Mn/Co ratio derived from the XRF analysis. In the case of the Fe–Co sample, a doubling of the (311) diffraction peak (35.59° and 36.59°), revealed the existence of two different sub-structures of the Fe–Co spinel. Based on Rietveld refinement, and the application of an empirical Vegard-type correlation published elsewhere,109 they can be assigned to an inverse spinel with x = 1.41, and a normal spinel x = 0.73. The simultaneous occupation of A and B-sites by iron in FeCo2O4 has been revealed before by Mössbauer studies,97,110 and found to the sensitive to the preparation conditions.111,112
Although nickel is known to preferentially occupy octahedral sites in opposition to Cu, which prefers tetrahedral coordination,113 the Rietveld refinement for the Ni and Cu-doped samples revealed the formation of the spinels with the hetero-cations partially entering into the tetrahedral and octahedral sites, without significant changes in the position of the oxygen anions, as reported previously.114 In both samples, the segregation of 17.7% of CuO and a smaller one of NiO (6.5%) were found, in agreement with TEM observations (Fig. 2). In the case of the Zn-substituted spinel sample, the hetero-cations were located essentially in the tetrahedral positions,115 and a small fraction (4.1%) was segregated in the form of the corresponding mixed Co, Zn monoxide impurity phases (Table 1).
The RS, IR, XPS and XRD results show that the examined mixed spinels can be divided into two categories, depending on the oxygen u parameter value. This is controlled by the average ionic radii (<rA> and <rB>) of the cations in the tetrahedral (A) and octahedral (B) sites, (u = (<rA> − <rB>)/[(1 +  )a] + 1.058/(1 +
)a] + 1.058/(1 +  )), and is modified upon cobalt spinel doping. The Cr, Mn and Fe-containing spinels with rM > rCo and the oxygen parameter u < 0.2625 (critical value for the centric oxygen atom location with equal distances from the A and B metals116), exhibit elongated octahedral and compressed tetrahedral sites. They are labelled hereafter as type-B (since the alien cation tends to occupy the octahedral positions), whereas the Co and Ni, Cu, Zn-doped spinels with rM ∼ rCo that exhibit u ∼ 0.2625, and the resultant slight deformation of the octahedra, are labelled hereafter as type-A (since the dopant cations tend also to be present in the tetrahedral positions). The B-type mixed spinels have much higher lattice parameter in comparison to the bare Co3O4, in a contrast to the A-type spinels (Table 1). The latter samples exhibit segregated minor NiO, CuO and ZnO phases, apart from the dominant spinel phase, which results from the destabilization of the oxygen 2p band center (lifting of its energy toward the Fermi level) upon doping.68
)), and is modified upon cobalt spinel doping. The Cr, Mn and Fe-containing spinels with rM > rCo and the oxygen parameter u < 0.2625 (critical value for the centric oxygen atom location with equal distances from the A and B metals116), exhibit elongated octahedral and compressed tetrahedral sites. They are labelled hereafter as type-B (since the alien cation tends to occupy the octahedral positions), whereas the Co and Ni, Cu, Zn-doped spinels with rM ∼ rCo that exhibit u ∼ 0.2625, and the resultant slight deformation of the octahedra, are labelled hereafter as type-A (since the dopant cations tend also to be present in the tetrahedral positions). The B-type mixed spinels have much higher lattice parameter in comparison to the bare Co3O4, in a contrast to the A-type spinels (Table 1). The latter samples exhibit segregated minor NiO, CuO and ZnO phases, apart from the dominant spinel phase, which results from the destabilization of the oxygen 2p band center (lifting of its energy toward the Fermi level) upon doping.68
For a complex structure of mixed spinel catalysts with the alien cations entering into both the octahedral and tetrahedral positions, we used the function (Φ), which gauges the modification of the Fermi energy position due to the incorporation of the 3d dopants, as a unifying descriptor of their average electronic properties related to redox behavior (Fig. 4).
 | ||
| Fig. 4 The work function of the synthetized mixed spinels along with the inserted dependence of the work function on their mean electronegativities. | ||
The measured Φ values of the mixed spinels plotted in the order of the increasing number of dn electrons of the alien cations (from d5 to d10) exhibit a maximum for Ni–Co with a shoulder for the Mn–Co and Fe–Co samples. The most pronounced lowering of the work function takes place for the Cr, Mn, Fe cations, with fewer dn electrons than Co3+ (n < 6), incorporated preferentially in the octahedral sites (B-type spinels). A global volcano shape of the work function changes for the materials doped with various 3d metals was previously reported,117 and it may be approximately rationalized in terms of the spinel mean electronegativity variation, calculated as described elsewhere.118 This is illustrated in the inset in Fig. 4, implying that the measured work function values assess quite well the collective chemical potential of the electrons (average electron electronegativity) in the mixed spinel catalyst, and in that way also indirectly assess their redox behavior. The deviations observed in the correlation plot for Cu–Co and Ni–Co may be associated with the partial segregation of those spinels. Nevertheless, the resultant changes in the Fermi level position of these spinel|oxide junctions as a whole are adequately reflected by the measured work function values.
3.2. Catalytic CO-PROX performance of the mixed spinels
H2-TPR measurements were conducted for the evaluation of the catalysts' reducibility (stability) in the temperature range of the CO-PROX reaction. Inspection of Fig. S4† shows that the reduction onset (measured by T5%) for the B-type spinels starts at T > 300 °C, i.e., above the temperature window of the CO-PROX reaction (marked in green). However, for the A-type spinels (except Co3O4) the temperature of the incipient H2 reduction slightly overlaps with that of the CO-PROX window. These results show that the B-type spinels (Cr–Co, Mn–Co and Fe–Co) are more redox-stable under the strongly reductive conditions in the temperature window of the CO-PROX reaction, whereas in the case of the A-type spinels (Ni–Co, Cu–Co and Zn–Co) some reduction may be expected at the late stages of the reaction. The H2-TPR results referring to extended bulk reduction do not preclude, however, the participation of the surface oxygen (Osurf2−) in the reversible redox processes constituting the CO-PROX reaction according to the Mars–van Krevelen mechanism.The catalytic performance of the synthesized mixed spinels was next examined in the CO-PROX reaction, complemented by the reference sole CO and sole H2 oxidation under the same conditions. The obtained results shown in Fig. 5 reveal the pronounced effects of the dopant nature and the reaction mixture (H2 + CO and CO or H2 alone) on the course of the CO and H2 oxidation. Indeed, in the standard PROX process conditions, the CO conversion profiles (Fig. 5a1) are distinctly shifted toward higher temperatures, and their spread is narrowed (ΔT = 54 °C) in comparison to the conversion profiles of the sole CO oxidation (ΔT = 80 °C) shown in Fig. 5a2; notably, they also appear in a different sequence. Analogous, yet more pronounced changes can be distinguished in the case of H2 conversion (Fig. 5b1 and b2). The temperature spread of the corresponding profiles decreases from ΔT = 80 °C (oxidation of H2 alone) down to ΔT = 32 °C (H2 conversion in the presence of CO), and the shift toward the higher temperatures is significantly higher than that observed during the CO oxidation. The sequence of the profiles also changes, with the most spectacular behavior observed for the Fe–Co catalyst, which is the most active in the sole H2 oxidation, and the least active in the presence of CO. These effects are concisely illustrated in the corresponding parity plots (Fig. S5a and b†), which show that in the case Fe–Co, Ni–Co and Co3O4, the discrepancy in the CO oxidation results mainly from the temperature shift of the corresponding conversion profiles, whereas for the H2 oxidation it results from lowering of the overall catalyst activity.
 | ||
| Fig. 5 Conversion profiles of CO (a1) and H2 (b1) oxidation in the PROX reaction over the mixed spinel catalysts along with the conversion profiles of single CO (a2) and single H2 (b2) oxidation. | ||
Noting the significant differences in the specific surface area of the investigated catalysts (Table S1†), to evaluate the specific effect of the dopant nature on the activity, the conversion curves (Fig. 5) were translated into the corresponding specific oxidation activities (conversion rates) and the results are shown in Fig. 6.
 | ||
| Fig. 6 The conversion rates of CO (a) and H2 (b) oxidation along with the O2 conversion rate in the PROX reaction (c). | ||
Analysis of the sequence of the conversion rate profiles revealed that the replacement of the cobalt cations in both tetra- and octahedral positions influences the specific activities of the catalysts in the CO and H2 oxidation. With the apparent exceptions of Fe and Zn, the reactivity sequence in both oxidation processes is roughly preserved. For the CO oxidation, the A-type (Cu–Co and Ni–Co) catalysts exhibit higher activity in comparison to the B-type catalysts (Cr–Co, Mn–Co, Fe–Co). In the case of H2 oxidation, this tendency is partially smeared out (Fig. 6b). The associated profiles of the O2 conversion rates (Fig. 6c) exhibit more similarity to the CO than the H2 conversion, in accordance with the catalysts' selectivities.
In order to evaluate the stability of the catalysts in the CO-PROX reaction, four consecutive runs were carried out for the parent Co3O4. The results shown in Fig. S6† reveal negligible deviations in the consecutive catalytic activities, implying that the catalyst surface remains effectively restored after each cycle. From the QMS profiles and the parity analysis of the CO versus CO2 evolution with the reaction temperature, we recognized that only slight CO methanation takes place at the highest temperatures, not exceeding 6–10% (see Fig. S7†). This finding remains in an accordance with the previous studies on the dry CO-PROX reaction over cobalt spinel catalysts.37,119
The selectivity variation with the temperature is shown in Fig. 7 for both types of spinel catalysts.
 | ||
| Fig. 7 The selectivity variation as a function of temperature for the B-type (a) and A-type mixed spinels (b). | ||
Inspection of the results indicates that the selectivity curves pass through broad maxima in the temperature range 150–200 °C, except for the Zn–Co sample, for which the selectivity maximum is significantly flattened and moved toward high temperatures (Fig. 7). Doping of the cobalt spinel results in the lowering of the selectivity in the low temperatures, but above 200 °C, the Mn–Co and then Fe–Co catalysts become the most selective. The superior initial selectivity observed for the Co3O4 catalyst results from the delay of the onset of H2 oxidation with respect to CO oxidation in this temperature range (see Fig. 5a1 and b1). The overall decrease in the selectivity occurs earlier for the A-type spinels (T > 180 °C) than for the B-type spinels (T > 200 °C), and is associated with enhanced hydrogen oxidation by the Osurf2− species, and then with slowly evolving CO methanation (see Fig. S7†). The latter does not exceed 10% of the full CO conversion to CO2. The intriguing opposite behavior of the selectivity that increases with the temperature found for the Zn–Co catalyst suggests that early surface hydroxylation and higher temperature methanation may be responsible, as suggested by low initial selectivity and the largest deviation between the CO and CO2 parity among all the investigated catalyst (see Fig. S7†).
The influence of the dopants on the H2 and CO oxidation activity and selectivity exhibited distinct regular trends when correlated with the work function values of the investigated spinels (Fig. 8) for temperatures selected from the range where carbonate contamination is ceasing. The structural discrepancies between the A- and B-type spinels appear to be reflected in how their activity and selectivity depend on the work function variation. As can be seen in Fig. 8a and b, the H2 and CO oxidation activities (conversion rates) are well correlated with the work function of the A- and B-type catalysts, respectively. The decrease in Φ has a stronger impact on the H2 oxidation in the case of the A-type spinels.
 | ||
| Fig. 8 Variation in the PROX H2 oxidation activity (a), CO oxidation activity (b) and selectivity (c) plotted against the work function of the spinel catalysts. | ||
The response of the CO oxidation activity to the varying Φ tends to deviate from linearity, achieving a minimum for Mn and a maximum for Co below 210 °C, and shift to Fe and Cu, respectively, above this temperature. Interestingly, similar work function values translate into dramatically different activities for the Cu and Mn catalyst of different u values. Thus, in the presented plots, the distinct generic catalytic behaviors of the A- and B-type spinels are also revealed. The exception is provided by the Zn–Co sample, exhibiting higher H2 than CO oxidation ability, which may be related to the non-redox character of the Zn2+ cations and the enhanced basicity, caused by the lifting of the oxygen 2p band center above the metal 3d center (see below). In contrast to the catalyst activity, the selectivity increases with the increasing work function of the A- and B-type catalysts including Zn–Co (Fig. 8c). For the A-type spinels, there is a rapid drop in the selectivity on passing from Cu into Zn-doped catalysts, resulting from the reversal of the H2 and CO activities in comparison to the other spinels. The latter contrasts with the highest selectivity of the Mn–Co catalyst, despite nearly the same value of the work function as that of Zn–Co. Thus, localization of the dopant in the tetrahedral or octahedral positions, and its redox (Mn) or non-redox (Zn) character, as well as the u-value, play an important role in the tradeoff between CO and H2 for the reactive oxygen species, which is discussed below in more detail.
The catalytic activity and selectivity patterns were then analyzed in terms of the intrinsic electronic structure of the related spinel catalysts. Due to the significant segregation of CuO (17.7%), the catalytic behavior of the Cu–Co sample cannot be assigned entirely to the intrinsic electronic properties of the spinel moiety. Therefore, the performance of this catalyst, though indicated on the proposed graphs, was excluded from establishing structure-reactivity correlations. It should be noted, however, that it does not preclude the correlation of its activity/selectivity with the work function (see Fig. 8) because the latter being an experimentally determined parameter, gauges the average electronic properties of the whole CuO|Cu0.92Co2.08O4 heterojunction.
Taking into account that in the CO and H2 oxidation reactions, apart from the Osurf2− anions (MvK mechanism), Oads intermediates (LH mechanism) may also be involved in the CO-PROX reaction,120,121 we examined the influence of the oxygen 2p and metal 3d band centers on the activity and selectivity of the investigated mixed spinels. This attempt was inspired by previous literature reports about the role of this descriptor in the energetics of the oxygen vacancy formation in oxide materials,122 and in oxidation reactions over spinel catalysts.44 This parameter can further be tuned by taking into account the difference between the average electronegativity of the metals constituting the particular mixed spinel and oxygen, Δ|<χM> − χO| = Δχ, as discussed elsewhere.122 The latter term refers to the explicit inclusion of the effect of charge transfer between the metal and oxygen atoms. High values of Δχ enhance the energy of the oxygen vacancy formation since for the oxygen release the metal-to-oxygen charge transfer has to be reversed. As a result, by regression analysis, the following form of a heuristic descriptor, EO2p + kΔχ with k ∼ 1.9, was proposed. In this formula, the Pauling electronegativity values were translated into the energy eV units using the simple relation χ(eV) = 2.976χ(Pauling unit) + 0.615.123 The (EO2p + kΔχ) descriptor was found to be suitable for analyzing the selectivity of the catalysts.
For elucidation of the catalyst activity in the CO and H2 oxidation during the PROX process, we found that the difference in the energy of the metal 3d band (EM3d) and the oxygen 2p band (EO2p) centers, ΔEM–O = (EM3d − EO2p), proposed by Sun et al.124 may be used for the establishment of the volcano-type relationships between the CO and H2 conversion rates (rCO and rH2) and the ΔEM–O values (Fig. 9). The corresponding EO2p and M3d values used for the construction of this plot have been calculated previously, elsewhere,122 and are listed in Table S4,† along with the Pauling electronegativities for convenience.
 | ||
| Fig. 9 Conversion rate of H2 (a) and CO oxidation (b) during the CO-PROX reaction over the mixed spinel catalysts plotted against the ΔEM–O parameter. | ||
Generally, both the CO and H2 oxidation rates show clear maxima for the most active Co3O4 spinel, yet the shapes of the resultant volcano curves are distinctly asymmetric. For the B-type spinels, the dependence of rCO and rH2 on ΔEM–O exhibited a certain similarity, in contrast to the A-type spinels, where much larger discrepancies in both oxidation rates can be observed. The observed volcano-shapes for CO and H2 revealed that the oxidation properties of the mixed spinels vary in a non-monotonous way with the relative position of the 3d band centers with respect to the O2p center, providing useful guidelines for the optimization of the spinel structure toward the highest activity. The catalytic performance of the Cu–Co sample, for the already mentioned sizable segregation, does not follow this general trend. The reason for such odd behavior may be related to the high intrinsic activity of the segregated CuO for the CO and H2 oxidation reported previously for bulk and supported copper oxide.125–127
As already stated, the catalysts' selectivities, tend to be correlated with the heuristic (EO2p + kΔχ) parameter, as illustrated in Fig. 10.
 | ||
| Fig. 10 Plot of catalyst selectivity versus the (EO2p + kΔχ) parameter for the investigated mixed spinels. The points associated with the segregated Cu–Co sample are indicated by the red open circle. | ||
The observed general trend implies that the lifting of the oxygen 2p-band center toward the Fermi level was induced by doping and favors the hydrogen oxidation over the carbon monoxide, accounting for the enhancement of the selectivity for the B-type spinels and the decrease in the selectivity in the case of the A-type mixed spinels (above 190–200 °C). The inclusion of the kΔχ term has a meaningful effect on the extent of the correlation, mitigating the scattering of the points in Fig. 10. As a result, the spinel electronic structure, epitomized by the position of the metal 3d and oxygen 2p band centers, controls its activity in CO and H2 oxidation in a volcano-type fashion, whereas the selectivity appears to be more sensitive to the position of the O2p center with respect to the Fermi level, and the M–O bond ionicity in a meaningful fashion. The extent of the correlation increases with the increasing temperature, in accordance with the enhancement of the role of the electroprotic H2 activation on the catalyst surface (H2 + Osurf2− → OHsurf−) in controlling the catalyst selectivity. The unique behavior of the Zn–Co spinel results from the lifting of the O2p center above the 3d band center of the octahedral redox Co3+ cations,68 in contrast to the remaining mixed spinels (Table S4†). This favors the formation of oxygen vacancies in the hypoxic conditions, and also CO methanation (see Fig. S7†). Alternatively, oxidation of H2 may occur via the interaction with Oads intermediates produced directly upon the dissociation of the O2 adspecies. This channel dominates at low temperatures (T < 190–200 °C), and is favored by the high position of the metal 3d band center (low work function values), accounting for the higher selectivity of the A-type spinels in comparison to the B-type ones in this temperature region of the CO-PROX reaction. Indeed, the 3d band center lift favors the activation of dioxygen into superoxo and peroxo intermediates via interfacial electron transfer, which has been previously elucidated by us via comprehensive DFT modeling for the (100) and (111) surfaces of Co3O4.45,128 Noteworthily, there is also a dramatically different impact of the heteroatom nature and its localization on the specific molecular aspects of O2, CO and H2 activation, manifested by Zn vs. Mn dopants in the most spectacular way, which is currently a subject of further research, along with isotopic CO-PROX and sole H2 and CO oxidation using 18O2 as an oxidant. Preliminary isotopic results strongly support the operation of a mixed Mars–van Krevelen and Langmuir–Hinshelwood mechanism in the preferential oxidation of CO in the H2-abundant conditions, proposed in this study for the interpretation of the CO-PROX sensitivity to the electronic structure of the mixed spinel catalysts.
Conclusions
A series of cobalt spinel catalysts doped with d-metals (Cr, Mn, Fe, Ni, Cu, Zn) was synthesized and thoroughly characterized by XRD, RS, IR, SEM, TEM, XRF, XPS techniques. Depending on the u parameter value (oxygen centricity), two types of mixed spinel catalyst with distinctly different catalytic behaviors in the CO-PROX reaction can be distinguished. The A-type spinels (Co, Ni, Cu) feature u < 0.2625 and higher activity in CO and H2 oxidation in comparison to the B-type spinels (Cr, Fe, Mn) with u ∼ 0.2625. A selectivity switch around T ∼ 180 °C was observed, with the A-type spinels being more selective at the lower temperatures and the B-type spinels at the higher temperatures. The unique CO-PROX performance of the Zn–Co spinel results from the lifting of the O2p center above the 3d band center of the octahedral redox Co3+ cations, in contrast to the remaining spinels. The work function of the mixed spinel was found to be a suitable concise parameter in accounting for the catalyst activity and selectivity, nicely corroborating the proposed categorization of the spinel catalysts into A- and B-types. Heuristic descriptors, EO2p + kΔ|<χM> − χO| and ΔEM–O = (EM3d − EO2p), based on the position of the oxygen 2p and the metal 3d band centers allow for the simple rationalization of the selectivity and activity of the mixed spinel catalysts in terms of their intrinsic electronic properties.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
Authors would like to acknowledge the funding awarded by the Polish National Science Center (decision number 2017/27/B/ST4/01155). Camillo Hudy was partially supported by the EU Project POWR.03.02.00-00-I004/16.Notes and references
- T. V. Choudhary and D. W. Goodman, Catal. Today, 2003, 44, 224 Search PubMed.
- E. D. Park, D. Lee and H. C. Lee, Catal. Today, 2009, 139, 280–290 CrossRef CAS.
- H. Wang, H. Zhu, Z. Qin, G. Wang, F. Liang and J. Wang, Catal. Commun., 2008, 9, 1487–1492 CrossRef CAS.
- J. Saavedra, T. Whittaker, Z. Chen, C. J. Pursell, R. M. Rioux and B. D. Chandler, Nat. Chem., 2016, 8, 584–589 CrossRef CAS PubMed.
- G. Landi, A. Di Benedetto and L. Lisi, Appl. Sci., 2018, 8, 13–32 Search PubMed.
- B. Viswanathan, Catal. Rev.: Sci. Eng., 1992, 34, 337–354 CrossRef CAS.
- N. Bion, F. Epron, M. Moreno, F. Mariño and D. Duprez, Top. Catal., 2008, 51, 76–88 CrossRef CAS.
- P. Jing, X. Gong, B. Liu and J. Zhang, Catal. Sci. Technol., 2020, 10, 919–934 RSC.
- A. Martinez-Arias, D. Gamarra, M. Fernandez-Garcia, A. Hornes, P. Bera, Z. Koppany and Z. Schay, Catal. Today, 2009, 143, 211–217 CrossRef CAS.
- A. Davó-Quiñonero, E. Bailon-Garcia, S. López-Rodríguez, D. Lozano, M. Garcia-Melchor, F. C. Herrera, E. Pellegrin, C. Escudero and A. Bueno-López, ACS Catal., 2020, 10, 6532–6545 CrossRef.
- M. Navlani-García, M. Martis, D. Lozano-Castelló, D. Cazorla-Amorós, K. Mori and H. Yamashita, Catal. Sci. Technol., 2015, 5, 364–371 RSC.
- A. Manasilp and E. Gulari, Appl. Catal., B, 2002, 37, 17–25 CrossRef CAS.
- A. Wootsch, C. Descorme and D. Duprez, J. Catal., 2004, 225, 259–266 CrossRef CAS.
- G. K. Bethke and H. H. Kung, Appl. Catal., A, 2000, 195, 43–53 CrossRef.
- S. Chayaporn, C. Thunyaratchatanon and A. Luengnaruemitchai, Res. Chem. Intermed., 2020, 46, 4173–4192 CrossRef CAS.
- Y. F. Han, M. J. Kahlich, M. Kinne and R. J. Behm, Phys. Chem. Chem. Phys., 2002, 4, 389–397 RSC.
- H. Wakita, Y. Kani, K. Ukai, T. Tomizawa, T. Takeguchi and W. Ueda, Appl. Catal., A, 2005, 283, 53–61 CrossRef CAS.
- Z. Qu, S. Zhou, W. Wu, C. Li and X. Bao, Catal. Lett., 2005, 101, 21–26 CrossRef CAS.
- S. Dey and G. C. Dhal, Mater. Today Chem., 2019, 14, 100180 CrossRef CAS.
- H. F. Wang, R. Kavanagh, Y. Y. L. Guo, Y. Y. L. Guo, G. Lu and P. Hu, J. Catal., 2012, 296, 110–119 CrossRef CAS.
- Z. Li, H. Wang, X. Wu, Q. Ye, X. Xu, B. Li and F. Wang, Appl. Surf. Sci., 2017, 403, 335–341 CrossRef CAS.
- S. Royer and D. Duprez, ChemCatChem, 2011, 3, 24–65 CrossRef CAS.
- Z. Zhao, M. M. Yung and U. S. Ozkan, Catal. Commun., 2008, 9, 1465–1471 CrossRef CAS.
- F. Mariño, C. Descorme and D. Duprez, Appl. Catal., B, 2005, 58, 175–183 CrossRef.
- G. Avgouropoulos, T. Ioannides, H. K. Matralis, J. Batista and S. Hocevar, Catal. Lett., 2001, 73, 33–40 CrossRef CAS.
- A. Martínez-Arias, M. Fernández-García, J. Soria and J. C. Conesa, J. Catal., 1999, 182, 367–377 CrossRef.
- L. Zhong, M. Barreau, V. Caps, V. Papaefthimiou, M. Haevecker, D. Teschner, W. Baaziz, E. Borfecchia, L. Braglia and S. Zafeiratos, ACS Catal., 2021, 11, 5369–5385 CrossRef CAS.
- S. S. Maluf and E. M. Assaf, Catal. Commun., 2011, 12, 703–706 CrossRef CAS.
- P. V. Gosavi and R. B. Biniwale, Int. J. Hydrogen Energy, 2012, 37, 3958–3963 CrossRef CAS.
- C. A. Chagas, E. F. De Souza, M. C. N. A. De Carvalho, R. L. Martins and M. Schmal, Appl. Catal., A, 2016, 519, 139–145 CrossRef CAS.
- T. Valdés-Solís, I. López and G. Marbán, Int. J. Hydrogen Energy, 2010, 35, 1879–1887 CrossRef.
- L. Lukashuk, K. Föttinger, E. Kolar, C. Rameshan, D. Teschner, M. Hävecker, A. Knop-Gericke, N. Yigit, H. Li, E. McDermott, M. Stöger-Pollach and G. Rupprechter, J. Catal., 2016, 344, 1–15 CrossRef CAS.
- F. Zasada, J. Janas, W. Piskorz, M. Gorczyńska and Z. Sojka, ACS Catal., 2017, 7, 2853–2867 CrossRef CAS.
- M. P. Yeste, H. Vidal, A. L. García-Cabeza, J. C. Hernández-Garrido, F. M. Guerra, G. A. Cifredo, J. M. González-Leal and J. M. Gatica, Appl. Catal., A, 2018, 552, 58–69 CrossRef CAS.
- P. T. A. Santos, H. L. Lira, L. Gama, F. Argolo, H. M. C. Andrade and A. C. F. M. Costa, Mater. Sci. Forum, 2010, 660, 771–776 Search PubMed.
- A. Elmhamdi, L. Pascual, K. Nahdi and A. Martínez-Arias, Appl. Catal., B, 2017, 217, 1–11 CrossRef CAS.
- T. M. Nyathi, N. Fischer, A. P. E. York and M. Claeys, ACS Catal., 2020, 10, 11892–11911 CrossRef CAS.
- K. Omata, T. Takada, S. Kasahara and M. Yamada, Appl. Catal., A, 1996, 146, 255–267 CrossRef CAS.
- Q. Guo and Y. Liu, Appl. Catal., B, 2008, 82, 19–26 CrossRef CAS.
- M. Jin, Z. Li, W. Piao, J. Chen, L. Y. Jin and J. M. Kim, Catal. Surv. Asia, 2017, 21, 45–52 CrossRef CAS.
- Z. Zhao, X. Lin, R. Jin, Y. Dai and G. Wang, Catal. Commun., 2011, 12, 1448–1451 CrossRef CAS.
- G. Grzybek, K. Ciura, J. Gryboś, P. Indyka, A. Davó-Quiñonero, D. Lozano-Castelló, A. Bueno-Lopez, A. Kotarba and Z. Sojka, J. Phys. Chem. C, 2019, 123, 20221–20232 CrossRef CAS.
- W. Shen, Res. Chem. Intermed., 2021, 47, 195–209 CrossRef CAS.
- T. Wang, Y. Sun, Y. Zhou, S. Sun, X. M. Hu, S. Xi, Y. Du, Y. Yang and Z. J. Xu, ACS Catal., 2018, 8, 8568–8577 CrossRef CAS.
- F. Zasada, J. Grybos, E. Budiyanto, J. Janas and Z. Sojka, J. Catal., 2019, 371, 224–235 CrossRef CAS.
- L. Zhong, T. Kropp, W. Baaziz, O. Ersen, D. Teschner, R. Schlogl, M. Mavrikakis and S. Zafeiratos, ACS Catal., 2019, 9, 8325–8336 CrossRef CAS.
- J. Jansson, A. E. C. Palmqvist, E. Fridell, M. Skoglundh, L. Osterlund, P. Thorm and V. Langer, J. Catal., 2002, 397, 387–397 CrossRef.
- P. T. A. Santos, A. C. F. M. Costa, R. H. G. A. Kiminami, H. M. C. Andrade, H. L. Lira and L. Gama, J. Alloys Compd., 2009, 483, 399–401 CrossRef CAS.
- A. Varma, A. S. Mukasyan, A. S. Rogachev and K. V. Manukyan, Chem. Rev., 2016, 116, 14493–14586 CrossRef CAS PubMed.
- U. Zavyalova, F. Girgsdies, O. Korup, R. Horn and R. Schlo, J. Phys. Chem. C, 2009, 113, 17493–17501 CrossRef CAS.
- F. Deganello and A. K. Tyagi, Prog. Cryst. Growth Charact. Mater., 2018, 64, 23–61 CrossRef CAS.
- A. Choya, B. Rivas, J. I. Gutierrez-Ortiz and R. Lopez-Fonseca, Catalysts, 2022, 12, 87 CrossRef CAS.
- S. Wójcik, G. Ercolino, M. Gajewska, C. W. M. Quintero, S. Specchia and A. Kotarba, Chem. Eng. J., 2019, 377, 120088 CrossRef.
- U. Zavyalova, P. Scholz and B. Ondruschka, Appl. Catal., A, 2007, 323, 226–233 CrossRef CAS.
- S. L. González-Cortés and F. E. Imbert, Appl. Catal., A, 2013, 452, 117–131 CrossRef.
- S. Specchia, E. Finocchio, G. Busca and V. Specchia, in Handbook of Combustion, Wiley-VCH, 1st edn, 2010 Search PubMed.
- T. Baidya, T. Murayama, P. Bera, O. V. Safonova, P. Steiger, N. K. Katiyar, K. Biswas and M. Haruta, J. Phys. Chem. C, 2017, 121, 15256–15265 CrossRef CAS.
- CasaXPS Processing Software, http://www.casaxps.com/.
- J. Zieliński, React. Kinet. Catal. Lett., 1981, 17, 69–75 CrossRef.
- P. S. Barbato, S. Colussi, A. Di Benedetto, G. Landi, L. Lisi, J. Llorca and A. Trovarelli, J. Phys. Chem. C, 2016, 120, 13039–13048 CrossRef CAS.
- E. J. Verwey and P. W. Haayman, J. Chem. Phys., 1947, 15, 181–187 CrossRef CAS.
- B. Lavina, G. Salviulo and D. Della Giusta, Phys. Chem. Miner., 2002, 29, 10–18 CrossRef CAS.
- C. Yang and A. Grimaud, Catalysts, 2017, 7, 149 CrossRef.
- F. Zasada, J. Gryboś, P. Indyka, W. Piskorz, J. Kaczmarczyk and Z. Sojka, J. Phys. Chem. C, 2014, 118, 19085–19097 CrossRef CAS.
- D. Kaczorowski, E. Murashova, Z. Kurenbaeva and A. Gribanov, J. Alloys Compd., 2019, 802, 437–444 CrossRef CAS.
- S. R. Gawali, A. C. Gandhi, S. S. Gaikwad, J. Pant, T. S. Chan, C. L. Cheng, Y. R. Ma and S. Y. Wu, Sci. Rep., 2018, 8, 1–12 CAS.
- K. Narita, R. Yuge, S. Kuroshima, M. Tabuchi, K. Doumae, H. Shibuya, N. Tamura and M. Tsuji, Electrochim. Acta, 2018, 290, 577–585 CrossRef CAS.
- Y. Duan, S. Sun, Y. Sun, S. Xi, X. Chi, Q. Zhang, X. Ren, J. Wang, S. Jun, H. Ong, Y. Du, L. Gu, A. Grimaud and Z. J. Xu, Adv. Mater., 2019, 1807898, 2–9 Search PubMed.
- M. Bouchard and A. Gambardella, J. Raman Spectrosc., 2010, 41, 1477–1485 CrossRef.
- S. W. da Silva, F. Nakagomi, A. Franco Jr, V. K. Garg, A. C. Oliveira and P. C. Morais, J. Nanopart. Res., 2012, 14, 798 CrossRef.
- C. M. Julien and M. Massot, Mater. Sci. Eng., B, 2003, 100, 69–78 CrossRef.
- T. Yu, Z. X. Shen, Y. Shi and J. Ding, J. Phys.: Condens. Matter, 2002, 14, 37 CrossRef.
- M. A. Laguna-Bercero, M. L. Sanjuán and R. I. Merino, J. Phys.: Condens. Matter, 2007, 19, 18 CrossRef PubMed.
- J. Preudhomme and P. Tarte, Spectrochim. Acta, Part A, 1971, 27, 1817–1835 CrossRef CAS.
- J. Preudhomme and P. Tarte, Spectrochim. Acta, Part A, 1972, 28, 69–79 CrossRef CAS.
- V. D'Ippolito, G. B. Andreozzi, D. Bersani and P. P. Lottici, J. Raman Spectrosc., 2015, 46, 1255–1264 CrossRef.
- D. Lenaz and V. Lughi, Phys. Chem. Miner., 2013, 40, 491–498 CrossRef CAS.
- S. Y. Chazhengina, Z. P. Rybnikova and S. A. Svetov, Geol. Ore Deposits, 2016, 58, 628–635 CrossRef.
- F. Giovannelli, V. Marsteau, M. Zaghrioui, C. Autret and F. Delorme, Adv. Powder Technol., 2017, 28, 1325–1331 CrossRef CAS.
- F. Kovanda, T. Rojka, J. Dobešová, V. Machovič, P. Bezdička, L. Obalová, K. Jirátová and T. Grygar, J. Solid State Chem., 2006, 179, 812–823 CrossRef CAS.
- A. Gaur, M. A. Mohiddon and V. M. Sglavo, J. Eur. Ceram. Soc., 2018, 38, 4543–4552 CrossRef CAS.
- F. Arena, G. Trunfio, J. Negro, B. Fazio and L. Spadaro, Chem. Mater., 2007, 19, 2269–2276 CrossRef CAS.
- T. L. Phan, N. X. Nghia and S. C. Yu, Solid State Commun., 2012, 152, 2087–2091 CrossRef CAS.
- C. W. Tang, C. Bin Wang and S. H. Chien, Thermochim. Acta, 2008, 473, 68–73 CrossRef CAS.
- Z. Chen, C. X. Kronawitter and B. E. Koel, Phys. Chem. Chem. Phys., 2015, 17, 29387–29393 RSC.
- J. Wang, R. Gao, D. Zhou, Z. Chen, Z. Wu, G. Schumacher, Z. Hu and X. Liu, ACS Catal., 2017, 7, 6533–6541 CrossRef CAS.
- Z. Y. Tian, P. H. T. Ngamou, V. Vannier, K. Kohse-Höinghaus and N. Bahlawane, Appl. Catal., B, 2012, 117–118, 125–134 CrossRef CAS.
- B. C. Babu, G. Wang, B. Yan, Q. Yang and A. P. Baker, Ceram. Int., 2018, 44, 938–946 CrossRef CAS.
- H. W. Nesbitt and D. Banerjee, Am. Mineral., 1998, 83, 305–315 CrossRef CAS.
- P. H. T. Ngamou, M. E. Ivanova, C. Herwartz, N. Lühmann, A. Besmehn, W. A. Meulenberg, J. Mayer and O. Guillon, RSC Adv., 2015, 5, 82717–82725 RSC.
- Y. Wu and R. Holze, Electrochemical Energy Conversion and Storage, John Wiley & Sons, 2021 Search PubMed.
- O. A. Bulavchenko, T. N. Afonasenko, A. V. Ivanchikova, V. Y. Murzin, A. M. Kremneva, A. A. Saraev, V. V. Kaichev and S. V. Tsybulya, Inorg. Chem., 2021, 60, 16518–16528 CrossRef CAS PubMed.
- B. Djurfors, J. N. Broughton, M. J. Brett and D. G. Ivey, Acta Mater., 2005, 53, 957–965 CrossRef CAS.
- J. F. Moulder, Handbook of X-ray Photoelectron spectroscopy: a reference book of standard scpectra for identification and interpretation of xps data, Physical Electronics Division, 1992 Search PubMed.
- H. Chen and J. Wang, Chemosphere, 2019, 234, 14–24 CrossRef CAS PubMed.
- M. Fantauzzi, F. Secci, M. Sanna Angotzi, C. Passiu, C. Cannas and A. Rossi, RSC Adv., 2019, 9, 19171–19179 RSC.
- G. Maniak, P. Stelmachowski, J. J. Stanek, A. Kotarba and Z. Sojka, Catal. Commun., 2011, 15, 127–131 CrossRef CAS.
- M. Prabu, K. Ketpang and S. Shanmugam, Nanoscale, 2014, 6, 3173–3181 RSC.
- M. Cheng, H. Fan, Y. Song, Y. Cui and R. Wang, Dalton Trans., 2017, 46, 9201–9209 RSC.
- M. N. Iliev, P. Silwal, B. Loukya, R. Datta, D. H. Kim, N. D. Todorov, N. Pachauri and A. Gupta, J. Appl. Phys., 2013, 114, 3 CrossRef.
- A. Białas, K. Rugała, C. Czosnek, G. Mordarski and J. Gurgul, Catalysts, 2020, 10, 1–14 CrossRef.
- A. L. Rosa-Toro, R. Berenguer, C. Quijada, F. Montilla, E. Morallon and J. L. Vazquez, J. Phys. Chem. B, 2006, 110, 24021–24029 CrossRef PubMed.
- Z. Sojka and K. Klier, J. Electron Spectrosc. Relat. Phenom., 1992, 60, 155–174 CrossRef CAS.
- B. Chi, H. Lin and J. Li, Int. J. Hydrogen Energy, 2008, 33, 4763–4768 CrossRef CAS.
- F. Severino, J. L. Brito, J. Laine, J. L. G. Fierro and A. López Agudo, J. Catal., 1998, 177, 82–95 CrossRef CAS.
- A. V. Fetisova and M. V. Kuznetsov, J. Appl. Spectrosc., 2009, 76, 523–527 CrossRef.
- T. H. Dolla, D. G. Billing, C. Sheppard, A. Prinsloo, E. Carleschi, B. P. Doyle, K. Pruessner and P. Ndungu, RSC Adv., 2018, 8, 39837–39848 RSC.
- P. L. Meena, R. Kumar and K. Sreenivas, Int. J. Phys., Chem. Math. Sci., 2014, 3, 7–17 CAS.
- N. Bahlawane, P. H. T. Ngamou, V. Vannier, T. Kottke, J. Heberle and K. Kohse-Höinghaus, Phys. Chem. Chem. Phys., 2009, 11, 9224–9232 RSC.
- C. D. Spencer and D. Schroeer, Phys. Rev. B: Solid State, 1974, 9, 3658–3665 CrossRef CAS.
- M. Takahashi and M. E. Fine, J. Appl. Phys., 1972, 43, 4205–4216 CrossRef.
- P. A. Smith, C. D. Spencer and R. P. Stillwell, J. Phys. Chem. Solids, 1978, 39, 107–111 CrossRef CAS.
- D. Pyke, K. K. Mallick, R. Reynolds and A. K. Bhattacharya, J. Mater. Chem., 1998, 8, 1095–1098 RSC.
- B. Nandan, M. C. Bhatnagar and S. C. Kashyap, J. Phys. Chem. Solids, 2019, 129, 298–306 CrossRef CAS.
- S. Wójcik, G. Grzybek, P. Stelmachowski and Z. Sojka, Catalysts, 2020, 10, 41 CrossRef.
- M. Avezac and A. Zunger, Phys. Rev. Lett., 2010, 105, 11–14 Search PubMed.
- M. Yoshitake, J. Vac. Sci. Technol., A, 2014, 32, 061403 CrossRef.
- J. P. Jolivet, Metal Oxide Chemistry and Synthesis: From Solution to Solid State, John Wiley & Sons, 2000 Search PubMed.
- T. M. Nyathi, N. Fischer, A. P. E. York, D. J. Morgan, G. J. Hutchings, E. K. Gibson, P. P. Wells, C. R. A. Catlow and M. Claeys, ACS Catal., 2019, 9, 7166–7178 CrossRef CAS PubMed.
- S. A. Singh and G. Madras, Appl. Catal., A, 2015, 504, 463–475 CrossRef CAS.
- L. Lukashuk, N. Yigit, R. Rameshan, E. Kolar, D. Teschner, M. Hävecker, A. Knop-Gericke, R. Schlögl, K. Föttinger and G. Rupprechter, ACS Catal., 2018, 8, 8630–8641 CrossRef CAS PubMed.
- A. Derni, A. M. Holder, R. P. O'Hayre, C. B. Musgrave and V. Stevanovic, J. Phys. Chem. Lett., 2015, 6, 1948–1953 CrossRef PubMed.
- G. Campet, J. Portier and M. A. Subramanian, Mater. Lett., 2004, 58, 437–438 CrossRef CAS.
- Y. Sun, H. Liao, J. Wang, B. Chen, S. Sun, S. J. H. Ong, S. Xi, C. Diao, Y. Du, J. O. Wang, M. B. H. Breese, S. Li, H. Zhang and Z. J. Xu, Nat. Catal., 2020, 3, 554–563 CrossRef CAS.
- J.-H. Park, J. H. Cho, K. Cho, T. W. Lee, H. S. Han and C.-H. Shin, Korean J. Chem. Eng., 2012, 29, 1151–1157 CrossRef CAS.
- G. Avgouropoulos, T. Ioannides, H. K. Matralis, J. Batista and S. Hocevar, Chem. Eng. J., 2011, 176–177, 14–21 CrossRef CAS.
- Z. Liu, Z. Wu, X. Peng, A. Binder, S. Cha and S. Dai, J. Phys. Chem. C, 2014, 118, 27870–27877 CrossRef CAS.
- F. Zasada, W. Piskorz, J. Janas, J. Gryboś, P. Indyka and Z. Sojka, ACS Catal., 2015, 5, 6879–6892 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2cy00388k |
| This journal is © The Royal Society of Chemistry 2022 |