
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectroreduction of NO3− on tubular porous Ti electrodes†
Piotr M.
Krzywda
 ab,
Ainoa
Paradelo Rodríguez
a,
Lukas
Cino
a,
Nieck E.
Benes
b,
Bastian T.
Mei
a and
Guido
Mul
*a
ab,
Ainoa
Paradelo Rodríguez
a,
Lukas
Cino
a,
Nieck E.
Benes
b,
Bastian T.
Mei
a and
Guido
Mul
*a
aPhotocatalytic Synthesis Group, Faculty of Science & Technology of the University of Twente, PO Box 217, Enschede, The Netherlands. E-mail: g.mul@utwente.nl
bMembrane Science and Technology Cluster, Faculty of Science & Technology of the University of Twente, PO Box 217, Enschede, The Netherlands
First published on 4th April 2022
Abstract
Inefficient fertilizer use in agriculture causes nitrate runoff, polluting rivers and streams. This pollution can be mitigated by partially converting nitrate into ammonia – rebalancing the composition to ammonium nitrate, and allowing recycling of fertilizer. Here, we present efficient electrochemical conversion of nitrate (50 mM) to ammonia in acidic electrolyte using tubular porous Ti electrodes. A high faradaic efficiency (FE) of 58% and partial current density to ammonia of −33 mA cm−2 at −1 V vs. RHE were achieved in the absence of inert gas purge. Additionally, we reveal that hydroxylamine is formed, as well as NO and N2O by spontaneous decomposition of nitrite, as has been determined by EC-MS analysis. The effective increase in local mass transport by introducing a flow of inert gas exiting the wall of the hollow fiber electrode results in an unprecedently high partial current density to ammonia of ∼−75 mA cm−2, while maintaining a faradaic efficiency to ammonia of up to 45%. This concept facilitates nitrate conversion at high FE even at low concentrations, and holds promise for development to practical scale if electrochemical potential and exiting gas flow rate are well controlled.
Introduction
The efficiency of food production in the agricultural sector depends on the efficient utilization of nitrogen based fertilizers.1 The production of fertilizer includes the industrial synthesis of NH3 by the Haber–Bosch process, which largely contributes to the global carbon footprint.2 Alternative approaches which allow for a more environmentally friendly, green ammonia synthesis are explored,3 and electrochemical routes appear to be of particular interest for small-scale, renewable energy-based production facilities. Although N2 is the most attractive resource for electrochemical ammonia production due to its abundancy,4 the stability of the N2 molecule limits the electrochemical conversion rates currently achieved, if any.5 Waste streams containing NOx have recently been considered as alternative feedstock for green NH3 synthesis, such as nitric oxide6,7 and nitrate.8–10Focusing on nitrate, various sources are available11,12 and among others utilization of agricultural runoff might be of interest. Over-fertilization combined with low nitrogen utilization efficiencies in agriculture13 result in fertilizer release to the groundwater, mostly in the form of nitrates. Ammonium nitrate produced from collected (possibly concentrated) and partially reduced nitrate can thus be considered as recycled fertilizer.14 Alternatively, NO3− obtained from direct oxidation of nitrogen by an electrochemical15–18 or plasma (catalytic)19–23 approach might be available as resource for ammonia production. It has been proven that plasma is potentially viable for electricity-based HNO3 production,24 which can be consecutively converted to ammonia in one process.25,26 A decentralized, small-scale ammonium nitrate production unit for production of carbon-free, green fertilizer also appears feasible. Still, electroreduction of nitrate is complex,27 and suitable electrode materials and configurations for efficient electrochemical production of ammonia need to be developed. Moreover, research on the electroreduction of nitrate has been primarily performed for purification of drinking water, targeting selective reduction to nitrogen in neutral electrolyte conditions,28 even though the kinetics of the reaction are more favourable in either acidic or basic pH conditions.29
In general, Cu has been identified as a suitable electrode for nitrate reduction to ammonia,30 yet in acidic electrolyte electrode stability is insufficient (e.g. by dissolution of Cu in (diluted) nitric acid solution31). Titanium, which is available at low cost,32 has been reported as one of the elements offering high stability in various process conditions, and moreover allows operation in an extended electrochemical window due to poor activity for hydrogen evolution. Efficient removal of nitrate was reported in acidic conditions using a Ti cathode and a Pt anode,33 and preferential ammonia formation rather than N2 formation was reported.32 Recently, metallic Ti plates were extensively studied, and the best NH3 selectivity was observed at high nitrate concentration and in strongly acidic pH conditions, which result in high abundance of both, NO3− and H+ at the Ti surface.29 DFT calculations suggest that a combination of Ti and graphitic carbon nitride is potentially interesting for efficient formation of ammonia.34 Finally, TiO2 nanotubes with a high concentration of oxygen vacancies (TiO2−x) were reported to produce NH3 with 85% faradaic efficiency in neutral pH,35 further improved to 92% by additional functionalization with Pd.36 Still high faradaic efficiencies are only observed at low to moderate current densities.
Low current densities are most likely caused by mass transport limitations of the reactant, especially when nitrate is present in low concentration.37,38 A low concentration of nitrate close to the electrode surface results in preferential hydrogen production, and clearly high mass transport rates are required to improve process efficiency. Specific catalyst design,39 reactor design,40 increasing the NO3− concentration,29 or inducing efficient convection in the cell have been investigated to mitigate mass transfer limitations.
Herein, we report the use of Ti based hollow fiber electrodes for electrochemical nitrate reduction in acidic electrolyte, which were shown to enable efficient supply of reactant in aqueous solutions to the electrode surface when introducing inert, or reactant containing gas flow.6,41–43 We will compare a case of conventional mixing by gas introduction through a separate sparger, with conditions of efficient mixing near the solid–liquid interface by introducing an inert gas flow through the porous wall of the electrode. We will demonstrate that low concentrations of nitrate (50 mM KNO3) can be converted to ammonia at high partial current densities, and that the concept of gas flow induced mixing significantly improves nitrate conversion. We also reveal the formation of hydroxylamine, and provide unprecedented detail in the solution chemistry by explaining advanced EC-MS measurements.
Experimental
Electrode preparation and characterization
Ti hollow fibers were prepared by dry-wet spinning according to a previously reported method.44 In brief, Ti powder was mixed with polyethersulfone (PES) and N-methylpyrrolidone (NMP) to form a homogeneous suspension. The spinning mixture was pressed through a spinneret with water as a bore liquid. Fibers were thermally treated at 800 °C in order to remove the polymer and to form the metallic hollow fiber. Details on the suppliers of the various chemicals can be found in the ESI.†Hollow fiber electrode assemblies were prepared using silver epoxy glue to ensure electrical contact to a Swagelok stainless steel tube (Fig. S1†). The assembly was covered with two-compartment adhesive glue to prevent contact of silver or the stainless steel tube with the electrolyte. To ensure homogeneous distribution of gas through the fibers, the open-end of the fiber was also covered with adhesive. Electrodes were characterized using scanning electron microscopy (SEM) (JSM-6010LA, JEOL system) and X-ray diffraction (XRD, Bruker Phaser D2). For EC-MS experiments a Ti disc (∅ 5 mm) was polished, following the routine of cleaning provided by Pine Research for rotating disc electrodes.
Electrochemical measurements
All electrochemical measurements were carried out at room temperature using a BioLogic VSP potentiostat. A gas tight, H-type cell (a Nafion 117 membrane was used for compartment separation) was used for linear scan voltammetry and chronoamperometry experiments. A Pt mesh and Ag/AgCl (3 M NaCl, BASi) were used as counter and reference electrode, respectively. Measured potentials were corrected for the solution resistance (iR drop) and converted to potentials vs. RHE using:| ERHE = EAg/AgCl + 0.059pH + EAg/AgCl0 |
Stability measurements were performed for 4 h. The electrolyte was refreshed after 3 h if no-flow was used, and every 1 h for experiments performed in flow-through mode using an Ar flow of 20 ml min−1. The electrochemical surface area (ECSA) of the hollow fiber electrodes was measured before and after the stability test by the capacitance method. Cyclic voltammograms in the purely capacitive potential were measured at variable scan rates from 20 to 100 mV s−1 in Ar saturated 0.1 M KClO4. Product detection and quantification (for calibration curves, see Fig. S3–S5) is described in detail in the ESI.†
To further analyse the product composition EC-MS measurements were performed using a SpectroInlets (Copenhagen, Denmark) system, with He as carrier gas at a flow rate of 1 ml min−1. Chronoamperometry was performed at different potentials. Potentials were applied for 30 s. Afterwards the system was allowed to rest at OCV conditions in between potential pulses. A Ag/AgCl (sat. KCl, CH Instrument) and a Pt mesh were used as reference and counter electrode respectively. Counter and working compartments were separated by a borosilicate filter, which in combination with the long distance between the electrodes prevents product diffusion from one to the other electrode. The schematic representation of the cell design is shown in Fig. S2.† Decomposition experiments using the EC-MS set-up were performed without the electrode assembled. Here, the chip, integrated with the EC-MS inlet, was covered with the electrolyte and a solution of interest was dropped into the electrolyte to determine the decomposition products.
Results and discussion
Ti electrode characterization
Compared to widely employed flat electrodes, hollow fiber electrodes allow for gas supply through the porous structure of the wall into the electrolyte. The gas distribution is largely dependent on the porosity of the wall. SEM images (Fig. 1) of a representative Ti hollow fiber reveal the uniform pore distribution over the entire length of the fiber, which should lead to a homogeneous gas distribution (see also Fig. S6†). The XRD pattern shows that the hollow fiber is composed solely of metallic Ti.44 | ||
| Fig. 1 SEM images of a Ti hollow fiber electrode. (a) The cross section at different magnifications and (b) the surface of the fiber. (c) XRD pattern obtained from the Ti hollow fiber electrode. | ||
NO3− electroreduction on a Ti electrode
Linear scan voltammetry performed in acidic conditions in 0.1 M HClO4 at different nitrate concentrations is shown in Fig. 2a.
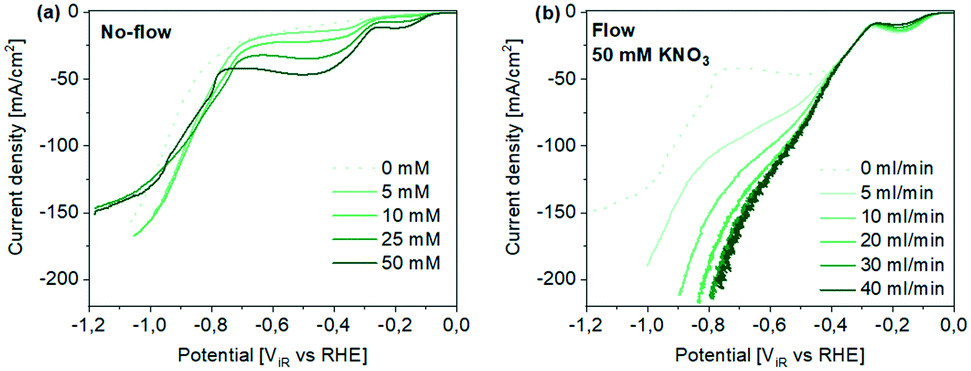 | ||
| Fig. 2 Linear scan voltammetry using Ti hollow fiber electrodes. (a) The influence of nitrate concentration (KNO3) in “no-flow” conditions and (b) the influence of Ar flow rate on current density at 50 mM KNO3. | ||
In contrast to the onset potential in neutral conditions, the onset potential for NO3− electroreduction is already observed at −0.05 V vs. RHE, and significantly higher current densities can be achieved (to compare see Fig. S8†), indicating faster kinetics of NO3− reduction in acidic conditions.29 With increasing KNO3 concentration, two plateau-like regions are observed before the hydrogen evolution occurs at potentials <−0.8 ViRvs. RHE (Fig. 2a). The first plateau region starting at ∼−0.1 ViRvs. RHE has previously been assigned to the formation of NO2,6 formed in a 1 e− transfer reaction from NO3− (see Table S1†). Afterwards in the potential range between −0.3 ViR and ∼−0.8 ViRvs. RHE, a second mass-transport limited region is observed, which becomes more obvious at increasing nitrate concentrations. Besides nitrate, we also suggest that proton transport becomes limiting at relatively high nitrate concentrations, considering that the reduction of NO3− to ammonia requires 9 H+ (see Table S1†). In fact it was previously reported that H+ transport limitations in NO3− electroreduction on a flat Ti electrode in acidic electrolyte can occur already at ∼−25 mA cm−2, in nitrate concentrations of 0.1 M.29 Presumably hydrogen evolution (HER) starts to compete at potentials <−0.8 V vs. RHE, as indicated by the current increase also observed in the absence of NO3−. Finally, linear sweep voltammetry also reveals the diffusion limitation of proton reduction at potentials below approx. −1 ViRvs. RHE.
Flow-through experiments using Ar as an inert sparging gas were subsequently performed at various flow rates and a constant concentration of nitrate (50 mM) (Fig. 2b). At moderate flow rates of 5 mL min−1, a significant increase in diffusion limited current is obtained in the potential range between −0.3 ViR and ∼−0.8 ViRvs. RHE as shown in Fig. 2b, while increasing the flow to 20 mL min−1 further enhances the current density, to unprecedented values for Ti hollow fibres, as high as 200 mA cm−1. Likely, higher flow rates induce additional (smaller) pores between the sintered Ti particles to open,6 inducing more efficient, homogeneous mixing at the solid–liquid interface. It should be mentioned that the extent of enhancement of current density by gas flow, is a function of the nitrate concentration. Specifically at lower NO3− concentrations, gas flow affects the system to a lower extent (see Fig. S9†). In blank experiments and at the lowest nitrate concentrations (1 mM KNO3) only the HER seems to be affected by introduction of a gas flow, as a result of the mitigation of proton transport limitations and likely improved hydrogen bubble release. Thus, the observed impact of gas flow through the porous wall of the hollow fiber confirms that transport of both, NO3− and H+ to the electrode surface is significantly improved by local mixing (see Fig. S10† for a schematic representation).
Generally, a stable current density was obtained throughout the measurement, independent of the applied potential (see Fig. 3a and S11†). An increase in current density was observed by increasing the Ar flow rate, which is in agreement with the LSV study. Only for measurements performed at a high flow rate, a slight decrease in current density with time was noticed, which can be assigned to the depletion of nitrate in the electrolyte. Reactant depletion is confirmed by NO3− conversion calculations reported in Fig. S12.† Moreover, to support the hypothesis that flow-through conditions facilitate effective mixing, chronoamperometry at −0.6 V vs. RHE was performed using vigorous magnetic stirring. The similarity in current density with “no-flow” and stirred conditions, and the significantly higher current densities obtained in purged conditions, are evidence for the highly effective mixing at the electrode–electrolyte interface by gas flow exiting the pores of the hollow fibre structure (Fig. S13†).
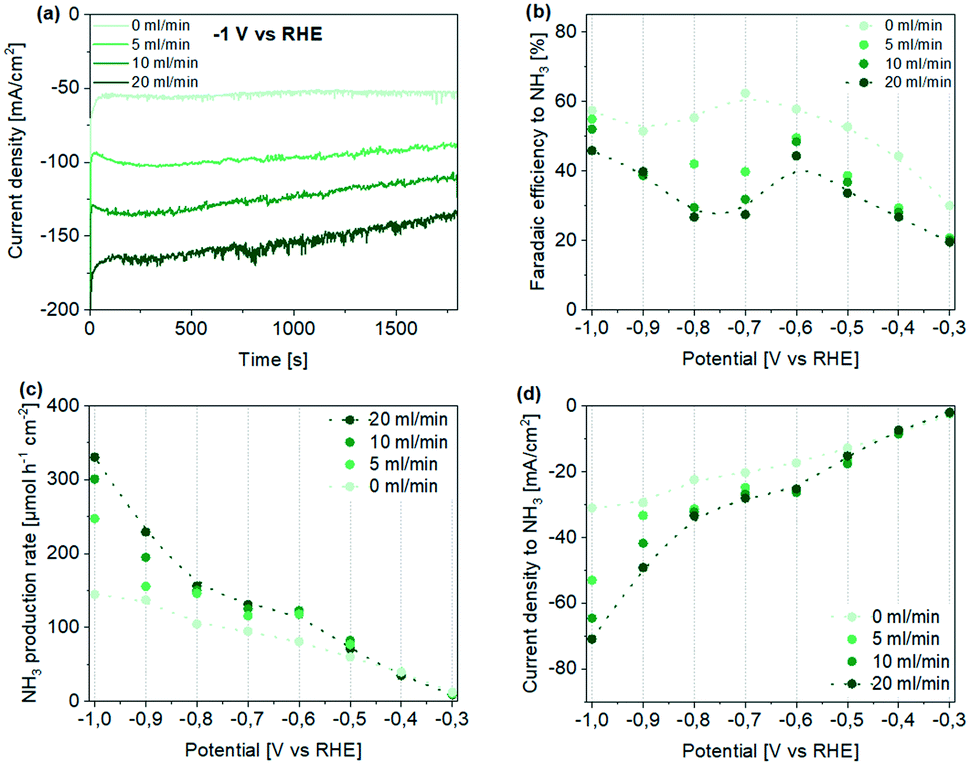 | ||
| Fig. 3 Performance of Ti hollow fiber electrodes for the electrochemical reduction of NO3− to NH3 in 0.1 M HClO4 using 50 mM KNO3. (a) Obtained current density profiles during chronoamperometry at −1 V vs. RHE at different Ar flow rates. (b–d) Potential- and Ar flow rate-dependent (b) faradaic efficiency, (c) production rate, and (d) partial current density to ammonia. Note that only ammonia in the catholyte was quantified here (see also Fig. S15 and S16† for measurements including ammonia quantified in the anolyte). Dotted lines are meant for guidance only. | ||
Quantification of the ammonia concentrations in the catholyte compartment revealed that the current efficiency to NH3 is generally increasing with more negative potential (for quantification of ammonia in the anolyte, which agrees with the trend in concentration obtained for the catholyte compartment, see ESI†). Yet, we like to emphasize two observations revealed in Fig. 3b. First, a decrease in faradaic efficiency is observed after maximizing in the potential range of ∼−0.6 to −0.7 V vs. RHE (Fig. 3b), in particular in flow-through conditions. Second, in the absence of flow, the highest FE of ∼65% was reached (at −0.7 V vs. RHE), while in flow-through conditions a lower FE of ∼50% was obtained (at −0.6 V vs. RHE), decreasing by approx. 20% in the range of −0.6 to ∼−0.75 V, to again increase at potentials <−0.8 V vs. RHE. The drop in FE is also associated with the shoulder in the current potential profile observed during LSV measurements in no flow-conditions (see Fig. 2b dotted line). We will provide an explanation for these observations in the following paragraph, providing information on trends in other products. Now we like to state that even though the FE in flow-through conditions is lower than in the absence of flow, the NH3 production rate, as well as partial current density to ammonia, are significantly larger, especially at more negative potentials, reaching production rates of 330 μmol h−1 cm−2 and partial current densities of −70 mA cm−2 respectively (Fig. 3c and d). To the best of our knowledge, these obtained faradaic efficiencies, as well as partial current densities to ammonia (based on geometrical surface area) are the highest reported for Ti electrodes used for the electroreduction of nitrate to ammonia. Moreover, in our study low nitrate concentrations (50 mM) are used, which generally result in even lower conversion efficiency.29
 | ||
| Fig. 4 Total faradaic efficiency of liquid products in no-flow and flow (20 ml min−1) configuration after chronoamperometry in 0.1 M HClO4 with 50 mM KNO3. See Fig. S17† for measurements at 5 and 10 ml min−1, which fit a trend in production composition from no-flow to a flow of 20 ml min−1. | ||
In flow conditions, the total FE of liquid phase products is dependent on potential (Fig. 4, right, compare to Fig. 3b). In the potential range between −0.3 and −0.6 V vs. RHE the ammonia FE increases, and the hydroxylamine FE is rather constant. The strongest attenuation of the FE is observed at −0.7 V and −0.8 V, and a significant reduction in FE for both hydroxylamine and NH3 is obtained. At −0.9 V and −1.0 V, the FE towards NH3 increases again, as accompanied by a slight increase in FE for hydroxylamine.
Moreover, in contrast to the outlined trend in the FE towards ammonia, in flow-through conditions we observe that the faradaic efficiencies for nitrite (NO2−) and hydroxylamine (NH2OH) are generally higher. We propose that the changes in selectivity in flow-through conditions are caused by local pH changes. As a general rule, although it might be catalyst dependent, selectivity to NO2– (ref. 33, 48 and 49) and NH2OH (ref. 50 and 51) in nitrate electroreduction is more favourable at higher pH, while ammonia formation is more favourable in an acidic environment. Thus, at the higher current density in flow-through conditions, the pH will locally increase, thereby influencing the reaction selectivity.37,47 Another possible explanation of selectivity change can be found in the adsorption strength of intermediates of the reduction of nitrate on the catalyst surface. External factors such as intensive mixing induced by gas flow can weaken the strength of these intermediates, such as nitrite or hydroxylamine, and these are therefore formed without consecutive reduction to ammonia. It also seems that a locally acidic pH cannot be sustained by flow induced proton transport, as a consequence of the enhancement in current density by the favourable transport of nitrate. This is likely related to the previously mentioned reaction stoichiometry (9 moles of protons and 1 mole of nitrate are needed for production of 1 mole of NH3 and 3 moles of H2O).
Considering that closing the electron balance is not feasible when only liquid phase products are considered, an attempt was made to detect gaseous products using online electrochemical mass-spectrometry (EC-MS).52 Here, considering the specifications of the EC-MS, a Ti disc electrode instead of an electrode in hollow fiber geometry was used. Mass spectra of the selected signals recorded during chronoamperometry at different potentials are shown in Fig. 5. After recording the background signal, potential pulses were applied (starting from −0.3 V vs. RHE) for 30 s, which is sufficient to observe formation of products with low vapour pressure without favouring their excessive accumulation in the small volume electrochemical cell. Ammonia and hydroxylamine were not detected in EC-MS experiments due to their non-volatile nature at low pH.
 | ||
| Fig. 5 In situ EC-MS on Ti disc (a) during chronoamperometry in 0.1 M HClO4 with 50 mM KNO3 at different potentials. (b and c) Product distribution obtained for decomposition reactions using solutions containing NO2− and NH2OH. Note that the increase of signals at m/z: 28, 33, and 44 is due to the addition of a non-deaerated solution as evidenced by the addition of 0.1 M HClO4 (b) where NO signal does not increase. | ||
Nitric oxide (m/z: 30) is observed almost throughout the entire potential-range, peaking at −0.6 V vs. RHE, similar to the potential at which the FE towards ammonia shows a minimum in the flow-through experiments shown in Fig. 3b. Please note that the mass signal for NO is significantly larger than the signal of any other volatile nitrogen products. NO is not observed at the highest applied potentials (>−0.9 V), at which hydrogen evolution is dominant, in line with the reported LSV measurements performed with Ti hollow fiber electrodes (Fig. 2), and the generally expected high overpotential for H2 evolution on Ti. Similarly, m/z: 28 and m/z: 44 (both assigned to N2O) were observed with the highest peak intensity at −0.7 V vs. RHE. Considering that during bulk electrolysis nitrogen is not detected (in preliminary experiments), and the signal intensity ratio between m/z: 44 and 28 matches the ratio expected for N2O (according to the NIST database), the appearance of the signal at m/z: 28 originates from fragmentation of N2O in the mass spectrometer. Thus N2, frequently reported in the literature as a product of nitrate reduction, is not observed here.33,53 Surprisingly, a potential dependent increase in the signal at m/z: 46 was also detected, which can be assigned to traces of NO2.
The detected gas phase products are in agreement with the formation of nitrite from nitrate, followed by consecutive chemistry of nitrite. Nitrite has a rich homogeneous chemistry in the electrolyte, which can be summarized by reactions (1)–(5).47,54Reactions (1)–(3) show that NO2− (HNO2 in acid) decomposes, thereby releasing NO and NO2via disproportionation reactions (2) and (3). Since an increase in FE to NO2− was observed using the hollow fiber electrode as a function of increasing flow rate, it is expected that disproportion is increased in flow-through conditions, partially explaining the ‘missing electrons’ in the balance reported above.
| H+ + NO2− ↔ HNO2 | (1) |
| 2HNO2(aq) ↔ NO(aq) + NO2(aq) + H2O | (2) |
| 3HNO2(aq) ↔ HNO3 + 2NO + H2O | (3) |
| 2NO2 + H2O → HNO2 + H+ + NO3− | (4) |
| NH3OH+ + NO2− → N2O + 2H2O | (5) |
In addition to the disproportion reaction of nitrite, the reaction between nitrite and hydroxylamine (which was also detected in electrolysis experiments) will result in N2O formation in acid environment (reaction (5)).55,56 In Fig. 5c MS signals do not change when NH2OH solution is added, but after the addition of NO2− both, the NO and N2O signals rise, the former again in agreement with reaction (2), and the latter confirming the occurrence of reaction (5). Note that the increase in the m/z: 28 is not observed, due to the high background signal arising from air, exposed to the solution by the open cell configuration used in these experiments.
Although the electrochemical formation of any gaseous products cannot be excluded (see Table S1†), it is evident by the performed analysis that homogeneous reactions of electrochemically formed nitrite likely explain the ‘missing electrons’ in the electron balance. Most importantly, the formation of gaseous products might explain the significant drop in faradaic efficiency observed at ∼−0.7 V and −0.8 V vs. RHE in flow-through conditions (Fig. 4). We again note that this drop is in agreement with the (peak) formation rates of NO and N2O as observed by EC-MS analysis (Fig. 5). Therefore, not only formation, but also decomposition of NO2− (and NH2OH) is expected to be enhanced in this potential window by introducing flow. We suggest that flow-through configurations not only promote transport of protons and nitrate towards the electrode, but also of nitrite and NH2OH away from the electrode. Rather than consecutive electrochemistry towards ammonia, homogeneous chemistry in solution is promoted. As such the decrease in hydroxylamine FE at −0.7 and −0.8 V vs. RHE (see Fig. 4) is observed because of its reaction with nitrite forming N2O.
At more negative potentials (−0.9 and −1 V vs. RHE) formation of NO2− is less dominant, and thus the observed faradaic efficiency to NH2OH increases, additionally causing an overall increase in total faradaic efficiency to liquid products.
Finally, it appears to be important to address the increase in faradaic efficiency to ammonia for potentials more negative than −0.6 V vs. RHE. A similar behaviour was observed on Ti plate electrodes beyond −0.75 V vs. RHE at high nitrate concentrations (0.4 M NO3−), but was not discussed in detail.29 We suggest that interaction of nitrate with electrochemically formed H2 might be responsible for the increase in FE observed in the H2 evolution regime. It is known that reduction of nitrate in aqueous solutions can occur in the presence of hydrogen over a variety of catalysts57,58 and leads to the formation of ammonia.59
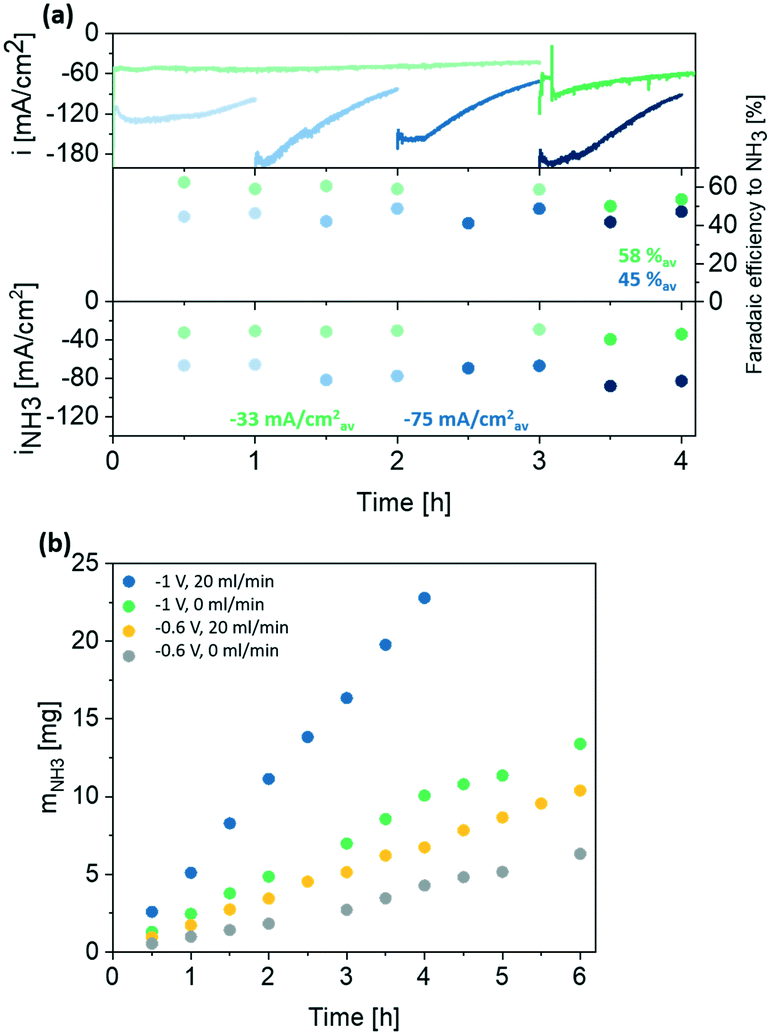 | ||
| Fig. 6 (a) Stability test in 0.1 M HClO4 with 50 mM KNO3 at −1 V vs. RHE with 0 ml min−1 (green) and 20 ml min−1 (blue) flow rate showing overall current density as well as faradaic efficiency and partial current density to NH3. (b) NH3 mass increase in stability test in different conditions. | ||
Independent on the applied potential, a decrease in current density over time is observed in flow-through conditions due to depletion of nitrate (see also Fig. 3 and S12†). Therefore, the electrolyte was refreshed periodically. Despite small variations in current density after introduction of fresh nitrate, the faradaic efficiency to ammonia was stable over the period tested, reaching ∼58% and 45% at no-flow and flow-through conditions (20 ml min−1 Ar) respectively (on average during 4 h). Moreover, a linear increase in mNH3 was observed for all conditions tested (Fig. 6b) and the electrochemically active surface area (ECSA) remained the same before and after the stability test (Fig. S19†). Also SEM and XRD characterization do not reveal any significant deterioration of the electrode structure and composition (see Fig. S20 and S21†). Most importantly, a remarkable partial current density to ammonia of ∼−75 mA cm−2 was maintained over 4 h of electrolysis used with 50 mM KNO3 in acidic electrolyte and polarized at −1 V vs. RHE (Table S4†). We assign the improvement in comparison to previous studies to efficient mass transport of NO3− to the electrode surface. However, we note that the geometrical surface area was used to calculate current density, which is standard practice nowadays. For comparison, partial current densities to ammonia based on the actual electrochemically active surface area are shown in Fig. S22.† Still the faradaic efficiency reported here (58% at −1 V vs. RHE in no flow conditions) exceeds values reported earlier obtained for similar electrolyte compositions and reactor configurations where FEs to ammonia of <20% are reported (Table S4†). Given this significant increase, we speculate that electrode preparation is of importance. Here, Ti hollow fibers were used that undergo a thermal treatment likely having a positive influence on catalytic activity. Moreover, phase pure Ti is used in this study while contaminations likely in form of TiC and/or TiN can be observed in other reports.29 We emphasize that phase purity of Ti electrodes will be of interest for further investigation. Finally, we like to highlight that in this work nitrate waste streams of low nitrate concentration were targeted, yet higher nitrate concentrations will potentially further increase selectivity towards ammonia.
Conclusions
Using Ti hollow fiber electrodes for electroreduction of nitrate in 50 mM KNO3 at −1 V vs. RHE, a faradaic efficiency of 58% towards ammonia is achieved in acidic conditions, with a performance stability of over 4 h. Other products formed are hydroxylamine, NO and N2O, the latter formed by homogeneous disproportionation chemistry of nitrite. Optimizing mass transport by introduction of an exiting flow of inert gas, enhances the partial current density to ∼−75 mA cm−2 with a FE of ∼45%. We have revealed the overall, potential and gas flow dependent trends in product formation can be explained by 1) local pH changes affecting proton-coupled electron reactions to ammonia, nitrite and hydroxylamine, 2) homogenous reactions of nitrite and hydroxylamine to NO and N2O, promoted by gas flow induced favourable liquid mixing, and finally 3) catalytic hydrogenation of nitrate resulting in an overall increase in liquid product formation at the most negative potentials investigated.The reported current densities for electrochemical formation of ammonia are among the highest ever reported for (Ti) hollow fibre electrodes, which approach the values needed for practical implementation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work took place within the framework of the Institute of Sustainable Process Technology, co-funded with subsidy from the Topsector Energy by the Ministry of Economic Affairs and Climate Policy, The Netherlands.References
- J. Lim, C. A. Fernández, S. W. Lee and M. C. Hatzell, ACS Energy Lett., 2021, 3676–3685 CrossRef CAS.
- K. H. R. Rouwenhorst, P. M. Krzywda, N. E. Benes, G. Mul and L. Lefferts, Ullmann's Encycl. Ind. Chem., 2020, pp. 1–20 Search PubMed.
- K. H. R. Rouwenhorst, P. M. Krzywda, N. E. Benes, G. Mul and L. Lefferts, in Techno-Economic Challenges of Green Ammonia as an Energy Vector, 2020, pp. 41–83 Search PubMed.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C. P. Chen, M. R. Smith and T. W. Hamann, Chem. Rev., 2020, 120, 5437–5516 CrossRef CAS PubMed.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh, B. A. Rohr, M. J. Statt, S. J. Blair, S. Mezzavilla, J. Kibsgaard, P. C. K. Vesborg, M. Cargnello, S. F. Bent, T. F. Jaramillo, I. E. L. Stephens, J. K. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- P. M. Krzywda, A. Paradelo Rodríguez, N. E. Benes, B. T. Mei and G. Mul, ChemElectroChem, 2022, 9, e202101273 CAS.
- J. Long, S. Chen, Y. Zhang, C. Guo, X. Fu, D. Deng and J. Xiao, Angew. Chem., Int. Ed., 2020, 59, 9711–9718 CrossRef CAS PubMed.
- G. F. Chen, Y. Yuan, H. Jiang, S. Y. Ren, L. X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 5, 605–613 CrossRef CAS.
- R. Daiyan, T. Tran-Phu, P. Kumar, K. Iputera, Z. Tong, J. Leverett, M. H. A. Khan, A. A. Esmailpour, A. Jalili, M. Lim, A. Tricoli, R.-S. Liu, X. Lu, E. Lovell and R. Amal, Energy Environ. Sci., 2021, 14, 3588–3598 RSC.
- P. Li, Z. Jin, Z. Fang and G. Yu, Energy Environ. Sci., 2021, 14, 3522–3531 RSC.
- Y. Zhang, F. Li, Q. Zhang, J. Li and Q. Liu, Sci. Total Environ., 2014, 490, 213–222 CrossRef CAS PubMed.
- S. Roy, M. S. Hegde and G. Madras, Appl. Energy, 2009, 86, 2283–2297 CrossRef CAS.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- L. F. Greenlee, Nat. Energy, 2020, 5, 557–558 CrossRef CAS.
- M. Kuang, Y. Wang, W. Fang, H. Tan, M. Chen, J. Yao, C. Liu, J. Xu, K. Zhou and Q. Yan, Adv. Mater., 2020, 32, 1–7 Search PubMed.
- S. Han, C. Wang, Y. Wang, Y. Yu and B. Zhang, Angew. Chem., 2021, 133, 4524–4528 CrossRef.
- C. Dai, Y. Sun, G. Chen, A. C. Fisher and Z. J. Xu, Angew. Chem., Int. Ed., 2020, 59, 9418–9422 CrossRef CAS PubMed.
- M. Anand, C. S. Abraham and J. K. Nørskov, Chem. Sci., 2021, 12, 6442–6448 RSC.
- B. S. Patil, N. Cherkasov, J. Lang, A. O. Ibhadon, V. Hessel and Q. Wang, Appl. Catal., B, 2016, 194, 123–133 CrossRef CAS.
- F. Jardali, S. Van Alphen, J. Creel, H. Ahmadi Eshtehardi, M. Axelsson, R. Ingels, R. Snyders and A. Bogaerts, Green Chem., 2021, 23, 1748–1757 RSC.
- Y. Yu, C. Wang, Y. Yu, Y. Huang, C. Liu, S. Lu and B. Zhang, J. Mater. Chem. A, 2020, 8, 19623–19630 RSC.
- E. Vervloessem, M. Aghaei, F. Jardali, N. Hafezkhiabani and A. Bogaerts, ACS Sustainable Chem. Eng., 2020, 8, 9711–9720 CrossRef CAS.
- H. Patel, R. K. Sharma, V. Kyriakou, A. Pandiyan, S. Welzel, M. C. M. Van De Sanden and M. N. Tsampas, ACS Energy Lett., 2019, 4, 2091–2095 CrossRef CAS.
- K. H. R. Rouwenhorst, F. Jardali, A. Bogaerts and L. Lefferts, Energy Environ. Sci., 2021, 14, 2520–2534 RSC.
- L. Li, C. Tang, X. Cui, Y. Zheng, X. Wang, H. Xu, S. Zhang, T. Shao, K. Davey and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 14131–14137 CrossRef CAS PubMed.
- L. Hollevoet, F. Jardali, Y. Gorbanev, J. Creel, A. Bogaerts and J. A. Martens, Angew. Chem., Int. Ed., 2020, 59, 23825–23829 CrossRef CAS PubMed.
- Y. Wang, C. Wang, M. Li, Y. Yu and B. Zhang, Chem. Soc. Rev., 2021, 50, 6720–6733 RSC.
- M. Duca and M. T. M. Koper, Energy Environ. Sci., 2012, 5, 9726–9742 RSC.
- J. M. McEnaney, S. J. Blair, A. C. Nielander, J. A. Schwalbe, D. M. Koshy, M. Cargnello and T. F. Jaramillo, ACS Sustainable Chem. Eng., 2020, 8, 2672–2681 CrossRef CAS.
- Q. Hu, Y. Qin, X. Wang, Z. Wang, X. Huang, H. Zheng, K. Gao, H. Yang, P. Zhang, M. Shao and C. He, Energy Environ. Sci., 2021, 14, 4989–4997 RSC.
- M. N. Desai and S. S. Rana, Anti-Corros. Methods Mater., 1973, 20, 8–10 CrossRef CAS.
- A. S. Fajardo, P. Westerhoff, C. M. Sanchez-Sanchez and S. Garcia-Segura, Appl. Catal., B, 2021, 281, 119465 CrossRef CAS.
- J. Yao, Y. Mei, T. Yuan, J. Chen, H. Pan and J. Wang, J. Electroanal. Chem., 2021, 882, 115019 CrossRef CAS.
- H. Niu, Z. Zhang, X. Wang, X. Wan, C. Shao and Y. Guo, Adv. Funct. Mater., 2020, 2008533, 1–8 Search PubMed.
- R. Jia, Y. Wang, C. Wang, Y. Ling, Y. Yu and B. Zhang, ACS Catal., 2020, 10, 3533–3540 CrossRef CAS.
- Y. Guo, R. Zhang, S. Zhang, Y. Zhao, Q. Yang, Z. Huang, B. Dong and C. Zhi, Energy Environ. Sci., 2021, 14, 3938–3944 RSC.
- S. Garcia-Segura, M. Lanzarini-Lopes, K. Hristovski and P. Westerhoff, Appl. Catal., B, 2018, 236, 546–568 CrossRef CAS.
- P. H. van Langevelde, I. Katsounaros and M. T. M. Koper, Joule, 2021, 5, 290–294 CrossRef.
- W. Sun, H. Ji, L. Li, H. Zhang, Z.-K. Wang, J.-H. He and J.-M. Lu, Angew. Chem., Int. Ed., 2021, 60, 22933–22939 CrossRef CAS PubMed.
- C. Yan, S. Kakuturu, A. H. Butzlaff, D. M. Cwiertny, S. Mubeen and C. J. Werth, ACS ES&T Engg, 2021, 1, 204–215 Search PubMed.
- I. Merino-Garcia, J. Albo, P. Krzywda, G. Mul and A. Irabien, Catal. Today, 2020, 346, 34–39 CrossRef CAS.
- R. Kas, K. K. Hummadi, R. Kortlever, P. De Wit, A. Milbrat, M. W. J. Luiten-Olieman, N. E. Benes, M. T. M. Koper and G. Mul, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- X. Wei, D. Vogel, L. Keller, S. Kriescher and M. Wessling, ChemElectroChem, 2020, 7, 4679–4684 CrossRef CAS.
- R. P. H. Jong, P. M. Krzywda, N. E. Benes and G. Mul, RSC Adv., 2020, 10, 31901–31908 RSC.
- X. Zou, X. Huang, A. Goswami, R. Silva, B. R. Sathe, E. Mikmeková and T. Asefa, Angew. Chem., Int. Ed., 2014, 53, 4372–4376 CrossRef CAS PubMed.
- C. T. Dinh, A. Jain, F. P. G. de Arquer, P. De Luna, J. Li, N. Wang, X. Zheng, J. Cai, B. Z. Gregory, O. Voznyy, B. Zhang, M. Liu, D. Sinton, E. J. Crumlin and E. H. Sargent, Nat. Energy, 2019, 4, 107–114 CrossRef CAS.
- I. Katsounaros, Curr. Opin. Electrochem., 2021, 28, 100721 CrossRef CAS.
- X. Xing, D. A. Scherson and C. Mak, J. Electrochem. Soc., 1990, 137, 2166–2175 CrossRef CAS.
- S. Taguchi and J. M. Feliu, Electrochim. Acta, 2008, 53, 3626–3634 CrossRef CAS.
- Z. Wang, D. Richards and N. Singh, Catal. Sci. Technol., 2021, 705–725 RSC.
- E. Pérez-Gallent, M. C. Figueiredo, I. Katsounaros and M. T. M. Koper, Electrochim. Acta, 2017, 227, 77–84 CrossRef.
- D. B. Trimarco, S. B. Scott, A. H. Thilsted, J. Y. Pan, T. Pedersen, O. Hansen, I. Chorkendorff and P. C. K. Vesborg, Electrochim. Acta, 2018, 268, 520–530 CrossRef CAS.
- B. P. Dash and S. Chaudhari, Water Res., 2005, 39, 4065–4072 CrossRef CAS PubMed.
- V. Rosca, M. Duca, M. T. DeGroot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- F. T. Bonner, J. Kada and K. G. Phelan, Inorg. Chem., 1983, 22, 1389–1391 CrossRef CAS.
- A. A. Bothner-By and L. Friedman, J. Chem. Phys., 1952, 20, 5473–5475 CrossRef.
- O. S. G. P. Soares, J. J. M. Órfão and M. F. R. Pereira, Catal. Lett., 2008, 126, 253–260 CrossRef CAS.
- C. J. Werth, C. Yan and J. P. Troutman, ACS ES&T Engg, 2021, 1, 6–20 Search PubMed.
- L. Wei, D. J. Liu, B. A. Rosales, J. W. Evans and J. Vela, ACS Catal., 2020, 10, 3618–3628 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy00289b |
| This journal is © The Royal Society of Chemistry 2022 |