
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ DRIFT studies on N2O formation over Cu-functionalized zeolites during ammonia-SCR†
Ghodsieh
Isapour
a,
Aiyong
Wang
 b,
Joonsoo
Han
b,
Yingxin
Feng
c,
Henrik
Grönbeck
c,
Derek
Creaser
b,
Louise
Olsson
b,
Magnus
Skoglundh
a and
Hanna
Härelind
*a
b,
Joonsoo
Han
b,
Yingxin
Feng
c,
Henrik
Grönbeck
c,
Derek
Creaser
b,
Louise
Olsson
b,
Magnus
Skoglundh
a and
Hanna
Härelind
*a
aDepartment of Chemistry and Chemical Engineering, Division of Applied Chemistry Competence Centre for Catalysis, Chalmers University of Technology, Gothenburg, Sweden. E-mail: hanna.harelind@chalmers.se
bDepartment of Chemistry and Chemical Engineering, Division of Chemical Engineering, Competence Centre for Catalysis, Chalmers University of Technology, Gothenburg, Sweden
cDepartment of Physics, Division of Chemical Physics, Competence Centre for Catalysis, Chalmers University of Technology, Gothenburg, Sweden
First published on 3rd May 2022
Abstract
The influence of the zeolite framework structure on the formation of N2O during ammonia-SCR of NOx was studied for three different copper-functionalized zeolite samples, namely Cu-SSZ-13 (CHA), Cu-ZSM-5 (MFI), and Cu-BEA (BEA). The evolution of surface species during the SCR reaction at different temperatures was monitored with step-response experiments using in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) at different reaction conditions. Also, density functional theory (DFT) calculations were performed to assist the interpretation of the experimental results. The DRIFTS results indicate that NO+ and nitrate species are the main products formed during NO oxidation, and NO appears to adsorb on both Cu-Lewis and Al-Lewis acid sites. The DFT calculations for NO adsorption on the SSZ-13 sample reveal adsorption at Brønsted acid sites with similar adsorption energies but with a slight difference in NO+ stretching vibrations in the DRIFT spectra. Within the standard SCR reaction, in the O–H stretching region, the number of NH3 molecules adsorbed on the Brønsted acid sites is higher for the small-pore size sample compared to the medium- and large-pore zeolites. The obtained DRIFTS results for nitrate species are supported by DFT calculations by simulating the IR spectra of mobile and framework bound nitrate species, which both have a signature at 1604 cm−1 associated with the O–N bond on NO3−. It is revealed that N2O is produced in a higher amount at lower temperatures for all three samples irrespective of the NO/NO2 ratio. Furthermore, the obtained results from both DRIFTS studies and flow reactor experiments show the higher formation of N2O for the large-pore zeolite compared to the medium- and small-pore zeolite.
1. Introduction
Selective catalytic reduction using ammonia as a reducing agent (NH3-SCR) is a widely used technique to abate NOx (NO and NO2) emissions from both stationary and mobile sources, such as diesel engines.1,2 In the SCR reaction, however, unwanted byproducts like nitrous oxide (N2O), can be formed. This is a problem particularly as N2O is a strong greenhouse gas and causes depletion of the ozone layer.3–5 Although the annual emissions of N2O are much lower than those of CO2, the global warming potential (GWP) of N2O is 310 times higher than that of CO2.3,5 The concentration of N2O in the atmosphere has consistently increased due to natural and anthropogenic activities by 0.2% per year.3 Owing to the mentioned reasons, it is important to minimize N2O formation as a by-product in the SCR reaction.Nowadays, this can be done by using zeolites with various pore-size that are ion-exchanged with transition metal cations like Cu and Fe. The zeolite framework structure plays a decisive role in determining the properties of copper-functionalized zeolite catalysts for NH3-SCR. In general, small-pore (8-membered ring windows) zeolite catalysts (such as Cu-SSZ-13), represent higher durability during hydrothermal aging conditions, premier N2 selectivity with less N2O formation, and excellent hydrocarbon tolerance, compared to medium (10-membered ring windows) or large-pore (12-membered ring windows) zeolites such as Cu-ZSM-5 and Cu-BEA, respectively.1,6,7 Both the exchanged transition metal cations and the Brønsted acid sites are important for the SCR reactions.1,8 Zhang and Yang have analyzed the N2O formation pathways in NH3-SCR over different catalysts (Cu-ZSM-5, Fe-ZSM-5, Cu-SAPO-34, Fe-SAPO-34, Cu-SSZ-13, and Fe-SSZ-13), and the results indicated that for most of the Cu catalysts, the main N2O formation pathway at higher temperatures is the non-selective catalytic oxidation of NH3.2 Non-selective behavior takes place when other products (specifically N2O) are formed rather than N2, e.g. during standard or fast SCR reaction conditions or when the converted NO/NH3 molar ratio is lower than 1.2
In the temperature range 250–350 °C, the N2O formation partially stems from the decomposition of ammonium nitrate (AN).9 Furthermore, NH3-SCR over Cu-BEA, Cu-SAPO-34, and Cu-SSZ-13 has been investigated by Leistner et al.6 Various reactions like NO oxidation, ammonia oxidation, standard SCR, fast SCR, and NO2-SCR were studied to investigate the influence of the zeolite type on the N2O formation. The obtained results proposed that N2O formation takes place on different copper sites or proceeds via different mechanisms depending on the temperature. They observed that increasing the temperature above 300 °C resulted in a higher N2O formation for Cu/SAPO-34 followed by Cu/SSZ-13 and Cu-BEA samples. The results indicate a higher formation of ammonium nitrate at low temperatures and its subsequent decomposition at higher temperatures. To sum up, the choice of type of zeolite/zeotype and reaction temperature affects the activity and selectivity of the different steps in the SCR process.
Density functional theory (DFT) calculations have also been used to identify the fundamental reaction pathways of the SCR reaction.10,11 Recently, Feng et al.8 proposed a reaction mechanism at low temperatures for N2O formation over the small-pore zeolite Cu-CHA based on DFT calculations and related the N2O formation to H2NNO decomposition over (NH3)2Cu–OOH–Cu(NH3)2 complexes. This can explain why the formation of N2O increases with increasing copper loading at low temperatures.
The present work aims at investigating the influence of the zeolite framework on the formation of N2O during ammonia SCR of NOx. For this purpose, three different zeolite samples with varying framework structures: Cu-SSZ-13 (CHA), Cu-ZSM-5 (MFI), and Cu-BEA (BEA) are exposed to NH3-SCR reaction conditions in step-response experiments where the evolution of surface species simultaneously is followed by in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). The samples are exposed to gas sequences corresponding to standard and fast SCR reaction conditions in the temperature range of 130–200 °C. In addition, flow reactor studies are performed to evaluate the N2O formation during NH3-SCR reaction conditions. Furthermore, the binding energy of different surface species during NH3 adsorption are together with vibrational properties obtained by density functional theory calculations.
2. Experimental
2.1. Sample preparation
Three different copper-functionalized zeolite samples, Cu-SSZ-13, Cu-ZSM-5, and Cu-BEA were prepared according to the following procedure. The synthesis of Na-SSZ-13 with a Si/Al molar ratio of 12 was performed via sol–gel and hydrothermal crystallization in an autoclave. First, 1 M NaOH was prepared by mixing NaOH (Sigma-Aldrich, >98% anhydrous) in Milli-Q water (18 MΩ cm). Then, the structure-directing agent (SDA) (25% solution of SDA 2825, Sachem, ZeoGen), aluminum hydroxide (Sigma-Aldrich, 50.0–57.5% Al (as Al2O3) basis, reagent grade), and fumed silica (Sigma-Aldrich, S5130-500G, 0.007 μm, standard grade) were subsequently added to the solution, which was stirred vigorously until a homogenized gel was formed. The obtained gel was transferred to a 125 ml Teflon-lined stainless-steel autoclave, sealed off, and heated to 160 °C for 96 h under stirring to accomplish hydrothermal synthesis. The autoclave was subsequently cooled to room temperature. In this synthesis method, continuous stirring is crucial to achieving a uniform Na-SSZ-13 sample. The obtained slurry was washed three times with Milli-Q water, and the solid/liquid mixture was separated by centrifugation. The resulting precipitate was dried at 110 °C overnight and calcined at 600 °C to remove all SDA. The prepared powder was then dissolved in Milli-Q water and ammonium nitrate (Sigma-Aldrich, 98%, hemi(pentahydrate)), and nitric acid (Sigma-Aldrich, 69%), were added to adjust the pH. The solution was stirred for 1 h (700 rpm) at 80 °C and thereafter washed with Milli-Q water and centrifuged. This step was repeated two times to remove all sodium from the sample. The remaining precipitate was kept at 110 °C overnight to obtain NH4-SSZ-13.For the preparation of the Cu-SSZ-13 sample, incipient wetness impregnation was used to functionalize the zeolite with copper. First, a copper nitrate solution (Sigma-Aldrich, 98%) was added to the specific amount of NH4-SSZ-13 and then, 3 g of ethanol was added to the mixture. The slurry was stirred for 15 min and then kept at room temperature overnight and thereafter calcined. The calcination of the Cu-SSZ-13 sample was carried out in two steps. The temperature was first increased from room temperature to 600 °C with a rate of 2 °C min−1 and kept constant for 6 h, and then increased to 750 °C and kept constant for 2 h after which the sample was cooled to room temperature. The other two zeolites (NH4-ZSM-5; Si/Al = 11.5, NH4-BEA; Si/Al = 12.5) were commercially available (Zeolyst international, CBV; 2314 and CP; 814E, respectively), and the functionalization with copper was performed using the same method as used for Cu-SSZ-13. The only difference was the calcination temperature for these two zeolites, which was carried out in one step at 600 °C for 8 h. All samples were degreened under 400 ppm NO, 400 ppm NH3 and 10% O2 at 200 °C, before being used in the DRIFTS studies.
For the flow reactor experiments, the prepared powder samples were coated on honeycomb-structured cordierite monolith substrates (400 cpsi). Before wash-coating, the monolith substrates were cut to a dimension of 20 mm in length and 15 mm in diameter, and subsequently heated in air at 600 °C for 2 h to eliminate any contaminations. After cooling to room temperature, the monolith substrates were coated with a slurry including a solid phase composed of the powder sample and boehmite binder (Dispersal P2) with a mass ratio of 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 and a liquid phase containing equal amounts of Milli-Q water and ethanol. The monolith was coated by dipping into the slurry to fill all the channels thoroughly. Thereupon, to remove the excess slurry and hinder channel blockage, the coated monolith was dried at 80 °C for 2 min with a heating gun. The process of wash-coating and drying was continued until reaching the aimed loading of wash-coat (∼300 mg). Moreover, the coated monoliths were weighed after heating at 500 °C for 1 min and finally calcined in air at 500 °C for 2 h, starting at room temperature with a heating rate of 2 °C min−1.
:5 and a liquid phase containing equal amounts of Milli-Q water and ethanol. The monolith was coated by dipping into the slurry to fill all the channels thoroughly. Thereupon, to remove the excess slurry and hinder channel blockage, the coated monolith was dried at 80 °C for 2 min with a heating gun. The process of wash-coating and drying was continued until reaching the aimed loading of wash-coat (∼300 mg). Moreover, the coated monoliths were weighed after heating at 500 °C for 1 min and finally calcined in air at 500 °C for 2 h, starting at room temperature with a heating rate of 2 °C min−1.
2.2. Sample characterization
Powder X-ray diffraction (XRD) measurements were performed using a Siemens diffractometer D5000 operating at 40 kV and 40 mA with Cu Kα radiation (λ = 1.5418 Å). Data were collected with 2θ ranging from 5 to 50° using a step size of 0.02. Scanning electron microscopy (SEM) images were obtained using an LEO Ultra 55 SEM equipped with an energy dispersive X-ray (EDX) system (Oxford Inca). A Tristar 3000 (Micromeritics) instrument was used to obtain N2 adsorption and desorption isotherms at 77 K. All fresh powder samples were degassed at 250 °C for 10 h under the flow of N2. The Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods were applied to determine the specific surface area and the pore volume, respectively, which were obtained from the t-plot method and by measuring the multilayer adsorption of N2. Although the BET method is beneficial for determining specific surface areas, it can be confusing due to the difficulty of forming multilayers on the internal surface of microporous materials such as small-pore zeolites. Elemental analysis of the samples was performed using inductively coupled plasma sector field mass spectrometry (ICP-SFMS) by ALS Scandinavia AB in Luleå, Sweden.
In situ DRIFTS was used to follow the evolution of surface species on the samples during reaction conditions. The DRIFTS measurements were performed with a VERTEX70 spectrometer (Bruker), equipped with liquid nitrogen cooled mercury cadmium telluride (MCT) detector with the bandwidth of 600–12000 cm−1, and a high-temperature stainless steel reaction chamber (Harrick Scientific Products Inc.) with CaF2 windows. The measurement of all spectra was done between 400 and 4000 cm−1 with a spectral resolution of 1 cm−1 and the reported spectra are obtained by an average of 90 scans. The approximate amount of sample loaded into the reaction chamber was 100 mg. The samples were pretreated in 10 vol% O2, with Ar as a carrier gas, for 1 h at 500 °C to remove impurities from the surface of the sample, and subsequently cooled to reaction temperature in Ar. Background spectra were taken under Ar at 200, 180, 150, and 130 °C. Gases were introduced to the reaction chamber by individual mass flow controllers (HiTech), and the total flow was 100 ml min−1 (balanced with Ar). The evolution of surface species on the samples was measured at the mentioned temperatures during step-response experiments with 15 min length for each step, as shown in Table 1. In the first step, the sample was exposed to NO at 200 °C for 15 min using a feed consisting of 10 vol% O2, 400 ppm NO, and Ar as balance. During the NO exposure, IR spectra were collected by varying exposure time using the related background spectra taken before the introduction of NO. In the second step, the sample was exposed to a full standard NH3-SCR feed by introducing 400 ppm NH3 in the presence of 400 ppm NO, 10 vol% O2 and Ar for 15 min. The experiment continued by switching off the NO feed for 15 min in the third step and then switching on the NO feed again to have a second full standard NH3-SCR feed for 15 min in the fourth step. Directly following, the sample was exposed to 200 ppm NO, 200 ppm NO2 and 10 vol% O2 for 15 min in the fifth step before the addition of NH3 in the sixth step for having a full fast NH3-SCR feed for 15 min. This step was followed by the final seventh step where the sample was exposed to 400 ppm NH3 in Ar for 15 min. The outlet gas composition was continuously analyzed by mass spectrometry (MS) using a Hidden Analytical, HPS-20 QIC instrument following the m/z ratios 17 (NH3), 28 (N2 and CO), 30 (NO), 32 (O2), 40 (Ar), 44 (N2O and CO2) and 46 (NO2). The experimental procedure is summarized in Table 1.
| Feed composition | Exposure time | |
|---|---|---|
| 1. NO oxidation | 400 ppm NO, 10 vol% O2 | 15 min |
| 2. Standard SCR | 400 ppm NO, 400 ppm NH3, 10 vol% O2 | 15 min |
| 3. NH3 adsorption | 400 ppm NH3, 10 vol% O2 | 15 min |
| 4. Standard SCR | 400 ppm NO, 400 ppm NH3, 10 vol% O2 | 15 min |
| 5. NO2 adsorption | 200 ppm NO, 200 ppm NO2, 10 vol% O2 | 15 min |
| 6. Fast SCR | 200 ppm NO, 200 ppm NO2, 400 ppm NH3, 10 vol% O2 | 15 min |
| 7. NH3 adsorption | 400 ppm NH3, 10 vol% O2 | 15 min |
The quantification of DRIFTS data for the formation of N2O, NO+ and nitrate species was done by integrating the attributed peak area in the wavenumber range of 2237 cm−1with a shoulder at 2208 cm−1, 2050–2150 cm−1, and 1530–1700 cm−1, respectively, at the steady-state of the reaction.
2.3. Flow reactor experiments
After wash-coating, the monolith samples were degreened in standard SCR conditions for 2 h at 500 °C, starting at room temperature with a heating rate of 20 °C min−1. The catalytic activity was measured in a flow reactor consisting of a horizontal quartz tube placed in a heating coil and covered with an isolating material, which was equipped with several Bronkhorst mass flow controllers and a controlled evaporation and mixing (CEM) system for controlling the supply of gases and dosing water vapor, respectively. An FTIR spectrometer (MKS Multigas 2030 HS) was used to analyze the reactor outlet gas concentrations. A LabVIEW interface was used for controlling the setup system. The temperature of the sample and the inlet gas flow were measured by a type-K thermocouple placed in the central channel of the monolith sample and 15 mm upstream of the catalyst in the gas phase, respectively. For all experiments, a total flow of 1200 ml min−1 (gas hourly space velocity of 20400 h−1) was used with Ar as a balance to attain constant total flow. The pretreatment of the samples was performed in the presence of 10 vol% O2 in Ar for 20 min at 500 °C before the standard and fast SCR reaction.
The standard NH3-SCR experiments were carried out isothermally at 130, 150, 180, and 200 °C by exposing the sample to 400 ppm NH3, 400 ppm NO, 10 vol% O2 in Ar for 1 h. Then, the reactor temperature was increased stepwise to 200 °C with a 20 °C increment and kept constant at each step for 30 min, to measure the NOx conversion and N2O formation. Further on, fast SCR experiments were performed by exposing the sample to 400 ppm NH3, 200 ppm NO, 200 ppm NO2, 10 vol% O2 in Ar for 1 h. The experimental procedure was the same as for standard SCR.
2.4. DFT calculations
Spin-polarized density functional theory calculations were carried out using the Vienna ab initio simulation package (VASP).12–15 The projector augmented wave (PAW)16,17 method was used to describe the interaction between the valence electrons and the core and the valence electrons were expanded with a plane wave basis set up to a cutoff energy of 480 eV. The number of valence electrons treated in the calculations was Cu(11), Si(4), Al(3), O(6), N(5), and H(1). Only the gamma point was used in the k-point sampling.Exchange–correlation effects were described using the gradient-corrected Perdew–Burke–Ernzerhof (PBE) functional,18 augmented Grimme-D3 corrections to describe van der Waals interactions,19,20 and a Hubbard-U term for Cu 3d. Based on previous work, the U-parameter was set to 6 eV.21 The convergence criterion in the self-consistent-field cycles was set to 1 × 10−5 eV and the structures were relaxed until the force on each atom was lower than 0.02 eV Å−1. Frequency analyses were performed on the optimized models. The Zenodo program was used to obtain the vibrational intensities.22 A rhombohedral unit cell, including 12 tetrahedral Si-sites was used to describe the chabazite structure. The lattice parameters were fixed experimentally determined values (α = β = γ = 94.2 Å, a = b = c = 9.42 Å). To model Cu-exchanged SSZ-13 or H-SSZ-13, one or two Si atoms in the six-membered ring of the zeolite cage were substituted by Al giving a Si/Al ratio of 11 or 5. The chosen Si/Al ratios are within the experimental range23–25 and therefore, a reasonable model for the Cu-SSZ-13 material.
3. Results and discussion
3.1. Catalyst characterization
Table 2 displays for the powder samples, the Cu loading, Si/Al ratio, and Cu/Al ratio, obtained from the ICP-SFMS analysis, and the specific surface area and specific pore volume, obtained from the nitrogen sorption analysis. The Cu content, Si/Al ratio and Cu/Al ratio are 2.07 ± 0.08 wt%, 10.8 ± 0.9 and 0.30 ± 0.04, respectively, which is close to the target values. The specific pore volume is significantly higher for the H-SSZ-13 and Cu-SSZ-13 samples compared to the samples based on ZSM-5 or BEA, which is owing to the characteristic large cage void in the CHA-type framework.26 A decrease in specific surface area is observed upon Cu loading as compared to the zeolites in the H-form (almost 20% of surface area loss). This is due to the presence of Cu species, such as Cu2+ and [Cu (OH)]+ in the cages of the zeolite framework and consequently less amount of N2 can adsorb.27 However, the obtained results do not represent a diverse transformation of the microporous structure upon copper ion exchange.26,27| Sample | Cu content (wt%) | Si/Al ratio | Cu/Al ratio | Specific surface area (m2 g−1) | Specific pore volume (cm3 g−1) |
|---|---|---|---|---|---|
| Cu-SSZ-13 | 1.99 | 11.4 | 0.28 | 540 | 0.23 |
| Cu-ZSM-5 | 2.13 | 10.6 | 0.28 | 323 | 0.12 |
| Cu-BEA | 2.10 | 9.94 | 0.34 | 508 | 0.15 |
| H-SSZ-13 | — | 10.0 | — | 674 | 0.30 |
| H-ZSM-5 | — | 11.3 | — | 425 | 0.16 |
| H-BEA | — | 11.5 | — | 631 | 0.19 |
Fig. S1 (see ESI†) shows SEM images of the Cu-SSZ-13, Cu-ZSM-5, and Cu-BEA powder samples. The O, Al, Si, and Cu content of the crystals obtained from the SEM-EDX analysis is summarized in Table S1.† Based on EDX results, although the Si/Al ratio is close to the ICP-SFMS results (11.8, 12.2, 11.8, for Cu-SSZ-13, Cu-ZSM-5, and Cu-BEA, respectively), the Cu/Al ratio (0.198, 0.27, 0.29, for Cu-SSZ-13, Cu-ZSM-5, and Cu-BEA, respectively) is lower compared to the ICP-SFMS analysis, which can be due to the fact that only a small area is analyzed in the EDX measurements. The results obtained from the XRD analysis of the powder samples (Fig. S2†) provide the diffraction patterns for the parent zeolite samples which are characteristic for H-SSZ-13, H-ZSM-5, and H-BEA (JCPDS no.; 52-0784,28 89-142129 and 48-0038,30 respectively) samples. Furthermore, the diffractograms show that the structure of the samples is maintained after functionalization with copper and there is no distinct intensity increase or broadening of the diffraction peaks upon adding Cu to the parent zeolites (H-type). There are no diffraction peaks of crystalline CuO (CuO: highest peaks at ca. 2theta = 35.6 and 38.8),31,32 which indicates that copper is well dispersed in the Cu-functionalized samples or it is in amorphous form. The other possibility is that the amount of CuO is too low to be detected by the diffractometer.
3.2. Evolution of surface species during NH3-SCR step response experiments
The interaction of feed gas species (i.e., NO, NO2 and NH3) with the surface of the copper-functionalized samples was followed by in situ DRIFTS during NH3-SCR step response experiments. Fig. 1 shows the IR spectra in the 2300–1300 cm−1 region after exposing the Cu-SSZ-13, Cu-ZSM-5, and Cu-BEA samples to 400 ppm NO and 10% O2 for 15 min at 200, 180, 150, and 130 °C. The spectra in the entire wavenumber region for the experiment performed at 200 °C are shown in Fig. S4.† Starting with the spectrum for the Cu-SSZ-13 sample obtained at 200 °C, an absorption band around 2156 cm−1 can be observed. This band becomes more intense when the exposure temperature is decreased and evolves to two absorption peaks, positioned at 2156 with a shoulder at 2185 cm−1, at 130 °C. Based on the literature, the peak at 2156 cm−1 has been attributed to N–O stretching vibrations of NO adsorbed on Cu-Lewis acid sites (Cu2+) over Cu-based zeolites.9,33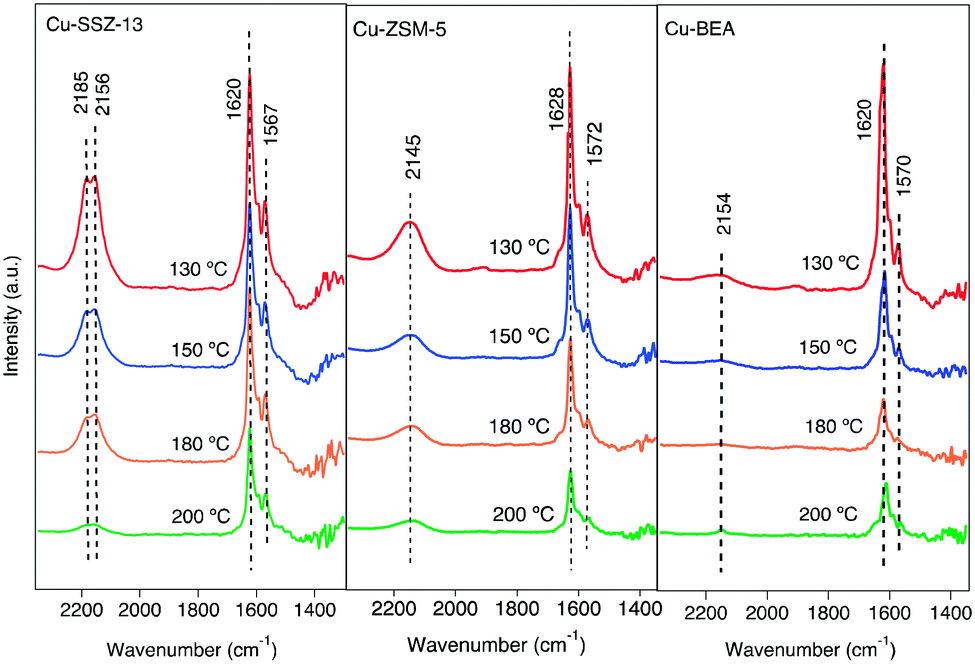 | ||
| Fig. 1 In situ DRIFT spectra after exposing the Cu-SSZ-13, Cu-ZSM-5, and Cu-BEA samples to 400 ppm NO and 10% O2 for 15 min at 200, 180, 150, and 130 °C. | ||
An alternative interpretation suggests the possibility of two cationic positions existing in the zeolite.9 Kwak et al.34 observed the presence of cationic sites of Cu2+ ions in Cu-SSZ-13 with different reducibility, which can have interaction with NO. The interaction can happen either inside the six-membered rings or in the super cages in front of the six-membered ring.
For the Cu-ZSM-5 sample, the corresponding spectra show a less intense band centered at 2145 cm−1, which also becomes more intense when the exposure temperature is decreased. The Cu-BEA sample shows a weak band at 2154 cm−1, which becomes only slightly more intense with decreasing temperature. By moving from a small pore-zeolite to a large-pore zeolite, it is clearly seen that the intensity of the peak attributed to NO adsorbed on Cu2+ decreases.
Comparing the parent zeolites reveals peaks at 2196 and 2089 cm−1 for H-SSZ-13, attributed to N–O stretching vibrations of NO adsorbed on Al–O–Al sites (Al-Lewis acid sites). DRIFTS spectra during NO oxidation at 200 °C for H-type zeolites are shown in Fig. S5.† The results for N–O stretching vibrations of NO+ for the H-ZSM-5 and H-BEA samples show a single peak at 2162 and 2195 cm−1, respectively, which also can be related to NO adsorbed on Brønsted acid sites. All samples show two absorption peaks around 1620 and 1570 cm−1. Both peaks become more intense with decreasing exposure temperature. These peaks are commonly attributed to N–O stretching vibrations of surface nitrate species.35,36 The band around 1620 cm−1 could be characterized as symmetric O–N–O–N–O vibrations, and the one around 1570 cm−1 is attributed to N–O vibrations from adsorbed asymmetric N2O3 species.37
The exact identification of peaks in the nitrate region is not straightforward as the different nitrate species (i.e., monodentate nitrates, bridging monodentate, chelating, and bridging bidentate nitrates) have overlapping vibrations. Wang et al.35 investigated the interaction between NO and Cu-SAPO-34 and observed three peaks in the 1650–1500 cm−1 wavenumber region after the exposure to NO and O2. These peaks were assigned to surface nitrate and nitro species. Furthermore, Chen et al.7 compared the catalytic reaction for NO oxidation over Cu-CHA and Cu-BEA during NH3-SCR reaction conditions and recognized one group of peaks in the 1550–1600 cm−1 region that was attributed to different configurations of surface nitrates.
As can be seen from Fig. 1, the intensities of the bands decrease with increasing temperature. Since comparing the behavior of different samples based on the intensity of the absorption peaks is difficult, the intensity ratio of these two distinct bands (nitrate, nitrite/NO+) has been analyzed and an interesting trend emerged, which is depicted in Fig. 2. It is clear that all three samples represent various behavior at low and high temperatures. At 130 °C, the peak ratio is higher for the Cu-BEA compared to the Cu-ZSM-5 and Cu-SSZ-13 samples, which decreases by increasing temperature. The Cu-ZSM-5 sample shows an increase from 130 °C to around 165 °C and then proceeds through a maximum and eventually (at 200 °C) reaches a similar ratio as for 130 °C. However, for the Cu-SSZ-13 sample, an increasing trend starts from 130 °C and reaches its maximum at 200 °C. Changes in the peak ratio attributed to nitrate or nitrite/NO+ species by increasing temperature can likely be owing to a change in the relative abundance of NO+ with increasing temperature. The large-pore zeolite sample (Cu-BEA) shows a decrease in this ratio, as compared to the small-, and medium-pore zeolites (Cu-CHA and Cu-MFI). This indicates that at low temperature, NO+ is stabilized to a higher degree in the small-pore zeolite than in the medium- and large-pore zeolites, and by increasing temperature the relative NO+ value decreases for small-pore zeolite and increases for large-pore zeolite. This can probably be owing to the higher stability of NO+ at low temperature for SSZ-13 catalyst and its higher stability for the large-pore zeolite at the higher temperature, whereas Cu-ZSM-5 shows maximum NO+ coverage at intermediate temperature. The values for the peak intensity of two distinct peaks in Fig. 1 (NO+ ∼2150 and nitrate species ∼1624) have been evaluated individually and are shown in supplementary information at Fig. S3(a) and (b).†
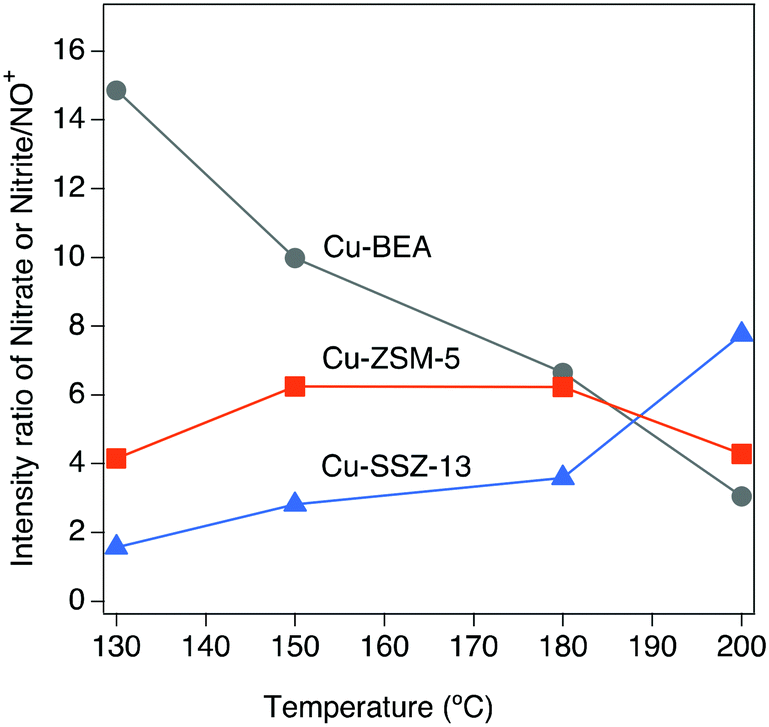 | ||
| Fig. 2 The effect of temperature on the intensity ratio of nitrate or nitrite/NO+ during exposure of the Cu-SSZ-13, Cu-ZSM-5, and Cu-BEA samples to 400 ppm NO and 10% O2 for 15 min at 200, 180, 150, and 130 °C. | ||
In addition, in Fig. S4,† a weak negative band is observed for Cu-SSZ-13 at 3653 cm−1, which likely is caused by the depletion of Brønsted acid sites (Si–OH–Al) due to the interaction of NO+ with protons.23,35,38
The addition of 400 ppm NH3 to the 400 ppm NO + 10% O2 feed (experiment 2, Table 1) resulted in the development of a series of absorption peaks for all three samples, which is shown in Fig. S6† for the O–H stretching (a), N–H stretching (b) and N–H bending (c) wavenumber regions. The evolution of peaks versus time is shown in Fig. S7.† Adsorbed ammonia provides information on surface acid sites and their corresponding acidity.39 For all samples, IR features appear at two different regions related to ammonia species after the sample was exposed to 400 ppm NH3, 400 ppm NO, 10% O2 in Ar for 15 min. For adsorbed ammonia, the bands at high wavenumbers (higher than 3000 cm−1) are related to N–H stretching vibrations, and the bands at lower wavenumbers are attributed to N–H bending vibrations. The negative peaks at 3579 and 3608 cm−1 after ammonia adsorption for Cu-SSZ-13 are assigned to O–H stretching vibrations from Brønsted acid sites (–Al–O(H)–Si–) in the zeolite framework, respectively (Fig. 3a). An analogous trend is observed for the Cu-ZSM-5 and Cu-BEA samples (Fig. 3a) for the O–H stretching vibrations from Brønsted acid sites with the values at 3604 and 3606 cm−1, respectively. The results indicate two types of Brønsted acid sites for Cu-SSZ-13 and one type for Cu-ZSM-5 and Cu-BEA.
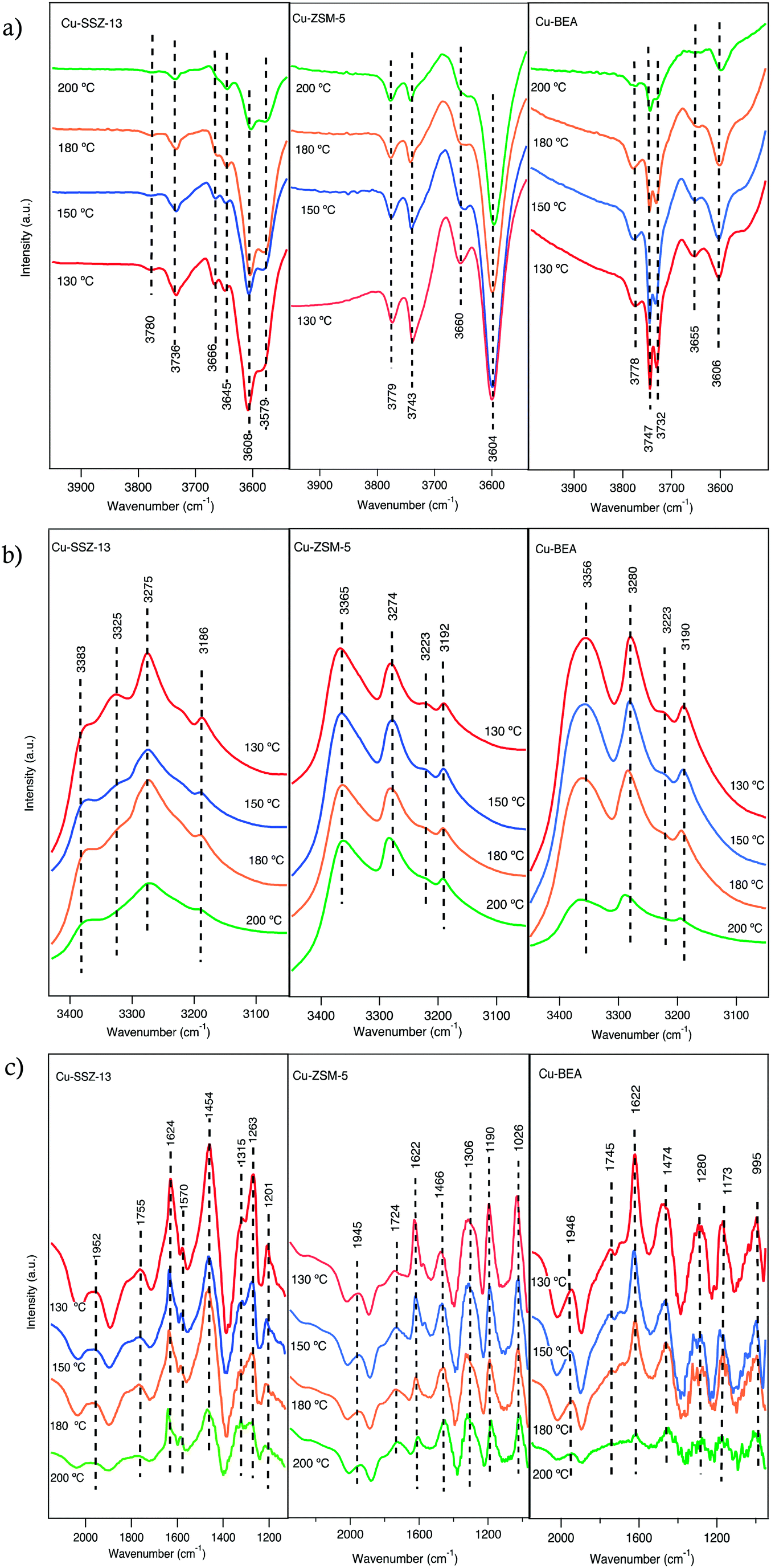 | ||
| Fig. 3 In situ DRIFT spectra after exposing the Cu-SSZ-13, Cu-ZSM-5 and Cu-BEA samples to 400 ppm NO, 400 ppm NH3 and 10% O2 for 15 min at 200, 180, 150 and 130 °C. The fig. (a–c) show different wavenumber regions. | ||
Another type of OH group can appear at wavenumbers above 3700 cm−1, which are attributed to O–H stretching vibrations of Al (OH) or Si (OH) surface species.35,40 The evolution of negative bands at 3736 cm−1 for Cu-SSZ-13 and 3743 cm−1 for Cu-ZSM-5 and 3732 and 3747 cm−1 for Cu-BEA samples upon exposure to NH3 can be attributed to the adsorption of NH3 on Si–OH groups.35 Hence, both surface silanol and structural OH groups in the zeolite framework are consumed or blocked by the adsorption of ammonia.40,41 The other bands at 3780, 3779, and 3778 cm−1 for Cu-SSZ-13, Cu-ZSM-5, and Cu-BEA, respectively, are related to adsorption of NH3 on Al (OH) sites.42
Another group of positive bands observed at 3186 and 3383 cm−1 in the N–H stretching region (Fig. 3b) and positive bands at 1624 and 1263 cm−1 in the N–H bending region (Fig. 3c), can be attributed to ammonia coordinated to Lewis acid sites (copper ions),43,44 while the peaks at 3275 and 1454 cm−1 features NH4+ species adsorbed on Brønsted acid sites.39,45–47 Hence, the peaks that are attributed to the same ammonia species, during NH3 addition, should represent the same trend of increasing or decreasing intensities of related peaks. This implies that while the intensities of the peaks in the N–H stretching region increase over time, the peaks related to N–H bending vibrations should also show an increasing trend (see Fig. S6†). In addition, the peaks at 2152, 2183, 1624, and 1570 cm−1 (Fig. S6†) decrease by the introduction of ammonia, which can be due to that surface NOx species mainly is formed on Brønsted acid sites where NH3 likely adsorbs strongly and can form NH4+ ions and alternatively also can be due to the reaction between NO species and ammonia.46 Simultaneously, adsorbed ammonia species are observed in the N–H stretching vibration region, at 3365 and 3274 cm−1 for Cu-ZSM-5 and at 3356 and 3280 cm−1 for Cu-BEA. Furthermore, adsorbed ammonia visible as peaks at 3192 and 3190 cm−1, for Cu-ZSM-5 and Cu-BEA, respectively are formed. In the N–H bending region, the peaks at 1466 and 1474 cm−1 for Cu-ZSM-5 and Cu-BEA, respectively, attributed to ammonium ions adsorbed on Brønsted acid sites and the emerged peaks at 1622 cm−1 for Cu-ZSM-5 and Cu-BEA attributed to ammonium ions adsorbed on Lewis acidic sites (see Fig. S6†). Comparing the results for the small- to large-pore zeolites at high temperatures shows that the intensity of the peaks related to nitrate and nitrite species is lower in the large-pore zeolite, which can be due to the lower stability of nitrate and ammonia species in the large-pore zeolites compared to medium and small-pore zeolites. The values for the peak intensity for nitrate species (around 1624 cm−1), are shown in S3(b).†
Ma et al.48 investigated NH3-SCR over Cu-SSZ-13 and Cu-SAPO-34 and attributed absorption bands at 1493 and 1449 cm−1 to N–H bending vibrations of NH4+ on Brønsted acid sites. In addition, bands at 1617 and 1270 cm−1 were assigned to N–H bending vibrations of NH3 adsorbed on Lewis acid sites. Also, NH3-SCR over zeolites has been studied by Simons et al.,49 and almost the same observations of the vibrational bands at 1450 and 1610 cm−1 were reported. They recognized that the band at 1450 cm−1 is the most pronounced signal of the whole spectra and corresponds to deformation vibrations of NH4+ as NH3 species are bonded to the Brønsted acid sites by the formation of NH4+. The band at 1610 cm−1 is attributed to N–H bending vibrations of ammonia on Lewis acid sites. These Lewis acid sites can either be related to the transition metal ions exchanged into the porous structure of the zeolite50 or can originate from Al species located at the external surface of the zeolite.35,40,50,51 In addition, Simons et al.49 proposed that broadband at 2700–2300 cm−1 is owing to stretching vibrations of weakly adsorbed ammonia that is not bonded directly to the acidic centers of the zeolite's lattice but alternatively attached to the strongly bonded ammonia as a secondary coordination layer.
Comparing the spectra for the samples exposed to standard SCR conditions at four different temperatures (Fig. 3a), reveals a trend with increasing intensity for the peaks in the O–H stretching region with decreasing temperature. Furthermore, for Cu-SSZ-13, the intensity of the IR bands for adsorbed NH3 at 1624 cm−1 (Fig. 3c) diminishes at 200 °C, while the peak at 1454 cm−1 (Fig. 3c), assigned to NH4+ ions, is still detectable at this temperature. Hence, it can be concluded that at high temperatures, the NH4+ ions are far more stable on Brønsted acid sites (–Al–O(H)–Si–) than on the Lewis acid sites (copper ions). Compared to Cu-SSZ-13, similar trends for ammonia adsorption are also observed during the adsorption process on Cu-ZSM-5 and Cu-BEA up to 180 °C, whereas a further increase in the temperature results in the disappearance of these two peaks. These results indicate that the ammonia-derived species adsorbed on Cu-ZSM-5 and Cu-BEA correspond to the adsorbed species on Cu-SSZ-13. Similarly, as for Cu-SSZ-13, ammonia adsorbs on both Lewis and Brønsted acidic sites in Cu-ZSM-5 and Cu-BEA, however, ammonia is less stable on Lewis acidic sites than on Brønsted acid sites and desorbs easily at 200 °C.45 It is important to emphasize that the SCR reaction is complex in terms of reaction stoichiometry, as three reactants need to be activated and the nature of the catalytically active Cu species is crucial. From the low- to high-temperature reaction regimes,24,52 the intensity of the peaks in the O–H stretching region decreases with increasing temperature between 130 and 200 °C which demonstrates the temperature dependency of the nature of the active Cu sites. The NH3-SCR reaction advances through reduction and oxidation steps.53 Copper ions are reduced from Cu(II) to Cu(I), while Cu(I) is reoxidized to Cu(II) in the reduction and oxidation step, respectively. It has been shown that under standard NH3-SCR reaction conditions at low temperatures, the Cu ions are solvated by NH3.54–56 Two NH3 ligands are needed for the solvation of Cu(I) forming a linear Cu(NH3)2+ complex, while for Cu(II) several types of four-fold coordinated complexes can form, such as Cu(NH3)42+, Cu(OH)(NH3)3+, and complexes containing a mixture of NH3 and NOx ligands.53,57 Also, above 250 °C, Cu ions coordinated to oxygen in the zeolite framework.55 Also, Gao et al.23 demonstrate that at low temperatures, the involvement of two Cu sites is essential for the standard SCR reaction, and the SCR reaction rate varies roughly linearly with Brønsted acid density. Furthermore, it should be considered that, O2 dissociation is an essential step in the NH3-SCR reaction,52,54,58 and its adsorption is advanced owing to the presence of Cu(NH3)2+ pairs.53,59 It is proposed that, in the NH3-SCR reaction first O2 is adsorbed on the surface and prior to the NH3 adsorption step, NO is adsorbed on the surface of zeolite as NO+, and the activation of the NH3 molecule occurs by its coordination to NO+, and the proton in the N–H bond is conveyed to oxygen to form an OH− and a nitrosamide (H2NNO) species. Two nitrosamide species desorb from the Cu sites and decompose on Brønsted acid sites to form N2 and H2O. NO+ is formed by adsorption of NO on the Cu2+ sites and through the reduction of Cu(II) to Cu(I). During the NH3-SCR reaction, HONO can form and react with gas-phase NH3 and form an ammonium nitrite complex, which subsequently can decompose to N2 and H2O. Another HONO species can form on the Cu+ site where NO+ and OH− already are absorbed. Coordination of an NH3 molecule to the NO+ species results in the formation of HONO and H2NNO species due to the transfer of a proton to the nitrite species.53
Upon the addition of NO2 (200 ppm) to the inlet gas composition (Fig. 4), a decreasing intensity trend appears for the peaks in both N–H stretching and bending vibration region over time, which indicates that NO2 consumes accumulated NH3 from the previous standard SCR reaction step, i.e., indicating a higher reactivity of NO2 compared to NO.
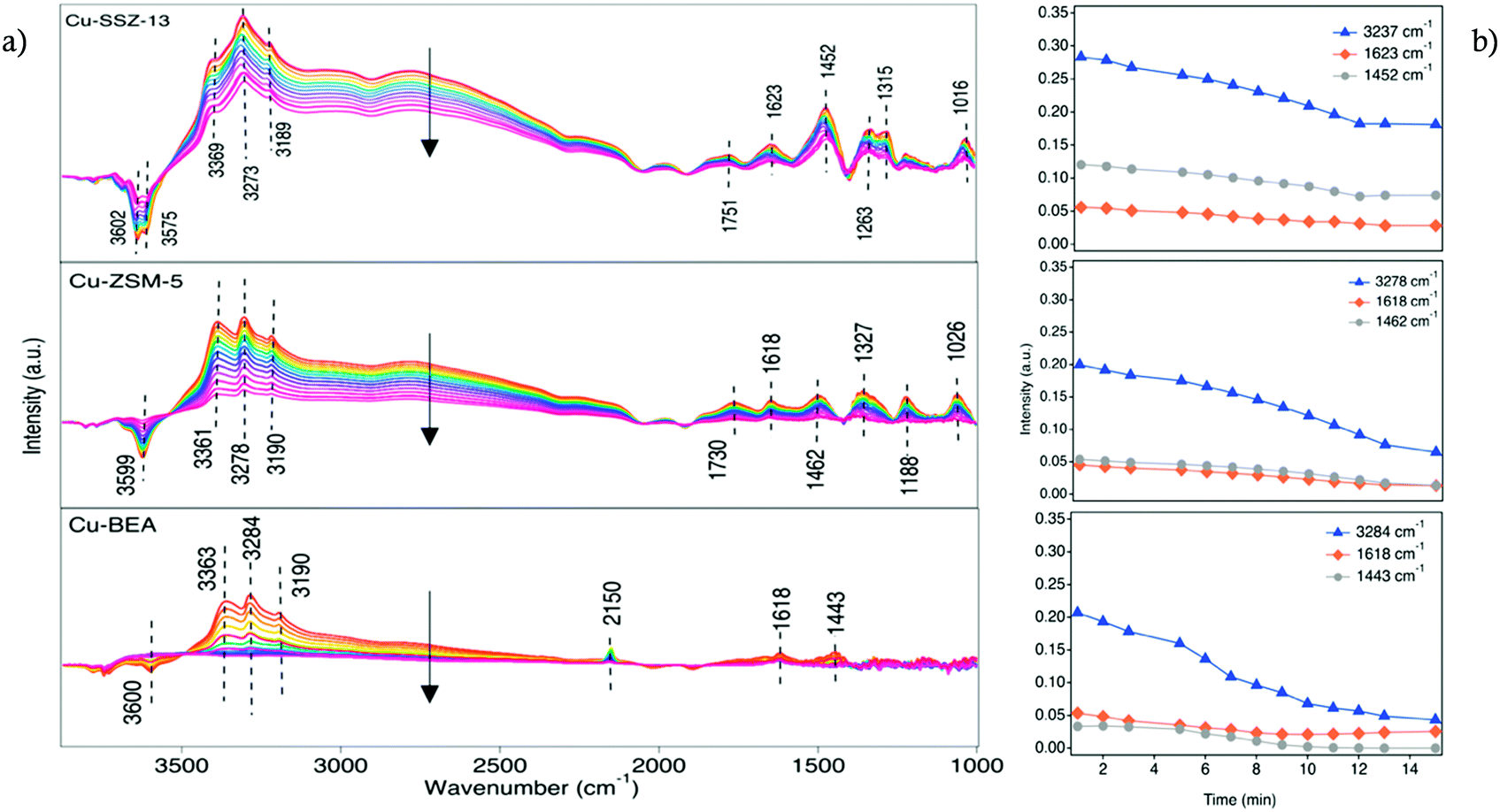 | ||
| Fig. 4 a) In situ DRIFT spectra during exposure of the Cu-SSZ-13, Cu-ZSM-5, and Cu-BEA samples to 200 ppm NO, 200 ppm NO2 and 10% O2 at 200 °C, b) the intensity of three different peaks versus time. | ||
A distinct rate of NH3 consumption is seen for species adsorbed on both Lewis and Brønsted acid sites in the series of DRIFT spectra. The intensity of the peak related to NH3 species adsorbed on the copper sites at 1623 cm−1 decreases with time after the exposure to NO2. On the other hand, the band at 1452 cm−1, indicating NH3 adsorbed on Brønsted acid sites, does not decrease in intensity to a large extent. While NH3 on Lewis acid sites is consumed by NO2, nitrate species start to form on Lewis acid sites by the interactions with NO2. According to the DRIFTS results for all three samples, it can be seen that the trend with decreasing intensity of the peaks related to the NH3 species is more pronounced for the large-pore zeolite compared to the medium and small-pore ones. This indicates that NO2 consumes NH3, accumulated in the previous step of the experiment and the degree of consumption seems to be connected to the framework of the zeolite. It can be assumed that small and medium pore-zeolite accumulates more ammonia in its cages compared to large-pore zeolite, and the consumption of trapped ammonia needs a higher amount of NO2 and a longer reaction time. On the other hand, the intensity of the peaks at the N–H stretching region for the Cu-BEA sample decreases to almost zero within the same reaction time, which can be due to the less amount of ammonia trapped in large cages and consequently its faster consumption compared to other two zeolites. Chen et al.7 reported that the formation of NO+ decreases with time upon exposure to NO2 and O2 and it has been revealed that after 6 minutes of exposure, the intensity of the NO+ peak decreased to about 2/3 of its initial size. Also, at the same time, the intensity of the peaks related to nitrate species increased slightly. Also, Zhu et al.60 observed that under fast NH3-SCR reaction conditions, the various reactivity of NH4NO3 with NO leads to the distinguished NO2 effects on N2O formation for Cu-SSZ-13 and Cu-SSZ-39. For instance, for Cu-SSZ-13, the slow reaction between NH4NO3 and NO results in accumulation of NH4NO3 and blocking of the pores of the zeolite, and subsequently inhibition of the SCR activity by NO2.
These results are in agreement with NH3-TPD experiments carried out in the flow reactor to quantify the NH3 storage (data reported in our recent publication).61 From the integrated peak area of TPD profiles, it is observed that the NH3 storage is roughly in the same range for Cu-SSZ-13 (0.756 mmol g−1) and Cu-ZSM-5 (0.791 mmol g−1) followed by Cu-BEA (0.626 mmol g−1). Based on the discussed DRIFTS results in Fig. 3), it is clear that in the Cu-SSZ-13 and Cu-ZSM-5 samples, NH3 is strongly bonded to the OH Brønsted acid sites rather than to the silanol groups, but in the case of Cu-BEA NH3 is mainly bonded weakly to silanol groups. This facilitates NH3 consumption with NO2 in large-pore zeolites and results in a fast-decreasing trend in N–H stretching and bending region compared to medium- and small-pore zeolites.
3.3. Formation of N2O during standard and fast SCR reaction conditions
The N2O formation is evaluated by kinetic tests in the flow reactor during both standard and fast SCR reaction conditions (original data are shown in our previous work).61 Further details regarding the experimental descriptions are given in section 2.3.In Fig. 5, the N2O concentration is shown as a function of catalyst temperature for Cu-SSZ-13, Cu-ZSM-5, and Cu-BEA during both standard and fast SCR reaction conditions at 130, 150, 180, and 200 °C. The results reveal a clear trend for low and high temperatures. As can be seen, the N2O formation is higher for Cu-BEA compared to Cu-ZSM-5 and Cu-SSZ-13 for the entire temperature range.
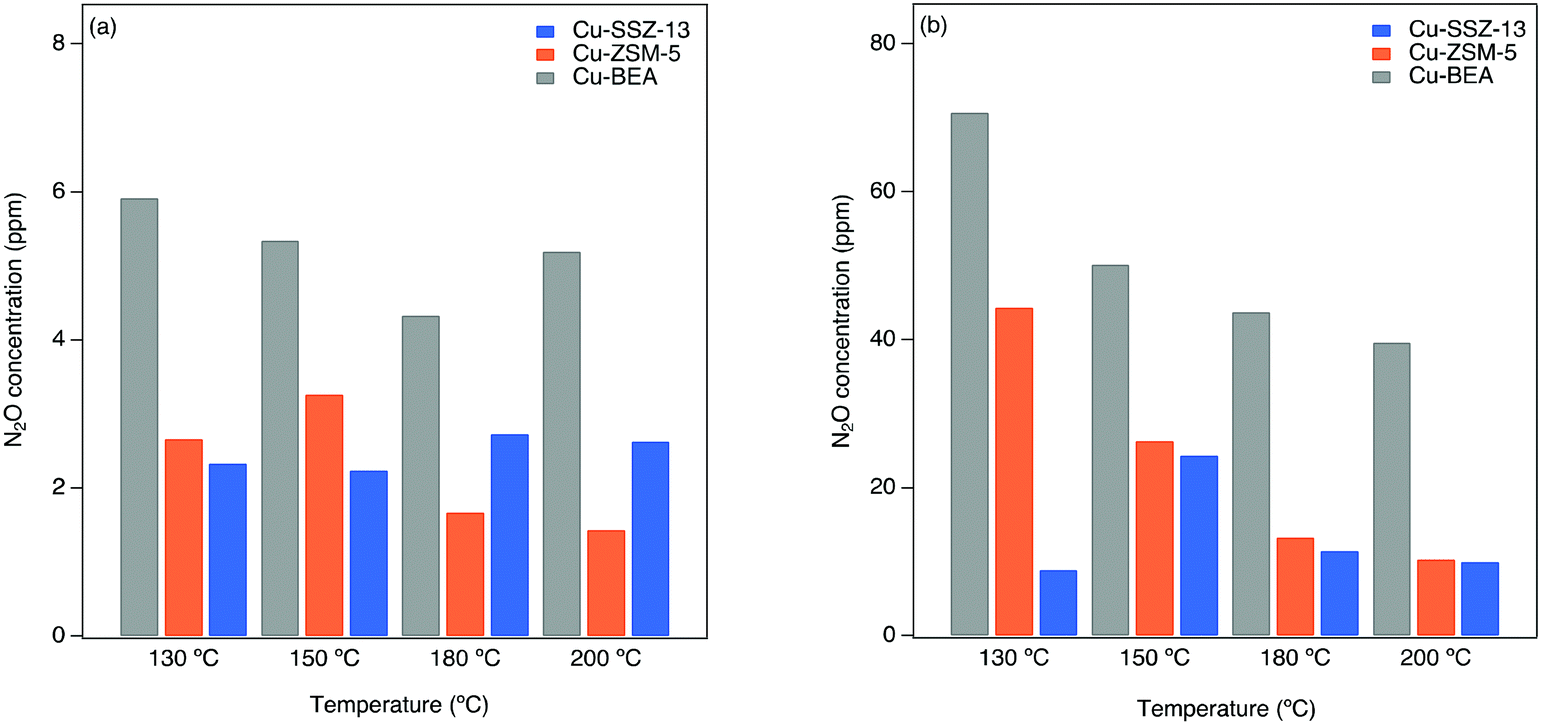 | ||
| Fig. 5 N2O concentration as a function of catalyst temperature within a) standard (400 ppm NO, 400 ppm NH3, 10% O2) and b) fast SCR (200 ppm NO, 200 ppm NO2, 400 ppm NH3, 10% O2) reaction over Cu-based zeolites. | ||
During fast SCR reaction conditions, the formation of N2O is much higher for all samples compared to standard SCR reaction conditions. It is noteworthy that the presence of NO2 has a considerable effect on the SCR activity at lower temperatures,1,62 which is likely due to the higher activity of NO2 compared to O2 to reoxidize Cu(I) to Cu(II).In Fig. 5, the highest amount of N2O during fast SCR conditions is observed for Cu-BEA, followed by Cu-ZSM-5 and Cu-SSZ-13.
As mentioned earlier, the N2O formation during the SCR reaction at 250–280 °C proceeds partly via the decomposition of NH4NO3, which is an intermediate formed on the zeolite when ammonia reacts with the surface nitrate species during NH3-SCR reaction conditions.2,63,64 It is also proposed by Lee et al.65 that over Cu-ZSM-5 and Cu-SSZ-13 catalyst, the N2O yield is due to the decomposition of NH4NO3. The same was also reported by Devadas et al.,66 for low-temperature N2O formation over Fe-ZSM-5. On the contrary, Feng et al.8 recently proposed that N2O formation at low temperatures over Cu-CHA is connected to H2NNO decomposition over (NH3)2Cu–OOH–Cu(NH3)2 complexes.
The ammonium nitrate (AN) TPD results over the samples indicate that AN has a shorter resistance time in large-pore zeolites, which results in a continuous decomposition of AN to N2O.61 These results are in agreement with the DRIFTS measurements during NO2 adsorption (step 5 in Table 1). As discussed in section 3.2, Fig. 4, for the large-pore zeolite all pre-adsorbed NH3 can be consumed by the introduced NO2. This can lead to higher and faster AN formation and release during the SCR reaction and the formation of more N2O by its decomposition. Furthermore, based on DRIFTS studies on NO oxidation (depicted in Fig. 2), at low temperatures, the nitrate/NO+ ratio is higher for Cu-BEA compared to Cu-ZSM-5 and Cu-SSZ-13.
3.4. Effect of NO/NO2 ratio on the evolution of surface species
To investigate the effect of the NO/NO2 ratio on the formation of N2O, a series of experiments was carried out at three different bed temperatures (220, 200, 180 °C) for the Cu-SSZ-13, Cu-ZSM-5, and Cu-BEA samples with varying NO/NO2 ratio from 4:0, 1:1, 1:1.5, 1:3, 1:4 to 0:4. The results for the integrated peak area of N2O (2237 cm−1 with a shoulder at 2208 cm−1) and NO+ (2150 cm−1) species at the steady-state of the reaction are shown in Fig. 6. The peak position for N2O has been measured by DRIFTS on both KBr and diamond powder and compared with the gas phase N2O obtained from Nist (the symmetric NN and NO stretch is at 2202 and 1270 cm−1, respectively).67 The related spectra for the peak position of N2O are shown in Fig. S10.† Based on the obtained results from related peak integration (original spectra are shown in Fig. S9(a–c)†), even though there is N2O formation in the presence of only NO or NO2, it is clear that for higher N2O formation the presence of both NO and NO2 is required. It is also obvious from reactor studies (data is shown in section 3.3) that a higher amount of N2O is formed during the fast SCR reaction conditions compared to standard SCR.
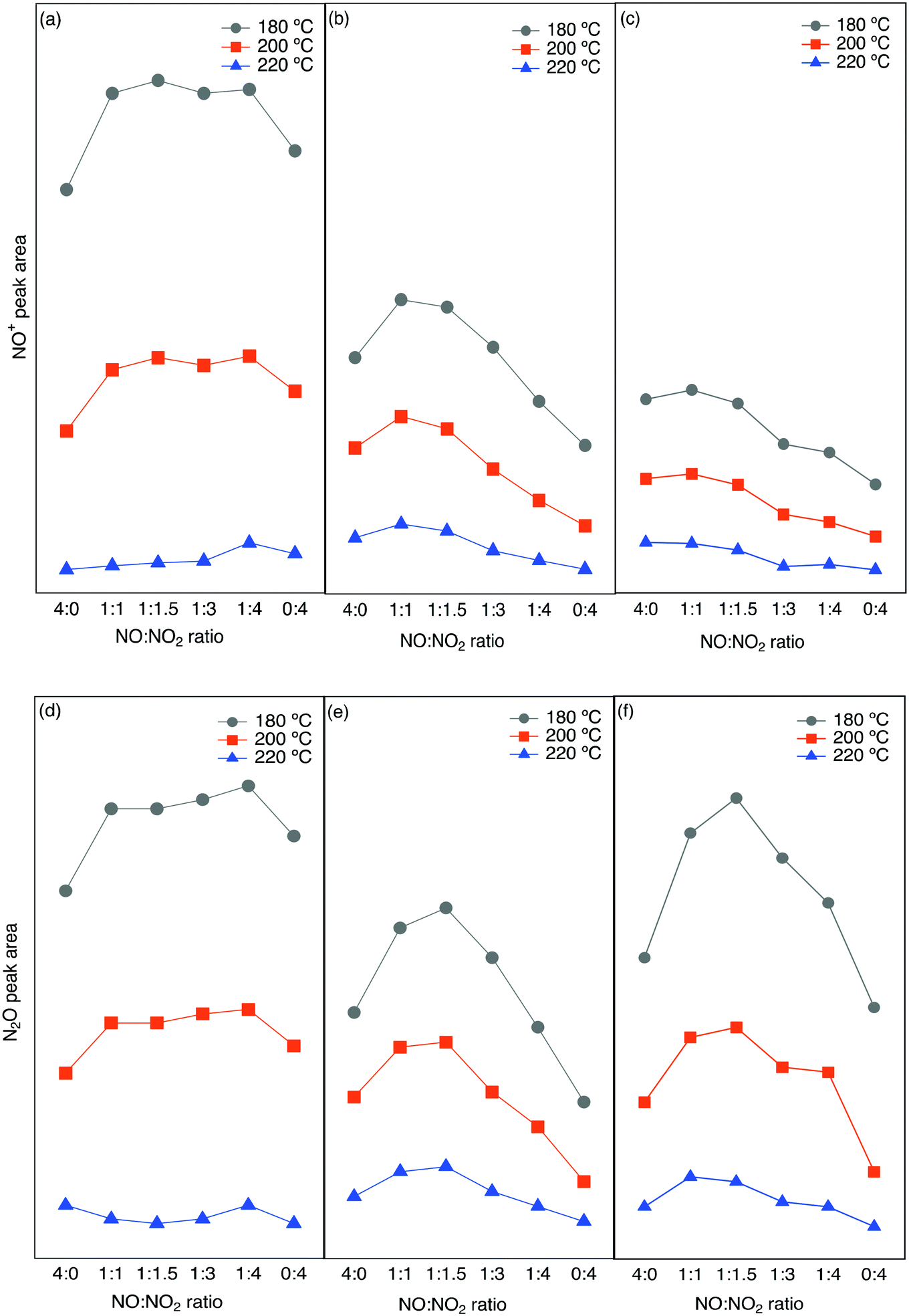 | ||
| Fig. 6 Integrated peak area from in situ DRIFTS experiments for NO+ (a–c) and N2O (d–f) for different NO/NO2 ratios at three different temperatures over (a and d) Cu-SSZ-13, (b and e) Cu-ZSM-5 and (c and f) Cu-BEA. | ||
The integrated N2O and NO+ peak area are plotted as a function of the NO/NO2 ratio, and it clearly shows a higher peak area for the small-pore zeolite sample compared to the other two samples. Comparing the N2O peak area for the three samples shows that for the small-pore Cu-SSZ-13 sample, at all three temperatures the N2O peak area gradually increases from NO/NO2 = 0:4 to 1:4 and decreases again, while for the medium (Cu-ZSM-5) and large-pore (Cu-BEA) samples the N2O peak area reaches a maximum at NO/NO2 = 1:1.5. A further increase in the NO/NO2 ratio results in a decline in N2O formation for the catalysts. The same trend is seen for the Cu-ZSM-5 and Cu-BEA samples at all three temperatures. Furthermore, the highest value for the NO+ peak area for Cu-SSZ-13 appears at a NO/NO2 ratio of 1:1.5, whereas Cu-ZSM-5 and Cu-BEA show the highest peak area for the 1:1 ratio. At 180 °C, however, N2O formation on Cu-SSZ-13 increases slightly when the NO/NO2 ratio increases from 0:4 to 1:4 and starts to decrease again at a higher ratio. For Cu-BEA, the value of NO+ reveals an increasing trend with an increased ratio. Almost the same trend can be seen for the NO+ peak as well, which can be due to the relation between the amount of NO+ and N2O formation. For Cu-SSZ-13, at all temperatures, the peak area for N2O formation is the highest at NO/NO2 ratio of 1:4. For the other two zeolites, the highest amount is seen for the ratio of 1:1 for all three temperatures. Furthermore, NO+ and N2O formation are higher at a lower temperature, which can be due to the coverage of NO+ that decreases with increasing temperature. Based on the study by Liu et al.68 on the effect of NOx ratio on the N2O formation, the N2O amount is different below and above 250 °C. Below 250 °C there should be maximum N2O formation with less NO2 as is in line with our study. The reason for this can be the formation of a higher amount of ammonium nitrate with high NO2 concentration and consequently blocking of the surface, which results in lower conversion and thereby a lower N2O formation. At higher temperatures, no surface blocking occurs and the maximum N2O formation is achieved for 100% NO2.
The integrated peak area attributed to nitrate species (1530–1700 cm−1) has been plotted versus the NO/NO2 ratio in Fig. 7. A similar trend is seen for all three zeolites, with the highest value of nitrate species at low temperature and low NO2 concentrations. At an NO/NO2 ratio of 4:0, which is the standard SCR reaction condition, the highest peak area can be related to the lower reaction rate. On the other hand, by the addition of NO2 with a NO/NO2 ratio of 1:1, the fast SCR reaction proceeds, consuming the surface nitrate species giving rise to a decrease in the related peak area.
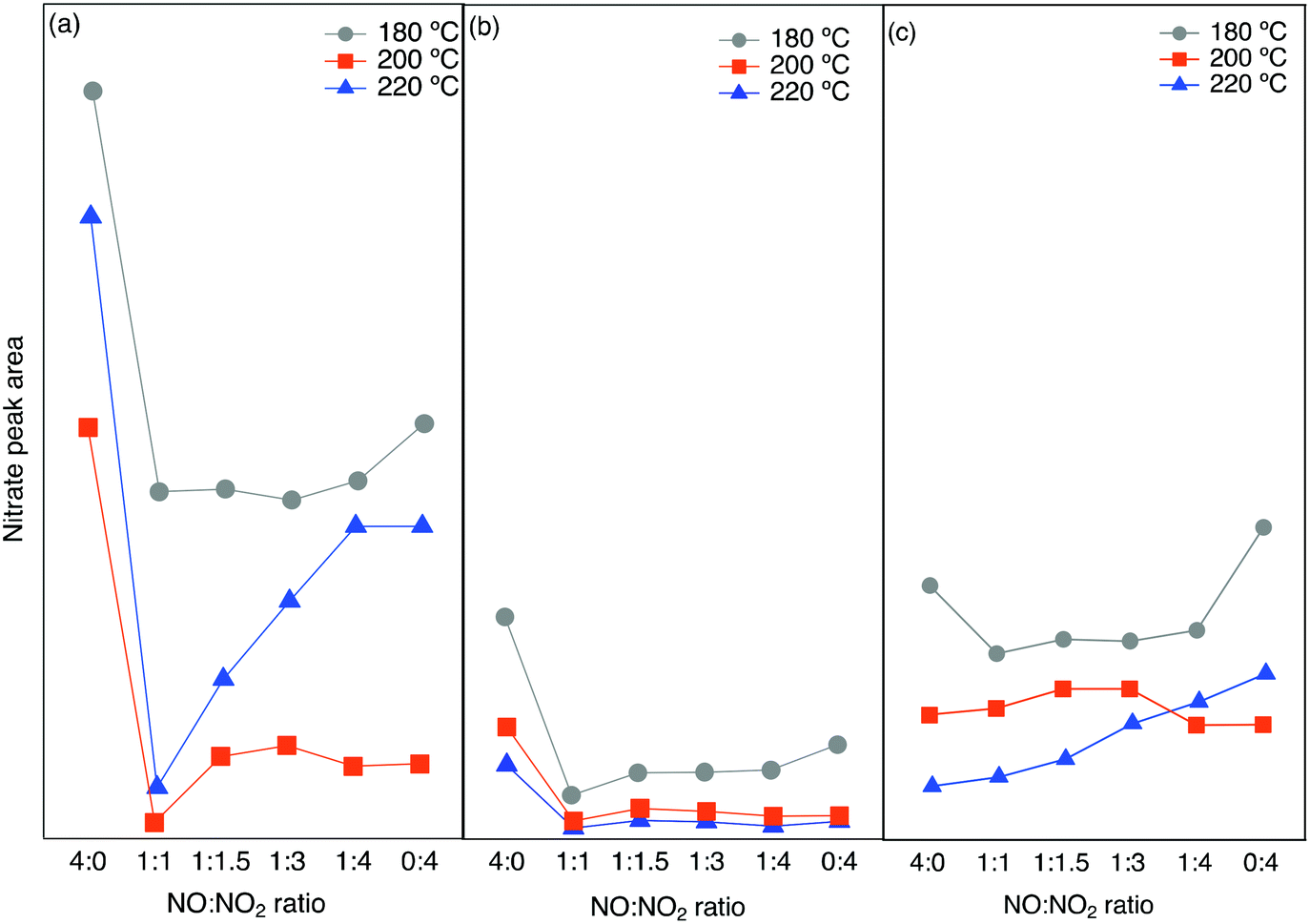 | ||
| Fig. 7 Integrated peak area from in situ DRIFTS experiments for nitrate species for different NO/NO2 ratios at three different temperatures over a) Cu-SSZ-13, b) Cu-ZSM-5 and c) Cu-BEA. | ||
The addition of more NO2 gradually increases the related peak area. This is in line with the lowest amount of NO+ and N2O formation when we have the highest nitrate species.
In summary, the zeolite framework structure and reaction temperature have an influence on the formation of undesired by-products during ammonia-SCR of NOx. The results from DRIFTS and flow reactor experiments reveal that NO+ and nitrate species are the major surface species during NO oxidation. Evaluating the effect of various NOx-ratio shows the higher formation of NO+ and N2O species at lower temperatures and higher N2O formation for the large-pore zeolite (Cu-BEA) compared to medium- (Cu-ZSM-5) and small-pore (Cu-SSZ-13), based on the results from DRIFTS studies and flow reactor experiments.
3.5. DFT calculations
DFT calculations were performed to assist the assignments of DRIFTS results. Among the three studied zeolites, SSZ-13 has emerged as the preferred system for NH3-SCR and is, therefore, considered in the DFT calculations.To obtain a reference, the wavenumber of the N–O stretch vibration for NO and NO+ in the gas phase was calculated, resulting in 1923 and 2405 cm−1, respectively. The calculated wavenumbers are higher than the experimental corresponding values by a factor of 1.03, which is consistent with previous DFT studies using the same approach.69
Previous work suggests that the peak at 2156 cm−1 should be attributed to the N–O stretching vibration of NO+ adsorbed on Lewis acid sites.9 However, Hadjiivanov et al.33 assign a peak at 2132 cm−1 to NO adsorbed on H-ZSM-5. As the relationship between the NO wavenumber and the adsorption site remains unclear, the wavenumber of N–O stretching vibration of NO on potential sites in SSZ-13 was calculated, see Fig. 9. NO adsorbed at Lewis acid site has an adsorption energy of 0.6 eV and a stretching vibration at 1926 cm−1, which indicates that NO at this adsorption site is neutral. However, when NO occupies Al-Lewis acid sites (O–Al–O), the wavenumber is shifted to 2060 cm−1 and the adsorption energy is increased to 0.81 eV. As a comparison, NO adsorption over SSZ-13 in the absence of Cu-sites was also investigated. There are four types of Brønsted acid sites in SSZ-13 with one Al on the six-membered ring. The structure is shown in Fig. 8. The adsorption energy for NO adsorbed on the different sites, is high and similar, being about 2 eV (within 0.1 eV) and the wavenumber for the stretching vibration is in the range from 2047 to 2074 cm−1. Comparing the results for NO and NO+ in the gas phase indicates that the peak around 2150 cm−1 should be associated with NO adsorbed on Cu-Lewis acid sites (Cu2+). Moreover, the similar adsorption energies and slightly different frequencies for NO adsorbed on Al-Lewis acid sites (O–Al–O) close to the Cu-Lewis sites could explain the double absorption peaks in the DRIFT spectra (Fig. 1).
 | ||
| Fig. 8 Four types of Brønsted acid sites in SSZ-13. | ||
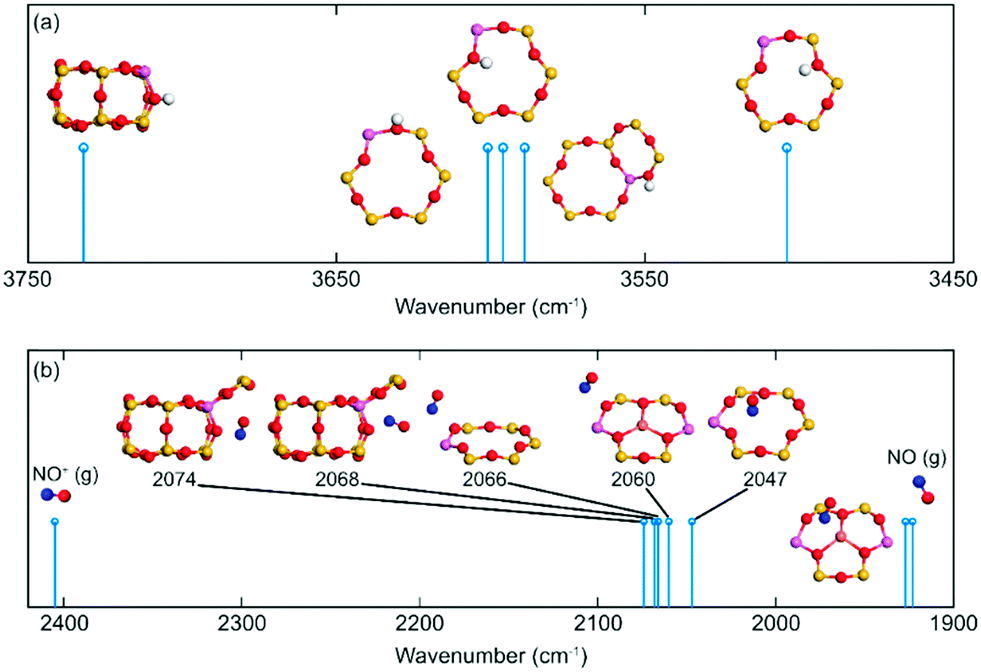 | ||
| Fig. 9 Simulated vibrations for (a) O–H and (b) N–O on different CHA sites. Atomic color codes: H (white), N (blue), O (red), Al (pink), and Si (yellow). | ||
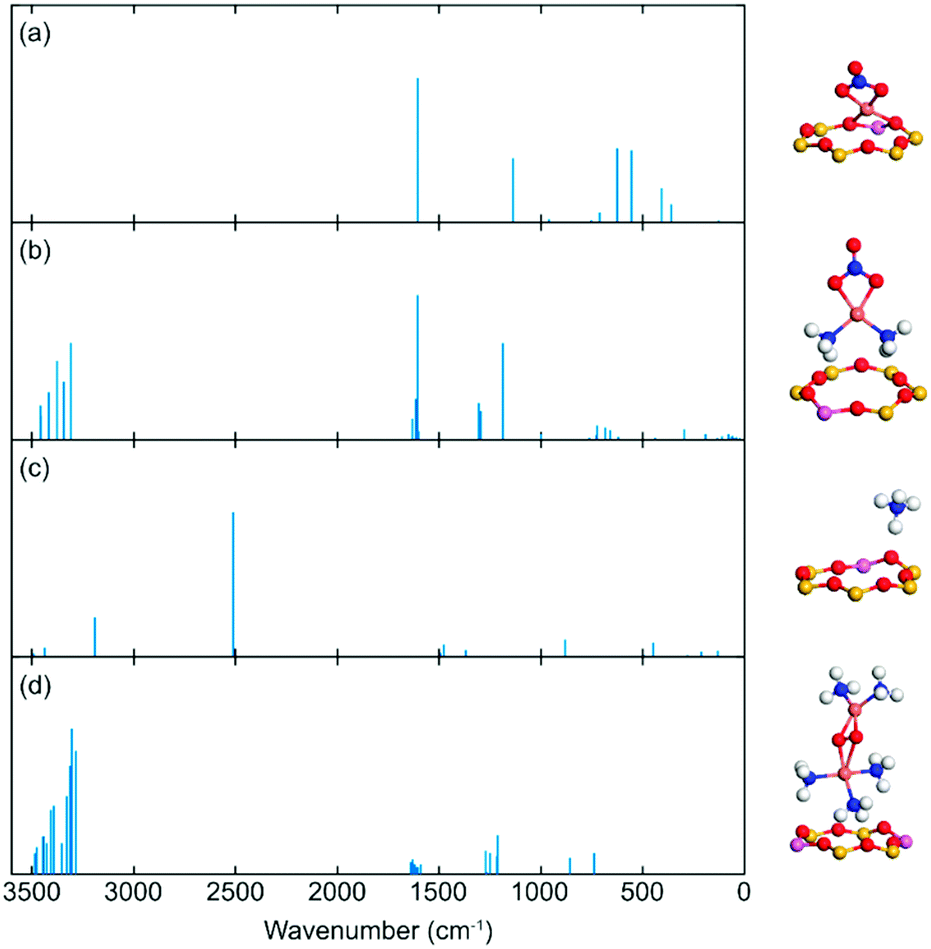 | ||
| Fig. 10 Simulated IR vibrations for different species adsorbed on different Cu-CHA and CHA sites. (a) [CuNO3]+ complex, (b) [Cu(NH3)2NO3]+ complex, (c) NH4+, and (d) [Cu2(NH3)5(O2)]2+ complex. Atomic colour codes: H (white), N (blue), O (red), Al (pink), Si (yellow) and Cu (bronze). | ||
The negative peaks after ammonia adsorption in the high wavenumbers region shown in Fig. 3(a) were considered to originate from O–H stretching vibrations from Brønsted acid sites in the zeolite framework. It is noteworthy that SSZ-13 has a more complex multiple peak profile than the other two zeolites. Thus, the O–H frequency of H on different sites has been calculated to compare with the experimental results. The data indicate that the position of the stretching vibration for O–H depends on the adsorption site. O–H stretching vibrations of Si (OH) gives a relatively low wavenumber (3504 cm−1), whereas the Al (OH) surface species results in a wavenumber at about 3600 cm−1. The highest calculated wavenumber of O–H on Brønsted acid sites is 3732 cm−1. Al (OH) is the preferred adsorption site with adsorption energy that is about 0.7 eV higher than Si (OH). This is probably the reason why the measured wavenumber is concentrated mainly at 3579 and 3608 cm−1.
The DRIFTS results indicate that nitrate species are the main product formed during the NO oxidation step, which contributed to the peak at about 1620 cm−1, being the N–O stretching vibrations of surface nitrate species. It is noteworthy that this peak appears independently of whether ammonia is present or not in all three zeolites. To study this, the IR spectra of framework nitrate ([CuNO3]+) and mobile nitrate species ([Cu(NH3)2NO3]+) have been calculated (Fig. 10). Both cases show a peak of 1604 cm−1 which is associated with a O–N stretching vibrations in NO3−. This result is an illustration of the 1620 and 1624 cm−1 peaks of Cu-SSZ-13 in Fig. 1 and 3.
Previous spectroscopy and first-principles calculations have demonstrated that Cu(I) is preferably present in the form of linear Cu(NH3)2+ under low-temperature SCR reaction conditions. The linear complex has also been proven to be a critical site for O2 activation by forming Cu-peroxo ([Cu2(NH3)4(O2)]2+). In the recent work of Negri et al.,70 the strong adsorption of extra NH3 on Cu-peroxo has been reported. Based on the measurement conditions, we calculated the IR spectra of [Cu2(NH3)5(O2)]2+ complex for reference. The stretching vibrations of the N–H bond are in the 3283 to 3484 cm−1 range, whereas the bending wavenumbers are between 1589 to 1638 cm−1 and 1213 to 1272 cm−1, respectively. These peaks are consistent with the measurements.43,44 The DFT calculations of NH3 adsorption on Brønsted acid sites (NH4+) shows high-intensity signatures at 3191, 2512, and 1492 cm−1, which are in good agreement with the DRIFTS results in Fig. S8.†
4. Conclusions
The influence of the zeolite framework structure on the formation of N2O during ammonia SCR of NOx has been studied for three different zeolite samples with varying framework structures: Cu-SSZ-13 (CHA), Cu-ZSM-5 (MFI), and Cu-BEA (BEA). The results show that the formation of N2O is closely connected with the structure of the zeolite, feed gas composition, and temperature. At lower temperatures, the formation of NO+ species is higher for the Cu-SSZ-13, Cu-ZSM-5, and Cu-BEA samples and starts to decrease when increasing the temperature to 200 °C. In addition, comparing the intensity ratio of nitrate or nitrite/NO+ for the samples at different temperatures reveals that at low temperature, this value is higher for Cu-BEA compared to the two other zeolites, which decreases by increasing temperature. This can be owing to the higher stabilization of NO+ in the small-pore zeolite than in the zeolites with medium and large-pores at low temperatures. Furthermore, by adding NH3, i.e., resulting in standard SCR reaction, absorption peaks corresponding to N–H stretching and bending vibrations appear, and at the same time, negative peaks appear in the O–H stretching region, indicating blocking/replacement of surface OH-groups by NH3. Interestingly, when NH3 is removed and NO2 is added to the feed, the N–H stretching and bending vibrations show a trend with decreasing intensity, with the decrease being more pronounced for increasing pore size. It is likely that ammonia surface species, accumulated during the previous step, are consumed by NO2 and the degree of consumption seems to be related to the framework of the zeolite. Also, the weakly bonded NH3 on silanol groups in Cu-BEA accelerates its consumption by adding NO2 and can be the result of a higher intensity decrease in the N–H stretching and bending vibration region compared to the other two zeolite samples. Additional experiments with the Cu-based zeolites, at different NO/NO2 ratios during SCR reaction conditions show that the presence of both NO and NO2 is essential for N2O formation, with the highest formation for an NO/NO2 ratio of 1:1. Based on the obtained results, it can be concluded that both reaction temperature and framework structure of the zeolite have a direct influence on the N2O formation. The results from both DRIFTS studies and flow reactor experiments illustrate higher N2O formation for the large-pore zeolite compared to medium- and small-pore zeolites. At low temperatures, this can be due to a higher formation of ammonium nitrate over the Cu-BEA zeolite and its subsequent decomposition to N2O.
In addition, density functional theory calculations for Cu-SSZ-13 and H-SSZ-13 reveals that NO can be adsorbed at different Al–O–Al sites with similar adsorption energies and N–O stretching vibrations, which rationalizes the emergence of the double absorption peaks in the DRIFT spectrum. Furthermore, in the O–H stretching region, a low wavenumber is observed for Si (OH) compared to the value for Al (OH) surface species. Also, simulating the IR spectra of the DRIFTS results for mobile nitrate species and framework nitrate provides a peak around 1604 cm−1 attributed to the O–N bond in NO3−. In general, the N2O formation during the NH3-SCR reaction depends on the emergence of various intermediates, and the surface species and follows different mechanistic pathways at low and high temperatures. It seems that adsorbed NO and formed nitrate species during NO oxidation can be two of the main contributors and initiators for the N2O formation and it seems that both Cu-Lewis (Cu2+) and Al-Lewis (Al–O–Al) sites can provide adsorption sites for the formed NO+.
Author contributions
Ghodsieh Isapour: investigation – conducting a research and investigation process, specifically performing the experiments, or data/evidence collection. Visualization – preparation, creation, and/or presentation of the published work, specifically visualization/data presentation. Writing the original draft – preparation, creation, and/or presentation of the published work, specifically writing the initial draft (including substantive translation). Aiyong Wang: conceptualization – ideas; formulation or evolution of overarching research goals and aims. Joonsoo Han: investigation – conducting a research and investigation process, specifically performing the experiments, or data/evidence collection for the flow reactor section. Yingxin Feng: formal analysis – application of statistical, mathematical, computational, or other formal techniques to analyze or synthesize study data. Writing the original draft – writing the initial draft for the DFT calculation section. Henrik Grönbeck: conceptualization – ideas; formulation or evolution of overarching research goals and aims. Derek Creaser: conceptualization – ideas; formulation or evolution of overarching research goals and aims. Louise Olsson: conceptualization – ideas; formulation or evolution of overarching research goals and aims. Magnus Skoglundh: conceptualization – ideas; formulation or evolution of overarching research goals and aims. Supervision – oversight and leadership responsibility for the research activity planning and execution, including mentorship external to the core team. Hanna Härelind: conceptualization – ideas; formulation or evolution of overarching research goals and aims. Supervision – oversight and leadership responsibility for the research activity planning and execution, including mentorship external to the core team.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been performed within the Competence Centre for Catalysis, which is hosted by the Chalmers University of Technology and financially supported by the Swedish Energy Agency, Chalmers, and the member companies; AB Volvo, ECAPS AB, Johnson Matthey AB, Preem AB, Scania CV AB, and Umicore Denmark ApS. The calculations have been performed at C3SE (Göteborg) through a SNIC grant and additional financial support was obtained from the Swedish Energy Agency project 47110-1.Notes and references
- H. Kubota, C. Liu, T. Toyao, Z. Maeno, M. Ogura, N. Nakazawa, S. Inagaki, Y. Kubota and K. I. Shimizu, ACS Catal., 2020, 10, 2334–2344 CrossRef CAS.
- D. Zhang and R. T. Yang, Energy Fuels, 2018, 32, 2170–2182 CrossRef CAS.
- M. J. Kim, S. J. Lee, I. S. Ryu, M. W. Jeon, S. H. Moon, H. S. Roh and S. G. Jeon, Mol. Catal., 2017, 442, 202–207 CrossRef CAS.
- T. Nobukawa, M. Yoshida, K. Okumura, K. Tomishige and K. Kunimori, J. Catal., 2005, 229, 374–388 CrossRef CAS.
- J. Pérez-Ramírez, F. Kapteijn, K. Schöffel and J. A. Moulijn, Appl. Catal., B, 2003, 44, 117–151 CrossRef.
- K. Leistner, O. Mihai, K. Wijayanti, A. Kumar, K. Kamasamudram, N. W. Currier, A. Yezerets and L. Olsson, Catal. Today, 2015, 258, 49–55 CrossRef CAS.
- H. Y. Chen, M. Kollar, Z. Wei, F. Gao, Y. Wang, J. Szanyi and C. H. F. Peden, Catal. Today, 2019, 320, 61–71 CrossRef CAS.
- Y. Feng, T. V. W. Janssens, P. N. R. Vennestrøm, J. Jansson, M. Skoglundh and H. Grönbeck, J. Phys. Chem. C, 2021, 125(40), 21975–21987 CrossRef.
- M. P. Ruggeri, I. Nova, E. Tronconi, J. A. Pihl, T. J. Toops and W. P. Partridge, Appl. Catal., B, 2015, 166–167, 181–192 CrossRef CAS.
- H. Liu, C. You and H. Wang, Chem. Eng. J., 2020, 382, 122756 CrossRef CAS.
- M. Anstrom, N. Y. Topsøe and J. A. Dumesic, J. Catal., 2003, 213, 115–125 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- R. A. Vargas-Hernández, J. Phys. Chem. A, 2020, 124, 4053–4061 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Allouche, J. Comput. Chem., 2012, 32, 174–182 CrossRef PubMed.
- L. Y. Isseroff and E. A. Carter, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 1–7 CrossRef.
- D. Karhánek, dakarhanek/VASP-infrared-intensities: VASP-infrared-intensities, DOI:10.5281/zenodo.3930989.
- F. Gao, N. M. Washton, Y. Wang, M. Kollár, J. Szanyi and C. H. F. Peden, J. Catal., 2015, 331, 25–38 CrossRef CAS.
- F. Gao, E. D. Walter, M. Kollar, Y. Wang, J. Szanyi and C. H. F. Peden, J. Catal., 2014, 319, 1–14 CrossRef CAS.
- C. Paolucci, A. A. Parekh, I. Khurana, J. R. Di Iorio, H. Li, J. D. Albarracin Caballero, A. J. Shih, T. Anggara, W. N. Delgass, J. T. Miller, F. H. Ribeiro, R. Gounder and W. F. Schneider, J. Am. Chem. Soc., 2016, 138, 6028–6048 CrossRef CAS PubMed.
- J. Liang, J. Su, Y. Wang, Z. Lin, W. Mu, H. Zheng, R. Zou, F. Liao and J. Lin, Microporous Mesoporous Mater., 2014, 194, 97–105 CrossRef CAS.
- X. Wang, A. Shishkin, F. Hemmingsson, M. Skoglundh, F. J. Martinez-Casado, L. Bock, A. Idström, L. Nordstierna, H. Härelind and P. A. Carlsson, RSC Adv., 2018, 8, 36369–36374 RSC.
- Y. S. Ryou, J. Lee, S. J. Cho, H. Lee, C. H. Kim and D. H. Kim, Appl. Catal., B, 2017, 212, 140–149 CrossRef CAS.
- S. K. Jesudoss, J. J. Vijaya, K. Kaviyarasu, L. J. Kennedy, R. Jothi Ramalingam and H. A. Al-Lohedan, RSC Adv., 2018, 8, 481–490 RSC.
- A. R. Do Nascimento, G. P. De Figueredo, E. M. F. Silva, M. A. F. Melo, D. M. A. Melo and M. J. B. De Souza, Rev. Virtual Quim., 2017, 9, 1570–1582 CrossRef CAS.
- B. Chen, R. Xu, R. Zhang and N. Liu, Environ. Sci. Technol., 2014, 48, 13909–13916 CrossRef CAS PubMed.
- C. M. Chanquía, K. Sapag, E. Rodríguez-Castellón, E. R. Herrero and G. A. Eimer, J. Phys. Chem. C, 2010, 114, 1481–1490 CrossRef.
- K. Hadjiivanov, J. Saussey, J. L. Freysz and J. C. Lavalley, Catal. Lett., 1998, 52, 103–108 CrossRef CAS.
- J. H. Kwak, H. Zhu, J. H. Lee, C. H. F. Peden and J. Szanyi, Chem. Commun., 2012, 48, 4758–4760 RSC.
- D. Wang, L. Zhang, K. Kamasamudram and W. S. Epling, ACS Catal., 2013, 3, 871–881 CrossRef CAS.
- M. P. Ruggeri, A. Grossale, I. Nova, E. Tronconi, H. Jirglova and Z. Sobalik, Catal. Today, 2012, 184, 107–114 CrossRef CAS.
- A. Penkova, K. Hadjiivanov, M. Mihaylov, M. Daturi, J. Saussey and J. C. Lavalley, Langmuir, 2004, 20, 5425–5431 CrossRef CAS PubMed.
- M. Iwasaki and H. Shinjoh, Phys. Chem. Chem. Phys., 2010, 12, 2365–2372 RSC.
- H. Y. Chen, Z. Wei, M. Kollar, F. Gao, Y. Wang, J. Szanyi and C. H. F. Peden, J. Catal., 2015, 329, 490–498 CrossRef CAS.
- L. Wang, W. Li, G. Qi and D. Weng, J. Catal., 2012, 289, 21–29 CrossRef CAS.
- H. Zhu, J. H. Kwak, C. H. F. Peden and J. Szanyi, Catal. Today, 2013, 205, 16–23 CrossRef CAS.
- R. Hajjar, Y. Millot, P. P. Man, M. Che and S. Dzwigaj, J. Phys. Chem. C, 2008, 112, 20167–20175 CrossRef CAS.
- K. I. Hadjiivanov, Catal. Rev.: Sci. Eng., 2000, 42, 71–144 CrossRef CAS.
- F. Giordanino, P. N. R. Vennestrøm, L. F. Lundegaard, F. N. Stappen, S. Mossin, P. Beato, S. Bordiga and C. Lamberti, Dalton Trans., 2013, 42, 12741–12761 RSC.
- S. Elzey, A. Mubayi, S. C. Larsen and V. H. Grassian, J. Mol. Catal. A: Chem., 2008, 285, 48–57 CrossRef CAS.
- M. Wallin, C. J. Karlsson, A. Palmqvist and M. Skoglundh, Top. Catal., 2004, 30–31, 107–114 CrossRef.
- F. Giordanino, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. V. Soldatov, P. Beato, S. Bordiga and C. Lamberti, J. Phys. Chem. Lett., 2014, 5, 1552–1559 CrossRef CAS PubMed.
- L. Ma, Y. Cheng, G. Cavataio, R. W. McCabe, L. Fu and J. Li, Appl. Catal., B, 2014, 156–157, 428–437 CrossRef CAS.
- T. Simons, P. Chen, D. Rauch, R. Moos and U. Simon, Sens. Actuators, B, 2016, 224, 492–499 CrossRef CAS.
- S. Brandenberger, O. Kröcher, A. Wokaun, A. Tissler and R. Althoff, J. Catal., 2009, 268, 297–306 CrossRef CAS.
- J. Dědeček, L. Čapek, P. Sazama, Z. Sobalík and B. Wichterlová, Appl. Catal., A, 2011, 391, 244–253 CrossRef.
- F. Gao, D. Mei, Y. Wang, J. Szanyi and C. H. F. Peden, J. Am. Chem. Soc., 2017, 139, 4935–4942 CrossRef CAS PubMed.
- L. Chen, T. V. W. Janssens, P. N. R. Vennestrøm, J. Jansson, M. Skoglundh and H. Grönbeck, ACS Catal., 2020, 10, 5646–5656 CrossRef CAS.
- T. V. W. Janssens, H. Falsig, L. F. Lundegaard, P. N. R. Vennestrøm, S. B. Rasmussen, P. G. Moses, F. Giordanino, E. Borfecchia, K. A. Lomachenko, C. Lamberti, S. Bordiga, A. Godiksen, S. Mossin and P. Beato, ACS Catal., 2015, 5, 2832–2845 CrossRef CAS.
- K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C. Lamberti, P. Beato, H. Falsig and S. Bordiga, J. Am. Chem. Soc., 2016, 138, 12025–12028 CrossRef CAS PubMed.
- L. Chen, H. Falsig, T. V. W. Janssens and H. Grönbeck, J. Catal., 2018, 358, 179–186 CrossRef CAS.
- E. Borfecchia, C. Negri, K. A. Lomachenko, C. Lamberti, T. V. W. Janssens and G. Berlier, React. Chem. Eng., 2019, 4, 1067–1080 RSC.
- C. Paolucci, I. Khurana, A. A. Parekh, S. Li, A. J. Shih, H. Li, J. R. Di Iorio, J. D. Albarracin-caballero, A. Yezerets, J. T. Miller, W. N. Delgass, F. H. Ribeiro, W. F. Schneider and R. Gounder, Science, 2017, 903, 898–903 CrossRef PubMed.
- X. Wang, L. Chen, P. N. R. Vennestrøm, T. V. W. Janssens, J. Jansson, H. Grönbeck and M. Skoglundh, ChemCatChem DOI:10.1002/cctc.202100253.
- N. Zhu, Y. Shan, W. Shan, Y. Sun, K. Liu, Y. Zhang and H. He, Environ. Sci. Technol., 2020, 54, 15499–15506 CrossRef CAS PubMed.
- J. Han, A. Wang, G. Isapour, H. Härelind, M. Skoglundh, D. Creaser and L. Olsson, Ind. Eng. Chem. Res., 2021, 60, 17826–17839 CrossRef CAS.
- M. Koebel, G. Madia and M. Elsener, Catal. Today, 2002, 73, 239–247 CrossRef CAS.
- J. M. Fedeyko, B. Chen and H. Y. Chen, Catal. Today, 2010, 151, 231–236 CrossRef CAS.
- O. Mihai, C. R. Widyastuti, S. Andonova, K. Kamasamudram, J. Li, S. Y. Joshi, N. W. Currier, A. Yezerets and L. Olsson, J. Catal., 2014, 311, 170–181 CrossRef CAS.
- J. H. Lee, Y. J. Kim, T. Ryu, P. S. Kim, C. H. Kim and S. B. Hong, Appl. Catal., B, 2017, 200, 428–438 CrossRef CAS.
- M. Devadas, O. Kröcher, M. Elsener, A. Wokaun, N. Söger, M. Pfeifer, Y. Demel and L. Mussmann, Appl. Catal., B, 2006, 67, 187–196 CrossRef CAS.
- https://webbook.nist.gov/cgi/cbook.cgi?ID=C10024972&Units=SI&Type=IR-SPEC&Index=1#IR-SPEC .
- B. Liu, D. Yao, F. Wu, L. Wei, X. Li and X. Wang, Ind. Eng. Chem. Res., 2019, 58, 20516–20527 CrossRef CAS.
- R. Ramprasad, W. F. Schneider, K. C. Hass and J. B. Adams, J. Phys. Chem. B, 1997, 101, 1940–1949 CrossRef CAS.
- C. Negri, T. Selleri, E. Borfecchia, A. Martini, K. A. Lomachenko, T. V. W. Janssens, M. Cutini, S. Bordiga and G. Berlier, J. Am. Chem. Soc., 2020, 142, 15884–15896 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy00247g |
| This journal is © The Royal Society of Chemistry 2022 |