
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly efficient and selective aqueous phase hydrogenation of aryl ketones, aldehydes, furfural and levulinic acid and its ethyl ester catalyzed by phosphine oxide-decorated polymer immobilized ionic liquid-stabilized ruthenium nanoparticles†
S.
Doherty
 *a,
J. G.
Knight
*a,
T.
Backhouse
a,
T. S. T.
Tran
a,
R.
Paterson
a,
F.
Stahl
a,
H. Y.
Alharbi
a,
T. W.
Chamberlain
*b,
R. A.
Bourne
b,
R.
Stones
b,
A.
Griffiths
b,
J. P.
White
b,
Z.
Aslam
b,
C.
Hardare
c,
H.
Daly
c,
J.
Hart
d,
R. H.
Temperton
d,
J. N.
O'Shea
d and
N. H.
Rees
e
*a,
J. G.
Knight
*a,
T.
Backhouse
a,
T. S. T.
Tran
a,
R.
Paterson
a,
F.
Stahl
a,
H. Y.
Alharbi
a,
T. W.
Chamberlain
*b,
R. A.
Bourne
b,
R.
Stones
b,
A.
Griffiths
b,
J. P.
White
b,
Z.
Aslam
b,
C.
Hardare
c,
H.
Daly
c,
J.
Hart
d,
R. H.
Temperton
d,
J. N.
O'Shea
d and
N. H.
Rees
e
aNewcastle University Centre for Catalysis (NUCAT), School of Chemistry, Bedson Building, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: simon.doherty@ncl.ac.uk; Tel: +44 (0)191 208 6537
bInstitute of Process Research & Development, School of Chemistry and School of Chemical and Process Engineering, University of Leeds, Woodhouse Land Leeds, LS2 9JT, UK
cSchool of Chemical Engineering and Analytical Sciences, The University of Manchester, The Mill, Sackville Street Campus, Manchester, M13 9PL, UK
dSchool of Physics & Astronomy, University of Nottingham, Nottingham, NG7 2RD, UK
eInorganic Chemistry Laboratory, University of Oxford, South Parks Road, Oxford OX1 3QR, UK
First published on 7th April 2022
Abstract
Impregnation of phosphine-decorated styrene-based polymer immobilized ionic liquid (PPh2-PIIL) with ruthenium(III) trichloride resulted in facile reduction of the ruthenium to afford Ru(II)-impregnated phosphine oxide-decorated PIIL (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh2PIIL). The derived phosphine oxide-decorated polymer immobilized ionic liquid-stabilized RuNPs (RuNP@OPPh2-PIILS) catalyse the highly efficient and selective aqueous phase reduction of the carbonyl group in aryl and heteroaryl ketones and aldehydes, including furfural, as well as the hydrogenation of levulinic acid and its ethyl ester to afford γ-valerolactone (GVL). While this is the first report of RuNPs stabilized by a phosphine oxide-modified support, there appear to be several recent examples of similar serendipitous oxidations during the synthesis of RuNPs by impregnation of a phosphine-decorated polymer with ruthenium trichloride; as these were either misinterpreted or not recognised as such we have carried out a detailed characterization and evaluation of this system. Reassuringly, RuNP@OPPh2-PIILS generated from phosphine oxide-decorated polymer immobilized ionic liquid (OPPh2-PIIL) impregnated with ruthenium trichloride is as efficient as that prepared directly from RuCl3 and PPh2-PIIL. Incorporation of PEG into the polymer support improved catalyst performance and the initial TOF of 2350 h−1 obtained for the aqueous phase hydrogenation of acetophenone is among the highest to be reported for a ruthenium nanoparticle-based catalyst. Under optimum conditions, RuNP@OPPh2-PEGPIILS recycled ten times with only a minor reduction in activity and no detectable change in selectivity. High yields and excellent selectivities were also obtained for hydrogenation of the CO across a range of substituted aryl and heteroaryl ketones. Complete hydrogenation of the aromatic ring and CO could also be achieved by increasing the pressure and temperature accordingly. The same system also catalyzes the aqueous phase hydrogenation of furfural under mild conditions with an initial TOF of 3160 h−1 as well as the selective hydrogenation of levulinic acid and its ethyl ester to γ-valerolactone (GVL); reaction times for the latter could be reduced quite significantly by addition of either butyric acid or Amberlyst H-15.
PPh2PIIL). The derived phosphine oxide-decorated polymer immobilized ionic liquid-stabilized RuNPs (RuNP@OPPh2-PIILS) catalyse the highly efficient and selective aqueous phase reduction of the carbonyl group in aryl and heteroaryl ketones and aldehydes, including furfural, as well as the hydrogenation of levulinic acid and its ethyl ester to afford γ-valerolactone (GVL). While this is the first report of RuNPs stabilized by a phosphine oxide-modified support, there appear to be several recent examples of similar serendipitous oxidations during the synthesis of RuNPs by impregnation of a phosphine-decorated polymer with ruthenium trichloride; as these were either misinterpreted or not recognised as such we have carried out a detailed characterization and evaluation of this system. Reassuringly, RuNP@OPPh2-PIILS generated from phosphine oxide-decorated polymer immobilized ionic liquid (OPPh2-PIIL) impregnated with ruthenium trichloride is as efficient as that prepared directly from RuCl3 and PPh2-PIIL. Incorporation of PEG into the polymer support improved catalyst performance and the initial TOF of 2350 h−1 obtained for the aqueous phase hydrogenation of acetophenone is among the highest to be reported for a ruthenium nanoparticle-based catalyst. Under optimum conditions, RuNP@OPPh2-PEGPIILS recycled ten times with only a minor reduction in activity and no detectable change in selectivity. High yields and excellent selectivities were also obtained for hydrogenation of the CO across a range of substituted aryl and heteroaryl ketones. Complete hydrogenation of the aromatic ring and CO could also be achieved by increasing the pressure and temperature accordingly. The same system also catalyzes the aqueous phase hydrogenation of furfural under mild conditions with an initial TOF of 3160 h−1 as well as the selective hydrogenation of levulinic acid and its ethyl ester to γ-valerolactone (GVL); reaction times for the latter could be reduced quite significantly by addition of either butyric acid or Amberlyst H-15.
Introduction
The selective reduction of carbonyl compounds is a fundamental transformation that has been widely studied due to its importance as a tool in synthesis as well as the hydrogenative transformation of biomass-derived substrates such as furfural, 2-hydroxymethylfurfuraldehyde and levulinic acid and its esters as the products are key platform molecules for the production of renewable fuels and value-added chemicals.1,2 While a host of transition metal nanoparticle-based catalysts have been reported to catalyze the reduction of carbonyl compounds, ruthenium appears to be the metal of choice as it has an unrivalled selectivity for hydrogenation of the CO bond in the presence of other reducible fragments.3 To this end, there are numerous recent examples of systems with encouraging activity and selectivity profiles including ultrasmall RuNPs stabilized by porous solids,4 carbon materials,5 polymers,6 β-cyclodextrins7 and ionic liquids.8 Other systems with good performance profiles for the hydrogenation of levulinic acid or its esters to γ-valerolactone9 include RuNPs immobilized on either acid-functionalized mesoporous carbon,10a sulfonic acid-modified reduced graphene oxide,10b commercial sulfonic acid ion exchange resin,11 acidic zirconium-containing spherical mesoporous silica,12 mesoporous TiO2,13 RuNP supported on sulfonic acid functionalized UiO-16,14 or the chromium-based MOF MIL-101,15 UiO-66 nanocrystals impregnated with RuNPs and POMs16 as well as Ru–hexadecyl(2-hydroxyethyl)dimethylammonium dihydrogen phosphate colloids deposited on TiSi2O6.17 These systems act as bifunctional catalysts with the RuNPs promoting the hydrogenation step while the acid catalyzes dehydration of the intermediate γ-hydroxyvaleric acid into γ-valerolactone. Efficient hydrogenation of levulinic acid to γ-valerolactone has also been achieved in the absence of acid with ruthenium nanoparticles supported on reduced graphene oxide18a and amino-modified silica (RuNP–NH2–α-Al2O3);18b the efficacy of these systems was attributed to the formation of highly dispersed ruthenium centers with an electron-rich state.18c High activity and selectivity for the conversion of levulinic acid to γ-valerolactone catalyzed by few layer graphene-supported RuNPs compared with ruthenium-loaded on traditional supports was attributed to the greater metallic content; this system was also stable with respect to aggregation and recycled efficiently whereas Ru/C showed continuous deactivation.19
While ionic liquids can act as both stabilizer and solvent for a range of transition metal nanoparticles,20a–c the primary stabilization resulting from weak electrostatic interactions does not provide sufficient stabilization against aggregation to less active/selective species under the conditions of catalysis.20d–f Additional practical limitations associated with the use of ionic liquids to stabilize nanoparticles include leaching of the IL under continuous flow operation or during work-up, high cost per unit volume as a solvent and their high viscosity and associated mass transfer limitations. Despite these drawbacks, nanoparticle ionic liquid-based systems have been developed that exhibit potentially beneficial effects that could guide catalyst design.21a For example, RuNPs stabilized by ionic liquid grafted on silica (RuNP@SIILP) catalyze the hydrogenation of aromatic ketones and aldol condensation adducts of furfuryl derivatives with preferential selectivity for CC, arenes and heteroarenes over the CO bond.21b Interestingly, partial substitution of iron for ruthenium resulted in a dramatic switch in selectivity such that FeRuNP@SIILP (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) was highly selective for the CO bond and hydrogenation of the arene was suppressed; this was explained by a more polarized activation of hydrogen on the FeRu surface than on pure Ru as well as by activation of the CO bond on the more oxophilic FeRu surface. A triphasic RuNP ionic liquid-based catalyst for the hydrogenation of levulinic acid to γ-valerolactone facilitated product recovery and recycled efficiently with no loss in activity, however, reactions were conducted at high temperature and activity was significantly lower than commercial Ru/C.22
:3) was highly selective for the CO bond and hydrogenation of the arene was suppressed; this was explained by a more polarized activation of hydrogen on the FeRu surface than on pure Ru as well as by activation of the CO bond on the more oxophilic FeRu surface. A triphasic RuNP ionic liquid-based catalyst for the hydrogenation of levulinic acid to γ-valerolactone facilitated product recovery and recycled efficiently with no loss in activity, however, reactions were conducted at high temperature and activity was significantly lower than commercial Ru/C.22
As the electrostatic stabilization provided by ionic liquids is often not sufficient to prevent aggregation, metal binding heteroatom donors have been incorporated into the ionic liquid on the basis that an additional covalent interaction would supplement the electrostatic stabilization and improve the long-term stability of the nanoparticles.23a However, there is now a convincing body of evidence that heteroatom donors23b or molecularly modified surfaces21a can also modify the activity and/or selectivity of a NP catalyst. For example, phosphine-functionalized ionic liquid-stabilized RuNPs exhibit solvent dependent switchable chemoselectivity for the hydrogenation of aromatic ketones and aldehydes; reactions in [BMIM][BF4] gave high selectivity for hydrogenation of the carbonyl group while reactions in water resulted in complete hydrogenation of the arene and the carbonyl.24 Reactions conducted under the same conditions but in the absence of phosphine were much less selective in both solvents, which confirmed the role the phosphine in tuning selectivity. Other relevant studies with RuNPs generated in the presence of a phosphine or N-heterocyclic carbene have shown that activity and/or selectivity in the hydrogenation of aromatic ketones can be tuned but that hydrogenation of the arene generally competes with the carbonyl.21a,b,25 The activity and selectivity profiles of sulfonated diphosphine-stabilized RuNPs as catalysts for the hydrogenation of acetophenone has been reported to be influenced by the presence of cyclodextrin, which enhances activity by acting as a phase transfer catalyst and modifies selectivity by forming an inclusion complex between the cyclodextrin and diphosphine at the RuNP surface.26 The chemoselectivity of gold nanoparticles as a catalyst for the hydrogenation of the carbonyl group in α,β-unsaturated aldehydes improved quite dramatically in the presence of a secondary phosphine oxide.27 Platinum nanoparticles stabilized on triphenylphosphine-modified silica exhibited a markedly higher chemoselectivity in the hydrogenation of acetophenone than its unmodified counterpart; this was attributed to an increase in the surface electron density by the σ-donor phosphine.28 Triphenylphosphine cross-linked in the nanopores of FDU-12 has been used to modulate the surface electronic properties of PdNPs and thereby tune their performance as a catalyst for the hydrogenation of aryl ketones such that PdNP@PPh3FDU-12 gave >99% selectivity for the alcohol while its non-phosphine containing counterpart PdNP@FDU-12 only gave 63% selectivity.29 A recent DFT study investigated the role of phosphine-decorated PdNPs in the decarbonylation of fatty acids and found that monodentate phosphines passivate the surface while the flexibility of their bidentate counterparts allows transient cavities to be created that enhance selectivity and prevent catalyst deactivation.30 Most recently, ruthenium nanoparticles incorporated into phosphine-modified nanoreactors catalyze the hydrogenation of benzoic acid whereas the corresponding unmodified nanoreactor was completely inactive; the efficacy of the phosphine-modified system was attributed to preferential absorption of the benzoic acid on the electron-rich RuNP surface.31 However, ligand effects are not restricted to phosphine-based donors as amines have also been shown to modify the performance of Pt, Pd and Ru nanoparticles as catalysts for the hydrogenation of carbonyl compounds and nitroarenes by modifying surface electronic structure and/or steric properties.32
We have recently been exploring the concept of heteroatom donor-modified polymer immobilized ionic liquid-stabilized nanoparticles (MNP@HAD-PIILS) on the basis that covalent attachment of an ionic liquid to a polymer would combine the well-documented advantages of ionic liquids such as catalyst stabilization, facile catalyst activation and enhancements in rate and/or selectivity with those of a solid support.33a–g While the heteroatom donor was initially incorporated to supplement the weak electrostatic stabilization provided by the PIIL, the studies described above suggest that heteroatom-modified PIILs could enable the surface electronic structure and steric environment to be modified and/or NP size and morphology to be controlled and, as such, could prove to be versatile and tunable supports for developing new catalyst technology. Our initial foray in this area has already demonstrated that PEG-modified phosphine-decorated PIIL-stabilized palladium nanoparticles are remarkably selective catalysts for the aqueous phase hydrogenation of α,β-unsaturated carbonyl compounds33c and nitroarenes33d as well as the Suzuki–Miyaura cross-coupling in aqueous media33e while the corresponding gold system is a multiselective solvent-dependent catalyst for the reduction of nitroarenes to N-phenylhydroxylamine, azoxyarene and the corresponding aniline.33f
We have now extended our study to investigate the efficacy of phosphine-decorated polymer-immobilized ionic liquid-stabilized ruthenium nanoparticles as catalysts for the hydrogenation of aryl ketones and aldehydes as well as biomass-derived carbonyl compounds to explore whether the heteroatom donor modifies selectivity and/or activity. Although RuNPs appear to have unprecedent selectivity for hydrogenation of the CO bond in the presence of other functional groups, the selective hydrogenation of aromatic and heteroaromatic ketones to the corresponding aromatic alcohol under mild conditions is extremely challenging as reduction of the arene often competes with or is more rapid than that of the CO bond, resulting in complete hydrogenation of the substrate. Moreover, while a guiding principle to achieve selective reduction of ketones is polarized hydrogen activation3c the factors that control selectivity are not yet fully understood. Our initial design principle was based on the use of a PEG-modified phosphine-decorated polymer immobilized ionic liquid, reasoning that the phosphine and ionic liquid fragments would function as ligands and modifiers to influence NP formation as well as selectivity and stability while the PEG unit would improve the dispersibility of the catalyst in water and facilitate aqueous phase hydrogenation. To this end, there is now considerable literature precedent that water is the solvent of choice for the ruthenium-catalyzed hydrogenation of carbonyl compounds, particularly biomass-derived platform molecules rich in carbonyl groups.3a,b,34
While our initial hypothesis was to establish whether a phosphine-based HAD influences the efficacy of RuNPs as catalysts for hydrogenation, we inadvertently discovered that impregnation of phosphine-decorated PIILs with ruthenium trichloride resulted in rapid reduction of the ruthenium(III) with concomitant formation of phosphine oxide. Thus, we became interested in exploring whether the phosphine oxide could act as a hemilabile ligand to supplement stabilization of the nanoparticles by the ionic liquid while allowing facile access of the substrate to the catalyst surface by virtue of the lability of the Ru⋯OPAr3 interaction as well as facilitate polarized activation of the hydrogen to achieve selective reduction of the CO bond in aromatic ketones. To this end, the corresponding phosphine oxide-decorated polymer immobilized ionic liquid stabilized RuNPs (RuNP@OPPh2-PEGPIILS) catalyze the aqueous phase hydrogenation of aromatic ketones, aldehydes, furfural and levulinic acid and its ethyl ester with remarkable efficacy and selectivity. The high selectivity of this system is quite exceptional when compared with phosphine-functionalized ionic liquid-stabilized RuNPs24 as well as various RuNP/phosphine combinations25b–e,26 as the former gave complete reduction of the arene and carbonyl in water while the latter systems typically gave mixtures of products due to competing hydrogenation of the carbonyl and arene. Complimentary studies are currently underway to prepare the corresponding phosphine decorated PIIL, PPh2-PEGPIILS, to compare the selectivity of phosphine- and phosphine oxide-based polymer immobilized ionic liquid stabilized RuNPs as catalysts for the hydrogenation of aryl ketones and thereby understand the role of the phosphine versus the phosphine oxide in NP formation and controlling selectivity. While this is the first report of the use of a phosphine oxide modified polymer to stabilize RuNPs, there appear to be several recent examples of serendipitous oxidation of phosphine-decorated supports during the preparation of RuNPs, by impregnation with ruthenium trichloride, that were either not recognized as such or misinterpreted in the publication.31,35 The most recent of these are microenvironment engineered RuNPs incorporated into phosphine-modified silica nanoreactors31 and RuNPs stabilized by phosphorus-rich cross-linked network polymers.35 Herein, we report detailed characterization of a range of phosphine oxide-decorated PIIL stabilized RuNPs together with comparative evaluation of their efficacy as catalysts for the hydrogenation of a range of carbonyl-based substrates.
Results and introduction
Synthesis and characterization of polymer immobilized ionic liquid-stabilized ruthenium nanoparticles
Our initial attempt to prepare the phosphine-decorated polymer-immobilized ionic liquid-stabilized RuNPs (RuNP@PPh2-PIILS) involved impregnation of the corresponding PIIL 1a and 1a.PEG with ruthenium trichloride, to afford a 1:1 ratio of ruthenium to phosphine, followed by reduction with sodium borohydride. However, the presence of a single broad signal at ca. δ 27 ppm in the solid state 31P NMR spectra of the products provided the first indication that they were the corresponding phosphine oxide-based PIIL-stabilized RuNPs, 2a and 2a.PEG (Fig. 1), as there was no evidence for free phosphine at δ −6.8 ppm in the 31P NMR spectrum. In hindsight, it is entirely reasonable that impregnation of either PPh2-PIIL (1a) or PPh2-PEGPIIL (1a.PEG) with ruthenium trichloride would result in facile oxidation of the phosphine with concomitant reduction to a ruthenium(II) species as RuCl2(PPh3)3 is prepared by reaction of ruthenium trichloride with an excess of triphenylphosphine, which generates triphenylphosphine oxide as the by-product.36 However, while the solid state 31P NMR spectra of an authentic sample of OPPh2-PIIL (1a′) and its derived ruthenium nanoparticles, RuNP@OPPh2-PIILS (2a′) support this interpretation, as both contain a single broad signal at δ 27, a recent report that a triarylphosphine-coordinated ruthenium cluster (Ar3P–Ru) also appears in this region37 introduced an element of uncertainty to this interpretation. With the aim of addressing unequivocally whether the resonances for a RuNP–PAr3 interaction would coincide with that for Ar3PO, samples of RuNP@PPh2-PIILS and RuNP@PPh2-PEGPIILS were prepared by hydrogenation of a THF mixture of [Ru(COD)(COT)] with either PPh2-PIIL (1a) or PPh2-PEGPIIL (1a.PEG), following a previously documented literature protocol.38 This Ru(0)-based precursor was chosen as it generates RuNPs under extremely mild conditions and would avoid deleterious oxidation of the phosphine that occurs in the presence of ruthenium trichloride. The solid state 31P NMR spectra of the resulting RuNP@PPh2-PIILP and RuNP@PPh2-PEGPIILP both contain a major signal at ca. δ −7 ppm, associated with uncoordinated phosphine, together with a minor signal at ca. δ 27 ppm, which can now be confidently attributed to Ru-coordinated triarylphosphine on the basis that the 31P NMR spectra of 1a and 1a.PEG showed no evidence for the corresponding oxide immediately prior to generation of the corresponding Ru(COD)(COT)-derived RuNPs. However, as the 1:1 stoichiometry of ruthenium to phosphine used for the preparation of 2a and 2a.PEG should only result in oxidation of only one half of the phosphine the absence of a signal in the region of associated with uncoordinated phosphine suggests that the resonance at δ 27 ppm most likely corresponds to a mixture of phosphine oxide-based RuNP@OPPh2-PIILP and RuNP@PPh2-PIILP; the latter containing a triaryl-coordinated ruthenium (RuNP–PAr3). The presence of a strong PO stretching band at 1129 cm−1 in the IR spectrum of 2a confirmed the presence of oxide and the red-shift of this band compared with 1181 cm−1 for an authentic sample of phosphine oxide-decorated polymer OPPh2-PIILP (1a′) is consistent with a Ru⋯OPAr3 interaction;39a,d for comparison the PO stretching frequency of triphenylphosphine oxide has been well-documented and appears at 1190 cm−1.39e,f Interestingly, while van Leeuwen has previously reported that generation of RuNPs from [Ru(COD)(COT)] in the presence of a triarylphosphine results in hydrogenation of the aromatic rings to afford cyclohexyl-based phosphines,37 there was no evidence for hydrogenation of the PPh2 groups in 1a or 1a.PEG. The 13C CP/MAS NMR spectra of 2a and 2a.PEG contain characteristic signals between δ 123 and 145 ppm associated with the imidazolium ring and the aromatic carbon atoms as well as additional signals at higher field which correspond to the methyl group attached to the imidazolium ring and the aliphatic carbon atoms of the polystyrene backbone; resonances for the methylene groups of the PEG unit in 2a.PEG appear at δ 70 ppm while the terminal methoxy fragment appears at δ 58 ppm (see ESI† for full details).
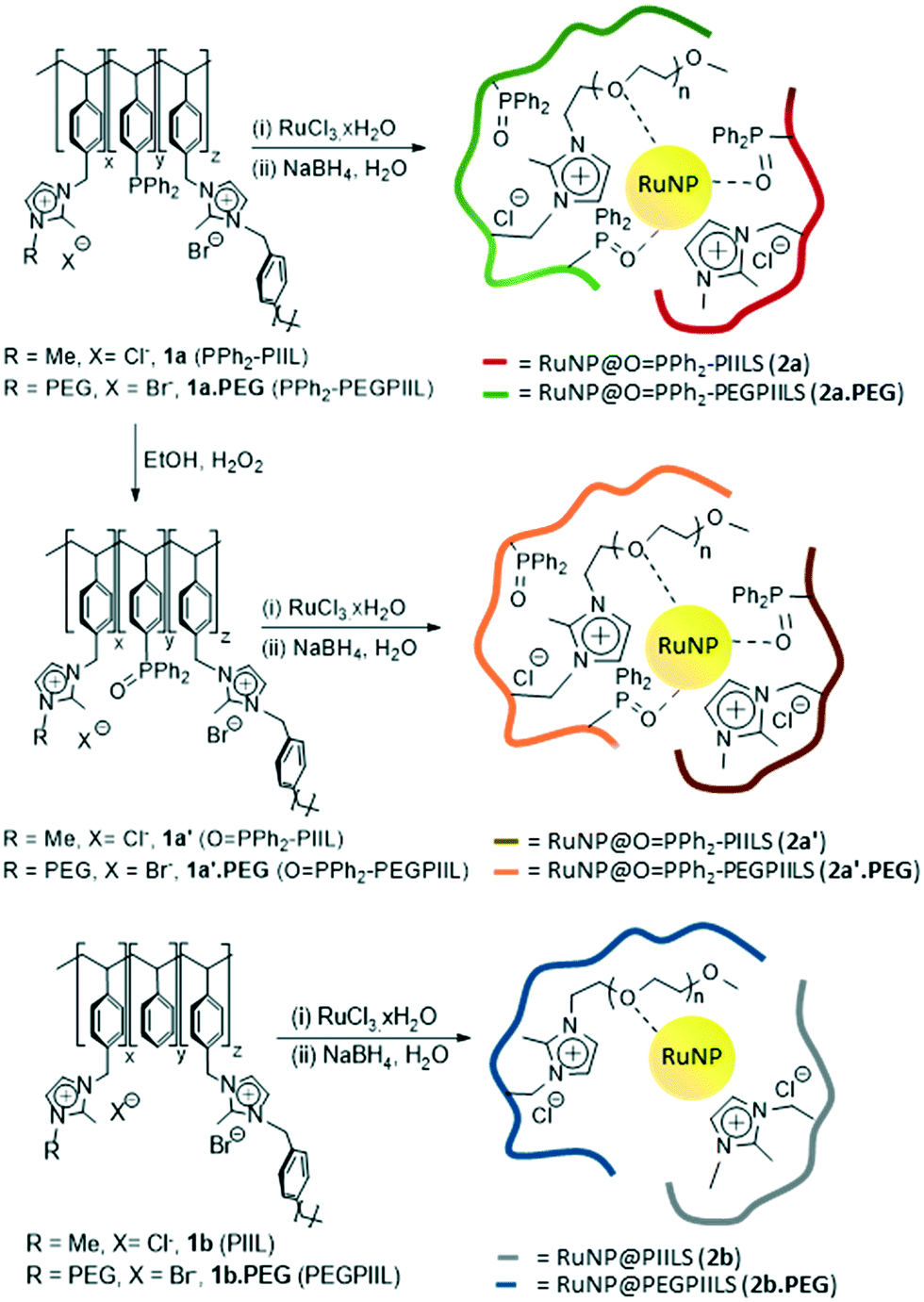 | ||
| Fig. 1 Synthesis and composition of polymers 1a, 1a.PEG, 1a′, 1a′.PEG, 1b and 1b.PEG and the corresponding PIIL-stabilized ruthenium nanoparticles 2a (red), 2a.PEG (green), 2a′ (brown), 2a′.PEG (orange), 2b (grey) and 2b.PEG (blue). | ||
Having established that 2a and 2a.PEG are in fact generated by sodium borohydride reduction of a Ru(II)/phosphine oxide-decorated PIIL-based precursor via serendipitous oxidation during impregnation of 1a/1a.PEG with ruthenium trichloride, the corresponding phosphine oxide decorated PIILs, OPPh2PIIL (1a′) and OPPh2-PEGPIIL (1a′.PEG), were prepared, impregnated with ruthenium trichloride and the resulting Ru(III)/phosphine oxide-based precursors reduced in situ to afford RuNP@OPPh2-PIILS (2a′) and RuNP@OPPh2-PEGPIILS (2a′.PEG). As expected the IR, 31P and 13C NMR spectroscopic data for 2a′ and 2a′.PEG are similar to those for 2a and 2a.PEG as both contain phosphine oxide-decorated PIIL. The ruthenium loadings of 2a and 2a.PEG and 2a′ and 2a′-PEG were determined to be 0.28–0.47 mmol g−1 by ICP-OES which corresponds to ruthenium loadings between 2.8–4.7 wt%.
Surface characterization of the RuNP-based catalysts was undertaken by X-ray photoelectron spectroscopy (XPS) with analysis of the P 2p and Ru 3p regions performed. The Ru 3p region was analyzed due to the overlap of the C 1s and Ru 3d regions. No change in the P 2p region could be detected and all the catalyst contained a broad peak 131.9–132.2 eV due to overlap of the 2p3/2 and 2p1/2 bands associated with the phosphine oxide fragment.40a For catalyst 2b, where the RuNP was only stabilised by the PIILP backbone, a Ru 3p3/2 peak was found at 462.9 eV which is assigned to RuO2, with a satellite feature fitted at 464.8 eV (Table S4 and Fig. S49†).40b The presence of RuO2 species could be attributed to a degree of surface oxidation of the pre-formed metallic Ru nanoparticles. For the catalysts containing OPPh2 (2a and 2a′) the Ru 3p3/2 peak position was shifted to lower binding energies (462.3 and 462.1 eV for 2a and 2a′ respectively) relative to the Ru 3p peak position of 462.9 eV for catalyst 2b (Table S4 and Fig. S17 and S37†). The shift to lower binding energy upon addition of P-containing ligands suggests electron transfer from the P-ligands to the RuNPs. The PEGylated catalyst 2b.PEG exhibited a smaller shift in the binding energy for the Ru 3p3/2 peak, which was observed at 462.6 eV, suggesting interaction of P-ligands with the Ru could more significantly alter the surface electronic properties of the nanoparticles (Fig. S54†). Addition of OPPh2 (or OPPh2 and PEG) resulted in the largest shift (−0.9 eV) in binding energy of the Ru 3p3/2 peak relative to the RuNP@PIILP catalyst which was found at 462.0 eV for 2a′.PEG (Fig. S44†).
ATR-IR spectroscopy of CO adsorption (10% CO/Ar, at room temperature) was performed over catalysts 2a and 2a′ and 2b to probe the oxidation states and chemical environment of the Ru nanoparticles upon introduction of the OPPh2 ligand (2a and 2a′) to the RuNP@PIILP catalyst (2b). The Ru–CO spectra for catalysts 2a, 2a′ and 2b (Fig. 2) exhibited bands in the 2000–1800 cm−1 region which are attributed to CO adsorption on hydrated metallic Ru species.39g–i The Ru–CO spectra for catalyst 2b exhibited bands at 2014, 1990, 1960, 1942, 1906 and 1890 cm−1 indicating the presence of several different Ru–CO species; however, as a result of a number of overlapping bands in this spectrum definitive assignment of the Ru–CO species is not possible. Moreover, these assignments are debated in the literature due to the number of linear and multicarbonyl adsorption modes which are possible on Ru of different oxidation states and coordination sites.39g,j–m The spectra of CO adsorbed on catalysts 2a and 2a′ showed a significant reduction in intensity of the bands at 2014, 1960, 1942 and 1890 cm−1 which is attributed to blocking of sites on the RuNP surface by the P-containing ligands. Ligand interactions with metal nanoparticles blocking sites for CO adsorption has also been reported for Pt and Pd nanoparticle SCILL catalysts39n and amine capped colloidal Pt particles on metal oxide supports where the amine ligand (dodecylamine) blocked terrace sites at room temperature.39o
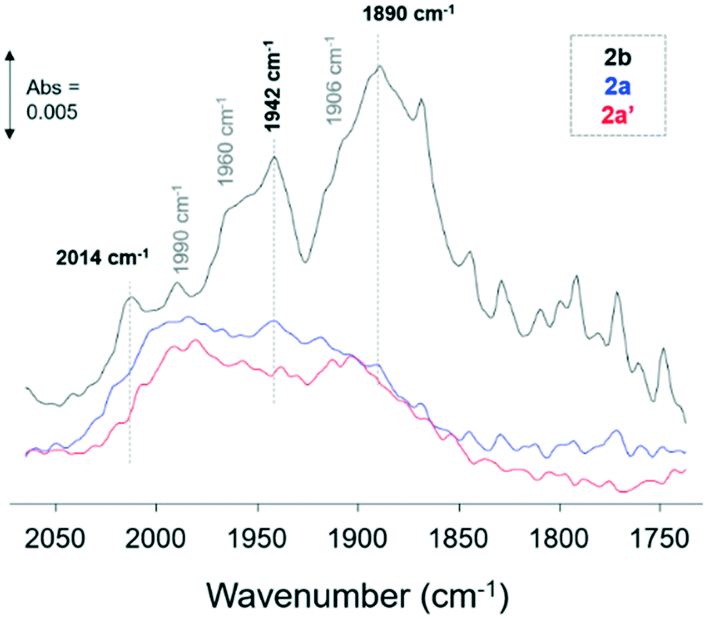 | ||
| Fig. 2 ATR spectra of CO (10% CO in Ar) adsorbed on catalysts 2a and 2a′ and 2b at RT. | ||
Interestingly, CO adsorption on catalysts 2a and 2a′ gave comparable spectra with Ru–CO bands at 1990 and 1907 cm−1 and a weak band at 2062 cm−1 which was observed to form at higher coverage of CO (Fig. S56†). These bands are tentatively assigned to the low frequency, (LF) (1990 cm−1), mid frequency (MF) (1907 cm−1) and high frequency (HF) (2062 cm−1) bands of CO adsorbed on metallic Ru particles. The LF band is attributed to atop linear adsorption of CO and the MF and HF bands to multicarbonyl species.39g,p The Ru nanoparticles in catalyst 2a were prepared from Ru(II)/phosphine oxide decorated PIIL, albeit via serendipitous oxidation during impregnation of 1a with ruthenium trichloride, and in catalyst 2a′ from phosphine oxide-decorated PIIL and the comparable Ru–CO spectra observed show similar RuNP oxidation states and environment in line with the NMR and XPS results for these catalysts.
The ATR spectra for CO adsorption over catalyst 2a.PEG are shown in Fig. S57† (after exposure to NaBH4/ethanol solution as for 2a, 2a′ and 2b). With the addition of PEG, loss of bands at 1990 and 1906 cm−1 were observed which were the predominant Ru–CO bands observed for catalysts 2a and 2a′. The Ru–CO bands for catalyst 2a.PEG were observed at 2049, 1972 and 1942 cm−1 which are again assigned to HF, MF and LF Ru–CO bands, respectively. Catalyst 2a.PEG was also treated with a flow of H2 saturated water at 50 °C, to simulate the reaction conditions for the hydrogenation of acetophenone, before introduction of CO. The Ru–CO bands obtained following H2 treatment were comparable to the spectra obtained after NaBH4 reduction (Fig. S58†). The Ru environment of the catalyst was not influenced by the nature of the reductive treatment with the bands assigned to CO adsorbed on metallic Ru. The XPS spectra indicated RuO2 species but facile reduction to metallic Ru nanoparticles in H2/water at 50 °C (reaction conditions) may indicate that metallic Ru is part of the active phase of the catalyst.
TEM micrographs of 2a and 2a.PEG revealed that the ruthenium nanoparticles are near monodisperse with average diameters of 1.59 ± 0.42 nm and 1.76 ± 0.35 nm, respectively while the nanoparticles in 2a′ and 2a′.PEG have average diameters of 1.54 ± 0.37 nm and 1.32 ± 0.30, respectively; the former is similar to that for 2a while the latter are slightly smaller than those in 2a.PEG. Representative micrographs and associated distribution histograms based on >100 particles are shown in Fig. 3. HRTEM images analysis of the lattice spacing of the RuNPs in 2a confirm it is metallic Ru and HAADF and STEM imaging and EDX mapping confirms the presence of the chloride anions in the vicinity of the Ru nanoparticles and the presence of P in the polymer support (see Fig. S19–S24 in the ESI† for full details).41 The mean diameters of the nanoparticles in benchmark catalyst 2b (1.81 ± 0.51 nm) and 2b.PEG (1.50 ± 0.40 nm), respectively, fall within the range for their phososphine oxide-based counterparts (see ESI† for TEM micrographs of 2b and 2b.PEG). For comparison, RuNPs stabilized by phosphine-functionalized ionic liquids have slightly larger average mean diameters of 2.2 nm,24a although these systems were generated by hydrogenation of RuO2 rather than sodium borohydride-mediated reduction of Ru(III) chloride or derived Ru(II) species. Similarly ionic liquid-stabilized RuNPs generated by hydrogenation of Ru(allyl)2(COD) or Ru(COD)(COT) also have slightly larger average mean diameters of 2.70 ± 0.20 nm and 3.50 nm, respectively.8a,25
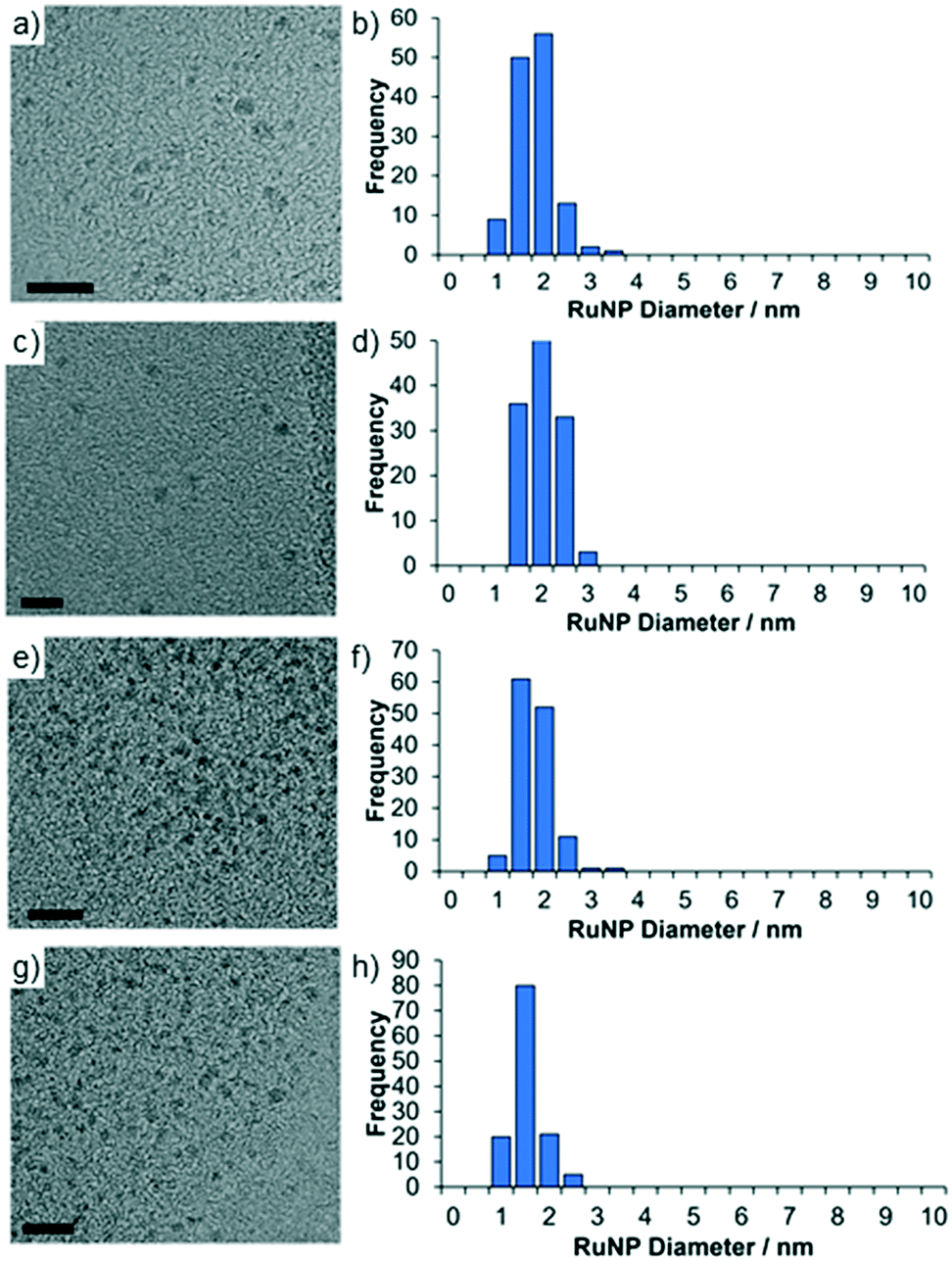 | ||
| Fig. 3 HRTEM images and corresponding size distributions determined by counting >100 particles of (a and b) 2a, (c and d) 2a.PEG, (e and f) 2a′ and (g and h) 2a′.PEG. Mean particle diameters are 1.59 ± 0.42 nm (2a), 1.76 ± 0.35 nm (2a.PEG), 1.54 ± 0.37 nm (2a′) and 1.32 ± 0.30 nm (2a′.PEG). Black scale bars are 10 nm. | ||
Hydrogenation of ketones and aldehydes
The hydrogenation of ketones and aldehydes was initially targeted as selective reduction of the carbonyl group is a key transformation in synthesis as well as the conversion of biomass-derived substrates such as furfural, levulinic acid (LA) and alkyl levulinates to platform molecules; as such they were considered ideal candidates for preliminary optimization studies. Preliminary catalytic studies focused on comparing the performance of 2a, 2a.PEG, 2a′ and 2a′.PEG as catalysts for the hydrogenation of acetophenone in order to establish whether in situ formation of phosphine oxide during impregnation or the use of pre-formed phosphine oxide influences the efficacy of the derived RuNPs as well as to identify the most efficient system for further optimization. Initial reactions were conducted in water with a catalyst loading of 0.1 mol% for 3 h at 50 °C under 70 psi of hydrogen, full details of which are summarized in Table 1. Under these conditions, 2a.PEG proved to be most efficient giving 67% conversion with 85% selectivity for 1-phenylethanol (entries 1–4). Comparative catalyst testing against 2b and 2b.PEG also revealed that removal of the phosphine oxide was detrimental to catalyst performance as both systems only gave 64% selectivity for 1-phenylethanol under the same conditions (entries 5 and 6). At this stage we can only speculate about the origin of the differences in catalyst efficacy but note that while 2a and 2a′ and their PEGylated counterparts have seemingly similar compositions there are two key differences. Firstly the phosphine oxide in 2a/2a.PEG is generated in situ whereas 2a′/2a′.PEG are generated from preformed phosphine oxide-decorated PIILP and, secondly, 2a/2a.PEG are generated by reduction of an in situ formed Ru(II) species whereas 2a′/2a′.PEG are generated from Ru(III). For comparison, 0.1 mol% Ru/C (5 wt%) catalyzed the hydrogenation of acetophenone under the same conditions but only reached 39% conversion with 63% selectivity for 1-phenylethanol after 3 h (entry 7). As catalyst 2a′.PEG was the most efficient system tested, it was taken forward for further optimization.|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Base | Conv.b (%) | Select.c (%) |
| a Reaction conditions: 1 mmol of acetophenone, 0.1 mol% Ru in 2a, 2a.PEG, 2a′, 2a′.PEG, 2b, 2b.PEG and 5 wt% Ru/C, 13 mL solvent, 10 mol% base, 3 h, 50 °C, 70 psi H2. b Conversions determined by 1H NMR spectroscopy using 1,3-dinitrobenzene as internal standard and gas chromatography using decane as internal standard. Average of at least three runs. c Selectivity for 1-phenylethanol = [% 1-phenylethanol/(% 1-phenylethanol) + (% 1-cyclohexylethanol) + (% 1-cyclohexylethanone)] × 100%. | |||||
| 1 | 2a | Water | — | 48 | 70 |
| 2 | 2a.PEG | Water | — | 52 | 71 |
| 3 | 2a′ | Water | — | 48 | 69 |
| 4 | 2a′.PEG | Water | — | 67 | 85 |
| 5 | 2b | Water | — | 44 | 64 |
| 6 | 2b.PEG | Water | — | 55 | 64 |
| 7 | Ru/C | Water | — | 39 | 63 |
| 8 | 2a′.PEG | EtOH/H2O | — | 68 | 96 |
| 9 | 2a′.PEG | EtOH | — | 9 | 92 |
| 10 | 2a′.PEG | Toluene | — | 4 | >99% |
| 11 | 2a′.PEG | 2Me-THF | — | 3 | >99% |
| 12 | 2a′.PEG | EtOH/H2O | K2CO3 | 95 | 96 |
| 13 | 2a′.PEG | EtOH/H2O | Na2CO3 | 78 | 91 |
| 14 | 2a′.PEG | EtOH/H2O | NaOAc | 81 | 93 |
| 15 | 2a′.PEG | EtOH/H2O | KOH | 77 | 93 |
A survey of the performance of 2a′.PEG in selected solvents revealed that reactions conducted in a 1:1 mixture of water and ethanol resulted in a substantial improvement in selectivity to 96% (entry 8) and while high selectivity for 1-phenylethanol was also obtained in 2-Me-THF, ethanol and toluene conversions were significantly lower (entries 9–11). A more detailed study of catalyst performance as a function of the ethanol-water composition revealed the 1:1 mixture to be optimum as conversions dropped quite dramatically with increasing ethanol content while selectivity dropped as the water content increased (see ESI† for details). Interestingly, addition of base to the catalytic reaction mixture resulted in a marked improvement in conversion such that the same reaction conducted in the presence of 10 mol% potassium carbonate, under otherwise identical conditions, reached 95% conversion with 96% selectivity for 1-phenylethanol compared with 67% conversion and the same selectivity in the absence of base. Addition of other bases such as sodium carbonate, sodium acetate and potassium hydroxide also gave an improvement in conversion, albeit less than the 27% increase obtained with potassium carbonate (entries 12–15). A comparable increase in conversion and a slight increase in selectivity was also obtained in the presence of Sc(OTf)3, however, potassium carbonate was identified as the base of choice for all further studies as it is inexpensive, readily available and more environmentally benign. The composition profile as a function of the amount of base revealed that only 10 mol% was required to achieve optimum conversion and selectivity as higher loadings resulted in a dramatic drop in conversion. To this end, the influence of basic additives on the chemoselective hydrogenation of aryl ketones is well-documented.42 For example, a low conversion for the hydrogenation of acetophenone was obtained with RuNPs embedded in a siloxane matrix in the absence of base6a while phosphine-functionalized ionic liquid stabilized RuNPs were completely inactive in the absence of base but reached 77% conversion with 99% selectivity to 1-phenylethanol in the presence of 1-butyl-2,3-dimethylimidazolium hydroxide.24a Interestingly, the latter system only gave 100% selectivity for 1-phenylethanol in [BMIM][BF4] while reactions conducted in water were much faster and resulted in complete reduction to 1-cyclohexylethanol with 100% selectivity.
A study of the composition as a function of pressure for the hydrogenation of acetophenone catalyzed by 0.1 mol% 2a′.PEG in a 1:1 mixture of water and ethanol at room temperature for a reaction time of 45 min revealed that conversions increased with increasing pressure and ultimately plateaued at ca. 400 psi (Fig. 4). However, this increase in conversion was accompanied by a drop in selectivity due to the formation of 1-cyclohexylethanol resulting from hydrogenation of the aromatic ring of 1-phenylethanol. While this profile indicates mass transfer limited dissolution of hydrogen in the reaction solvent, high conversions could be obtained at 50 °C under a hydrogen pressure as low as 70 psi and as such all further reactions and substrate screening were conducted at this pressure by extending the reaction time where necessary.
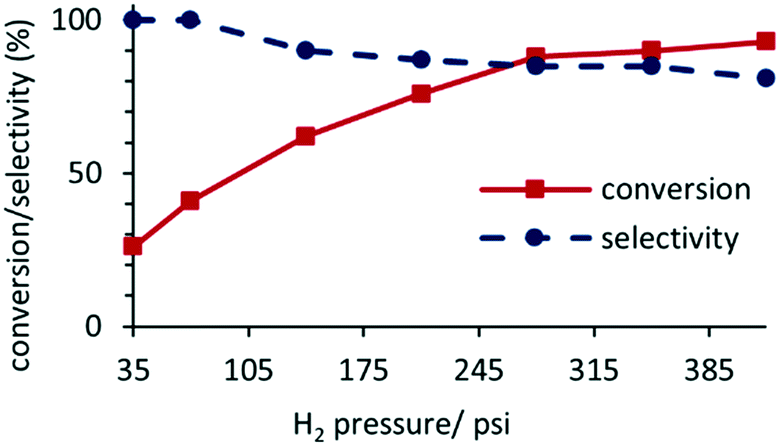 | ||
| Fig. 4 Reaction profile and selectivity for 1-phenylethanol (1-PE) as a function of pressure for the hydrogenation of acetophenone in a water–ethanol mixture (1:1) at 50 °C using 0.1 mol% 2a′.PEG and 10 mol% K2CO3. | ||
A reduction in the catalyst loading to 0.05 mol% also resulted in an increase in activity such that a conversion of 43% with 100% selectivity for 1-phenylethanol was obtained after only 1 h at 50 °C and 70 psi of hydrogen; this corresponds to a TOF of 860 h−1 (measured as moles of product per mole catalyst per hour). The efficacy of 2a′.PEG was further tested by reducing the catalyst loading to 0.005 mol% and the conversion of 47% with 100% selectivity for 1-PE obtained after 4 h corresponds to an initial TOF of 2350 h−1, which is probably more representative of the potential intrinsic turnover rate of the catalyst; full conversion to 1-PE (96%) and 1-CE (4%) was obtained by extending the reaction time to 15 h. A map of the conversion and selectivity as a function of temperature revealed 50 °C to be the optimum temperature as selectivity for 1-PE dropped to 90% at 94% conversion when the reaction temperature was raised to 70 °C. Even though reaction times were shorter at this temperature, 50 °C represented the best compromise between reaction time and selectivity.
The high selectivity for 1-phenylethanol obtained under such mild conditions is quite remarkable and in sharp contrast to RuNPs stabilized by triphenylphosphine, which favored reduction of the aromatic ring over the ketone in THF,25b as well as long chain NHC-stabilized RuNPs that catalyzed complete reduction to 1-cyclohexylethanol in hexane.25a However, a selectivity of 84% for 1-phenylethanol with a TOF of 4 h−1 has been reported for the hydrogenation of acetophenone in water with RuNPs stabilized by a sulfonated diphosphine.26 This improved to a selectivity of 91% and a TOF of 28 h−1 in the presence of 5.0 equivalents of cyclodextrin due to formation of an inclusion complex between the cyclodextrin and diphosphine at the RuNP surface. While an increase in pressure accelerated the hydrogenation, the selectivity profile showed a marked dependence on the phosphine to cyclodextrin ratio. For example, reactions conducted under 10 bar of hydrogen in the absence of cyclodextrin resulted in complete hydrogenation to afford 1-cyclohexylethanol with a TOF of 28 h−1 whereas catalyst enriched with 5.0 equivalents of cyclodextrin was more active, with a TOF of 944 h−1, but less selective as it gave a mixture of 1-cyclohexylethanone, 1-phenylethanol and 1-cyclohexylethanol.
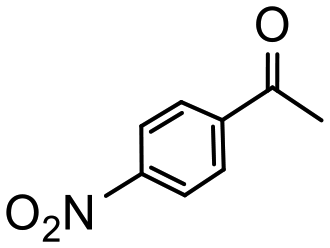
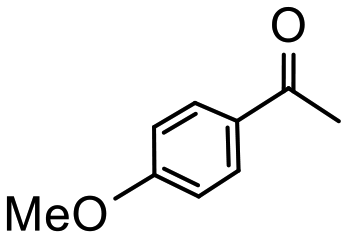
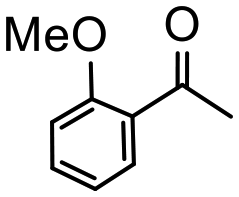
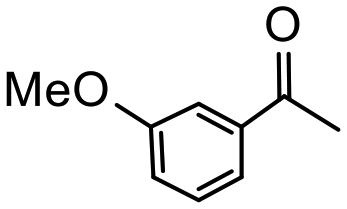
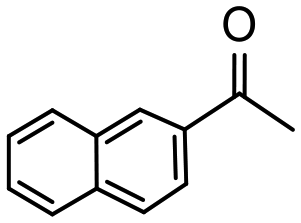
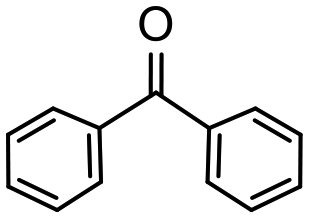
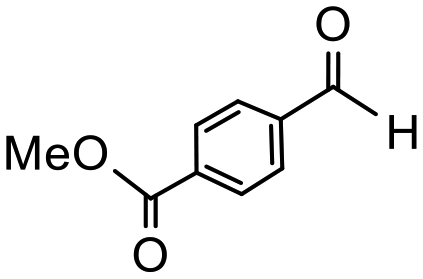
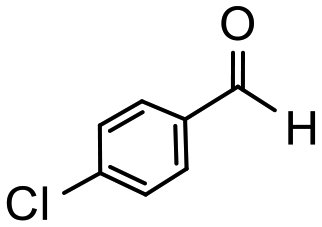
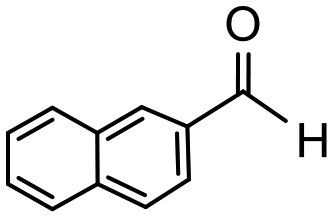
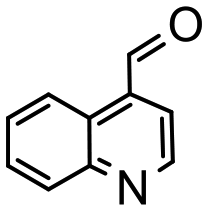
Having established optimum conditions and obtained an encouraging conversion profile for the benchmark hydrogenation of acetophenone, catalyst testing was extended to a range of substituted aromatic ketones and aldehydes to explore and assess the scope and efficacy of 2a′.PEG (Table 2). Good conversions were obtained for acetophenone substituted at the 4-position with electron-withdrawing group such as methyl ester, chloro or fluoro, which were reduced to the corresponding aryl ethanol with high selectivity (entries 1–4). To this end, while the hydrogenation of methyl 4-acetylbenzoate reached 70% conversion after 4 h with 98% selectivity for methyl 4-(1-hydroxyethyl)benzoate, with only a trace amount of the corresponding substituted cyclohexylethanol (entry 1), hydrogenation of 4-nitroacetophenone resulted in chemoselective reduction of the nitro group to afford 4-aminoacetophenone as the sole product at 66% conversion after 3 h (entry 5). While complete conversion was obtained by extending the reaction time to 4 h, the selectivity dropped to 86% due to hydrogenation of the ketone in 4-aminoacetophenone to afford 1-(4-aminophenyl)ethan-1-ol. Under the same conditions, hydrogenation of 4-bromoacetophenone occurred with facile competing hydrodehalogenation to afford a mixture of acetophenone, 1-(4-bromophenyl)ethan-1-ol and 1-phenylethanol at 63% conversion (entry 6). At this stage, we tentatively suggest that 1-phenylethanol forms via hydrogenation of acetophenone generated by hydrodehalogenation of 4-bromoacetophenone, as well as by competing hydrodehalogenation of 1-(4-bromophenyl)ethan-1-ol on the basis that hydrogenation of a commercial sample resulted in 69% conversion to 1-phenylethanol under the same conditions. Surprisingly, 4-acetylbenzonitrile showed no evidence for reduction even after a reaction time of 10 h (entry 7), which is most likely due to deactivation or poisoning of the catalyst as the large excess of nitrile donor-based substrate would saturate the surface-active ruthenium atoms and prevent access of the substrate. To this end, 2a′.PEG pretreated with 1 mmol of benzonitrile for 5 min was completely inactive for the hydrogenation of acetophenone whereas a conversion of 98% was obtained under the same conditions in the absence of benzonitrile. In addition, a competition reaction containing 4-acetylbenzonitrile and acetophenone catalyzed by 2a′.PEG only gave 3% conversion to 1-phenylethanol after 3 h compared with a conversion of 98% obtained in the absence of 4-acetylbenzonitrile. At this stage, we are not sure whether this catalyst inhibition is due to modification of the surface reactivity by coordination of the heteroatom donor-based substrate or saturation of the active sites, which would prevent substrate binding, and further studies will need to be conducted to distinguish between these processes. High conversions and good selectivities were obtained for the electron rich substrates 3- and 4-methoxyacetophenone while the sterically more demanding 2-substituted isomer gave a slightly lower yield but with excellent selectivity; the yield improved to 96% after extending the reaction time to 6 h (entries 8–10). A significantly longer reaction time was required to achieve high conversions for 2-acetonaphthone, benzophenone and isobutyrophenone, which are sterically more challenging substrates, however, good conversions with 100% selectivity for the aromatic alcohol were achieved for each substrate when the reaction time was extended to 24 h (entry 11–13). The same protocol was extended to the hydrogenation of heteroaromatic ketones including 3- and 4-acetylpyridine, which gave 98% and 94% conversion, respectively, to the corresponding alcohol, both with 100% selectivity, as well as 2-acetylthiophene and 2-acetylfuran both of which achieved high conversion with 100% selectivity after only 6 h (entries 14–17). High conversions were also obtained for the hydrogenation of a range of aryl aldehydes substituted with electron-donating and electron-withdrawing groups with each giving 100% selectivity for the corresponding alcohol in short reaction times (Table 3, entries 1–6). The sterically demanding substrates 2-naphthaldehyde and quinoline-4-carboxaldehyde both achieved reached 90% conversion with 100% selectivity after 8 h (Table 3, entries 7 and 8).
| Entry | Substrate | Time (h) | Conv.b (%) | Selectc (%) | TOFd (h−1) |
|---|---|---|---|---|---|
|
a Reaction conditions: 1 mmol of substrate, 0.1 mol% 2a′.PEG, 13 mL ethanol/water (1:1), 10 mol% K2CO3, time, 50 °C, 70 psi H2.
b Conversions determined by 1H NMR spectroscopy using 1,3-dinitrobenzene as internal standard or gas chromatography using decane as internal standard. Average of three runs.
c Selectivity for arylalcohol = [% arylalcohol/(% arylalcohol) + (% cyclohexylalcohol) + (% cyclohexylethanone)] × 100%.
d TOF moles of product per mole of catalyst per hour based on total ruthenium content.
e Selectivity for 4-aminoacetophenone.
f Selectivity for 1-phenylethanol resulting from hydrodebromination.
|
|||||
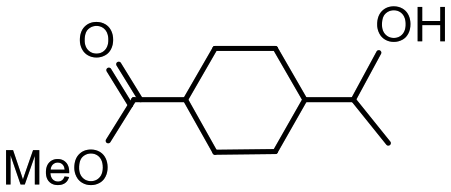
| 1 |
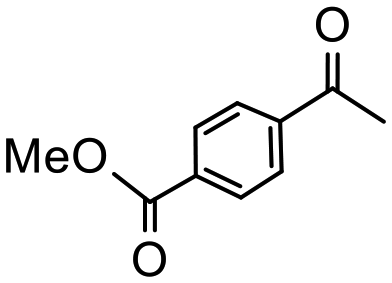
|
4 | 70 | 98 | 175 |
| 2 |
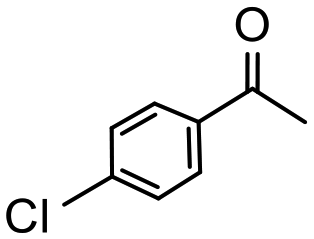
|
4 | 92 | 96 | 230 |
| 3 |
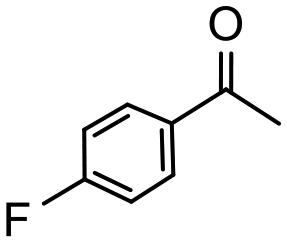
|
4 | 96 | 100 | 240 |
| 4 |
|
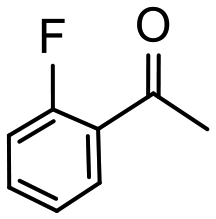
4 | 99 | 98 | 248 |
| 5 |
|
3 | 66 | 100e | 220 |
| 6 |
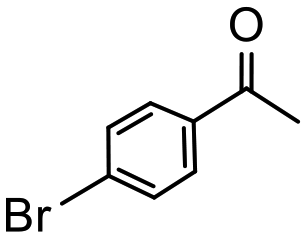
|
4 | 64 | 37f | 160 |
| 7 |
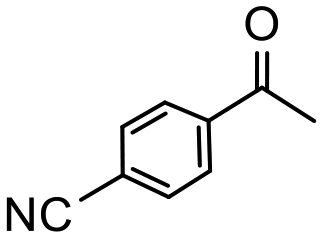
|
4 | 0 | 0 | 0 |
| 8 |
|
4 | 97 | 88 | 243 |
| 9 |
|
4 | 83 | 100 | 208 |
| 10 |
|
4 | 95 | 100 | 238 |
| 11 |
|
24 | 96 | 100 | 40 |
| 12 |
|
24 | 45 | 100 | 19 |
| 13 |
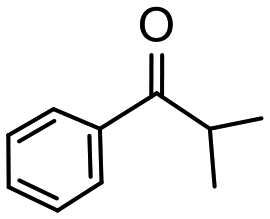
|
24 | 57 | 100 | 24 |
| 14 |
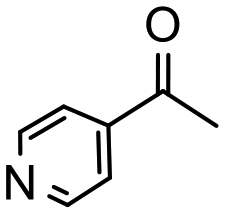
|
4 | 98 | 100 | 245 |
| 15 |
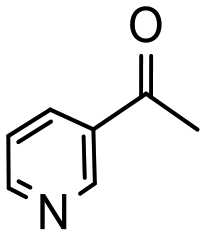
|
4 | 96 | 100 | 240 |
| 16 |
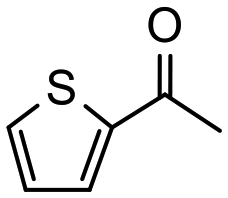
|
4 | 84 | 100 | 210 |
| 17 |

|
4 | 79 | 100 | 198 |
| Entry | Substrate | Time (h) | Conv.b (%) | Selectc (%) | TOFd (h−1) |
|---|---|---|---|---|---|
|
a Reaction conditions: 1 mmol of substrate, 0.1 mol% 2a′.PEG, 13 mL ethanol/water (1:1), 10 mol% K2CO3, time, 50 °C, 70 psi H2.
b Conversions determined by 1H NMR spectroscopy using 1,3-dinitrobenzene as internal standard or gas chromatography using decane as internal standard. Average of three runs.
c Selectivity for arylalcohol = [% arylalcohol/(% arylalcohol) + (% cyclohexylalcohol) + (% cyclohexanecarbaldehyde)] × 100%.
d TOF moles of product per mole of catalyst per hour based on total ruthenium content.
|
|||||
| 1 |
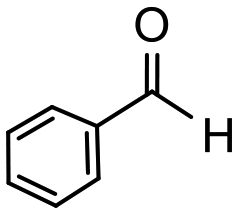
|
2 | 98 | 100 | 490 |
| 2 |
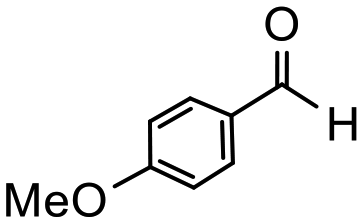
|
6 | 90 | 100 | 150 |
| 3 |
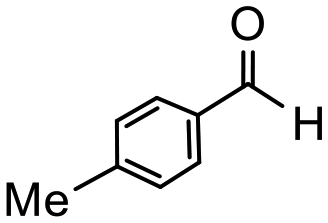
|
4 | 87 | 100 | 218 |
| 4 |
|
3 | 73 | 100 | 243 |
| 5 |
|
2 | 80 | 100 | 400 |
| 6 |
|
8 | 90 | 100 | 113 |
| 7 |
|
8 | 90 | 100 | 113 |
The presence of trace amounts of 1-cyclohexylethanol (1-CE) that were consistently obtained during catalyst optimization and evaluation together with several recent reports of RuNP-catalyzed hydrogenation of aromatic compounds25c,43 prompted us to investigate the conditions required to achieve complete reduction of acetophenone to 1-cyclohexylethanol with the aim of comparing the efficacy of 2a′.PEG against previously reported systems. Using the studies described above as a lead, the reduction of acetophenone was monitored as a function of time in a 1:1 mixture of water and ethanol at 70 °C under 400 psi of hydrogen using 0.1 mol% 2a′.PEG and the composition quantified by 1H NMR spectroscopy. The resulting composition-time profile in Fig. 5 shows rapid consumption of acetophenone with concomitant formation of 1-phenylethanol as the major species after only 10 min (64%) together with a minor amount of 1-cyclohexylethanol (24%). Longer reaction times resulted in hydrogenation of 1-phenylethanol to afford 1-cyclohexylethanol, which was obtained in quantitative yield after 70 min. A comparison using 0.1 mol% Ru/C (5 wt%) as the benchmark, under otherwise identical conditions, gave 100% conversion but only 31% selectivity for 1-cyclohexylethanol after 3 h. A reduction of the catalyst loading to 0.005 mol% resulted in 84% conversion with 80% selectivity for 1-cyclohexylethanol after 4 h, which corresponds to an initial TOF of 3400 h−1 (moles of 1-cyclohexylethanol per mol of catalyst per hour based on total ruthenium content). The same protocol was also extended to the reduction of 4-methoxyacetophenone, 4-hydroxyacetophenone, 4- and 2-fluoroacetophenone and methyl 4-acetylbenzoate, as representative examples of electron-rich and electron-poor substrates; gratifyingly, each gave the corresponding substituted 1-cyclohexylethanol with high selectivity in relatively short reaction times (Table 4). Although the vastly disparate conditions reported for hydrogenation of aryl ketones in the literature hampers a meaningful or credible comparison with existing systems, 2a′.PEG appears to compete with or outperform most reported RuNP-based catalysts.
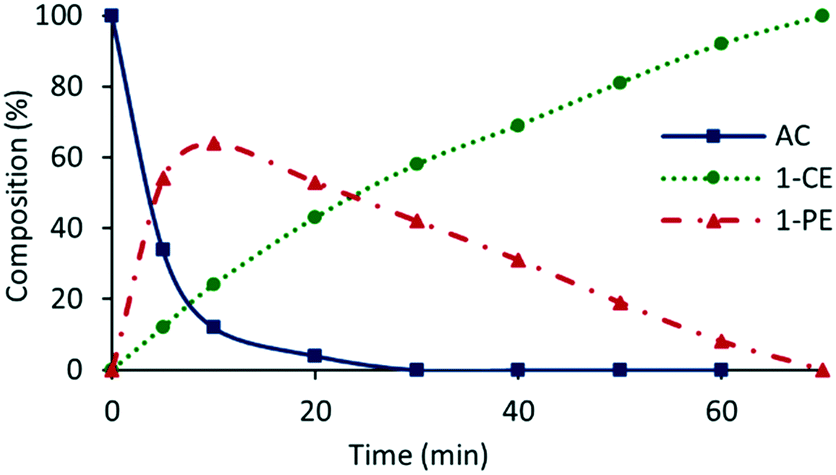 | ||
| Fig. 5 Reaction profile as a function of time for the hydrogenation of acetophenone (AC) to 1-phenylethanol (1-PE) and 1-cyclohexylethanol (1-CE) in a 1:1 mixture of water and ethanol at 70 °C under 400 psi of hydrogen using 0.1 mol% 2a′.PEG. | ||
|
a Reaction conditions: 1 mmol of substrate, 0.1 mol% 2a′.PEG,13 mL ethanol/water (1:1), 10 mol% K2CO3, time in parenthesis after conversion, 70 °C, 400 psi H2.
b Conversions determined by 1H NMR spectroscopy using 1,3-dinitrobenzene as internal standard. Average of three runs.
c Selectivity for the cyclohexylethanol product [% cyclohexylethanol/(% arylethanol) + (% cyclohexylethanol) + (% cyclohexylethanone)] × 100%.
|
||
|---|---|---|
| Product |
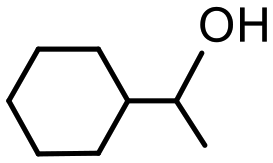
|
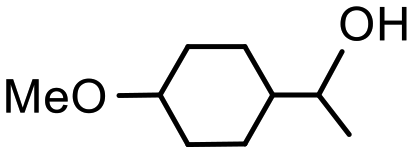
|
| Conversionb | 100% (70 min) | 99% (70 min) |
| Selectivityc | 100% | 79% |
| Product |
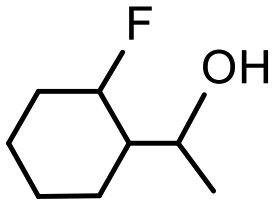
|
|
| Conversionb | 100% (70 min) | 100% (70 min) |
| Selectivityc | 84% | 62% |
| Product |
|
|
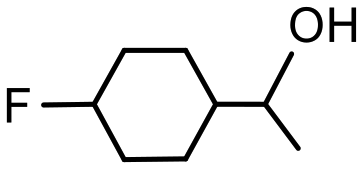
| Conversionsb | 100% (70 min) | 100% (70 min) |
| Selectivityc | 57% | 100% |
For example, the initial TOF of 3400 h−1 obtained in water/ethanol with 2a′.PEG at 70 °C under 400 psi hydrogen is a substantial improvement on that of 60 h−1 for RuNP@[C12MIM][BTA] (120 °C, 1740 psi H2)8a and 24 h−1 obtained with NHC-stabilized RuNPs (80 °C, THF, 580 psi H2)25c and higher than 2600 h−1 obtained with poly-N-vinylpyrrolidone-stabilized RuNPs in cyclohexane (80 °C, 580 psi H2).43h Several systems have been reported to operate close to room temperature but with markedly lower TOFs including methylated β-cyclodextrin-capped RuNPs (TOF of 30 h−1, 290 psi H2),7a RuNP-ligated by a cholesterol-derived NHC (TOF of 1 h−1, THF, 70 psi H2),43i RuNP stabilized by PPh3 (TOF = 5 h−1, THF, 290 psi H2),25b water soluble RuNPs stabilized by a sulfonated diphosphine cyclodextrin combination (TOF of 89 h−1, 145 psi H2)26 and alkyl sulfonated diphosphine-stabilized RuNPs (TOF of 5 h−1, 145 psi H2).25e
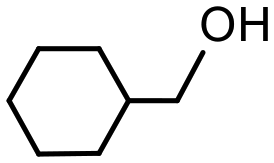
As high conversions and selectivities were obtained for the hydrogenation of ketones and aldehydes in water, as well as a water/ethanol mixture, a series of recycle experiments were conducted on the basis that aqueous phase catalysis would facilitate separation and recovery via a straightforward extraction protocol. Recycle experiments were conducted using benzaldehyde as the substrate and extracting the product and unreacted substrate into ethyl acetate before recharging the aqueous catalyst solution with a further portion of benzaldehyde and re-pressurizing the reactor with hydrogen. The data in Fig. 6a is encouraging as the catalyst retained good activity and complete selectivity for ten cycles conducted over two days with only a minor drop in conversion between runs 7 to 10. A minor increase in conversion from 77% to 85% after the first run as well as an additional increase after storing the spent catalyst under hydrogen overnight may be due to an increase in the number of active metallic ruthenium sites resulting from reduction of surface ruthenium oxide and/or a morphological change. However, further surface studies on the catalyst before and after reaction using a combination of TEM, XPS and X-ray absorption fine structure analysis to profile the composition and morphology as a function of treatment will be required to establish the origin of this enhancement. To this end, Hutchings has demonstrated that treatment of commercial Ru/C catalyst under the conditions required for hydrogenation of lactic acid reduces the ruthenium and changes the morphology from raft-like to more active small discrete nanoparticles.5d Analysis of the organic phase collected after each reuse revealed that the ruthenium content was below the detection limit of ICP-OES indicating that the drop in conversion is unlikely to be due to leaching of the catalyst during extraction and may well result from adhesion of the catalyst to the surface of the reactor and glassware during separation and recovery. TEM analysis of the aqueous phase remaining after the 10th run revealed that the ruthenium nanoparticles remained monodisperse with a mean diameter of 1.31 ± 0.42 nm compared with 1.32 ± 0.30 nm for a freshly prepared sample of 2a′.PEG (Fig. 6b and c). While further studies are clearly required, the stable profile obtained for reuse of 2a′.PEG suggests that this system may well be sufficiently robust for integration into a scale-up continuous flow process.33c
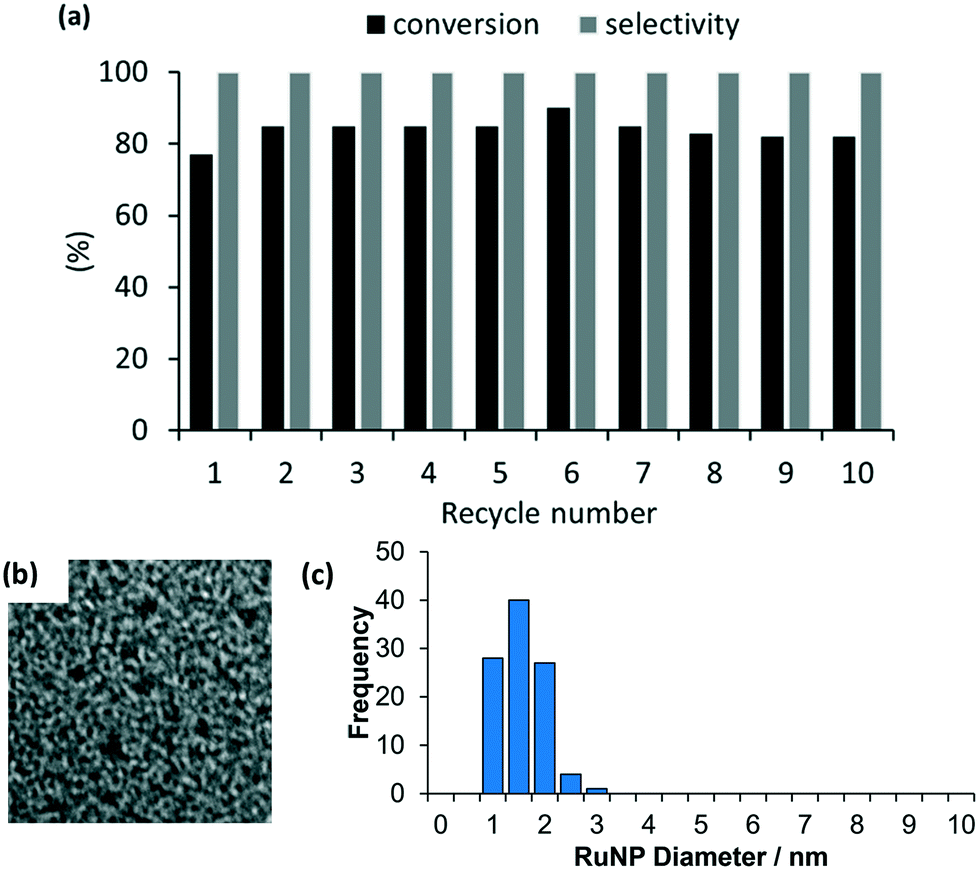 | ||
| Fig. 6 (a) Recycle study for the hydrogenation of benzaldehyde in water catalyzed by 2a′.PEG and (b) HRTEM image of catalyst after 10th run and (c) particle size distribution for RuNP@OPPh2-PEGPIILP (2a′.PEG) after 10 cycles revealing an average NP diameter of 1.31 ± 0.42 nm. Scale bars are 5 nm (white). | ||
The heterogenous nature of the catalyst was examined by conducting a hot filtration test in which the hydrogenation of acetophenone catalyzed by 0.1 mol% 2a′.PEG was allowed to reach 50% conversion (time 45 min) after which the reaction was stopped by venting the reactor. The reaction mixture was filtered through a 45-micron syringe filter and the filtrate subjected to another hydrogenation cycle. Analysis of the reaction mixture after a further 3 h revealed that the conversion remained at 50% (Fig. 7, blue line); for comparison, the corresponding conversion-time profile for a reaction conducted under the same conditions without filtration reached completion after 240 min (Fig. 7, red line), which is a strong indication that the active species is heterogeneous. In a complimentary hot filtration test, a hydrogenation conducted under the same conditions was allowed to reach completion after which the reaction mixture was filtered through a syringe filter (0.45 μm) and an additional portion of acetophenone added to the filtrate before repressurizing the reactor with hydrogen. There was no change in the conversion even after an additional 4 h under the same reaction conditions which lends further support for catalysis by a heterogeneous system.
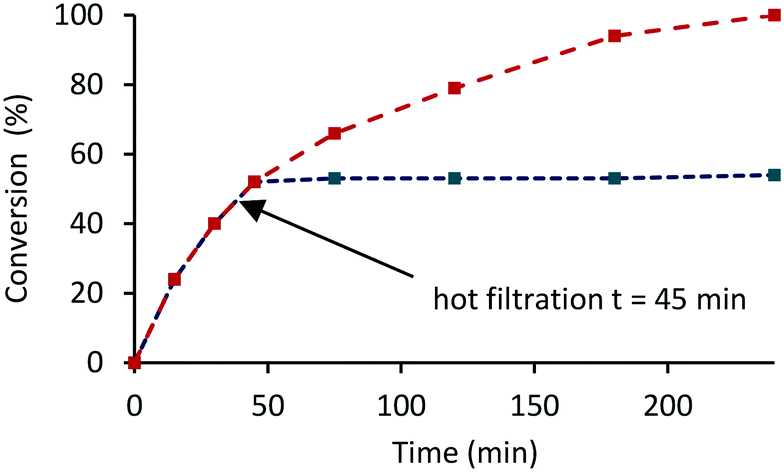 | ||
| Fig. 7 Hot filtration experiment for the hydrogenation of acetophenone catalysed by 0.1 mol% 2a′.PEG at 50 °C under 70 psi of hydrogen showing that the reaction has been quenched after filtration. Red line – hydrogen evolution in the presence of 2a′.PEG; blue line – hydrogen evolution in the presence of 2a′.PEG with filtration after t = 45 min. | ||
At this stage we tentatively suggest that the high selectivity for hydrogenation of the CO bond in aryl and heteroaryl ketones to afford the corresponding aromatic alcohol could be due to either (i) hemilabile coordination of the phosphine oxide to the NP surface preventing arene coordination while allowing the ketone to coordinate and/or (ii) the phosphine oxide causing polarized activation of the hydrogen on the NP surface as shown in Fig. 8a–c and d–f, respectively. As such, it should be possible to control factors that influence catalyst selectivity such as the interaction of the substrate with the NP surface and/or the activation of the hydrogen by modifying the ionic and steric environment of the polymer or introducing functional groups to affect the hydrogen activation step.
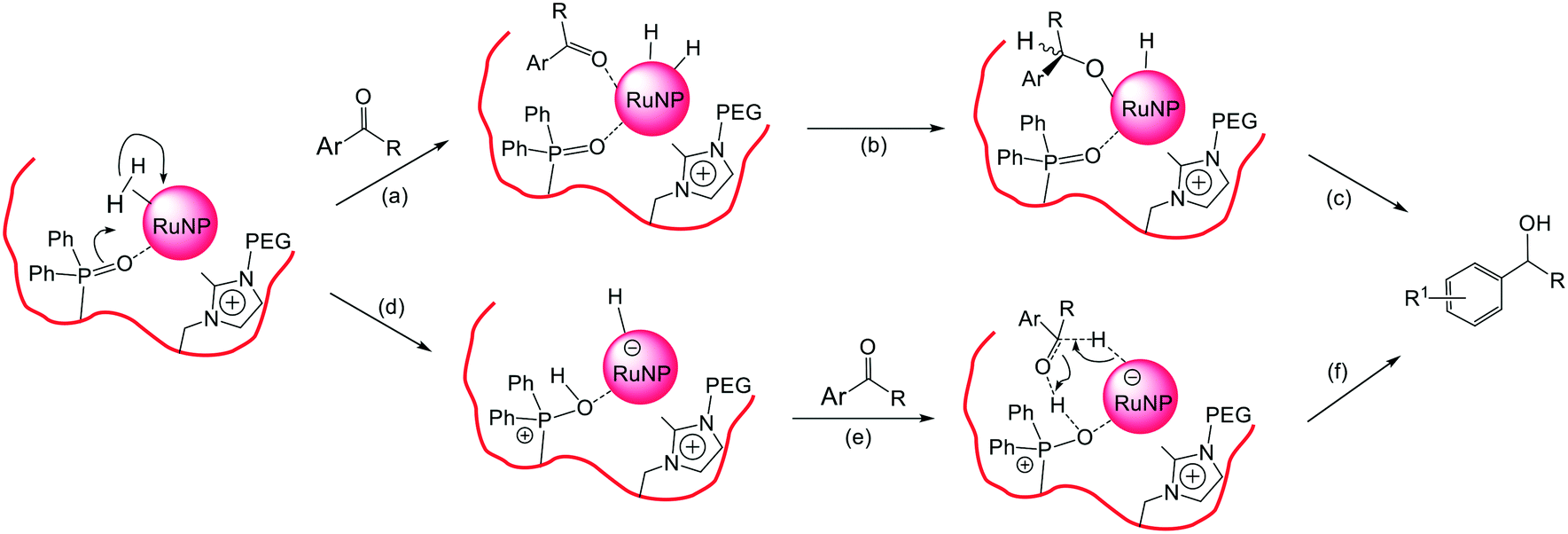 | ||
| Fig. 8 Proposed key steps in the pathway for the selective hydrogenation of the CO bond in aryl ketones catalyzed by either (a–c) selective coordination of the carbonyl group to the NP surface and (d–f) phosphine oxide induced polarized activation of hydrogen on the NP surface. | ||
The efficacy of 2a′.PEG for the hydrogenation of acetophenone under mild conditions and the promising recycle studies prompted us to explore the practicality for scale-up. Under the optimum conditions identified above, 0.1 mol% 2a′.PEG catalyzed the hydrogenation of 10 mmol of acetophenone (0.2 M) in a 1:1 mixture of ethanol and water to afford 92% conversion with 97% selectivity for 1-phenylethanol after 4 h; this compares favourably with the 95% conversion and 96% selectivity obtained during optimization. The group now conducts the synthesis of 2a′.PEG on a larger scale (4–5 mmol) with no deleterious effect on performance as this system forms part of the group's catalyst portfolio and is routinely tested in our catalyst screening programme. To this end, 2a′.PEG has been identified as an extremely efficient catalyst for the hydrolytic evolution of hydrogen from sodium borohydride,33g the selective reduction of nitroarenes to N-phenylhydroxylamine and the selective hydrogenation of quinoline and its derivatives.
Hydrogenation of furfural, levulinic acid and ethyl levulinate
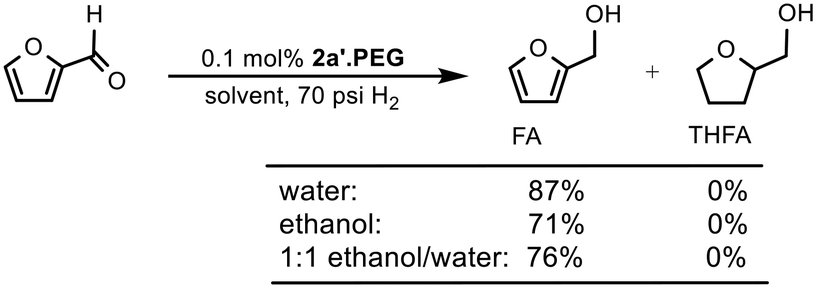
Having demonstrated the efficacy of 2a′.PEG as a catalyst for the selective hydrogenation of aryl ketones and aldehydes, the substrate range was extended to include key sustainable biomass-derived substrates such as furfural, levulinic acid and its ethyl ester as the products are important bio-derived platform chemicals and transportation fuels.2,44 Selective hydrogenation of furfural affords furfuryl alcohol, which is an intermediate for the production of plasticizers, dispersing agents, lubricants and resins45 while hydrogenation of levulinic acid and its esters affords γ-valerolactone, which is a green solvent, perfume additive, fuel additive and a raw material in the production of biofuels, fine chemicals, pharmaceuticals, green polymers and resins.3b,46Relying on the optimum conditions identified above as a lead, a solvent screen using 0.1 mol% 2a′.PEG as catalyst revealed that reactions conducted in water, ethanol and a 1:1 water–ethanol mixture gave conversions of 87%, 71% and 76%, respectively, with 100% selectivity for furfuryl alcohol (FA) after 3 h at 50 °C under 70 psi of hydrogen (Table 5). In comparison, reactions conducted in organic solvents such as toluene and 2-Me-THF gave markedly lower conversions (see Table S2 in the ESI†). In contrast to the hydrogenation of ketones, addition of potassium carbonate had no noticeable effect on the conversion. The variation in conversion as a function of pressure for the aqueous phase hydrogenation of furfural catalyzed by 0.1 mol% 2a′.PEG mirrored that for the hydrogenation of acetophenone in that conversions increased with increasing pressure and ultimately plateaued at ca. 400 psi. However, in stark contrast to acetophenone, there was no evidence for hydrogenation of the furan ring to afford tetrahydrofurfuryl alcohol (THFA) (see Fig. S1 in the ESI†). Comparative catalyst testing revealed that similar conversions were obtained with 2a, 2a.PEG, 2a′ and 2a′.PEG while modification by removal of the phosphine oxide resulted in a marked drop in activity as 2b and 2b.PEG only gave 56% and 58% conversion, respectively. Gratifyingly, 2a′.PEG was significantly more active than commercial Ru/C (5 wt%) which only reached 21% conversion with 100% selectivity for furfuryl alcohol after 3 h. The efficacy of 2a′.PEG was further tested by reducing the catalyst loading to 0.005 mol% and the conversion of 79% obtained after 5 h corresponds to a TON of 15800 and a TOF of 3160 h−1.
The high selectivity for furfuryl alcohol is consistent with several recent reports of RuNP-catalyzed hydrogenation of furfural including cyclodextrin-based RuNPs,7c ultra-small RuNPs on porous supports,4 a RuNP-coated polyethersulfone membrane,6b RuNPs supported on reduced graphene oxide46 and RuNPs stabilized on an acid MOF material (RuNP@MIL-10)48 or an alumina-based MOF (RuNP@Al-MIL-53-BDC).49 With the exception of the RuNP-polyethersulfone catalytic membrane, which gave an unprecedented TOF of 48000 h−1, 2a′.PEG outperforms each of these systems, in the majority of cases by quite some margin. For example, the initial TOF of 3160 h−1 obtained with 2a′.PEG in water is significantly higher than 237 h−1 for silica-supported RuNPs at 100 °C,50 137 h−1 for graphene-modified RuNPs at 20 °C,18a and a marked improvement on 16 h−1 for cyclodextrin-based RuNPs at 30 °C,7c,b,51 30 h−1 for RuNPs on porous supports at 40 °C,4 29 h−1 with RuNPs supported on reduced graphene oxide at 20 °C,47 20 h−1 for RuNPs supported on an alumina-based MOF at 20 °C,49 7 h−1 for RuNPs supported on zirconium-based MOFs at 20 °C,52 and 13 h−1 for Rh and Pt-nanoparticles stabilized by phosphine-functionalized silica at 80 °C.53
Ruthenium also appears to be the metal of choice for the selective hydrogenation of levulinic acid (LA) and its esters to γ-valerolactone (GVL) due to its exceptional selectivity for hydrogenation of the carbonyl group without reducing other unsaturated functional groups.5f,14,17 The major pathway for this transformation appears to involve selective hydrogenation of the ketone to afford 4-hydroxypentanoic acid (4-HPA) which undergoes lactonisation to afford GVL (Scheme 1). The efficacy of 2a′.PEG as a catalyst for the hydrogenation of ketones, aldehydes and furfural prompted us to extend its application to the hydrogenation of levulinic acid as commercial catalysts suffer from deactivation and poor reusability and durability. To this end, there are already numerous reports of RuNP-based systems with encouraging reaction credentials for the hydrogenation of LA to GVL. For example, UiO-66-derived Ru/ZrO2@C,5f few layer graphene-supported RuNPs,19,5c cyclodextrin-based polymer stabilized RuNPs,7d Ru–N-heterocyclic carbene-derived RuNPs,54 RuNPs supported on TiO2,34 RuNPs on mixed magnesium lanthanum oxide,55 and Ru(OH)2 supported on high surface area anatase.56 Other recently developed catalysts with promising reaction profiles include bifunctional systems containing RuNPs stabilized by an acid-modified support designed for the nanoparticles to catalyze the hydrogenation step and the acid to facilitate the dehydration-lactonisation.10–12,27a
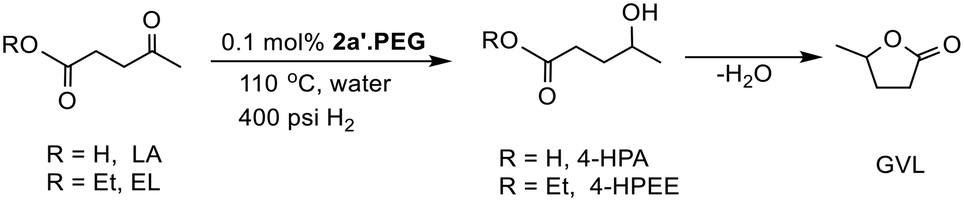 | ||
| Scheme 1 Conversion of levulinic acid (R = H, LA) and ethyl levulinate (R = Et, EL) to GVL via hydrogenation followed by lactonization. | ||
Variation of the catalyst loading, reaction temperature, solvent, and hydrogen pressure rapidly established that good conversions of levulinic acid could be obtained in water at 110 °C under 400 psi of hydrogen using 0.1 mol% of 2a′.PEG (see Table S3 in the ESI†). Under these conditions, complete conversion to GVL (77%) and 4-HPA (23%) was obtained after only 4 h, while GVL was obtained as the sole product when the reaction time was extended to 8 h. for comparison, 0.1 mol% of commercial Ru/C (5 wt/%) only reached 41% conversion with 95% selectivity for GVL after 8 h. In stark contrast to the enhancement in conversion obtained upon addition of a base in the hydrogenation of aryl ketones, addition of 10 mol% K2CO3 resulted in a marked reduction in activity as the conversion only reached 58% to afford a mixture of 4-HPA (15%) and GVL (43%). Reduction of the catalyst loading to 0.005 mol% gave 61% conversion to a mixture of 4-HPA (5%) and GVL (56%) after 5 h, which corresponds to an initial TOF of 2440 h−1; furthermore, complete conversion to GVL could be obtained by extending the reaction time to 20 h.
With the aim of comparing the efficacy of 2a′.PEG against existing systems, the hydrogenation of LA was further investigated by monitoring the composition as a function of time in water at 110 °C with a catalyst loading of 0.1 mol%. The resulting profile, presented graphically in Fig. 9a, shows rapid consumption of LA with concomitant formation of GVL in the early stages of the reaction while there was a significant build-up of 4-HPA after 2 h, which was gradually consumed to afford GVL as the sole product after 8 h. This profile is consistent with facile intramolecular lactonization of 4-HPA as it is generated in the initial stages of the reaction after which it becomes rate limiting when all the LA has been consumed;57 this may well be attributed to depletion of the acidic substrate which catalyzes the lactonization step. To this end, a series of reactions conducted under the same conditions in the presence of one equivalent of either a commercial sulfonated cation exchange resin (Amberlyst H15, 4.7 meq g−1 dry weight) or butyric acid showed no evidence for build-up of 4-HPA and gave 100% conversion to GVL after only 4 h, consistent with rapid lactonization over the entire reaction. Moreover, 10 mol% loading of Amberlyst was enough to prevent build-up of 4-HPA as GVL was also obtained as the sole product after only 4 h, which is clear from the composition-time profile in Fig. 9b. As the addition of base had a dramatic effect on the conversion and selectivity profile, the hydrogenation of LA was monitored as a function of time under the same conditions but in the presence of 10 mol% K2CO3. The resulting composition-time profile in Fig. 9c reveals a marked difference compared with the profile in the absence of base (Fig. 9a) as both the hydrogenation step and lactonization of the resulting 4-HPA appear to be inhibited. Under these conditions, the conversion of LA only reached 57% after 8 h to afford a mixture of 4-HPA (15%) and GVL (42%). While the influence of the base on the lactonization step clearly results from neutralization of the acid, the influence of the base on the hydrogenation step is less easy to rationalize especially since base enhances the hydrogenation of aryl ketones.
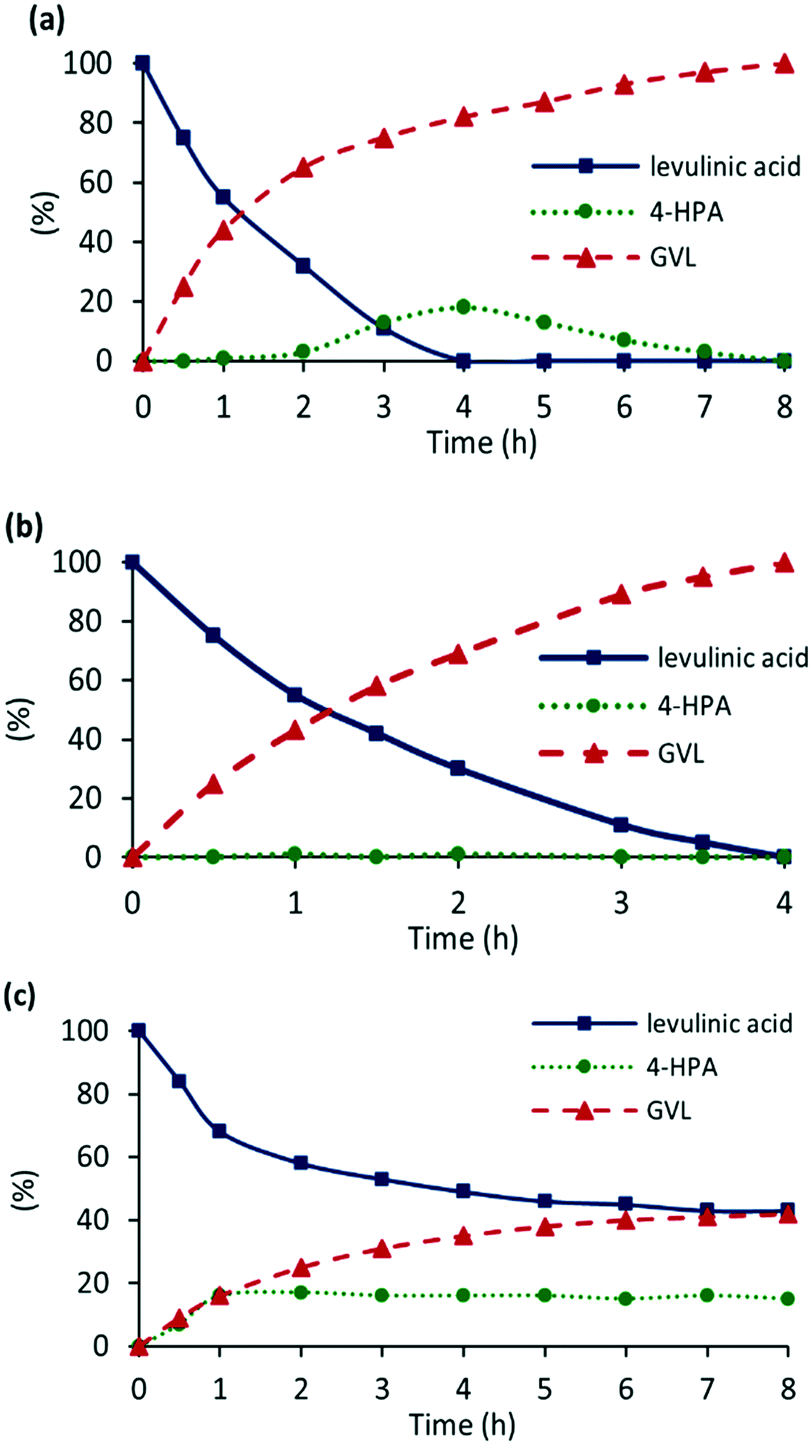 | ||
| Fig. 9 Reaction composition as a function of time for the hydrogenation of levulinic acid (1.0 mmol) in water at 110 °C under 400 psi of hydrogen using 0.1 mol% of 2a′.PEG (a) in the absence of base, (b) in the presence of 10 mol% Amberlyst H-15 and (c) in the presence of 10 mol% K2CO3. | ||
A survey of the literature reveals that 2a′.PEG either competes with or outperforms many existing RuNP-based systems. For example, the initial TOF of 2440 h−1 obtained with 2a′.PEG in water at 110 °C under 400 psi of hydrogen is significantly higher than 612 h−1 for Ru/ZrO2@C at 140 °C,5f 679 h−1 for ultrafine RuNPs immobilized on few-layer graphene at 80 °C,5c 661 h−1 for RuNPs supported on sulfonic acid-modified reduced graphene at 50 °C,10b 1440 h−1 for Ru/H-β at 200 °C (ref. 13b) and a substantial improvement on 374 h−1 obtained with Ru–NHC derived RuNPs at 130 °C,54 210 h−1 for cyclodextrin-based polymer-assisted RuNPs,7d 102 h−1 for RuNP@DOWEX at 70 °C,11 48 h−1 for RuNPs confined in zirconium-containing spherical mesoporous silica at 70 °C,12 100 h−1 for RuNP supported on acid-functionalized mesoporous carbon at 70 °C,10a and 365 h−1 obtained with few-layer graphene-supported RuNPs at room temperature.19 There have been reports of systems with more efficient reaction profiles including RuNP–NH2–γ-Al2O3 and RuNP@TiO2, which gave TOFs of 3355 h−1 and 7676 h−1, respectively.18b,34 The efficacy of the former was attributed to the highly dispersed nature of the ruthenium centers with electron rich states while the latter was due to the promoting effect of water, which participated in the hydrogenation of the CO group. The composition of these systems will inform the design of more active nanoparticle-based polymer-immobilized ionic liquid-supported catalysts as the system is modular and amenable to modification and functionalization.
As alkyl levulinates can be obtained by acid-catalyzed esterification of cellulose feed the efficacy of 2a′.PEG as a catalyst for the hydrogenation of ethyl levulinate (EL) was examined to compare with LA. Under the optimum conditions identified above, 2a′.PEG gave 100% conversion of EL to a mixture of GVL (62%) and 4-hydroxypentanoic acid ethyl ester (38%) after 8 h whereas LA gave quantitative conversion to GVL in the same time. However, addition of 10 mol% K2CO3 as base resulted in a dramatic improvement to afford a quantitative yield of GVL. The key difference between reactions conducted in the absence and presence of base are evident from a comparison of the composition-time profiles shown in Fig. 10a and b. In the absence of base (Fig. 10a), the concentration of 4-hydroxypentanoic acid ethyl ester (4-HPEE) increases to 52% during the first 3 h, indicating that the hydrogenation of EL also occurs via a two-step hydrogenation–lactonization sequence. Fig. 10a also reveals that formation of GVL is markedly slower than for the corresponding hydrogenation of LA and that the poor selectivity for GVL after 8 h in the absence of base is due to the persistence of the 4-HPEE, as lactonization is the limiting step. As expected, addition of base accelerated the lactonization to afford GVL as the sole product after 8 h, even though the hydrogenation step appears to be slightly slower than in the absence of base (Fig. 10b).
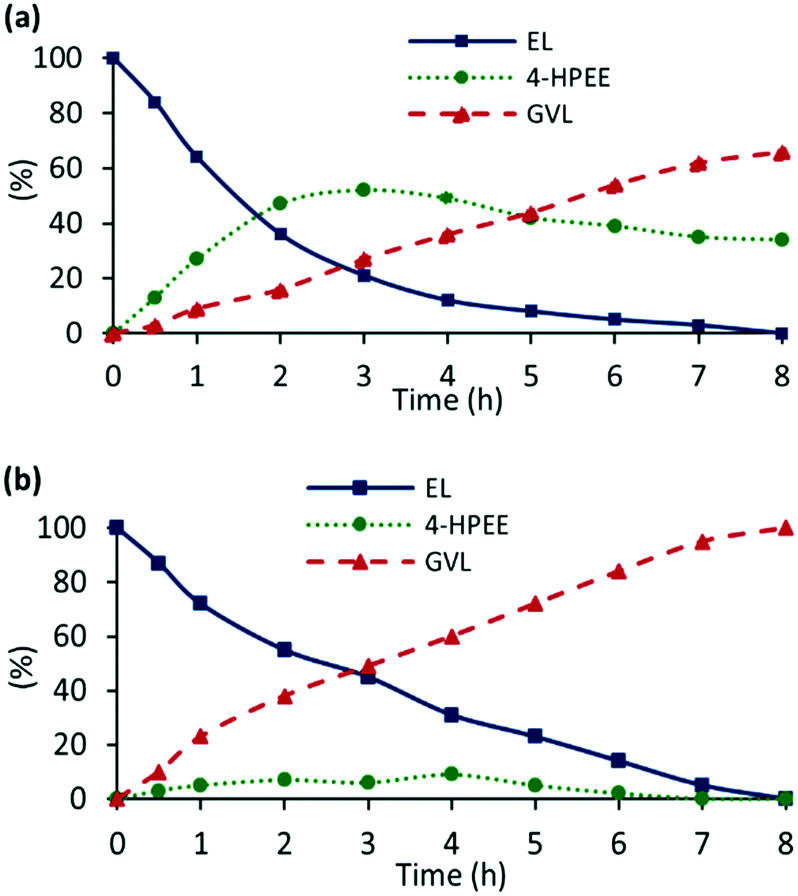 | ||
| Fig. 10 Reaction composition as a function of time for the hydrogenation of ethyl levulinate (1.0 mmol) in water (13 mL) at 110 °C under 400 psi of hydrogen using 0.1 mol% of 2a′.PEG (a) in the absence of base and (b) in the presence of 10 mol% K2CO3. | ||
Enhancement in the RuNP-catalysed production of GVL by assisting the lactonization step either by addition of base or by functionalization of the nanoparticle support with acid has been reported. For example, ruthenium hydroxide supported on high surface area anatase showed a significant improvement in the GVL yield in the presence of heterogeneous bases such as MgO and hydrotalcite whereas homogeneous bases appeared to suppress the catalytic transfer hydrogenation step.56 Dual, bifunctional catalysts based on RuNPs supported in sulfonic acid-modified MOFs (UiO-66 and MIL-101) also gave a much higher yield of GVL than the corresponding system without acid, which gave 4-hydroxypentanoic acid methyl ester as the major product.14,15 Similarly, catalyst based on RuNPs and a polyoxometalate integrated within UiO-66 was markedly more selective for GVL than RuNP@UiO-66 because of an enhancement in the lactonization step.16
A preliminary reusability study was undertaken for the hydrogenation of levulinic acid to GVL to assess the robustness and longevity of 2a′.PEG, with the aim of establishing its suitability for integration into a continuous flow system. The practical problems associated with recovering a small amount of catalyst (0.5 mol%, 2.1 mg) by filtration prevented us from undertaking a conventional recycle experiment. Therefore, a reuse experiment was conducted by extracting the product and unreacted starting material with ethyl acetate before recharging the aqueous phase with a further portion of levulinic acid and Amberlyst. Reactions were run for 2.5 h to obtain conversions that would also allow any changes in catalyst activity to be identified. Under these conditions, 2a′.PEG recycled ten times over three days with only a negligible drop in conversion from 85% to 84% and no detectable loss in selectivity (Fig. 11). The successful reuse of 2a′.PEG is extremely encouraging and suggests that this class of catalyst may be suitable for scale-up studies using a continuous flow system.
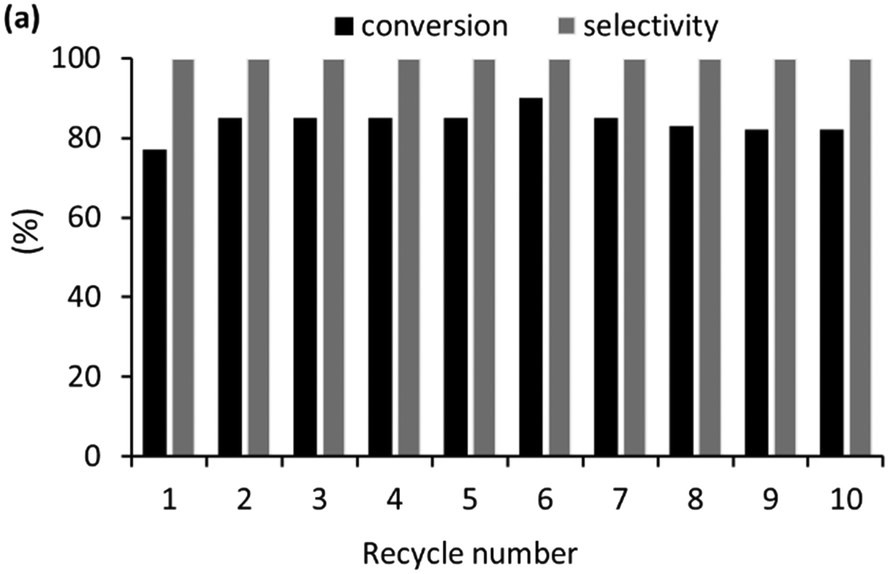 | ||
| Fig. 11 Reuse study for the aqueous phase hydrogenation of levulinic acid catalysed by 0.5 mol% of 2a′.PEG at 110 °C for 4 h under 400 psi hydrogen with 1 equiv. of Amberlyst H-15 as additive. | ||
Conclusions
Impregnation of phosphine-decorated polymer immobilized ionic liquids with ruthenium trichloride during the preparation of ruthenium nanoparticles resulted in serendipitous oxidation to afford phosphine oxide and ruthenium(II) species. The derived phosphine oxide-decorated PIIL-stabilized RuNPs (RuNP@OPPh2PIILS) are remarkably active and selective catalysts for the aqueous phase hydrogenation of a wide range of aryl and heteroaryl ketones and aldehydes to either the corresponding aryl alcohol or the cyclohexyl-based alcohol as well as the conversion of levulinic acid and its ethyl ester to GVL. Moreover, catalysts generated by reduction of pre-formed phosphine oxide-decorated PIIL impregnated with ruthenium trichloride are as efficient as that generated from ruthenium trichloride and either PPh2PIILP or PPh2PEGIILP which simplified catalyst preparation, handling and storage. The initial TOFs of 2350 h−1 and 3160 h−1 for the hydrogenation of acetophenone to 1-phenylethanol and furfural to furfuryl alcohol are among the highest to be reported for these transformations catalyzed by RuNPs in aqueous media. Batch recycle experiments on the hydrogenation of benzaldehyde were extremely encouraging as the activity–selectivity profile for RuNP@OPPh2PEG-PIILP was relatively stable over ten runs with only a minor reduction in conversion between runs 7 and 10. A stable profile was also obtained over a ten run recycle study on the hydrogenation of levulinic acid to GVL, which suggest that this system may well be suitable for integration into a continuous flow reactor platform. Even though this is the first report of the use of a phosphine oxide modified support in combination with ruthenium nanoparticles, there have been recent examples of the use of a phosphine-modified polymer to synthesize phosphine-stabilized RuNPs which appear to have been misinterpreted as we believe that the resulting catalysts contain a phosphine oxide derived from reduction of impregnated ruthenium trichloride, in much the same manner as described herein. We are currently exploring the synthesis of the corresponding phosphine-decorated polymer immobilized ionic liquid-stabilized RuNPs to explore the influence of the phosphine heteroatom donor on catalyst performance and to undertake a comparison with their phosphine oxide-based counterparts. Additional surface studies are also underway to unequivocally establish the composition of the nanoparticles and the nature/existence of any nanoparticle⋯OPPh2 interactions and its influence on surface properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. P. thanks the EPSRC Centre for Doctoral Training in Renewable Energy Northeast Universities (‘ReNU’) EP/S023836/1 for a studentship. We gratefully acknowledge Newcastle University for financial support (T. S. T. T.) and Taibah University, Saudi Arabia (Scholarship to H. Y. A.). This research was also supported through a studentship supported by the EPSRC Centre for Doctoral Training in Molecules to Product (EP/SO22473/1). The authors greatly acknowledge their support of this work. We also thank Dr Kathryn White for the SEM images (Faculty of Medical Sciences, Newcastle University). This article is dedicated to the memory of Elwood, Jake and Ella, may they rest in peace.Notes and references
- H.-U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steiner and M. Studer, Adv. Synth. Catal., 2003, 345, 103–151 CrossRef CAS.
- (a) T. Werpy and G. Peterson, Top Value-Added Chemicals from Biomass, U.S. Department of Energy, Pacific Northwest National Laboratory, National Renewable Energy Laboratory, Office of Biomass Program, Richland, WA, 2004 Search PubMed; (b) D. R. Dodds and R. A. Gross, Science, 2007, 218, 1250–1251 CrossRef PubMed; (c) M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–546 RSC; (d) K. Yan, T. Lafleur, X. Wu, J. Chai, G. Wu and X. Xie, Chem. Commun., 2015, 51, 6984–6987 RSC; (e) I. J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef PubMed; (f) I. T. Hovarth, H. Mehdi, V. Fabos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238–242 RSC; (g) D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC; (h) J. C. Serrano-Ruiz, R. Luque and A. Sepulveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC.
- (a) For an up-to-date and highly informative review of catalysis with colloidal ruthenium see A. R. Axet and K. Philippot, Chem. Rev., 2020, 120, 1085–1145 CrossRef PubMed; (b) C. Michel and P. Gallezot, ACS Catal., 2015, 5, 4130–4132 CrossRef CAS; (c) L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS PubMed.
- Z. Zhang, J. Song, Z. Jiang, Q. Meng, P. Zhang and B. Han, ChemCatChem, 2017, 9, 2448–2452 CrossRef CAS.
- (a) F. Leng, I. C. Gerber, M. R. Axet and P. Serp, C. R. Chim, 2018, 21, 346–353 CrossRef CAS; (b) Z. Jiang, G. Lan, X. Liu, H. Tang and Y. Li, Catal. Sci. Technol., 2016, 6, 7259–7266 RSC; (c) H. Tao, J. Ding, C. Xie, Y. Gao, J. Song and Z. Sun, Nanotechnology, 2018, 29, 075708 CrossRef PubMed; (d) S. Iqbal, S. A. Kondrat, D. R. Jones, D. C. Schoenmakers, J. K. Edwards, L. Lu, B. R. Yeo, P. P. Wells, E. K. Gibson, D. J. Morgan, C. J. Kiely and G. J. Hutchings, ACS Catal., 2015, 5, 5047–5059 CrossRef CAS; (e) Y. Ang, Z. Rong, Y. Wang and J. Qu, J. Catal., 2016, 333, 8–16 CrossRef; (f) W. Co, W. Luo, H. Ge, Y. Su, A. Wang and T. Zhang, Green Chem., 2017, 19, 2201–2211 RSC.
- (a) J. Gmeiner, S. Behrens, B. Spliethoff and O. Trapp, ChemCatChem, 2016, 8, 571–576 CrossRef CAS; (b) G. Gagnato, A. Figoli, F. Galiano and A. Sanna, J. Mater. Chem. A, 2018, 6, 4955–4965 RSC; (c) A. Indra, P. Maitry, S. Bhaduri and G. K. Lahir, ChemCatChem, 2013, 5, 322–330 CrossRef CAS.
- (a) N. T. T. Chau, S. Handjani, J.-P. Guegan, M. Guerrero, E. Monflier, K. Philippot, A. Denicourt-Nowicki and A. Roucoux, ChemCatChem, 2013, 5, 1497–1503 CrossRef CAS; (b) R. Herbois, S. Noel, B. Leger, S. Tilloy, S. Menuel, A. Addad, B. Martel, A. Ponchel and E. Monflier, Green Chem., 2015, 7, 2444–2454 RSC; (c) S. Noel, D. Bourbiaux, N. Tabary, A. Ponchel, B. Martel, E. Monflier and B. Leger, Catal. Sci. Technol., 2017, 7, 5982–5992 RSC; (d) M. Chen, Q. Dong, W. Ni, X. Zhao, Q. Gu, G. Tang, D. Li, W. Ma and Z. Hou, ChemistrySelect, 2017, 2, 10537–10545 CrossRef CAS.
- (a) J. Julis, M. Hölscher and W. Leitner, Green Chem., 2010, 12, 1634–1639 RSC; (b) K. L. Luska, P. Migowski and W. Leitner, Green Chem., 2012, 2, 1403–1409 Search PubMed.
- For a timely, highly relevant, and insightful review on the Ruthenium-Catalyzed Hydrogenation of Renewable Biomass-Derived Levulinic Acid in Aqueous Media, see A. Seretis, P. Diamantopoulou, I. Thanou, P. Tzevelekidis, C. Fakas, P. Lilas and G. Papadogianakis, Front. Chem., 2020, 8, 221 CrossRef CAS PubMed.
- (a) A. Villa, M. Schiavoni, C. E. Chan-Thaw, P. F. Fulvio, R. T. Mayes, S. Dai, K. L. More, G. M. Veith and L. Prati, ChemSusChem, 2015, 8, 2520–2528 CrossRef CAS PubMed; (b) Y. Wang, Z. Rong, Y. Wang, T. Wang, Q. Du, Y. Ang and J. Qu, ACS Sustainable Chem. Eng., 2017, 5, 1538–1548 CrossRef CAS.
- (a) C. Moreno-Marrodan and P. Barbaro, Green Chem., 2014, 16, 3434–3438 RSC; (b) Y. Yao, Z. Wang, S. Zhao, D. Wang, Z. Wu and M. A. Zhang, Catal. Today, 2014, 234, 245–250 CrossRef CAS.
- Y. Kuwahara, Y. Magatani and H. Yamashita, Catal. Today, 2015, 258, 262–269 CrossRef CAS.
- (a) U. Mandi, N. Salam, S. K. Kundu, A. Bhaumik and S. K. M. Islam, RSC Adv., 2016, 6, 73440–73449 RSC; (b) W. Luo, U. Deka, A. M. Beale, E. R. van Eck, P. C. A. Bruijnincx and B. M. Weckhuysem, J. Catal., 2013, 301, 175–186 CrossRef CAS.
- Z. Lin, X. Cai, Y. Fu, W. Zhu and F. Zhang, RSC Adv., 2017, 7, 44082–44088 RSC.
- Z. Lin, M. Luo, Y. Zhang, X. Wu, Y. Fu and F. Zhang, Appl. Catal., A, 2018, 563, 54–63 CrossRef CAS.
- C. Cai, Q. Xu, G. Tu, Y. Fu, F. Zhang and W. Zhu, Front. Chem., 2019, 7, 42, DOI:10.3389/fchem.2019.00042.
- D. Albani, Q. Li, G. Vilé, S. Mitchel, N. Almora-Barrios, P. T. White, N. López and J. Pérez-Ramírez, Green Chem., 2017, 19, 2361–2370 RSC.
- (a) J. Tan, J. Cui, X. Cui, T. Deng, X. Li, Y. Zhu and Y. Li, ACS Catal., 2015, 5, 7379–7384 CrossRef CAS; (b) J. Tan, J. Cui, G. Ding, T. Deng, Y. Zhu and Y.-W. Li, Catal. Sci. Technol., 2016, 6, 1469–1475 RSC; see also (c) Y. Wang, Z. Rong, Y. Wang, P. Zhang, Y. Wang and J. Qu, J. Catal., 2015, 329, 95–106 CrossRef CAS.
- C. Xiao, T.-W. Goh, Z. Qi, S. Goes, K. Brashler, C. Perez and W. Huang, ACS Catal., 2016, 6, 593–598 CrossRef CAS.
- (a) J. D. Scholten, B. C. Leal and J. Dupont, ACS Catal., 2012, 2, 184–200 CrossRef CAS; (b) S. Doherty, in Catalysis in Ionic Liquids: From Catalysis Synthesis to Applications, RSC Catalysis Series, ed. C. Hardacre and V. Parvulescu, The Royal Society of Chemistry, 2014, pp. 44–308 Search PubMed; (c) P. Migowski, K. L. Luska and W. Leitner, in Nanocatalysts in Ionic Liquids, ed. M. H. G. Prechtl, Wiley VCH, Weinheim, 2016 Search PubMed; (d) C. Janiak, Z. Naturforsch., 2013, 68, 1059–1089 CrossRef CAS; (e) A. S. Pensado and A. A. H. Pádua, Angew. Chem., Int. Ed., 2011, 50, 8683–8687 ( Angew. Chem. , 2011 , 123 , 8842–8846 ) CrossRef CAS PubMed; (f) A. Kraynov and T. E. Müller, Concepts for the Stabilization of Metal Nanoparticles in Ionic Liquids, Applications of Ionic Liquids in Science and Technology, ed. S. Handy, IntechOpen, September 22nd 2011, DOI:10.5772/22111, Available from: https://www.intechopen.com/books/applications-of-ionic-liquids-in-science-and-technology/concepts-for-the-stabilization-of-metal-nanoparticles-in-ionicliquids.
- (a) For an insightful account on metal nanoparticles immobilised on molecularly modified surfaces for controlled hydrogenation and hydrogenolysis see: A. Bordet and W. Leitner, Acc. Chem. Res., 2021, 54, 2144–2157 CrossRef CAS PubMed; (b) K. L. Luska, A. Bordet, S. Tricard, I. Sinev, W. Grünert, B. Chaudret and W. Leitner, ACS Catal., 2016, 6, 3719–3726 CrossRef CAS.
- M. Selva, M. Gottardo and A. Perosa, ACS Sustainable Chem. Eng., 2012, 1, 180–189 CrossRef.
- (a) K. L. Luska and A. Moores, ChemCatChem, 2012, 4, 1534–1546 CrossRef CAS; (b) For a comprehensive and highly informative review on the impact of ligands on heterogeneous nanocatalysis see L. Lu, Z. Zou and B. Fang, ACS Catal., 2021, 11, 6020–6058 CrossRef CAS.
- (a) H. Jiang and X. Zheng, Catal. Sci. Technol., 2015, 5, 3728–3734 RSC; (b) Z. Wang and H. Jiang, RSC Adv., 2015, 5, 34622–34629 RSC.
- (a) L. M. Martinez-Prieto, A. Ferry, L. Rakers, C. Richter, P. Lacante, K. Philippot, B. Chaudret and F. Glorius, Chem. Commun., 2016, 52, 4768–4771 RSC; (b) J. L. Castelbou, E. Breso-Femenia, P. Blondeau, B. Chaudret, S. Castillon, C. Claver and C. Godard, ChemCatChem, 2014, 6, 3160–3168 CrossRef; (c) D. Gonzalez-Galvez, P. Lara, O. Rivada-Wheelaghan, S. Conejero, B. Chaudret, K. Philippot and P. W. N. M. van Leeuwen, Catal. Sci. Technol., 2013, 3, 99–105 RSC; (d) M. J. L. Tschan, O. Diebolt and P. W. N. M. van Leeuwen, Top. Catal., 2014, 57, 1054–1065 CrossRef CAS; (e) M. Guerro, A. Roucoux, A. Denicourt-Nowicki, H. Monflier, V. Colliere, K. Fajerwerg and K. Philippot, Catal. Today, 2012, 183, 34–41 CrossRef.
- M. Guerrero, Y. Coppel, N. T. T. Chau, A. Roucoux, A. Denicourt-Nowicki, E. Monflier, H. Bricout, P. Lecante and K. Philippot, ChemCatChem, 2013, 5, 3802–3811 CrossRef CAS.
- (a) I. Cano, A. M. Chapman, A. Urakawa and P. W. M. N. van Leeuwen, J. Am. Chem. Soc., 2014, 136, 2520–2528 CrossRef CAS PubMed; (b) N. Almora Barrios, I. Cano, P. W. M. N. van Leeuwen and N. Lopez, ACS Catal., 2017, 7, 3949–3954 CrossRef CAS; (c) I. Cano, M. A. Huertos, A. M. Chapman, G. Buntkowsky, T. Gutmann, P. B. Groszewicz and P. W. M. N. van Leeuwen, J. Am. Chem. Soc., 2015, 137, 7718–7727 CrossRef CAS PubMed.
- S. Jayakumar, A. Modak, M. Guo, H. Li, X. Hu and Q. Yang, Chem. – Eur. J., 2017, 23, 7791–7797 CrossRef CAS PubMed.
- M. Guo, H. Li, Y. Ren, X. Ren, Q. Yang and C. Li, ACS Catal., 2018, 8, 6476–6485 CrossRef CAS.
- M. A. Ortuño and N. López, ACS Catal., 2018, 8, 6138–6145 CrossRef.
- X. Ren, M. Guo, H. Li, C. Li, L. Yu, J. Liu and Q. Yang, Angew. Chem., Int. Ed., 2019, 58, 14625–14630 CrossRef.
- Nitroarenes: (a) M. R. Axet, A. Conejero and I. C. Gerber, ACS Appl. Nano Mater., 2018, 1, 5885–5894 CrossRef CAS; (b) G. Chen, C. Xu, X. Huang, J. Ye, L. Gu, G. Li, Z. Tang, B. Wu, H. Yang, Z. Zho, Z. Zhou, G. Fu and N. Zheng, Nat. Mater., 2016, 15, 564–569 CrossRef CAS PubMed; (c) E. H. Boymans, P. T. Witte and D. Vogt, Catal. Sci. Technol., 2015, 5, 176–183 RSC; (d) J. Mondal, S. K. Kundu, W. K. H. Ng, R. Singuru, P. Borah, P. H. Hirao, Y. Zhao and A. Bhaumik, Chem. – Eur. J., 2015, 21, 19016–19027 CrossRef CAS PubMed Carbonyls (e) Z. Guo, C. Xiao, R. V. Maligal-Ganesh, L. Zhou, T. W. Goh, X. Li, D. Tesfagaber, A. Thiel and W. Huang, ACS Catal., 2014, 4, 1340–1348 CrossRef CAS; (f) I. Schrader, J. Warneke, J. Backenkohler and S. Kunz, J. Am. Chem. Soc., 2015, 137, 905–912 CrossRef CAS PubMed Alkynes (g) S. G. Kwon, G. Krylova, A. Sumer, M. M. Schwartz, E. E. Bunel, C. L. Marshall, S. Chattopadhyay, B. Lee, J. Jellinek and E. V. Shevchenko, Nano Lett., 2012, 12, 5382–5388 CrossRef CAS PubMed; (h) W. Long, N. A. Brunelli, S. A. Didas, E. W. Ping and C. W. Jones, ACS Catal., 2013, 3, 1700–1708 CrossRef CAS; (i) F. P. da Silva, J. L. Fiorio and L. M. Rossi, ACS Omega, 2017, 2, 6014–6022 CrossRef CAS PubMed Arenes (j) M. Guo, C. Li and Q. Yang, Catal. Sci. Technol., 2017, 7, 2221–2227 RSC.
- For a recent insightful review, describing the use of covalently supported ionic liquids in catalysis see: (a) F. Giacalone and M. Gruttadauria, ChemCatChem, 2016, 8, 664–684 CrossRef CAS; (b) K. L. Luska, P. Migowskia and W. Leitner, Green Chem., 2015, 17, 3195–3206 RSC; (c) S. Doherty, J. G. Knight, T. Backhouse, T. E. Abood, H. Al-shaikh, I. J. S. Fairlamb, R. A. Bourne, T. W. Chamberlain and R. Stones, Green Chem., 2017, 19, 1635–1641 RSC; (d) S. Doherty, J. G. Knight, T. Backhouse, A. Bradford, F. Saunders, R. A. Bourne, T. W. Chamberlain, R. Stones, A. Clayton and K. R. J. Lovelock, Catal. Sci. Technol., 2018, 8, 1454–1467 RSC; (e) S. Doherty, J. G. Knight, T. Backhouse, E. Abood, H. Al-shaikh, A. R. Clemmet, J. R. Ellison, R. A. Bourne, T. W. Chamberlain, R. Stones, N. J. Warren, I. J. S. Fairlamb and K. R. J. Lovelock, Adv. Synth. Catal., 2018, 360, 3716–3731 CrossRef CAS; (f) S. Doherty, J. G. Knight, T. Backhouse, R. J. Summers, E. Abood, W. Simpson, W. Paget, R. A. Bourne, T. W. Chamberlain, R. Stones, K. R. J. Lovelock, J. M. Seymour, M. A. Isaacs, C. Hardacre, H. Daly and N. H. Rees, ACS Catal., 2019, 9, 4777–4791 CrossRef CAS; (g) S. Doherty, J. G. Knight, H. Y. Alharbi, R. Paterson, C. Wills, C. Dixon, L. Šiller, T. W. Chamberlain, A. Griffiths, S. M. Collins, K. Wu, M. D. Simmons, R. A. Bourne, K. R. J. Lovelock and J. M. Seymour, ChemCatChem, 2022, 14, e202101752 CrossRef CAS.
- J. Tan, J. Cui, T. Deng, X. Cui, G. Ding, Y. Zhu and Y. Li, ChemCatChem, 2015, 7, 508–512 CrossRef CAS.
- X. Cai, J. Nie, G. Yang, F. Wang, C. Ma, C. Lu and Z. Chen, Mater. Lett., 2019, 240, 80–83 CrossRef CAS.
- (a) T. A. Stephenson and G. Wilkinson, J. Inorg. Nucl. Chem., 1966, 28, 945–956 CrossRef CAS; (b) P. S. Hallman, T. A. Stephenson and G. Wilkinson, Inorg. Synth., 1970, 12, 237 CAS.
- D. González-Galvez, P. Nolis, K. Philippot, B. Chaudret and P. W. N. M. van Leeuwen, ACS Catal., 2012, 2, 317–321 CrossRef.
- K. Philippot and B. Chaudret, C. R. Chim, 2003, 6, 1019–1034 CrossRef CAS.
- (a) R. A. Zinger and R. H. Hedges, J. Phys. Chem., 1961, 65, 1132–1138 CrossRef; (b) M. J. Frazer, W. Gerrard and R. Twaits, J. Inorg. Nucl. Chem., 1963, 25, 637–640 CrossRef CAS; (c) D. M. L. Goodgame and F. A. Cotton, J. Chem. Soc., 1961, 3735–3741 RSC; (d) R. D. Bannister, W. Levason, M. E. Light and G. Reid, Polyhedron, 2018, 154, 259–262 CrossRef CAS; (e) G. B. Deacon and J. H. S. Green, Spectrochim. Acta, Part A, 1968, 24, 845–852 CrossRef CAS; (f) M. Hallman and S. Pinchas, J. Chem. Soc., 1958, 3264–3267 RSC; (g) S. Y. Chin, C. T. Williams and M. D. Amiridis, J. Phys. Chem. B, 2006, 110, 871–882 CrossRef CAS PubMed; (h) A. S. Baird, K. M. Kross, D. Gottschalk, E. A. Hinson, N. Wood and K. A. Layman, J. Phys. Chem. C, 2007, 111(38), 14207–14214 CrossRef CAS; (i) H. W. Chen, Z. Zhong and J. M. White, J. Catal., 1984, 90, 119–126 CrossRef CAS; (j) B. R. A. Dalla Betta, J. Phys. Chem., 1975, 79, 2519–2525 CrossRef; (k) K. Hadjiivanov, J.-C. Lavalley, J. Lamotte, F. Mauge, J. Saint-Just and M. Che, J. Catal., 1998, 176, 415–425 CrossRef CAS; (l) F. Schröder, D. Esken, M. Cokoja, M. W. E. van den Berg, O. I. Lebedev, G. Van Tendeloo, B. Walaszek, G. Buntkowsky, H.-H. Limbach, B. Chaudret and R. A. Fischer, J. Am. Chem. Soc., 2008, 130, 6119–6130 CrossRef PubMed; (m) A. Duteil, R. Queau, B. Chaudret, R. Mazel, C. Roucau and J. S. Bradley, Chem. Mater., 1993, 5, 341–347 CrossRef CAS; (n) M. Sobota, M. Happel, M. Amende, N. Paape, P. Wasserscheid, M. Laurin and J. Libuda, Adv. Mater., 2011, 23, 2617–2621 CrossRef CAS PubMed; (o) M. Siemer, G. Tomaschun, T. Klüner, P. Christopher and K. Al-Shamery, ACS Appl. Mater. Interfaces, 2020, 12, 27765–27776 CrossRef CAS PubMed; (p) D. Gottschalk, E. A. Hinson, A. S. Baird, H. L. Kitts and K. A. Layman, J. Phys. Chem. C, 2010, 114, 4950–4960 CrossRef CAS.
- (a) G. Biagiotti, V. Langè, C. Ligi, S. Caporali, M. Muniz-Miranda, A. Flis, K. M. Pietrusiewicz, G. Ghini, A. Brandi and S. Cicchi, Beilstein J. Nanotechnol., 2017, 8, 485–493 CrossRef CAS PubMed; (b) D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- F. Ye, H. Liu, J. Yang, H. Cao and J. Yang, Dalton Trans., 2013, 42, 12309–12316 RSC.
- T. Ohkuma, H. Ooka, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 2675–2676 CrossRef CAS.
- (a) A. Maximov, A. Zolotukhina, V. Murzin, E. Karakhanov and E. Rosenberg, ChemCatChem, 2015, 7, 1197–1210 CrossRef CAS; (b) M. H. G. Prechtl, M. Scariot, J. D. Scholten, G. Mochado, S. R. Teixeira and J. Dupont, Inorg. Chem., 2008, 47, 8995–9001 CrossRef CAS PubMed; (c) A. Gual, M. R. Azet, K. Philippot, B. Chaudret, A. Denicourt-Nowicki, A. Roucoux, S. Castillion and C. Claver, Chem. Commun., 2008, 2759–2761 RSC; (d) Y. Morioka, A. Matsuoka, K. Binder, B. R. Knappett, A. E. H. Wheatley and H. Naka, Catal. Sci. Technol., 2016, 6, 5801–5805 RSC; (e) A. Dwivedi, R. K. Rai, K. Gupta and S. K. Singh, ChemCatChem, 2017, 9, 1930 CrossRef CAS; (f) Y. Ma, Y. Huang, Y. Cheng, L. Wang and X. Li, Appl. Catal., A, 2014, 484, 154–160 CrossRef CAS; (g) E. Breso-Femenia, B. Chaudret and S. Castillon, Catal. Sci. Technol., 2015, 5, 2741–2751 RSC; (h) F. Lu, J. Liu and J. Xu, Adv. Synth. Catal., 2006, 348, 857–861 CrossRef CAS; (i) L. Rakers, L. M. Martinez-Prieto, A. M. Lopez-Vinasco, K. Philippot, P. W. N. M. van Leeuwen, B. Chaudret and F. Glorious, Chem. Commun., 2018, 54, 7070–7073 RSC.
- (a) D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC; (b) M. Benson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 4, 1827–1870 Search PubMed; (c) G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- J.-P. Lange, E. van der Heide, J. van Buijtenen and R. Price, ChemSusChem, 2012, 5, 150–166 CrossRef CAS PubMed.
- (a) F. Liguori, C. Moreno-Marrodan and P. Barbaro, ACS Catal., 2015, 5, 1882–1894 CrossRef CAS; (b) F. D. Pileidis and M. M. Titirici, ChemSusChem, 2016, 9, 562–582 CrossRef CAS PubMed; (c) J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed; (d) E. I. Gurbuz, J. M. R. Gallo, D. M. Alonso, S. G. Wettstein, W. Y. Yin and J. A. Dumesic, Angew. Chem., Int. Ed., 2013, 52, 1308–1312 CrossRef.
- C. Ramirez-Barria, M. Isaacs, K. Wilson, A. Guerrero-Ruiz and I. Rodriquez-Ramos, Appl. Catal., A, 2018, 563, 177–184 CrossRef CAS.
- R. Fang, H. Liu, R. Luque and Y. Li, Green Chem., 2015, 17, 4183–4188 RSC.
- J. Yang, J. Ma, Q. Yuan, P. Zhang and Y. Guan, RSC Adv., 2016, 6, 92299–92304 RSC.
- L. J. Durndell, G. Zou, W. Shangguan, A. F. Lee and K. Wilson, ChemCatChem, 2019, 11, 3927–3932 CrossRef CAS.
- A. Cocq, B. Léger, S. Nöel, H. Bricout, F. Djedaïni-Pilard, S. Tilloy and E. Monflier, ChemCatChem, 2020, 12, 1013–1018 CrossRef CAS.
- Q. Yuan, D. Zhang, L. van Haadel, F. Ye, T. Xue, E. J. M. Hensen and Y. Guan, J. Mol. Catal. A: Chem., 2015, 406, 58–64 CrossRef CAS.
- J. L. Catelbou, K. C. Szeto, W. Barakat, N. Merle, C. Godard, M. Taoufik and C. Claver, Chem. Commun., 2017, 53, 3261–3264 RSC.
- B. Y. Tay, C. Wang, P. H. Phua, L. P. Stubbs and H. V. Huynh, Dalton Trans., 2016, 3558–3563 RSC.
- V. S. Jaya, M. Sudhakar, S. N. Kumar and A. Venugopal, RSC Adv., 2015, 5, 9044–9049 RSC.
- Y. Kuwahara, W. Kaburagi and T. Fujitani, RSC Adv., 2014, 4, 45848–45855 RSC.
- A. Abdelrahman, A. Heyden and J. Q. Bond, ACS Catal., 2014, 4, 1171–1181 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of monomers, co-polymers 1a′ and 1a′.PEG and polymer immobilised ionic liquid stabilised RuNPs 2a, 2a.PEG, 2a′, 2a′.PEG, 2b and 2b.PEG, TEM images, FT-IR traces and X-ray photoelectron spectra for 2a.PEG, 2a′.PEG, and 2b.PEG. See DOI: https://doi.org/10.1039/d2cy00205a |
| This journal is © The Royal Society of Chemistry 2022 |