
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemoenzymatic deracemization of lisofylline catalyzed by a (laccase/TEMPO)-alcohol dehydrogenase system†
Paweł Borowiecki *a,
Aleksandra Rudzkaa,
Tamara Reiterb and
Wolfgang Kroutilb
*a,
Aleksandra Rudzkaa,
Tamara Reiterb and
Wolfgang Kroutilb
aLaboratory of Biocatalysis and Biotransformation, Department of Drugs Technology and Biotechnology, Faculty of Chemistry, Warsaw University of Technology, Koszykowa St. 75, 00-662 Warsaw, Poland. E-mail: pawel.borowiecki@pw.edu.pl
bInstitute of Chemistry, University of Graz, NAWI Graz, BioTechMed Graz, Field of Excellence BioHealth, Heinrichstrasse 28, 8010 Graz, Austria
First published on 16th May 2022
Abstract
Lisofylline (LSF) is a synthetic methylxanthine active agent exhibiting potent anti-inflammatory and immunomodulatory properties; therefore, it has been widely investigated as a promising drug candidate for treating various autoimmune disorders, including type 1 diabetes. In this study, we report on developing a sequential chemoenzymatic one-pot two-step deracemization protocol for racemic LSF. This task was accomplished in a stereo-complementary manner via a tandem bi-enzymatic oxidation–reduction reaction sequence composed of (i) non-selective chemoenzymatic aerobic oxidation of LSF to pentoxifylline (PTX) catalyzed by commercially available laccase from Trametes versicolor (LTv) and 2,2,6,6-tetramethylpiperidinyloxy radical (TEMPO) as a redox mediator, and (ii) stereoselective bioreduction of in situ formed PTX to give enantiomeric LSF, which was catalyzed by home-made lyophilized E. coli cells harboring overexpressed alcohol dehydrogenases (ADHs) with complementary stereospecificity. Firstly, a multi-step optimization procedure of LTv/TEMPO-catalyzed oxidation of LSF allowed achieving dramatic improvement of the conversion rates from an initial 16% up to 95%, demonstrating the high synthetic potency of this method compared to traditional chemical reactions requiring toxic oxidants used in stoichiometric amounts. In turn, separate stereoselective bioreductions of PTX using recombinant ADHs from Rhodococcus ruber (E. coli/ADH-A) and Lactobacillus kefiri (E. coli/LK-ADH Prince) furnished both LSF enantiomers (>99% ee) with high 93–94% conversion and in 65–67% yield range, respectively. The coupling of the above-mentioned chemoenzymatic steps afforded both antipodes of LSF on a preparative scale (0.16 mmol of racemic LSF) in the range of 56–67% yield and 94% ee depending on the employed ADHs.
1. Introduction
Lisofylline (LSF) is a powerful pharmaceutical molecule whose strong anti-inflammatory and immunomodulatory properties1 are useful in the effective prevention and treatment of various autoimmune disorders, such as inflammatory bowel disease (i.e., Crohn's disease),2 insulin-dependent type 1 diabetes,3 bronchial asthma,4etc., as well as in attenuating the side effects (i.e., neutropenic infections) of chemotherapy of cancers.5 In the case of LSF, pharmacokinetic and pharmacodynamic studies revealed that the desired biological activities reside only in the (R)-enantiomer, which is, by the way, several hundred-fold more potent at inhibiting the activity of inflammatory cytokines than its parent molecule, pentoxifylline (PTX).6 Due to the essential pharmacological significance of (R)-LSF, the elaboration of asymmetric methods of its synthesis has attracted significant attention from synthetic laboratories. Among biocatalytic attempts worth mentioning are (R)-stereoselective bioreduction of PTX catalyzed by microbial whole cells of Saccharomyces cerevisiae (wine yeast)7 or resting cells of Lactobacillus kefiri DSM 205878 as well as purified (R)-specific alcohol dehydrogenase (ADH) isolated from Lactobacillus kefiri (LK-ADH)9 (Fig. 1).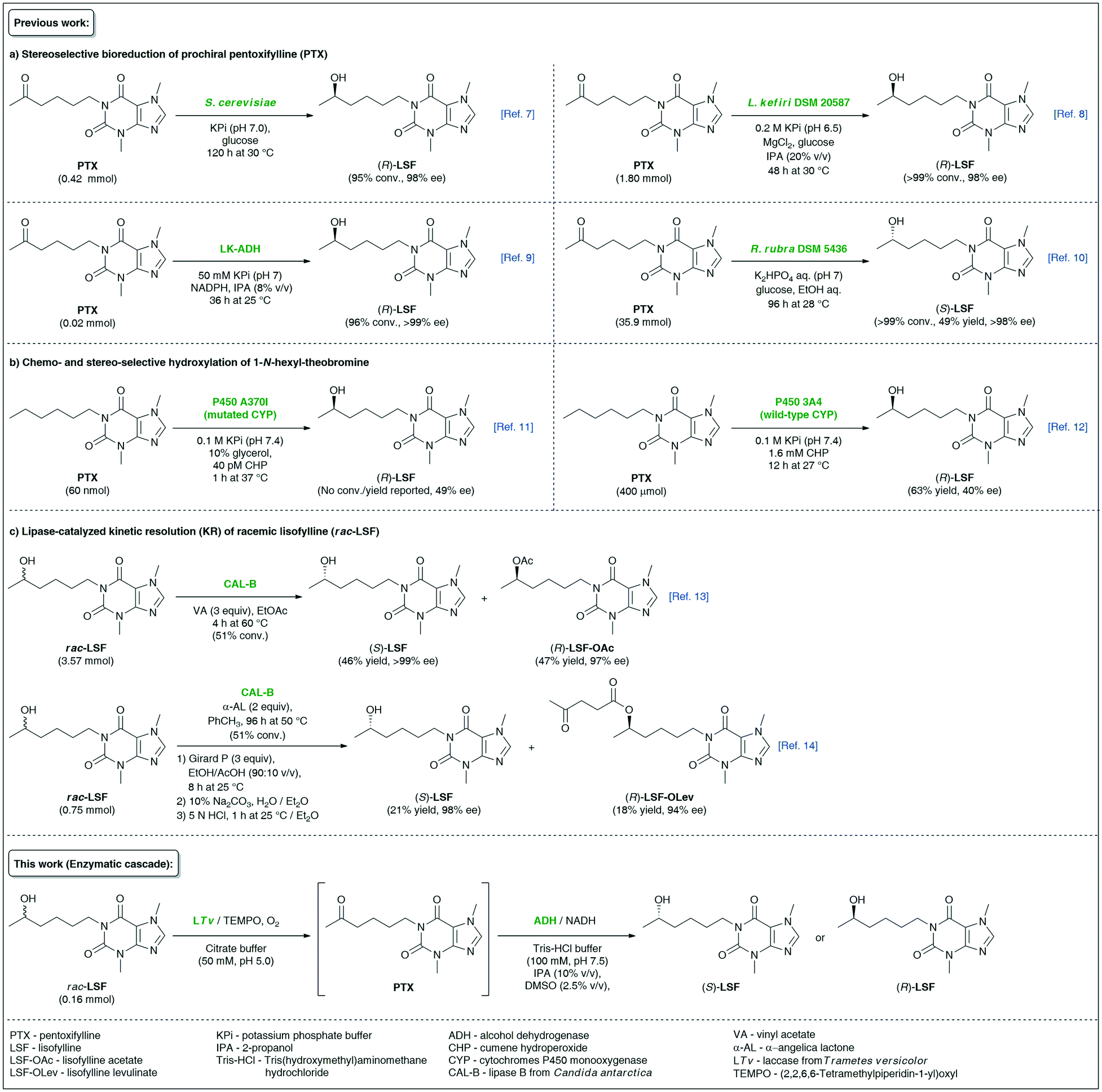 | ||
| Fig. 1 Overview of different biocatalytic strategies for the synthesis of enantiomeric lisofylline (LSF). | ||
Alternative enzymatic approaches relying on the microbial bioreduction of PTX using cell cultures of Rhodotorula rubra DSM 5436 exhibiting Prelog stereoselectivity10 or chemo- and stereoselective hydroxylation of 1-N-hexyl-theobromine catalyzed by wild-type11 or engineered human cytochrome P450 3A4 monooxygenases12 seem to be less attractive from the viewpoint of undesired absolute configuration or low enantiomeric excess values of the obtained non-racemic LSF, respectively. With the aim to expand the synthetic toolbox for the synthesis of the titled API, we have recently developed two independent variants of lipase-catalyzed kinetic resolution (KR) of racemic LSF by either using a classic transesterification approach with vinyl acetate as acyl donor13 or utilizing α-angelica lactone as a nonconventional irreversible acylating agent for chromatography-free KR.14 However, enzymatic KR of sec-alcohols suffers from low reaction yields, which renders the use of this approach unsustainable for industrial applications.
Biocatalytic cascades represent an attractive strategy in the synthesis of optically active compounds.15 This assumption stems from the fact that one-pot combinations of sequential catalytic reactions improve step economy, lower production costs, and generate less waste to be recycled. In this context, elaboration of novel synthetic methodologies that would efficiently couple sequential non-selective oxidation of racemic alcohols with biocatalytic stereoselective reductions of the in situ generated ketones through one-pot cascades under mild conditions has received considerable attention.16 Initially, deracemizations of sec-alcohols via a biocatalytic oxidation–reduction sequence have been achieved mainly using whole microbial cells,17 enantiocomplementary ADHs18 or single ADH-catalyzed biohydrogen transfer mediated by α-chloro ketones.19 Furthermore, a combination of organocatalytic oxidation of racemic alcohols catalyzed by Ir-complex,20 sodium hypochlorite (NaOCl) and 2-azaadamantane N-oxyl (AZADO),21 Zr-beta zeolite22 or iodine/(2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO)23 system with ADH-catalyzed asymmetric bioreduction of the formed ketones have appeared as interesting alternatives to obtain optically pure hydroxyl compounds. In the last few years, a relatively novel catalytic system composed of laccase from Trametes versicolor (LTv), stable nitroxyl radical TEMPO, and the respective ADHs has been successfully applied in sustainable deracemization of various benzylic,24 allylic,25 and propargylic26 alcohols using a linear one-pot bio-redox cascade strategy. This methodology was developed on the basis of a pioneering paper published by Fabbrini et al.,27 which was the first that reported on chemoenzymatic oxidation of alcohols with oxygen catalyzed by laccase from Trametes versicolor and mediated by TEMPO. However, the LTv/TEMPO-ADH system, despite being proven useful for deracemization of a broad spectrum of activated sec-alcohols, is still underexploited toward unactivated substrates, such as aliphatic, alicyclic and/or heteroaromatic hydroxy derivatives. Therefore, extending the substrate scope for more demanding compounds in terms of oxidation of their secondary hydroxyl groups remains a challenging task for (bio)organic chemists.
In continuation of our interest in the syntheses of enantiomerically pure APIs via biocatalytic methodologies,28 we report here an unprecedented synthetic strategy toward both enantiomers of lisofylline (LSF) achieved by the employment of tandem catalysis based on one-pot reversed redox transformations mediated by the LTv/TEMPO-ADH system in an aqueous medium supplemented with organic co-solvents and under mild conditions.
2. Results and discussion
In chemoenzymatic one-pot redox-driven deracemization of secondary alcohols using laccase/TEMPO and ADH as the conjugated catalysts, the non-selective oxidation of the respective racemate seems to be a more demanding process from the synthetic point of view (Fig. 2).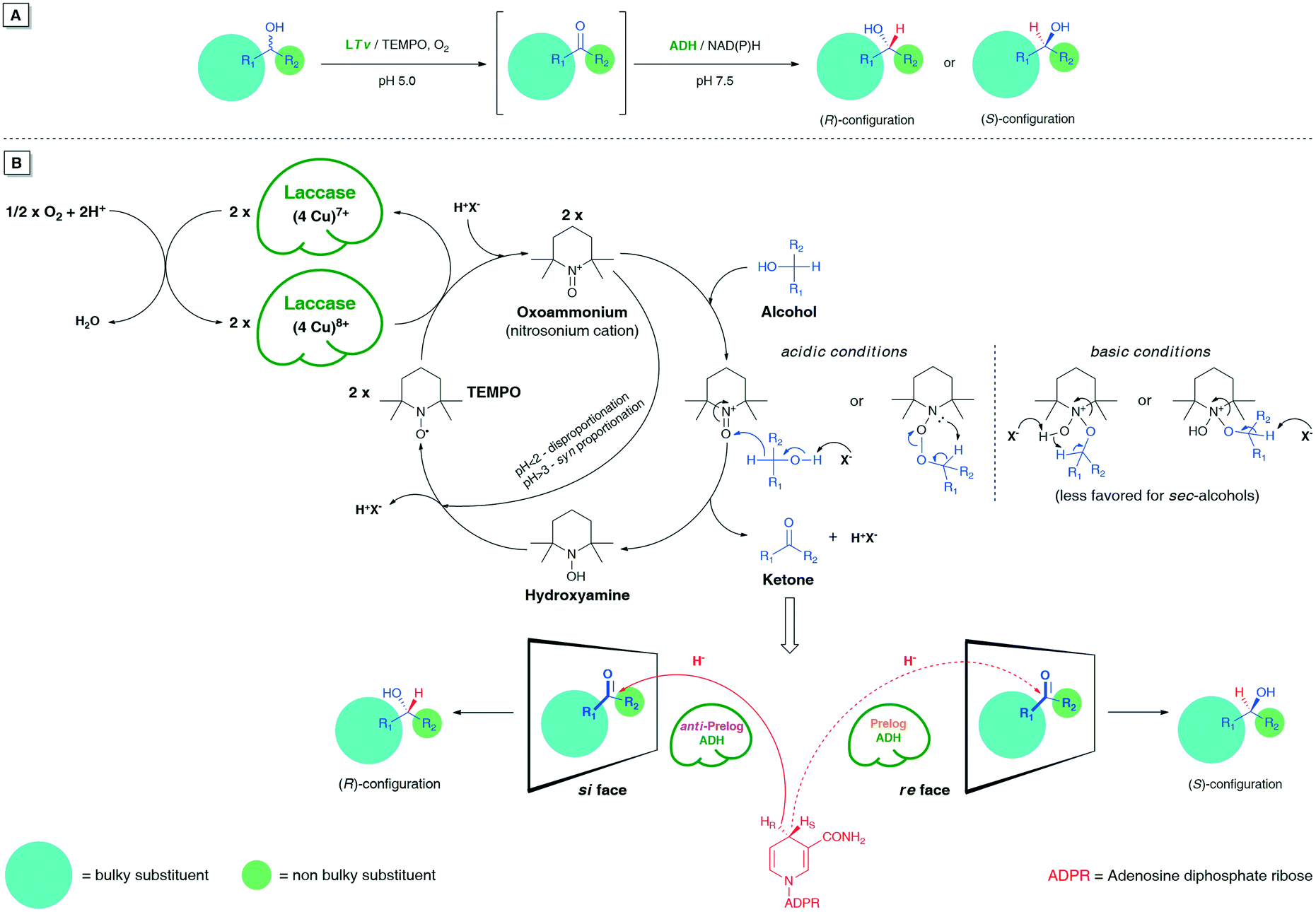 | ||
| Fig. 2 (A) Schematic representation of LTv/TEMPO-ADH-mediated linear deracemization of sec-alcohols. (B) A plausible mechanism of laccase/TEMPO-mediated non-selective oxidation of sec-alcohols using air/O2 as the oxidant followed by stereoselective bioreduction of the ketone intermediates. | ||
It is especially true when one desires to obtain optically pure compounds with this methodology; then, an oxidative step must proceed in >99% yield to provide quantitative amounts of prochiral ketone intermediate for the subsequent ADH-catalyzed bioreduction. Otherwise, each 1% less conv. prompts a 1% drop in an enantiomeric excess of the isolated non-racemic product even if the ADH-catalyzed reaction step remains fully stereoselective.
We commenced our study by optimizing the LTv/TEMPO-catalyzed non-selective oxidation of lisofylline (rac-1) using air/oxygen as an oxidant. In the first step, we examined the addition of various water-miscible and water-immiscible organic solvents, within which the employed substrate could form a homogenous solution, and gave rise to monophasic (in the case of DMSO, 1,4-dioxane, MeOH, EtOH, CH3CN, acetone, THF, 2-PrOH) and/or biphasic (in the case of EtOAc, CH2Cl2, tert-amyl alcohol, PhCH3) reaction systems with an oxygen-saturated citrate buffer (Table 1), respectively. The total volume of the reaction medium was set at a 1.6 mL level, while the minimum amount of the supplemented organic co-solvent (10–50% (v/v) in ratio to citrate buffer) relied strictly on the solubility of rac-1. Moreover, to minimize limitations concerning mass transfer issues from one side and to avoid the splitting of the reactant and catalysts around the vial walls from the other side, a gentle magnetic stirring (150 rpm) was applied in all cases. The solvent optimization showed that oxidation of rac-1 is favored in acetone, EtOAc, THF, CH2Cl2, tert-amyl alcohol, and PhCH3, which indicates that no significant effects from the polarity of the solvents are expected to play a decisive role in this process. The highest conversion for rac-1 was observed in acetone and EtOAc. In contrast, the most detrimental effect on the activity of the laccase/TEMPO system exhibited low-molecular alcohols (MeOH, EtOH, and 2-PrOH) and highly polar DMSO. This observation can be either rationalized by strong promotion of protein unfolding, leading to denaturation of the enzyme in the presence of DMSO, or attributed to an inhibition mechanism caused by alcohols. Moreover, the short-chain alcohols used in a large molar excess can also compete with the substrate rac-1 to be oxidized by TEMPO-oxoammonium species. In turn, despite a high concentration of acetone (50% (v/v)), laccase from Trametes versicolor turned out to be significantly less vulnerable to inactivation effects when compared to the rest of the polar solvents (i.e., DMSO, 1,4-dioxane, MeOH, EtOH, and CH3CN). This is quite an interesting result as in the case of many other studied fungal laccases, the complete inhibition of their catalytic activity was observed mainly at 50% organic co-solvent concentration.29 Since the most efficient oxidation of rac-1 has been found in an aqueous citrate buffer/acetone mixture, this medium was chosen for further investigations. We arbitrarily omitted using EtOAc in later experiments as this solvent formed a biphasic system with an aqueous phase and is less volatile than acetone, which during the reaction progress is easily eliminated via evaporation.
|
|
|||
|---|---|---|---|
| Entry | Co-solventa,b (log P) | Co-solvent amountc (% v/v) | Conv.d (%) |
| a Reaction conditions: rac-1 (23 mg, 0.08 mmol, 50 mM final conc.), T. versicolor laccase (LTv, 7 mg, 4.6 U), TEMPO (4.1 mg, 33% mol), oxygenated citrate buffer (50 mM, pH 5.0), co-solvent (15–50% (v/v)) 1.6 mL final volume, 16 h, 30 °C, stirring in an open-to-air test vial (150 rpm, magnetic stirrer). b Logarithm of the partition coefficient of a given solvent between n-octanol and water according to ChemBioDraw Ultra 13.0 software indications. c Minimal amount of co-solvent in which a homogenous solution of the substrate rac-1 could be formed. d Conversion values (%) (i.e., consumption of substrate rac-1) were determined by GC analyses after derivatization of the crude mixture with N,O-bis(trimethylsilyl)acetamide (BSA) as a silylating reagent. e Not detected. | |||
| 1 | DMSO (−1.49) | 20 | N.D.e |
| 2 | 1,4-Dioxane (−0.31) | 30 | 9 |
| 3 | MeOH (−0.27) | 30 | N.D.e |
| 4 | EtOH (0.07) | 50 | N.D.e |
| 5 | CH3CN (0.17) | 50 | 5 |
| 6 | Acetone (0.20) | 50 | 16 |
| 7 | EtOAc (0.29) | 20 | 16 |
| 8 | 2-PrOH (0.38) | 50 | N.D.e |
| 9 | THF (0.40) | 20 | 12 |
| 10 | CH2Cl2 (1.01) | 15 | 13 |
| 11 | tert-Amyl alcohol (1.09) | 20 | 13 |
| 12 | PhCH3 (2.52) | 50 | 10 |
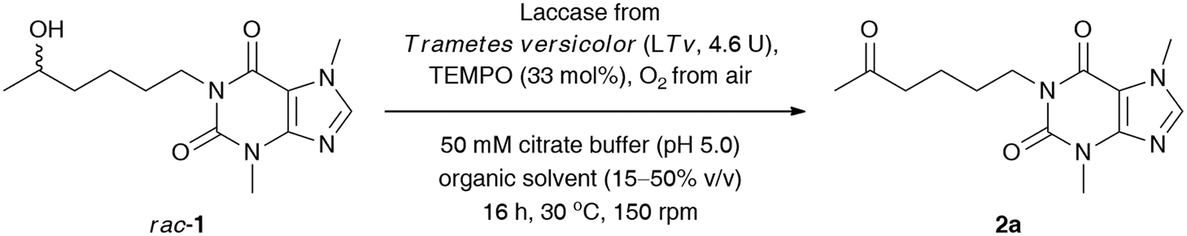
Next, to improve the ecological footprint of the process, the influence of the amount of acetone (10–50% (v/v)) on the % conv. of rac-1 was examined (Table 2). In addition, the low oxidation rates of rac-1 observed in previous experiments forced us to extend the reaction time up to 96 h. It turned out that 20% (v/v) acetone was beneficial for the efficiency of the LTv/TEMPO-mediated oxidation of rac-1, leading to a 2.5-fold improvement of the % conversion. It is a somehow interesting result as lower than 50% (v/v) acetone concentrations resulted in the formation of suspensions due to the limited solubility of lisofylline (rac-1). In this case, the improvement in the reaction rates might be twofold: acetone deactivates the enzyme and/or the substrate rac-1 exhibits inhibitory potency toward laccase. In turn, lower than 20% (v/v) co-solvent concentration decreased the solubility of rac-1 to such an extent that the reaction progress ceased.
|
|
||
|---|---|---|
| Entry | Amount of acetonea (% v/v) | Conv.b (%) |
| a Reaction conditions: rac-1 (23 mg, 0.08 mmol, 50 mM final conc.), T. versicolor laccase (LTv, 7 mg, 4.6 U), TEMPO (4.1 mg, 33% mol), oxygenated citrate buffer (50 mM, pH 5.0), acetone (10–50% (v/v)) 1.6 mL final volume, 96 h, 30 °C, stirring in an open-to-air test vial (150 rpm, magnetic stirrer). b Conversion values (%) (i.e., consumption of substrate rac-1) were determined by GC analyses after derivatization of crude mixture with N,O-bis(trimethylsilyl)acetamide (BSA) as a silylating reagent. | ||
| 1 | 10 | 30 |
| 2 | 20 | 40 |
| 3 | 30 | 38 |
| 4 | 40 | 26 |
| 5 | 50 | 28 |
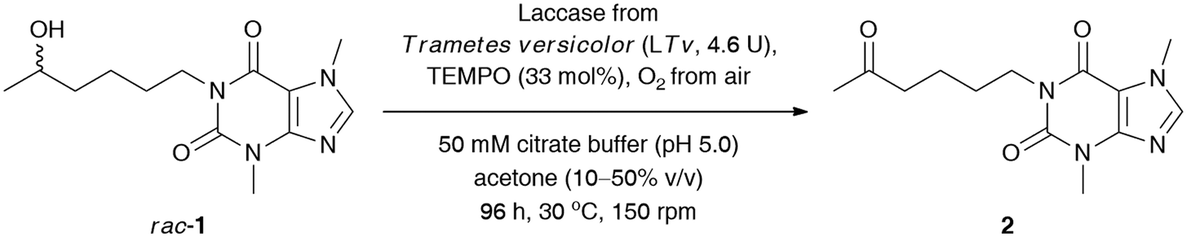
Since laccases (EC 1.10.3.2) are multi-copper-containing oxidases,30 we found it crucial for this study to investigate the effect of the presence of Cu2+ ions on the catalytic activity of T. versicolor laccase and indirectly on the whole LTv/TEMPO system (for details, see the ESI†). Moreover, inspired by the reported stimulatory effect of Cu2+, Zn2+, Fe2+, and Mg2+ on the laccases' activity,31 we also decided to determine the influence of different double-positive metal ions on the % conv. of the LTv/TEMPO-catalyzed oxidation of rac-1. In this regard, the reaction mixtures performed in citrate buffer (50 mM, pH 5.0) and acetone (20% (v/v)) for 24 h at 30 °C were supplemented with 1 mM final conc. of MgSO4, FeCl2·4H2O, FeSO4·7H2O, CoCl2·6H2O, ZnSO4·7H2O, or CuSO4·5H2O. The control reactions were assayed without added metal ions and in two variants with aerial oxygen as well as under an O2 atmosphere incorporated through an O2-filled balloon. The results indicated that in major cases, there was no positive influence of metal ions on the activity of the LTv/TEMPO system in the tested reactions. Since the previous optimizations were unsuccessful in finding significant enhancements on bio-oxidation of rac-1 and still a relatively high quantity of unreacted substrate remained in the reaction mixture, therefore, the reoxidation of the crude product for the subsequent 24 h through the addition of LTv and TEMPO was necessary to improve the conversion (Table 3, entry 1). Disappointingly, such a single treatment failed to give the expected results, so further studies were carried out toward a sequential increase of the amount of both laccase and oxyradical mediator. It is noteworthy that for this purpose, three separate reactions were carried out with sequential addition of laccase (7 mg) and TEMPO (4.1 mg) after every 24 h, without changing the initial volume of the medium (Table 3, entries 2–4). Analysis of the data presented in Table 3 revealed that in order to obtain a reasonably high conversion of rac-1 (>95%), the oxidation process had to be extended to 120 h, and the overall amounts of laccase and TEMPO had to be increased to 14.5 U mL−1 and 165 mol%, respectively.
|
|
||||
|---|---|---|---|---|
| Entry | Laccasea (U mL−1) | TEMPO (mol%) | t (h) | Conv.b (%) |
| a Reaction conditions: rac-1 (23 mg, 0.08 mmol, 50 mM final conc.), T. versicolor laccase (LTv, 7 mg, 4.6 U, ca. 2.9 U mL−1) and TEMPO (4.1 mg, 33% mol) – each portion added sequentially after 24 h, TEMPO (4.1 mg, 33% mol) – each portion added sequentially after 24 h, oxygenated citrate buffer (50 mM, pH 5.0) and acetone (20% v/v) 1.6 mL final volume, 48–120 h, 30 °C, stirring in an open-to-air test vial (150 rpm, magnetic stirrer). b Conversion values (%) (i.e., consumption of substrate rac-1) were determined by GC analyses after derivatization of the crude mixture with N,O-bis(trimethylsilyl)acetamide (BSA) as a silylating reagent. | ||||
| 1 | 5.8 | 66 | 48 | 55 |
| 2 | 8.7 | 99 | 72 | 82 |
| 3 | 11.6 | 132 | 96 | 87 |
| 4 | 14.5 | 165 | 120 | 95 |
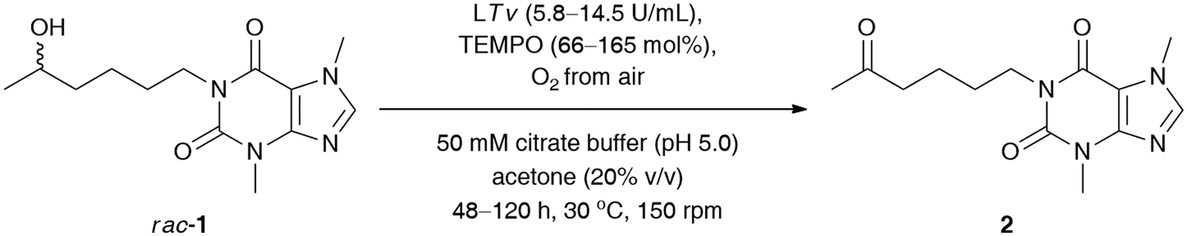
Next, our efforts were devoted to evaluating the efficiency of the laccase-mediated oxidation step performed on a larger scale of the employed substrate rac-1 (0.16 mmol) (Table 4). The best result in terms of the achieved % conv. was obtained in the reaction conducted for 120 h with T. versicolor laccase (14.5 U mL−1) and 165 mol% TEMPO. However, higher isolated yield for the desired ketone 2 was detected when the oxidation of rac-1 was performed with T. versicolor laccase (11.6 U mL−1) and 132 mol% TEMPO, and stopped after 96 h. This can be attributed to lower contamination from redundant laccase and TEMPO co-catalysts, and thus fewer drawbacks were encountered during purification of the crude product via preparative column chromatography. Once again, further extension of the reaction time failed to achieve quantitative conversions of rac-1.
|
|
||||
|---|---|---|---|---|
| Entry | LTv (U mL−1)/TEMPOa (mol%) | t (h) | Conv.b (%) | Yieldc (%) |
| a Reaction conditions: rac-1 (46 mg, 0.16 mmol, 50 mM final conc.), T. versicolor laccase (LTv, 14 mg, 9.2 U, ca. 2.9 U mL−1) and TEMPO (8.2 mg, 33% mol) – each portion added sequentially after 24 h, oxygenated citrate buffer (50 mM, pH 5.0), acetone (20% (v/v)) 3.2 mL final volume, 72–144 h, 30 °C, stirring in an open-to-air test vial (150 rpm, magnetic stirrer). b Conversion values (%) (i.e., consumption of substrate rac-1) were determined by GC analyses after derivatization of the crude mixture with N,O-bis(trimethylsilyl)acetamide (BSA) as a silylating reagent. c Isolated yield after column chromatography packed with SiO2. d rac-1 (46 mg, 0.16 mmol, 50 mM final conc.), T. versicolor laccase (LTv, 14 mg, 9.2 U, ca. 2.9 U mL−1) and TEMPO (8.2 mg, 33% mol) – each portion added sequentially after 48 h (i.e., the second portion was added after 48 h, and the third portion was added after 96 h), oxygenated citrate buffer (50 mM, pH 5.0), acetone (20% (v/v)) 3.2 mL final volume, 120 h, 30 °C, stirring in an open-to-air test vial (150 rpm, magnetic stirrer). | ||||
| 1 | 8.7/99 | 72 | 60 | 48 |
| 2 | 11.6/132 | 96 | 80 | 75 |
| 3 | 14.5/165 | 120 | 95 | 68 |
| 4 | 17.4/198 | 144 | 93 | 63 |
| 5 | 8.7/99d | 120 | 86 | 81 |
| 6 | 14.5/33 | 120 | 62 | 56 |
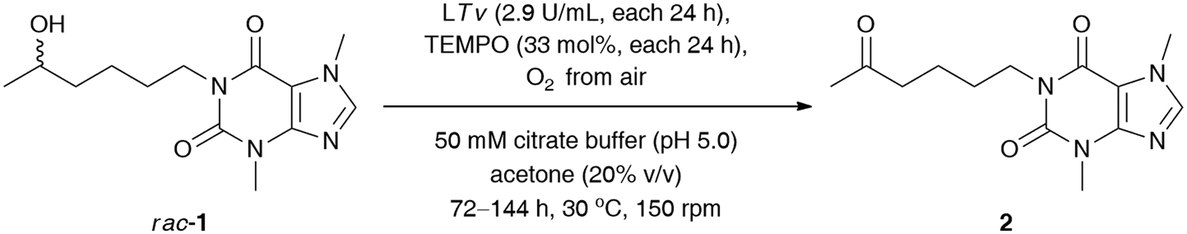
This phenomenon can be rationalized by the so-called suicide inactivation of laccases, which is observed at high substrate conversions when the reactive groups in the protein (or the associated glycosyl moieties located on the periphery of the enzyme) are oxidized by the accumulated oxoammonium cation.32 Therefore, we decided to extend our optimizations by investigating the effect of a lower concentration of TEMPO on the sequential oxidation of rac-1 (Table 4, entries 5 and 6). In the first case, the amounts of TEMPO and laccase have been reduced by half with respect to previous conditions. In the second attempt, we used only a catalytic amount of the mediator (33 mol%) coupled with laccase sequentially added in 24 h intervals at the same level as before (14.5 U mL−1 in total). Unfortunately, both trials revealed that it was not the TEMPO that was responsible for the deactivation of laccase. Therefore, the oxidation of rac-1 required stoichiometric amounts of the mediator as well as increased amounts of laccase for the efficient regeneration of the oxoammonium species of TEMPO, which is crucial for the oxidation step.
In the next step, stereoselective reduction of prochiral pentoxifylline (2) catalyzed by whole-cell biocatalysts was investigated to obtain optically pure lisofylline (non-rac-1). This task was accomplished using either wild-type microbial strains or recombinant alcohol dehydrogenases overexpressed in Escherichia coli cells (E. coli/ADHs) (Table 5). All the employed wild-type microorganisms (Table 5, entries 1–8) have been prepared by using standard cultivation conditions and lyophilized.33 In turn, the E. coli/ADH biocatalysts that originated from Ralstonia sp. (E. coli/RasADH34), Sphingobium yanoikuyae (E. coli/SyADH35), Rhodococcus ruber (E. coli/ADH-A36), Lactobacillus brevis (E. coli/LB-ADH37) and Lactobacillus kefiri (E. coli/Lk-ADH-Lica,38E. coli/LkADH,39E. coli/Lk-ADH Prince40) have been prepared as indicated in the appropriate literature. The catalytic behavior of the above-mentioned panel of wild-type microorganisms and E. coli/ADHs (10 mg of dried cells) was tested in bioreduction processes of 2 (10 mM final conc.) under standard conditions [i.e., 100 mM Tris-HCl buffer (pH 7.5) in the presence of NADH (0.5 mM) with 2.5% (v/v) DMSO due to the low solubility of 2 in aqueous medium, without air access for 48 h at 30 °C and 250 rpm] together with an in situ NAD(P)H regeneration system. Due to economic demands and the fact that the “cellular machinery” possesses metabolic pathways responsible for the efficient regeneration of nicotinamide cofactors, we decided to use an excess of isopropanol (2-PrOH) as a sacrificial co-substrate. Moreover, all the recombinant ADHs employed herein have already been confirmed to catalyze ketone reduction and cofactor recycling simultaneously in the presence of high concentrations of 2-PrOH (even up to 80% (v/v) in the case of E. coli/ADH-A41) as a reducing agent via hydrogen transfer. Therefore, in all the studied reactions, 10% (v/v) 2-PrOH was used with respect to Tris-HCl buffer, which additionally enhanced the solubility of substrate 2 in the aqueous phase. In turn, supplemental glucose (20 mM final conc.) was chosen to act as a carbon source and alternative cofactor-recycling system for NAD(P)H via the Embden−Meyerhof−Parnas (EMP) metabolic pathway.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Biocatalysta | Strain | Additive | Conv.b (%) | eepc (%) (Config.d) |
a Reaction conditions: 2 (10 mM final conc.), lyophilized biocatalyst (10 mg), 20 mM glucose, 0.5 mM NADH, 0.1 M Tris-HCl buffer (pH 7.5)/2-PrOH (500 μL, 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, v/v), DMSO (2.5% (v/v)), 48 h, 30 °C, 250 rpm (laboratory shaker). b Conversion values (%) (i.e., consumption of substrate 2) were determined by GC analyses after derivatization of the crude mixture with N,O-bis(trimethylsilyl)acetamide (BSA) as a silylating reagent. c Determined for non-rac-1 by chiral HPLC analysis using a Chiralcel AD-H column. d Absolute configuration of optically active lisofylline (non-rac-1) was established by comparison of HPLC picks elution order with enantiomeric standards. The major enantiomer is shown in parentheses. e Not detected. f Not applicable because there was no detectable conversion. :10, v/v), DMSO (2.5% (v/v)), 48 h, 30 °C, 250 rpm (laboratory shaker). b Conversion values (%) (i.e., consumption of substrate 2) were determined by GC analyses after derivatization of the crude mixture with N,O-bis(trimethylsilyl)acetamide (BSA) as a silylating reagent. c Determined for non-rac-1 by chiral HPLC analysis using a Chiralcel AD-H column. d Absolute configuration of optically active lisofylline (non-rac-1) was established by comparison of HPLC picks elution order with enantiomeric standards. The major enantiomer is shown in parentheses. e Not detected. f Not applicable because there was no detectable conversion. |
|||||
| 1 | Komagataella phaffi/Pichia pastoris | ATCC 76273 | 20 mM glucose | N.D.e | N.A.f |
| 2 | Pseudomonas sp. | DSM 6978 | 20 mM glucose | N.D.e | N.A.f |
| 3 | Arthrobacter sp. | DSM 7325 | 20 mM glucose | 83 | 80 (R) |
| 4 | Isolate Actinomyces sp. SRB-AN040 | FCC025 | 20 mM glucose | 54 | 97 (S) |
| 5 | Isolate Actinomyces sp. SRB-AN053 | FCC027 | 20 mM glucose | 92 | >99 (S) |
| 6 | Isolate Actinomyces sp. ARG-AN024 | FCC014 | 20 mM glucose | 93 | >99 (S) |
| 7 | Isolate ARG-AN025 | FCC015 | 20 mM glucose | 94 | 99 (S) |
| 8 | Isolate USA-AN012 | FCC021 | 20 mM glucose | 92 | 99 (S) |
| 9 | E. coli/RasADH | — | 20 mM glucose | 95 | 97 (S) |
| 10 | — | — | 95 | 97 (S) | |
| 11 | E. coli/SyADH | — | 20 mM glucose | 87 | >99 (S) |
| 12 | — | — | N.D.e | N.A.f | |
| 13 | E. coli/ADH-A | — | 20 mM glucose | 98 | 99 (S) |
| 14 | — | — | 98 | >99 (S) | |
| 15 | E. coli/LB-ADH | — | 20 mM glucose | 41 | 86 (R) |
| 16 | — | — | 37 | 39 (R) | |
| 17 | E. coli/Lk-ADH-Lica | — | 20 mM glucose | 98 | 53 (R) |
| 18 | — | — | 97 | 60 (R) | |
| 19 | E. coli/Lk-ADH | — | 20 mM glucose | 96 | 90 (R) |
| 20 | — | — | 97 | 81 (R) | |
| 21 | E. coli/Lk-ADH prince | — | 20 mM glucose | 98 | 96 (R) |
| 22 | — | — | 98 | >99 (R) | |
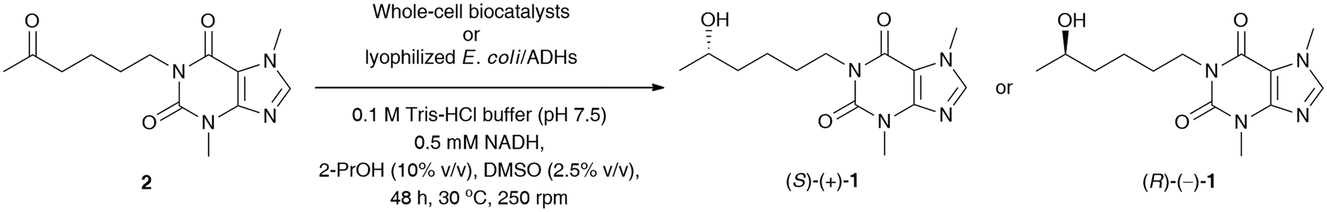
The results of this screening showed that among the tested wild-type strains, four out of eight are capable of (S)-stereoselectively reducing pentoxifylline (2), giving access to (S)-(+)-1 in 92–94% conv. and enantiopure form (from 99% ee to >99% ee) (Table 5, entries 5–8). In contrast, only one strain from Arthrobacter sp. exhibited anti-Prelog selectivity; however, (R)-(−)-1 was obtained in moderate 83% conv. and 80% ee. When analyzing the reactions catalyzed by E. coli/ADHs one could observe that the following enzyme preparations from Ralstonia sp. (E. coli/RasADH), Sphingobium yanoikuyae (E. coli/SyADH), and Rhodococcus ruber (E. coli/ADH-A) displayed Prelog selectivity, while in contrast, ADHs from Lactobacillus brevis (LB-ADH) and Lactobacillus kefiri (Lk-ADHs) exhibited the reversed stereopreference toward 2, leading to the anti-Prelog product (R)-(−)-1. The best results in terms of % conv. and % ee values were obtained in the reactions conducted without supplementation of glucose and catalyzed by both E. coli/ADH-A and E. coli/Lk-ADH Prince, which allowed isolation of optically pure (S)-(+)-1 and (R)-(−)-1 with high 98% conversions. To our surprise, we found that E. coli/SyADH, E. coli/LB-ADH, and E. coli/Lk-ADH required supplementation of 20 mM glucose to retain the desired catalytic activity and/or selectivity. In the case of E. coli/LB-ADH and E. coli/Lk-ADH, from moderate to significant drops in enantiomeric excess of (R)-(−)-1 (9–47 Δ% ee) were detected without a detrimental influence on the conversions (Table 5, entries 15 vs. 16 and 19 vs. 20), respectively. In sharp contrast, E. coli/SyADH turned inactive without glucose since even traces of bioreduction product were not observed (Table 5, entry 12). The effect of sugar additives has already been investigated for ADHs42 and is suggested to be responsible for compacting these enzymes into a protein globule, which streamlines the efficiency of the hydride transfer crucial for catalysis and decreases the activation energy. It is also worth underlining that although most of the tested E. coli/ADH preparations overexpress NADPH-dependent alcohol dehydrogenases (except E. coli/ADH-A), we decided to supplement the reaction medium with a catalytic amount of a cheaper and more stable NADH cofactor than its phosphorylated counterpart. Surprisingly, this manipulation did not deteriorate the outcome of the bioreductions of 2.
Based on preliminary screening, the feasibility of the ADH-catalyzed bioreductions of 2 was further demonstrated through up-scaling of these reactions to 0.16 mmol of the substrate (Scheme 1). Semi-preparative scale stereo-complementary bioreductions of 2 using (S)-selective E. coli/ADH-A or (R)-selective E. coli/Lk-ADH Prince furnished both LSF enantiomers, (S)-(+)-1 and (R)-(−)-1, in 65–67% isolated yield at 93–94% conv., depending on the applied ADH.
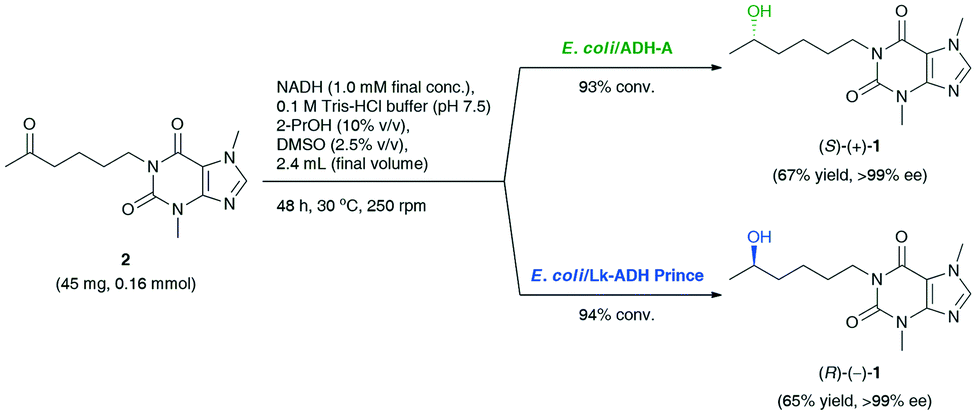 | ||
| Scheme 1 Stereo-complementary bioreductions of pentoxifylline (2) catalyzed by E. coli/ADH-A or E. coli/Lk-ADH Prince, affording enantiomerically pure lisofylline [(S)-(+)-1 and (R)-(−)-1]. | ||
Finally, the last task was to combine the afore-optimized mechanistically reversed redox transformations into stereo-complementary ‘one-pot two-step’ biocatalytic cascades. The synthetic value of the novel protocol, giving the possibility to access both optical antipodes of the title API, was demonstrated on a preparative scale (Scheme 2). The coupling of T. versicolor laccase (14.5 U mL−1) and 165 mol% TEMPO [both added sequentially in portions every 24 h: LTv (14 mg, 9.2 U) and TEMPO (8.2 mg, 33 mol%)] with the respective ADH (15 mg mL−1) and preservation of the appropriate reaction conditions for both the oxidation (pH 5.0, magnetic stirring, 150 rpm) and the bioreduction (pH 7.5, orbital shaking, 250 rpm) steps afforded (S)-(+)-1 in 56% yield and 94% ee in the case of E. coli/ADH-A, and (R)-(−)-1 in 31% yield and 92% ee in the case of E. coli/Lk-ADH Prince. The decreased reaction yield for LTv/TEMPO-E. coli/Lk-ADH Prince system might be due to a partial deactivation of the alcohol dehydrogenase in the presence of a high concentration of TEMPO mediator. To verify this hypothesis, we proceeded with an additional experiment and checked if the preparative-scale E. coli/Lk-ADH Prince-catalyzed bioreduction of 2 was affected by the elevated amount of TEMPO (41 mg, 0.26 mmol). The results of this trial proved that TEMPO was detrimental to the employed ADH, and thus the bioreduction proceeded less efficiently to afford (R)-(−)-1 in only 29% isolated yield at 47% conv., but without erosion in enantiomeric excess (>99% ee). However, the subsequent experimental validation also concerned measuring the pH value of the final reaction medium of the one-pot/two-step deracemization procedure. It turned out that the addition of 0.1 M Tris-HCl buffer (pH 7.5) to the concentrated reaction's residue consisting of 50 mM citrate buffer (pH 5.0) gave a mildly acidic mixture with pH 6.2.
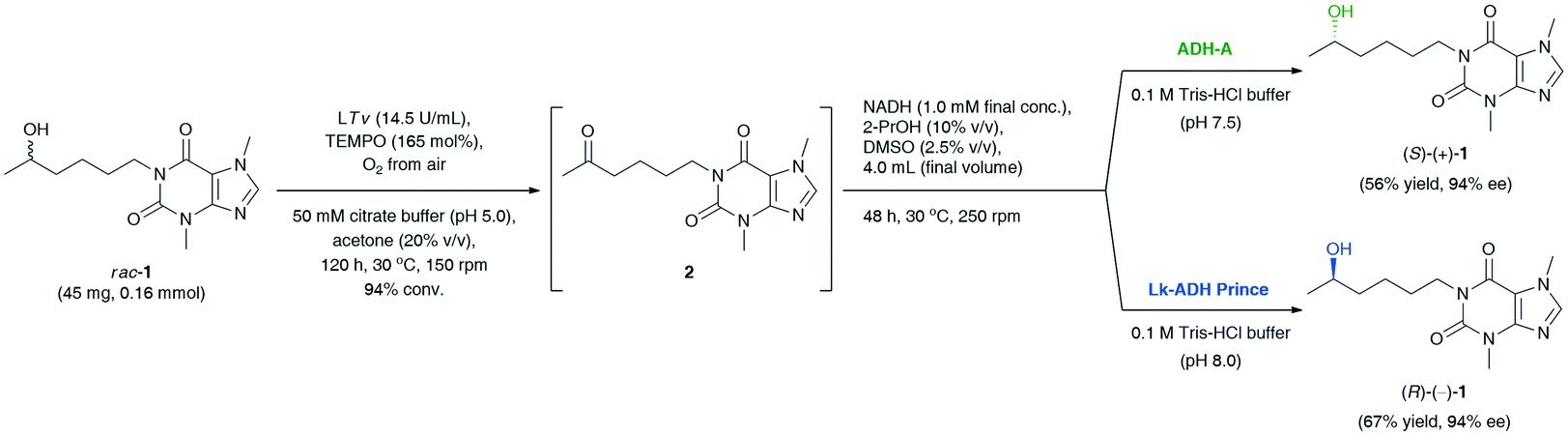 | ||
| Scheme 2 Deracemization of racemic lisofylline (rac-1) catalyzed by T. versicolor laccase/TEMPO-alcohol dehydrogenase system. | ||
To avoid a negative interaction of the acidic medium with E. coli/LK-ADH Prince, we have optimized its pH by a proper dilution of the reaction mixture with 0.1 M Tris-HCl buffer (pH 8.0), leading to a pH adjustment of ca. 7.5. This time, the bioreduction step was not affected by the pH of the medium and the TEMPO mediator, and thus the yield of the whole process improved significantly. Finally, the application of modified reaction conditions of LTv/TEMPO-E. coli/Lk-ADH Prince-mediated deracemization of rac-1 resulted in the isolation of (R)-(−)-1 in 67% yield with 94% ee.
3. Conclusions
In this work, an attempt was made to synthesize optically active lisofylline in both enantiomeric forms (S)-(+)-1 and (R)-(−)-1 through a straightforward one-pot/two-step redox-driven chemoenzymatic-deracemization route employing laccase from Trametes versicolor coupled with TEMPO as a redox mediator, and the subsequent combination of two recombinant alcohol dehydrogenases (E. coli/ADH-A or E. coli/Lk-ADH Prince) as biocatalysts for an asymmetric hydrogen transfer bioreduction of the in situ generated pentoxifylline (2). In this regard, a complex screening of the optimal reaction conditions for (laccase/TEMPO)-mediated non-selective oxidation of racemic lisofylline (rac-1) was performed to obtain pentoxifylline (2) in high 95% conv. and 68% yield. Next, a series of recombinant ADHs were screened in stereoselective reduction of prochiral ketone 2 to afford both enantiomers of LSF. Finally, asymmetric bioreductions were integrated with the elaborated oxidation step. Applying the (LTv/TEMPO)-E. coli/ADH-A system afforded (S)-lisofylline in 56% yield and 94% ee, while employing the LTv/TEMPO-E. coli/Lk-ADH Prince system provided (R)-lisofylline in 67% yield and 94% ee. One-pot deracemization of rac-1 revealed that E. coli/Lk-ADH Prince was more sensitive toward TEMPO than E. coli/ADH-A, which rationalizes the lower yield in this case. The chemoenzymatic approach presented herein constitutes a valuable extension of the synthetic repertoire to synthesize challenging pharmaceuticals in high enantiomeric enrichment and completes our journey toward non-racemic lisofylline. Although this attempt is the first example of the employment of the LTv/TEMPO-ADH catalytic system toward deracemization of unactivated sec-alcohols, further optimization of these cascades and expansion of the substrate scope to understand the full potential of laccase/TEMPO-ADH tandem catalysis is required to make it a direct approach in fruitful biotransformations.4. Experimental
4.1. Materials and methods
Laccase from Trametes versicolor (LTv, 0.66 U mg−1) was purchased from Sigma-Aldrich (Cat. No.: 38429). 2,2,6,6-Tetramethyl-1-piperidinyloxy, free radical (TEMPO) was purchased from Sigma-Aldrich (Cat. No.: 214000). β-Nicotinamide adenine dinucleotide, disodium salt, hydrate, 95+%, reduced form (NADH) was purchased from Across Organics (Cat. No.: 271100010). All other reagents for chemical transformations and product isolation/purification were obtained from commercial sources and used as received without additional pretreatment. Citrate buffer (pH 5, 50 mM) was saturated with molecular oxygen by bubbling it for 30 min prior to being used in the chemoenzymatic oxidation experiments. The chromatographic (GC) analyses were performed using an Agilent Technologies 6890 N instrument equipped with a flame ionization detector (FID) and fitted with an HP-50+ (30 m) semipolar column (50% phenyl–50% methylpolysiloxane); helium (2 mL min−1) was used as carrier gas; retention times (tR) are given in minutes under these conditions. The GC method conditions for the resolution of the respective compounds rac-1, 2, and rac-3 are shown in Table S2 (see the ESI†). The enantiomeric excesses (% ee) of bioreduction products were determined by HPLC analysis performed on a Shimadzu CTO-10ASV chromatograph equipped with an STD-20A UV detector and/or a Shimadzu Nexera-i (LC-2040C 3D) instrument equipped with a photodiode array detector (PAD) using a Chiralpak AD-H (4.6 mm × 250 mm, coated on 5 μm grain size silica gel, from Daicel Chemical Ind., Ltd.) chiral column equipped with a respective pre-column (4 mm × 10 mm, 5 μm); the HPLC analyses were executed in an isocratic and isothermal (25 °C) mode; the method conditions for the resolution of racemic lisofylline (rac-1) are shown in Table S3 (see the ESI†). Optical rotations ([α]) were measured with a PolAAr 32 polarimeter in a 2 dm long cuvette using the sodium D line (λ = 589 nm). 1H NMR (500 MHz) and 13C NMR (126 MHz) spectra were recorded on a Varian NMR System 500 MHz spectrometer. The 1H and 13C chemical shifts (δ) are reported in parts per million (ppm) relative to the solvent signals [CDCl3, δH (residual CHCl3) 7.26 ppm, δC 77.16 ppm]. Chemical shifts are presented as s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet), and br s (broad singlet); coupling constants (J) are reported in hertz. Raw spectroscopic data were processed with ACD/NMR Processor Academic Edition (Product Version: 12.01). Mass spectrometry was recorded on a Q Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer, ESI source: electrospray with spray voltage 4.00 kV for FTMS analysis; all samples were prepared by dilution of MeOH (0.5 mL) and additives of mixtures of CH3CN/MeOH/H2O (50:25:25, v/v/v) + 0.5% formic acid (HCOOH) each.
4.2. Synthetic procedures
:10 v/v) mixture. The obtained solid was additionally quenched with Et2O (2 × 1 mL) and dried under reduced pressure to afford the desired ketone 2 (203 mg, 0.73 mmol, 73%) as a white solid.
3,7-Dimethyl-1-(5-oxohexyl)-3,7-dihydro-1H-purine-2,6-dione (2). Mp 108–111 °C (Et2O) [lit.43 104–106 °C (CHCl3)]; Rf [CHCl3/acetone (90
:10, v/v)] 0.15; 1H NMR (500 MHz, CDCl3): δ 1.58–1.70 (m, 4H), 2.12 (s, 3H), 2.48 (t, J = 7.1 Hz, 2H), 3.55 (s, 3H), 3.96 (s, 3H), 3.99 (t, J = 7.1 Hz, 2H), 7.49 (s, 1H); 13C NMR (126 MHz, CDCl3): δ 21.1, 27.5, 29.8, 30.0, 33.7, 40.9, 43.3, 107.7, 141.5, 148.8, 151.6, 155.4, 208.8; IR (Nujol): νmax = 2924, 1660, 1456; FTMS (ESI-TOF) m/z: [M + H]+ Calcd for C13H19N4O3+ m/z: 279.14517, found 279.14504; GC [260 (const.)]: tR = 9.75 min.
:5, v/v) as an eluent to afford the desired silylated lisofylline rac-3 (102 mg, 0.30 mmol, 69%) as a white solid.
3,7-Dimethyl-1-{5-[(trimethylsilyl)oxy]hexyl}-2,3,6,7-tetrahydro-1H-purine-2,6-dione (rac-3). Mp 119–123 °C (CHCl3/MeOH); Rf [CHCl3/MeOH (95:5, v/v)] 0.30; 1H NMR (500 MHz, CDCl3): δ 0.08 (s, 9H), 1.11 (d, J = 6.1 Hz, 3H), 1.26–1.52 (m, 4H), 1.57–1.72 (m, 3H), 3.56 (s, 3H), 3.71–3.79 (m, 1H), 3.97 (d, J = 0.5 Hz, 3H), 3.99 (s, 2H), 7.48 (d, J = 0.5 Hz, 1H); 13C NMR (126 MHz, CDCl3): δ 0.4, 23.6, 24.0, 28.2, 29.8, 33.7, 39.4, 41.5, 68.6, 107.8, 141.5, 148.9, 151.6, 155.4; IR (Nujol): νmax = 2924, 1656; FTMS (ESI-TOF) m/z: [M + H]+ Calcd for C16H29N4O3Si+m/z: 353.20034, found 353.20027; GC [260 (const.)]: tR = 7.28 min.
4.2.3.2 Method B (effect of acetone amount). In an open-to-air test tube, TEMPO (4.1 mg, 33 mol%) was added to a suspension of racemic lisofylline (rac-1, 23 mg, 0.08 mmol, 50 mM final conc.) in a monophasic mixture of oxygen-saturated 50 mM citrate buffer (pH 5)/acetone (10–50% (v/v)), for a total volume of 1.6 mL. The mixture was magnetically stirred for a few minutes to dissolve all the reagents, and then the laccase from Trametes versicolor (LTv, 7 mg, 4.6 U) was added. Afterward, the reaction mixture was gently stirred (150 rpm) in the presence of aerial O2 for 96 h at 30 °C. The rest of the procedure was carried out in analogy to method A. For additional data, see also Table 2.
4.2.3.3 Method C (effect of metal ions). In an open-to-air test tube, TEMPO (4.1 mg, 33 mol%) and the respective inorganic salt (1 mM final conc.) were added to a suspension of racemic lisofylline (rac-1, 23 mg, 0.08 mmol, 50 mM final conc.) in a monophasic mixture of oxygen-saturated 50 mM citrate buffer (pH 5)/acetone (20% (v/v)), for a total volume of 1.6 mL. The mixture was magnetically stirred for a few minutes to dissolve all the reagents, and then the laccase from Trametes versicolor (LTv, 7 mg, 4.6 U) was added. Afterward, the reaction mixture was gently stirred (150 rpm) in the presence of aerial O2 and/or with additional O2 purging (bubbling oxygen) for 24 h at 30 °C. The rest of the procedure was carried out in analogy to method A and method B. For additional data, see Table S1 in the ESI.†
4.2.3.4 Method D (effect of the reaction time and the laccase/TEMPO amount). In an open-to-air test tube, TEMPO (4.1 mg, 33 mol%) and laccase from Trametes versicolor (LTv, 7 mg, 4.6 U) were added sequentially every 24 h to a suspension of racemic lisofylline (rac-1, 23 mg, 0.08 mmol, 50 mM final conc.) in a mixture of oxygenated 50 mM citrate buffer (pH 5)/acetone (20% (v/v)), for a total volume of 1.6 mL. The reaction mixture was gently stirred (150 rpm) in the presence of aerial O2 for 48–120 h at 30 °C. The rest of the procedure was carried out in analogy to methods A–C. For additional data, see also Table 3.
:5, v/v) as an eluent, thus affording the desired ketone 2 (31 mg, 0.11 mmol, 68% yield, 95% conv.) as a white solid. The spectroscopic data for 2 were essentially the same as for the chemical standard obtained in the reaction with the PCC reagent (see protocol 4.2.1.). The values of % conv. were assigned using the established protocol employing BSA as a derivatization agent and GC. For additional data, see also Table 4.:10:2.5, v/v/v). In general, biotransformations were conducted in glass vials (V = 1.5 mL) without air access for 48 h at 30 °C using a laboratory shaker (250 rpm) in a final volume of 0.5 mL. After this time, each reaction was stopped by extracting the content of the vial with EtOAc (3 × 1 mL), the combined organic phase was dried over anhydrous MgSO4, the filtrate was additionally centrifuged (5 min, 6000 rpm), and finally the supernatant was transferred into a separate HPLC vial and concentrated under vacuum. The oil residue in one of the vials was used to determine % conv. by using GC analysis after derivatization of the crude mixture with BSA (see protocol 4.2.5.1 below), and the other oil residue (2 mg) was re-dissolved in HPLC-grade 2-PrOH (1.5 mL) and analyzed by HPLC on a chiral stationary phase to establish enantiomeric excesses of optically active alcohols. For additional data, see also Table 5. Note: lyophilized cells of the appropriate biocatalysts were rehydrated at first by suspending them in an aqueous buffer solution of 0.1 M Tris-HCl (pH 7.5) (337.5 μL), NADH (50 μL) added from a 5 mM stock solution prepared by dissolving the cofactor (3.55 mg) in 0.1 M Tris-HCl (pH 7.5, 1 mL), and in the case of wild-type microorganisms, glucose (50 μL) was also added from a 0.2 M stock solution prepared by dissolving carbohydrate (36 mg) in 0.1 M Tris-HCl (pH 7.5, 1 mL). After 30 min of incubation of the whole-cell biocatalysts at 30 °C (250 rpm), the reaction was initiated by the addition of ketone 2 (50 μL) from a 0.1 M stock solution prepared by dissolving substrate 2 (27.8 mg) in 2-PrOH (1 mL) and supplemented with DMSO (12.5 μL).
4.2.5.1 Derivatization of the samples for GC analyses with BSA as silylation reagent.
To a vial containing oil residue after enzymatic reactions, a solution of N,O-bis(trimethylsilyl)acetamide (BSA, 15 mg, 71.3 μmol, 18 μL) in CH2Cl2 (100 μL) was added in one portion. After 20 min of vigorous vortexing of the reaction mixture at room temperature, an aliquot of the sample was directly analyzed using GC.
:5, v/v) as an eluent. The obtained yellowish solid was further purified by dissolving it in CH2Cl2 (0.5 mL) and adding a few drops of EtO2 until the solution became turbid. Next, the mixture was placed in a freezer and cooled at −18 °C for 30 min until a white solid precipitated. Subsequent filtration of the solid and drying under high vacuum afforded the desired optically active products: (S)-(+)-1 [30 mg, 67% isolated yield, >99% ee, [α]D24.5 = +7.5 (c 1.00, CHCl3) {lit.13 [α]D24.0 = +7.5 (c 1.00, CHCl3)}] in the case of E. coli/ADH-A, and (R)-(−)-1 (29 mg, 65% isolated yield, >99% ee, [α]D24.5 = −5.0 (c 1.00, CHCl3) {lit.9 [α]D25.0 = −8.75 (c 0.40, CHCl3)}] in the case of E. coli/Lk-ADH Prince.:5, v/v) as an eluent. The obtained yellowish solid was further purified by dissolving it in a small amount of CH2Cl2 (0.5 mL) and precipitating it with EtO2. After cooling the solution at −18 °C for 30 min, a fine precipitate was filtered off using a Pasteur pipette plugged with cotton wool and dried under a high vacuum to afford the desired optically active products as white solids: (S)-(+)-1 (26 mg, 56% isolated yield, 94% ee) in the case of E. coli/ADH-A, and (R)-(−)-1 (30 mg, 67% isolated yield, 94% ee) in the case of E. coli/Lk-ADH Prince.Author contributions
P. B.: conceptualization, methodology, validation, investigation, formal synthesis and analysis, data curation and processing, writing – original draft, writing – review & editing & revision, visualization, supervision, project administration, funding acquisition; A. R.: investigation, formal synthesis and analysis; T. R.: investigation, preparation of biocatalysts; W. K.: writing – original draft, proofreading.Conflicts of interest
The authors declare no competing financial interest. All authors have approved the final version of the manuscript. There are no conflicts to declare.Acknowledgements
This work was funded by the National Science Center (NCN) of Poland grant “SONATA 15” (Grant No. 2019/35/D/ST4/01556). Statutory support by the Department of Chemistry, Warsaw University of Technology (WUT), is also acknowledged. A. R. acknowledges financial support from the IDUB project (‘Scholarship Plus’ program for Ph.D. students). The University of Graz and the Field of Excellence BioHealth are acknowledged for financial support.Notes and references
- (a) A. Świerczek, E. Wyska, S. Bas, M. Wojciechowska and J. Młynarski, Naunyn-Schmiedeberg's Arch. Pharmacol., 2017, 390, 1047–1059 CrossRef PubMed; (b) A. Jankowska, A. Świerczek, G. Chlon-Rzepa, M. Pawłowski and E. Wyska, Curr. Med. Chem., 2017, 24, 673–700 CrossRef CAS PubMed.
- T. C. Peterson, M. R. Peterson and J. M. Raoul, Eur. J. Pharmacol., 2011, 662, 47–54 CrossRef CAS PubMed.
- (a) J. S. Striffler and J. L. Nadler, Metabolism, 2004, 53, 290–296 CrossRef CAS PubMed; (b) M. Chen, Z. Yang, R. Wu and J. L. Nadler, Endocrinology, 2002, 143, 2341–2348 CrossRef CAS PubMed; (c) Z. D. Yang, M. Chen, R. Wu, M. McDuffie and J. L. Nadler, Diabetologia, 2002, 45, 1307–1314 CrossRef CAS PubMed.
- K. Wojcik-Pszczola, K. Hincza, D. Wnuk, D. Kadziolka, P. Koczurkiewicz, M. Sanak, Z. Madeja, E. Pekala and M. Michalik, Acta Biochim. Pol., 2016, 63, 437–442 CrossRef CAS PubMed.
- P. de Vries and J. W. Singer, Exp. Hematol., 2000, 28, 916–923 CrossRef CAS.
- D. Bleich, S. Chen, S. L. Bursten and J. L. Nadler, Endocrinology, 1996, 137, 4871–4877 CrossRef CAS PubMed.
- E. Pękala and T. Wójcik, Acta Pol. Pharm., 2007, 64, 109–113 Search PubMed.
- (a) E. Pękala, A. Godawska-Matysik and D. Zelaszczyk, Biotechnol. J., 2007, 2, 492–496 CrossRef PubMed.
- E. Pękala and D. Żelaszczyk, Sci. Pharm., 2009, 77, 9–17 CrossRef.
- W. Aretz, H. Furrer, U. Gebert and H.-J. Hinze, US Pat., US5710272A, 1998; Search PubMed; W. Aretz, H. Furrer, U. Gebert and H.-J. Hinze, DE Pat., DE3942872A1, 1991 Search PubMed.
- A. T. Larsen, E. M. May and K. Auclair, J. Am. Chem. Soc., 2011, 133, 7853–7858 CrossRef CAS PubMed.
- P. Schiavini, K. J. Cheong, N. Moitessier and K. Auclair, ChemBioChem, 2017, 18, 248–252 CrossRef CAS PubMed.
- P. Borowiecki, B. Zdun and M. Dranka, Mol. Catal., 2021, 504, 111451 CrossRef CAS.
- M. Poterała and P. Borowiecki, ACS Sustainable Chem. Eng., 2021, 9, 10276–10290 CrossRef.
- (a) E. T. Hwang and S. Lee, ACS Catal., 2019, 9, 4402–4425 CrossRef CAS; (b) J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed; (c) S. P. France, L. J. Hepworth, N. J. Turner and S. L. Flitsch, ACS Catal., 2016, 7, 710–724 CrossRef; (d) R. Sigrist, B. Z. da Costa, A. J. Marsaioli and L. G. de Oliveira, Biotechnol. Adv., 2015, 33, 394–411 CrossRef CAS PubMed; (e) J. Muschiol, C. Peters, N. Oberleitner, M. D. Mihovilovic, U. T. Bornscheuer and F. Rudroff, Chem. Commun., 2015, 51, 5798–5811 RSC; (f) R. C. Simon, N. Richter, E. Busto and W. Kroutil, ACS Catal., 2013, 4, 129–143 CrossRef; (g) E. Ricca, B. Brucher and J. H. Schrittwieser, Adv. Synth. Catal., 2011, 353, 2239–2262 CrossRef CAS; (h) S. F. Mayer, W. Kroutil and K. Faber, Chem. Soc. Rev., 2001, 30, 332–339 RSC.
- (a) G. de Gonzalo and C. E. Paul, Curr. Opin. Green Sustainable Chem., 2021, 100548 CrossRef; (b) J. Fan, Y. Peng, W. Xu, A. Wang, J. Xu, H. Yu, X. Lin and Q. Wu, Org. Lett., 2020, 22, 5446–5450 CrossRef CAS PubMed; (c) M. Hall and A. S. Bommarius, Chem. Rev., 2011, 111, 4088–4110 CrossRef CAS PubMed; (d) J. H. Schrittwieser, J. Sattler, V. Resch, F. G. Mutti and W. Kroutil, Curr. Opin. Chem. Biol., 2011, 15, 249–256 CrossRef CAS PubMed.
- C. C. Gruber, I. Lavandera, K. Faber and W. Kroutil, Adv. Synth. Catal., 2006, 348, 1789–1805 CrossRef CAS.
- (a) S. A. Nafiu, M. Takahashi, E. Takahashi, S. M. Hamdan and M. M. Musa, Catal. Sci. Technol., 2020, 10, 8213–8218 RSC; (b) C. E. Paul, I. Lavandera, V. Gotor-Fernández, W. Kroutil and V. Gotor, ChemCatChem, 2013, 5, 3875–3881 CrossRef CAS; (c) W. Kroutil, C. Voss and C. Gruber, Synlett, 2010, 2010, 991–998 CrossRef; (d) C. V. Voss, C. C. Gruber, K. Faber, T. Knaus, P. Macheroux and W. Kroutil, J. Am. Chem. Soc., 2008, 130, 13969–13972 CrossRef CAS PubMed; (e) C. V. Voss, C. C. Gruber and W. Kroutil, Angew. Chem., Int. Ed., 2008, 47, 741–745 CrossRef CAS PubMed; (f) I. Lavandera, A. Kern, V. Resch, B. Ferreira-Silva, A. Glieder, W. M. Fabian, S. de Wildeman and W. Kroutil, Org. Lett., 2008, 10, 2155–2158 CrossRef CAS PubMed.
- F. R. Bisogno, I. Lavandera, W. Kroutil and V. Gotor, J. Org. Chem., 2009, 74, 1730–1732 CrossRef CAS PubMed.
- F. G. Mutti, A. Orthaber, J. H. Schrittwieser, J. G. de Vries, R. Pietschnig and W. Kroutil, Chem. Commun., 2010, 46, 8046–8048 RSC.
- E. Liardo, N. Ríos-Lombardía, F. Morís, J. González-Sabín and F. Rebolledo, Eur. J. Org. Chem., 2018, 2018, 3031–3035 CrossRef CAS.
- J. M. Carceller, M. Mifsud, M. J. Climent, S. Iborra and A. Corma, Green Chem., 2020, 22, 2767–2777 RSC.
- D. Méndez-Sánchez, J. Mangas-Sánchez, I. Lavandera, V. Gotor and V. Gotor-Fernández, ChemCatChem, 2015, 7, 4016–4020 CrossRef.
- K. Kędziora, A. Díaz-Rodríguez, I. Lavandera, V. Gotor-Fernández and V. Gotor, Green Chem., 2014, 16, 2448–2453 RSC.
- (a) J. Albarrán-Velo, V. Gotor-Fernández and I. Lavandera, Mol. Catal., 2020, 493, 111087 CrossRef; (b) L. Martínez-Montero, V. Gotor, V. Gotor-Fernández and I. Lavandera, ACS Catal., 2018, 8, 2413–2419 CrossRef.
- S. González-Granda, D. Méndez-Sánchez, I. Lavandera and V. Gotor-Fernández, ChemCatChem, 2019, 12, 520–527 CrossRef.
- M. Fabbrini, C. Galli, P. Gentili and D. Macchitella, Tetrahedron Lett., 2001, 42, 7551–7553 CrossRef CAS.
- (a) P. Borowiecki, M. Młynek and M. Dranka, Bioorg. Chem., 2021, 106, 104448 CrossRef CAS PubMed; (b) M. Poterała, M. Dranka and P. Borowiecki, Eur. J. Org. Chem., 2017, 2017, 2290–2304 CrossRef; (c) P. Borowiecki, D. Paprocki, A. Dudzik and J. Plenkiewicz, J. Org. Chem., 2016, 81, 380–395 CrossRef CAS PubMed; (d) P. Borowiecki, D. Paprocki and M. Dranka, Beilstein J. Org. Chem., 2014, 10, 3038–3055 CrossRef PubMed.
- A. Singhal, G. Choudhary and I. S. Thakur, Prep. Biochem. Biotechnol., 2012, 42, 113–124 CrossRef CAS PubMed.
- For the review articles on laccases see: (a) A. C. Sousa, L. O. Martins and M. P. Robalo, Molecules, 2021, 26, 3719 CrossRef CAS PubMed; (b) T. Itoh and Y. Takagi, ACS Sustainable Chem. Eng., 2020, 9, 1443–1458 CrossRef; (c) K. Agrawal and P. Verma, Heliyon, 2020, 6, e03972 CrossRef PubMed; (d) C. Romero-Guido, A. Baez and E. Torres, Catalysts, 2018, 8, 223 CrossRef; (e) A. Mayer and R. C. Staples, Phytochemistry, 2002, 60, 551–565 CrossRef CAS PubMed; (f) A. I. Yaropolov, O. V. Skorobogat'ko, S. S. Vartanov and S. D. Varfolomeyev, Appl. Biochem. Biotechnol., 1994, 49, 257–280 CrossRef CAS.
- K. N. Niladevi, N. Jacob and P. Prema, Process Biochem., 2008, 43, 654–660 CrossRef CAS.
- R. A. Sheldon, Front. Chem., 2020, 8, 132 CrossRef CAS PubMed.
- (a) W. Stampfer, K. Edegger, B. Kosjek, K. Faber and W. Kroutil, Adv. Synth. Catal., 2004, 346, 57–62 CrossRef CAS; (b) W. Krenn, I. Osprian, W. Kroutil, G. Braunegg and K. Faber, Biotechnol. Lett., 1999, 21, 687–690 CrossRef CAS; (c) W. Kroutil, M. Mischitz and K. Faber, J. Chem. Soc., Perkin Trans. 1, 1997, 3629–3636 RSC.
- I. Lavandera, A. Kern, B. Ferreira-Silva, A. Glieder, S. de Wildeman and W. Kroutil, J. Org. Chem., 2008, 73, 6003–6005 CrossRef CAS PubMed.
- I. Lavandera, A. Kern, V. Resch, B. Ferreira-Silva, A. Glieder, W. M. Fabian, S. de Wildeman and W. Kroutil, Org. Lett., 2008, 10, 2155–2158 CrossRef CAS PubMed.
- K. Edegger, C. C. Gruber, T. M. Poessl, S. R. Wallner, I. Lavandera, K. Faber, F. Niehaus, J. Eck, R. Oehrlein, A. Hafner and W. Kroutil, Chem. Commun., 2006, 2402–2404 RSC.
- (a) K. Niefind, J. Müller, B. Riebel, W. Hummel and D. Schomburg, J. Mol. Biol., 2003, 327, 317–328 CrossRef CAS PubMed; (b) K. Tauber, M. Fuchs, J. H. Sattler, J. Pitzer, D. Pressnitz, D. Koszelewski, K. Faber, J. Pfeffer, T. Haas and W. Kroutil, Chem. – Eur. J., 2013, 19, 4030–4035 CrossRef CAS PubMed.
- P. Borowiecki, N. Telatycka, M. Tataruch, A. Żądło-Dobrowolska, T. Reiter, K. Schühle, J. Heider, M. Szaleniec and W. Kroutil, Adv. Synth. Catal., 2020, 362, 2012–2029 CrossRef CAS.
- (a) A. Weckbecker and W. Hummel, Biocatal. Biotransform., 2009, 24, 380–389 CrossRef; (b) C. V. Voss, C. C. Gruber and W. Kroutil, Angew. Chem., Int. Ed., 2008, 47, 741–745 CrossRef CAS PubMed; (c) C. V. Voss, C. C. Gruber, K. Faber, T. Knaus, P. Macheroux and W. Kroutil, J. Am. Chem. Soc., 2008, 130, 13969–13972 CrossRef CAS PubMed.
- (a) M. A. Emmanuel, N. R. Greenberg, D. G. Oblinsky and T. K. Hyster, Nature, 2016, 540, 414–417 CrossRef CAS PubMed; (b) A. Gohel, D. Smith, B. Wong, J. Sukumaran, W. L. Yeo and S. J. Collier, WO Pat., WO2012142302, 2012 Search PubMed.
- B. Kosjek, W. Stampfer, M. Pogorevc, W. Goessler, K. Faber and W. Kroutil, Biotechnol. Bioeng., 2004, 86, 55–62 CrossRef CAS PubMed.
- (a) A. E. Wilcox, M. A. LoConte and K. M. Slade, Biochemistry, 2016, 55, 3550–3558 CrossRef CAS PubMed; (b) D. Spickermann, S. Kara, I. Barackov, F. Hollmann, U. Schwaneberg, P. Duenkelmann and C. Leggewie, J. Mol. Catal. B: Enzym., 2014, 103, 24–28 CrossRef CAS; (c) O. Miyawaki, G.-L. Ma, T. Horie, A. Hibi, T. Ishikawa and S. Kimura, Enzyme Microb. Technol., 2008, 43, 495–499 CrossRef CAS.
- Z. Han, S. L. Bonnet and J. H. van der Westhuizen, Tetrahedron, 2008, 64, 2619–2625 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy00145d |
| This journal is © The Royal Society of Chemistry 2022 |