
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Automated flow and real-time analytics approach for screening functional group tolerance in heterogeneous catalytic reactions†
Kevin
Simon
 ab,
Peter
Sagmeister
ab,
Rachel
Munday
c,
Kevin
Leslie
c,
Christopher A.
Hone
*ab and
C. Oliver
Kappe
*ab
ab,
Peter
Sagmeister
ab,
Rachel
Munday
c,
Kevin
Leslie
c,
Christopher A.
Hone
*ab and
C. Oliver
Kappe
*ab
aCenter for Continuous Flow Synthesis and Processing (CCFLOW), Research Center Pharmaceutical Engineering (RCPE), Inffeldgasse 13, 8010 Graz, Austria. E-mail: christopher.hone@rcpe.at; oliver.kappe@uni-graz.at; Web: http://goflow.at
bInstitute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, A-8010 Graz, Austria
cChemical Development, Pharmaceutical Technology & Development, Operations, AstraZeneca, Macclesfield, UK
First published on 10th February 2022
Abstract
Heterogeneous hydrogenation reactions are widely used in synthesis, and performing them using continuous flow technologies addresses many of the safety, scalability and sustainability issues. However, one of the main potential drawbacks is catalyst deactivation by substrate inhibition. The efficient and rapid evaluation of functional group/heterocycle tolerance of an heterogeneous hydrogenation reaction using an automated continuous flow and real-time analytics platform is achieved. The information obtained is important for benchmarking catalytic reactions, particularly in the preparation of complex molecules. The methodology is applied to an aromatic nitro group reduction using hydrogen gas and palladium on alumina (Pd/Al2O3) as a heterogeneous catalyst. The system utilizes catalytic static mixer (CSM) technology within a thermoregulated shell-and-tube reactor. The flow approach is configured to collect large datasets for a catalytic system with different additives (12 in total). The generation of data in real-time for complex reaction mixtures is a significant challenge. The flow setup integrates inline FT-IR and online UHPLC as orthogonal analytical methods for rapid data acquisition and for quantification of the main chemical species (substrate, product and all the additives). Thus, the change in the reaction outcome is assessed along with the stability of the additive to the reaction conditions. In particular, advanced data analysis models (partial least squares regression) were used to quantify the chemical species by FT-IR in real time. Our approach facilitates the generation of quantitative data early in development to understand the sensitivity of the reaction performance in the presence of different functional groups and heterocycles, greatly reducing experimental effort.
Introduction
Synthetic organic chemistry is becoming an increasingly data-driven science.1 There is an ever growing demand for synthetic methods that enable the exploration of diverse and novel molecular property space.2 Despite the advances made in organic chemistry, the synthetic toolkit available to discovery chemists remains small. When a reaction methodology is newly developed or modified then chemists perform a substrate scope to demonstrate the tolerance to specific functional groups (Fig. 1a).3 A substrate scope provides information regarding the steric, electronic and functional group tolerance to a particular set of reaction conditions. However, a substrate scope is limited by the ability to access certain molecules and is generally restricted to small substrates. Many of the compounds within a substrate scope are at least bifunctional in nature and their corresponding precursor compounds often need to be synthesized. The secondary functionality influences the reactivity of the main primary reactive center through steric and (stereo)electronic effects. Consequently, it is difficult to delineate the influence to reaction rate by a secondary functional group or the stability of this secondary functionality to the reaction conditions. This applies even more for when larger and more flexible substrates are considered, which is typical for natural products and biologically active compounds.4 Primarily, substrate scopes are designed to showcase the strengths of a new methodology rather than illustrate the limitations.5 Authors often do not include lower-yielding examples due to the perception from reviewers that this then demonstrates the reaction is less synthetically valuable. Consequently, there is generally a lack of data regarding the reaction performance outside of the idealized conditions. It is our view, along with others in the chemistry community, that it is equally important to demonstrate the functionalities which are unstable to the reaction conditions, or inhibit a reaction, as it is to demonstrate the ones that work successfully.2–6 All of this information is important to identify pitfalls and develop future strategies that address the limitations of existing reaction methodologies, and with data unavailable it is difficult for a field to progress forward. | ||
| Fig. 1 (a) Traditional substrate scope of a reaction methodology; (b) robustness (intermolecular) screening developed by Collins and Glorius; (c) methodology outlined in this paper for an automated continuous flow and real-time analytics platform for the screening of catalytic reactions. | ||
In response to the limitations associated with performing a traditional substrate scope, Collins and Gloris pioneered a so-called robustness screen, also termed intermolecular reaction screening. A robustness screen is the rapid evaluation of synthetic organic methodology against different functional groups and heterocycles.7,8 The screening approach evaluates the tolerance of reaction conditions to specific secondary functional groups or chemical motifs, a so-called “additive”, and the compatibility of this functionality to the reaction conditions (Fig. 1b).9,10 The same reaction conditions are performed in the presence of one molar equivalent of additive in a batch vessel. Subsequently, the reaction performance is recorded after a predetermined reaction time for the different measured responses (conversion, yield and additive remaining).11 Robustness screening has been applied to a range of different organic reactions by other groups, including Pd-catalyzed fluorination,12 allylic C–H acetoxylation,13O-arylation of phenols,14 and hetero-Diels–Alder reactions.15 The robustness screening methodology facilitates the investigation of a potentially wider range of structures than can be typically assessed with a traditional substrate scope, since substrate preparation is not required. Indeed, the robustness approach is inherently simple and labor saving, as commercially available additives can give an indication to the likelihood of a more complex molecule working successfully. Overall, it can help users early in development to identify if a methodology is likely to work, and the extent that re-optimization might be necessary.
The main limitation of the robustness screening approach is that it does not give information regarding steric or electronic properties of potential substrates, which would be gained via a traditional substrate scope. The methodology is applied to a single set of reaction conditions, and measurement of the reaction components is obtained for a single time endpoint. Robustness screens can also be considered as competition experiments, as the relative reaction rates can be obtained when studying a reaction at partial conversion.16 Richardson et al. extended the approach to a high throughput screening (HTS) format to screen different reaction conditions for different additives.17 Thus, the HTS format enabled the identification and optimization of conditions which displayed high functional group tolerance. An aspect not considered within the existing approach developed by Collins and Glorius is the change in the responses measured over operation time due to changes in catalyst performance. This is particularly important in the case of heterogeneous catalysis to ensure that a system will be stable for a sufficient period of time to be industrially-viable.
In heterogeneous catalysis, the catalyst is present as a solid and the reactants are present as liquids or gases. The catalyst is dispersed on a support, such as carbon, metal oxide or other inorganic material, or covalently bonded with a linker.18 Heterogeneous catalysis has the benefit that it prevents the product from becoming contaminated by the catalyst because it is in a different phase, assuming leaching of the catalyst does not occur.19 Furthermore, heterogeneous catalysts are relatively easy to reuse and recycle when compared to their homogeneous counterparts. However, in order to be useful heterogeneous catalysts need to display sufficient stability to the reaction constituents and conditions, and thus for long-term applicability and scale-up to be realized.20 The poisoning of a catalyst can be grouped into two main categories: reversible or irreversible. From the perspective of process developers, they are interested in identifying reaction conditions that display high and consistent catalytic activity and product selectivity.21 Currently, there exists no standard method for the benchmarking of heterogeneous catalyst reaction performance and stability towards different organic molecules, namely additives.
Continuous processing is now established as an enabling technology for performing catalytic reactions.22 The technology is particularly valuable for handling hazardous chemistry, multiphase transformations, reaction telescoping, and for the opportunity for process integration and automation.23 There is ever growing pressure by regulatory agencies, such as the US Food and Drug Administration (FDA), to implement continuous processing for pharmaceutical manufacture.24 This pressure is reflected by the potential improvement in yield, quality, sustainability, safety, energy usage and cost facilitated by continuous manufacturing. Flow reactors provide precise control over the reaction parameters, such as temperature, pressure and residence time, and the parameters can be continuously varied.25 There is high potential for automation for flow reactors, since the input conditions are easily altered through manipulation of the liquid and gas flow rates.26 Furthermore, the reaction temperature and pressure can also be rapidly changed due to the small reactor unit size used in development. Flow reactors can be readily coupled to process analytical technology (PAT)26–30 to implement automated design of experiments (DoE),27 self-optimizing and machine-learning systems,28 kinetic profiling,29 and process control.30 There is a reliance on chromatographic analytical techniques, such GC and HPLC, to generate this data. These are highly-resolved methods which can be easily calibrated, but require longer acquisition times in the range of minutes. In-line process analytics, such as Fourier-transform infrared spectroscopy (FT-IR), ultraviolet-visible spectroscopy (UV-vis) and Raman spectroscopy, provide real-time data, in the range of seconds. However, individual quantification of reaction components can be challenging due to overlapping peaks in the spectra. This limitation can be overcome through the utilization of more powerful data processing techniques, such as indirect hard modeling (IHM), partial least squares (PLS) regression and neural networks.30 By using an orthogonal approach, whereby two or more analytical techniques are used simultaneously, more reaction components can be precisely determined and quantified. Automation for unified data collection, enables the use of principal component analysis (PCA) or other data mining technologies on these large datasets.
This article presents a novel automated continuous flow approach for the rapid generation of quantitative data regarding the influence of additives on heterogeneous catalytic reaction performance, and also the influence of the reaction conditions on the additives. Continuous flow is a more rigorous method to investigating catalyst stability than the investigation in batch.20 A single computer communicates with all aspects of the integrated flow and analysis platform (pumps, sensors, thermostats, mass flow controllers and analysis). The system measures the change in the responses over operation time, with and without additive at partial and full conversion (Fig. 1c). The flow methodology is designed to provide consistent and quantitative detection of numerous species throughout operation. We believe this methodology will serve as a benchmarking approach for testing the robustness of heterogeneous catalyst towards different functional groups/heterocycles and vice versa.
Results and discussion
The fully automated additive screening approach is demonstrated on the reduction of ortho-nitrotoluene (1) to the corresponding ortho-toluidine (2) using methanol (MeOH) as solvent and hydrogen (H2) gas as reductant (Scheme 1). The intermediates include azo and azoxy compounds, arising from condensation of the nitroso and hydroxylamine intermediates. Nitro reductions are routine reactions in continuous flow, typically using a packed-bed reactor, whereby the gas and liquid phases flow through the catalyst bed.31 Despite their widespread use, they remain highly challenging to handle due to their inherent multiphasic and highly exothermic nature, and also because of the propensity of catalyst inhibition or leaching to occur. Moreover, optimization can be scale-dependent due to the catalyst particle sizing, to balance reactivity against pressure drop, and also reproducible catalyst packing can be difficult to achieve.32 Nevertheless, performing hydrogenation reactions in continuous flow offers significant advantages over batch in terms of improved safety, reduced manufacturing footprint, higher throughput and increased sustainability through a more efficient use of the precious metal catalyst.33 The recent progress made in 3D printing and coating technologies has resulted in the development of catalytically-coated static mixers (CSMs) to overcome many of the limitations of using packed-bed reactors.34–37 There is almost no pressure drop through the use of these supports, therefore they are relatively easy to scale by increasing the number of CSMs in parallel or series.35,36 The hydrogenation in this study was performed using a tube-in-shell reactor with rectangular channels (Ehrfeld, Miprowa), with bespoke palladium on alumina (Pd/Al2O3) CSMs. The static mixers are manufactured by selective laser melting and then coated with Pd/Al2O3via a slurry coating technique.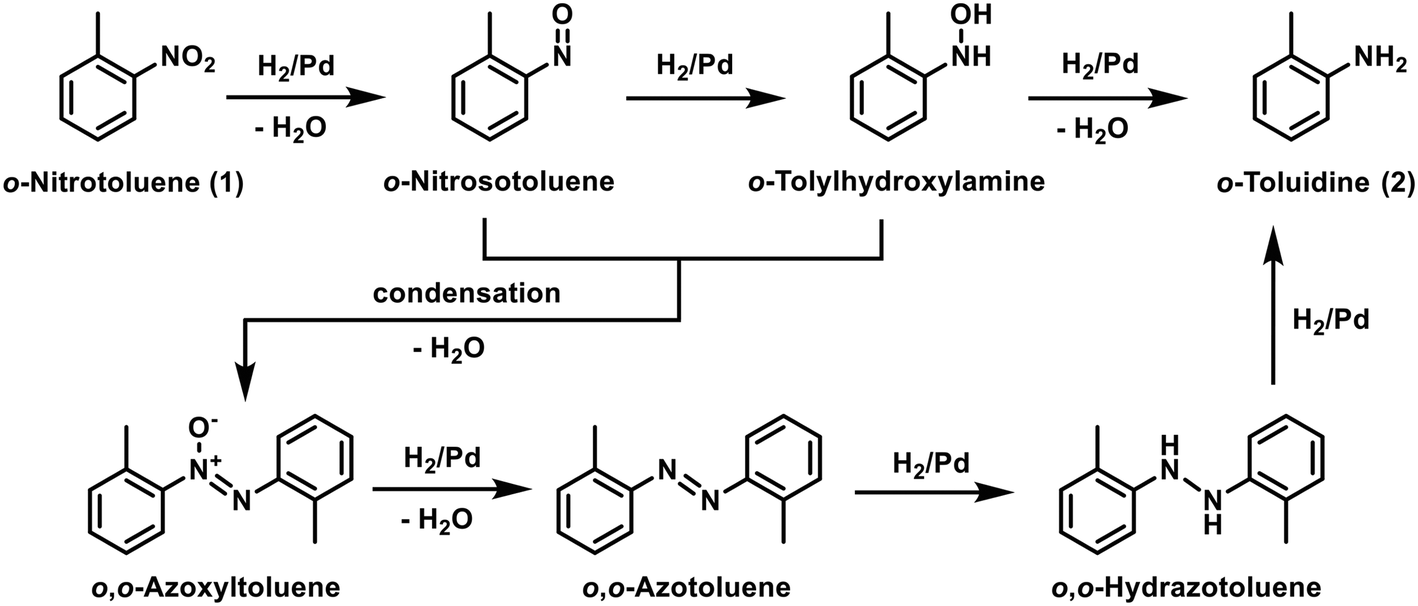 | ||
| Scheme 1 The hydrogenation of ortho-nitrotoluene (1) to the corresponding ortho-toluidine (2) using palladium on alumina, showing all the chemical species, including the intermediates in the reaction. | ||
A schematic of the flow platform utilized in this study is shown in Scheme 2 (see also Fig. S1 and section 1 in the ESI†). The hydrogenation was performed using a continuous flow reactor system with rectangular channels (Ehrfeld Mikrotechnik, Miprowa), equipped with a bespoke palladium on alumina (Pd/Al2O3) CSM (CSIRO/precision plating).37 There was a single “blank” static mixer (300 mm length) prior to the segment of the channel containing a Pd/Al2O3 CSM (150 mm length). A total of 7 Pd/Al2O3 CSMs were used throughout the project. The internal void volume of the segment containing the Pd/Al2O3-coated CSM corresponded to ∼1.7 mL. H2 gas was produced using a commercial H2 generator (ThalesNano Energy, H-Genie) which has an integrated mass flow controller (MFC) for the controlled introduction of the gas into the flow system. The gas flow rate was measured in the units of mLn min−1, where n represents measurement under standard conditions, i.e., Tn = 25 °C, Pn = 1.01 bar. The three liquid feeds were: (i) ortho-nitrotoluene (1) and biphenyl as internal standard in MeOH; (ii) additive in MeOH, and; (iii) MeOH for dilution. The liquid feeds were introduced by high performance liquid chromatography pumps (HPLCs, Knauer, Azura P4.1S). The system pressure was controlled by a back pressure regulator (BPR, Equilibar, Zero Flow) which was linked to a pressurized nitrogen supply, with automated electronic regulation (Bronkhorst, EL-PRESS). The pressure was also measured at a sensor after the reactor. The temperature of the reactor was controlled using a thermostat (Huber, CC-304) and was monitored at two different points for each process stream and thermal fluid stream. A simple gas–liquid separator was constructed and connected to a FT-IR probe (Mettler Toledo, ReactIR 15) for real-time monitoring. The reactor was also connected for online ultra high performance liquid chromatography (UHPLC) sampling (Shimadzu, Nexera X2). The entire setup was monitored and controlled using a HighTec Zang LabManager and LabVision software. FT-IR data were processed and visualized in real-time by Process Link software (S-PACT) (Fig. S21†). The setup has built-in safety measures to automatically shut down in case of leakage or over-pressure.
 | ||
| Scheme 2 Schematic representation of the automated flow reactor and analytics setup used for the hydrogenation (P and T = pressure and temperature sensors; HE = heat exchanger). | ||
Prior to performing the additive screening, we performed preliminary reaction screening to determine the ideal operating space for the study (section 4.1, ESI†) using a two-pump setup. A study was performed examining the influence of concentration, hydrogen equivalents, temperature and equivalents of water. The results demonstrated that it would be possible to reach higher conversion by operating at higher concentration. The hydrogen equivalents were increased from 3.3, 4.95 and 6.6 equiv. Increasing from 3.3 to 4.95 equiv. resulted in a moderate rise in conversion and yield, but no further increase was provided at 6.6 equiv. (Fig. S22†). The temperature was increased from 80 to 120 °C in steps of 20 °C, to determine its influence on reaction rate and stability. Temperature appeared to have a small positive linear influence on the reaction, both in terms of increasing conversion and yield (Fig. S23†). In a previous study, we had observed that hydrogenation reactions can be very sensitive to water.35 The CSM was exposed to an increasing quantity of water in a third input feed from 0 to 5 equiv. Surprisingly, a consistent performance with no loss of activity or visible degradation was observed (Fig. S37†).
The additives (12 in total) used in the study were comprised of commonly occurring functional groups and heterocycles (Fig. 2): (i) aliphatic amines, morpholine (A1), N-methylmorpholine (A2), piperazine (A3), 1,4-diazabicyclo[2.2.2]octane (DABCO) (A4) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (A5); (ii) the reaction product, o-toluidine (2, A6); (iii) N-heterocycles, N-methylimidazole (A7) and 3,5-lutidine (A8); (iv) sulfur-containing compounds, thiophenol (A9) and thioanisole (A10); and (v) benzoaromatic compounds, toluene (A11) and 2-chlorotoluene (A12). One molar equivalent of additive was investigated with respect to the ortho-nitrotoluene (1) at two concentration levels (0.5 M and 0.1 M).
 | ||
| Fig. 2 List of additives. | ||
We were interested in developing an approach for the benchmarking of heterogeneous catalytic systems. The robustness screening developed by Collins and Glorius focused on reaction endpoints. There are three key stages to our workflow: (1) selection of experiments to generate understanding; (2) application of the experimental design in the flow reactor and collection of data; and (3) evaluation of the data in terms of confidence, and then utilization of data to make informed decisions. The methodology focused on addressing the following points: (i) if the particular functional group/heterocycle (additive) is stable to the conditions; (ii) if the additive causes retardation of rate, drop in yield or catalyst poisoning; and (iii) if the system is stable over operation time. Moreover, one set of conditions is selected to achieve partial conversion (below the maximum equilibrium level) to avoid operation within an “excess catalyst regime”, thus any small drop in reaction rate is immediately observed, otherwise the catalyst could be partially deactivated whilst conversion remained quantitative. A second set of conditions was used at full conversion to measure performance at conditions which are more typical of process operation. The collection of multiple points at constant input conditions enables an assessment of the stability of the system over operation time. The collection of multiple data points for each set of conditions increases the confidence of the user in terms of the reliability of the measurement and develops an understanding of the stability of the system. The final set of conditions at both concentration levels were a replicate of the initial conditions, to determine whether any change had occurred over the duration. Bruijnincx, Heeres and co-workers investigated the influence of possible impurities, such as formic acid, sulfuric acid, furfural, 5-hydroxymethylfurfural, humins, and sulfur-containing amino acids, on catalyst performance for a continuous flow hydrogenation.38 Benchmark continuous-flow experiments were performed for extended times on-stream to show the deactivation profiles. However, in this contribution the introduction of additive was performed manually and analysis of starting material and product was performed offline with no monitoring of the impurity (additive).
The benefit of automation is that large valuable datasets can be rapidly collected and processed with minimal effort and intervention from the user. Recent efforts have demonstrated that transient flow data can be used to quickly generate experimental data.29 To rapidly collect experimental data, we developed an automated flow ramp protocol (Fig. 3). A script was written to fully automate the control of the HPLC pumps and H2 mass flow controller (Fig. S19†). Initially, the flow reactor was operated at 0.5 M substrate 1 concentration to ensure the flow platform and analytics were operating as anticipated. Subsequently, the system was switched to solvent to save on material consumption, whilst the reactor was heating to the desired temperature and the H2 flow started. Based on the preliminary screening, the flow ramp was configured to cover conditions which afforded partial and full conversion of substrate 1. A set of conditions (0.5 M of substrate 1, total liquid flow rate 2 mL min−1, H2 = 74 mL min−1 = 3.3 equiv.) were selected to achieve partial conversion (between 60 to 80%) of substrate 1. A second set of conditions (0.1 M of substrate 1, total liquid flow rate 2 mL min−1, H2 = 20 mL min−1 = 4.5 equiv.) used to achieve quantitative conversion of substrate 1. The temperature, pressure and contact time were fixed (T = 100 °C, p = 20 bar, contact time = ∼51 s). The relative ratio of the pump flow rate of solvent (pump 3) to substrate (pump 1) were varied to obtain the desired concentration of substrate 1. On the introduction of one molar equivalent of additive, the relative ratio of the pump flow rates for solvent (pump 3) to additive (pump 2) were varied to maintain the desired concentration of substrate 1. The measurements without additive (before and after) were performed at both concentrations of substrate 1 to observe any change to the system and thus acted as “built-in” control experiments. This enabled us to account for any change in the catalyst condition at the start of the run and so we could monitor changes in the catalyst for the duration of the study (see section 1.2.5, ESI† for more information regarding the performance of the catalysts used in the study). The flow rate of H2 was controlled to provide 3.3 and 4.5 equiv. of H2 (20 mL min−1 minimum for the MFC) relative to the substrate 1 at 0.5 M and 0.1 M respectively. A change in flow rate produces a very unstable flow regime for a short time. This instability was observed by the FT-IR measurements, which showed that it took approximately between 7 to 8 min for the system to fully stabilize after a change. The contact time (contact time = void volume of reactor containing active catalyst ÷ total flow rate) corresponded to ∼51 s. The experimental data collected for each run corresponded to ∼300 min.
 | ||
| Fig. 3 Changes in flow rates over time during the run. Volumetric flow rate for pump 1 (1 in MeOH), pump 2 (additive in MeOH), and pump 3 (MeOH). UHPLC measurements were acquired at 7 min intervals. The flow rate of H2 corresponded to 3.3 and 4.5 equiv. of H2 relative to the substrate at 0.5 M and 0.1 M respectively. All steps carried out at a jacket temperature of 100 °C and a backpressure of 20 bar. The flow ramp was extended for additives requiring a 10 min UHPLC method (flow ramp method B, see Fig. S20†). | ||
Prior to running the additive screening experiments, it was necessary to develop robust analytical methods for monitoring. Reactions which form mixtures of compounds are a constant challenge to chemists. The ideal reaction scenario is that the starting material is fully consumed and the reaction completes with product exclusively formed. The common reality is starting material is consumed, product is formed, impurities also form, and the reaction stalls or overreacts. We selected to use FT-IR and UHPLC as two orthogonal techniques to facilitate the monitoring of the reaction system. We argue, that there are significant benefits in having one real-time method and one chromatographic method to enable even more insight into the reaction. For instance, many of the additives do not have a distinctive characteristic peak (e.g. no chromophore) by UHPLC, therefore FT-IR was essential. A further benefit is that if there is an intermittent disturbance which disrupts the UHPLC measurement, then the extent of this disturbance is observed by the FT-IR, otherwise we would need to wait for the next UHPLC measurement. These two analytical methods act as two complementary orthogonal approaches for studying a complex mixture in real time. Certainly there is extra experimental effort required for setting up the calibration, but this effort is minimal given the additional information it provides. The calibration methods ensured that in most instances that absolute values for substrate 1, product 2 and additive could be determined.
An FT-IR spectrum was acquired every 15 s for the flow experiments. PLS regression modelling was performed using the software PEAXACT 5.3 (S-PACT). Initially, IHM was attempted, but PLS regression was selected due to improved performance. The product was difficult to observe by FT-IR due to low peak intensity and overlap with many of the additives used and water (Fig. S9†). The models were generated by using calibration mixtures comprised of additive, substrate and product at 5 different levels. The PLS regression models were further refined by using a selection of the data obtained by UHPLC analysis during the experiments (see section 2, ESI†). The root mean square errors of cross-validation (RMSEcv) were found to be low, considering the difficulty of quantification. The RMSEcv of substrate 1, product 2, and additives were found to be below 4.8, 11.3 and 13.7 mM respectively, which is <3% when considering a 500 mM concentration. Furthermore, the variation between measurements was found to be very low, so no additional filter or averaging of the data was required. This set of PLS models provided useful concentration predictions for all 14 of the examined species. Furthermore, the limit of detection (LOD) and limit of quantification (LOQ) by FTIR was determined for all the additives (Table S2†). The LOD is the lowest amount of analyte that can be detected but not necessarily quantified. The LOQ is the lowest amount of analyte that can be quantitatively determined with a suitable precision and accuracy.
Online UHPLC measurements were enabled by using a 10 nL sample injector (Vici, Cheminert Nanovolume). A fast gradient method was developed, which enabled injection every 7 min. In the cases which used toluene (A11) or 2-chlorotoluene (A12) as additive then a 10 min gradient method was developed to provide satisfactory separation. The substrate 1, product 2, 3,5-lutidine (A8), toluene (A11) and 2-chlorotoluene (A12) were calibrated against biphenyl as an internal standard at five different concentration levels to enable precise quantification. For the components that could be analyzed by UHPLC, an analytical error of <2% was assessed, when there was no issue caused by the analysis, such as overlapping peaks.
The flow ramp was performed separately in the presence of different additives, and the yield of the product 2 as well as the amount of additive and starting material 1 were monitored by FT-IR and UHPLC (Fig. 4). Gratifyingly, the reaction proceeded in the presence of amine containing aliphatic compounds (A1 to A5), but the rate of reaction was retarded. Morpholine (A1) itself was stable to the reaction conditions. In the case of N-methylmorpholine (A2) and piperazine (A3), there was a small decrease in the measured additive concentration in both cases. The retardation in reaction rate is demonstrated by the drop in conversion and yield, but the mass balance is maintained when considering the peaks for intermediate compounds. We attempted to identify the identity of the main intermediate observed, with a retention time of 1.98 min by UHPLC. The peak was well-defined, albeit with some tailing. Therefore we believe the peak represents a single compound. Liquid chromatography mass spectrometry (LC-MS) and gas chromatography mass spectrometry (GC-MS) were both investigated, however both were inconclusive. Thus, we could not confirm the identity of the intermediate in the manuscript, see section 2.2.2 in the ESI† for more discussion. We tentatively propose the hydroxylamine as the intermediate based on our experience in a previous nitration study.37 The formation of the intermediate compounds was elevated when using piperazine (A3) as additive, and was measured GC-MS analysis (Fig. S17†). Therefore showing that increasing the contact time in these instances would result in increased yields. Interestingly, the inhibition of the reaction rate was more pronounced in the cases of DABCO (A4) and DBU (A5) as additives. DBU (A5) caused a drop in reaction rate even when the substrate 1 was at 0.1 M concentration. Nevertheless, DABCO (A4) and DBU (A5) were highly tolerant to the reaction conditions. The measurement by UHPLC was difficult for DBU (A5) as additive, because the additive peak partially overlapped with the product peak. We observed by real-time FT-IR analysis that additive A5 appeared to stick to the catalyst for some time even after the additive feed had been switched-off, therefore the flow ramp at these conditions was extended through manual intervention to observe if catalyst performance could be recovered. A5 displays reversible adsorption to the catalyst, since the catalytic activity can be slowly restored when additive is no longer introduced. The use of elevated temperature would also help to improve catalytic activity. Furthermore, the influence appears to be less pronounced at higher H2 equivalents and lower substrate concentration levels. When using the product (A6, 2), ortho-toluidine, as additive the reaction proceeded successfully, but the conversion and yield slightly dropped due to product inhibition, which is anticipated at higher concentrations.
 | ||
| Fig. 4 Robustness screening experiments. Fixed conditions: total liquid flow rate = 2 mL min−1, T = 100 °C, p = 20 bar, contact time = ∼51 s. Partial conversion conditions: 0.5 M of substrate 1, H2 = 74 mL min−1 = 3.3 equiv. Full conversion conditions: 0.1 M of substrate 1, H2 = 20 mL min−1 = 4.5 equiv. | ||
The screening reactions with N-heterocycles, 1-methylimidazole (A7) and 3,5-lutidine (A8), show that the reaction is tolerant to the two heterocycles assessed, but there was a drop in reaction rate. Interestingly, 1-methylimidazole (A7) causes a drop even when at 0.1 M of substrate. Importantly though, starting material still remains suggesting that the additive only retards the rate of reaction rather than inhibits product formation. 3,5-Lutidine (A8) proceeded with small drop in yield. Unsurprisingly, the reaction was not successful in the presence of more challenging sulfur containing compounds, thiophenol (A9), thioanisole (A10), thus demonstrating these compounds would not work for this transformation and are highly detrimental to the catalyst. Sulfur compounds are known to poison metal catalysts.39 Nevertheless, both additives A9 and A10 were stable to the reaction conditions. Even by using a longer method, thiophenol (A9) came very close in retention time to the substrate 1 by UHPLC, therefore FT-IR proved to be better for monitoring. When poisoned with thioanisole (A10) then some of the catalyst could be regenerated under standard conditions, however; the same was not the case for thiophenol (A9), which appears to be irreversible. In the case of A10, even though a slow improvement in yield is observed when additive was no longer introduced, it appears that it would be difficult to restore the catalyst to its initial activity.
A longer UHPLC method was developed for toluene (A11) and 2-chlorotoluene (A12) to enable clear separation of the compounds. There is no characteristic peak for toluene (A11) by FT-IR, therefore UHPLC was more reliable for monitoring this additive. The reaction proceeds in a similar manner for toluene (A11) and 2-chlorotoluene (A12) as in the absence of additive. In the case of 2-chlorotoluene (A12), a ∼20% drop in the additive was observed due to hydrodechlorination to form toluene. Experimental flow ramps were also performed using a Pd-electroplated CSM and a Pt/Al2O3 CSM to compare the performance. In both these cases, no hydrodechlorination was observed (Fig. S38†). There is a significantly larger surface area for the Pd/Al2O3 when compared to both the electroplated-Pd and Pt/Al2O3, thus accounting for its higher catalytic activity.37,40
When considering the long-term stability of such a process, it is vital to ensure that no leaching of the catalyst is occurring. Inductively coupled plasma-mass spectrometry (ICP-MS) measurements were made to assess the leaching of Pd and Al from the CSM. ICP-MS measurements were carried out on two of the fractions collected and compared with measurements of the input feeds for the substrate 1 and additive, when using piperazine (A3) and thiophenol (A9) as additives. For both Al and Pd, contents in all samples were determined to be below 50 parts per billion, thus demonstrating that there was no leaching from the reactor over the duration of the runs, in agreement with previous work using this type of CSM.37
An alternative approach to visualize the data is shown in Table 1. The screening showed that the reaction is tolerant to amine functionality, additives A1–A8, albeit always with a drop in reaction rate. Sulfur compounds, A9 and A10, resulted in the poisoning of the catalyst, thus the conditions are intolerant to this class of compounds as the conversion and yield continuously dropped throughout their addition. Pd/Al2O3 resulted in hydrodechlorination of 2-chlorotoluene (A12), but this additive could be tolerated through the use of Pt/Al2O3 or Pd-electroplated. The table also clearly demonstrates the benefit of applying two orthogonal analytical techniques as in certain cases FT-IR is better for quantification and in others UHPLC.
| Table shows the effect of one molar of a given additive when compared to the start of the run at conditions. Conditions: 0.5 M of substrate 1, total liquid flow rate = 2 mL min−1, H2 = 74 mL min−1 = 3.3 equiv., T = 100 °C, p = 20 bar, contact time = ∼51 s. The change is based on the average of 4 number of measurements before the introduction of additive and then the average of 4 number of measurements during the additive addition (any outliers were excluded). Color coded grading helps the assessment of the data: green (0 to 10% decrease), amber (11 to 20% decrease), red (more than 20% decrease). Unless otherwise stated, the drop in conversion of 1 and yield 2 are based on UHPLC data, and drop in additive are based on FT-IR data.a Assessed by UHPLC data.b Assessed by FT-IR. The drop in conversion and yield are approximated for the sulfur containing compounds as the values were continuously changing. |
|---|

|
In order to demonstrate the longer-term stability of the reactor system, experiments were performed at partial conversion over a longer time period than during the additive experiments. The long run experiments performed were: (i) for the duration of 480 min (8 h) without additive; and (ii) for piperazine as an additive for (5 h), including 60 min without additive before and 120 after additive introduction. Fig. 5 shows that the performance is similar to that observed in the screening experiments. Fig. 5a also shows the temperature and pressure measurements from the sensors within the system. Noticeably, the long run experiments demonstrate that there was a very slow retardation of the catalyst to the reaction conditions over time. In the absence of additive the concentration of substrate 1 increased from 0.19 mol L−1 to 0.22 mol L−1 over the 8 h, corresponding to a change of less than 5 mmol L−1 h−1. A decrease in performance can be attributed to catalyst inhibition over time by the reaction species. We have previously demonstrated that by operating at higher temperature the inhibition by reaction species can be reduced.37 It is important to stress that we deliberately selected to operate the system at conditions where any change will be observed as this gives the most valuable insight into the reaction system.
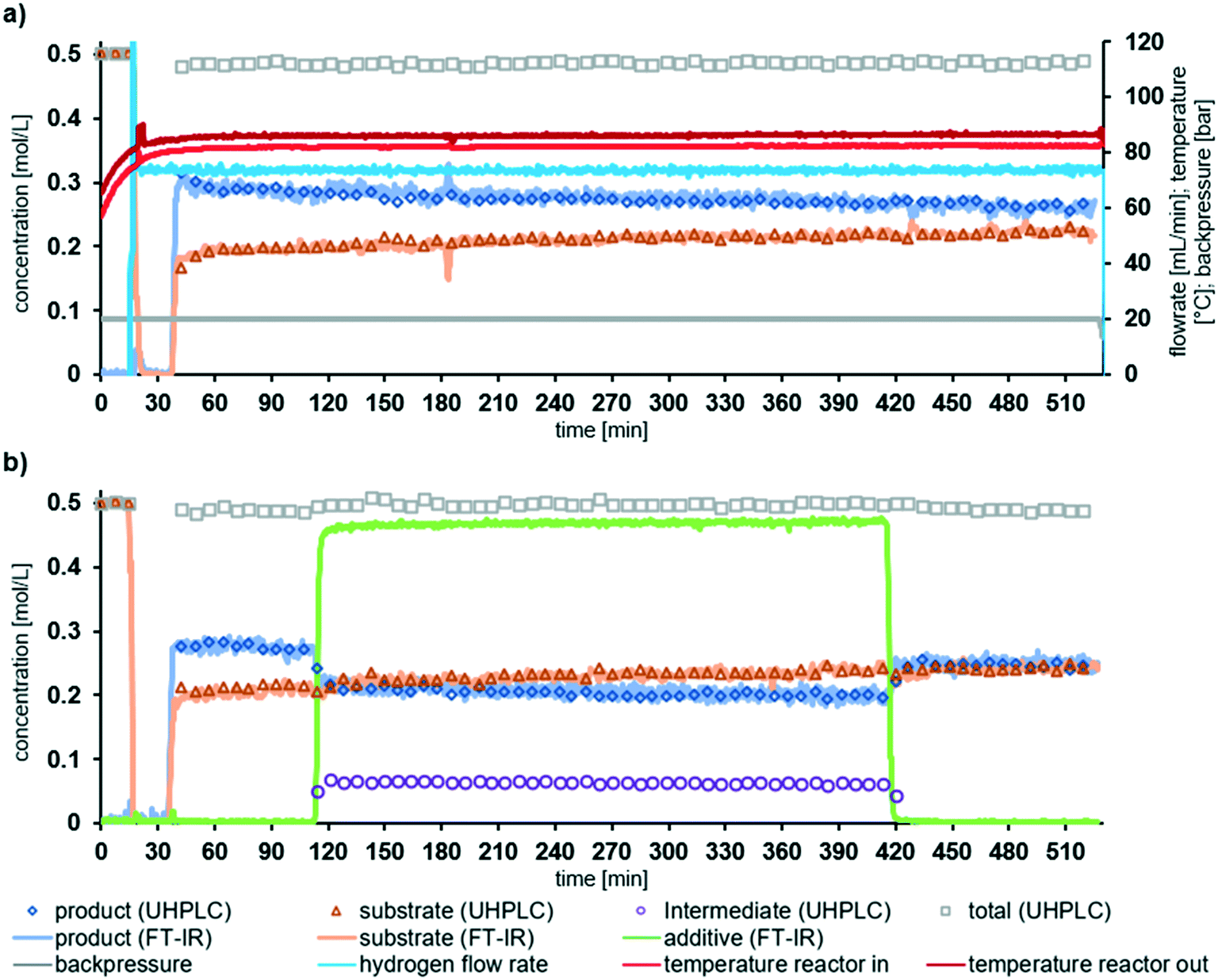 | ||
| Fig. 5 Long run experiments. (a) 8 h long run without additive, including additional measured data (temperature, backpressure and flow rate); (b) 8 h long run, with one molar equiv. of piperazine for 5 h. Conditions: 0.5 M of substrate 1, total liquid flow rate = 2 mL min−1, H2 = 74 mL min−1 = 3.3 equiv., T = 100 °C, p = 20 bar, contact time = ∼51 s. | ||
Conclusion and outlook
We have developed an efficient methodology that evaluates the tolerance and stability of an heterogeneous catalyst hydrogenation system to a variety of functional groups/heterocycles. The approach further develops the robustness screening approach pioneered by Collins and Glorius. Flow hydrogenations offer improved sustainability when compared to batch procedures. However, one of the main potential drawbacks is catalyst deactivation. The approach recognises this limitation and measures the influence that certain additives have on reducing activity of the catalyst. However, in some instances the partial or full regeneration of the catalyst activity is possible overcoming a potential drawback in the technology. A further challenge relating to heterogeneous catalysis is the stability of the catalyst to the reaction conditions over time, which the continuous flow approach considers. The methodology is expeditious to implement, since the data collection and analysis is fully automated.The approach can be readily extended to other common moieties or functional groups by preparing feed solutions using commercially available chemicals that are representative of these structural components. The methodology can also be used to screen additives when questions arise regarding the compatibility of a reagent from a previous step in a reaction telescope.41 One of the limitations as previously described by Collins and Glorius does still apply, this type of bimolecular screening strategy does not show intramolecular electronic and steric effects. Thus, care should be taken when drawing conclusions. We further demonstrated that the approach can also be applied to screen different catalysts for a fixed additive.
The analysis of complex mixtures is complicated, often requiring multiple analytical techniques to observe all reaction species. We implemented two orthogonal analytical techniques (FT-IR and UHPLC) within the platform to enable the rapid collection of an information-rich dataset. PLS regression was implemented to quantity components in real time, with data acquired every 15 s. Online UHPLC data were collected at 7 or 10 min intervals. The analysis does require careful calibration which does result in extra experimental effort, but this effort is small when compared to the gain from the quantitative measurements obtained for the responses. Ongoing work within our laboratories is investigating the development of a fully self-calibrating system. The screening approach could be readily extended to other analytical techniques. To gain further mechanistic insight into the catalyst inhibition, temperature programmed desorption/reaction (TPD/TPR) studies could be implemented. TPD/TPR analysis is an approach for obtaining information about quantities and binding properties of adsorbed species on a surface. Further advances in the approach could be achieved by incorporating additional analytical tools for TPD/TPR analysis within the continuous flow setup.42
In summary, the approach facilities the generation of valuable data that can be utilized for evaluating new methodologies, synthesis planning and in the design of better catalysts. There is great value in the collection of negative results, especially in an era of machine-learning and artificial intelligence. We are convinced that the implementation of new catalyst benchmarking approaches will be embraced by the organic chemistry and catalysis community, and thus accelerate advances made in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The CCFLOW Project (Austrian Research Promotion Agency FFG No.862766) was funded through the Austrian COMET Program by the Austrian Federal Ministry for Climate Protection, Environment, Energy, Mobility, Innovation, and Technology (BMK), the Austrian Federal Ministry for Digital and Economic Affairs (BMDW), and by the State of Styria (Styrian Funding Agency SFG). We thank Prof. W. Goessler (Graz University) for performing ICPMS analysis.References
- (a) W. L. Williams, L. Zeng, T. Gensch, M. S. Sigman, A. G. Doyle and E. V. Anslyn, ACS Cent. Sci., 2021, 7, 1622–1637 CrossRef CAS PubMed; (b) I. W. Davies, Nature, 2019, 570, 175–181 CrossRef CAS PubMed; (c) S. Caron and N. M. Thomson, J. Org. Chem., 2015, 80, 2943–2958 CrossRef CAS PubMed.
- (a) D. J. Foley, A. Nelson and S. P. Marsden, Angew. Chem., Int. Ed., 2016, 55, 13650–13657 CrossRef CAS PubMed; (b) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed.
- (a) T. Gensch and F. Glorius, Science, 2016, 352, 294–295 CrossRef CAS PubMed; (b) L. Pitzer, F. Schäfers and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 8572–8576 CrossRef CAS PubMed.
- (a) A. G. Atanasov, S. B. Zotchev and V. M. Dirsch, Nature, 2021, 20, 200–216 CAS; (b) A. Nadin, C. Hattotuwagama and I. Churcher, Angew. Chem., Int. Ed., 2012, 51, 1114–1122 CrossRef CAS PubMed.
- T. Nguyen, Chem. Eng. News, 2017, 95, 16–18 Search PubMed.
- C. A. Hone and C. O. Kappe, Chemistry - Methods, 2021, 1, 454–467 CrossRef.
- K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597–601 CrossRef CAS PubMed.
- For similar approaches to the robustness screening approach, see: (a) A. DeAngelis, M. T. Taylor and J. M. Fox, J. Am. Chem. Soc., 2009, 131, 1101–1105 CrossRef CAS PubMed; (b) S. A. Enthaler, Catal. Sci. Technol., 2011, 1, 104–110 RSC; (c) S. A. Enthaler, ChemCatChem, 2011, 3, 666–670 CrossRef CAS; (d) S. Enthaler and M. Weidauer, Catal. Lett., 2011, 141, 833–838 CrossRef CAS; (e) D. W. Stephan, S. Greenberg, T. W. Graham, P. Chase, J. J. Hastie, S. J. Geier, J. M. Farrell, C. C. Brown, Z. M. Heiden, G. C. Welch and M. Ullrich, Inorg. Chem., 2011, 50, 12338–12348 CrossRef CAS PubMed.
- For a discussion, see: I. Churcher, Nat. Chem., 2013, 5, 554–555 CrossRef CAS PubMed.
- For a review of robustness screening, see: (a) K. D. Collins and F. Glorius, Acc. Chem. Res., 2015, 48, 619–627 CrossRef CAS PubMed; (b) For selected examples of robustness screening from Collins and Glorius, see: K. D. Collins and F. Glorius, Tetrahedron, 2013, 69, 7817–7825 CrossRef CAS; (c) K. D. Collins, T. Gensch and F. Glorius, Nat. Chem., 2014, 6, 859–871 CrossRef CAS PubMed; (d) K. D. Collins, A. Rühling, F. Lied and F. Glorius, Chem. – Eur. J., 2014, 20, 3800–3805 CrossRef CAS PubMed; (e) T. Gensch, M. Teders and F. Glorius, J. Org. Chem., 2017, 82, 9154–9158 CrossRef CAS PubMed.
- For detailed information to how to perform a robustness screening, see: K. D. Collins and F. Glorius, Nat. Protoc., 2014, 9, 1348–1353 CrossRef CAS PubMed.
- H. G. Lee, P. J. Milner and S. L. Buchwald, Org. Lett., 2013, 15, 5602–5605 CrossRef CAS PubMed.
- H. A. Malik, B. L. H. Taylor, J. R. Kerrigan, J. E. Grob, K. N. Houk, J. Du Bois, L. G. Hamann and A. W. Patterson, Chem. Sci., 2014, 5, 2352–2361 RSC.
- Y. Tan, J. M. Muñoz-Molina, G. C. Fu and J. C. Peters, Chem. Sci., 2014, 5, 2831–2835 RSC.
- S. Billiet, K. De Bruycker, F. Driessen, H. Goossens, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Nat. Chem., 2014, 6, 815–821 CrossRef CAS PubMed.
- D.-T. D. Tang, K. D. Collins, J. B. Ernst and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 1809–1813 CrossRef CAS PubMed.
- J. Richardson, J. C. Ruble, E. A. Love and S. A. Berritt, J. Org. Chem., 2017, 82, 3741–3750 CrossRef CAS PubMed.
- M. Campanati, G. Fornasari and A. Vaccari, Catal. Today, 2003, 77, 299–314 CrossRef CAS.
- (a) S. Vásquez-Céspedes, R. C. Betori, M. A. Cismesia, J. K. Kirsch and Q. Yang, Org. Process Res. Dev., 2021, 25, 740–753 CrossRef; (b) D. Cantillo and C. O. Kappe, ChemCatChem, 2015, 19, 2030–2050 Search PubMed.
- C. Hammond, Green Chem., 2017, 19, 2711–2728 RSC.
- S. Hîbner, J. G. Vries and V. Farina, Adv. Synth. Catal., 2016, 358, 3–25 CrossRef.
- (a) K. Masuda, T. Ichitsuka, N. Koumura, K. Sata and S. Kobayashi, Tetrahedron, 2018, 74, 1705–1730 CrossRef CAS; (b) R. Munirathinam, J. Huskens and W. Verboom, Adv. Synth. Catal., 2015, 357, 1093–1123 CrossRef CAS; (c) A. Tanimu, S. Jaenicke and K. Alhooshani, Chem. Eng. J., 2017, 327, 792–821 CrossRef CAS.
- For general reviews, see: (a) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed; (b) M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio and S. V. Ley, Chem. Soc. Rev., 2016, 45, 83–117 RSC; (c) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed; (d) R. Gérardy, N. Emmanuel, T. Toupy, V. Kassin and N. N. Tshibalonza, Eur. J. Org. Chem., 2018, 2301–2351 CrossRef.
- S. L. Lee, T. F. O'Connor, X. Yang, C. N. Cruz, S. Chatterjee, R. D. Madurawe, C. M. V. Moore, L. X. Yu and J. Woodcock, J. Pharm. Innov., 2015, 10, 191–199 CrossRef.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed.
- (a) C. P. Breen, A. M. K. Nambiar, T. F. Jamison and K. F. Jensen, Trends Chem., 2021, 3, 373–386 CrossRef; (b) M. Trobe and M. D. Burke, Angew. Chem., Int. Ed., 2018, 57, 4192–4214 CrossRef CAS PubMed; (c) R. W. Epps, A. A. Volk, M. Y. S. Ibrahim and M. Abolhasani, Chem, 2021, 7, 2541–2545 CrossRef CAS; (d) M. A. Morin, W. P. Zhang, D. Mallik and M. G. Organ, Angew. Chem., 2021, 60, 20606–20626 CrossRef CAS PubMed.
- P. Sagmeister, J. D. Williams, C. A. Hone and C. O. Kappe, React. Chem. Eng., 2019, 4, 1571–1578 RSC.
- (a) A. D. Clayton, J. A. Manson, C. J. Taylor, T. W. Chamberlain, B. A. Taylor, G. Clemens and R. A. Bourne, React. Chem. Eng., 2019, 4, 1545–1554 RSC; (b) C. Mateos, M. J. Nieves-Remacha and J. A. Rincón, React. Chem. Eng., 2019, 4, 1536–1544 RSC; (c) D. Cortés-Borda, E. Wimmer, B. Gouilleux, E. Barré, N. Oger, L. Goulamaly, L. Peault, B. Charrier, C. Truchet, P. Giraudeau, M. Rodriguez-Zubiri, E. Le Grognec and F.-X. Felpin, J. Org. Chem., 2018, 83, 14286–14299 CrossRef PubMed; (d) A. M. Schweidtmann, A. D. Clayton, N. Holmes, E. Bradford, R. A. Bourne and A. A. Lapkin, Chem. Eng. J., 2018, 352, 277–282 CrossRef CAS; (e) M. I. Jeraal, S. Sung and A. A. Lapkin, Chemistry - Methods, 2021, 1, 71–77 CrossRef.
- (a) J. S. Moore and K. F. Jensen, Angew. Chem., Int. Ed., 2014, 53, 470–473 CrossRef CAS PubMed; (b) C. A. Hone, N. Holmes, G. R. Akien, R. A. Bourne and F. L. Muller, React. Chem. Eng., 2017, 2, 103–108 RSC; (c) S. Mozharov, A. Nordon, D. Littlejohn, C. Wiles, P. Watts, P. Dallin and J. M. Girkin, J. Am. Chem. Soc., 2011, 133, 3601–3608 CrossRef CAS PubMed; (d) K. C. Aroh and K. F. Jensen, React. Chem. Eng., 2018, 3, 94–101 RSC; (e) C. J. Taylor, M. Booth, J. A. Manson, M. J. Willis, G. Clemens, B. A. Taylor, T. W. Chamberlain and R. A. Bourne, Chem. Eng. J., 2021, 413, 127017 CrossRef CAS.
- P. Sagmeister, R. Lebl, I. Castillo, J. Rehrl, J. Kruisz, M. Sipek, M. Horn, S. Sacher, D. Cantillo, J. D. Williams and C. O. Kappe, Angew. Chem., Int. Ed., 2021, 60, 8139–8148 CrossRef CAS PubMed.
- (a) T. Yu, J. Jiao, P. Song, W. Nie, C. Yi, Q. Zhang and P. Li, ChemSusChem, 2020, 13, 2876–2893 CrossRef CAS PubMed; (b) M. Orlandi, D. Brenna, R. Harms, S. Jost and M. Benaglia, Org. Process Res. Dev., 2018, 22, 430–445 CrossRef CAS; (c) M. Irfan, T. N. Glasnov and C. O. Kappe, ChemSusChem, 2011, 4, 300–316 CrossRef CAS PubMed.
- (a) C. Yang, A. R. Teixeira, Y. Shi, S. C. Born, H. Lin, Y. L. Song, B. Martin, B. Schenkel, M. P. Lachegurabi and K. F. Jensen, Green Chem., 2018, 20, 886–893 RSC; (b) A. Carangio, L. J. Edwards, E. Fernandez-Puertas, J. F. Hayes, M. M. Kucharski, G. W. Rutherford, K. M. P. Wheelhouse and G. D. Williams, Org. Process Res. Dev., 2020, 24, 1909–1915 CrossRef CAS.
- (a) D. Dallinger and C. O. Kappe, Curr. Opin. Green Sustain. Chem., 2017, 7, 6–12 CrossRef; (b) L. Rogers and K. F. Jensen, Curr. Opin. Green Sustain. Chem., 2019, 21, 3481–3498 CAS; (c) A. J. Blacker, J. R. Breen, R. A. Bourne and C. A. Hone, The Growing Impact of Continuous Flow Methods on the Twelve Principles of Green Chemistry, in Green and Sustainable Medicinal Chemistry: Methods, Tools and Strategies for the 21st Century Pharmaceutical Industry, Royal Society of Chemistry, Cambridge, U.K., 2016, ch. 12, pp. 140–155 Search PubMed; (d) F. Ferlin, D. Kanari and L. Vaccaro, Green Chem., 2020, 22, 5937–5955 RSC.
- (a) J. Gardiner, X. Nguyen, C. Genet, M. D. Horne, C. H. Hornung and J. Tsanaktsidis, Org. Process Res. Dev., 2018, 22, 1448–1452 CrossRef CAS; (b) A. Avril, C. H. Hornung, A. Urban, D. Fraser, M. Horne, J.-P. Veder, J. Tsanaktsidis, T. Rodopoulos, C. Henry and D. R. Gunasegaram, React. Chem. Eng., 2017, 2, 180–188 RSC; (c) C. H. Hornung, X. Nguyen, A. Carafa, J. Gardiner, A. Urban, D. Fraser, M. D. Horne, D. R. Gunasegaram and J. Tsanaktsidis, Org. Process Res. Dev., 2017, 21, 1311–1319 CrossRef CAS; (d) M. Kundra, T. Grall, D. Ng, Z. Xie and C. H. Hornung, Ind. Eng. Chem. Res., 2021, 60, 1989–2002 CrossRef CAS; (e) M. Kundra, Y. Zhu, X. Nguyen, D. Fraser, C. H. Hornung and J. Tsanaktsidis, React. Chem. Eng., 2022, 7, 284–296 RSC.
- R. Lebl, Y. Zhu, D. Ng, C. H. Hornung, D. Cantillo and C. O. Kappe, Catal. Today, 2022, 338, 55–63 CrossRef.
- Y. Zhu, B. B. M. Sultan, X. Nguyen and C. Hornung, J. Flow Chem., 2021, 11, 515–523 CrossRef CAS.
- The continuous flow and analytics setup has previously been described, see: R. Lebl, S. Bachmann, P. Tosatti, J. Sedelmeier, K. Püntener, J. D. Williams and C. O. Kappe, Org. Process Res. Dev., 2021, 25, 1988–1995 CrossRef CAS.
- H. C. Genuino, H. H. van de Bovenkamp, E. Wilbers, J. G. M. Winkelman, A. Goryachev, J. P. Hofmann, E. J. M. Hensen, B. M. Weckhuysen, P. C. A. Bruijnincx and H. J. Heeres, ACS Sustainable Chem. Eng., 2020, 8, 5903–5919 CrossRef CAS.
- L. J. Hoyos, M. Primet and H. Praliaud, J. Chem. Soc., Faraday Trans., 1992, 88, 113–119 RSC.
- Y. Hashimoto, Y. Uemichi and A. Ayame, Appl. Catal., A, 2005, 287, 89–97 CrossRef CAS.
- (a) J. García-Lacuna, T. Fleiß, R. Munday, K. Leslie, A. O'Kearney-McMullan, C. A. Hone and C. O. Kappe, Org. Process Res. Dev., 2021, 25, 947–959 CrossRef; (b) J. Britton and C. L. Raston, Chem. Soc. Rev., 2017, 46, 1250–1271 RSC.
- For a proof-of-concept microfabricated flow system with an integrated capillary leak to a mass spectrometer for high sensitivity temperature programmed desorption, see: U. J. Quaade, S. Jensen and O. Hansen, Rev. Sci. Instrum., 2004, 75, 3345–3347 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2cy00059h |
| This journal is © The Royal Society of Chemistry 2022 |