
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From tandem to catalysis – organic solvent nanofiltration for catalyst separation in the homogeneously W-catalyzed oxidative cleavage of renewable methyl 9,10-dihydroxystearate†
Johanna
Vondran
 *,
Marc
Peters
,
Alexander
Schnettger
,
Christian
Sichelschmidt
and
Thomas
Seidensticker
*
*,
Marc
Peters
,
Alexander
Schnettger
,
Christian
Sichelschmidt
and
Thomas
Seidensticker
*
TU Dortmund University, Department for Biochemical and Chemical Engineering, Laboratory of Industrial Chemistry, Emil-Figge-Straße 66, 44227 Dortmund, Germany. E-mail: thomas.seidensticker@tu-dortmund.de
First published on 25th April 2022
Abstract
Feasibility of oxidative cleavage of methyl oleate in a homogeneous reaction, facilitating the subsequent recovery of the catalyst from a single phase, is a challenge. Using the high molecular catalyst phosphotungstic acid (2880 Da) as an affordable catalyst offers potential for membrane separation. To gain insight into side-reactions, the intermediate methyl 9,10-dihydroxystearate was first applied as a model substrate. Thus, the stability of the intermediate methyl 9,10-epoxystearate and the vicinal diol was significantly improved under reaction conditions. Oxidative cleavage of the vicinal diol as a stable intermediate is very promising reaching an overall selectivity of 90% and a selectivity towards the cleavage carboxylic acids of 80%, considering dilution and acidity as the most important parameters. Retention of the catalyst via organic solvent nanofiltration was investigated and we retained 94% of the catalyst in the monophasic system as the first step towards a process concept for a product purification or catalyst recycling strategy.
Introduction
High oleic sunflower oil contains a large amount of oleic acid, from which methyl oleate can be obtained in comparatively high concentrations for a technical grade product (ca. 91.5%). Therefore, it is an interesting renewable raw material for the chemical industry. An established process is ozonolysis, resulting in double bond cleavage yielding pelargonic acid and azelaic acid, mono methyl azelate, respectively.1 Both products exhibit an odd hydrocarbon chain length of nine carbon atoms, which are naturally rare.2 However, especially azelaic acid is of high industrial interest as it is used for the synthesis of nylon 6.9 or further esters and polyesters for elastomers, plasticizers, adhesives or cosmetics.3 Pelargonic acid is used for example for the production of herbicides,4 ester lubricants or cosmetics.5 So far, ozonolysis is the most economic method for double bond cleavage of unsaturated fatty acids and fatty acid methyl esters, industrially processed by Emery Oleochemicals, Croda Sipo and P2 Science.6 However, this technology has several drawbacks: the process is highly energy-consuming,6 the intermediates cause the risk of spontaneous decomposition and explosion and the instability of ozone requires its dilution in air or oxygen.7Alternative routes have been in focus of research.8 In terms of green chemistry, especially green oxidants avoiding the formation of toxic byproducts are of interest. Hence, oxygen or hydrogen peroxide are green oxidants resulting in the formation of water as the only byproduct. All other oxidants, such as NaIO4 or KMnO4, will result in the formation of more hazardous and toxic waste. Additionally, the active oxygen content of oxygen is determined to be 50 wt%, since in most cases, only one oxygen atom is transferred to the substrate. Using hydrogen peroxide, this value is only reduced to 47 wt% whilst selectivity is often better under even milder conditions.9 Thus, hydrogen peroxide is widely used for various oxidations, one of which is oxidative cleavage.8 Thereby, catalysts are of need to activate hydrogen peroxide and also to control selectivity of the oxidized products. For this purpose, especially transition metal catalysts have been investigated.10 Amongst several homogeneous transition metal catalysts, offering the advantage of low mass transfer limitations and high reaction rates, tungsten has proven to be efficient in the oxidative cleavage of unsaturated fatty acids and methyl esters whilst being cheap and less toxic compared to other transition metal catalysts, particularly those based on Ru and Os.8
First application of an efficient W-catalyst in oxidation was described by Venturello et al. for epoxidation, which is the first intermediate step of oxidative cleavage (Fig. 1).11 As in many following works (Table 1), the system was biphasic using an organic alkene and aqueous hydrogen peroxide as oxidant (Table 1, entry 1). To enhance mass transfer, and a reaction respectively, the addition of a phase transfer agent was required. The highly effective catalyst allowed for short reaction times and therefore short contact times between the two phases. Consequently, ring-opening hydrolysis was prevented and selectivity towards the epoxide was increased.
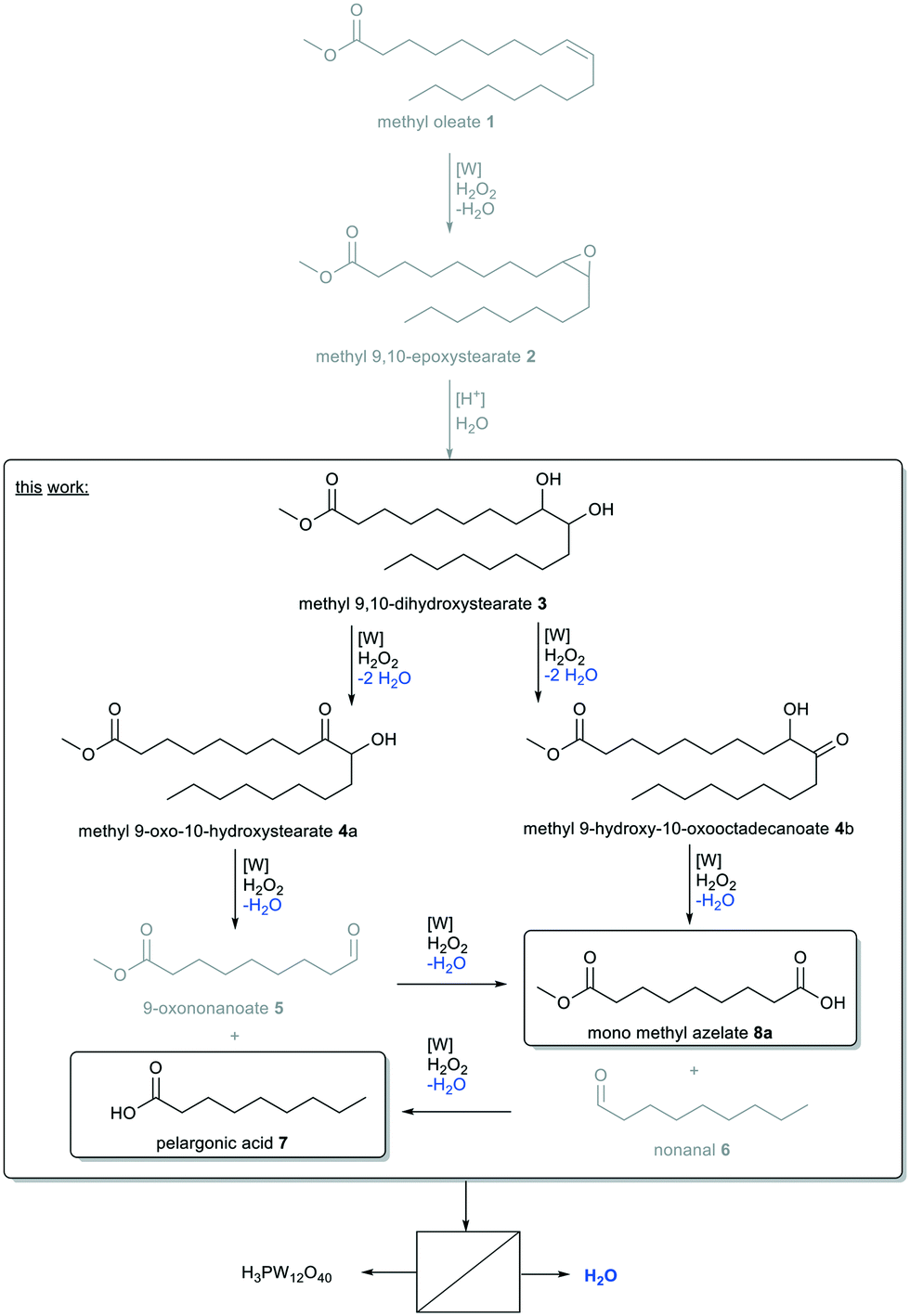 | ||
| Fig. 1 Hydrolytic reaction pathway for oxidative cleavage of methyl oleate 1. | ||
| Reaction | Catalyst | Additive | Solvent | Oxidant | Products | ||
|---|---|---|---|---|---|---|---|
| AA = azelaic acid, MO = methyl oleate, OA = oleic acid, PA = pelargonic acid, NHPI = N-hydroxyphthalimide. | |||||||
| 1 | Epox. of cyclohexene | Na2WO4 + H3PO4 | Quaternary ammonium or phosphonium salts | — | H2O2 | Y cyclohexene oxide = 88% (ref. 11) | Biphasic |
| 2 | Ox. cleavage of MO | H3PW12O40 | Methyl trioctyl ammonium chloride (Aliquat 336) | — | H2O2 | Y AA = 91%, Y PA = 72% (ref. 12) | |
| 3 | Ox. cleavage of OA | H3PW12O40 | Cetyl pyridinium chloride/bromide | — | H2O2 | Y AA/PA = 80% (ref. 2 and 13) | |
| 4 | Dihydroxylation of MO (1), ox. cleavage of diol (2) | (1) H2WO4, (2) Co(Oac)2 | — | — | (1) H2O2, (2) air | Y AA = 80%; Y PA = 77% (ref. 6 and 14) | |
| 5 | Two-step ox. cleavage of OA | H2WO4 | — | — | (1) H2O2, (2) NaOCl | Y AA = 45%, Y PA = 34% (ref. 15) | |
| 6 | Ox. cleavage of MO | H2WO4 | — | — | H2O2 | Y AA = 91%; Y PA = 69% (ref. 16) | |
| 7 | Dihydroxylation of MO | H3PW12O40 | — | — | H2O2 | Y methyl 9,10-dihydroxystearate = 99% (ref. 17) | |
| 8 | Ox. cleavage of MO | Ex situ prepared from H3PW12O40 and quaternary ammonium salt | — | H2O2 | Y AA = 83%, Y PA = 84% (ref. 18) | ||
| 9 | Oxidative cleavage of OA | Methyltrioctylammonium tetrakis(oxodiperoxotungsto)phosphate | — | H2O2 | Y AA = 79%, Y PA = 82% (ref. 19) | ||
| 10 | Ox. cleavage of MO | Dipyridinium-phosphotungstate | — | H2O2 | Y cleavage carboxylic acids = 89% (ref. 20) | ||
| 11 | Ox. cleavage of olive oil | H2WO4 | — | H2O2 | Y AA = 71%; Y PA = 50% (ref. 21) | ||
| 12 | Epox. of OA | Ex situ prepared peroxo phosphotungstate | Aliquat 336 | — | H2O2 | Y epoxide = 88%,22Y epoxide = 91% (ref. 18) | |
| 13 | Ox. cleavage of methyl 9,10-epoxystearate | WO3 + SnO2 | — | H2O2 | Y < 80% (nonanal and methyl 9-oxononanoate)23 | Het. | |
| 14 | Ox. cleavage of MO | WO3 | Na2[Sn(OH)6] (ref. 24) | t-BuOH | H2O2 | Y AA = 89%, Y PA = 65% (ref. 25) | |
| 15 | Epox. of 1-octene | H3PW12O40 | Cetyl pyridinium chloride | CHCl3 | H2O2 | X = 82%; S(cyclooctene oxide) >98% (ref. 26) | Bi. |
| 16 | Epox. of 1-octene | H3PW12O40 | Cetyl pyridinium chloride | t-BuOH | H2O2 | X = 58%; S 1,2-octanediol = 60% (ref. 26) | Hom. |
| 17 | Ox. cleavage of OA | WO3 nanoparticles | — | t-BuOH | H2O2 | Y AA = 23%, Y PA = 38% (ref. 25) | Het. |
| 18 | Ox. cleavage of cyclohexene | H2WO4, 20 mol% | — | t-BuOH | H2O2 | Y adipic acid = 81% (ref. 27) | Homogeneous |
| 19 | Ox. cleavage of MO | H2WO4 | — | t-BuOH | H2O2 + O2 | Y AA = 93%, Y PA = 5% (ref. 12) | |
| 20 | Ox. cleavage of 1-octene | Tris(cetylpyridinium) 12-tungstphosphate | t-BuOH | H2O2 | Y heptanoic acid = 45% (ref. 26) | ||
| 21 | Dihydroxylation of OA/MO (1), autoxidation of diols (2) | (1) H2WO4, (2) Co(acac)3 + NHPI | — | t-BuOH | (1) H2O2, (2) O2 | Y carboxylic acid < 20% (ref. 28) | |
In contrast, under homogeneous reaction conditions, induced through the addition of tert-butanol as semi-polar solvent, the main product from oxidation of 1-octene was found to be 1,2-octanediol (Table 1, entry 16).26 The approach towards epoxidation in a biphasic, W-catalyzed system was successfully extended to oleic acid (Table 1, entry 12)18,22 and also transferred to the oxidative cleavage of several alkenes. In contrast to epoxidation, hydrolysis of the epoxide is desired regarding the pathway of the full oxidative cleavage (Fig. 1, methyl oleate as model substrate). Thus, the reaction solution can be homogenized using tert-butanol (Table 1, entry 18–21).26 However, in terms of green chemistry, the use of either a solvent or a phase transfer agent is sufficient to enhance mass transfer, avoiding the other to reduce the use of auxiliary agents. As outlined by Ishii and Ogawa,27 the use of a solvent is sufficient for oxidative cleavage of cyclohexene, but the catalyst loading was ineconomically high (Table 1, entry 18).
In another homogeneously catalyzed approach using tungstic acid without phase transfer agent but with tert-butanol as solvent for the oxidative cleavage of methyl oleate, 93% azelaic acid but only 5% pelargonic acid were obtained (Table 1, entry 19). In contrast, the best result was achieved using a phase transfer agent in the absence of any solvent (Table 1, entry 2).12 Regarding the stoichiometry of the oxidative cleavage, 1 mol of alkene, and diol respectively, contributes to the formation of 1 mol of carboxylic acids each. As outlined, especially in the absence of a phase transfer agent, the difference in yield of azelaic acid and pelargonic acid in some systems is remarkable (Table 1, entries 5, 6, 11, 17, 19) and to the best of our knowledge has not been further investigated.
To increase selectivity, also two-step approaches have been in focus of research. The first step of dihydroxylation is followed by oxidative cleavage of the diol. Often, different catalysts are used for the two steps and hydrogen peroxide is used for dihydroxylation only (Table 1, entries 4, 5, 21). One of those approaches is implemented industrially, carried out by Matrica (Table 1, entry 4), reaching yields of up to 80%. The implementation of a two-stage process can be advantageous, if intermediates undergo side-reactions under reaction conditions of a tandem process, which is often a compromise of reaction conditions for each single step.
However, such tandem-catalytical procedures, using a W-catalyst and phase transfer agent for oxidative cleavage of oleic acid/methyl oleate were also subject of several patents.12,29,30 A drawback of this technique is the catalyst being dissolved in both phases, resulting in challenging separation from the products also hindering its potential recycling.31
Using a solid, heterogeneous catalyst (Table 1, entries 13, 14, 17) facilitates catalyst separation and recycling. Heterogeneous tungsten oxide nanoparticles were shown efficient for oxidative cleavage of oleic acid in yields of 75% over four recycling runs in tert-butanol.24 Starting from methyl 9,10-epoxystearate, oxidative cleavage towards the aldehydes nonanal and methyl 9-oxononanoate was feasible, allowing for ten recycling runs with yields up to 80% using a catalytic system comprising tungsten oxide and mesoporous SnO2 allowed for ten recycling runs yielding 80% of the aldehydes nonanal (Table 1, entry 13).
Zhang et al. found sodium stannate to be stabilizing hydrogen peroxide, which is relevant due to decomposition of hydrogen peroxide at high temperature and in the presence of transition metals, resulting in a yield of 89% azelaic acid using a heterogeneous tungsten oxide catalyst for oxidative cleavage. However, the yield of pelargonic acid was limited to 65% (Table 1, entry 14), similarly as in biphasic approaches in the absence of a phase transfer agent as aforementioned.25
Recently, and contrary to the findings described above, phosphotungstic acid was applied in a biphasic system for efficient, quantitative dihydroxylation of methyl oleate in the absence of solvent and phase transfer agent (Table 1, entry 7). The authors claim the reaction pathway via direct dihydroxylation, without formation of epoxide as an intermediate. However, full oxidative cleavage was not feasible using this system.17
To the best of our knowledge, no other study addressed W-POM-catalyzed oxidative cleavage of methyl oleate/oleic acid with hydrogen peroxide in a homogeneous reaction system without the use of a phase transfer catalyst or a secondary oxidant. However, as we described above, a biphasic system can limit hydrolysis of the epoxide. Since the formation of diol is a desired intermediate step on the pathway of oxidative cleavage towards mono methyl azelate and pelargonic acid, our interest is to investigate a homogeneous reaction system using a solvent to prevent mass transfer limitations. Thereby, no phase transfer agent will be added. As outlined above, reaching an overall high selectivity seems challenging using tungsten catalysts in oxidative cleavage of methyl oleate and large differences in yields of the two cleavage products are not uncommon. Furthermore, side-reactions resulting in higher molecular weight products are described, for example due to acetalization and esterification, especially in the presence of diols.6,18,32 For a better understanding of side-reactions and to improve selectivity, we also focused on oxidative cleavage of the intermediate methyl 9,10-dihydroxystearate and admit that a two-stage process might be more promising than a tandem reaction in this case. So our first aim is to identify a homogeneous reaction system at which selectivity can be controlled under mild reaction conditions. Thereby, the key towards a more sustainable and economical process would be the subsequent separation of the catalyst to avoid contamination of the product.
A special feature of the W-POM phosphotungstic acid H3PW12O40 is its high molecular weight of 2880 Da, arousing interest in organic solvent nanofiltration (OSN) to reach catalyst separation. OSN membranes allow for catalyst recycling under reaction conditions and are highly energy-efficient compared to different separation techniques like distillation.33–35 In 2006 Chowdhury et al. investigated a mesoporous γ-alumina membrane to separate W-POM from an aqueous and a toluene-based media. In both cases, more than 97% of the POM could be recovered.36 The idea was further developed by Evonik Degussa GmbH, investigating membrane filtration as recovery technique for a catalyst made from sodium tungstate dihydrate and phosphoric acid. Since this catalyst system was proven to be efficient for epoxidation of fatty acid esters, epoxidized fatty acid esters were added to the test system containing catalyst, hydrogen peroxide and a phase transfer agent. After phase separation, both the organic and aqueous phases were investigated towards catalyst recovery via membrane filtration. Depending on the membrane material, close to 100% W were retained from the organic phase and up to 94% from the aqueous phase.31 However, this approach includes a phase separation unit and two following membrane setups, resulting in a more complex separation and higher investment as well as operating costs.
The aim of the present work is first the development of a homogeneous reaction system that still allows for high selectivity towards the cleavage products pelargonic acid and mono methyl azelate. As described above, especially the formation of equimolar amounts of the carboxylic acids seems challenging in a homogeneous system. Additionally, overall selectivity towards cleavage products is also often limited. Since the oxidative cleavage of methyl oleate includes several intermediates, our focus will be on the comparison of a tandem-catalyzed oxidation, starting from methyl oleate, and the direct oxidation of the vicinal diol to gain insight into potential side-reactions and especially ways to avoid them. If we are able to control and improve selectivity, the approach of recovery of phosphotungstic acid via organic solvent nanofiltration seems very promising to develop an efficient protocol for a holistic improvement of W-catalyzed oxidative cleavage towards mono methyl azelate and pelargonic acid.
Results and discussion
Selection of solvent
For our first experiments, we had to choose a semi-polar solvent to homogenize the reaction solution of non-mixable aqueous hydrogen peroxide and organic methyl oleate. We chose tert-butanol, since it had been described in other examples of homogenized reaction systems for oxidative cleavage using W-catalysts.12,24,26,27,37 Furthermore, we chose acetonitrile, since it was described as a suitable co-solvent in the W-catalyzed epoxidation of cyclohexene38 and in the Ru-catalyzed epoxidation of methyl oleate.39,40 The catalyst loading was initially set to 5 mol% since we expect higher conversion at higher catalyst loading. The temperature was set to 100 °C and reactions were performed in glass pressure tubes, allowing for such high temperature without evaporation/reflux of solvent. From the stoichiometry of the oxidative cleavage of methyl oleate, four equivalents of hydrogen peroxide would be necessary for the formation of pelargonic acid and mono methyl azelate. We used an excess of hydrogen peroxide (7 eq. in total) since decomposition of hydrogen peroxide might occur at reaction temperature. The amount of solvent allows for a reaction in a homogeneous solution. The results of our solvent screening are shown in Fig. 2. | ||
| Fig. 2 Selection of solvent for oxidative cleavage of methyl oleate 1. Reaction conditions: methyl oleate 1 (0.1 g, 0.3 mmol), H3PW12O40 (5 mol%), H2O2 (50 wt%, 7 eq.), acetonitrile (0.36 g), 32T = 100 °C, t = 6 h. Conversion (X) and yield (Y) determined via GC-FID-analysis with dibutyl ether as internal standard. | ||
With tert-BuOH, only traces of cleavage products could be obtained. In contrast, with acetonitrile as solvent, yields were much higher and the cleavage products are obtained in an equimolar ratio, considering azelaic acid 8b as the product of ester hydrolysis, which is very promising for further optimization of reaction conditions. Thus, acetonitrile is our solvent of choice for all following investigations.
Catalyst loading
In our initial solvent screening, we applied a relatively high catalyst loading of 5 mol%. Although tungsten is a rather cheap transition metal, a reduction of catalyst loading is desired to save resources and increase catalyst productivity. Both can be further improved when it comes to recycling of the catalyst at a later stage of our study. In contrast, increasing the catalyst loading might result in increased conversion. Hence, we carried out a screening of catalyst loading, including a blind experiment without catalyst (Fig. 3). Thereby, we also prolonged the reaction time to 24 h, to ensure high conversion even at lower catalyst loadings.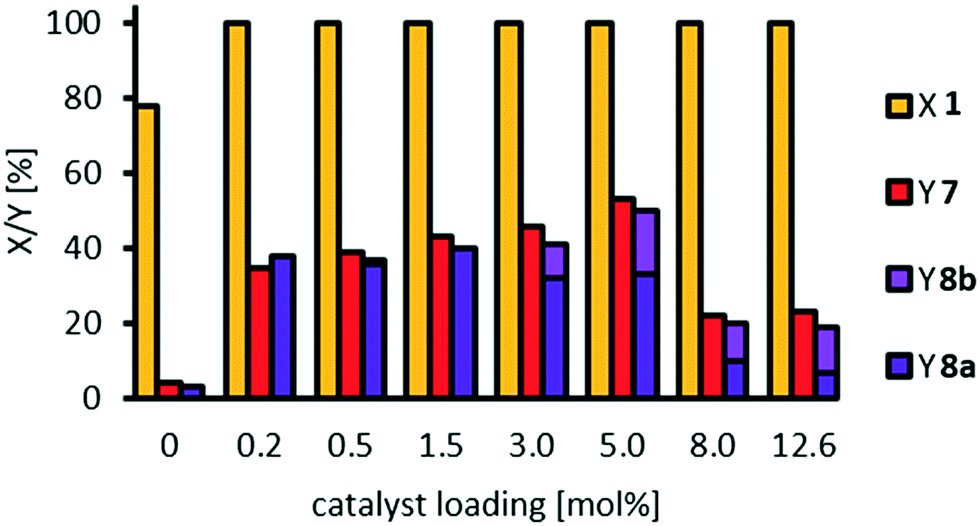 | ||
| Fig. 3 Influence of catalyst loading on oxidative cleavage of methyl oleate 1. Reaction conditions: methyl oleate 1 (0.1 g, 0.3 mmol), H3PW12O40, H2O2 (50 wt%, 7 eq.), acetonitrile (0.36 g), T = 100 °C, t = 24 h. Conversion (X) and yield (Y) determined via GC-FID-analysis with dibutyl ether as internal standard. | ||
Quantitative conversion of methyl oleate 1 is reached at any catalyst loading. Even without catalyst, 80% of 1 are converted, so that we assume that a side-reaction occurs. However, selectivity towards the cleavage products pelargonic acid 7 and mono methyl azelate 8a/azelaic acid 8b is rather low, reaching a maximum of 50% with 5 mol% tungsten catalyst. Azelaic acid 8b is formed only at catalyst loadings of 3 mol% or higher due to hydrolysis of the methyl ester under acidic conditions. Other products are not detected. At catalyst loadings of more than 8 mol%, yields are decreased. Therefore, we assume some side reaction towards higher molecular oligomers (see ESI† for MS-spectra of side-product as indicator). For a better understanding of those, and also to enlighten which intermediates or product are involved in side-reactions, we monitored the reaction over time (Fig. 4) using a low catalyst loading for a rather slow reaction, starting not only from methyl oleate but also from the intermediates.
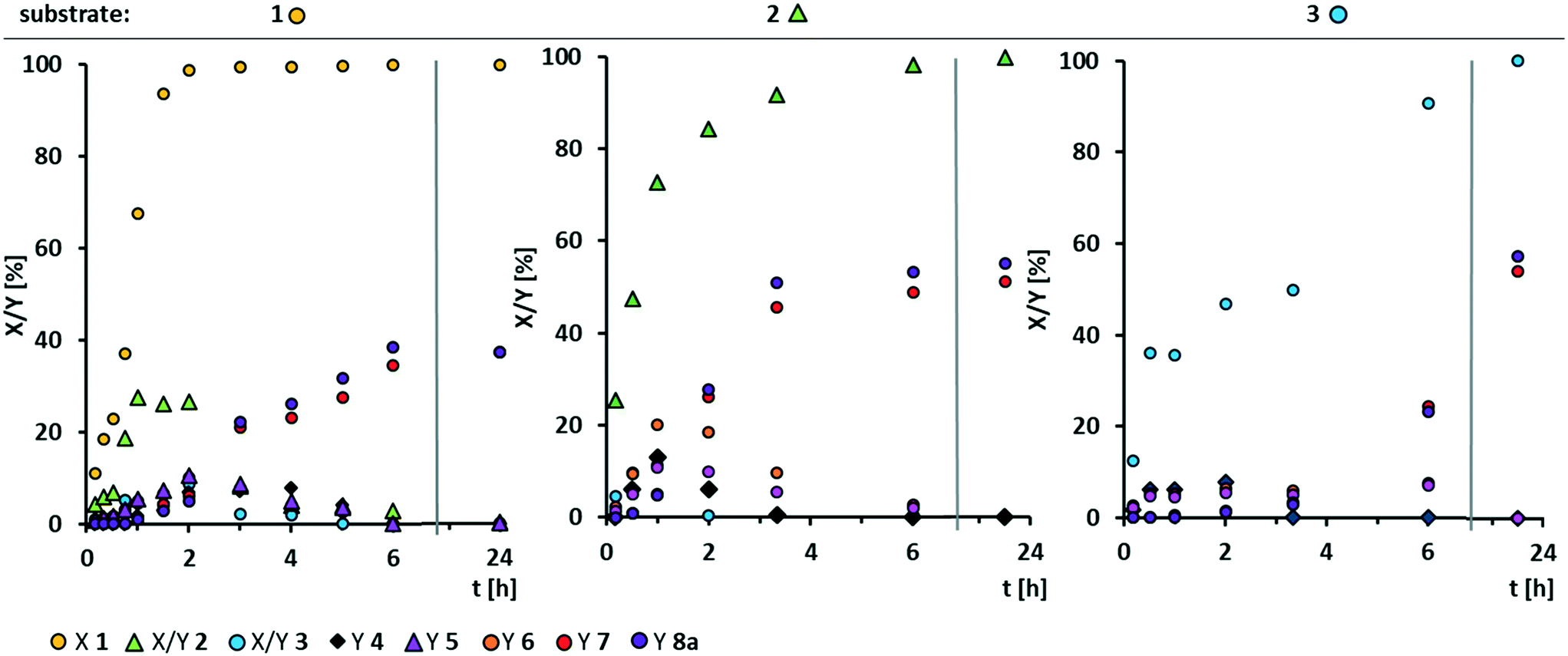 | ||
| Fig. 4 Conversion of substrate and yield of respective products over time in homogeneously W-catalyzed oxidative cleavage starting from methyl oleate 1 (left), methyl 9,10-epoxystearate 2 (middle) and methyl 9,10-dihydroxystearate 3 (right). Reaction conditions: substrate 1 (left)/2 (middle)/3 (right) (0.3 mmol, ∼17 wt%), H3PW12O40 (0.2 mol%), H2O2 (50 wt%, 7 eq.), acetonitrile (0.36 g), T = 100 °C. Conversion (X) and yield (Y) determined via GC-FID-analysis with dibutyl ether as internal standard. | ||
Even at a low catalyst loading of 0.2 mol%, full conversion of methyl oleate 1 is reached after 2 h. However, total selectivity is below 40% during the whole reaction. Methyl 9,10-epoxystearate 2 is an intermediate that leads to the formation of cleavage products. The total reaction time is 6 h. Regarding oxidative cleavage of methyl 9,10-epoxystearate 2, full conversion is not reached within the first 3 h. Thus, the reaction is slower than oxidative cleavage of methyl oleate 1 and selectivity is slightly increased to 50%. Also 9-oxononanoate 5 and nonanal 6 are detected and lead to the formation of carboxylic acids. Surprisingly, neither in oxidative cleavage of methyl oleate 1, nor in oxidative cleavage of the first intermediate, epoxide 2, the diol 3 is obtained in significant amounts. The reason could be either a very fast oxidation of diol 3 to the acyloin 4 or a very fast occurring side-reaction at low concentration of the diol. However, conversion thereof is rather slow as seen from the conversion-time plot of oxidative cleavage of methyl 9,10-dihydroxystearate 3. Therefore, we assume that the diol is involved in the side-reaction that results in non-GC-detectable higher molecular products.
Additionally, selectivity towards carboxylic acids 7 and 8a is very low during the first 6 h. After 24 h, full conversion and a total selectivity of almost 60% is reached. The aldehyde-intermediates 5 and 6 are found only in traces. From the reaction profile, we assume there must be another intermediate which is not GC-detectable but leads to the desired products. However, increasing the reaction time to 48 h does not result in increased yields which may also result from decomposition of hydrogen peroxide. Thus, under the here chosen conditions, the carboxylic acids 7 and 8a are stable (no change in yield between 6 and 24 h), so that we assume that they can be excluded from any side-reaction. Instead, we assume the diol to be involved in a side-reaction.
Since investigation of reaction temperature, H2O2 concentration, H2O2 equivalents and continuous feed of H2O2, concentration of methyl oleate and additional water did not result in the desired optimization of oxidative cleavage of methyl oleate (see ESI,† Fig. S1–S6), we now applied the diol 3 as substrate to gain further insights into a potential side-reaction thereof and to be able to optimize the yield of cleavage carboxylic acids. Thereby, the reaction system is simplified, since the epoxide and methyl oleate, which are not present, cannot take part in any side-reaction.
Oxidative cleavage of methyl 9,10-dihydroxystearate 3
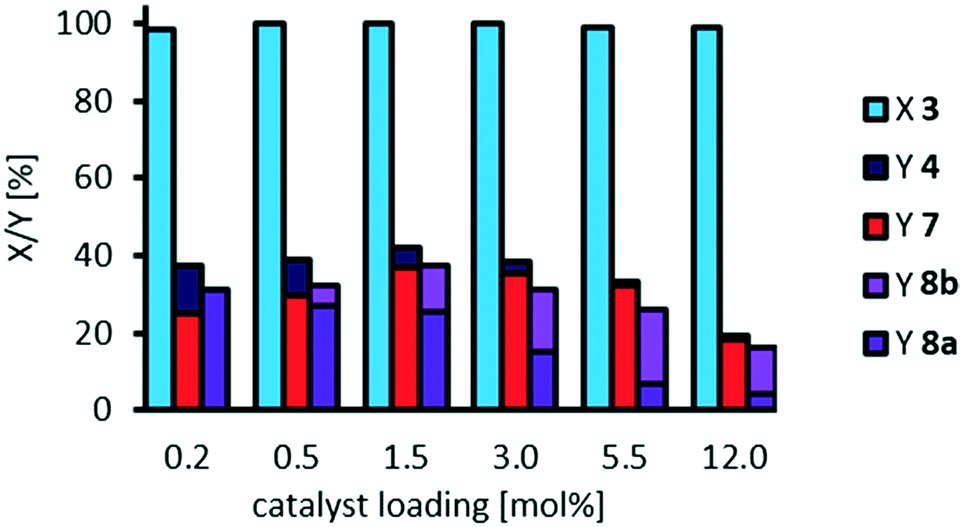 | ||
| Fig. 5 Influence of catalyst loading on oxidative cleavage of methyl 9,10-dihydroxystearate 3. Reaction conditions: 3 (0.1 g, 0.3 mmol, ∼7 wt%), H3PW12O40, H2O2 (35 wt%, 10 eq.), acetonitrile (1.02 g), T = 100 °C, t = 24 h. Conversion (X) and yield (Y) determined via GC-FID-analysis with dibutyl ether as internal standard. | ||
Similar to our study on oxidative cleavage of methyl oleate 1, conversion of methyl 9,10-dihydroxystearate 3 is excellent, whilst selectivity towards the cleavage products pelargonic acid 7 and mono methyl azelate 8a/azelaic acid 8b is limited to 35% and even decreased at a very high catalyst loading of 12 mol%. Additionally, the higher dilution did not result in increased selectivity. The acyloin 4 is first detected as an intermediate especially at low catalyst loading.
Stability of methyl 9,10-dihydroxystearate 3 and cleavage products 7 and 8b
Since phosphotungstic acid is a superacid with a pKa = −6.07 in acetonitrile,41 we assume that undesired side-reactions may occur due to the highly acidic reaction solution. Therefore, we investigated stability of intermediates 2 and 3 and products 7 and 8a in the presence of 5 mol% of the tungsten catalyst and also in the presence of additional sodium hydroxide as a base (Fig. 6). Assuming that 1 mol of methyl 9,10-dihydroxystearate 3 can be oxidized to 1 mol of pelargonic acid 7 and mono methyl azelate 8a each, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 mixture of methyl 9,10-dihydroxystearate 3/pelargonic acid 7 and mono methyl azelate 8a was prepared. To create realistic reaction conditions but still exclude an oxidative reaction, hydrogen peroxide was not applied in these experiments but replaced with an equivalent mass of water. Acetonitrile and phosphotungstic acid were added as in a standard oxidative cleavage reaction.
:1:1 mixture of methyl 9,10-dihydroxystearate 3/pelargonic acid 7 and mono methyl azelate 8a was prepared. To create realistic reaction conditions but still exclude an oxidative reaction, hydrogen peroxide was not applied in these experiments but replaced with an equivalent mass of water. Acetonitrile and phosphotungstic acid were added as in a standard oxidative cleavage reaction.
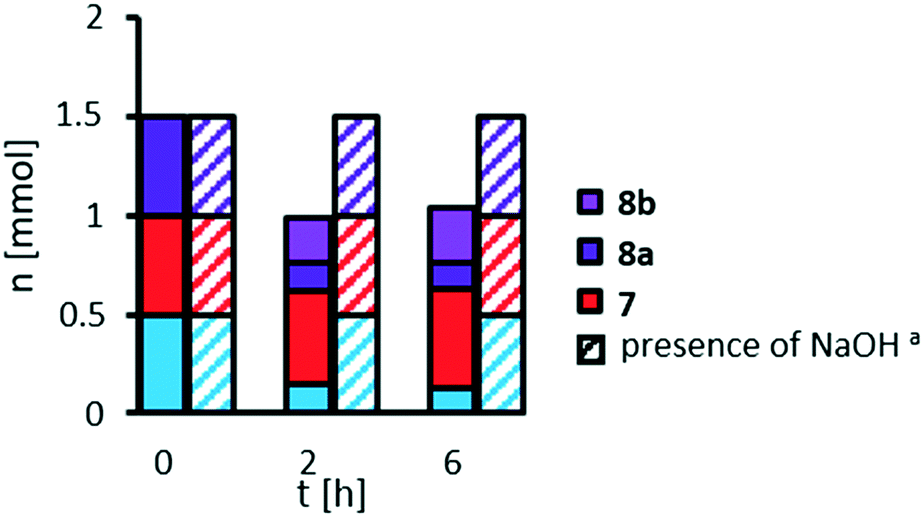 | ||
| Fig. 6 Stability of methyl 9,10-dihydroxystearate 3 and oxidative cleavage products 7 and 8a in absence and presence (striped) of NaOH. Reaction conditions: methyl 9,10-dihydroxystearate 3 (0.17 g, 0.5 mmol), H3PW12O40 (0.144 g; 0.05 mmol), H2O (0.97), acetonitrile (3.14 g), T = 100 °C. aNaOH (2 M, 0.1 g). Molar amounts determined via GC-FID-analysis with dibutyl ether as internal standard. | ||
Whilst pelargonic acid 7 is stable under reaction conditions, mono methyl azelate 8a is hydrolyzed to azelaic acid 8b, as already seen in previous studies on variation of catalyst loading. Furthermore, methyl 9,10-dihydroxystearate 3 is not stable in the absence of a base. We repeated the experiment also with methyl 9,10-epoxystearate 2 instead of the diol (see ESI,† Fig. S9) coming to a similar result: the cleavage products 7 and 8a are stable, methyl 9,10-epoxystearate 2 is not, since it is hydrolyzed towards methyl 9,10-dihydroxystearate 3 which in turn is not stable. However, by adding sodium hydroxide as a base, hydrolysis of the epoxide 2, for which hydrogen peroxide is not required, was completely inhibited and no side-reaction was observed. Under similar conditions, starting from a mixture of methyl oleate 1, pelargonic acid 7 and mono methyl azelate 8a, methyl oleate 1 is selectively hydrolyzed to oleic acid (70% after 6 h), but other side-products are not observed.
Addition of sodium hydroxide as a base
Since the addition of sodium hydroxide results in increased stability of methyl 9,10-dihydroxystearate 3, we also investigated sodium hydroxide as an additive in the oxidative cleavage (Fig. 7). | ||
| Fig. 7 Oxidative cleavage of methyl 9,10-dihydroxystearate 3 depending on molar ratio of NaOH:catalyst. Reaction conditions: methyl 9,10-dihydroxystearate 3 (0.1 g, 0.3 mmol), H3PW12O40 (5 mol%), H2O2 (35 wt%, 10 eq.), acetonitrile (1.02 g), NaOH (2 M), T = 100 °C, t = 24 h. Conversion (X) and yield (Y) determined via GC-FID-analysis with dibutyl ether as internal standard. | ||
Hydrolysis of mono methyl azelate 8a to azelaic acid 8b is significantly reduced from a NaOH:catalyst ratio of 3.5 or higher. Furthermore, selectivity towards cleavage products 7 and 8a is increased up to 77% at a ratio of 6.0 to 7.0. We repeated this study at a lower catalyst loading of 3 mol% and found the same optimal ratio of 7.1 resulting in 80% of the desired carboxylic acids as cleavage products (see ESI,† Fig. S10) and 11% of acyloin 4. At higher NaOH:catalyst ratios, the yield of cleavage products is decreased whilst yield of acyloin 4 is increased. This could also be an effect of hydrogen peroxide decomposition, which is faster at a higher pH and is also in accordance with our findings on decomposition depending on catalyst loading (see ESI,† Fig. S12). Further investigation on catalyst loading will be discussed later on. In addition, increasing the concentration of NaOH also results in increasing the pH, so that decomposition of the catalyst must be considered, as the pH dependent stability of Keggin-type heteropolyacids is well known.42 Anionic structures such as [P2W21O71]6−, [PW11O39]7−, [P2W18O62]7− are described at a pH of 5.4, whilst the structure [PW12O40]3− is only described at a pH of 1.43 In our optimized system, the pH is increased to around 5, so that we assume the catalyst species differs in its structure. Thus, further increase in pH might result in the formation of inactive catalyst species.
Decomposition of the catalyst must also be considered regarding a potential separation of the catalyst via organic solvent nanofiltration (OSN). Retention of the catalyst strongly depends on its molecular size. At a pH > 8.3, phosphotungstic acid completely decomposes to PO43− and WO42−.43 However, for a total neutralization of the acid, 27 eq. of base are required.41 At such high pH, we also assume a very fast decomposition of hydrogen peroxide. At a pH below 7.3, decomposition may occur but the catalyst still has a very high molecular weight,43 so that retention of the catalyst via organic solvent nanofiltration still offers potential.
Since tungsten is a suitable transition metal for oxidative cleavage, but phosphotungstic acid seems to be too acidic to inhibit side-reactions, we also applied tungstic acid (Fig. 8), which has a pKa of 3.8.
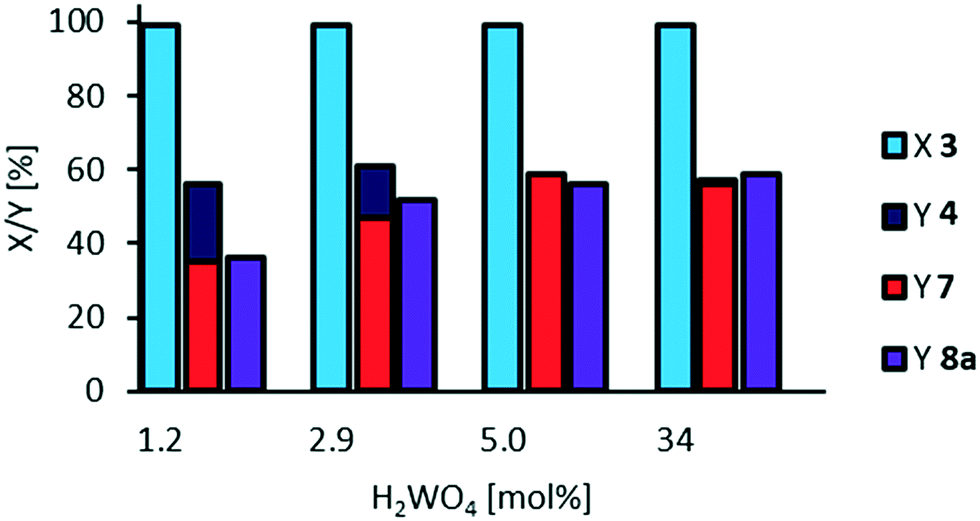 | ||
| Fig. 8 Oxidative cleavage of methyl 9,10-dihydroxystearate 3 with tungstic acid. Reaction conditions: methyl 9,10-dihydroxystearate 3 (0.1 g, 0.3 mmol), H2WO4, H2O2 (35 wt%, 10 eq.), acetonitrile (1.02 g), T = 100 °C, t = 24 h. Conversion (X) and yield (Y) determined via GC-FID-analysis with dibutyl ether as internal standard. | ||
As expected, hydrolysis of mono methyl azelate 8a does not occur even at a very high catalyst loading. However, selectivity towards cleavage products 7 and 8a and acyloin 4 is still limited to nearly 60%. Additionally, tungstic acid has a molar mass of 250 Da, whilst phosphotungstic acid has a molar mass of 2880 Da. Therefore, considering membrane separation to recover the catalyst, we decided to continue using phosphotungstic acid as the catalyst. Interestingly, Na2WO4 exhibits only poor catalytic activity (see ESI,† Fig. S12).
Dilution/concentration of substrate
With our optimized NaOH:catalyst ratio of 7:1 we also investigated the effect of dilution at a catalyst loading of 3 mol% (resulting in the highest yield of cleavage carboxylic acids of 80%), since one could assume oligomerization to be inhibited at higher dilution (Fig. 9).
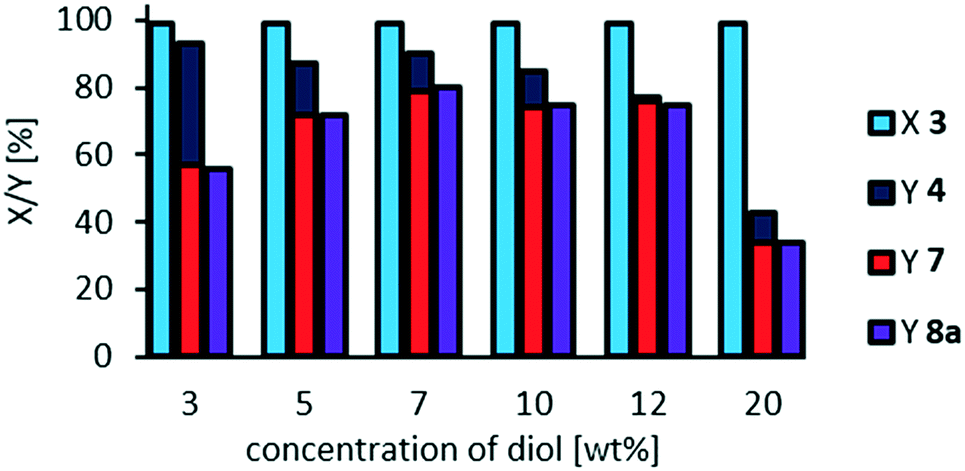 | ||
| Fig. 9 Effect of dilution on side-reactions and oxidative cleavage of methyl 9,10-dihydroxystearate 3. Reaction conditions: methyl 9,10-dihydroxystearate 3 (0.1 g, 0.3 mmol), H3PW12O40 (3 mol%), H2O2 (35 wt%, 10 eq.), acetonitrile (0.04–2.5 g), NaOH (2 M, 0.031 g), T = 100 °C, t = 24 h. Conversion (X) and yield (Y) determined via GC-FID-analysis with dibutyl ether as internal standard. | ||
Since an overall selectivity of 92% is obtained at a higher dilution, and lower concentration of the substrate respectively, we assume that side-reactions such as a potential oligomerization, can be inhibited. However, selectivity towards the desired carboxylic acids 7 and 8a is limited to 60%, which could be due to faster decomposition of hydrogen peroxide compared to oxidative cleavage at this concentration.
The optimum substrate concentration is found to be 7 wt%, reaching an overall selectivity of 91% at a selectivity towards carboxylic acids of 80%. Higher substrate concentrations result in a decreased selectivity, especially at a concentration of 20 mol%, at which the reaction system is not homogeneous: at reaction temperature, a yellowish upper phase and a clear lower phase are present. Therefore, following reactions are carried out at a concentration of methyl 9,10-dihydroxystearate 3 of 7 wt%, as also previously used.
Amount of oxidant
For oxidative cleavage of methyl 9,10-dihydroxystearate 3 a stoichiometric amount of three equivalents of hydrogen peroxide would be of need. So far, we used 10 equivalents. Finally, we investigated a potential reduction of the amount of oxidant (Fig. 10).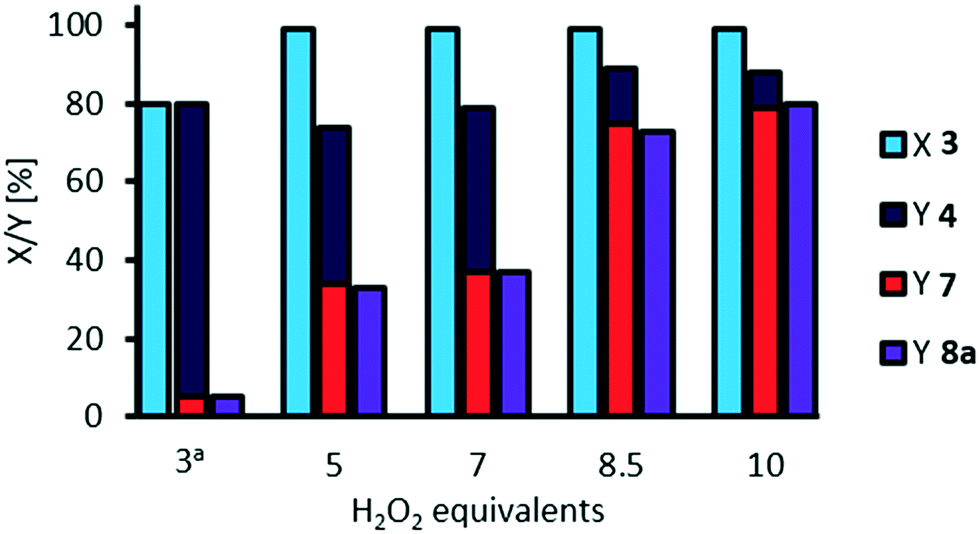 | ||
| Fig. 10 Influence of H2O2 equivalents on oxidative cleavage of methyl 9,10-dihydroxystearate 3. Reaction conditions: methyl 9,10-dihydroxystearate 3 (0.1 g, 0.3 mmol), H3PW12O40 (3 mol%), H2O2 (35 wt%), acetonitrile (1.02 g), NaOH (2 M, 0.031 g), T = 100 °C, t = 24 h; at = 3 h; H2O (0.06 g). Conversion (X) and yield (Y) determined via GC-FID-analysis with dibutyl ether as internal standard. | ||
The equivalents of H2O2 can be reduced to 8.5 without significant loss in yield of cleavage carboxylic acids. However, further reduction to 7 or 5 equivalents results in a decreased yield of 35% of carboxylic acids 7 and 8a and 40% of the acyloin 4, indicating that decomposition of H2O2 is too fast so that the oxidant is not present for further oxidation. In those experiments, a solid was observed during the reaction which we assumed is sodium hydroxide, since the amount of water is reduced as the amount of aqueous hydrogen peroxide is reduced. We repeated the experiments adding water to “refill” to an amount of water that is present in the experiments using 10 equivalents of hydrogen peroxide (see ESI,† Fig. S13). Thus, the solid was solved and the yield of cleavage products 7 and 8a was increased to 60% using 7 equivalents of oxidant. However, using 5 equivalents of oxidant, the presence of water did not result in any improvement, since competing decomposition of hydrogen peroxide is faster than oxidative cleavage. Interestingly, at 3 equivalents hydrogen peroxide, a shorter reaction time of 3 h and in the presence of additional water to solve NaOH, the acyloin 4 is obtained with a selectivity of 94%. From this procedure, the acyloin 4 was also isolated as a yellow oil (see ESI†).
Reaction sequence of oxidative cleavage of methyl 9,10-dihydroxystearate 3
In all our experiments on oxidative cleavage of methyl 9,10-dihydroxystearate 3, the corresponding aldehyde intermediates were only detected in traces of less than 2%. To clarify how these are involved in the reaction sequence, we monitored oxidative cleavage under our optimized reaction conditions (Fig. 11). | ||
| Fig. 11 Yield of respective products over time in oxidative cleavage of methyl 9,10-dihydroxystearate 3. Reaction conditions: methyl 9,10-dihydroxystearate 3 (0.1 g, 0.3 mmol), H3PW12O40 (3 mol%), H2O2 (35 wt%, 10 eq.), acetonitrile (1.02 g), NaOH (2 M, 0.031 g), T = 100 °C. Yield (Y) determined via GC-FID-analysis with dibutyl ether as internal standard. | ||
Methyl 9,10-dihydroxystearate 3 is converted very fast. The acyloin 4 is formed as an intermediate with a maximum yield of 37% after 30 min. The aldehydes 9-oxononanoate 5 and nonanal 6 are detected only in traces of less than 2% during the reaction, indicating that oxidation of aldehydes under those conditions is very fast. Thus, we also investigated oxidation of nonanal 6 (0.6 mmol; 5 eq. H2O2), resulting in 83% yield of pelargonic acid 7 after 30 min. In contrast, oxidation of the acyloin 4 is rate-determining. The reaction sequence of oxidative cleavage of methyl 9,10-dihydroxystearate 3via the acyloin 4 towards an aldehyde and acid, and the latter being further oxidized, is also in accordance with the literature-known reaction sequence of oxidative cleavage of shorter chain vicinal diols in the absence of a solvent using phosphotungstic acid44 or tungstic acid.27
Since the acyloin 4 is an intermediate, we also investigated oxidative cleavage of the acyloin 10-hydroxy-9-octadecanone resulting in a conversion of 58% after 70 min at excellent selectivity towards pelargonic acid 7, and 67% after 2 h respectively, which also correlates to the reaction profile of oxidative cleavage of methyl 9,10-dihydroxystearate 3.
Transfer to cleavage of methyl oleate 1
After optimization of oxidative cleavage of methyl 9,10-dihydroxystearate 3, we transferred the optimized conditions to oxidative cleavage of methyl oleate 1 (Fig. 12).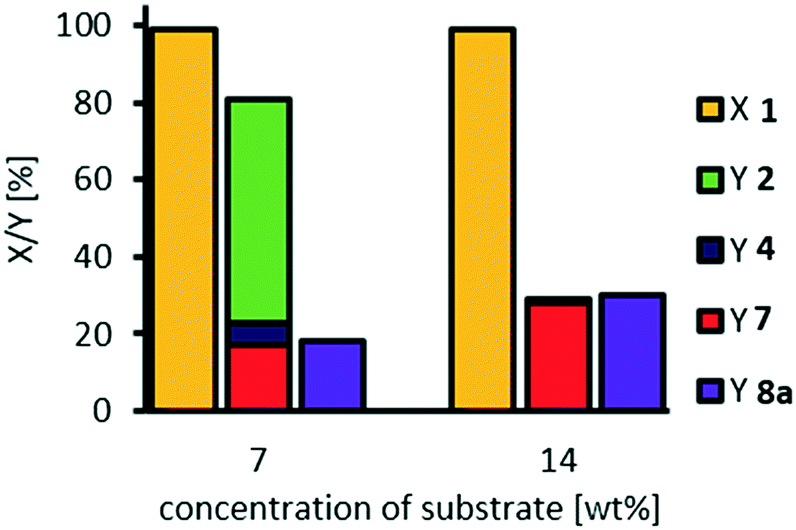 | ||
| Fig. 12 Transfer of optimized conditions to oxidative cleavage of methyl oleate 1. Reaction conditions: methyl oleate 1 (0.1 g, 0.3 mmol), H3PW12O40, H2O2 (35 wt%, 10 eq.), acetonitrile (0.25 g/1.02 g), NaOH (2 M, NaOH: H3PW12O40 7:1), T = 100 °C. Conversion (X) and yield (Y) determined via GC-FID-analysis with dibutyl ether as internal standard. | ||
In accordance with our study on stability of methyl 9,10-epoxystearate 2, hydrolysis of the epoxide 2 is inhibited under our optimized reaction conditions (7 wt% of substrate and catalyst loadings of 3 mol%). Consequently, the diol 3 is not detected in those experiments. However, since we detect traces of the acyloin 4, at least traces of diol 3 must have been formed, resulting in the desired formation of acyloin 4 and in an undesired side-reaction towards oligomers. Thus, we have shown that direct oxidative cleavage of methyl 9,10-dihydroxystearate 3 is superior compared with a tandem oxidative cleavage of methyl oleate 1 under these conditions. We also carried out this experiment at lower dilution, and concentration of methyl oleate 1 of 14 wt% respectively, which is similar to the concentration we used in our very first experiments. Interestingly, the yield of cleavage products 7 and 8a is significantly increased, while the overall selectivity decreases, proving that the reaction system is very complex and sensitive to minor changes.
Catalyst loading at NaOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :catalyst ratio of 7:1 in the oxidative cleavage of methyl 9,10-dihydroxystearate 3
:catalyst ratio of 7:1 in the oxidative cleavage of methyl 9,10-dihydroxystearate 3
Finally, with our optimized reaction conditions and a NaOH:catalyst ratio of 7:1 we now again investigated the influence of catalyst loading on oxidative cleavage of methyl 9,10-dihydroxystearate 3 (Fig. 13). Although phosphotungstic acid is relatively cheap, a lower catalyst loading is more economic and the productivity, measured as turnover number, can be significantly increased, when decreasing the catalyst loading, as long as conversion is kept constant.
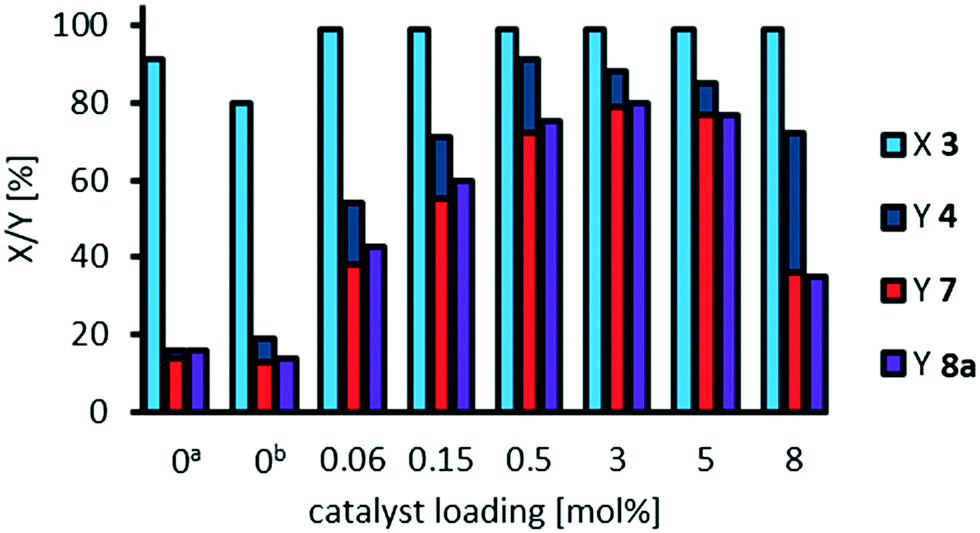 | ||
| Fig. 13 Effect of catalyst loading on oxidative cleavage of methyl 9,10-dihydroxystearate 3 at optimized NaOH:H3PW12O40 ratio of 7:1. Reaction conditions: methyl 9,10-dihydroxystearate 3 (0.1 g, 0.3 mmol), H3PW12O40, H2O2 (35 wt%, 10 eq.), acetonitrile (1.02 g), NaOH (2 M, NaOH:H3PW12O40 7:1), T = 100 °C, t = 24 h; ano NaOH, bNaOH (2 M, 0.007 g, 23 mol%); conversion (X) and yield (Y) determined via GC-FID-analysis with dibutyl ether as internal standard. | ||
By reducing the catalyst loading to 0.5 mol%, we could still obtain yields of cleavage carboxylic acids 7 and 8a of 65%, while overall selectivity is 95%. Thereby, the turnover number (regarding the cleavage products) is increased nearly 5-fold from 27 to 130. By further decrease in catalyst loading to 0.15 mol%, and 0.06 mol% respectively, the TON is increased up to 387, and 633 respectively, regarding cleavage carboxylic acids. For comparison, we calculated a TON of 180 in the oxidative cleavage of methyl oleate using the catalytic system comprising of an amphiphilic dipyridinium-phosphotungstate described by Moores et al. (Table 1, entry 10)20 and a TON of 11 regarding azelaic acid in the homogeneously catalyzed oxidative cleavage of methyl oleate (Table 1, entry 19).12 Regarding oxidative cleavage of methyl 9,10-epoxystearate as an intermediate, a TON of 47 is described towards the aldehydes nonanal and methyl 9-oxononanoate (Table 1, entry 13).23 Thus, we herein report a very high TON in the homogeneously W-catalyzed oxidative cleavage of methyl 9,10-dihydroxystearate, resulting in a great potential for catalyst recycling.
At a catalyst loading of 0.5 mol%, we investigated if the catalyst would be maintained active after a reaction to be potentially recycled after separation. Upon the replenishment of methyl 9,10-dihydroxystearate 3 and hydrogen peroxide after 24 h, 60% yield of cleavage products were obtained after another 24 h, showing that the catalyst still exhibits catalytic activity. However, the composition of reaction solution is slightly changed (due to the presence of products). Thus, we conclude that separation of the catalyst is relevant not only in terms of obtaining a pure product, but also to eventually recycle the homogeneous catalyst.
Screening of suitable membranes for OSN separation
After successful optimization of the oxidative cleavage reaction, we focused on the investigation of catalyst separation via organic solvent nanofiltration. The main goal was to identify a membrane rejecting more than 90% of the catalyst in a single-stage separation. Furthermore, the role of water has to be considered, since its formation results from the application of aqueous hydrogen peroxide as the oxidant, as a byproduct from oxidation and as a sideproduct from decomposition of excess hydrogen peroxide. Hence, a suitable membrane would be more selective towards water and retain the organic solvent to prevent water accumulation during a potential recycling of the retentate. Therefore, two commercially available membranes were screened (Evonik DuraMem 500 and AMS Nanopro S-3012) in a miniplant setup (see ESI,† Fig. S16 and S17). Thereby, a model feed was applied containing acetonitrile as an organic solvent, water, assuming complete decomposition/conversion of hydrogen peroxide and the catalyst complex in a stable composition. Catalyst concentration was set to 0.5 wt%, corresponding to a catalyst loading of 0.5 mol% under optimized conditions. The results for the retention are presented in Fig. 14. For the retention of the catalyst, AMS Nanopro S-3012 performed best with an average retention of 94.6% for tungsten and 94.0% for phosphorous compared to Evonik DuraMem 500 with an average retention of only 68.1% for tungsten and 70.8% for phosphorous. Furthermore, the ratio between tungsten and phosphor remained nearly constant for both membranes throughout the samples. Therefore, it can be concluded that decomposition of the catalyst complex was no issue.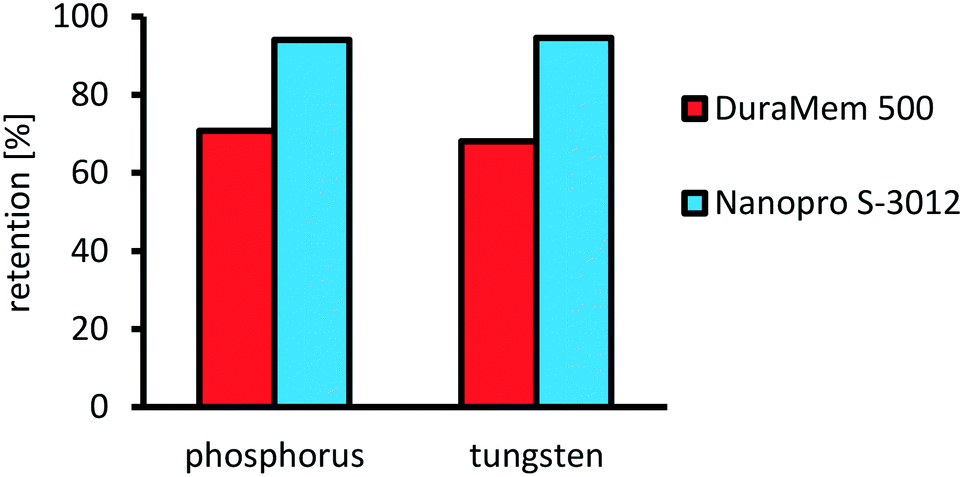 | ||
Fig. 14 Retention for DuraMem 500 and Nanopro S-3012. Reaction conditions: H3PW12O40 (0.5 wt%, 1.32 g), acetonitrile (300 g), water (90 g), T = 40 °C, Δp = 40 bar, ![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) feed = 25 ml min−1, fpump = 25 Hz, nstirrer = 200 min−1; retention determined via ICP-OES. feed = 25 ml min−1, fpump = 25 Hz, nstirrer = 200 min−1; retention determined via ICP-OES. | ||
Comparing the fluxes (Fig. 15), the membrane DuraMem 500 outperforms the membrane Nanopro S-3012 with an average flux after conditioning of 0.6 vs. 0.4 g cm−2 h−1. Since flux determines the membrane area in a continuously operated process, DuraMem 500 would be in favor here.
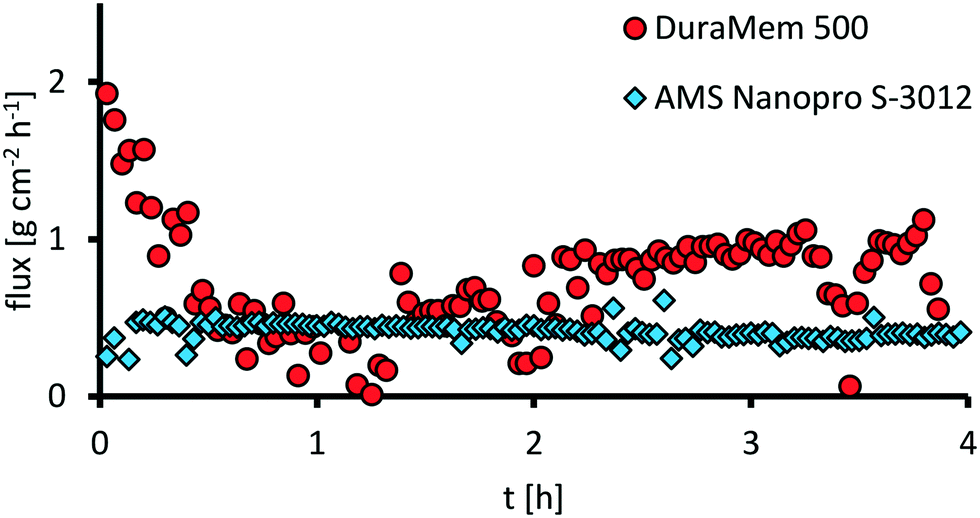 | ||
| Fig. 15 Flux over time for Evonik DuraMem 500 and AMS Nanopro S-3012 after 4 h of conditioning. Reaction conditions: H3PW12O40 (0.5 wt%, 1.32 g), acetonitrile (300 g), water (90 g), T = 40 °C, Δp = 40 bar, feed = 25 ml min−1, fpump = 25 Hz, nstirrer = 200 min−1; mass flow determined via mass flow meter. | ||
Given the low retentions of the Evonik DuraMem 500, AMS Nanopro S-3012 is chosen for further investigation, though.
Change of flux and water content over time with AMS Nanopro S-3012
In a following long-time experiment, the permeate was discharged (see ESI,† section 6), and therefore the feed was further concentrated. The water content in the permeate and retentate was measured over a time of 45 h. AMS Nanopro S-3012 is selective towards the byproduct water, which permeates preferably out of the system. Since the membrane area in the experiment was limited to 14 cm2, only a water content of 14.3% in the retentate was reached after 45 h (Fig. 16). For a continuous process, larger membrane areas would be required.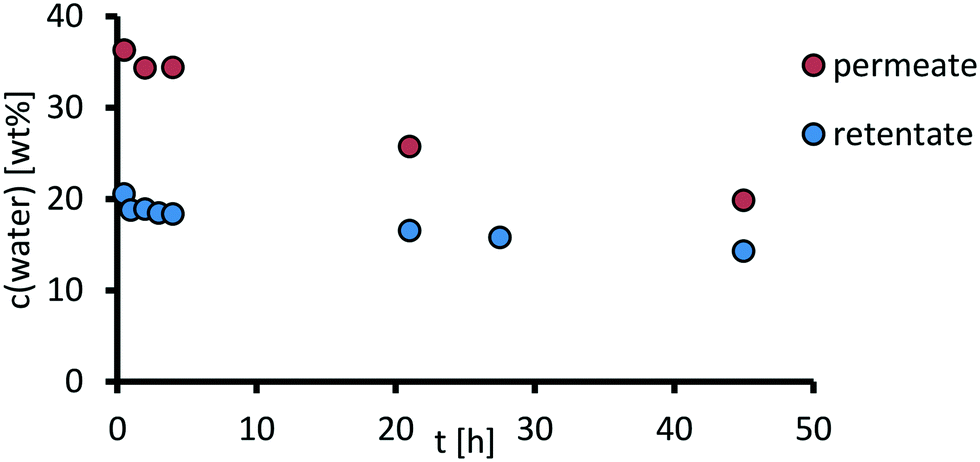 | ||
| Fig. 16 Decrease of water content in the retentate in a long time experiment with the AMS Nanopro S-3012. Reaction conditions: H3PW12O40 (0,5 wt%, 1,29 g), acetonitrile (303 g), water (90 g), T = 40 °C, Δp = 40 bar, feed = 25 ml min−1, fpump = 25 Hz, nstirrer = 200 min−1; composition determined via GC-TCD-analysis with methanol as internal standard. | ||
Notably, the retention of the catalyst complex after 45 h was still around 85% with a slightly decreasing flux of 0.19 g cm−2 h−1 which is expected due to less water in the feed, demonstrating the stability of the separation system as the first proof of concept for the catalyst recycling in a homogenously catalyzed monophasic oxidative cleavage reaction. Further studies should focus on combining a water-discharging membrane and a membrane that allows products to permeate.
Conclusion
We have shown that homogeneously W-catalyzed oxidative cleavage towards pelargonic acid and mono methyl azelate is most promising if carried out starting from methyl 9,10-dihydroxystearate instead of forcing a tandem-catalytic reaction from methyl oleate. This is due to the contradiction between a low pH, that is necessary for hydrolysis of methyl 9,10-epoxystearate towards methyl 9,10-dihydroxystearate and a high pH, that is necessary to prevent side-reactions and thus allow for oxidative cleavage. To the best of our knowledge, the described reaction system allows for the highest reported overall selectivity (90%) and selectivity towards cleavage carboxylic acids (80%) using an affordable, homogeneous W-catalyst at a moderate catalyst loading of 3 mol%. The addition of sodium hydroxide helps to inhibit the aforementioned acid-catalyzed side-reactions, allowing for oxidation towards the acyloin and finally oxidative cleavage thereof. Since oxidative cleavage of methyl oleate is via epoxidation and hydrolysis of the epoxide, the cleavage products cannot be obtained quantitatively since hydrolysis is inhibited in the presence of a base and epoxidation occurs as the main reaction. However, other methods are superior for selective epoxidation. To achieve these results, a compromise had to be found between acidity and dilution, which affects the decomposition of hydrogen peroxide and side-reactions of the vicinal diol. Lowering the catalyst loading to 0.5 mol% does not affect overall selectivity, but catalyst productivity towards cleavage products is increased 5-fold. By further decrease in catalyst loading, we were able to obtain a very high catalyst productivity in the homogeneously W-catalyzed oxidative cleavage of methyl 9,10-dihydroxystearate, reaching a TON of 633 regarding carboxylic acids. Additionally, the catalyst is remained active after the reaction, offering the potential for catalyst recycling in future works. Thereby, organic solvent nanofiltration seems very promising. We were able to retain above 94% of the catalyst from a model mixture of solvent/water using an AMS Nanopro S-3012 membrane as only one stage of separation, which is very promising to obtain a pure product. In the same step, the membrane was able to let water, resulting from the application of aqueous hydrogen peroxide and as a byproduct, permeate more selectively. This offers the prospect of the development of a holistic recycling approach. Therefore, the homogeneously, phosphotungstic acid catalyzed synthesis route towards pelargonic acid and mono methyl azelate is an efficient and sustainable reaction system with the potential to outperform competing reaction systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are very thankful to the German Federal Ministry of Food and Agriculture (Bundesministerium für Ernährung und Landwirtschaft), represented by the FNR (Fachagentur Nachwachsende Rohstoffe) for financial support of the young research group “Renewlysis” (project number 2219NR355). J. V. is thankful for a Ph.D. scholarship from Fonds der Chemischen Industrie. M. P. thanks the networking program ‘Sustainable Chemical Synthesis 2.0’ (SusChemSys 2.0) for the support and fruitful discussions across disciplines. We also thank Prof. Vogt for critical questioning and helpful discussion and Iris Henkel for ICP-OES measurements.References
- R. G. Ackman, M. E. Retson, L. R. Gallay and F. A. Vandenheuvel, Can. J. Chem., 1961, 39, 1956 CrossRef CAS.
- A. Godard, P. de Caro, S. Thiebaud-Roux, E. Vedrenne and Z. Mouloungui, J. Am. Oil Chem. Soc., 2013, 90, 133 CrossRef CAS.
- D. J. Anneken, S. Both, R. Christoph, G. Fieg, U. Steinberner and A. Westfechtel, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000, p. 102 Search PubMed.
- M. Kilian and C. Marienhagen, (Bayer Cropscience AG), WO2015/004086A1, 2015.
- H. Noureddini and M. I. Rempe, J. Am. Oil Chem. Soc., 1996, 73, 939 CrossRef CAS.
- A. Soutelo-Maria, J.-L. Dubois, J.-L. Couturier and G. Cravotto, Catalysts, 2018, 8, 464 CrossRef.
- M. D. Lundin, A. M. Danby, G. R. Akien, P. Venkitasubramanian, K. J. Martin, D. H. Busch and B. Subramaniam, AIChE J., 2017, 63, 2819–2826 CrossRef CAS.
- A. Enferadi Kerenkan, F. Béland and T.-O. Do, Catal. Sci. Technol., 2016, 6, 971 RSC.
- A. Goti and F. Cardona, in Green Chemical Reactions, ed. P. Tundo and V. Esposito, Springer, Dordrecht, London, 2008, pp. 191–212 Search PubMed.
- P. Spannring, P. C. A. Bruijnincx, B. M. Weckhuysen and R. J. M. Klein Gebbink, Catal. Sci. Technol., 2014, 4, 2182 RSC.
- C. Venturello, E. Alneri and M. Ricci, J. Org. Chem., 1983, 48, 3831 CrossRef CAS.
- U. Razdan, M. Shaiwale and G. Padmanaban, (Solvay SA), WO2018/104208A1, 2018.
- Z. P. Pai, A. G. Tolstikov, P. V. Berdnikova, G. N. Kustova, T. B. Khlebnikova, N. V. Selivanova, A. B. Shangina and V. G. Kostrovskii, Russ. Chem. Bull., 2005, 54, 1847 CrossRef CAS.
- A. Bieser, G. Borsotti, F. Digioia, A. Ferrari and A. Pirocco, (Novamont S.P.A), WO2011/080297A1, 2011.
- Z. Masyithah and A. Ginting, Orient. J. Chem., 2018, 34, 1249 CAS.
- V. Benessere, M. E. Cucciolito, A. de Santis, M. Di Serio, R. Esposito, F. Ruffo and R. Turco, J. Am. Oil Chem. Soc., 2015, 92, 1701 CrossRef CAS.
- N. Araji, G. Chatel, A. Moores, F. Jérôme and K. de Oliveira Vigier, New J. Chem., 2020, 44, 11507 RSC.
- T. B. Khlebnikova, Z. P. Pai, L. A. Fedoseeva and Y. V. Mattsat, React. Kinet. Catal. Lett., 2009, 98, 9 CrossRef CAS.
- E. Antonelli, R. D'Aloisio, M. Gambaro, T. Fiorani and C. Venturello, J. Org. Chem., 1998, 63, 7190 CrossRef CAS PubMed.
- L. C. de La Garza, K. de Oliveira Vigier, G. Chatel and A. Moores, Green Chem., 2017, 19, 2855 RSC.
- M. Melchiorre, V. Benessere, M. E. Cucciolito, C. Melchiorre, F. Ruffo and R. Esposito, ChemistrySelect, 2020, 5, 1396 CrossRef CAS.
- I. V. Kozhevnikov, G. P. Mulder, M. C. Steverink-de Zoete and M. G. Oostwal, J. Mol. Catal. A: Chem., 1998, 134, 223 CrossRef CAS.
- M. Lu, L. Peng, Q. Xie, N. Yang, H. Jin, Z. Wu, Y. Nie, X. Liu, X. Lu and J. Ji, Green Chem., 2019, 21, 560 RSC.
- A. Enferadi-Kerenkan, A. Gandon and T. O. Do, Dalton Trans., 2018, 47, 1214 RSC.
- X. Li, J. C. P. Syong and Y. Zhang, Green Chem., 2018, 20, 3619 RSC.
- Y. Ishii, K. Yamawaki, T. Ura, H. Yamada, T. Yoshida and M. Ogawa, J. Org. Chem., 1988, 53, 3587 CrossRef CAS.
- T. Oguchi, T. Ura, Y. Ishii and M. Ogawa, Chem. Lett., 1989, 857 CrossRef CAS.
- M. A. Oakley, S. Woodward, K. Coupland, D. Parker and C. Temple-Heald, J. Mol. Catal. A: Chem., 1999, 150, 105 CrossRef CAS.
- Y. Kon, K. Sato, H. Yazawa, C. Ohto and Y. Okamura, (Toyota Jidosha Kabushiki Kaisha), US2010/210873A1, 2010.
- R. Kadyrov, (Evonik Industries AG), WO2015/036342A1, 2010.
- H. Wiederhold, M. Grass, B. Woldt, F. S. Boeck, M. Priske, A. Boam and P. Kreis, (Evonik Degussa GmbH), WO2015/177009A2, 2015.
- A. Köckritz, M. Blumenstein and A. Martin, Eur. J. Lipid Sci. Technol., 2010, 112, 58 CrossRef.
- P. Marchetti, M. F. Jimenez Solomon, G. Szekely and A. G. Livingston, Chem. Rev., 2014, 114, 10735 CrossRef CAS PubMed.
- M. Priske, M. Lazar, C. Schnitzer and G. Baumgarten, Chem. Ing. Tech., 2016, 88, 39 CrossRef CAS.
- P. Vandezande, L. E. M. Gevers and I. F. J. Vankelecom, Chem. Soc. Rev., 2008, 37, 365 RSC.
- S. R. Chowdhury, P. T. Witte, D. H. A. Blank, P. L. Alsters and J. E. ten Elshof, Chem. – Eur. J., 2006, 12, 3061 CrossRef CAS PubMed.
- A. Enferadi-Kerenkan, A. S. Ello and T.-O. Do, Ind. Eng. Chem. Res., 2017, 56, 10639 CrossRef CAS.
- T. R. Amarante, M. M. Antunes, A. A. Valente, F. A. A. Paz, M. Pillinger and I. S. Gonçalves, Inorg. Chem., 2015, 9690 CrossRef CAS PubMed.
- A. Behr, N. Tenhumberg and A. Wintzer, Eur. J. Lipid Sci. Technol., 2012, 114, 905 CrossRef CAS.
- J. Vondran, J. Pela, D. Palczewski, M. Skiborowski and T. Seidensticker, ACS Sustainable Chem. Eng., 2021, 9, 11469 CrossRef CAS.
- K. M. Reddy, N. Lingaiah, P. S. Sai Prasad and I. Suryanarayana, J. Solution Chem., 2006, 35, 407 CrossRef CAS.
- A. Kondinski and T. N. Parac-Vogt, Front. Chem., 2018, 6, 346 CrossRef PubMed.
- Z. Zhu, R. Tain and C. Rhodes, Can. J. Chem., 2003, 81, 1044 CrossRef CAS.
- M. R. C. Venturello, J. Org. Chem., 1986, 51, 1599 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1cy02317a |
| This journal is © The Royal Society of Chemistry 2022 |