
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the chemoselectivity of the Pd-catalysed hydrogenation of pyridinecarbonitriles: an efficient and simple method for preparing pyridyl- or piperidylmethylamines
Krisztina
Lévay
a,
János
Madarász
b and
László
Hegedűs
 *a
*a
aDepartment of Organic Chemistry and Technology, Faculty of Chemical Technology and Biotechnology, Budapest University of Technology and Economics, Műegyetem rkp. 3, H-1111 Budapest, Hungary. E-mail: hegedus.laszlo@vbk.bme.hu
bDepartment of Inorganic and Analytical Chemistry, Faculty of Chemical Technology and Biotechnology, Budapest University of Technology and Economics, Műegyetem rkp. 3, H-1111 Budapest, Hungary
First published on 7th March 2022
Abstract
An effective method for the chemoselective, liquid-phase heterogeneous catalytic hydrogenation of some pyridinecarbonitriles [4-, 3- or 2-pyridinecarbonitrile (4PN, 3PN, 2PN)] to the corresponding pyridyl- or piperidylmethylamines over a Pd/C catalyst was developed. Using our process, not only can an adequate primary amine selectivity to the desired pyridine derivatives [4-, 3- or 2-(aminomethyl)pyridine (4PA, 3PA, 2PA)] be achieved, but it has also been proved to be effective for the selective preparation of piperidylmethylamines [4-, 3- or 2-(aminomethyl)piperidine (4PIPA, 3PIPA, 2PIPA)] by further hydrogenation of the pyridine ring in addition to the nitrile group. The essence of this method is that the synthesis can be fine-tuned by simply adjusting the amount of acidic additive (H2SO4) based on whether the product to be prepared is pyridyl- or piperidylmethylamine. Complete conversions were obtained under mild conditions (30–50 °C, 6 bar), in all cases, but the very high selectivity to 4PIPA or 4PA (98 and 93%, respectively) decreased to 76% (3PIPA) and 72% (3PA), as well as 10% (2PIPA) and 57% (2PA) by changing the position of the nitrile group in the pyridine ring. The possible reasons for the diverse primary amine selectivities observed in the hydrogenation of the constitutional isomers of pyridinecarbonitriles were confirmed by quantum chemical calculations (DFT). Adsorption energy profiles regarding the interactions between the nitrile starting materials, imine intermediates or amine products and palladium were computed to clarify the selectivity changes.
Introduction
Primary amines are valuable intermediates in the plastic, pharmaceutical or herbicide industries. There are several possibilities for their production, such as, but not limited to, ammonia alkylation, reduction of nitro compounds, or reductive amination of oxo substances.1,2 Heterogeneous catalytic hydrogenation of (hetero)aromatic or aliphatic (di)nitriles is the most widely used industrial method, especially in the pharmaceutical industry.1Previously, we developed a selective and technologically feasible liquid-phase heterogeneous catalytic hydrogenation process for converting nitriles to primary amines over supported Pd catalysts, in a mixture of two immiscible solvents (e.g. water/dichloromethane) and in the presence of a medium acidic additive (NaH2PO4) obtaining complete conversion, high isolated yield and selectivity (>90%).3 Accordingly, benzonitrile was reduced to benzylamine over 10% Pd/C with 90% isolated yield and 95% selectivity, under mild conditions (6 bar, 30 °C).3 When this method was used for the hydrogenation of benzyl cyanide to 2-phenylethylamine4 and that of 3-phenylpropionitrile to 3-phenylpropylamine,5 lower primary amine selectivities (45 and 26%, respectively) and isolated yields (40 and 20%, respectively) were achieved. However, the Pt-catalysed hydrogenation of benzonitrile and its homologues, under these conditions, resulted in comparatively high selectivity to benzylamine (68%), 2-phenylethylamine (57%) or 3-phenylpropylamine (59%) accompanying by the complete conversion of nitriles.6
In this work, the extendibility of our previously reported method to the heterogeneous catalytic hydrogenation of some heteroaromatic nitriles, more precisely to that of 4-, 3- or 2-pyridinecarbonitrile (4PN, 3PN, 2PN) to the corresponding 4-, 3- or 2-(aminomethyl)pyridine (4PA, 3PA, 2PA) or 4-, 3- or 2-(aminomethyl)piperidine (4PIPA, 3PIPA, 2PIPA) over a readily available 10% Pd/C (Selcat Q) catalyst was examined. In general, both (aminomethyl)pyridines and -piperidines are valuable raw materials and intermediates for the preparation of pharma-ceuticals, agricultural chemicals or other products. For example, 2-(aminomethyl)piperidine is an essential building block for the production of flecainide acetate, which is used as an antiarrhythmic drug.7 In addition, 2-(aminomethyl)pyridine is a key intermediate in the synthesis of optically active ionic liquids containing nitrogen,8 or that of befiradol, a highly selective serotonin receptor agonist for the treatment of chronic pain,9 as well as it can serve as catalyst ligands bearing this scaffold in Cu and Pd complexes applied for Henry reaction10 or Suzuki-Miyaura coupling reaction.11 Furthermore, new phase-shifting (aminomethyl)pyridine-based solvents with high CO2 capture capacity (11–20 wt%) were reported, which readily bind CO2 to form a crystalline salt under both dry and wet conditions.12
Some recent reviews have provided comprehensive reports on novelties for the development of heterogeneous or homogeneous catalytic hydrogenation of nitriles catalysed by transition metals.13–20 In addition, general methods for the reduction of nitriles to primary amines and the selectivity problems have also been reported in detail.3–6 Herein, we focus on the hydrogenation of pyridinecarbonitriles.
In general, the preparation of (aminomethyl)piperidines by catalytic hydrogenation of pyridinecarbonitriles takes place in two steps. In the first one, (aminomethyl)pyridine is formed as an intermediate by the hydrogenation of the nitrile group to amine while the pyridine ring remains untouched. In a subsequent second hydrogenation step, the desired (aminomethyl)piperidine is formed by the saturation of the heteroaromatic ring. Both heterogenous or heterogenised catalysts, such as nickel,21,22 rhodium,23 palladium,24–26 cobalt,27–29 cobalt phosphide30 or platinum,31 as well as homogeneous ones, such as Ru(II),32–34 Fe(II),35,36 Co(III),37–39 Mn(I)40,41 or Ni(0)42 complexes, were typically applied in these reductions.
According to a patented process for the preparation of (aminomethyl)piperidines, the corresponding pyridinecarbo-nitrile was firstly converted to 2PA over RANEY® nickel, in benzene, by addition of NH3, at 95 °C and 40 bar.21 The product was obtained in 88% yield and 99% purity after distillation. Subsequently, it was hydrogenated to 2PIPA over a Rh/C catalyst, in water at 110 °C and 40 bar.23 Although this piperidine derivative was afforded in a very good yield (97%) and purity (99%) after distillation, the use of two different catalysts and the need to work in two different reaction media makes the process laborious and inexpedient. When a mesoporous Al2O3 supported Ni catalyst was applied for the hydrogenation of 4PN under relatively mild conditions (60 °C and 2.5 bar), in the presence of ammonia, the corresponding primary amine was obtained in good yield (92%). In addition, this catalyst could be reused five times without activity loss through convenient magnetic recovery.22 Using a 10% Pd/C catalyst, 4PN, 3PN or 2PN was converted to 4PA, 3PA or 2PA in acetic acid as a solvent, under very mild reaction conditions (room temperature, 1.5 bar) in very good yields (90–94%).25 The products were prepared as acetate salts, and no formation of secondary amines were observed. The Pd-catalysed transfer hydrogenation of 2PN or 3PN resulted in 2PA or 3PA in 51 and 72% yields, respectively, over 10% Pd/C, in the presence of formic acid and triethylamine (molar ratio = 3.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as a hydrogen donor, in tetrahydrofuran, at room temperature.26 It was highlighted by the authors that the selective reduction of these types of substrates, especially 2PN, is known to be complicated. Likewise, in the hydrogenation of 2PN to 2PA, a nanostructured catalytic material composed by cobalt nanoparticles, magnesium oxide and a biowaste-derived carbon matrix was used in isopropyl alcohol, in the presence of triethylamine and water.29 Using this catalyst system, complete conversion of 2PN was achieved at 130 °C and 40 bar, after 24 h accompanied by a relatively good selectivity to 2PA (70%). Whereas, under the same conditions, with other precious metal catalysts based on Pd, Pt or Ru, much lower selectivities (1–49%) and mainly overhydrogenated products were obtained. The particular problem of this reduction was explained by the more preferred hydrolysis to picolinamide due to an anchimeric assistance of the heterocyclic nitrogen atom adjacent to the nitrile group. An efficient, new magnetic carbon nanotubes supported Pt(II) catalyst system was also applied for the selective hydrogenation of 4PN in water, using NaBH4, at 95 °C and atmospheric pressure to give 4PA in a very good yield (98%) after 30 min.31 These methods, however, have some drawbacks: using ammonia, prolonged reaction time, and applying special reaction conditions (high pressure and temperature).
:1) as a hydrogen donor, in tetrahydrofuran, at room temperature.26 It was highlighted by the authors that the selective reduction of these types of substrates, especially 2PN, is known to be complicated. Likewise, in the hydrogenation of 2PN to 2PA, a nanostructured catalytic material composed by cobalt nanoparticles, magnesium oxide and a biowaste-derived carbon matrix was used in isopropyl alcohol, in the presence of triethylamine and water.29 Using this catalyst system, complete conversion of 2PN was achieved at 130 °C and 40 bar, after 24 h accompanied by a relatively good selectivity to 2PA (70%). Whereas, under the same conditions, with other precious metal catalysts based on Pd, Pt or Ru, much lower selectivities (1–49%) and mainly overhydrogenated products were obtained. The particular problem of this reduction was explained by the more preferred hydrolysis to picolinamide due to an anchimeric assistance of the heterocyclic nitrogen atom adjacent to the nitrile group. An efficient, new magnetic carbon nanotubes supported Pt(II) catalyst system was also applied for the selective hydrogenation of 4PN in water, using NaBH4, at 95 °C and atmospheric pressure to give 4PA in a very good yield (98%) after 30 min.31 These methods, however, have some drawbacks: using ammonia, prolonged reaction time, and applying special reaction conditions (high pressure and temperature).
In this paper, the influences of acidic additives, solvents, catalyst amount and temperature on the isolated yield and selectivity to the desired primary amines, more specifically the pyridyl- or piperidylmethylamines (4PA, 3PA, 2PA or 4PIPA, 3PIPA, 2PIPA) and the conversion of the corresponding nitriles (4PN, 3PN, 2PN) are elucidated. In addition, adsorption energy profiles related to the interactions between the nitrile starting materials, imine intermediates or amine products and palladium were compared by quantum chemical calculations using a density functional theory (DFT) approach.
Experimental
Materials
The 10% palladium on carbon catalyst (Selcat Q)43 was supplied by Szilor Ltd. (Budapest, Hungary). Starting materials, such as 4-pyridinecarbonitrile (98%), 3-pyridinecarbonitrile (98%) and 2-pyridinecarbonitrile (99%), were purchased from Sigma-Aldrich (St. Louis, USA). Dichloromethane (p.a.), toluene (p.a.), ethyl acetate (p.a.), hexane (p.a.), tert-butyl methyl ether (p.a.), H2SO4 (98%), H3PO4 (85%), HCl (37%) and NaH2PO4·H2O (p.a.) were received from Merck (Darmstadt, Germany).Hydrogenations
The hydrogenation reactions were carried out in a 0.5 L BEP 280 (Büchi) glass autoclave (Büchi AG, Uster, Switzerland) equipped with a magnetically driven turbine stirrer (speed: 1800 rpm) and an automatic gas flow controlling and measuring unit (Büchi bpc 6010). The initial rate values (v0) were calculated from the hydrogen consumption and conversion curves according to the eqn (1):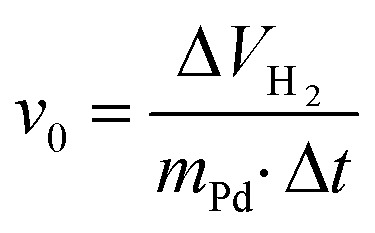 | (1) |
The reactor containing the given pyridinecarbonitrile, 10% Pd/C catalyst, acidic additive and solvents, was flushed with nitrogen (3×) and hydrogen (3×), then charged with hydrogen to 6 bar and the temperature was adjusted to 30 or 50 °C. After completion of the H2-uptake, the reaction mixture was cooled down to room temperature and the catalyst was removed by filtration. Then, the organic phase was separated and evaporated in vacuum. The residue was analysed GC–MS, and based on these results, the amount of by-products and unreacted pyridinecarbonitriles can be deduced. The aqueous fraction (pH ≈ 2–6.5) was also evaporated in vacuum, and the desired primary amines were obtained as sulphate, phosphate or hydrochloride salts. Since both (aminomethyl)pyridines and -piperidines are highly volatile in their free base form, they were just released from their salts, when the analytical samples were prepared. Their spectroscopic data are the following: 4PIPA1H NMR (CDCl3, 500 MHz) δ ppm 1.38–1.53 (m, 5H, CH2CHCH2), 2.35 (br s, 3H, NH and NH2), 2.94 (d, J = 5.0 Hz, 2H, CH2NH2), 3.40–3.47 (m, 4H, CH2NHCH2); 13C NMR (CDCl3, 125 MHz) δ ppm 25.7 (CH2CHCH2), 31.6 (CH2CHCH2), 43.4 (CH2NHCH2), 43.7 (CH2NH2); GC–MS m/z (rel%) 114(27), 96(20), 84(39), 69(32), 56(100). 4PIPA·H2SO4 mp.: 285–287 °C (decomp.). 4PA1H NMR (CDCl3, 500 MHz) δ ppm 1.79 (br s, 2H, NH2), 3.88 (s, 2H, CH2NH2), 7.24 (d, J = 5.0 Hz, 2H, Pyr-CHCCH), 8.52 (d, J = 5.0 Hz, 2H, Pyr-CHNCH); 13C NMR (CDCl3, 125 MHz) δ ppm 45.4 (CH2NH2), 122.0 (Pyr-CHCCH), 149.8 (Pyr-CHNCH), 151.9 (Pyr-CHCCH); GC–MS m/z (rel%) 108(12), 107(28), 80(100), 51(13). 4PA·0.5H2SO4 mp.: 162–168 °C (decomp.). 3PIPA1H NMR (CDCl3, 500 MHz) δ ppm 1.54–1.90 (m, 5H, CHCH2CH2), 2.61 (br s, 3H, NH and NH2), 2.88 (d, J = 5.0 Hz, 2H, CH2NH2), 3.16–3.32 (m, 4H, CH2NHCH2); 13C NMR (CDCl3, 125 MHz) δ ppm 24.3 (CHCH2CH2), 30.3 (CHCH2CH2), 32.5 (CHCH2CH2), 45.2 (CH2NH2), 53.3 (NHCH2CH2), 53.5 (NHCH2CH); GC–MS m/z (rel%) 114(2), 97(98), 82(100), 68(28), 56(63). 3PA1H NMR (CDCl3, 500 MHz) δ ppm 2.72 (br s, 2H, NH2), 4.05 (s, 2H, CH2NH2), 7.29 (dd, J = 8.0 and 5.0 Hz, 1H, Pyr-NCHCH), 7.64 (d, J = 8.0 Hz, 1H, Pyr-CCHCH) 8.46 (d, J = 5.0 Hz, 1H, Pyr-NCHCH) 8.51 (s, 1H, Pyr-NCHC); 13C NMR (CDCl3, 125 MHz) δ ppm 44.1 (CH2NH2), 122.6 (Pyr-NCHCH), 134.5 (Pyr-CCHCH), 138.4 (Pyr-NCHC), 148.1 (Pyr-NCHCH), 148.9 (Pyr-NCHC); GC–MS m/z (rel%) 107(23), 80(100), 52(15). 2PIPA GC–MS m/z (rel%) 114(1), 84(100), 67(4), 56(32). 2PA1H NMR (CDCl3, 500 MHz) δ ppm 2.56 (br s, 2H, NH2), 3.94 (s, 2H, CH2NH2), 7.17 (dd, J = 8.0 and 5.0 Hz, 1H, Pyr-NCCH), 7.22–7.27 (m, 1H, Pyr-NCHCH) 7.64 (td, J = 8.0 and 2.0 Hz, 1H, Pyr-CHCHCH) 8.52 (d, J = 5.0 Hz, 1H, Pyr-NCHCH); 13C NMR (CDCl3, 125 MHz) δ ppm 47.7 (CH2NH2), 121.9 (Pyr-NCHCH), 122.3 (Pyr-CCHCH), 136.7 (Pyr-CHCHCH), 149.3 (Pyr-NCHCH), 159.4 (Pyr-NCCH); GC–MS m/z (rel%) 108(100), 80(95), 52(50).
The side products (Schemes 1 and 2, Fig. 5 and 6) were also detected and identified by GC–MS. Mass spectroscopic data of the typical ones are the following: 4-piperidinecarbonitrile (4PIPCN) m/z (rel%) 110(36), 109(39), 83(12), 68(5), 57(100); bis(4-pyridylmethyl)amine (B4PA) m/z (rel%) 199(3), 121(44), 107(19), 93(100), 79(14), 65(44), 51(24); 4-methylpiperidine (4MPPD) m/z (rel%) 99(64), 98(97), 84(30), 70(10), 56(100); 3-methylpiperidine (3MPPD) m/z (rel%) 99(97), 98(100), 84(91), 70(42), 56(79); 1,4,5,6-tetrahydropyridine-3-carbonitrile (THPCN) m/z (rel%) 108(93), 107(100), 93(23), 80(42), 68(49), 53(38); 3-piperidinecarbonitrile (3PIPCN) m/z (rel%) 110(33), 82(14), 70(6), 57(100), 54(36); 3-pyridinemethanol (3POH) m/z (rel%) 109(63), 108(79), 91(12), 80(100), 65(14), 53(36); bis(3-pyridylmethyl) ether (B3PE) m/z (rel%) 201(75), 200(53), 171(15), 145(10), 119(15), 109(100), 92(86), 82(39), 65(45), 54(14); bis(2-pyridylmethyl)amine (B2PA) m/z (rel%) 199(1), 121(8), 107(57), 93(100), 78(10), 65(23), 51(10); 2-pyridine-methanol (2POH) m/z (rel%) 109(46), 108(100), 80(54), 52(42); 2-piperidinemethanol (2PIPOH) m/z (rel%) 115(1), 108(13), 84(100), 80(16), 56(43); 2-(N-methylaminomethyl)piperidine (2MPIPA) m/z (rel%) 128(1), 125(30), 97(20), 84(100), 69(13), 56(23); bis(2-piperidylmethyl)amine (B2PIPA) m/z (rel%) 211(1), 167(4), 139(38), 125(19), 97(21), 84(100), 56(13). These analytical results are in accordance with the literature data.44
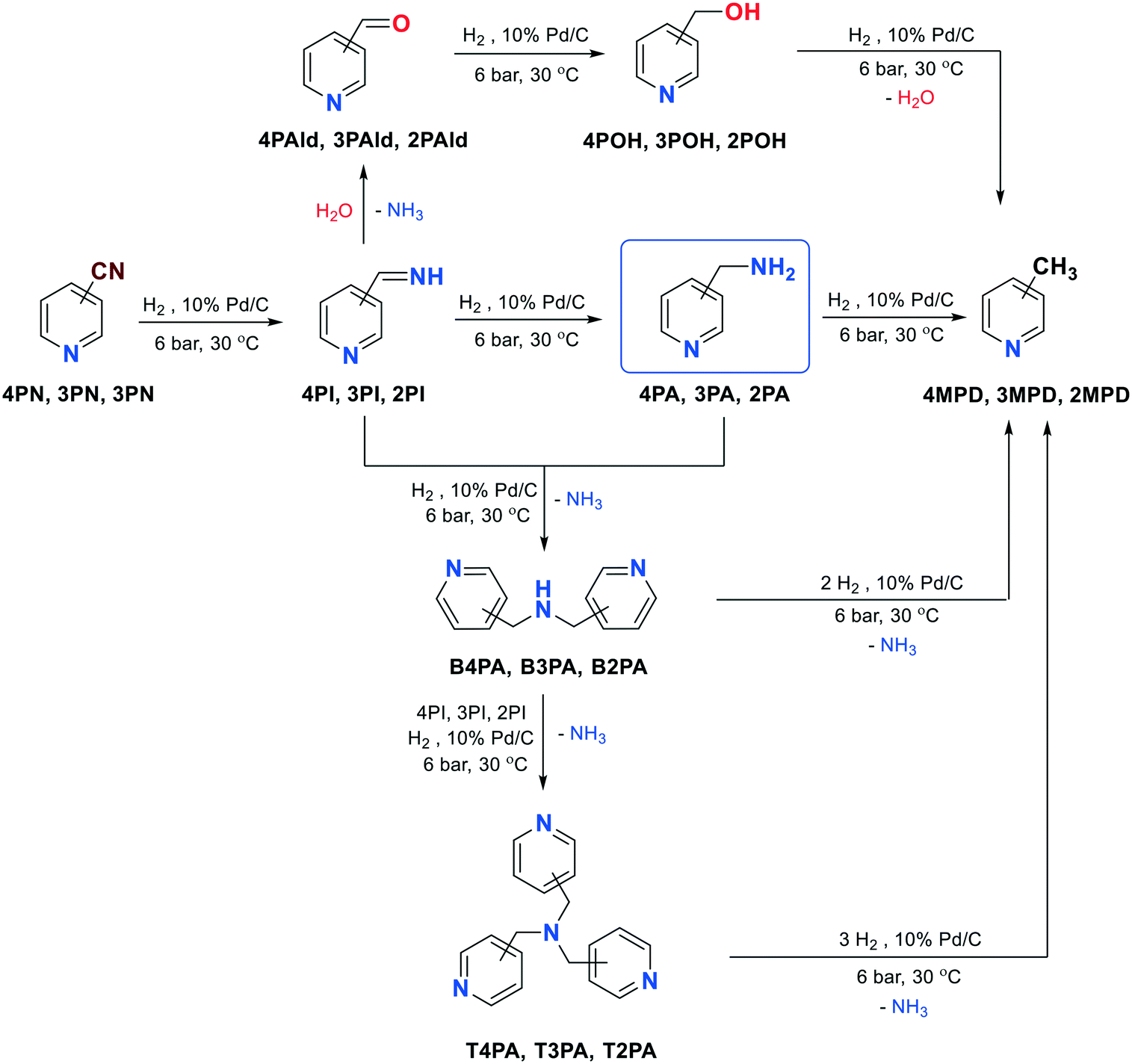 | ||
| Scheme 1 Possible reaction pathways for the hydrogenation of the nitrile group of 4-, 3- or 2-pyridinecarbonitrile (4PN, 3PN, 2PN). | ||
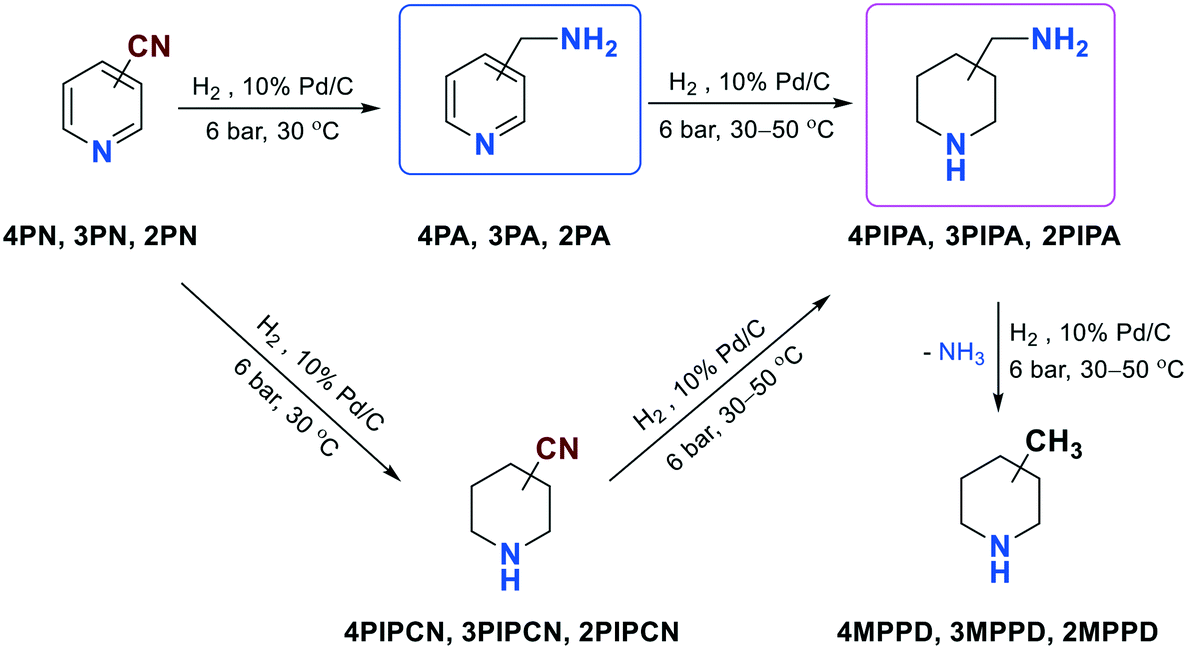 | ||
| Scheme 2 Other products and by-products in the hydrogenation of 4PN, 3PN or 2PN. | ||
Analytical procedures
The products and the by-products were analysed and identified by GC–MS, as well as 1H and 13C NMR measurements.GC–MS analyses were performed by an Agilent 7890A GC-system (7683 autosampler and 7683B injector) connected to an Agilent 5975C mass spectrometer using a Restek Rxi®-5Sil MS capillary column (15 m × 0.25 mm ID, 0.25 μm film). The temperature program was the following: 45 °C (0.5 min) to 300 °C at 50 °C/min, hold 1 min.
The NMR spectra were recorded in chloroform-D1 (CDCl3) on a Bruker DRX-500 spectrometer operating at 500 and 125 MHz, respectively. The chemical shift values (δ) are given relatively to that of δTMS.
Powder X-ray diffraction (XRD) measurements of 4PIPA·H2SO4 salt were performed on an X'pert Pro MPD multipurpose diffractometer (PANalytical B.v., the Netherlands) using Cu Kα radiation (λ = 0.15406 nm). The X-ray tube operated at 40 kV and 30 mA, and Ni foil as a β-filter, as well as an X'celerator detector was applied. Diffraction profiles were obtained in the range of 2θ = 4.01–45.42°. For purposes of indexing and simulated annealing structure solution the DASH software package,45 and a step size of 0.0167° and a counting time of 160 s were used.
Fourier transform infrared (FT–IR) spectra of the solid samples were measured by an Excalibur Series FTS 3000 (Biorad) FT–IR spectrophotometer in a KBr pastille, between 400 and 4000 cm−1, at a resolution of 4 cm−1.
Dispersion of the catalyst (D10% Pd/C = 0.50) was determined by O2-, H2- and CO-chemisorption measurements using an atmospheric flow system, as well as the usual stoichiometry for the adsorption of these gases on palladium was applied for the calculations.46,47
Computational methods
DFT calculations of the adsorption geometries and energies were carried out using the Quantum ESPRESSO software package.48 The exchange–correlation functional utilized was the Perdew-Burke-Ernzerhof (PBE) of generalized gradient approximation (GGA) with the plane wave ultrasoft pseudopotential approach.49 The energy cut-off of the plane wave basis set was 50 Ry and the convergence criterion of the self-consistent accuracy was adjusted to 1 × 10−6.A three-layer periodic slab with upper atoms allowed to relax was used to model the Pd with a (111) surface. A (4 × 4) supercell including a 10 Å vacuum slab was applied to study the adsorption of the reactant molecules (nitriles, imines or amines). The Monkhorst-Pack50 scheme k-point grid sampling of a (2 × 2 × 1) was used in all calculations over the entire Brillouin zone.
Adsorption energy (ΔEads) profiles were calculated according to eqn (2):
| ΔEads = Ereactant+Pd(111) − EPd(111) − Ereactant | (2) |
Results and discussion
Reaction pathways
Scheme 1 shows the possible reaction pathways for the hydrogenation of the nitrile group of 4PN, 3PN or 2PN.Basically, the formation of primary amines (4PA, 3PA or 2PA) takes place through imine intermediates, such as 4-, 3- or 2-(iminomethyl)pyridine (4PI, 3PI, 2PI). However, secondary amines, bis(4-pyridylmethyl)amine (B4PA), bis(3-pyridylmethyl)amine (B3PA) or bis(2-pyridylmethyl)amine (B2PA), can also be formed in the condensation reactions of the corresponding primary amines (4PA, 3PA or 2PA) and imines (4PI, 3PI or 2PI), along with ammonia elimination. Further addition of the imines to the secondary amines followed by hydrogenation can result in tertiary amines, such as tris(4-pyridylmethyl)amine (T4PA), tris(3-pyridylmethyl)amine (T3PA) or tris(2-pyridylmethyl)amine (T2PA). Moreover, 4-, 3- or 2-methylpyridine (4MPD, 3MPD, 2MPD) can also be formed in a hydrogenolytic step from both the primary, secondary and tertiary amines. Another side reaction may be the reaction of the imines with water during NH3 elimination, which results in the formation of 4-, 3- or 2-pyridinecarboxaldehyde (4PAld, 3PAld, 2PAld), which can be reduced to 4-, 3- or 2-pyridinemethanol (4POH, 3POH, 2POH), and its further hydrogenolysis may result in the formation of 4-, 3- or 2-methylpyridine (4MPD, 3MPD, 2MPD).
Nevertheless, saturation of the pyridine ring may cause an additional chemoselectivity problem (Scheme 2), which results in the formation of 4PIPA, 3PIPA or 2PIPA. In addition, 4PN, 3PN or 2PN can also be hydrogenated to 4-piperidinecarbonitrile (4PIPCN), 3-piperidinecarbonitrile (3PIPCN) or 2-piperidinecarbonitrile (2PIPCN), and further converted to 4PIPA, 3PIPA or 2PIPA. Finally, 4-, 3- or 2-methylpiperidine (4MPPD, 3MPPD, 2MPPD) may also be formed from 4PIPA, 3PIPA or 2PIPA in another hydrogenolytic step.
Hydrogenation of 4-pyridinecarbonitrile
| Entry | Acidic additive/4PN ratio (mol−1 mol−1) | Reaction time for complete conversion (h) | Product mixture | Selectivity to 4PIPA (%) | v 0 (NL H2 gPd−1 h−1) | |
|---|---|---|---|---|---|---|
| Isolated yield (%) | 4PIPA-Content (%) | |||||
| a Reaction conditions: 5.0 g (48.1 mmol) 4PN, 1.5 g 10% Pd/C (Selcat Q), 150 mL water and 50 mL dichloromethane, 30 °C, 6 bar. b 30–50 °C. c This phosphate-sulphate salt was very hygroscopic and difficult to handle. | ||||||
| 1b | 2.0 NaH2PO4 | 5.5 | 6 | 35.9 | 36 | 27.6 |
| 2b | 1.0 NaH2PO4/1.0 H2SO4 | 3.5 | –c | 97.8 | 93 | 122.0 |
| 3b | 1.0 HCl | 8.0 | 94 | 86.4 | 86 | 53.6 |
| 4 | 1.0 H2SO4 | 6.5 | 95 | 98.7 | 94 | 73.6 |
| 5b | 1.0 H3PO4 | 5.5 | 95 | 61.3 | 58 | 55.2 |
First, we wanted to adapt the previously well-tried reaction conditions and work-up procedure, which resulted in excellent primary amine selectivity, prepared yield with the complete conversion of benzonitrile in a Pd-catalysed hydrogenation.3 In the previously developed process, after completion of the reaction, the aqueous phase containing the primary amine was basified with 20% NaOH solution (pH> 13). Then, it was extracted with dichloromethane and the separated organic phase was evaporated in vacuum. In the presence of NaH2PO4, however, only poor isolated yield (6%) and primary amine selectivity (36%) were achieved in this hydrogenation. The reason for the poor isolated yield was the high volatility and the relatively good water solubility of this piperidine compound, so it is advantageous to prepare the product as a salt. Thus, several acidic additives were tested to find the ideal salt form of 4PIPA.
When a mixture of sulphuric acid and NaH2PO4 (1:1 molar ratio) was used, the reaction was fast (3.5 h) and the purity of the product (97.8% 4PIPA-content) was high, but the obtained salt was very hygroscopic and difficult to handle. The best isolated yield (95%) and primary amine selectivity (94%) were achieved in the presence of sulphuric acid. The obtained sulphate salt (4PIPA·H2SO4) proved to be very stable and easy to handle. Using hydrochloric or ortho-phosphoric acid, the isolated yields were excellent (94–95%), but the purity and selectivity values became appreciably lower (4PIPA-content: 86.4 and 61.3%, respectively; selectivity to 4PIPA: 86 and 58%, respectively). Moreover, in the presence of HCl, the longest reaction time (8 h) was observed. Since chloride ion is a mild catalyst poison,51 the slowing down of the reaction was presumably due to this effect.
The crystal structure of 4PIPA·H2SO4 salt was determined from its powder XRD profiles, using the build-in indexing and simulated annealing structure solution opportunities of DASH software package.45 The powder XRD measurements and the calculations allowed a crystal structure containing a diammonium-4PIPA dication (C5H9NH2+CH2NH3+) and a sulphate anion (SO42−) in a 1:1 molar ratio of ions, as shown in Fig. 1, and after proving the presence of ionic species by FT–IR spectroscopy (Fig. 2). The characteristic peaks at 1125 and 620 cm−1, respectively, can be assigned to the IR-active vibrations (ν3 and ν4) of the SO42− anion.52
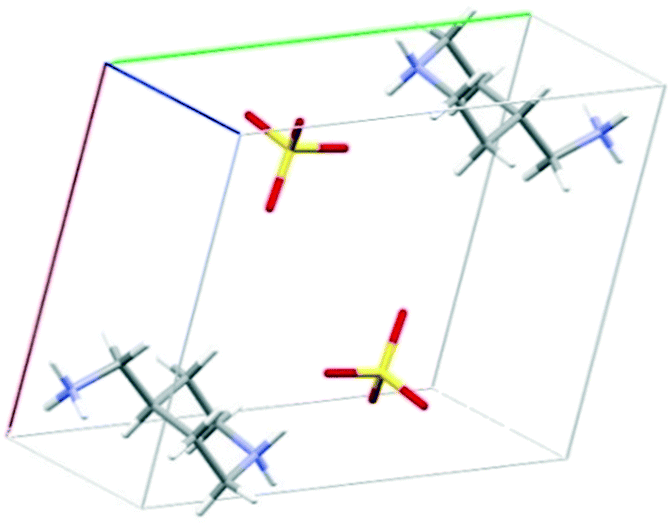 | ||
| Fig. 1 A unit cell of crystal structure for 4PIPA·H2SO4 [(4PIPA-H2)2+·(SO4)2−] created from powder XRD pattern by DASH program.45 | ||
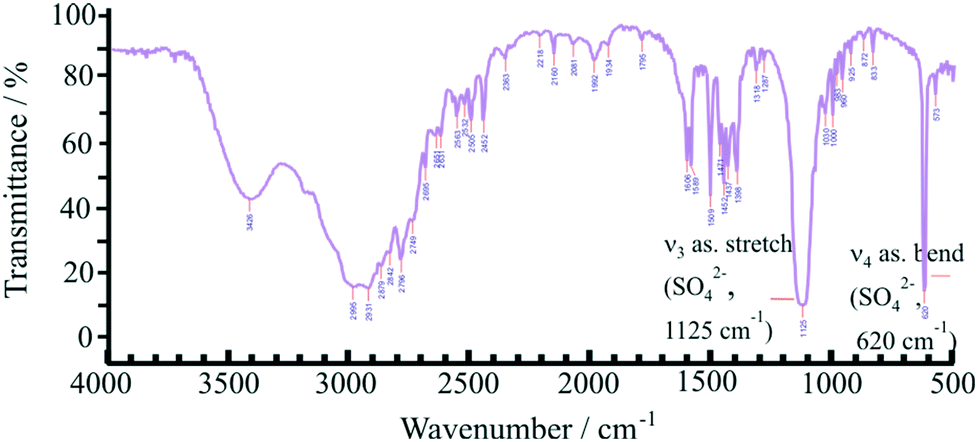 | ||
| Fig. 2 FT–IR spectrum of the salt 4PIPA·H2SO4. | ||
The results obtained by decreasing the amount of different acidic additives (1.0 → 0.5 mol mol−1) are summarised in Table 2. As seen, the hydrogenation was firstly carried out at 0.3 g g−1 catalyst/substrate ratio, in a dichloromethane/water solvent mixture, in the presence of sulphuric acid, at 30 °C and 6 bar, as these reaction conditions proved to be the best in previous experiments at 1.0 H2SO4/4PN molar ratio. However, the rate of hydrogen uptake slowed down after a very fast initial phase (0.3–0.4 h), so that the reaction was stopped after 0.7 h. The prepared sulphate salt contained mainly 4PA (76.7%), although 4PIPA and about 20% 4-piperidinecarbonitrile (4PIPCN) were also formed. Since the reaction was rapid, the next hydrogenations were performed at a lower catalyst/substrate ratio (0.1 g g−1), exclusively in water. As seen, the best results were again obtained by the addition of sulphuric acid. The conversion of 4PN was fast and complete after 0.3 h, while 4PIPA·0.5H2SO4 was isolated with excellent yield (98%) and selectivity (92%). Using hydrochloric acid, the reaction was slower (0.8 h), and the purity of the product, the isolated yield and the selectivity were also decreased (94.0 → 86.5%, 98 → 80%, as well as 92 → 69%). In this case, the product contained a significant amount of secondary amine (6.9%), more precisely bis(4-pyridylmethyl)amine (B4PA), as well as 4PIPCN (6.9%) was also formed. Applying ortho-phosphoric acid, the isolated yield (89%), the purity of product (86.3% 4PA-content) and the selectivity to 4PA (77%) were also diminished compared to the reaction using sulphuric acid. No secondary amine formation was observed in this reaction, the main by-product was 4PIPCN (12%).
| Entry | Acidic additives | Reaction time for complete conversion (h) | Product mixture | Selectivity to 4PA (%) | v 0 (NL H2 gPd−1 h−1) | |
|---|---|---|---|---|---|---|
| Isolated yield (%) | 4PA-Content (%) | |||||
| a Reaction conditions: 5.0 g (48.1 mmol) 4PN, 0.5 g 10% Pd/C (Selcat Q), 200 mL water, acidic additive/4PN molar ratio = 0.5, 30 °C, 6 bar. b 1.5 g 10% Pd/C (Selcat Q), 150 mL water and 50 mL dichloromethane. | ||||||
| 1b | H2SO4 | 0.7 | 94 | 76.7 | 74 | 71.1 |
| 2 | H2SO4 | 0.3 | 98 | 94.0 | 92 | 206.9 |
| 3 | HCl | 0.8 | 80 | 86.5 | 69 | 161.2 |
| 4 | H3PO4 | 0.4 | 89 | 86.3 | 77 | 200.4 |
Presumably, the more basic methylamine moiety (–CH2NH2) coordinates mainly with sulphuric acid, by decreasing its amount from 1.0 to 0.5 mol mol−1, thus the nitrogen of the pyridine ring is not protonated, which decreases the rate of ring saturation. Due to the much weaker basicity of pyridine nitrogen (pKa = 5.2) than that of methylamine moiety (pKa = 8.30),53 the pyridine ring remains mainly intact and 4PA is formed as a major product. On the contrary, in the presence of 1.0 mol mol−1 H2SO4, both basic nitrogen atoms are protonated resulting in the formation of fully saturated 4PIPA with excellent chemoselectivity (94%).
Overall it can be stated that sulphuric acid gave the best results in terms of isolated yield, selectivity and purity of both primary amines (4PIPA and 4PA).
The results of the hydrogenation of 4PN to 4PIPA in the mixtures of different organic solvents and water, over 10% Pd/C (0.3 g g−1 catalyst/substrate ratio), in the presence of H2SO4 (1.0 mol mol−1), at 30 °C and 6 bar are summarised in Table 3. As seen, the best results were obtained in a dichloromethane/ water solvent mixture (95% prepared yield, 98.7% 4PIPA-content and 94% selectivity to 4PIPA), but the complete conversion required a longer reaction time (5.5 h, Fig. 3). In toluene/water, the reduction of 4PN took place fast (3.5 h), and the prepared yield (96%) was excellent, whereas, both the purity of product (96.8%) and the selectivity to 4PIPA (93%) became lower. Using an ethyl acetate/water mixture, 95% prepared yield and 93% selectivity to 4PIPA were achieved, while in tert-butyl methyl ether/water these values were 96% and 90%, respectively, after 3.5–4.5 h. Applying a hexane/water mixture, the reaction was the fastest (2.5 h), but the purity of product (96.0%), the isolated yield (92%) and the selectivity (89%) were significantly lower compared to the other solvents. The main reason for this can be the fact that the starting material was insoluble in hexane, i.e. both the substrate and the product were present only in the aqueous phase. Only in water, without any organic solvents, the reaction was also fast (2.5 h) and the isolated yield (98%) was high, but the purity of product (70.0% 4PIPA-content) was moderate, due to the formation of 4-methylpiperidine (4MPPD) in a higher amount (23%) resulting in low primary amine selectivity (69%).
| Entry | Organic solvents | Reaction time for complete conversion (h) | Product mixture | Selectivity to 4PIPA (%) | v 0 (NL H2 gPd−1 h−1) | |
|---|---|---|---|---|---|---|
| Isolated yield (%) | 4PIPA-Content (%) | |||||
| a Reaction conditions: 5.0 g (48.1 mmol) 4PN, 1.5 g 10% Pd/C (Selcat Q), 150 mL water and 50 mL organic solvent, H2SO4/4PN molar ratio = 1.0, 30 °C, 6 bar. b 200 mL water. | ||||||
| 1 | Dichloromethane | 5.5 | 95 | 98.7 | 94 | 73.6 |
| 2 | Toluene | 3.5 | 96 | 96.8 | 93 | 122.9 |
| 3 | tert-Butyl methyl ether | 4.5 | 96 | 93.5 | 90 | 84.4 |
| 4 | Hexane | 2.5 | 92 | 96.0 | 89 | 169.0 |
| 5 | Ethyl acetate | 3.5 | 95 | 98.5 | 93 | 163.5 |
| 6b | — | 2.5 | 98 | 70.0 | 69 | 170.0 |
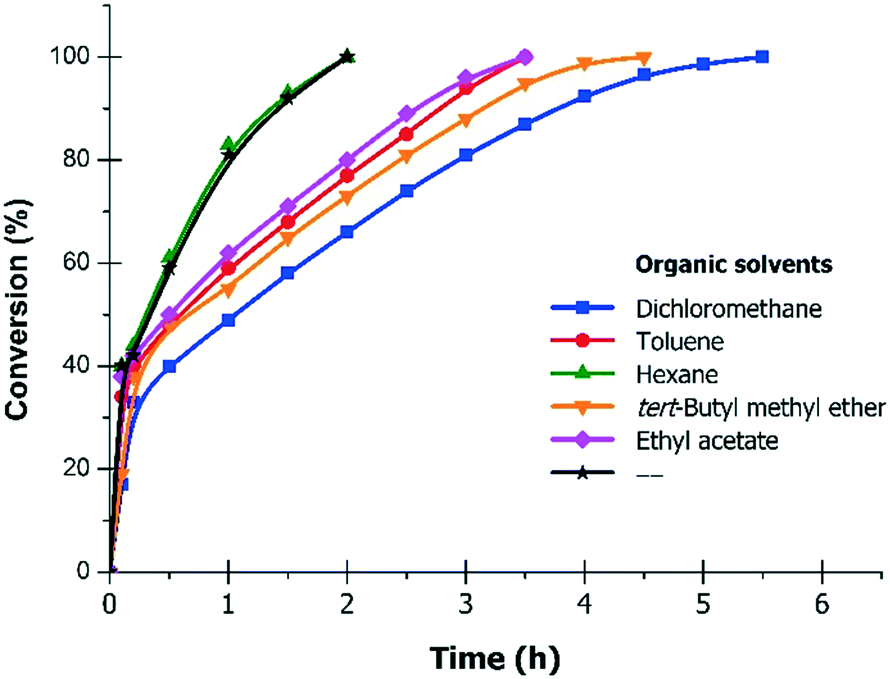 | ||
| Fig. 3 Conversion of 4PN to 4PIPAvs. time in the mixtures of different organic solvents and water. Reaction conditions: 5.0 g (48.1 mmol) 4PN, 1.5 g 10% Pd/C (Selcat Q), 250 mL water and 50 mL organic solvent, H2SO4/4PN molar ratio = 1.0, 30 °C, 6 bar. | ||
Based on our results, it can be concluded that it is advisable to use an organic solvent in the hydrogenation of 4PN to 4PIPA, and the best results were provided by dichloromethane as a solvent pair of water.
The influence of different organic solvents on the nitrile conversion, isolated yield and chemoselectivity was also investigated in the presence of 0.5 mol mol−1 H2SO4, i.e. in the hydrogenation of 4PN to 4PA, at 0.2 g g−1 catalyst/substrate ratio, at 30 °C and 6 bar. The results are summarised in Table 4.
| Entry | Organic solvents | Reaction time for complete conversion (h) | Product mixture | Selectivity to 4PA (%) | v 0 (NL H2 gPd−1 h−1) | |
|---|---|---|---|---|---|---|
| Isolated yield (%) | 4PA-Content (%) | |||||
| a Reaction conditions: 5.0 g (48.1 mmol) 4PN, 1.0 g 10% Pd/C (Selcat Q), 150 mL water and 50 mL organic solvent, H2SO4/4PN molar ratio = 0.5, 30 °C, 6 bar. b 200 mL water. | ||||||
| 1 | Dichloromethane | 2.0 | 95 | 78.0 | 74 | 73.6 |
| 2 | Toluene | 1.0 | 93 | 77.2 | 72 | 66.4 |
| 3 | tert-Butyl methyl ether | 0.5 | 91 | 84.9 | 78 | 132.5 |
| 4 | Hexane | 0.3 | 95 | 92.8 | 88 | 184.0 |
| 5 | Ethyl acetate | 0.7 | 93 | 85.2 | 79 | 119.2 |
| 6b | — | 0.2 | 94 | 93.2 | 88 | 233.6 |
In this case, the primary amine selectivity was lower (72–88%) than in the hydrogenation of 4PN to 4PIPA. Although good isolated yields (91–95%) were achieved and the reaction was fast (0.2–2.0 h), the purity of the product exceeded 90% when only water or a hexane/water mixture was applied (93.2 and 92.8% 4PA-content). In addition, the reduction of the nitrile group was the fastest in water, as well. The typical by-product was 4PIPCN (5.0–15.0%) in these reactions.
Accordingly, the best results in the chemoselective hydrogenation of the nitrile group beside the pyridine ring were obtained when the reaction was carried out exclusively in water. No significant improvements were observed in the isolated yields or the primary amine selectivities in the presence of water-immiscible organic solvents.
| Entry | Catalyst/substrate ratio (g g−1) | Reaction time for complete conversion (h) | Product | Selectivity to 4PIPA (%) | v 0 (NL H2 gPd−1 h−1) | |
|---|---|---|---|---|---|---|
| Isolated yield (%) | 4PIPA-Content (%) | |||||
| a Reaction conditions: 5.0 g (48.1 mmol) 4PN, 10% Pd/C (Selcat Q), 150 mL water and 50 mL dichloromethane, H2SO4/4PN molar ratio = 1.0, 30 °C, 6 bar. | ||||||
| 1 | 0.1 | 11.0 | 99 | 97.2 | 96 | 205.9 |
| 2 | 0.2 | 5.5 | 99 | 99.5 | 98 | 143.4 |
| 3 | 0.3 | 5.5 | 95 | 98.7 | 94 | 73.6 |
As seen, by decreasing the catalyst quantity (0.3 → 0.2 → 0.1 g g−1 ratio), the selectivity improved (94 → 98 → 96%), albeit the hydrogenation required longer reaction time (11.0 h) at 0.1 g g−1 ratio. The best results were achieved at 0.2 g g−1 catalyst/substrate ratio providing 98% primary amine selectivity and a complete conversion of 4PN after 5.5 h, while the product (99.5% 4PIPA-content) was isolated in 99% yield.
The influence of the amount of 10% Pd/C catalyst on selectivity and conversion in the hydrogenation of 4PN to 4PA in water, in the presence of 0.5 mol mol−1 H2SO4 was also examined. According to the results given in Table 6, contrary to that observed during the preparation of 4PIPA, the best ones were obtained at 0.1 g g−1 catalyst/substrate ratio. The product (4PA·0.5H2SO4) was isolated in 98% yield and it contained 94.1% 4PA resulting in a very good primary amine selectivity (93%). In addition, it took only 0.3 h to achieve complete conversion even using this small quantity of catalyst. These ratios are same or slightly higher than those (0.01–0.1 g g−1) typically used in the industry,56 this catalyst could also be applied economically on an industrial scale.
| Entry | Catalyst/substrate ratio (g g−1) | Reaction time for complete conversion (h) | Product mixture | Selectivity to 4PA (%) | v 0 (NL H2 gPd−1 h−1) | |
|---|---|---|---|---|---|---|
| Isolated yield (%) | 4PA-Content (%) | |||||
| a Reaction conditions: 5.0 g (48.1 mmol) 4PN, 10% Pd/C (Selcat Q), 200 mL water, H2SO4/4PN molar ratio = 0.5, 30 °C, 6 bar. | ||||||
| 1 | 0.1 | 0.3 | 98 | 94.1 | 93 | 206.9 |
| 2 | 0.2 | 0.2 | 94 | 93.2 | 88 | 233.6 |
| 3 | 0.3 | 0.1 | 94 | 90.3 | 85 | 246.8 |
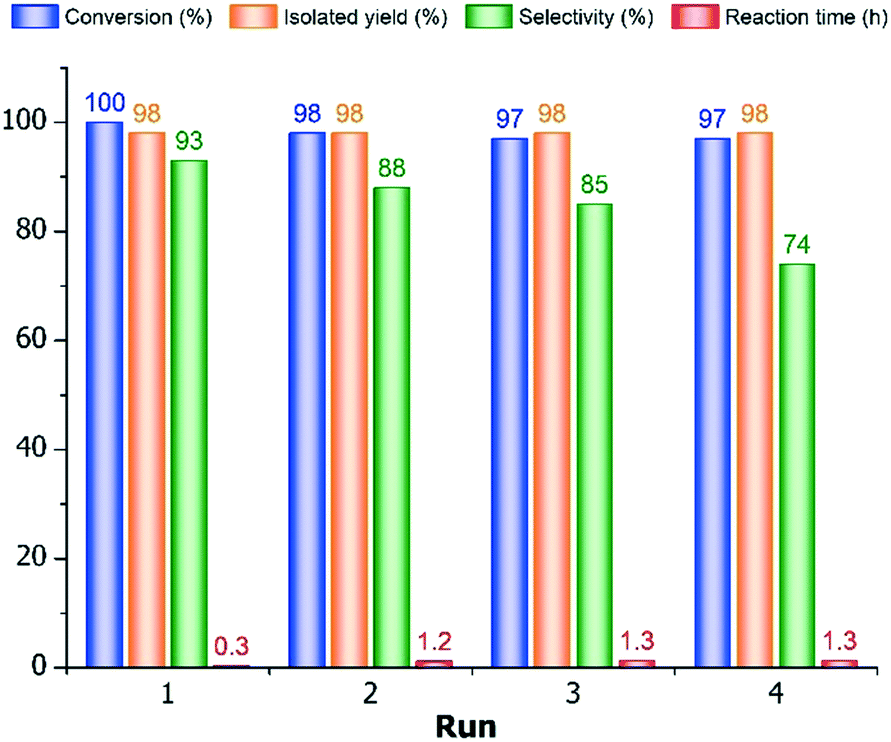 | ||
| Fig. 4 Effect of the reused 10% Pd/C catalyst on conversion, isolated yield, selectivity and reaction time in the hydrogenation of 4PN to 4PA. Reaction conditions: 5.0 g (48.1 mmol) 4PN, 0.5 g 10% Pd/C, 200 mL water, H2SO4/4PN molar ratio = 0.5, 30 °C, 6 bar. | ||
As seen, the conversion decreased slightly to 98% during even the second run, and it remained at this level after the third and fourth ones (97–97%), but the isolated yields were excellent (98%) in all cases. Whereas, the reaction time increased significantly from 0.3 h to 1.2–1.3 h during the 10% Pd/C recycling (runs 2–4). In addition, the 4PA-content of the product was also diminished (94.0 → 88.5 → 84.6 → 72.6%) after the multiple reuses of catalyst, which resulted in appreciable lower primary amine selectivities (93 → 88 → 85 → 74%). The reason for the diminution of the product purity was mainly due to the increase of amount of 4PIPCN (4.0 → 15.0%), similarly to the influence of different organic solvents, but the ratio of the secondary amine (B4PA) became also higher (0 → 5.0%) in the last catalytic run. Presumably, the observed selectivity decrease was due to the accumulation of the product and by-products on the surface of the used 10% Pd/C catalyst, because the reused catalyst was applied without any regenerative procedure, namely it was washed only with water. Thus, the product (4PA) already present on the catalyst can react easier with 4PI resulting in secondary amine in a higher amount, or the saturation of pyridine ring with untouching the nitrile group can also take place in a higher ratio due to the presence of more basic piperidine derivative (4PIPCN) in a higher amount to bind more acidic additive taken away from the wanted conversion (nitrile hydrogenation).
Accordingly, it is inexpedient to reuse the 10% Pd/C spent catalyst in this hydrogenation. Although the conversions were almost complete in all cases, it provided lower primary amine selectivity already in the second catalytic run.
Hydrogenation of 3-pyridinecarbonitrile
Next, it was investigated whether steric hindrances play a role in the primary amine selectivity. Thus, the experiments were continued by optimising the hydrogenation of the meta- substituted isomer of pyridinecarbonitrile (3PN).First, the method used for the selective hydrogenation of 4PN to 4PIPA or 4PA was also adapted for the reduction of 3PN. Thus, the amount of acidic additive (H2SO4) was adjusted according to whether the desired product was pyridyl- or piperidylmethylamine. It was found, however, that the saturation of the pyridine ring was extremely slow at 30 °C in this case, therefore the hydrogenation was carried out at 50 °C. Thus, 3PN was converted to 3PIPA over 10% Pd/C, in water/ dichloromethane, in the presence of 1.0 mol mol−1 H2SO4, and the product was also isolated as a sulphate salt (3PIPA·H2SO4) in 99% yield, with 76% selectivity and good purity (77.1% 3PIPA-content) after 4.5 h. (Table 7, entry 1). The main reason for the selectivity decrease compared to 4PIPA is that a hydrogenolytic step also took place in this case, and 3-methylpiperidine (3MPPD) was formed from the primary amine with a loss of ammonia.
| Entry | Organic solvents | Reaction time for complete conversion (h) | Product mixture | Selectivity to 3PIPA (%) | v 0 (NL H2 gPd−1 h−1) | |
|---|---|---|---|---|---|---|
| Isolated yield (%) | 3PIPA-Content (%) | |||||
| a Reaction conditions: 5.0 g (48.1 mmol) 3PN, 1.5 g 10% Pd/C (Selcat Q), 150 mL water and 50 mL organic solvent, H2SO4/3PN molar ratio = 1.0, 50 °C, 6 bar. b 200 mL water. | ||||||
| 1 | Dichloromethane | 4.5 | 99 | 77.1 | 76 | 38.7 |
| 2 | Toluene | 4.0 | 98 | 54.6 | 54 | 119.3 |
| 3 | Hexane | 1.0 | 90 | 43.0 | 39 | 164.5 |
| 4b | — | 1.0 | 93 | 58.8 | 55 | 166.9 |
Next, the effect of solvents was also investigated. The results of the reduction of 3PN to 3PIPA in the mixtures of water and various organic solvents, over 10% Pd/C (catalyst/substrate ratio = 0.3 g g−1), using sulphuric acid as additive (H2SO4/3PN molar ratio = 1.0), at 50 °C and 6 bar are shown in Table 7.
When a toluene/water solvent mixture was applied, the selectivity (54%) and the purity of product (54.6% 3PIPA-content) significantly decreased compared to those ones obtained in dichloromethane/water, but the reaction time for complete conversion (4.0 h) and the isolated yield (98%) were practically the same values. In water only, the reaction was very fast (1 h), as well as the selectivity to 3PIPA (55%) and the product purity (58.8% 3PIPA-content) were very similar to those achieved with toluene, 3PIPA·H2SO4 was isolated in lower yield (93%). Using a hexane/water mixture, the reaction was also the fastest (1 h), but the isolated yield (90%), the selectivity (39%) and the purity of product (43.0% 3PIPA-content) were appreciable lesser than those obtained by applying other solvents.
As seen, a significant decrease was observed in the primary amine selectivity (98 → 76%) compared to the para-substituted piperidine derivative (4PIPA). This was due to the formation of 3MPPD in a larger amount (22.9–57.0%) depending on the type of organic solvents used. Whereas, there is a similarity to the hydrogenation of 4PN to 4PIPA, namely it is expedient to apply an organic solvent in this reduction, and the best results were achieved in a dichloromethane/water solvent mixture.
The effect of organic solvents on the reduction of 3PN to 3PA was also investigated. The results of this hydrogenation in the mixtures of different organic solvents and water, over 10% Pd/C, at 0.2 g g−1catalyst/substrate ratio, in the presence of 0.5 mol mol−1 H2SO4, at 30 °C and 6 bar are summarised in Table 8.
| Entry | Organic solvents | Reaction time for complete conversion (h) | Product mixture | Selectivity to 3PA (%) | v 0 (NL H2 gPd−1 h−1) | |
|---|---|---|---|---|---|---|
| Isolated yield (%) | 3PA-Content (%) | |||||
| a Reaction conditions: 5.0 g (48.1 mmol) 3PN, 1.0 g 10% Pd/C (Selcat Q), 200 mL water and 50 mL organic solvent, H2SO4/3PN molar ratio = 0.5, 30 °C, 6 bar. b 200 mL water. | ||||||
| 1 | Dichloromethane | 2.0 | 94 | 77.0 | 72 | 36.1 |
| 2 | Toluene | 1.0 | 88 | 62.9 | 56 | 55.9 |
| 3 | tert-Butyl methyl ether | 1.2 | 81 | 62.8 | 51 | 55.6 |
| 4 | Hexane | 0.9 | 89 | 68.7 | 61 | 106.4 |
| 5 | Ethyl acetate | 1.0 | 87 | 66.7 | 58 | 69.0 |
| 6b | — | 0.8 | 91 | 69.1 | 63 | 111.2 |
Applying dichloromethane/water, the best isolated yield (94%), purity (77.0% 3PA-content) and selectivity to 3PA (72%) were achieved after a 2-hour reaction time. When a hexane/ water solvent mixture was used, 3PA·0.5H2SO4 was isolated in an appreciably lower yield (89%), selectivity (61%) and purity (68.7%) within 0.9 h. Using ethyl acetate or toluene as an organic solvent, the isolated yields (87 and 88%, respectively), the selectivities to 3PA (58 and 56%, respectively), the product purities (66.7 and 62.9% 3PA-content, respectively) and reaction time (1 h) were very similar to each other. In water only, the isolated yield (91%) was similar to that experienced in the presence of toluene, whereas both the purity of product (69.1% 3PA-content) and the selectivity to 3PA (63%) became significantly lower in the fastest reaction (0.8 h). When tert-butyl methyl ether was applied as a solvent pair of water, it provided the worst results, namely moderated isolated yield (81%), primary amine selectivity (51%) and purity (62.8% 3PA-content) were obtained. The main reason for the lower primary amine selectivity was the formation of larger amounts of by-products (Fig. 5), i.e. 1,4,5,6-tetrahydropyridine-3-carbonitrile (THPCN), 3-piperidinecarbonitrile (3PIPCN), 3-pyridinemethanol (3POH) and bis(3-pyridylmethyl) ether (B3PE), respectively, were formed in a ratio of 19.3–22.7% for THPCN, 7.5–9.6% for 3PIPCN, 2.2–18.2% for 3POH and 6.2–15.0% for B3PE.
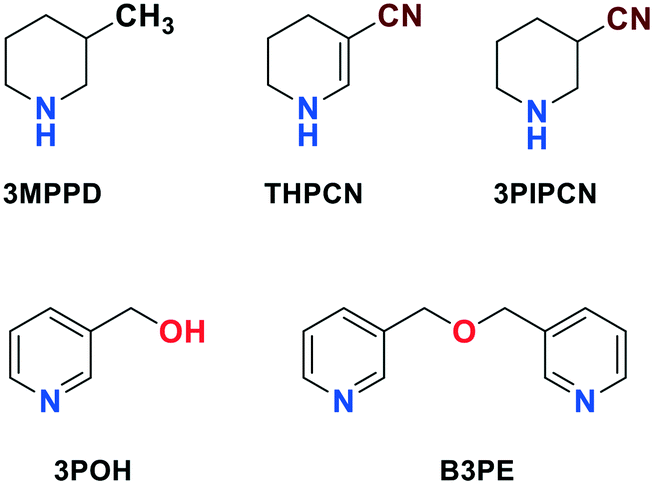 | ||
| Fig. 5 Typical by-products in the Pd-catalysed hydrogenation of 3PN to 3PIPA or 3PA. | ||
Accordingly, there was also a significant primary amine selectivity decrease (93 → 72%) in the hydrogenation of 3PN to 3PA (the meta-substituted pyridine derivative) compared to that of the para-substituted one (4PN to 4PA). However, it is favourable to apply an organic solvent in this reduction, contrary to the hydrogenation of 4PN to 4PA (vide supra), because the best results were obtained in the presence of dichloromethane beside water.
These significant differences in the primary amine selectivities can presumably be attributed to the dissimilar adsorption abilities of the nitrile starting materials, imine intermediates and amine products on palladium which will be discussed later (vide infra).
Hydrogenation of 2-pyridinecarbonitrile
Finally, the hydrogenation of the ortho-substituted isomer of pyridinecarbonitrile (2PN) was examined. First, the reduction of 2PN to 2PA was performed over 10% Pd/C, i.e. the conversion of the nitrile group to the amine one, applying the optimal reaction conditions found for 4PN to 4PA (0.1 g g−1 catalyst/substrate ratio, water, 0.5 mol mol−1 H2SO4, 30 °C and 6 bar).As seen in Table 9 (entry 1), the reduction of 2PN was complete after 1 h and the product (2PA·0.5H2SO4) was isolated in 93% yield, but the primary amine selectivity was moderate (54%) due to the formation of secondary amine, bis(2-pyridylmethyl)amine (B2PA), in a higher amount (41.4%). Thus, the amount of acidic additive was increased from 0.5 to 1.0 H2SO4/2PN molar ratio (Table 9, entry 2) to avoid forming secondary amine. Accordingly, 2PA·H2SO4 was obtained in a higher isolated yield (98%), but its purity (39.0% 2PA-content) and the primary amine selectivity (38%) remained still unsatisfactory. Although the amount of B2PA significantly decreased (41.4 → 4.0%), other by-product was also detected, namely 2-pyridinemethanol (2POH) was formed in a higher ratio (57.0%).
| Entry | Organic solvents | H2SO4/2PN ratio (mol−1 mol−1) | Reaction time for complete conversion (h) | Product mixture | Selectivity to 2PA (%) | v 0 (NL H2 gPd−1 h−1) | |
|---|---|---|---|---|---|---|---|
| Isolated yield (%) | 2PA-Content (%) | ||||||
| a Reaction conditions: 5.0 g (48.1 mmol) 2PN, 0.5 g 10% Pd/C (Selcat Q), 150 mL water and 50 mL organic solvent, 30 °C, 6 bar. b 200 mL water. | |||||||
| 1b | — | 0.5 | 1.0 | 93 | 58.0 | 54 | 195.0 |
| 2b | — | 1.0 | 0.3 | 98 | 39.0 | 38 | 433.2 |
| 3 | Dichloromethane | 1.0 | 0.8 | 98 | 58.1 | 57 | 256.4 |
| 4 | Toluene | 1.0 | 0.3 | 95 | 42.1 | 40 | 336.8 |
| 5 | tert-Butyl methyl ether | 1.0 | 0.3 | 95 | 43.4 | 41 | 331.2 |
| 6 | Hexane | 1.0 | 0.3 | 95 | 32.0 | 30 | 314.9 |
| 7 | Ethyl acetate | 1.0 | 0.3 | 95 | 50.7 | 48 | 334.1 |
Next, the influence of organic solvents was also investigated. As also seen in Table 9, the dichloromethane/ water solvent mixture provided the highest isolated yield (98%), selectivity to 2PA (57%) and purity (58.1% 2PA-content) after 0.8 h. Applying ethyl acetate, the product was isolated in 95% yield with moderate purity (50.7% 2PA-content) and primary amine selectivity (48%). Using a water/toluene mixture, the reduction of 2PN showed similar results (95% isolated yield and 40% selectivity to 2PA) to those obtained in the water/tert-butyl methyl ether one (95% and 41%, respectively). When a water/ hexane solvent mixture was applied, the hydrogenation of 2PN resulted in a high isolated yield (95%), but the selectivity (30%) and the purity of product (32.0% 2PA-content) were significantly weaker. In all cases, the low primary amine selectivity was due to the formation of 2POH in a higher amount (33.9–64.6%) presumably in consequence of the more acidic conditions (1.0 H2SO4/2PN molar ratio) causing more side reactions involving water (formation of aldehyde then alcohol).
When the hydrogenation of 2PN to 2PIPA, i.e. the conversion of the nitrile group and the pyridine ring to the corresponding saturated ones, was studied adapting the optimal reaction conditions found for 4PN to 4PIPA (0.3 g g−1 catalyst/substrate ratio, dichloromethane/water, 1.0 mol mol−1 H2SO4, 30 °C and 6 bar) high isolated yield (95%) was obtained, but the primary amine selectivity was very weak (10%). The complete reduction of 2PN required an appreciable longer reaction time (7 h) and higher temperature (50 °C), but the purity of 2PIPA·H2SO4 was very low (10.9%) because a complex product mixture containing mainly 2-piperidinemethanol (2PIPOH, 24.7%), 2-(N-methylaminomethyl)piperidine (2MPIPA, 19.4%) and bis(2-piperidylmethyl)amine (B2PIPA, 16.9%) was formed during this hydrogenation (Fig. 6) according to the GC–MS analysis. Therefore, it was not further examined.
 | ||
| Fig. 6 Typical by-products in the Pd-catalysed hydrogenation of 2PN to 2PIPA. | ||
Although moderate primary amine selectivity (57%) was achieved during the complete conversion of 2PN to 2PA over a 10% Pd/C (Selcat Q) catalyst, in water/dichloromethane, in the presence of 1.0 mol mol−1 H2SO4, at 30 °C and 6 bar, this result is much better than those reported previously by other research groups using precious metal catalysts, especially palladium.28
Theoretical considerations
In our previous studies concerning the Pd- or Pt-catalysed hydrogenation of nitriles,5,6 quantum chemical calculations (DFT approach) proved to be a very useful tool for modelling the adsorption of imine intermediates on palladium or platinum to elucidate the dissimilarities in the observed primary amine selectivities.Thus, we also performed similar quantum chemical calculations to explain the selectivity differences in the Pd-catalysed hydrogenation of these constitutional isomers of pyridinecarbonitriles. Adsorption energy profiles were computed to study the interactions between the nitrile starting materials, imine intermediates, amine products and other typical by-products and palladium. As known,57 the adsorption energies of molecules containing protonated N (e.g. ammonia, pyridine) are slightly higher by 0.1–0.2 eV (ca. 10–20 kJ mol−1) than that of nitrogen in free base form. Although our calculation results related to these pyridine and piperidine compounds in their base forms will provide barely different values, their trends and relative positions will remain practically the same.
First, the decrease in primary amine selectivity observed in the hydrogenation of meta-substituted pyridinecarbonitrile (3PN) was examined compared to the para-substituted compound (4PN). Fig. 7 shows the temperature-dependent by-product formation during the hydrogenation of 3PN. When the hydrogenation of 3PN to 3PA was carried out at 30 °C, the decrease in selectivity to 3PA (72%) was caused by the formation of 1,4,5,6-tetrahydropyridine-3-carbonitrile (THPCN), while at higher temperature (50 °C), during the reduction of 3PN to 3PIPA, the lower selectivity to 3PIPA (76%) was due to the formation of 3-methylpiperidine (3MPPD). The calculated most stable conformers of 3PN, 3PI, THPCN, 3PA, 3PIPA and 3MPPD are located on a Pd48 three-layer slab with a (111) surface, according to the energetically favoured adsorption mode of pyridine or piperidine (the heteroaromatic ring parallel to the surface) on Pd(111). To elucidate and confer these interactions, the energies of adsorption (ΔEads) of these compounds on palladium were also computed. As seen, the THPCN by-product formed at 30 °C (Route B) is more strongly adsorbed on the surface of palladium than the imine intermediate (3PI, Route A) (ΔEads = −72.73 and −44.08 kJ mol−1, respectively), which could explain why this by-product can remain stable and exist for a longer time at this temperature, but it can further be converted to the corresponding fully saturated amine (3PIPA) at 50 °C, similarly to that of 3PA. However, 3PIPA is very strongly adsorbed (ΔEads = −127.11 kJ mol−1) on Pd, thus there is a higher chance for occurring a deamination process providing 3-methylpiperidine (3MPPD) as another typical by-product.
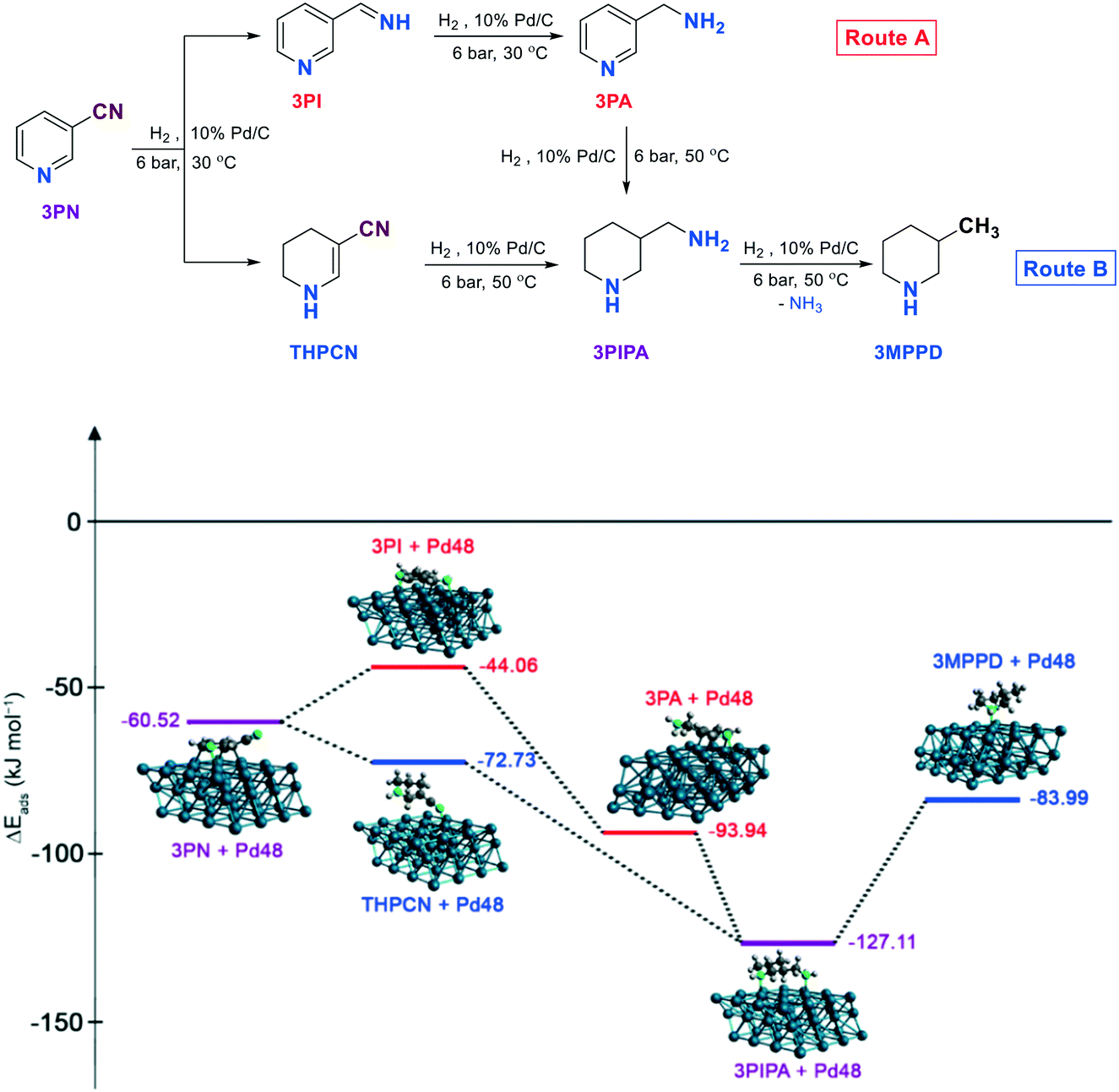 | ||
| Fig. 7 Computed adsorption energy (ΔEads) profiles for the hydrogenation of 3-pyridinecarbonitrile (3PN) to 3-(aminomethyl)pyridine (3PA) and/or 3-(aminomethyl)piperidine (3PIPA) on a Pd48 three-layer slab with a (111) surface. | ||
To clarify the lower selectivities to 2PA and 2PIPA, respectively, the energies of adsorption (ΔEads) of all compounds on palladium were also computed and their adsorption energy (ΔEads) profiles were created. As seen in Fig. 8, the most significant differences can be found in the ΔEads values of imines (4PI, 3PI, 2PI), (aminomethyl)pyridines (4PA, 3PA, 2PA) and (aminomethyl)piperidines (4PIPA, 3PIPA, 2PIPA), while there is no appreciable variation in the adsorption strength of the starting materials (4PN, 3PN, 2PN) on Pd comparing them to each other. Since 2PI and 2PA can adsorb very strongly on palladium (ΔEads = −124.02 and −121.18 kJ mol−1, respectively) due to the two adjacent nitrogen atoms and the pyridine ring positioned in the same plane, they could remain on the surface of Pd for a longer time, and thus the side reactions involving water (formation of aldehyde then alcohol derivatives) could take place more likely in the reaction mixture. Moreover, the adsorption of the fully saturated 2PIPA is weaker (ΔEads = −80.44 kJ mol−1) than that of its intermediates (2PI and 2PA), therefore its desorption is easier from the surface of the catalytically active metal and it can react with other by-products (e.g.2PAld) in the reaction mixture, as well.
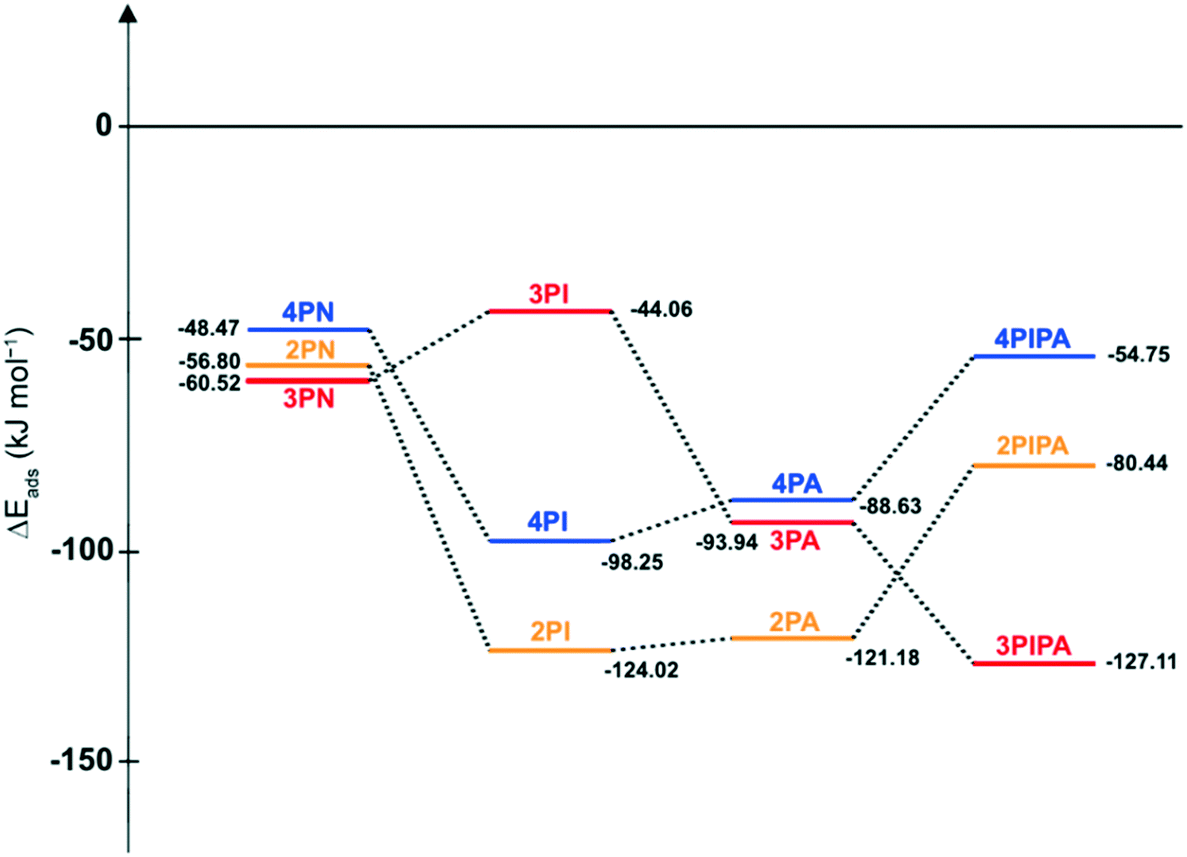 | ||
| Fig. 8 Comparing adsorption energy (ΔEads) profiles for the reduction of three constitutional isomers of pyridinecarbonitriles (4PN, 3PN, 2PN) to (aminomethyl)pyridines (4PA, 3PA, 2PA) or -piperidines (4PIPA, 3PIPA, 2PIPA) through imine intermediates (4PI, 3PI, 2PI) on a Pd48 cluster with a (111) surface. | ||
The very high selectivity to 4PA or 4PIPA can be attributed to the relatively strong adsorption strength of 4PI on Pd (ΔEads = −98.25 kJ mol−1) thus it could remain on the surface of the catalyst for a longer time and the possibility of by-product formation could be decreased. In addition, the relatively weaker adsorption interaction of 4PA or 4PIPA with Pd (ΔEads = −88.63 and −54.75 kJ mol−1, respectively) could hinder the taking place of other side reactions (e.g. deamination).
Based on the results of these DFT calculations, it can be concluded that the differences in the primary amine selectivities observed in the Pd-catalysed hydrogenation of these pyridinecarbonitriles were mainly due to the various adsorption interactions between the imine intermediates (4PI, 3PI, 2PI), amine products (4PA, 3PA, 2PA, 4PIPA, 3PIPA, 2PIPA), by-products (THPCN, 3MPPD) and palladium. Nevertheless, the more acidic water could also cause selectivity decrease in case of the hydrogenation of 2PN.
Conclusions
Three constitutional isomers of pyridinecarbonitriles (4-, 3- or 2-pyridinecarbonitrile) were hydrogenated to the corresponding pyridyl- or piperidylmethylamines over a 10% palladium on carbon catalyst, under mild reaction conditions (30–50 °C, 6 bar), in liquid phase adapting our slightly modified selective and industrially feasible method developed previously.3 By systematically changing the reaction conditions (acidic additives, solvents, temperature, catalyst amount), not only an appropriate primary amine selectivity to the desired pyridine derivatives (4PA, 3PA, 2PA) was achieved, but also the piperidylmethylamines were successfully prepared (4PIPA, 3PIPA) by further hydrogenation of the pyridine ring in addition to the nitrile group. Complete conversion was achieved in all cases, but the very high chemoselectivity to 4PIPA or 4PA (98 and 93%, respectively) decreased to 76% (3PIPA) and 72% (3PA), as well as 10% (2PIPA) and 58% (2PA) depending on the position of nitrile group in the pyridine ring. The differentiation between the products (pyridyl- or piperidylmethylamines) can be fine-tuned by simply adjusting the amount of acidic additive (0.5 or 1.0 H2SO4/nitrile molar ratio). The purity of products prepared as sulphate salts (4PIPA·H2SO4, 4PA·0.5H2SO4) was over 95% without applying any special procedures.Although the complete saturation of pyridine ring over Pd/C can take place under ambient conditions (atmospheric pressure, room temperature),58 but this requires the use of acetic acid as solvent and long reaction time (15 h). A relatively higher pressure (6 bar) is applied in our process, but this value is in the range (1–6 bar) that usually utilised in industry and provides a fast and complete reaction within 0.1–5.5 h depending on the catalyst amount. In addition, water is used mainly as a solvent, which contains a low amount of acidic additive (1.0 or 0.5 molar ratio related to nitriles).
To clear the reasons for the differences in the primary amine selectivities in these Pd-catalysed hydrogenations, high-level quantum chemical calculations were accomplished by using the DFT approach. The molecular modelling computations revealed that the diverse adsorption strengths of the imine intermediates, the amine products and other by-products (a partially saturated pyridinecarbonitrile or a deaminated piperidine derivative) could affect the selectivity of these reductions. The calculated structures of the conformers with minimal energy and the adsorption energy (ΔEads) profiles of these compounds on Pd(111) showed that observed selectivity differences were due to the various adsorption interaction between these N-containing heterocyclic derivatives and palladium, as well as the position of the nitrile group in the pyridine ring. In addition, the amount of acidic additive (H2SO4) can also influence the chemoselectivity in these hydrogenations. These distinctions can increase the possibility of by-products formation both on the catalyst surface and in the reaction mixture.
To extend this selective hydrogenation process for the conversion of heteroaromatic nitriles to primary amines over various platinum metal catalysts, further investigations are in progress.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research reported in this paper and carried out at BME has been supported by the NRDI Fund (TKP2020 NC, Grant No. BME-NC) based on the charter of bolster issued by the NRDI Office under the auspices of the Ministry for Innovation and Technology. This work was also connected to the scientific program of the “Talent care and cultivation in the scientific workshops at BME” project which was supported by the grant TÁMOP-4.2.2.B-10/1-2010-0009. The authors thank Dr. Antal Sárkány for determining dispersion of the catalyst, as well as Dr. Tibor Novák and Dr. Miklós Nyerges (Servier Research Institute, Hungary) for providing GC–MS analysis. The authors are also grateful to Dr. Tamás Kárpáti for his valuable advice concerning the molecular modelling calculations.References
- P. Roose, K. Eller, E. Henkes, R. Rossbacher and H. Höke, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2015, pp. 1–55 Search PubMed.
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, VCH, Weinheim, 1997 Search PubMed.
- L. Hegedűs and T. Máthé, Appl. Catal., A, 2005, 296, 209–215 CrossRef.
- L. Hegedűs, T. Máthé and T. Kárpáti, Appl. Catal., A, 2008, 349, 40–45 CrossRef.
- K. Lévay, K. D. Tóth, T. Kárpáti and L. Hegedűs, ACS Omega, 2020, 5, 5487–5497 CrossRef PubMed.
- K. Lévay, T. Kárpáti and L. Hegedűs, J. Ind. Eng. Chem., 2021, 101, 279–292 CrossRef.
- G. K. Andrikopoulos, S. Pastromas and S. Tzeis, World J. Cardiol., 2015, 7, 76–85 CrossRef PubMed.
- F. Fringuelli, F. Pizzo, S. Tortoioli and L. Vaccaro, J. Org. Chem., 2004, 69, 7745–7747 CrossRef CAS PubMed.
- S. Bollinger, H. Hübner, F. W. Heinemann, K. Meyer and P. Gmeiner, J. Med. Chem., 2010, 53, 7167–7179 CrossRef CAS PubMed.
- G. Blay, V. Hernández-Olmos and J. R. Pedro, Chem. Commun., 2008, 4840–4842 RSC.
- G. Grach, G. Pieters, A. Dinut, V. Terrasson, R. Medimagh, A. Bridoux, V. Razafimahaleo, A. Gaucher, S. Marque, J. Marrot, D. Prim, R. Gil, J. Giner Planas, C. Viñas, I. Thomas, J.-P. Roblin and Y. Troin, Organometallics, 2011, 30, 4074–4086 CrossRef CAS.
- D. Malhotra, J. P. Page, M. E. Bowden, A. Karkamkar, D. J. Heldebrant, V.-A. Glezakou, R. Rousseau and P. K. Koech, Ind. Eng. Chem. Res., 2017, 56, 7534–7540 CrossRef CAS.
- S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289–302 CrossRef CAS.
- D. B. Bagal and B. M. Bhanage, Adv. Synth. Catal., 2015, 357, 883–900 CrossRef CAS.
- K. Lévay and L. Hegedűs, Period. Polytech., Chem. Eng., 2018, 62, 476–488 Search PubMed.
- D. B. Bagal and B. M. Bhanage, in Science of Synthesis: Catalytic Reduction in Organic Synthesis, ed. J. G. de Vries, Thieme Verlag, Stuttgart, 2018, vol. 2, pp. 375–401 Search PubMed.
- A. M. Allgeier and S. K. Sengupta, in Hydrogenation: Catalysts and Processes, ed. S. D. Jackson, Walter de Gruyter, Berlin, Boston, 2018, pp. 107–154 Search PubMed.
- M. Feller, Phys. Sci. Rev., 2019, 4, 20180033 Search PubMed.
- K. Lévay and L. Hegedűs, Curr. Org. Chem., 2019, 23, 1881–1900 CrossRef.
- J. A. Garduño and J. J. García, ACS Catal., 2020, 10, 8012–8022 CrossRef.
- N. Toshinari and I. Yoshio, JP Pat., H06749, 1994 Search PubMed.
- J. Wang, Q. Tang, S. Jin, Y. Wang, Z. Yuan, Q. Chi and Z. Zhang, New J. Chem., 2020, 44, 549–555 RSC.
- I. Yoshio and N. Toshinari, JP Pat., H0670012, 1994 Search PubMed.
- P. E. Garrou, US Pat., 4159382, 1979 Search PubMed.
- J. F. J. Engbersen, A. Koudijs, M. H. A. Joosten and H. C. van der Plas, J. Heterocyclic Chem., 1986, 23, 989–990 CrossRef CAS.
- M. Vilches-Herrera, S. Werkmeister, K. Junge, A. Börner and M. Beller, Catal. Sci. Technol., 2014, 4, 629–632 RSC.
- F. Chen, C. Topf, J. Radnik, C. R. Kreyenschulte, H. Lund, M. Schneider, A.-E. Surkus, L. He, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8781–8788 CrossRef CAS PubMed.
- R. Ferraccioli, D. Borovika, A.-E. Surkus, C. Kreyenschulte, C. Topf and M. Beller, Catal. Sci. Technol., 2018, 8, 499–507 RSC.
- D. Formenti, R. Mocci, H. Atia, S. Dastgir, M. Anwar, S. Bachmann, M. Scalone, K. Junge and M. Beller, Chem. – Eur. J., 2020, 26, 15589–15595 CrossRef CAS PubMed.
- M. Sheng, S. Yamaguchi, A. Nakata, S. Yamazoe, K. Nakajima, J. Yamasaki, T. Mizugaki and T. Mitsudome, ACS Sustainable Chem. Eng., 2021, 9, 11238–11246 CrossRef CAS.
- S. J. Rezaei, H. Khorramabadi, A. Hesami, A. Ramazani, V. Amani and R. Ahmadi, Ind. Eng. Chem. Res., 2017, 56, 12256–12266 CrossRef.
- S. Enthaler, K. Junge, D. Addis, G. Erre and M. Beller, ChemSusChem, 2008, 1, 1006–1010 CrossRef CAS PubMed.
- Z. Lu and T. J. Williams, Chem. Commun., 2014, 50, 5391–5393 RSC.
- S. Saha, M. Kaur, K. Singh and J. K. Bera, J. Organomet. Chem., 2016, 812, 87–94 CrossRef CAS.
- C. Bornschein, S. Werkmeister, B. Wendt, H. Jiao, E. Alberico, W. Baumann, H. Junge, K. Junge and M. Beller, Nat. Commun., 2014, 5, 4111 CrossRef CAS PubMed.
- S. Chakraborty, G. Leitus and D. Milstein, Chem. Commun., 2016, 52, 1812–1815 RSC.
- A. Mukherjee, D. Srimani, S. Chakraborty, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2015, 137, 8888–8891 CrossRef CAS PubMed.
- Z. Shao, S. Fu, M. Wei, S. Zhou and Q. Liu, Angew. Chem., Int. Ed., 2016, 55, 14653–14657 CrossRef CAS PubMed.
- R. Adam, C. B. Bheeter, J. R. Cabrero-Antonino, K. Junge, R. Jackstell and M. Beller, ChemSusChem, 2017, 10, 842–846 CrossRef CAS PubMed.
- S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8809–8814 CrossRef CAS PubMed.
- S. Weber, L. F. Veiros and K. Kirchner, Adv. Synth. Catal., 2019, 361, 5412–5420 CrossRef CAS PubMed.
- A. A. Rodríguez, J. A. Garduño and J. J. García, New J. Chem., 2020, 44, 1082–1089 RSC.
- T. Máthé, A. Tungler and J. Petró, US Pat., 4361500, 1982 Search PubMed.
- NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg, MD, 20899, (accessed December 2021) Search PubMed.
- W. I. F. David, K. Shankland, J. van de Streek, E. Pidcock, W. D. S. Motherwell and J. C. Cole, J. Appl. Crystallogr., 2006, 39, 910–915 CrossRef CAS.
- A. Sárkány, Z. Zsoldos, F. Furlong, J. W. Hightower and L. Guczi, J. Catal., 1993, 141, 566–582 CrossRef.
- A. Sárkány, G. Stefler and J. W. Hightower, Appl. Catal., A, 1995, 127, 77–92 CrossRef.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, U. Umari and R. M. Wentzcovitch, Quantum ESPRESSO, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- M. Freifelder, Practical Catalytic Hydrogenation, Wiley, New York, 1971, p. 25 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds: Part A: Theory and Applications in Inorganic Chemistry, John Wiley, New York, 2009, p. 194 Search PubMed.
- F. Milletti, L. Storchi, L. Goracci, S. Bendels, B. Wagner, M. Kansy and G. Cruciani, Eur. J. Med. Chem., 2010, 45, 4270–4279 CrossRef CAS PubMed.
- P. N. Rylander, in Catalysis in Organic Syntheses, ed. W. H. Jones, Academic Press, New York, 1980, pp. 155–171 Search PubMed.
- D. J. Segobia, A. F. Trasarti and C. R. Apesteguía, Catal. Sci. Technol., 2014, 4, 4075–4083 RSC.
- P. E. Gunjal and V. V. Ranade, in Industrial Catalytic Processes for Fine and Specialty Chemicals, ed. S. S. Joshi and V. V. Ranade, Elsevier, Amsterdam, 2016, pp. 263–314 Search PubMed.
- S. Rangarajan and M. Mavrikakis, ACS Catal., 2016, 6, 2904–2917 CrossRef CAS.
- N. Tanaka and T. Usuki, Eur. J. Org. Chem., 2020, 2020, 5514–5522 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |