
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Squalene–polyethyleneimine–dynamic constitutional frameworks enhancing the enzymatic activity of carbonic anhydrase†
Dan-Dan
Su
ab,
Karim
Aissou
a,
Yan
Zhang
 c,
Virginie
Gervais
d,
Sebastien
Ulrich
*b and
Mihail
Barboiu
*a
c,
Virginie
Gervais
d,
Sebastien
Ulrich
*b and
Mihail
Barboiu
*a
aInstitut Européen des Membranes, Adaptive Supramolecular Nanosystems Group, University of Montpellier, ENSCM-CNRS, Place E. Bataillon CC047, Montpellier, F-34095, France. E-mail: mihail-dumitru.barboiu@umontpellier.fr
bInstitut des Biomolécules Max Mousseron (IBMM), CNRS, Université de Montpellier, ENSCM, Montpellier, France. E-mail: sebastien.ulrich@cnrs.fr
cKey Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, School of Pharmaceutical Sciences, Jiangnan University, 1800 Lihu Avenue, Wuxi, 214122, P.R. China
dUniversité Paris-Saclay, CEA, CNRS, Institute for Integrative Biology of the Cell (I2BC), 91198, Gif-sur-Yvette, France
First published on 25th April 2022
Abstract
Carbonic anhydrase is an essential enzyme that catalyzes the hydration/dehydration of carbon dioxide, which is highly relevant to carbon capture processes. However, its efficient encapsulation in host materials is of utmost importance for the system performance (durability, stability, and efficiency). In this paper we demonstrate that polyethyleneimine–polyethylene glycol squalene constitutional nanoparticles PEI–DCFs, synthesized via reversible imine/amino-carbonyl chemistry, are efficient host matrixes for bovine carbonic anhydrase (bCA) encapsulation. This system showed an impressive one-order-of-magnitude-improved catalytic proficiency (kcat/Km = 7396 M−1 s−1) as compared to bCA alone (kcat/Km = 504 M−1 s−1). This performance rivals the current state-of-the-art systems with equivalent amounts of the enzyme, even after heating for a prolonged period at 80 °C, translating into its direct application for enhancing carbon dioxide capture and conversion.
Introduction
The significant growth of carbon dioxide (CO2) emissions is a serious environmental and public health issue causing atmospheric pollution and the green-house effect. Many attempts have been made using electrocatalytic reduction, photosynthesis, chemical absorption and membrane transport methods to capture and store CO2,1,2 but so far the high costs and energy requirements restrict their effective application.3 On the other hand, a modern alternative rests on dynamic and adaptive materials which can be obtained by cheaper and more versatile covalent/supramolecular self-assembly processes – a proof-of-concept having already reported simultaneous CO2 capture and metal purification.4During the past decades, the use of carbonic anhydrases (CAs) for CO2 hydration and storage has attracted attention due to its high turnover rate, its environmentally benign properties and wide sources.5 CAs are already exploited in artificial systems for applications in CO2 hydration/dehydration,6,7 CO2 hydrogenation8 or CO2 conversion to methanol.9 A wide range of materials have been used for CA immobilization in order to improve its tolerance to hazardous conditions such as high temperature (up to 80–100 °C), high acidity, etc., but also to maintain its efficiency when embedded in artificial matrix hosts.10 Nanotechnology has the potential to address this problem. Indeed, multivalent binders can lead to interfacial interactions/stabilization with the enzyme while also promoting its encapsulation in nano-bioreactors or biomimetic protocells. The high surface-to-volume ratio and the controllable display of binding/reactive groups have been used in artificial11 and CA-based catalytic nanoparticles.12,13
The catalytic center of CAs contains a zinc-binding pocket and a hydrophobic binding pocket. In the catalytic cycle of CA, hydroxyl ions are produced under neutral pH conditions to generate bicarbonate upon reaction with CO2. Protons are therefore byproducts which are expelled out of the catalytic center in a rate-limiting step.14,15 While most of the host materials developed hitherto inhibit the catalytic activity of CAs either by altering the enzyme conformation or by hindering the access of the substrates to the catalytic center, a few recent systems showed an enhancement of catalytic activity instead. This peculiar behavior is best understood by the host facilitating the proton shuttle out of the active site.16 We previously reported interesting results in that direction using dynamic constitutional frameworks (DCFs) which adaptively stabilize a microenvironment through confining H-bonding or electrostatic interactions on the outer enzyme surface and play the role of multi-proton sponges, both essential for an effective catalytic activation mechanism.17,18 In this case, CA can be activated by simple addition of building blocks in aqueous solution, which are easily and modularly self-assembled via reversible covalent bonds on the external enzyme surface.19
On the other hand, solid absorbents, made with polyethyleneimine (PEI) loaded on different porous substrates, are promising materials for carbon dioxide capture.20 The gas absorption/transport mechanism is based on the same hydrolytic reaction as performed by CA, which may further convert CO2 into carbamic acid and/or bicarbonate at the secondary amine sites of PEI in the presence of water. This makes PEI one of the highly performing nanomaterials for CO2 capture from flue gas or air.
Herein, we report a novel strategy, aimed to be as simple and broadly applicable as possible relating to the encapsulation of bovine carbonic anhydrase (bCA) within a polyethyleneimine (PEI)–DCF nanoparticles with enhanced catalytic activity. This results in the production of scalable biomimetic DCF–bCA nanoparticles that remarkably outperform the classical bCA activity in solution and combine the extraordinary absorption capacity of PEI materials.20 This study leads to a greater fundamental understanding of how CA incorporation can be optimized at the nanoscale to simultaneously stabilize and activate the overall catalytic cycles of a DCF–PEI–bCA system operated under harsh conditions, as required by industrial CO2 capture processes. The relatively straightforward development of DCF–PEI–bCA nanoparticles is a very important and previously unreported strategy. They are operating synergistically to give enhanced catalytic turnovers, higher that those resulted from the sum of the former components that are relevant to their subsequent use in larger scale applications as absorbent materials or membranes.
Results and discussion
Design and synthesis
The preparation of DCFs was carried by self-assembly of the following components: (1) 1.3.5-benzenetrialdehyde, BTA as a core center, PEGylated-1,1′,2-tris-norsqualene constructed from (2) hydrophobic squalene aldehyde, SQ-CHO with a remarkable folded conformation in water,21 (3) hydrophilic poly(ethylene glycol)-bis-(3-aminopropyl)-terminated (Mn ∼ 1500) PEG1500, with a favorable solubility in water and used as an artificial biocompatible chaperone for bCA refolding and limiting aggregation22 and (4) different polyethyleneimines–PEIs: a) branched polyethyleneimine, bPEI800 (Mn ∼ 800) (12 PEI units), b) pentaethylene-hexamine, PEH (5 PEI units), and c) triethylenetetramine, TAA (3 PEI units) used as charged H-bonding heads interacting with the bCA surface and presumed to act as catalytic multivalent proton shuttles for bCA activation (Fig. 1). DCF–PEI, DCF–PEH and DCF–TAA were prepared via the aminocarbonyl/imine reversible chemistry between (1), (2), (3) and positively-charged bPEI800, PEH and TAA, respectively in order to quantify their role in the enhanced catalytic effects of DCF–bCA systems at room temperature and 80 °C. The 1H-NMR spectra are in accordance with the proposed structures (Fig. S1–S5†). | ||
| Fig. 1 a) Schematic representation of molecular components used to construct multivalent dynamic constitutional frameworks (DCFs) for bovine carbonic anhydrase (bCA) immobilization in hybrid nanoparticles. The four-component DCFs are synthesized step-wise through imine/amino-carbonyl chemistry, and different cationic headgroups (bPEI800, PEH, and TAA), shown in orange, were tested. b) Fluorescence spectra showing the quenching of bCA upon addition of increasing amounts of DCF–PEI (top) or bPEI800 (bottom). c) Stern–Volmer plots23 of relative fluorescence intensity versus cationic monomer concentration present in free and DCF bonded forms in solution, used for the determination of the Stern–Volmer constant (Ka) between bCA and DCFs/PEI monomer. | ||
Binding to bCA
The binding behaviors of DCFs or PEI monomers to bCA were first evaluated by monitoring the intrinsic fluorescence emission of bCA, at its maximum of emission λem = 343 nm, following the excitation at λex = 280 nm.23 An obvious decrease in fluorescence emission was observed, upon addition of the DCFs (DCF–PEI, DCF–PEH and DCF–TAA) onto an aqueous solution of bCA (Fig. 1b and S6†). From the Stern–Volmer analysis24 the observed Stern–Volmer constants of DCF–PEI, DCF–PEH and DCF–TAA with bCA were determined to be 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 052 M−1, 8400 M−1 and 7912 M−1 respectively (Fig. 1c). The superiority of DCF–PEI compared to DCF–PEH and DCF–TAA reveals that an increasing number of cationic groups results in a more effective quenching of the fluorescence emission of bCA, which can be a result of either a quenching triggered by the DCFs or by enforced structural changes of the enzyme upon binding to the DCFs. Compared with the DCFs, monomeric bPEI800, PEH and TAA show two orders of magnitude weaker effects, with Stern–Volmer constants of 201 M−1, 121 M−1 and 196 M−1, respectively, demonstrating the importance of multivalent presentation of positive PEIs on DCF surfaces.
052 M−1, 8400 M−1 and 7912 M−1 respectively (Fig. 1c). The superiority of DCF–PEI compared to DCF–PEH and DCF–TAA reveals that an increasing number of cationic groups results in a more effective quenching of the fluorescence emission of bCA, which can be a result of either a quenching triggered by the DCFs or by enforced structural changes of the enzyme upon binding to the DCFs. Compared with the DCFs, monomeric bPEI800, PEH and TAA show two orders of magnitude weaker effects, with Stern–Volmer constants of 201 M−1, 121 M−1 and 196 M−1, respectively, demonstrating the importance of multivalent presentation of positive PEIs on DCF surfaces.
Isothermal titration calorimetry (ITC) experiments were carried out in parallel (either by titrating the enzyme with ligands or by titrating ligands with bCA) and revealed marked differences (Table 1). While DCF–PEI, DCF–PEH, and bPEI800 were found to be similarly potent binders of bCA with association constants in a narrow range of 0.93–1.02 × 106 M−1, notable differences in thermodynamic behavior were observed. DCF–PEI with its cationic superiority leads to the most pronounced enthalpic contribution compared to DCF–PEH. DCF–TAA with the smallest cationic group gives the largest entropy change including contributions of the solvent entropy and the protein conformational entropy. The difference with the above fluorescence analysis indicates that binding (measured by ITC) is not correlated with the structural changes (monitored by intrinsic fluorescence measurements) of the enzyme. Altogether, these results show that the multivalency displayed by the DCF with the largest cationic group PEI (DCF–PEI) triggers the most drastic structural changes in bCA, through enthalpy- and entropy-driven binding, which may affect the catalytic efficiency of the enzyme. On the other hand, DCF–TAA with the smallest cationic group was found to be the most potent binder, with an association constant of 7.14 × 106 M−1 and a reaction mainly driven by entropy, but it does not trigger structural changes within the bCA enzyme according to the fluorescence assay.
| Samples | K d (μM) | K a (106 M−1) | ΔH (kcal mol−1) | −TΔS (kcal mol−1) |
|---|---|---|---|---|
| DCF–PEI | 1.04 ± 0.66 | 0.96 | −3.39 ± 0.32 | −4.77 |
| DCF–PEH | 1.07 ± 0.82 | 0.93 | −0.86 ± 0.10 | −7.28 |
| DCF–TAA | 0.14 ± 0.44 | 7.14 | −0.15 ± 0.04 | −9.22 |
| bPEI800 | 0.98 ± 0.84 | 1.02 | −0.22 ± 0.04 | −7.97 |
Nano-scale observation of DCFs–bCA conjugates
Self-assembly at the nanoscale of DCFs–bCA conjugates. Dynamic light scattering (DLS) was used to estimate the average diameter of nanoparticles formed after 12 h at room temperature in solutions of bCA in the presence of DCF–PEI, DCF–PEH and DCF–TAA. First, it is worth noting that there is no fluctuation in size for the DCFs alone, within a concentration range of 0.05 mM to 0.2 mM, with average sizes of 173.5 ± 0.2 nm, 127.0 ± 0.5 nm and 171.1 ± 1.6 nm for DCF–PEI, DCF–PEH and DCF–TAA, respectively (Table S3†). In the presence of bCA, the DLS results showed the formation of dimensionally similar nanoparticles (Fig. 2a–c). Thus, the addition of bCA does not affect the overall self-assembly in solution, occurring with the total inclusion of enzyme within bCA/DCF nanoparticles without affecting their size and shape. In contrast, the polyamine building blocks bPEI800, PEH, and TAA lead to the formation of much larger (>300 nm) and more polydisperse nanoparticles in the presence of bCA (Table S3†). | ||
| Fig. 2 DLS experiments showing the size distribution of colloidal aggregates in aqueous solutions of a) DCF–PEI with bCA, b) DCF–PEH with bCA and c) DCF–TAA with bCA. Representative AFM scans of d) DCF–PEI with bCA (1.5 × 1.5 μm; scale bar: 200 nm), e) DCF–PEH with bCA (1.5 × 1.5 μm; scale bar: 200 nm) and f) DCF–TAA with bCA (1.5 × 1.5 μm; scale bar: 200 nm). Representative TEM images of g) DCF–PEI, h) DCF–PEI with bCA and i) bPEI800 with bCA. The dense PEI–PEG regions are darker while the less dense grey brighter spots correspond to the SQ regions. The 5 nm spots in h) correspond to the bCA dispersed in the hydrophilic corona of PEG–PEI components. | ||
In order to further elucidate their aggregation behaviors, the bCA/DCF bioconjugates were imaged by atomic force microscopy (AFM) (Fig. 2d–f and S8†). The AFM topography images of DCF–PEI with bCA (Fig. 2d), DCF–PEH with bCA (Fig. 2e) and DCF–TAA with bCA (Fig. 2f) showed homogeneous particles over the entire samples, with no sign of aggregation. The apparent sizes of all samples were found to be below 200 nm, which is in good agreement with the DLS results, revealing the uniform particle distribution.
The transmission electronic microscopy TEM showed that the DCF–PEI (Fig. 2g) and DCF–PEI with bCA (Fig. 2h) had an aggregated morphology with an average overall size of 200 nm for all formulations. Similar results were obtained in previous studies on PEI decorated nanoparticles for gene delivery.25,26 Our results are in accordance with the DLS data, confirming the narrow particle size distribution of the aggregates, which was observed by TEM. DCF–PEI is obtained via aggregation of several discrete brighter and darker nanoparticles, most probably formed by segregation of squalene and PEG–PEI components, respectively, self-assembled via reversible covalent interactions at the interfaces between nanoparticles. Interestingly DCF–PEI exhibited a more complex core and shell morphology when bCA is added. The aggregates were composed of a darker central core of ∼50 nm and of a surrounding corona, in which well-defined and homogeneously distributed brighter nanoparticles can be identified (Fig. 2h). These white grey spots of ∼5 nm are hypothesized to be mostly composed of bCA for which the crystallographic data suggest an overall ellipsoid shape with the dimensions of 4 × 4 × 5 nm.27
The dark color of the bPEI800–bCA samples suggested that strong electrostatic aggregation occurred between bPEI800 and bCA, homogeneously distributed within dense nanoparticles. In contrast the DCF–PEI with the bCA sample showed that larger voids and bright less dense spots can be observed inside these hybrid samples when compared with the pristine PEI sample micrographs, where very few or no bright spots were seen.
The nano-structural distribution of dense and light regions within the DCF–bCA aggregates is essential to control the catalytic enzymatic performance, since they determine the pathways for the substrate flow and diffusion across the active nanoparticle.
Enzymatic activation activity of bCA
The esterase catalytic activity of cationic monomers alone/DCFs in the presence (Fig. S9†) or in the absence (Fig. S10†) of bCA was tested by monitoring the hydrolysis reaction of p-nitrophenyl acetate (NPA) in PBS buffer (pH 7.4) by UV-vis spectroscopy. The esterase activity through the hydrolysis reaction of NPA is directly related to CO2 hydration reaction catalyzed by both bCA and PEI systems.28 The time course of the absorbance at λ = 400 nm was monitored to estimate the catalytic activity relative to the reference activity of free bCA in solution at different concentrations of PEI monomers. Unless the concentration exceeds 0.1 mM, bPEI800 and DCF–PEI present negligible catalytic activity in the absence of bCA (Fig. 3a), which is in line with only a few examples in the literature reporting the catalytic activity of PEI itself28 on the CA stabilization within a PEI/dopamine protecting matrix.29 It is also similar to the literature data where only up to 43% of the primary amine of PEI is acetylated using acetic anhydride at 60 °C in anhydrous methanol.30 For further support, the 1H NMR analysis of aqueous solutions of PEI and NPA indeed showed the exclusive formation of p-nitrophenol (NP) and acetic acid which completely exclude the acetylation of PEI in aqueous solution at room temperature (Fig. S19†).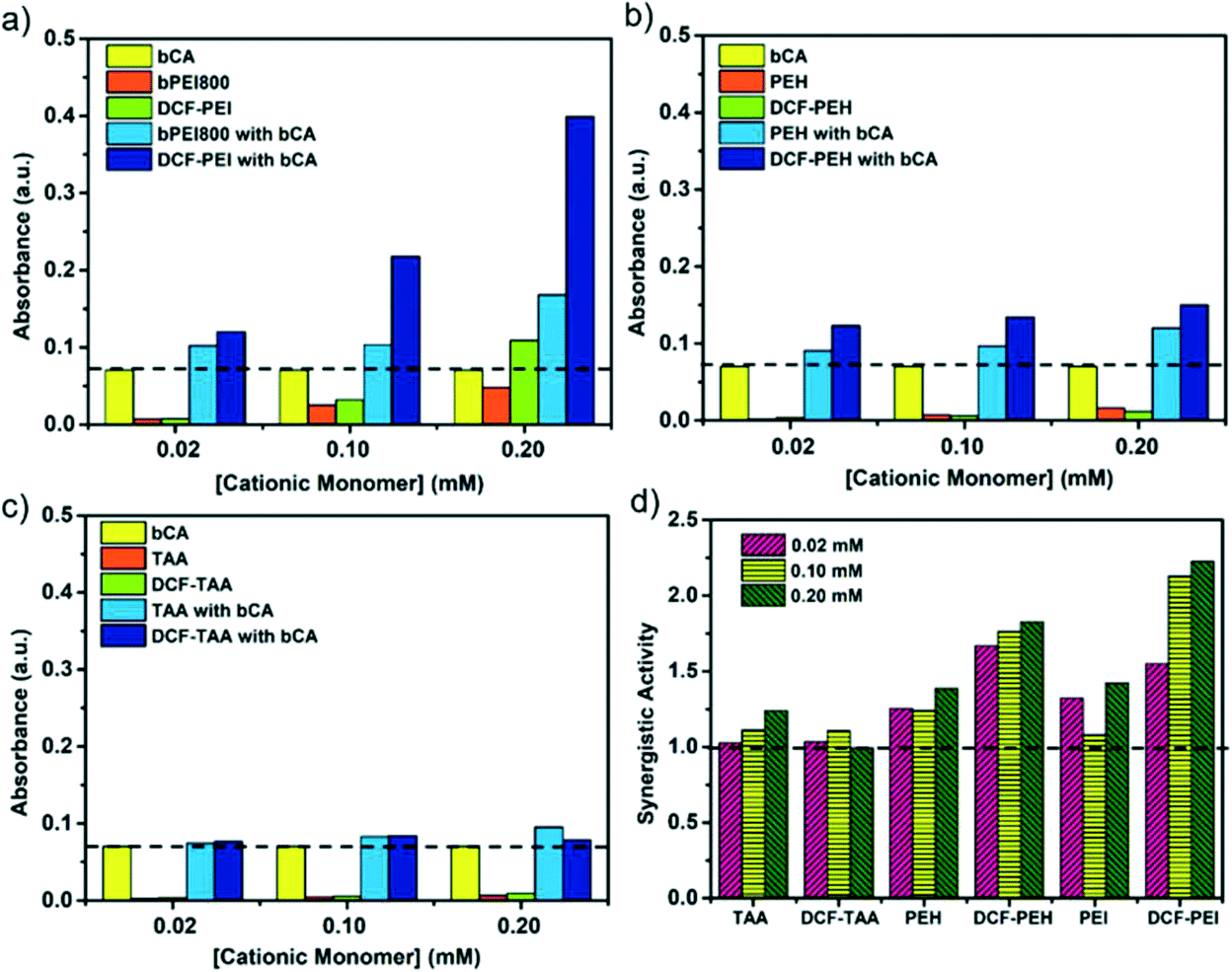 | ||
| Fig. 3 Hydrolysis of p-NPA, monitored by UV-vis absorption spectroscopy (A – absorbance measured at 4 min) with the addition of a) DCF–PEI, bPEI800, b) DCF–PEH, PEH, c) DCF–TAA, TAA to the solutions of bCA, used as a reference. d) Synergistic activity of all monomers/DCFs. | ||
We then hypothesized that combining an increasing number of PEI with bCA in hybrid bioconjugates would lead to strong synergistic improvement of the catalytic turnover and enzyme protection within a multicharged environment. When combined with bCA, important enhancements of catalytic activity were observed for DCF–PEI and bPEI800. In the presence of bCA, the multivalent DCF–PEI system reaches an important ∼5.8-fold increase of catalytic activity at 0.2 mM (Fig. 3a). Compared with that, bPEI800 reached a ∼2.4-fold increase in catalytic activity in the presence of bCA. In contrast, the enhancements of activity are very moderate when PEH, DCF–PEH, (Fig. 3b) and TAA, DCF–TAA (Fig. 3c) have been added to bCA, with a maximum of ∼1.8-fold increase in catalytic activity observed for DCF–PEH at 0.2 mM of PEH.
Synergistic activity was calculated by using the following equation: N = A1/(A0 + A2), where A1 is the absorbance measured with DCF or a cationic monomer with bCA at 4 min, A0 is the absorbance measured with CA alone at 4 min, and A2 is the absorbance measured with DCF or a monomer alone at 4 min (Fig. 3d). It is worthy to note that the synergistic activity of DCF–PEI and DCF–PEH is strongly dependent on the nature and the concentration of bPEI800 and PEH monomers and shows that the multivalent effect of enzyme encapsulation by DCFs and enhanced enzyme activity play a role in the overall catalytic process.
The catalytic activity was then quantified by determining Km, equal to the substrate concentration at half of the maximum rate Vmax, which were both determined by fitting the data with the Michaelis–Menten model. The observed ∼2-fold decrease of Km for DCF–PEI with bCA (Km = 2.08 mM) or bPEI800 with bCA (Km = 1.68 mM) as compared to bCA alone (Km = 3.13 mM) reveals the stronger affinity with the bCA enzyme under DCF confinement. In contrast, DCF–PEH, DCF–TAA, PEH, and TAA show much greater Km values (Table 2), meaning lower binding affinity which is in line with all results presented above. A proficiency constant (kcat/Km) is used to measure the catalytic efficiency of the enzymatic biohybrids. Since the proficiency constant reflects the affinity and catalytic ability of the enzyme to the substrate at the same time, it can be used to compare the catalytic efficiency of different enzymes for a specific substrate (Fig. 4a, Table 2). With kcat/Km values of 7396 and 6630 M−1 s−1 for DCF–PEI with bCA and bPEI800 with bCA, respectively, these two systems clearly stand out, being one order of magnitude more powerful catalysts than all others tested, including bCA alone. It was obvious that DCF/cationic monomer with bCA presents higher catalytic efficiency, with an estimated kcat range of 2.49 s−1 to 15.36 s−1 compared to the reaction rate k of DCF/monomer without bCA (Table S2†), emphasizing a significant catalytic effect. These results are in agreement with previous results, when the enzymatic activity of hCAII is slightly inhibited by its immobilization of PEI/polydopamine layers31 or by the absorption on positively charged polystyrene nanoparticles that do not inhibit the enzyme.32
| Samples | K m (mM) | k cat (s−1) | k cat/Km (M−1 s−1) |
|---|---|---|---|
| Enzyme DCF | Enzyme DCF | Enzyme DCF | |
| DCF–PEI | 2.08 | 15.36 | 7396 |
| DCF–PEH | 4.20 | 3.79 | 902 |
| DCF–TAA | 3.71 | 2.49 | 672 |
| Samples | Enzyme monomer | Enzyme monomer | Enzyme monomer |
|---|---|---|---|
| bPEI800 | 1.68 | 11.12 | 6630 |
| PEH | 3.36 | 2.77 | 826 |
| TAA | 3.31 | 3.24 | 978 |
| bCA | 3.13 | 1.58 | 504 |
 | ||
| Fig. 4 Estimation of Km of bCA enzymatic reaction based on the Michaelis–Menten equation for DCF and monomers in the a) presence of bCA. The concentration of p-NPA varied from 0.1 to 10 mM, and the concentration of DCFs/monomer was fixed to 0.2 mM/0.4 mM. b) and c) Kinetic hydrolysis experiments at 80 °C: blank = p-NPA in water at 25 °C (light magenta) and 80 °C (black), bCA at 25 °C (magenta) and 80 °C (grey); TAA (yellow dot), DCF–TAA (yellow), PEH (green dot), DCF–PEH (green), bPEI800 (light blue dot), DCF–PEI (light blue) at 80 °C and bCA at 80 °C with the addition of DCF–PEI at 180 s (red). | ||
The exceptional activation effects observed with DCF–PEI or bPEI800 with bCA strongly demonstrate the importance of a critical number of ethylene imine units and more importantly shed light on their multivalent presentation within DCFs. They can strongly increase the activity of bCA up to one order of magnitude under confinement within a dynamic microenvironment. Moreover, while direct interactions of monomeric PEI groups with the internal enzyme active site can lead to enzyme activation, the activation mechanism observed with nanometric hybrids is mostly related to the external binding of larger DCF–PEI. The multivalent network of PEI substituents mostly accelerates outwardly the proton-transfer step via amino groups and are equally conferring a dynamic crowding microenvironment for the enzyme activation.
The bCA aggregation driven by intermolecular hydrophobic interactions is one of the problems that hinder its use in real applications. The confinement within a protecting matrix may enhance its stability and disfavor its denaturation via aggregation which might occur at high temperature for example.33,34 The activation effect of bCA at 80 °C was tested by UV experiments with the time-course analysis at pH 7.4. The final concentration of DCF–PEI was 0.2 mM. As shown in Fig. 4b and c, the hydrolysis reaction of p-nitrophenyl acetate was promoted at 25 °C (black line) as compared to the reaction with bCA at 80 °C (grey line).
Interestingly, catalytic activation effects can be obtained by simple addition of bPEI800 (light blue dot) and DCF–PEI (light blue) in the absence of bCA. The addition of bPEI800 (blue dot) and DCF–PEI (blue line) components to a solution of bCA induce a 3- or 5-fold, increased enzymatic activity at 80 °C, respectively. The same activation effect was observed with the addition of DCF–PEI in an aqueous solution of bCA after 180 s reaction time at 80 °C (red line).
The particle size of bPEI800 with bCA and DCF–PEI with bCA evaluated through DLS increases to 1000–1200 nm when heated in aqueous solution at 80 °C (Fig. S16†). The same aggregation effects were observed by TEM. Heterogeneous dark and bright nanoparticles of DCF–PEI with bCA with an overall diameter of 800–1000 nm (Fig. S17†) are formed. Larger fractal aggregates were observed for bPEI800 with the bCA samples when heated at 80 °C (Fig. S18†). The multivalent hydrophilic bPEI800 and DCF–PEI cross-linked nanoparticles enable to activate the catalytic properties of the native bCA, through accelerated proton sponge effects of the external PEI groups. Moreover, at high temperature they strengthened significantly the structural stability, and effectively inhibited the formation of protein aggregate occurred to free bCA.
Conclusions
In conclusion, we have established that multivalent DCFs provide a possibility to enhance the bCA enzyme catalytic activity by changing the enzyme microenvironment. Moreover, those DCFs can dynamically interact and generate the optimal enzyme–DCF structure through adaptation, leading to the formation of nanoparticles that optimally encapsulate the enzyme, thereby 8-fold accelerating the overall catalytic activity of DCF–bCA hybrids compared with bCA alone. The results obtained in this work showed that multivalent presentation of bPEI800 groups within DCF–PEI platforms presents outstanding binding behaviors triggering structural changes within bCA, which results in an impressive improved catalytic proficiency of kcat/Km of 7396 M−1 s−1, more than one order of magnitude higher than that for bCA alone. The DCF–PEI with bCA aggregates was notably robust and able to provide catalytic activation effects after being exposed to relatively harsh temperatures. The observations implied their feasible deployment in real systems for CO2 capture where they would undergo cycles requiring materials to maintain high performance at 80–100 °C. This rationalization corroborates the results discussed above, suggesting that a direct perspective is to immobilize encapsulated and activated enzymes in a solid substrate for enhancing CO2 capture and conversion. The expected performances must be greater than simple enzyme immobilization under such confined conditions within hydrophobic–hydrophilic nanoparticles. Their stability must be preserved under wet conditions, so they must be used as solid sorbents or entrapped with a high loading content in the thin layers of membranes. The “facilitated transport” with the CO2 enzyme carriers would compensate for the low CO2 partial pressure difference over the membrane and increase the driving force. This leads to HCO3− transport under wet conditions in applications where water vapors are present: flue gas, space exploration, breathing, and fermentation.35Experimental
Materials and methods, Fig. S1 to S19, Tables S1 to S3, and supplementary references are provided in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
DS appreciates the scholarship as support from China Scholarship Council at the University of Montpellier, France. This work was supported by the M-ERA NET 2019 grant to MB via Agence Nationale de la Recherche ANR-20-MERA-0001-01, SMARTMATTER. YZ thanks financial support from the National Natural Science Foundation of China (NSFC) (22008090) and the Natural Science Foundation of Jiangsu Province (BK20180625). The ITC experiments were performed on the PIM Platform of I2BC (Platform for measurements of Interactions of Macromolecules).Notes and references
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- J. Septavaux, C. Tosi, P. Jame, C. Nervi, R. Gobetto and J. Le-claire, Nat. Chem., 2020, 12, 202–212 CrossRef CAS PubMed.
- J. K. J. Yong, G. W. Stevens, F. Caruso and S. E. Kentish, J. Chem. Technol. Biotechnol., 2015, 90, 3–10 CrossRef CAS.
- C. T. Supuran, Nat. Rev. Drug Discovery, 2008, 7, 168–181 CrossRef CAS PubMed.
- M. Barboiu, C. T. Supuran, L. Menabuoni, A. Scozzafava, F. Mincione, F. Briganti and G. Mincione, J. Enzyme Inhib., 1999, 15, 23–46 CrossRef CAS PubMed.
- R. K. Singh, R. Singh, D. Sivakumar, S. Kondaveeti, T. Kim, J. Li, B. H. Sung, B.-K. Cho, D. R. Kim, S. C. Kim, V. C. Kaliaet, Y.-H. P. J. Zhang, H. Zhao, Y. C. Kang and J.-K. Lee, ACS Catal., 2018, 8, 11085–11093 CrossRef CAS.
- X. Ji, Z. Su, P. Wang, G. Ma and S. Zhang, ACS Nano, 2015, 9, 4600–4610 CrossRef CAS PubMed.
- C. Molina-Fernández and P. Luis, J. CO2 Util., 2021, 47, 101475 CrossRef.
- L. J. Prins, Acc. Chem. Res., 2015, 48, 1920–1928 CrossRef CAS PubMed.
- M. Abbas, W. P. Lipiński, J. Wang and E. Spruijt, Chem. Soc. Rev., 2021, 50, 3690–3705 RSC.
- S. Huang, X. Kou, J. Shen, G. Chen and G. Ouyang, Angew. Chem., Int. Ed., 2020, 59, 8786–8798 CrossRef CAS PubMed.
- C. T. Supuran, Expert Opin. Drug Metab. Toxicol., 2016, 12, 423–431 CrossRef CAS PubMed.
- C. T. Supuran, Expert Opin. Drug Discovery, 2017, 12, 61–88 CrossRef CAS PubMed.
- D.-D. Su, Y. Zhang, S. Ulrich and M. Barboiu, ChemPlusChem, 2021, 86, 1500–1510 CrossRef CAS PubMed.
- Y. Zhang, M. Barboiu, O. Ramström and J.-H. Chen, ACS Catal., 2019, 10, 1423–1427 CrossRef.
- Y. Zhang, W.-X. Feng, Y.-M. Legrand, C. T. Supuran, C.-Y. Su and M. Barboiu, Chem. Commun., 2016, 52, 13768–13770 RSC.
- Y. Zhang, Y. C. Qi, S. Ulrich, M. Barboiu and O. Ramström, Mater. Chem. Front., 2020, 4, 489–506 RSC.
- X. Shen, H. Du, R. H. Mullins and R. R. Kommalapati, Energy Technol., 2017, 5, 822–833 CrossRef CAS.
- D. Desmaële, R. Gref and P. Couvreur, J. Controlled Release, 2012, 161, 609–618 CrossRef PubMed.
- J. L. Cleland, C. Hedgepeth and D. I. Wang, J. Biol. Chem., 1992, 267, 13327–13334 CrossRef CAS.
- Y. Chen and D. M. Barkley, Biochemistry, 1998, 37, 9976–9982 CrossRef CAS PubMed.
- N. J. B. Green, S. M. Pimblott and M. Tachiya, J. Phys. Chem., 1993, 97, 196–202 CrossRef CAS.
- S. S. Rohiwal, N. Dvorakova, J. Klima, M. Vaskovicova, F. Senigl, M. Slouf, E. Pavlova, P. Stepanek, D. Babuka, H. Benes, Z. Ellederova and K. Stieger, Sci. Rep., 2020, 10, 4619 CrossRef CAS PubMed.
- T.-H. Tran, S. Krishnan and M. M. Amiji, PLoS One, 2016, 11, e0152024 CrossRef PubMed.
- C. L. Lomelino, J. T. Andring and R. McKenna, Int. J. Med. Chem., 2018, 9419521 Search PubMed.
- L. Y. Zakharova, A. B. Mirgorodskaya, E. I. Yackevich, A. V. Yurina, V. V. Syakaev, S. K. Latypov and A. I. Konovalov, J. Chem. Eng. Data, 2010, 55, 5848–5855 CrossRef CAS.
- S. M. Gould and D. S. Tawfik, Biochemistry, 2005, 44, 5444–5452 CrossRef CAS PubMed.
- N. P. Gabrielson and D. W. Pack, Biomacromolecules, 2006, 7, 2427–2435 CrossRef CAS PubMed.
- J. Sun, C. Wang, Y. Wang, S. Ji and W. Liu, J. Appl. Polym. Sci., 2019, 136, 47784 CrossRef.
- A. Assarsson, I. Pastoriza-Santos and C. Cabaleiro-Lago, Langmuir, 2014, 30, 9448–9456 CrossRef CAS PubMed.
- M. Yan, Z. Liu, D. Lu and Z. Liu, Biomacromolecules, 2007, 8, 560–565 CrossRef CAS PubMed.
- M. Y. M. Abdelrahim, C. F. Martins, L. A. Neves, C. Capasso, C. T. Supuran, I. M. Coelhoso, J. G. Crespo and M. Barboiu, J. Membr. Sci., 2017, 528, 225–230 CrossRef CAS.
- M. Sandru, S. H. Haukebo and M. B. Hagg, Composite hollow fiber membranes for CO2 capture, J. Membr. Sci., 2010, 346, 172–186 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1cy02290c |
| This journal is © The Royal Society of Chemistry 2022 |