
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface ligands enhance the catalytic activity of supported Au nanoparticles for the aerobic α-oxidation of amines to amides†
Puranjan
Chatterjee
 ab,
Hsin
Wang
ab,
J. Sebastián
Manzano
ab,
Uddhav
Kanbur
ab,
Aaron D.
Sadow
ab and
Igor I.
Slowing
*ab
ab,
Hsin
Wang
ab,
J. Sebastián
Manzano
ab,
Uddhav
Kanbur
ab,
Aaron D.
Sadow
ab and
Igor I.
Slowing
*ab
aU.S. Department of Energy, Ames Laboratory, Ames, Iowa 50011-3020, USA
bDepartment of Chemistry, Iowa State University, Ames, Iowa 50011-3111, USA. E-mail: islowing@iastate.edu
First published on 4th February 2022
Abstract
The catalytic aerobic α-oxidation of amines in water is an atom economic and green alternative to current methods of amide synthesis. The reaction uses O2 as terminal oxidant, avoids hazardous reactants and gives water as the only byproduct. Here we report that the catalytic activity of silica-supported Au nanoparticles for the aerobic α-oxidation of amines can be improved by tethering pyridyl ligands to the support. In contrast, immobilization of thiol groups on the material gives activities comparable to Au supported on bare silica. Our studies indicate that the ligands affect the electronic properties of the Au nanoparticles and thereby determine their ability to activate O2 and mediate C–H cleavage in the amine substrate. The reaction likely proceeds via an Au catalyzed β-hydride elimination enabled by backdonation from electron-rich metal to the 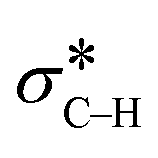 orbital. O2, which is also activated on electron-rich Au, acts as a scavenger to remove H from the metal surface and regenerate the active sites. The mechanistic understanding of the catalytic conversion led to a new approach for forming C–C bonds α to the N atoms of amines.
orbital. O2, which is also activated on electron-rich Au, acts as a scavenger to remove H from the metal surface and regenerate the active sites. The mechanistic understanding of the catalytic conversion led to a new approach for forming C–C bonds α to the N atoms of amines.
Introduction
Despite their wide application in high volume chemical commodities (e.g. plastics, antiblock agents, inks, pharmaceuticals, etc.)1 amides are typically produced through methods that have poor atom economy, use hazardous reagents such as H2SO4, hydrazoic acid, acid chlorides, thionyl chloride, or produce large quantities of waste like SO2, carboxylic acids or HCl.2–8 Therefore, there has been great interest in developing more environmentally friendly and atom economic processes for their synthesis.9,10The selective aerobic oxidation of amines is a promising approach to overcome these problems (eqn (1)).11–14 This reaction is catalysed by Au powder in the presence of silica or by supported Au nanoparticles, and gives water as the only byproduct.15–18 While supported Au nanoparticles (Au/CeO2 and Au/Al2O3) have a broad substrate scope at low O2 pressure, they have relatively low activity, with turnover numbers (TON) of ca. 25 for N-methylpyrrolidine over 24 h.19,20 Besides, weak interactions of the metal with the supports result in Au sintering, which leads to a drop in its catalytic activity. Large Au particles are less active than their smaller counterparts because the latter have higher proportions of undercoordinated sites.21–23 These sites are proposed to be the active centres in oxygen activation catalysis.24,25 Therefore, preventing Au nanoparticle aggregation is necessary for attaining high catalytic activity. The sintering of metal nanoparticles depends on their chemical potential and adhesion energy to the support.26,27 Modifying the surface of supports with organic ligands can enhance nanoparticle binding and decrease their chemical potential.28–30 The interaction of nanoparticles with surface organic ligands raises the activation barriers for migration and thereby prevents sintering via Ostwald ripening.31,32
Importantly, surface-immobilized organic ligands can also affect the catalytic properties of supported nanoparticles in a similar way in which organic ligands control the reactivity of homogeneous catalysts via electronic or steric effects.33–43 For instance, electron-donating ligands can increase the electron density of nanoparticles and modify their work function,44 to ultimately affect their electrochemical potential in redox processes. Herein, we explored the effect of silica-grafted organic ligands on the selective aerobic oxidation of amines to amides catalysed by supported Au nanoparticles. We modified the surface of mesoporous silica nanoparticles (MSN) with thiol or pyridyl moieties, used them as supports for Au nanoparticles, and determined their catalytic performance in the aerobic oxidation of amines to amides. We observed that while thiols provide strong interactions with the nanoparticles to prevent sintering, the supported Au has poor catalytic performance. In contrast, surface pyridyl species confer high activity (TON ca. 72 for N-methylpyrrolidine over 6 h) to the supported Au nanoparticles, while still averting sintering.
 | (1) |
Experimental section
Materials
Hexadecyltrimethylammonium bromide, tetraethyl-orthosilicate, ethylene carbonate, deuterium oxide, N-methylpyrrolidine, N-methylpiperidine, pyrrolidine, piperidine, N,N-dimethylbutylamine, butylamine, N,N-diisopropyl ethylamine, 4-tert-butyl benzylamine, diethylamine phenylacetylene, 4-(tert-butyl)-benzyl bromide, and 5,5-dimethyl-1-pyrrolidine N-oxide (DMPO) were purchased from Sigma-Aldrich. 1-Benzylpyrrolidine was purchased from ChemScene. 2-(4-Pyridylethyl)triethoxysilane and 3-mercaptopropyl trimethoxysilane were purchased from Gelest, Inc. Gold(III) chloride was purchased from Alfa Aesar. Toluene and sodium hydroxide were purchased from Fisher. All reagents were used without further purification.Specific surface area and porosimetry measurements
Textural properties of MSN, grafted MSNs and Au/R-MSNs were measured by nitrogen sorption isotherms at −196 °C in a Micromeritics Tristar Analyzer. The specific surface areas of the materials were calculated using the Brunauer–Emmett–Teller (BET) method and the pore widths with the Barrett–Joyner–Halenda (BJH) method. Pretreatment of the samples was done under N2 flow at 100 °C for 6 h.Elemental analysis
Organic group loadings were determined using a Perkin Elmer 2100 series II CHNS analyser. Acetanilide was used as a calibration standard with combustion and reduction temperatures 925 °C and 640 °C, respectively. All samples were prepared in triplicate.Powder X-ray diffraction (XRD)
X-ray diffraction patterns of the materials were collected on a Bruker Siemens D500 X-ray diffractometer using Cu Kα radiation (40 kV, 44 mA) over the 0–10 and 30–70 2θ°. The samples were finely ground and compacted on top of a polycarbonate holder for obtaining the patterns.Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS)
The infrared spectra of MSN, R-MSNs and Au/R-MSNs were collected on a Bruker Vertex 80 FT-IR spectrometer equipped with HeNe laser, photovoltaic MCT detector and OPUS software. A series of 64 scans with a resolution of 4 cm−1 were acquired to generate each spectrum in Kubelka–Munk mode. Samples were dried, finely ground and used neat for the measurements. For the high temperature experiments (100 °C), Au/PyEt-MSN (100 mg) was first impregnated with N-methylpyrrolidine (20 μL) and air-dried. The samples were then set in a Harrick Praying Mantis diffuse reflectance cell in a sealed ambient pressure chamber with ZnSe window. The spectra were collected every 5 min up to 30 min.Inductively coupled plasma-optical emission spectroscopy (ICP-OES)
The Au loadings of Au/R-MSN were determined on a Perkin Elmer Optima 2100 DV inductively coupled plasma-optical emission spectroscope. The concentrations of Au were determined by interpolation in a calibration curve produced using freshly prepared standards. For the sample preparation, ca. 5 mg of Au/R-MSN were mixed with 0.2 mL HF solution (38–51%) in a 15 mL polypropylene centrifuge tube and vortexed overnight to dissolve the silica. 1.8 mL of freshly prepared aqua regia was added to this mixture and vortexed for 3 h at high speed. The samples were diluted with 10 mL DI H2O (17.4 MΩ) and stirred for 1 h before analysing. Results are reported as an average of triplicates.Pulse chemisorption
CO chemisorption was performed in a Micromeritics Autochem II Analyzer. ca. 100 mg of catalyst was placed in a U-tube sample holder and the following sequence was applied: the material was pretreated at 100 °C by flowing H2 (10% in Ar) for 60 min to ensure the metal was fully reduced and then by flowing He for 60 min to remove excess surface bound H from the Au particles. The sample was then cooled to 80 °C under flowing He. The sample was then subjected to pulses of 10% CO in He. The % dispersion (D%) was of Au was determined as:| D% = (Sf × M × Vad × 100)/(m × W × Vm) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 414 mL mol−1) at STP.
414 mL mol−1) at STP.
Electron microscopy
Transmission electron microscopy (TEM) and high angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) were performed on a FEI Tecnai G2 F20 operating at 200 kV. The samples were prepared by placing 2–3 drops of dilute ethanol suspensions (ca. 0.1 mg mL−1) of materials on a lacey-carbon-coated copper grid followed by air-drying.Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC)
TGA–DSC was performed on Netzsch STA449 F1 instrument under air gas flow. The temperature was ramped at a rate of 10 K min−1 from 40 °C to 1000 °C. Approximately 9 mg of sample was placed in freshly cleaned alumina crucible for the run. The evolved gases were monitored by mass spectrometry using a range of m/z from 10 to 150. The data was analysed using Proteus software.Nuclear magnetic resonance (NMR) spectroscopy
The 1H NMR spectra of most products were collected in a Bruker Avance III 600 MHz instrument. 1H NMR spectra of products 1 and 16 and 13C NMR spectra of products 17, 18, 19, 20 and 21 were collected in a Bruker Avance NEO 400 MHz instrument.Electron paramagnetic resonance (EPR) spectroscopy
EPR spectra were collected on Bruker ELEXSYS E580 FT-EPR system. The spin trap DMPO (140.3 mg, 1.24 mmol) was dissolved in 1 mL toluene. 500 μL of this solution was mixed with the Au/PyEt-MSN or Au/MP-MSN catalysts (ca. 20 mg) exposed to air, and analysed by EPR. As controls, EPR spectra were collected for the original DMPO solution after exposure to air, and for a 500 μL aliquot of the DMPO solution mixed with ca. 20 mg Au/PyEt-MSN under N2 inside a glovebox. The sample tube prepared in the glovebox was sealed under N2 prior to taking it for EPR analysis.Mass spectrometry
Mass spectra were collected on an Agilent QTOF 6540 MS using electrospray ionization in positive ion mode or on an Agilent GC–MS instrument (7890A, 5975C) equipped with HP-5MS column using electron impact ionization.Mesoporous silica nanoparticle (MSN) synthesis
In a 5 L three-necked round bottom flask 5.10 g (14 mmol) of cetyltrimethylammonium bromide (CTAB) was dissolved in 2.4 L of deionized water. To this 17.5 mL of 2 M NaOH was added, and the reaction mixture was stirred at 550 rpm at 80 °C for 1 h. After this 25 mL (113 mmol) of tetraethylorthosilicate (TEOS) was added dropwise over a period of 10 min. The reaction mixture was further stirred for 2 h at 80 °C and 550 rpm. The product was then filtered and washed with abundant water and methanol and dried under vacuum at r.t. The dried product was then calcined at 550 °C for 6 h. BET surface area: 1320 m2 g−1, pore diameter: 2.6 nm, pore volume: 0.81 cm3 g−1. Powder XRD: (100) 2.46 2θ°, (110) 4.23 2θ°, (200) 4.89 2θ°.Grafting of MSN
In a typical procedure, 1.0 g of calcined MSN was suspended in 100 mL of toluene and then 3 mmol of organofunctional silane (2-(4-pyridylethyl)-triethoxysilane or 3-mercaptopropyl-trimethoxysilane) was added to the mixture. The mixture was then stirred at 500 rpm and heated at 100 °C for 12 h. The solid product was washed with water and methanol and dried under vacuum at r.t. overnight. The resulting products were designated PyEt-MSN and MP-MSN.Catalyst preparation
2.85 μmol of AuCl3 were dissolved in 50 mL of H2O (0.06 mM). This solution was then added to 1 g of MSN, PyEt-MSN or MP-MSN in a 100 mL round bottom flask (target loading 0.5 wt%). The mixture was heated to 60 °C and stirred at 500 rpm for 1 h. The catalyst was then separated by filtration and washed thoroughly with water. After overnight drying under vacuum, the catalyst was reduced in a tube furnace under H2 flow (15 mL min−1) for 2 h at 100 °C. Catalysts with higher Au loading (4–5 wt% Au) were prepared by the same method but using 22.8 μmol of AuCl3.Catalytic α-oxidation of amines
A 10 mL Pyrex glass crimp tube was charged with 0.25 mol% of Au/R-MSN (ca. 50 mg catalyst with 0.5 wt% Au) followed by addition of 2 mL of DI H2O. Then, 0.5 mmol of the amine was added, and the tube was plugged with a septum, crimped, and sealed. O2 was flown through the overhead space of the solution for 3 min at 40 mL min−1 at r.t. The reaction mixture was then stirred at 1100 rpm and 100 °C for 6 h. The catalyst was separated by centrifugation, and the supernatant was diluted with D2O and analysed by NMR using ethylene carbonate as internal standard. A 200 μL aliquot was drawn from the reaction mixture (2 mL) and combined with 400 μL D2O and 100 μL of D2O solution of ethylene carbonate standard (113 mM). The mixture was set in an NMR tube and the 1H NMR was collected. Because of the basic conditions, a portion of ethylene carbonate is dissociated to ethylene glycol. The mmol of product were calculated using the following equation:where IP is the integration of a reference peak in the product, IEC the integration of the ethylene carbonate protons (4.5 ppm), IEG the integration of the ethylene glycol protons (3.6 ppm), 4 is the number of protons in ethylene carbonate and ethylene glycol, HP is the number of protons giving the reference product peak (e.g. for pyrrolidone Fig. S7,† the reference peak 1 at 3.35 ppm corresponds to 2 protons, so HP is 2), mmol EC is the amount of ethylene carbonate standard added to the mixture, and 10 is the ratio of aliquot to reaction mixture volumes. Sample spectra (please see below) are provided in Fig. S7.†
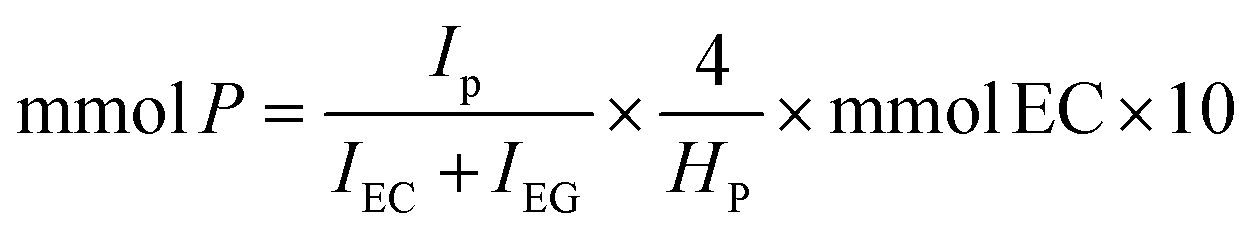
The turnover numbers (TON) for amide formation at any given time were calculated by dividing the mmoles of amide produced by the mmoles of Au used in the reaction (determined by ICP-OES).
Initial turnover frequencies (TOFini) for the catalytic α-oxidation of N-methylpyrrolidine
0.25 mol% Au/R-MSN (ca. 50 mg catalyst with 0.5 wt% Au) was added to a 10 mL Pyrex glass crimp tube, followed by addition of 2 mL of DI H2O. 0.5 mmol (52 μL) of N-methylpyrrolidine was then added and the tube was plugged with a septum and crimped to seal. O2 was flown through the overhead space of the solution for 3 min at 40 mL min−1 and r.t. The reaction mixture was then stirred at 1100 rpm and 85 °C for 0.5, 1, 1.5 or 2 h to obtain TON at different times. After each reaction time, the catalyst was separated from the solution by centrifugation, and the supernatant was diluted with D2O and analysed by NMR using ethylene carbonate as internal standard.The initial turnover frequencies (TOFini) were calculated as the slopes of the kinetic plots (time vs. TON).
Dehydrogenation of pyrrolidine to 5-(pyrrolidin-1-il)-3,4-dihydro-2H-pyrrole
0.25 mol% Au/PyEt-MSN (ca. 50 mg catalyst with 0.5 wt% Au) was added to a 10 mL Pyrex glass crimp tube followed by addition of 2 mL benzene. Then, 0.5 mmol (41 μL) of pyrrolidine was added, and the tube was crimped and sealed. O2 was then flown through the overhead space of the solution for 3 min at 40 mL min−1 at r.t. The reaction mixture was then stirred at 1100 rpm and 100 °C for 6 h. The catalyst was separated by centrifugation, and the supernatant was diluted with CDCl3 and analysed by NMR using ethylene carbonate as internal standard.Conversion of imine to amide
The 5-(pyrrolidin-1-il)-3,4-dihydro-2H-pyrrole product was mixed with 2 mL of H2O and heated at 100 °C for 6 h in presence of MSN (0.25 mol% Au). After catalyst centrifugation, the supernatant was diluted with D2O and analysed by NMR using ethylene carbonate as internal standard.Catalyst recycling
In a 10 mL Pyrex glass crimp tube 0.25 mol% Au/PyEt-MSN (ca. 50 mg catalyst with 0.5 wt% Au) was mixed with 2 mL DI H2O and 0.5 mmol N-methylpyrrolidine. The tube was then sealed and O2 was flown through the overhead space of the solution for 3 min at 40 mL min−1. The mixture was then stirred at 1100 rpm for 2 h at 85 °C. After 2 h the reaction mixture was filtered while hot and the catalyst was washed with H2O and acetone and dried under vacuum. The dried recovered catalyst was weighted and used for the subsequent runs. For each cycle the amount of amine used was adjusted so that the catalyst was 0.25 mol% of the amine. The volume of H2O was also adjusted to make the concentration of amine 250 mM. Each cycle was done in duplicate. After the last run the material was analysed by ICP-OES to obtain the Au loading in the spent catalyst.Synthesis of N,N-diethyl-4-(tert-butyl)-benzenemethanamine
0.6 mmol (110 μL) of 4-(tert-butyl)-benzyl bromide was mixed with 4 mL of toluene. 200 μL (1.1 mmol) of diisopropylethylamine and 0.72 mmol (76 μL) of diethylamine was added to the mixture. The reaction mixture was refluxed at 110 °C for 1 h. The reaction was quenched by the addition of H2O, the product was extracted with EtOAc and concentrated under vacuum. The product was purified on a silica column using a hexanes:EtOAc = 60:1 mixture as mobile phase. The product was identified by NMR spectroscopy and used as a substrate for α-oxidation.
Coupling of amines with phenylacetylenes
A 10 mL Pyrex glass crimp tube was charged with 4 mol% Au/PyEt-MSN (ca. 100 mg of 4 wt% Au) catalyst and 2 mL (18 mmol) of phenylacetylene. 0.5 mmol of amine (N-methylpyrrolidine, N-methylpiperidine or N,N-diisopropylethylamine) was then added to the mixture. The system was sealed and O2 was flown through the overhead space of the solution for 3 min at 40 mL min−1 at r.t. The reaction was stirred at 1100 rpm for 24 h at 100 °C. After the reaction, the catalyst was separated by centrifugation and the supernatant was concentrated under vacuum. The product was analysed by MS and NMR in CDCl3 using ethylene carbonate as internal standard. For the substituted phenylacetylenes the reactions utilized 2 mL benzene as solvent and 5 mmol of the corresponding acetylene, all other conditions remained the same as those with phenylacetylene.Results and discussion
Catalyst synthesis and characterization
To examine the effects of surface organic ligands on the behaviour of supported Au nanoparticle catalysts, we functionalized mesoporous silica nanoparticle (MSN) supports with groups containing thiol and pyridyl moieties (Scheme 1). Because of their strong interactions with metals,45–48 we expected –SH containing ligands would prevent Au nanoparticle aggregation.20,49,50 The ligand was incorporated by grafting 3-mercaptopropyltrimethoxysilane onto the support, and the resulting material was designated as MP-MSN. Because recent reports have suggested that increasing the electron density in Au nanoparticles enhances catalytic activity for aerobic oxidations51–53 we also selected pyridyl groups as surface ligands. We expected that their σ-donor character could give electron-enriched Au nanoparticles and thereby enhanced catalytic activity. To this end we grafted MSN with 2-(4-pyridyl)ethyltriethoxysilane and designated the resulting material as PyEt-MSN. | ||
| Scheme 1 Grafting of mesoporous silica nanoparticles (MSN) with mercaptopropyl- and 2-(4-pyridyl)ethyl-trimethoxysilanes to give MP-MSN and PyEt-MSN supports, respectively. | ||
N2 sorption and powder XRD analyses indicated that both MP-MSN and PyEt-MSN retained the high surface area and ordered hexagonal p6mm mesopore structure of the parent MSN material (Table S1, Fig. S1 and S2†). Elemental analyses confirmed presence of S and N in MP- and PyEt-MSN and provided estimates of group loadings at 1.42 ± 0.01 and 0.94 ± 0.01 mmol g−1, respectively. DRIFTS analysis of the functionalized materials provided additional evidence of organic ligand incorporation. Specifically, the spectrum of MP-MSN showed characteristic bands of S–H (2582 cm−1) and C–H (2900–2950 cm−1) stretches (Fig. S3†), and PyEt-MSN (Fig. S4†) displayed bands corresponding to aliphatic and aromatic C–H stretches (2827–3080 cm−1) and bends (1423–1456 cm−1). In addition, PyEt-MSN had a band at 1612 cm−1 assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretch and a small absorption at 1562 cm−1 suggesting a fraction of the pyridyl groups were protonated via acid–base reaction with surface silanols.54
N stretch and a small absorption at 1562 cm−1 suggesting a fraction of the pyridyl groups were protonated via acid–base reaction with surface silanols.54
Au was then incorporated into the materials via deposition–precipitation from an aqueous AuCl3 solution, followed by washing, drying and reduction of the powders at 100 °C under H2 fow (Fig. S5, S6 and Table S2†). ICP-OES analysis indicated that both functionalized materials adsorbed and retained the target 0.5 wt% of the metal. In contrast, the non-functionalized parent MSN was only able to hold 0.03 wt% Au. Because the organic groups in MP-MSN and PyEt-MSN are present in a large excess relative to the Au, (mole ratios estimated at 55:1 and 35:1 for Au/MP-MSN and Au/PyEt-MSN, respectively), we deduce that the metal nanoparticles have a significantly stronger interaction with the grafted ligands in MP-MSN and PyEt-MSN than with the surface silanol or siloxy bridges of the original support. Due to the low metal loading, no significant differences were observed between the DRIFT spectra of Au/MP-MSN and that of the MP-MSN support. A slight variation with respect to the support was only observed in the spectrum of Au/PyEt-MSN (Fig. S8†), where a shoulder at 1635 cm−1 appeared next to the 1612 cm−1 CN band, suggesting a fraction of the pyridyl groups were coordinating with the Au.54,55 This shoulder became an actual peak for materials at higher Au loading (3.9 wt% Au). An additional peak was also observed at 1504 cm−1 in Au/PyEt-MSN further suggesting pyridyl–Au coordination.56
Thermogravimetric analysis (TGA) of the materials revealed that the organic functionalities were stable up to 200 °C (Fig. S9†). Small mass losses were observed for Au/MP-MSN (3.6 wt%) and Au/PyEt-MSN (2.3 wt%) below this temperature, and MS detection of the evolved gases indicated they corresponded to elimination of physisorbed water.
Transmission electron microscopy (TEM) imaging of the materials (Fig. 1) revealed that the Au nanoparticles were smaller in Au/MP-MSN (<1 nm) than Au/PyEt-MSN (2.1 nm). The small particle sizes were consistent with high dispersions measured by CO pulsed chemisorption (52% and 44% for Au/MP-MSN and Au/PyEt-MSN, respectively) and with a lack of reflections corresponding to Au in the wide angle powder XRD patterns of the materials.57,58 We note that while the Au nanoparticles in non-functionalized Au/MSN sintered within 10 s of exposure to the electron beam of the TEM (Fig. S10†), no changes were observed for Au/PyEt-MSN or Au/MP-MSN during image acquisition.
 | ||
| Fig. 1 Electron micrographs of Au/MP-MSN a) TEM and b) STEM, and Au/PyEt-MSN c) TEM and d) STEM. | ||
Effects of surface groups on catalytic activity
We then evaluated the activities of the materials as catalysts for the selective aerobic oxidation of N-methyl pyrrolidine to N-methyl pyrrolidone (0.25 M amine in water, 0.25 mol% Au, 3 min O2 overhead flow, 85 °C). We calculated turnover numbers (TON) as mmoles of amide produced per mmol of Au in the catalyst. We derived initial turnover frequencies (TOFini) corresponding to initial rates from the slopes of the kinetic plots (time vs. TON, Fig. 2a). Interestingly, the activity of Au/PyEt-MSN (TOF: 10.2 h−1) was almost the triple of that of Au/MP-MSN (TOF: 3.5 h−1). We also tested the non-functionalized Au/MSN catalyst, which had a slightly better activity (TOF: 4.1 h−1) than Au/MP-MSN. Importantly, neither Au/PyEt-MSN nor Au/MP-MSN gave any indications of particle sintering or Au loss even after prolonged reaction times (24 h, Fig. S11†). In contrast, no metal was detected by ICP-OES and TEM on the spent Au/MSN, indicating all Au leached out of the non-functionalized material. | ||
| Fig. 2 a) Catalytic activities of Au/MP-MSN (grey circles), Au/PyEt-MSN (black circles) and Au/MSN (white squares) for the selective aerobic oxidation of N-methylpyrrolidine to N-methylpyrrolidone (conditions: 2 mL 0.25 M amine in H2O, 0.25 mol% Au, 85 °C, 3 min O2 overhead flow; TON = mmol of amide/mmol of Au). b) Au 4f and c) S 2p XP spectra of Au/MP-MSN, and d) Au 4f and e) N1s XP spectra of Au/PyEt-MSN. Discontinuous vertical line in b and d correspond to the position of the Au 4f7/2 peak in Au/MSN as a reference. | ||
Typically, activity decreases with increasing particle size.59,60 Thus, the higher activity of the larger Au nanoparticles in Au/PyEt-MSN cannot be attributed to particle size effects. This idea was further supported by comparing the activity of the original Au/MP-MSN with a new Au/MP-MSN with larger Au nanoparticles (1.6 nm, Fig. S12†). The new material was prepared using a higher metal loading (4.8 wt%, Table S2†). As expected the new Au/MP-MSN with larger metal nanoparticles was even less active than the original Au/MP-MSN (2 h TON of 5.8 for the original catalyst and 1.4 for the material with 1.6 nm Au particles, Fig. S13†). Therefore, the lower TOF for amine oxidations catalyzed by Au/MP-MSN compared to Au/PyEt-MSN is not due to particle size. Instead, we propose that the differences are caused by changes in the electronic properties of Au induced by the surface ligands.
For completeness, we also prepared an Au/PyEt-MSN with higher metal loading (3.9 wt%) to obtain larger metal nanoparticles (3.2 nm) than the original Au/PyEt-MSN (Fig. S14 and Table S2†). Consistently, the Au/PyEt-MSN material with the larger metal particles gave a TON of 30 after 6 h compared to the TON of 66 over the same timespan provided by the original Au/PyEt-MSN with 2.1 nm particles (0.25 M amine in water, 0.25 mol% Au, 3 min O2 overhead flow, 85 °C, Fig. S15†). We also note that increasing the PyEt– group loading from 1 to 1.4 mmol g−1 resulted in an increase in Au particle size to 2.5 nm, which led to a lower activity (TON of 44). In contrast, decreasing the PyEt– group loading to 0.64 mmol g−1 gave particles of similar size and the same catalytic activity as the original 0.94 mmol PyEt/g (Fig. S16†).
Recent reports have shown that increasing the electron density in Au benefits its catalytic activity for oxidation reactions.61–63 For instance, the interaction of electron donating species with the surface of the metal can modify its work function and polarize the surface-solvent interface to facilitate O2 activation.53 Electron-rich Au surfaces activate adsorbed O2via backdonation from the metal d to the O π* orbital to produce surface-bound superoxo species.61,64,65 We probed the electronic properties of the catalysts using XPS. The spectra revealed significant differences between the binding energies (BE) of their Au 4f7/2 electrons. While the Au 4f7/2 BE in Au/MP-MSN was higher than that of bulk Au0 (85.0 vs. 83.9 eV (ref. 66)) the peak in Au/PyEt-MSN was shifted to a much lower value (83.1 eV) (Fig. 2b and d), suggesting that in spite of the reduction treatment (2 h under H2 flow at 100 °C) Au had a cationic character in the former catalyst while the latter was electron-rich compared to the bulk metal. As a reference, the non-functionalized Au/MSN had an Au 4f BE centered at 83.9 eV, which is similar to that of bulk Au. These results indicate that surface ligands can control the electronic properties of the supported metal. We also observed small shifts in the S 2p and N 1s spectra of the ligands resulting from incorporation of Au into the supports. Importantly, these variations were complementary to the shifts observed in the corresponding Au 4f7/2 BE. For instance, the S 2p BE in Au/MP-MSN shifted to a slightly lower value (164.1 eV) than the original MP-MSN (164.2 eV, Fig. 2c). In contrast, the N 1s peak was slightly shifted towards higher BE (399.4 eV) compared to the parent PyEt-MSN (399.1 eV, Fig. 2e). Deconvolution of N 1s peak indicated that approximately 41% of the signal could be attributed to Au coordinated N (400.0 eV) (Fig. S17†).67 Taken together, the negative and positive shifts in Au and N BE indicate that the PyEt group donates electrons to the metal. Therefore, the higher activity of Au/PyEt-MSN can be attributed to electron donation of the ligand to the supported nanoparticles.
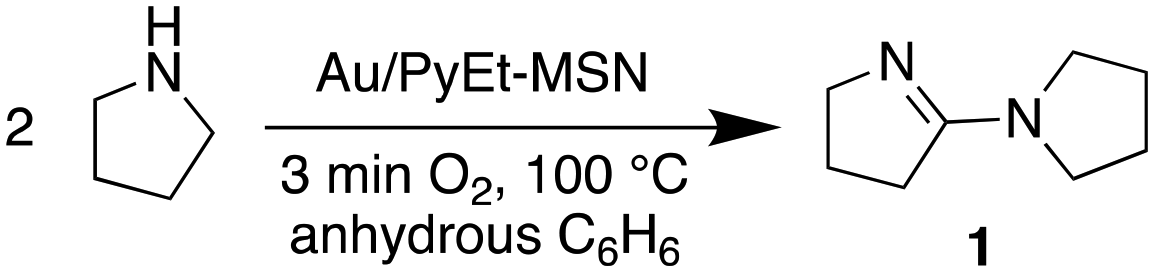 | (2) |
 | (3) |
Reaction mechanism
Our experiments suggest that the Au/PyEt-MSN catalysed conversion of amine to amide utilizes O2 as the terminal oxidant and water as oxygen donor. For instance, the Au/PyEt-MSN catalysed transformation of N-methyl pyrrolidine into N-methyl pyrrolidone gives a TON of 72 after 6 h under O2 but only a TON of 2 under N2 in the same timespan (0.25 M substrate in water, 0.25 mol% Au/PyEt-MSN, 100 °C, 3 min O2 or N2 overhead flow). While the Au/PyEt-MSN catalysed reaction of pyrrolidine under O2 in water gives pyrrolidone (6 h TON: 17), no amide is formed in anhydrous benzene. The reaction product in absence of water is 5-(pyrrolidin-1-il)-3,4-dihydro-2H-pyrrole (1) (6 h TON: 19, eqn (2)). Yet dissolution of the isolated product 1 in water at the same reaction temperature in presence of MSN gave pyrrolidone (eqn (3)). The product 1, however, resists hydrolysis to pyrrolidone in absence of MSN under the same conditions and reaction time, which indicates the silica support catalyses the hydrolysis step of the reaction (100 °C in water, 6 h reaction). Thus, the role of O2 must be exclusively to assist the amine dehydrogenation, and does not participate in the formation of the new CO bond. These results are consistent with previous reports of Au/Al2O3-catalysed amine oxidation using 18O2 and H218O that showed the O in the amide product is provided by the water solvent rather than the O2.19
While the reaction of pyrrolidine in anhydrous benzene gives product 1 and provides evidence for the formation of an imine intermediate, the tertiary amine N-methyl pyrrolidine does not give any product in absence of water. This lack of product formation cannot be attributed to low reactivity because N-methyl pyrrolidine is ca. 4 times more reactive than pyrrolidine in water under our reaction conditions (6 h TON: 72 vs. 17, respectively). Instead, the lack of product from the tertiary amine in benzene may be attributed to the reaction proceeding via an unstable iminium intermediate. Evidence for such intermediate is provided by DRIFTS analysis of Au/PyEt-MSN impregnated with N-methyl pyrrolidine. Heating the amine-impregnated catalyst in the DRIFTS cell to 100 °C results in the gradual emergence and growth of a band around 1578 cm−1, which can be attributed to formation of an iminium on the surface of the material (Fig. 3a).76–78 Co-impregnation of the material with the same substrate and water and heating to 100 °C results in the growth of a broad peak in the 1630 cm−1 to 1690 cm−1 region (Fig. 3b). As a reference, we collected the spectrum of N-methylpyrrolidone product adsorbed on the catalyst and observed two distinctive peaks centred at 1653 cm−1 and 1668 cm−1. This suggests that the species formed in the co-impregnation experiment is indeed the reaction product. These results support the idea that the reaction may proceed through a β-hydride elimination, i.e. coordination of the amine N to Au followed by H abstraction to give the imine or iminium intermediate.
 | ||
| Fig. 3 a) DRIFT spectra of N-methyl pyrrolidone impregnated Au/PyEt-MSN at 100 °C collected every 5 min (pink to dark blue spectra) showing the emergence and growth of a band at 1578 cm−1 attributed to iminium. The red spectrum is the non-impregnated catalyst. b) DRIFT spectrum of N-methyl pyrrolidine and water co-impregnated Au/PyEt-MSN at 100 °C (light blue). The spectra of reaction product N-methyl pyrrolidone (yellow) and the last spectrum from a) (dark blue) provided as reference. c) Au 4f XP spectra of Au/PyEt-MSN catalyst before (top) and after (bottom) exposing to O2. A 0.5 eV shift to higher binding energy is indicative of loss in electron density of the metal because of binding to O2. d) EPR spectrum of Au/PyEt-MSN exposed to O2 and treated with DMPO spin probe. The resulting spectrum is indicative of superoxide formation. | ||
On the other hand, the adsorption of O2 on electron-rich Au nanoparticles has been proposed to yield surface superoxo species.61,65 This process involves electron transfer from Au to the adsorbed O2 and induces a decrease in the electron density of the metal.68 Consistent with this information, XPS analysis of Au/PyEt-MSN catalyst treated with a stream of O2 results in a 0.5 eV shift of the Au 4f7/2 peak to higher binding energy compared to the original catalyst (83.6 eV vs. 83.1 eV, respectively, Fig. 3c). This shift suggests binding of oxygen to the metal. The nature of surface oxygen species was investigated by reacting Au/PyEt-MSN exposed to air with 5,5′-dimethylpyrroline N-oxide (DMPO) spin trap. EPR spectroscopy analysis confirmed the formation of a nitroxyl adduct resulting from addition of superoxide species to DMPO (Fig. 3d).69–71 Control experiments using 1) DMPO in air without catalyst gave only a weak signal, and 2) DMPO with the catalyst exposed to N2 gave only noise in the EPR spectra (Fig. S18†). These results strongly suggest the formation of superoxide upon treating the catalyst with O2. Furthermore, addition of superoxide quencher p-quinone72–74 to the Au/PyEt-MSN catalysed reaction of N-methyl pyrrolidine also led to a drop in conversion (6 h TON of 20) under our standard conditions, further suggesting that these reactive oxygen species take part in the process.
and induces a decrease in the electron density of the metal.68 Consistent with this information, XPS analysis of Au/PyEt-MSN catalyst treated with a stream of O2 results in a 0.5 eV shift of the Au 4f7/2 peak to higher binding energy compared to the original catalyst (83.6 eV vs. 83.1 eV, respectively, Fig. 3c). This shift suggests binding of oxygen to the metal. The nature of surface oxygen species was investigated by reacting Au/PyEt-MSN exposed to air with 5,5′-dimethylpyrroline N-oxide (DMPO) spin trap. EPR spectroscopy analysis confirmed the formation of a nitroxyl adduct resulting from addition of superoxide species to DMPO (Fig. 3d).69–71 Control experiments using 1) DMPO in air without catalyst gave only a weak signal, and 2) DMPO with the catalyst exposed to N2 gave only noise in the EPR spectra (Fig. S18†). These results strongly suggest the formation of superoxide upon treating the catalyst with O2. Furthermore, addition of superoxide quencher p-quinone72–74 to the Au/PyEt-MSN catalysed reaction of N-methyl pyrrolidine also led to a drop in conversion (6 h TON of 20) under our standard conditions, further suggesting that these reactive oxygen species take part in the process.
We note that while the electron-deficient Au/MP-MSN is still able to mediate the reaction, its activity does not change upon flowing the system with N2 instead of O2 (6 h TON is 6 regardless of flowing with N2 or O2). Besides, addition of benzoquinone to the system does not alter the activity of Au/MP-MSN, and reaction with DMPO after O2 treatment gives only background level EPR signal (Fig. S18d†). We also note that the Au/MP-MSN mediated reaction of N-methyl pyrrolidine reaches a maximum TON of 6 after 1.5 h and then the yield remains largely invariable at least until 6 h (in contrast, the same reaction catalysed by Au/PyEt-MSN gives a TON of 17 in 2 h that grows to 72 at 6 h). Thus, it appears that O2 is not involved in the reaction in presence of the electron deficient Au/MP-MSN, and that O2 does not participate in the early stages of the reaction catalysed by Au/PyEt-MSN. Instead, it is likely needed for catalyst turnover.
These results suggest that the catalytic process involves three fundamental components, specifically: the Au catalyst that mediates β-hydride elimination of the amine, water that provides O atoms for the amide product, and O2 that enables the catalytic turnover. These components can be integrated into a possible catalytic cycle that involves an initial Au mediated β-hydride elimination along with amine deprotonation to give an imine (i), which then undergoes hydrolysis to give a carbinolamine (ii). A second β-hydride elimination and NH deprotonation of the intermediate leads to an iminol that tautomerizes to the amide (iii). Finally, reactive oxygen species formed in the electron-rich Au scavenge H from the nanoparticle surface along with the protons from steps i and iii to give water and allow catalyst turnover (iv) (Scheme 2). Thus, the H atoms in the water molecule that attacks the imine intermediate are eventually transferred to the reactive oxygen species to give new water molecules as a by product. Importantly, while β-hydride elimination is favoured by backdonation from electron-rich metal centres to the  orbital, the process is less viable in electron poor Au.75 Therefore, the difference in performance between Au/PyEt-MSN and Au/MP-MSN can be attributed to their opposing effects on the electronic properties of the Au nanoparticles. These differences in electronic properties control two critical steps of the catalytic cycle: C–H bond cleavage and O2 activation. The more negative metal in Au/PyEt-MSN is more effective at inducing the cleavage of the C–H bond in the substrate to give an elimination product and activates O2 to enable catalytic cycling. In contrast, the electron poor metal in Au/MP-MSN is unable to activate O2 and behaves like a stoichiometric oxidant. We note that an additional effect of pyridyl moieties in Au/PyEt-MSN could be the promotion of nucleophilic water attack (step ii of the mechanism) by forming H-bonding interactions with water.
orbital, the process is less viable in electron poor Au.75 Therefore, the difference in performance between Au/PyEt-MSN and Au/MP-MSN can be attributed to their opposing effects on the electronic properties of the Au nanoparticles. These differences in electronic properties control two critical steps of the catalytic cycle: C–H bond cleavage and O2 activation. The more negative metal in Au/PyEt-MSN is more effective at inducing the cleavage of the C–H bond in the substrate to give an elimination product and activates O2 to enable catalytic cycling. In contrast, the electron poor metal in Au/MP-MSN is unable to activate O2 and behaves like a stoichiometric oxidant. We note that an additional effect of pyridyl moieties in Au/PyEt-MSN could be the promotion of nucleophilic water attack (step ii of the mechanism) by forming H-bonding interactions with water.
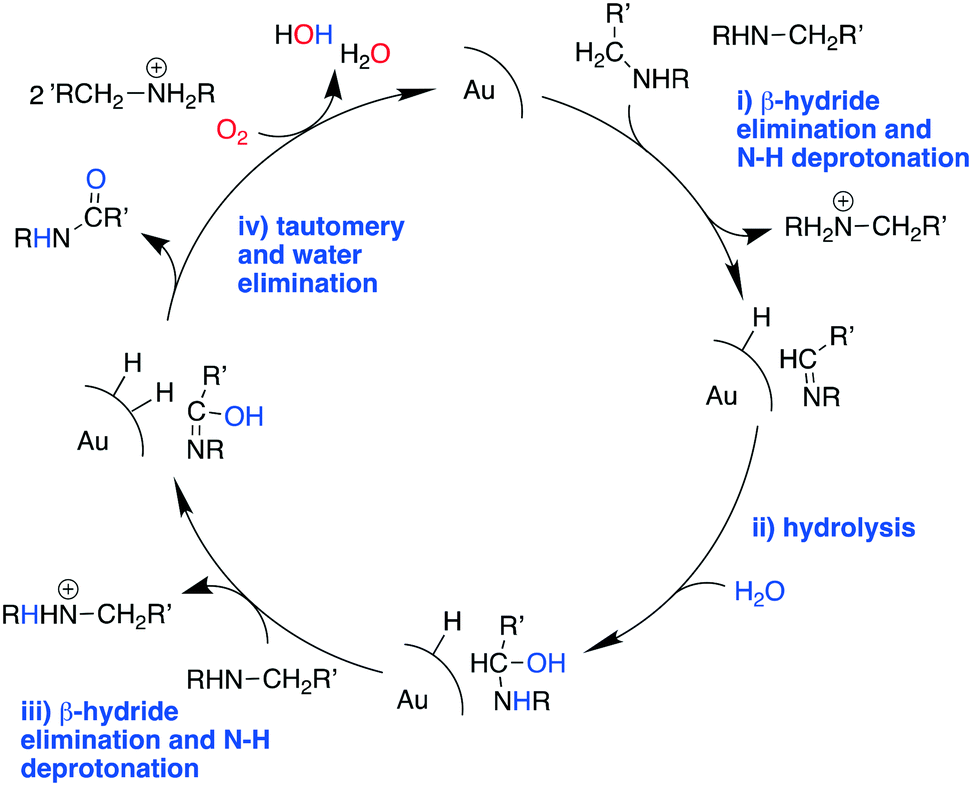 | ||
| Scheme 2 Possible reaction mechanism for the Au/PyEt-MSN catalysed oxidation of amines to amides. | ||
For completeness, we examined the recyclability of Au/PyEt-MSN. No significant drop in 2 h TON was observed over 5 recycling experiments (Fig. S19†). Furthermore, ICP-OES of the spent catalyst after the fifth cycle gave a 0.5 wt% Au indicating the pyridyl groups enabled retention of the metal, and XPS analysis of the spent catalyst indicated the metal remained electron rich (Au 4f7/2 peak centered at 83.5 eV compared to 83.9 eV of bulk Au) (Fig. S20†).
Effects of substrate structure on reactivity
The above results suggest that reaction rates should be favoured by the propensity of the substrates to undergo β-hydride elimination. Substrate structure-reactivity analysis indicates that catalytic rates are enhanced by increasing the degree of substitution of the amine (Table 1). The effect is likely due to electron donation by alkyl groups (i.e. inductive effect +I) that facilitates hydride formation. For example, under our standard conditions the reaction of the tertiary N,N-dimethyl butyl amine (2) gives a TON of 11 over 6 h, but no conversion is observed for the primary butyl amine (3) or the secondary diethyl amine (4) in the same timeframe (conditions: 100 °C, 0.25 mol% Au/PyEt-MSN, 3 min O2 overhead flow). The importance of electron donation by alkyl groups is further evidenced by the fact that the branched secondary t-butyl ethyl amine (5) is reactive (6 h TON of 26) but the linear secondary amine 4 is inert. Likewise, increasing the degree of α-alkyl substitution for tertiary amines leads to a consistent enhancement in reactivity: diethyl methyl amine (6) gives a TON of 25, triethyl amine (7) a TON of 45 and ethyl di-isopropyl amine (8) reaches TON of 91 over the same reaction time and conditions as above. It is noteworthy that benzylamines do not react (e.g. benzylamine (9) or t-butyl benzylamine (10)), which suggests that the electron withdrawing inductive effect (−I) of aryl groups inhibits hydride elimination. An exception however, are tertiary dialkyl benzylamines like diethyl p-(t-butyl)benzyl amine (11) (TON of 38) or N-benzylpyrrolidine (12) (TON of 24). Importantly, the amides resulting from the reactions of amines 11 and 12 have the oxygen bound to the non-benzylic positions. These results further suggest that the electron-withdrawing inductive effect of aryl groups prevents hydride elimination (and therefore amide formation) at the benzylic position but the electron-donating effect of alkyl groups enables the loss of hydride on the aliphatic side of the molecules. Likewise, reactions of N-methylated amines show selective oxidation of the groups other than the methyl (e.g. butyl or ethyl in 2 and 6, respectively). In contrast to the linear amines, cyclic secondary amines such as pyrrolidine (13) and piperidine (14) react well, the smaller cycle being more reactive than the latter (TON of 17 and 7, respectively). As expected, the corresponding N-methylated tertiary amines N-methylpyrrolidine (15) and N-methylpiperidine (16) are more reactive than the corresponding secondary amines, giving TON of 72 and 19, respectively. Overall, a clear trend is observed between the degree of alkyl substitution and the reactivity of the amine substrates. In contrast, the presence of additional functional groups with electron withdrawing inductive effect (−I) such as ether, alcohol, carboxylic acid, a second amine or bromine (Fig. S18†) inhibits the reaction. These results suggest that amine reactivity is favoured by the electron donation to the N atom.| Entry | Amine | Product | TON (6 h) |
|---|---|---|---|
| Reaction conditions: 0.5 mmol substrate, 0.25 mol% Au/PyEt-MSN (0.5 wt% Au), 2 mL H2O, 100 °C, 6 h, 3 min O2 overhead flow. | |||
| 2 |

|
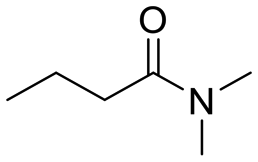
|
11 |
| 3 |
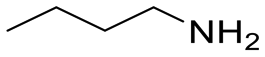
|
No reaction | N/A |
| 4 |

|
No reaction | N/A |
| 5 |
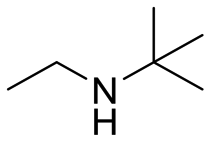
|

|
26 |
| 6 |

|
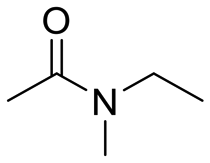
|
25 |
| 7 |

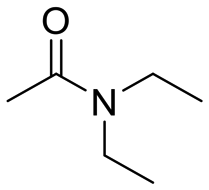
|
|
45 |
| 8 |

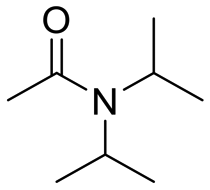
|
|
91 |
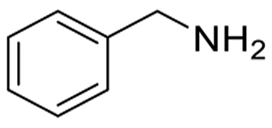
| 9 |
|
No reaction | N/A |
| 10 |

|
No reaction | N/A |
| 11 |

|

|
38 |
| 12 |
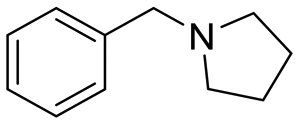
|

|
24 |
| 13 |
|

|
17 |
| 14 |

|

|
7 |
| 15 |

|

|
72 |
| 16 |

|
|
19 |
Replacing water with acetylenes to form new C–C bonds
Finally, since the results above and proposed mechanism suggest that the O atom introduced at the α-carbon of the product results from the nucleophilic attack of H2O, we considered that replacing water with another nucleophile could lead to other products. For example, as noted above, the reaction of pyrrolidine in absence of water gives an intermediate that is nucleophilically attacked by unreacted amine to give product 1 (eqn (2)). Therefore, we performed new reactions using neat phenylacetylene as reactant and solvent, with the expectation that deprotonated phenylacetylide could act as a nucleophile and attack the products of β-hydride elimination. Consistent with our hypothesis, we obtained 2-(phenylethynyl)-substituted amines 17–19 as products of the reactions of phenylacetylene with ethyl diisopropyl amine, N-methylpiperidine and N-methylpyrrolidine, respectively (Table 2, 4 mol% Au/PyEt-MSN, 100 °C, 24 h). We note that this reaction utilizes an excess of the alkyne, therefore, when attempting the conversion of N-methylpyrrolidine with 1,4-diethynyl benzene only the 1:1 coupling product 20 was obtained. Increasing the concentration of the amine inhibits the reaction. The conversion does not proceed with linear terminal alkynes such as 1-octyne under our conditions. Using p-MeO substituted phenylacetylene as reactant gives the coupling product 21, but p-CN and p-NH2 substituted phenylacetylenes give no conversion. A possible reason for the lack of reactivity of the latter is the propensity of nitrile and amine groups to bind to Au, which may decrease the number of sites available for N-methylpyrrolidine activation. This reaction provides a new approach for C–C bond formation at position α to amines, and we plan to further explore and develop it. To the best of our knowledge, this transformation has only been attained with organometallic reagents,79–82 therefore this is likely the first report of a heterogeneous catalyst giving this type of amine–alkyne coupling reaction.
| Entry | Amine | Acetylene | Product | TON (24 h) |
|---|---|---|---|---|
| a 0.5 mmol amine, 2 mL phenylacetylene, 4.0 mol% Au/PyEt-MSN (4.0 wt% Au), 3 min O2 flow, 24 h at 100 °C. b 0.5 mmol amine, 2 mL benzene (solvent), 5.0 mmol substituted phenylacetylene, 4.0 mol% Au/PyEt-MSN (4.0 wt% Au), 3 min O2 flow, 24 h at 100 °C. | ||||
| 17 |

|
|

|
3a |
| 18 |
|

|

|
4a |
| 19 |
|

|

|
4a |
| 20 |
|

|
|
2b |
| 21 |
|

|

|
2b |
Conclusions
In summary, organic ligands immobilized on a silica surface affect the electronic properties of supported Au nanoparticles. The effects of ligands are revealed by shifts in the X-ray photoelectron spectra of the metal. Thiol and pyridyl groups make the metal less and more electron-rich compared to Au on the non-modified support, respectively. These variations in electron density affect the catalytic performance of the metal in the aerobic oxidation of amines. Specifically, the electron-rich metal in Au/PyEt-MSN can backdonate electrons to the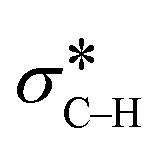 orbital in the substrate to induce C–H bond cleavage, while the electron-poor Au/MP-MSN has a more limited backdonation capacity. Furthermore, O2 is activated on the electron-rich Au/PyEt-MSN as a superoxide, which allows sustaining the catalytic cycle by removal of chemisorbed hydrides from the metal surface. In contrast, the electron poor thiol-ligated Au does not seem to interact with O2 and behaves as a stoichiometric oxidant rather than as a catalyst. Following the initial Au-catalysed β-hydride elimination, the intermediate imine undergoes a nucleophilic attack by water. A second β-hydride elimination followed by tautomerization yields the final amide. Based on this mechanism it is possible to introduce other groups on the α-carbon of amines by merely replacing water with a different nucleophile. An example of this is the reaction of phenylacetylene with amines, which in presence of Au/PyEt-MSN catalyst and O2 yields α-substituted alkynylamines. Overall, this is work demonstrates a green and atom-economic approach to transform amines into amides or α-substituted amines.
orbital in the substrate to induce C–H bond cleavage, while the electron-poor Au/MP-MSN has a more limited backdonation capacity. Furthermore, O2 is activated on the electron-rich Au/PyEt-MSN as a superoxide, which allows sustaining the catalytic cycle by removal of chemisorbed hydrides from the metal surface. In contrast, the electron poor thiol-ligated Au does not seem to interact with O2 and behaves as a stoichiometric oxidant rather than as a catalyst. Following the initial Au-catalysed β-hydride elimination, the intermediate imine undergoes a nucleophilic attack by water. A second β-hydride elimination followed by tautomerization yields the final amide. Based on this mechanism it is possible to introduce other groups on the α-carbon of amines by merely replacing water with a different nucleophile. An example of this is the reaction of phenylacetylene with amines, which in presence of Au/PyEt-MSN catalyst and O2 yields α-substituted alkynylamines. Overall, this is work demonstrates a green and atom-economic approach to transform amines into amides or α-substituted amines.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences, through the Ames Laboratory Catalysis Science program. The Ames Laboratory is operated for the U.S. Department of Energy by Iowa State University under Contract No. DE-AC02-07CH11358.Notes and references
- J. Kroschwitz, Kirk-Othmer Encyclopedia of Chemical Technology, Wiley, New York, 4th edn, 1991 Search PubMed.
- A. H. Blatt, Chem. Rev., 1933, 12, 215–260 CrossRef CAS.
- K. F. Schmidt, Ber. Dtsch. Chem. Ges. A, 1924, 57(4), 704–706 CrossRef.
- N. O. V. Sonntag, Chem. Rev., 1953, 52, 237–416 CrossRef CAS.
- A. Leggio, E. L. Belsito, G. De Luca, M. L. Di Gioia, V. Leotta, E. Romio, C. Siciliano and A. Liguori, RSC Adv., 2016, 6, 34468–34475 RSC.
- B. Thern, J. Rudolph and G. Jung, Tetrahedron Lett., 2002, 43, 5013–5016 CrossRef CAS.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- E. K. Nelson, J. Am. Chem. Soc., 1919, 41, 2121–2130 CrossRef CAS.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082–5103 RSC.
- W. Xu, Y. Jiang and H. Fu, Synlett, 2012, 23, 801–804 CrossRef CAS.
- J. R. Khusnutdinova, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2014, 136, 2998–3001 CrossRef CAS PubMed.
- J. W. Kim, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2008, 47, 9249–9251 CrossRef CAS PubMed.
- Y. Wang, H. Kobayashi, K. Yamaguchi and N. Mizuno, Chem. Commun., 2012, 48, 2642–2644 RSC.
- B. Zhu and R. J. Angelici, Chem. Commun., 2007, 2157–2159 RSC.
- L. Aschwanden, T. Mallat, F. Krumeich and A. Baiker, J. Mol. Catal. A: Chem., 2009, 309, 57–62 CrossRef CAS.
- B. Zhu, M. Lazar, B. G. Trewyn and R. J. Angelici, J. Catal., 2008, 260, 1–6 CrossRef CAS.
- H. Chen, C. Liu, M. Wang, C. Zhang, N. Luo, Y. Wang, H. Abroshan, G. Li and F. Wang, ACS Catal., 2017, 7, 3632–3638 CrossRef CAS.
- X. Jin, K. Kataoka, T. Yatabe, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2016, 55, 7212–7217 CrossRef CAS PubMed.
- T. O. Dairo, N. C. Nelson, I. I. Slowing, R. J. Angelici and L. K. Woo, Catal. Lett., 2016, 146, 2278–2291 CrossRef CAS.
- H. Tsunoyama, H. Sakurai and T. Tsukuda, Chem. Phys. Lett., 2006, 429, 528–532 CrossRef CAS.
- D. V. Jawale, E. Gravel, V. Geertsen, H. Li, N. Shah, R. Kumar, J. John, I. N. N. Namboothiri and E. Doris, Tetrahedron, 2014, 70, 6140–6145 CrossRef CAS.
- T. V. W. Janssens, B. S. Clausen, B. Hvolbæk, H. Falsig, C. H. Christensen, T. Bligaard and J. K. Nørskov, Top. Catal., 2007, 44, 15 CrossRef CAS.
- D. C. Lim, I. Lopez-Salido, R. Dietsche, M. Bubek and Y. D. Kim, Chem. Phys., 2006, 330, 441–448 CrossRef CAS.
- M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 454, 981–983 CrossRef CAS PubMed.
- Y. Dai, P. Lu, Z. Cao, C. T. Campbell and Y. Xia, Chem. Soc. Rev., 2018, 47, 4314–4331 RSC.
- J. A. Farmer and C. T. Campbell, Science, 2010, 329, 933 CrossRef CAS PubMed.
- S. Rana, S. Maddila, K. Yalagala and S. B. Jonnalagadda, Appl. Catal., A, 2015, 505, 539–547 CrossRef CAS.
- J. S. Manzano, H. Wang, T. Kobayashi, P. Naik, K. C. Lai, J. W. Evans and I. I. Slowing, Microporous Mesoporous Mater., 2020, 110276 CrossRef CAS.
- C. M. Crudden, M. Sateesh and R. Lewis, J. Am. Chem. Soc., 2005, 127, 10045–10050 CrossRef CAS PubMed.
- L. Xin, F. Yang, S. Rasouli, Y. Qiu, Z.-F. Li, A. Uzunoglu, C.-J. Sun, Y. Liu, P. Ferreira, W. Li, Y. Ren, L. A. Stanciu and J. Xie, ACS Catal., 2016, 6, 2642–2653 CrossRef CAS.
- R. van den Berg, T. E. Parmentier, C. F. Elkjær, C. J. Gommes, J. Sehested, S. Helveg, P. E. de Jongh and K. P. de Jong, ACS Catal., 2015, 5, 4439–4448 CrossRef CAS.
- C. Parise, B. Ballarin, D. Barreca, M. C. Cassani, P. Dambruoso, D. Nanni, I. Ragazzini and E. Boanini, Appl. Surf. Sci., 2019, 492, 45–54 CrossRef CAS.
- P. Wu, P. Bai, Z. Lei, K. P. Loh and X. S. Zhao, Microporous Mesoporous Mater., 2011, 141, 222–230 CrossRef CAS.
- P. Wu, P. Bai, K. P. Loh and X. S. Zhao, Catal. Today, 2010, 158, 220–227 CrossRef CAS.
- Z. Cao, J. S. Derrick, J. Xu, R. Gao, M. Gong, E. M. Nichols, P. T. Smith, X. Liu, X. Wen, C. Copéret and C. J. Chang, Angew. Chem., Int. Ed., 2018, 57, 4981–4985 CrossRef CAS PubMed.
- Y. Chen, Z. Guo, T. Chen and Y. Yang, J. Catal., 2010, 275, 11–24 CrossRef CAS.
- G. Chen, C. Xu, X. Huang, J. Ye, L. Gu, G. Li, Z. Tang, B. Wu, H. Yang, Z. Zhao, Z. Zhou, G. Fu and N. Zheng, Nat. Mater., 2016, 15, 564–569 CrossRef CAS PubMed.
- S. Kunz, Top. Catal., 2016, 59, 1671–1685 CrossRef CAS.
- K. Mori, S. Masuda, H. Tanaka, K. Yoshizawa, M. Che and H. Yamashita, Chem. Commun., 2017, 53, 4677–4680 RSC.
- X.-K. Wan, J.-Q. Wang, Z.-A. Nan and Q.-M. Wang, Sci. Adv., 2017, 3, e1701823 CrossRef PubMed.
- N. Ishito, K. Nakajima, Y. Maegawa, S. Inagaki and A. Fukuoka, Catal. Today, 2017, 298, 258–262 CrossRef CAS.
- L. Lu, S. Zou and B. Fang, ACS Catal., 2021, 11, 6020–6058 CrossRef CAS.
- P. Wang, D. Tanaka, S. Ryuzaki, S. Araki, K. Okamoto and K. Tamada, Appl. Phys. Lett., 2015, 107, 151601 CrossRef.
- A. I. Frenkel, S. Nemzer, I. Pister, L. Soussan, T. Harris, Y. Sun and M. H. Rafailovich, J. Chem. Phys., 2005, 123, 184701 CrossRef CAS PubMed.
- M. K. Corbierre and R. B. Lennox, Chem. Mater., 2005, 17, 5691–5696 CrossRef CAS.
- C. K. Yee, R. Jordan, A. Ulman, H. White, A. King, M. Rafailovich and J. Sokolov, Langmuir, 1999, 15, 3486–3491 CrossRef CAS.
- G. H. Woehrle, L. O. Brown and J. E. Hutchison, J. Am. Chem. Soc., 2005, 127, 2172–2183 CrossRef CAS PubMed.
- T. V. Choudhary and D. W. Goodman, Top. Catal., 2002, 21, 25–34 CrossRef CAS.
- J. P. Gabaldon, M. Bore and A. K. Datye, Top. Catal., 2007, 44, 253–262 CrossRef CAS.
- M. Okumura, Y. Kitagawa, M. Haruta and K. Yamaguchi, Chem. Phys. Lett., 2001, 346, 163–168 CrossRef CAS.
- S. Karanjit, K. Bobuatong, R. Fukuda, M. Ehara and H. Sakurai, Int. J. Quantum Chem., 2013, 113, 428–436 CrossRef CAS.
- Q. Gu, P. Sautet and C. Michel, ACS Catal., 2018, 8, 11716–11721 CrossRef CAS.
- M. I. Zaki, M. A. Hasan, F. A. Al-Sagheer and L. Pasupulety, Colloids Surf., A, 2001, 190, 261–274 CrossRef CAS.
- Q. Zhang, H. Su, J. Luo and Y. Wei, Tetrahedron, 2013, 69, 447–454 CrossRef CAS.
- S. Muratsugu, M. H. Lim, T. Itoh, W. Thumrongpatanaraks, M. Kondo, S. Masaoka, T. S. Andy Hor and M. Tada, Dalton Trans., 2013, 42, 12611–12619 RSC.
- Y. Liu, H. Tsunoyama, T. Akita, S. Xie and T. Tsukuda, ACS Catal., 2011, 1, 2–6 CrossRef CAS.
- Y. Kotolevich, O. Martynyuk, S. Martínez-González, H. Tiznado, A. Pestryakov, M. Avalos Borja, V. Cortés Corberán and N. Bogdanchikova, Fuel, 2019, 236, 589–597 CrossRef CAS.
- H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374–9375 CrossRef CAS PubMed.
- L. Alves, B. Ballesteros, M. Boronat, J. R. Cabrero-Antonino, P. Concepción, A. Corma, M. A. Correa-Duarte and E. Mendoza, J. Am. Chem. Soc., 2011, 133, 10251–10261 CrossRef CAS PubMed.
- F. Wang, W. Ueda and J. Xu, Angew. Chem., Int. Ed., 2012, 51, 3883–3887 CrossRef CAS PubMed.
- S. Nishimura, Y. Yakita, M. Katayama, K. Higashimine and K. Ebitani, Catal. Sci. Technol., 2013, 3, 351–359 RSC.
- M. L. Kimble, A. W. Castleman, R. Mitrić, C. Bürgel and V. Bonačić-Koutecký, J. Am. Chem. Soc., 2004, 126, 2526–2535 CrossRef CAS PubMed.
- A. Staykov, T. Nishimi, K. Yoshizawa and T. Ishihara, J. Phys. Chem. C, 2012, 116, 15992–16000 CrossRef CAS.
- H. Tsunoyama, N. Ichikuni, H. Sakurai and T. Tsukuda, J. Am. Chem. Soc., 2009, 131, 7086–7093 CrossRef CAS PubMed.
- A. Y. Klyushin, T. C. R. Rocha, M. Hävecker, A. Knop-Gericke and R. Schlögl, Phys. Chem. Chem. Phys., 2014, 16, 7881–7886 RSC.
- R. J. J. Jansen and H. van Bekkum, Carbon, 1995, 33, 1021–1027 CrossRef CAS.
- S. Laursen and S. Linic, Phys. Rev. Lett., 2006, 97, 026101 CrossRef PubMed.
- F. Su, S. C. Mathew, G. Lipner, X. Fu, M. Antonietti, S. Blechert and X. Wang, J. Am. Chem. Soc., 2010, 132, 16299–16301 CrossRef CAS PubMed.
- M. Yamada, K. D. Karlin and S. Fukuzumi, Chem. Sci., 2016, 7, 2856–2863 RSC.
- X.-Q. Deng, B. Zhu, X.-S. Li, J.-L. Liu, X. Zhu and A.-M. Zhu, Appl. Catal., B, 2016, 188, 48–55 CrossRef CAS.
- M.-T. Maurette, E. Oliveros, P. P. Infelta, K. Ramsteiner and A. M. Braun, Helv. Chim. Acta, 1983, 66, 722–733 CrossRef CAS.
- E. Lee-Ruff, Chem. Soc. Rev., 1977, 6, 195–214 RSC.
- O. Fónagy, E. Szabó-Bárdos and O. Horváth, J. Photochem. Photobiol., A, 2021, 407, 113057 CrossRef.
- F. Rekhroukh, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 11920–11929 CrossRef CAS PubMed.
- A. Comas-Vives, C. González-Arellano, A. Corma, M. Iglesias, F. Sánchez and G. Ujaque, J. Am. Chem. Soc., 2006, 128, 4756–4765 CrossRef CAS PubMed.
- K. M. Parida, S. Singha, P. C. Sahoo and S. Sahu, J. Mol. Catal. A: Chem., 2011, 342–343, 11–17 CrossRef CAS.
- G. R. Reddy, S. Balasubramanian and K. Chennakesavulu, J. Mater. Chem. A, 2014, 2, 15598–15610 RSC.
- M. Yamaguchi and I. Hirao, Tetrahedron Lett., 1983, 24, 1719–1722 CrossRef CAS.
- T. Murai, R. Toshio and Y. Mutoh, Tetrahedron, 2006, 62, 6312–6320 CrossRef CAS.
- A. Paul and D. Seidel, J. Am. Chem. Soc., 2019, 141, 8778–8782 CrossRef CAS PubMed.
- Y. Cui, W. Lin and S. Ma, Chem. Sci., 2019, 10, 1796–1801 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy02121d |
| This journal is © The Royal Society of Chemistry 2022 |