
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring graphene-supported Ru nanoparticles by functionalization with pyrene-tagged N-heterocyclic carbenes†
Adrián
García-Zaragoza‡
 a,
Christian
Cerezo-Navarrete‡
a,
Andrés
Mollar-Cuni
b,
Pascual
Oña-Burgos
a,
Jose A.
Mata
b,
Avelino
Corma
a and
Luis M.
Martínez-Prieto
*a
a,
Christian
Cerezo-Navarrete‡
a,
Andrés
Mollar-Cuni
b,
Pascual
Oña-Burgos
a,
Jose A.
Mata
b,
Avelino
Corma
a and
Luis M.
Martínez-Prieto
*a
aITQ, Instituto de Tecnología Química, Universitat Politècnica de València-Consejo Superior de Investigaciones Científicas, Av. de los Naranjos S/N 46022, Valencia, Spain. E-mail: luismiguel.martinez@csic.es
bInstitute of Advanced Materials (INAM), Universitat Jaume I, Avda. Sos Baynat S/N 12006, Castellón, Spain
First published on 27th December 2021
Abstract
The catalytic properties of graphene-supported ruthenium nanoparticles (Ru@rGO) have been finely tuned by modifying their metal surface with pyrene-tagged N-heterocyclic-carbene ligands (pyr-IMes). The nature and interaction modes of the pyr-IMes ligands on Ru@rGO were established by XPS, which were found as protonated carbenes, coordinated to the ruthenium surface and directly interacting with the graphene support. To evaluate the activity and selectivity of Ru@rGO functionalized with different equivalents of pyr-IMes (Ru@rGO/pyr-IMesn; n = 0, 0.2, 0.5, 0.8 or 1), we used acetophenone hydrogenation as a model reaction. The catalytic activity and selectivity are highly dependent on the NHC surface coverage degree. The higher the amount of surface NHC ligands, the lower the activity of the catalyst, but the higher the selectivity towards 1-phenylethanol (suppressing the hydrodeoxygenation side reaction at high surface coverages). The reactivity of the most interesting catalyst, Ru@rGO/pyr-IMes0.5, was evaluated in the hydrogenation of other molecules of interest, such as nitrobenzene, 5-hydroxymethylfurfural (HMF), quinoline or 1-methylindole, among others. Finally, by TEM analysis after catalysis we observed a clear correlation between the surface ligand coverage and the stability of the catalysts against sintering. It was then possible to control the reactivity and stability of graphene-supported Ru NPs by modifying their surface with pyr-IMes ligands.
Introduction
The catalytic use of metal nanoparticles (MNPs) for fine chemical synthesis has undergone an exponential growth since the early 90s.1 The interest is mainly due to the particular catalytic properties of MNPs, which combine the main advantages of homogeneous and heterogeneous catalysts.2 Generally, MNPs can be easily recycled and reused as heterogeneous catalysts, but with the characteristic high activity of the homogeneous ones. The high activity of MNPs is explained by their great number of available surface active sites due to their small size (1–100 nm) and high surface-to-volume ratio. However, this large number of surface atoms of different nature normally transforms the reactants in different ways, being difficult to precisely control the selectivity of MNPs. An efficient way to modulate the catalytic performance of MNPs is the use of organic ligands, which can modify the substrate–metal surface interactions during the catalysis and therefore change their activity and selectivity.3 Controlling the surface chemistry of MNPs by functionalization with ancillary ligands is a well-known strategy in colloidal catalysis.4 However, it has been much less employed in supported MNP catalysis, since traditionally it was supposed that surface ligands block active sites, decreasing the activity of the supported catalyst, which is a priori an undesired effect.5 All the same, as well as in organometallic chemistry, surface ligands are also able to modify the electronic and steric properties of supported MNPs and thus, to modulate their catalytic properties.6 Thus, the better stability/recyclability of supported MNPs compared to those of colloidal MNPs, together with the possibility of controlling the activity/selectivity through their surface modification with organic ligands, make functionalized supported MNPs promising catalysts for industrial processes, since they combine all the benefits of heterogeneous (stability and recyclability) and homogeneous (activity and selectivity) catalysis.Selective supported metal catalysts are highly important in the industrial synthesis of chemicals.7 Most industrial catalysts are MNPs supported on high surface area materials that maximize the metal surface exposure.8 A strong interaction between the MNPs and the support is crucial to obtain stable catalysts. Moreover, the reactivity of supported MNPs will not only depend on the particular electronic properties of the metal sites, but also on the chosen support, which, as well as surface ligands, can modify their reactivity.9 Therefore, the selection of an appropriate support with suitable properties (thermal stability, high surface area, basic/acidic sites, etc.) is fundamental for the stabilization of MNPs and to control their catalytic properties. In this line, high surface area two-dimensional (2D) structures such as reduced graphene oxide (rGO) are appealing supports for MNPs due to their specific electronic configuration that results in strong MNP–graphene interactions.10 As already mentioned, these metal–graphene interactions can also modify the electronic properties of the supported MNPs, changing their reactivity.11 In addition, the defect sites normally present in rGO are excellent anchoring points for MNPs, facilitating their generation and stabilization.12 All this, together with the recent advances in the synthesis of graphene materials, makes graphene-supported MNPs attractive heterogeneous catalysts for industrial applications.
Since the isolation of the first example of an N-heterocyclic carbene (NHC),13 this type of compound has been extensively used as an ancillary ligand in coordination chemistry.14 Due to their excellent electron donating properties, NHC ligands strongly coordinate to transition metals, being exceptional ligands for transition metal complexes.15 However, they are not only suitable ligands for the stabilization of organometallic complexes, but also effective MNP stabilizers.16 Indeed, during the last decade a great number of NHC-stabilized MNPs have been reported, including Au,17 Ir,18 Ru,19 Ni,20 Pd,21 or Pt22 NPs. Moreover, NHCs have been demonstrated to be an efficient tool to modify the size, stability, solubility and catalytic properties of MNPs. Depending on the molecular structure (e.g. backbone with long alkyl chains) or N-substituents (e.g. bulky groups) of the stabilizing NHC, the resulting MNPs will display different properties.16,23 Thanks to their versatility, NHC-stabilized MNPs have been successfully applied in numerous catalytic processes such as hydroboration,24 oxidation,23c deuteration25 and hydrogenation reactions.26 In the same way that NHCs can modify the catalytic properties of colloidal MNPs, they can be also used to functionalize supported MNPs and thereby modulate their surface chemistry.27 For example, in 2016, Glorius et al. reported how to control the selectivity of Ru/K–Al2O3 by functionalization with NHCs in the hydrogenation of phenylacetylene to ethylbenzene.28 Pieters et al. also controlled the selectivity of Ru NPs supported on carbon in the isotopic H/D exchange by modifying the surface metal with NHCs.29 However, the surface modification of supported MNPs with NHCs not only may improve their selectivity, but also is able to increase their stability. Recently, it has been shown that NHCs bearing a pyrene group can enhance the stability of graphene-supported MNPs via non-covalent π-interactions between the pyrene tag and the graphene layer.30
Herein, we present Ru NPs supported on reduced graphene oxide (Ru@rGO) functionalized with different equivalents (0.2, 0.5, 0.8 and 1 equiv.) of an NHC ligand containing a pyrene group (pyr-IMes). The obtained catalytic systems (Ru@rGO/pyr-IMes) have been fully characterized by state-of-the-art techniques. Additionally, it has been possible to modify their stability, activity and selectivity in the hydrogenation of different substrates (i.e. acetophenone, decanal, nitrobenzene, styrene, 3-methyl-2-cyclohexenone, quinoline, 1-methylindole and HMF) by means of the NHC surface coverage.
Results and discussion
Synthesis, characterization and surface studies
Ruthenium nanoparticles supported on rGO (Ru@rGO) functionalized with NHC ligands containing a pyrene tag were prepared by following a two-step synthetic route (Fig. 1). First, (i) Ru NPs were generated through an organometallic approach and directly immobilized onto reduced graphene oxide (rGO). More specifically, graphene supported Ru NPs (Ru@rGO) were generated by the controlled decomposition of Ru(COD)(COT) (COD: cyclooctadiene; COT: cyclooctatriene) under 3 bar H2 in a THF dispersion of rGO, according to a previously reported procedure.31 Then, (ii) the as-synthesized Ru@rGO was functionalized with different amounts of a pyrene-tagged N-heterocyclic carbene ligand (n = 0.2, 0.5, 0.8 or 1 equivalent relative to ruthenium),32 formed by the deprotonation of the corresponding imidazolium salt with KtBuO. Purification by washing with THF yields Ru NPs immobilized onto rGO functionalized with NHC ligands as black powders, namely Ru@rGO/pyr-IMesn. | ||
| Fig. 1 Two-step synthetic route followed for the synthesis and subsequent functionalization of Ru@rGO with pyr-IMes. | ||
Ruthenium contents in Ru@rGO and Ru@rGO/pyr-IMesn were determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis employing an optimized digestion method.31 In all cases, the ruthenium content was close to the theoretical value of 3 wt% (for more details see the Experimental section).
Transmission electron microscopy (TEM) analysis of the unmodified Ru@rGO revealed the presence of small spherical Ru NPs displaying low dispersion in size, good distribution on the graphene support and a mean diameter of 1.5 ± 0.5 nm (Fig. 2a). After their functionalization with 0.2, 0.5, 0.8 or 1 equivalent (equiv.) of pyr-IMes (Ru@rGO/pyr-IMesn), the morphology, size, dispersion and distribution of the graphene-supported Ru NPs did not significantly change (Fig. 2b; see the ESI,† Fig. S1–S3). A high-resolution TEM (HRTEM) micrograph of Ru@rGO (Fig. S4†) shows the presence of crystalline nanoparticles with a hexagonal close-packed (hcp) structure characteristic of bulk Ru(0). The Fourier transform analysis applied to this picture shows reflection planes at (102), (002) and (101).
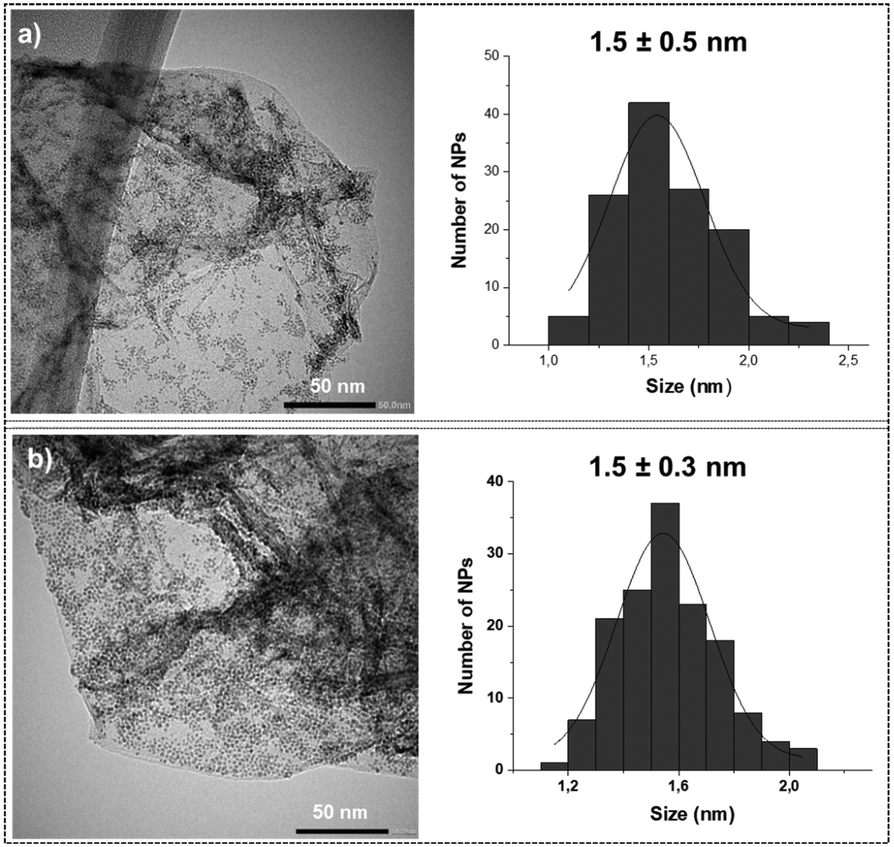 | ||
| Fig. 2 Comparative TEM images and size distribution histograms of (a) Ru@rGO and (b) Ru@rGO/pyr-IMes0.5. | ||
Since Raman spectroscopy is an established technique to investigate the quality of graphenic materials (defects, exfoliation degree, sp2 character, etc.), rGO, Ru@rGO and Ru@rGO/pyr-IMes0.5 were analyzed by this spectroscopic technique. In all recorded Raman spectra, two well-differenced domains were observed, one located between 1200 and 1700 cm−1 and another one from 2500 to 3250 cm−1 (see the ESI,† Fig. S5–S7). The first domain contains two bands of similar intensities at 1354 and 1595 cm−1, which correspond to D and G bands, respectively. A high ratio of the intensities of these bands (ID/IG) is related to a high percentage of defect sites, which are normally excellent anchoring points for MNP stabilization.12,33 In addition, comparing the ID/IG ratios before (rGO; Fig. S5†) and after the incorporation of Ru NPs (Ru@rGO or Ru@rGO/pyr-IMes0.5; Fig. S6 and S7†), a slight decrease from 1.54 to 1.50 was observed. This decrease in the ID/IG ratio is an indication of the increase of the sp2 character of Ru@rGO and Ru@rGO/pyr-IMes0.5, mainly due to the incorporation of Ru NPs. The second domain centered at ca. 3000 cm−1 corresponds to the 2D band and is related to a few graphene layers.
A series of surface studies were performed in order to better understand the coordination mode, location and dynamics of the pyrene-tagged N-heterocyclic carbenes at the MNP surface. First, to estimate the precise amount of pyr-IMes ligand incorporated into each one of the functionalized catalysts, a combined study involving elemental analysis (EA) and BET surface area determination was carried out. Once the available surface area of Ru@rGO was determined (331.8 m2 g−1), we calculated by EA the real amount of pyr-IMes ligand added onto rGO and thus, the actual number of equivalents of pyrene-tagged NHCs with which each catalyst has been functionalized (Table S1†). As a general trend, only half of the amount of NHC ligand added during the synthesis was finally incorporated into the functionalized catalysts.
Subsequently, a CO chemisorption analysis was performed to determine the accessible ruthenium surface of the unmodified Ru@rGO and the functionalized Ru@rGO/pyr-IMes0.5 and therefore, to evidence the coordination of the carbene ligand to the ruthenium surface. For the unmodified Ru@rGO, the CO uptake was 62.1 μg g−1, while for Ru@rGO/pyr-IMes0.5, the uptake of CO decreased to 35.9 μg g−1. The decrease in the number of available Ru sites on Ru@rGO/pyr-IMes0.5 is attributed to the coordination of pyr-IMes onto the Ru surface, which blocks the potential CO adsorption sites. Therefore, this partial decrease in the chemisorption of CO not only confirms the coordination of pyr-IMes to Ru NPs, but also demonstrates the presence of available active surface sites on the functionalized catalysts. With the aim of shedding some light on the nature and location of these active sites and to further investigate the coordination of pyr-IMes at the Ru surface, the unmodified and functionalized graphene-supported Ru NPs were studied by IR and solid-state MAS-NMR spectroscopy using CO as a probe molecule. However, due to their opacity and conductivity (which made the tuning of the NMR probe impossible), these analyses were unsuccessful. To circumvent this limitation, colloidal/unsupported Ru NPs stabilized with 0.2 and 0.5 equiv. of pyr-IMes (Ru/pyr-IMes0.2 and Ru/pyr-IMes0.5) were synthesized and fully characterized, including IR and MAS-NMR surface studies using CO as molecular probe. NHC ligand-stabilized Ru NPs were prepared by following the same organometallic approach shown in Fig. 1, but using the pyrene-tagged NHC ligands as a stabilizer (Fig. 3). Here, the non-supported Ru NPs are directly stabilized by the NHC ligands, as has been previously described.23b
 | ||
| Fig. 3 Synthesis of ligand-stabilized Ru/pyr-IMes NPs following the organometallic approach. | ||
The TEM and HRTEM images of Ru/pyr-IMes0.2 and Ru/pyr-IMes0.5 showed small, crystalline and monodispersed Ru NPs with a mean diameter of 1.4 ± 0.4 and 1.3 ± 0.4 nm, respectively (see the ESI,† Fig. S8–S10). The ICP analyses of Ru/pyr-IMes0.2 and Ru/pyr-IMes0.5 gave ruthenium contents of 55.7 and 33.2 wt%, respectively. It is worth mentioning that the ratio between the surface ruthenium atoms and coordinated NHC ligands (Ru(s)x/Ly) is not large enough to accommodate all the ligands on the Ru surface (steric hindrance). More specifically, Ru/pyr-IMes0.2 exhibits a Ru(s)x/Ly ratio of ca. 4.1, while the ratio for Ru/pyr-IMes0.5 is even lower, 1.4 (see the ESI,† Table S2). Therefore, we can assume that the remaining surface ligands are organized in a second coordination sphere by π–π stacking between the aromatic side groups of the pyrene-tagged NHCs. This type of second sphere of non-coordinated ligands has been previously observed in similar ligand-stabilized MNPs.22,34
Afterwards, the surface state of Ru/pyr-IMes0.2 and Ru/pyr-IMes0.5 was investigated by evaluating their capacity to bind CO by diffuse reflectance infrared Fourier transform (DRIFT) and 13C{1H} MAS NMR with 1H–13C cross-polarization (CP). Here the aim was to locate the available surface active sites of these NHC-stabilized Ru NPs. Fig. S11 and S12 (see the ESI†) show the DRIFT spectra of Ru/pyr-IMes0.2 and Ru/pyr-IMes0.5 before (blue) and after (red) exposure to CO (bubbling CO in a THF solution, for more details see the Experimental section). Interestingly, before the reaction with CO, both spectra already exhibited a weak and broad band at ca. 1900 cm−1, corresponding to adsorbed CO. The presence of this band is attributed to the decarbonylation of THF during the synthesis of the nanoparticles, as previously observed.23c,35 After the reaction with CO, both spectra showed an increase in intensity and a shift to higher frequency (ca. 2000 cm−1) of the aforementioned band. This confirmed the coordination of CO and the high availability of free Ru active sites at the NP surface, even in the presence of a large number of surface NHC ligands, as was previously determined by ICP (see the ESI,† Table S2).
The 13C{1H} CP-MAS NMR spectrum of Ru/pyr-IMes0.2 showed a number of distinct peaks characteristic of the pyrene-tagged NHC ligand (see the ESI,† Fig. S13a). The broad signal at ca. 130 ppm was assigned to the aromatic rings of the mesitylene and pyrene groups, together with the imidazole backbone. The peaks between 50 and 10 ppm were attributed to the CH2 group connected to the pyrene tag, as well as the methyl groups of the N-bonded mesitylene. After exposure of Ru/pyr-IMes0.2 NPs to 1 bar 13CO at room temperature (r.t.), the 13C{1H} CP-MAS NMR spectrum showed two new signals ascribed to adsorbed CO (Fig. S13b†). The broad and low-intensity resonance at ca. 230 ppm corresponds to 13CO coordinated in a bridging mode (COb), and the sharp and intense peak at ca. 200 ppm is due to 13CO adsorbed in a terminal mode (COt). It is well known that bridging CO molecules coordinate onto faces (or terraces) of nanoparticles and terminal ones onto their apexes and edges.36 Since most of the CO coordinates in a terminal mode, we can assume that the ruthenium faces are not accessible due to the π interactions between the aromatic rings of the pyrene and the ruthenium terraces, which hinders the coordination of CO in a bridging mode.37 Furthermore, the appearance of spinning sidebands (*) suggests that the terminal CO molecules are static on the surface. This lack of mobility of COt on the ruthenium surface can be explained by the coordination of NHC ligands near the active sites where CO molecules are adsorbed. The 13C{1H} CP-MAS NMR spectrum of Ru/pyr-IMes0.5 after exposure to 13CO showed similar resonances, but with a new peak at 204 ppm (see the ESI,† Fig. S14). This new resonance may correspond to the CO coordinated in a multi-terminal mode, as previously reported for Rh NPs.38 Again, we observed by CP-MAS NMR a great number of free active sites on these non-supported Ru NPs even at high surface ligand coverages.
The coordination of pyr-IMes onto the non-supported Ru/pyr-IMes0.5 NPs was also investigated by X-ray photoelectron spectroscopy (XPS), since it is a useful technique to study the binding mode of NHC surface ligands on MNPs.39 The N 1s area of Ru/pyr-IMes0.5 presents a binding energy (BE) of 399.1 eV (Fig. 4a, orange), which is shifted to low BE values compared to that of the corresponding imidazolium salt pyr-IMes·HBr (400.9 eV) (Fig. 4a, green). This decrease in the BE is due to a loss of electron density of the N atoms of pyr-IMes and indicates the direct coordination of the NHC ligand to the ruthenium surface. XPS analysis was also employed to investigate the coordination of the pyrene-tagged NHC ligand onto the graphene-supported Ru NPs functionalized with 0.5 equivalents of pyr-IMes (Ru@rGO/pyr-IMes0.5). For Ru@rGO/pyr-IMes0.5 the N 1s peak is centered at 399.6 eV and can be deconvoluted into three contributions with different BEs (Fig. 4b), corresponding to pyr-IMes coordinated to Ru (398.9 eV), interacting with rGO (399.8 eV), and protonated (401.2 eV). To confirm the BE of pyr-IMes which directly interacts with the rGO support (rGO/pyr-IMes), most probably by π-interactions between the pyrene tags of the NHC and the graphene layer, free NHC was added to a dispersion of rGO in THF and analyzed by XPS (for more details see the Experimental section). And, as was expected, the N 1s signal of rGO/pyr-IMes revealed a peak at 399.8 eV, which is located between the N 1s peaks of the corresponding imidazolium salt and the pyr-IMes ligand coordinated to ruthenium (Fig. 4a, blue). Therefore, using XPS we were able to evaluate the different nature and chemical environments of pyr-IMes ligands on the functionalized catalyst, Ru@rGO/pyr-IMes0.5: (i) protonated (400.9 eV), (ii) coordinated to ruthenium (399.1 eV) and (iii) interacting with the graphene support (399.8 eV) (Fig. 4). Furthermore, by analyzing their N 1s XPS areas after deconvolution, we can deduce that approximately 32% of the NHC ligand is coordinated to Ru, 45% interacts with the graphene support and 23% is protonated.
 | ||
| Fig. 4 X-ray photoelectron spectroscopy (XPS) spectra of (a) the N 1s signals of pyr-IMes·HBr (green), rGO/pyr-IMes (blue), Ru/pyr-IMes0.5 (orange) and (b) Ru@rGO/pyr-IMes0.5 (red). | ||
Moreover, the oxidation state of the ruthenium was also investigated by XPS upon analyzing the Ru 3p region of Ru/pyr-IMes0.5, Ru@rGO and Ru@rGO/pyr-IMes0.5 (Fig. S15†). The Ru 3p3/2 signal of the ligand-stabilized Ru/pyr-IMes0.5 was found at 462.2 eV, which after deconvolution showed two contributions, a main contribution at ca. 461.4 eV, attributed to Ru(0), and a minor one at ca. 463.8 eV, corresponding to RuO2 (Fig. S15a†). On the other hand, the Ru 3p3/2 signals of graphene-supported Ru NPs unmodified and modified with pyr-IMes were observed at 463.1 and 463.4 eV, respectively (see the ESI,† Fig. S15b and c). After deconvolution, we observed that most of the ruthenium was oxidized to Ru(IV). This is mainly due to the purification process of the graphene-supported Ru NPs, which is carried out in air (see the Experimental section). For this reason, a preliminary activation step (1 h at 150 °C and 50 bar H2) was necessary before catalysis. This activation step reduces most of the RuO2 to Ru(0), which is the active species in hydrogenation reactions.
Catalytic studies
First, to evaluate the influence of the pyrene-tagged NHC ligands on the activity and selectivity of the graphene-supported Ru NPs, the hydrogenation of acetophenone was used as a model reaction. This substrate is an interesting model molecule since it contains two potentially reducible functional groups (i.e. a ketone and a phenyl group), and the activity/selectivity of the functionalized catalysts herein prepared can be simply compared. Acetophenone (1) can be reduced to 1-cyclohexylethanol (4) through two pathways: (i) via 1-phenylethanol (2), hydrogenating first the ketone, and (ii) via 1-cyclohexylethanone (3), where the phenyl group is hydrogenated before (top of Table 1). Additionally, ethylbenzene (5) and ethylcyclohexane (6) can be also formed by hydrodeoxygenation (HDO) processes. By checking the selectivity of the non-functionalized Ru@rGO at total conversion (20 h, Table 1, entry 1), it can be seen that it mostly produces the over reduced product 4 (81.5%) together with 6 (18.5%), which comes from HDO processes. After 20 h reaction, Ru@rGO/pyr-IMes0.2 also shows complete conversion and a considerable amount of HDO products 5 (3.9%) and 6 (16.7%), but with a lower selectivity towards 4 (69.6%). Comparing the initial reaction rates of Ru@rGO and Ru@rGO/pyr-IMes0.2 (Fig. 5a and b), the unmodified Ru NPs show the highest activity. After 1 hour of reaction, Ru@rGO exhibits an acetophenone conversion of 98%, while the conversion by using Ru@rGO/pyr-IMes0.2 as catalyst is only 56%. The initial reaction rate for Ru@rGO/pyr-IMes0.5 slightly decreases in comparison to that for Ru@rGO/pyr-IMes0.2 (Fig. 5b and c); however, it was accompanied with an increase of the selectivity towards the fully hydrogenated product 4 and a significant drop in the formation of HDO products. More specifically, after 20 h, a conversion of ca. 98.6% was achieved with a selectivity towards 4 of 84.1%, producing only 8.9% of 6 (Table 1, entry 3). Upon increasing the number of equivalents of pyr-IMes up to 0.8 (Ru@rGO/pyr-IMes0.8), we find a marked decrease in the activity, together with a substantial increase in the selectivity towards 2, giving after 20 h reaction 63.6% 1-phenylethanol at 95.9% conversion (Fig. 5d; Table 1, entry 4). Finally, the graphene-supported Ru NPs with the highest surface coverage, Ru@rGO/pyr-IMes1, displays the lowest activity, but the highest selectivity towards 2 (98% selectivity at 60.2% conversion), practically avoiding the formation of HDO products (Fig. 5e; Table 1, entry 5). These results show a clear correlation between the surface coverage degree and the activity and selectivity of the functionalized catalysts. The higher the amount of surface pyr-IMes ligands, the lower the activity of the catalyst, but the higher the selectivity towards 2. In addition, at higher surface coverages the formation of HDO products is avoided. Both the decrease in the activity and the selectivity improvement can be explained by the blockage of the ruthenium active sites by pyr-IMes ligands. As we increase the number of equivalents of pyr-IMes, the amount of free ruthenium sites decreases, and therefore, the activity is negatively affected. However, this blockage of active sites also improves the selectivity of the functionalized catalysts, not only avoiding the undesired HDO side reactions (i.e.Ru@rGO/pyr-IMes0.5; Table 1, entry 3), but also hydrogenating selectively acetophenone to 1-phenylethanol at high surface coverages (i.e.Ru@rGO/pyr-IMes1; Table 1, entry 5). It is important to mention that the selective hydrogenation of aromatic ketones to form benzyl alcohols is normally catalyzed by homogeneous catalysts,40 since heterogeneous catalysts generally produce mixtures of 2, 3 and 4.41 Altogether, these findings demonstrate that it is possible to control the selectivity of graphene-supported Ru NPs during acetophenone hydrogenation by modifying their metal surface with pyrene-tagged N-heterocyclic carbene ligands.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Conversionb,c (%) | Selectivityb (%) | ||||
| 2 | 3 | 4 | 5 | 6 | |||
| a Reaction conditions: 0.5 mmol acetophenone, 0.5 mol% cat (0.0025 mmol Ru), 10 mL THF, 50 bar H2, 130 °C, 20 h. b Conversions and selectivities were determined by GC using dodecane as the internal standard and confirmed by GC-MS. c Metal-free rGO exhibited negligible activity in the hydrogenation of acetophenone under these reaction conditions. | |||||||
| 1 | Ru@rGO | >99 | 0.0 | 0.0 | 81.5 | 0.0 | 18.5 |
| 2 | Ru@rGO/pyr-IMes 0.2 | >99 | 1.1 | 8.7 | 69.6 | 3.9 | 16.7 |
| 3 | Ru@rGO/pyr-IMes 0.5 | 98.6 | 1.9 | 5.0 | 84.1 | 0.1 | 8.9 |
| 4 | Ru@rGO/pyr-IMes 0.8 | 95.9 | 63.6 | 9.8 | 21.7 | 4.2 | 0.7 |
| 5 | Ru@rGO/pyr-IMes 1 | 60.2 | 98.0 | 0.8 | 0.6 | 0.6 | 0.0 |
| 6 | Ru@rGO/ICy 1 | 98.4 | 0.0 | 9.6 | 77.2 | 3.7 | 9.5 |

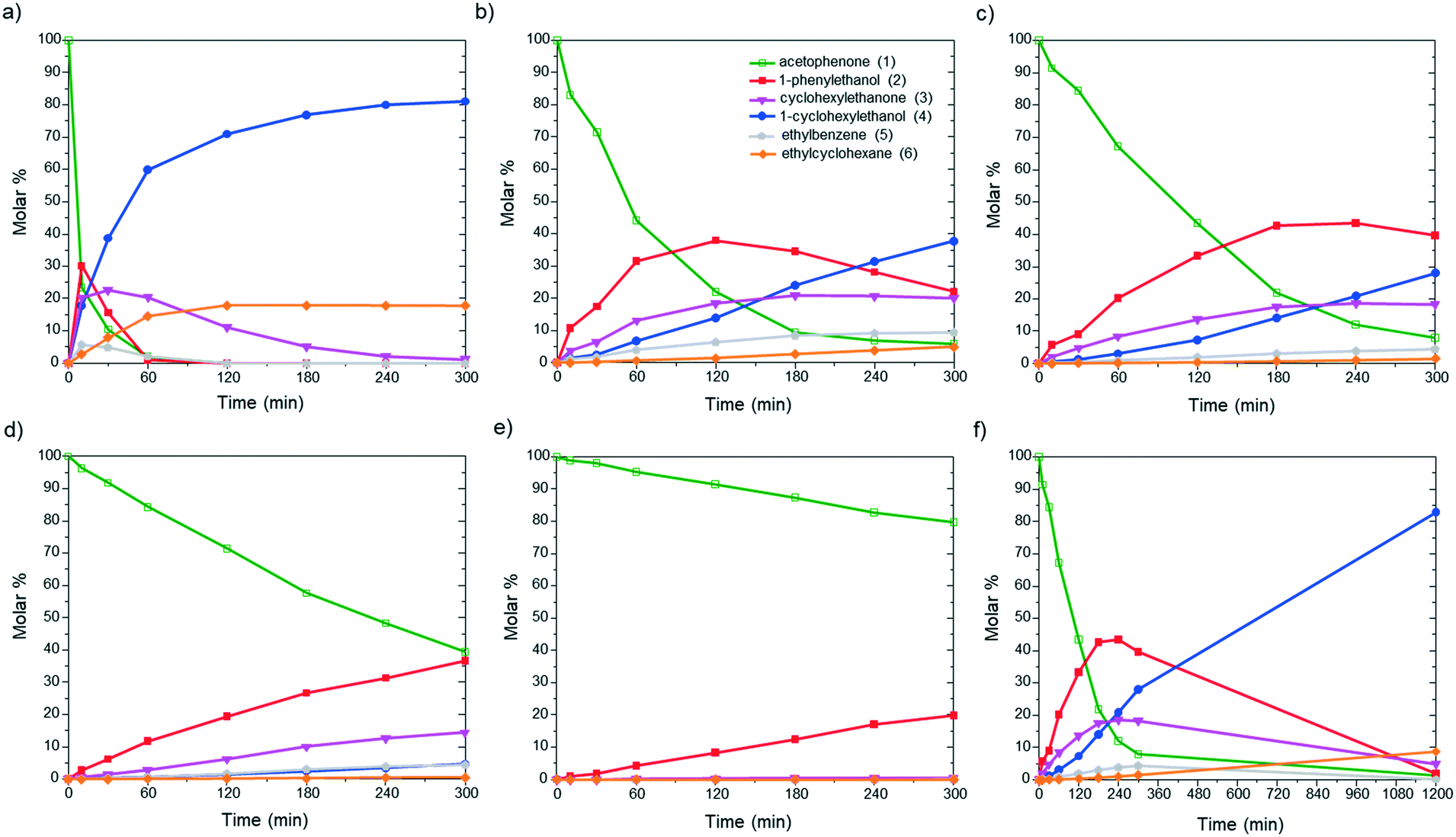 | ||
| Fig. 5 Hydrogenation of acetophenone using (a) Ru@rGO, (b) Ru@rGO/pyr-IMes0.2, (c and f) Ru@rGO/pyr-IMes0.5, (d) Ru@rGO/pyr-IMes0.8, and (e) Ru@rGO/pyr-IMes1 as catalysts. | ||
Among all the catalytic systems studied in the acetophenone hydrogenation reaction, Ru@rGO/pyr-IMes0.5 is the most attractive one since it presents the best compromise between activity and selectivity. This functionalized catalyst hydrogenates selectively acetophenone to 1-cyclohexylethanol, minimizing the generation of HDO products (Fig. 5f; Table 1, entry 3). Therefore, Ru@rGO/pyr-IMes0.5 was also tested in the hydrogenation of various substrates that contain other reducible functional groups such as aldehyde, vinyl, heterocyclic or nitro groups, under the same catalytic conditions (Table 2). Decanal (7) was selectively hydrogenated to 1-decanol (8) after 5 h reaction, without formation of the HDO product 9 (Table 2, entry 1). After 2 h, nitrobenzene (10) was also selectively converted to cyclohexylamine (12) (Table 2, entry 2). Similar reactivity was observed in the hydrogenation of 3-methyl-2-cyclohexenone (13) and quinoline (16). Both substrates, after 5 h, gave full conversion with high selectivity towards the corresponding over reduced product (Table 2, entries 3 and 4). On the other hand, at the same reaction time, styrene (20) only produced 43% ethylcyclohexane (22). Furthermore, we investigated the catalytic activity of Ru@rGO/pyr-IMes0.5 in other molecules of current interest such as 1-methylindole (23), which can be used as a liquid hydrogen carrier (LOHC), or 5-(hydroxymethyl)furfural (HMF; 27), a platform molecule derived from biomass. After 5 h reaction, Ru@rGO/pyr-IMes0.5 presented a 96% conversion of 23 with 90% selectivity towards the completely hydrogenated product 1-methyl-octahydroindole (22) (Table 2, entry 6). At longer reaction times (20 h), a complete conversion with 100% selectivity to 22 was achieved (Table 2, entry 7).These results are of high interest for hydrogen storage applications, since 1-methylindole is a promising LOHC due to its low melting point (−20 °C) and high hydrogen content (5.76 wt%).42 Due to the great potential of this functionalized catalyst, Ru@rGO/pyr-IMes0.5 has been also tested in the hydrogenation of an interesting biomass derived platform molecule, i.e. HMF (27). After 5 h of reaction, Ru@rGO/pyr-IMes0.5 showed a quantitative conversion with excellent selectivity towards 5-bis(hydroxymethyl)furan (BHMF) (28) (92%, Table 2, entry 8). HDO and hydrogenation products 29 and 30 were also formed, but in minute amounts.
| Entry | Substrates | Products | Time (h) | Conversionb (%) | Selectivityb (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 0.5 mmol substrates, 0.5 mol% cat (0.0025 mmol Ru), 10 mL THF, 50 bar H2, 130 °C. b Conversions and selectivities were determined by GC using dodecane as the internal standard and confirmed by GC-MS. | |||||
| 1 |
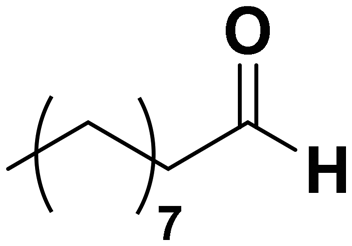 (7) (7) |
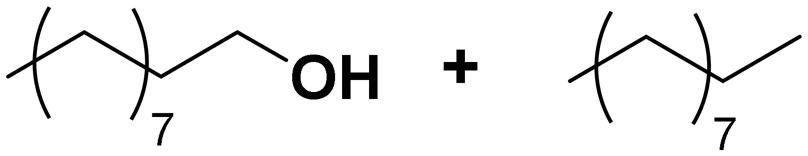 (8) (9) (8) (9) |
5 | >99 |
8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 = 100:0 :9 = 100:0 |
| 2 |
 (10) (10) |
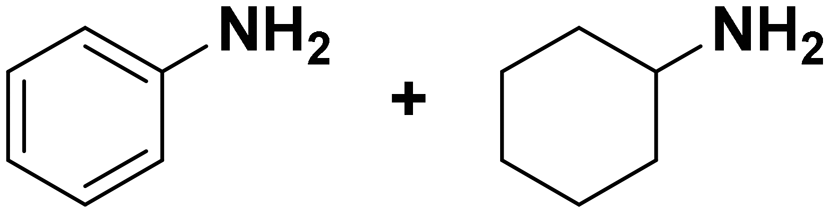 (11) (12) (11) (12) |
2 | >99 |
11:12 = 0:100 |
| 3 |
 (13) (13) |
 (14) (15) (14) (15) |
5 | >99 |
14:15 = 4:96 |
| 4 |
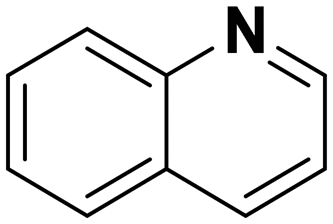 (16) (16) |
 (17) (18) (19) (17) (18) (19) |
5 | >99 |
17:18:19 = 0:9:91 |
| 5 |
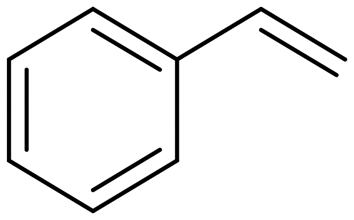 (20) (20) |
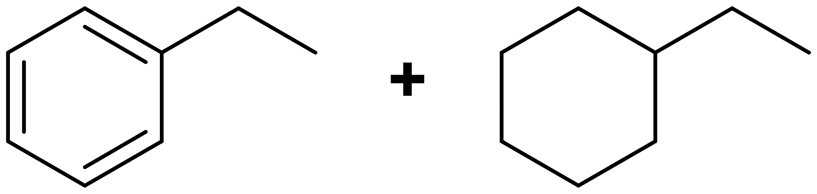 (21) (22) (21) (22) |
5 | >99 |
21:22 = 57:43 |
| 6 |
 (23) (23) |
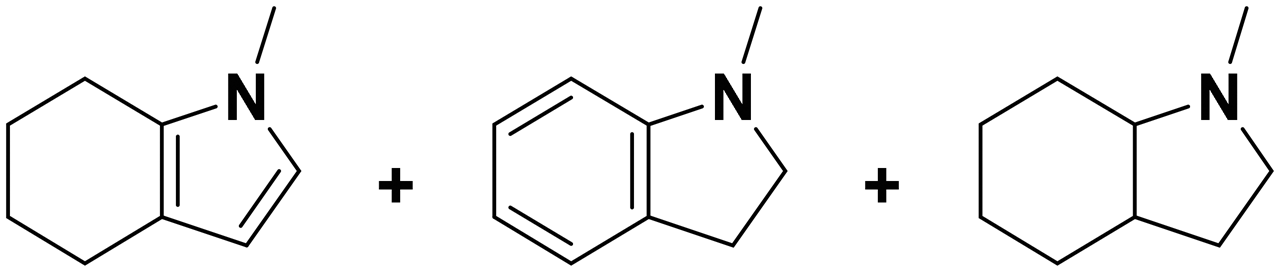 (24) (25) (26) (24) (25) (26) |
5 | 96 |
24:25:26 = 9:1:90 |
| 7 | 20 | >99 |
24:25:26 = 0:0:100 |
||
| 8 |
 (27) (27) |
 (28) (29) (30) (28) (29) (30) |
5 | >99 |
28:29:30 = 92:2:6 |
A series of experiments were performed to investigate the influence of the surface pyrene-tagged NHCs on the stability of graphene-supported Ru NPs. First, we analyzed by TEM the size and morphology of Ru NPs present on the unmodified and modified catalytic systems after a standard catalytic test (hydrogenation of acetophenone for 20 h at 50 bar H2 and 130 °C). The Ru NPs supported on Ru@rGO significantly increased their size from 1.5 to 2.3 nm (Table 3, entry 1). On the other hand, Ru NPs on Ru@rGO/pyr-IMes1 practically did not increase in size after catalysis (from 1.6 to 1.7 nm; Table 3, entry 5). The analysis after catalysis of the Ru NPs immobilized on the other three functionalized catalytic systems showed intermediate sizes. More specifically, we observed that the main diameter of the nanoparticles after catalysis decreases as the number of equivalents of pyr-IMes in the system increases (Table 3, entries 2–4). Therefore, there is an evident correlation between the amount of surface NHC ligands and the stability of the supported Ru NPs against sintering (Fig. S16–S20). The higher the surface coverage, the more resistant the catalyst is to sintering. This demonstrates the ability of the pyr-IMes ligands to increase the stability of graphene-supported MNPs, as previously reported for similar systems.30
| Entry | Catalyst | Size before catalysis (nm) | Size after catalysis (nm) |
|---|---|---|---|
| 1 | Ru@rGO | 1.5 ± 0.5 | 2.3 ± 0.5 |
| 2 | Ru@rGO/pyr-IMes 0.2 | 1.5 ± 0.4 | 2.1 ± 0.5 |
| 3 | Ru@rGO/pyr-IMes 0.5 | 1.5 ± 0.3 | 1.9 ± 0.3 |
| 4 | Ru@rGO/pyr-IMes 0.8 | 1.5 ± 0.7 | 1.8 ± 0.4 |
| 5 | Ru@rGO/pyr-IMes 1 | 1.5 ± 0.4 | 1.7 ± 0.4 |
| 6 | Ru@rGO/ICy 1 | 1.6 ± 0.5 | 2.3 ± 0.5 |
The influence of the pyrene tag on the activity/selectivity and stability of Ru@rGO/pyr-IMes was evaluated by using as a modifier another NHC ligand without polyaromatic groups on the N-substituents. Concretely, Ru@rGO was functionalized with 1 equiv. of an NHC with cyclohexyl groups on the N-substituents (1,3-dicyclohexylimidazol-2-ylidene; ICy) following the two-step synthetic route in Fig. 1 (Ru@rGO/ICy1) (Fig. S21†). In this case, the absence of the pyrene tag affected both the selectivity and stability of the resulting catalyst. After studying the reactivity of Ru@rGO/ICy1 during the hydrogenation of acetophenone, we observed that it was not possible to control the selectivity of the catalyst in the same way as with Ru@rGO/pyr-IMes1 (Fig. S22†). Here, even at high NHC surface coverage (1 equiv. of ICy), we observed the hydrogenation of both functional groups (carbonyl and phenyl), with the totally hydrogenated product 4 being the main product after 20 h (Table 1, entry 6). Probably, the lack of aromatic groups of ICy allows the interaction between the acetophenone and the faces of the Ru NPs and thus, the hydrogenation of phenyl groups. Already reported surface studies on colloidal Ru nanoparticles stabilized by ICy confirm the presence of free available faces.23b Contrary to the pyr-IMes ligands, which through π-interactions block the ruthenium terraces of Ru@rGO/pyr-IMes1, and make it highly selective in the hydrogenation of acetophenone (1) to 1-phenylethanol (2). The surface modification of Ru@rGO with ICy neither controls the formation of HDO side-products 5 and 6. By analyzing the size of Ru NPs present on Ru@rGO/ICy1 before and after the standard catalytic test (hydrogenation of acetophenone) by TEM (Table 3, entry 6; Fig. S23 and S24†), we observed a noticeable increase in size, from 1.5 to 2.3 nm, similar to that observed in the unmodified Ru@rGO. In addition, the nanoparticle distribution observed after catalysis is much less uniform (Fig. S24†). The absence of π-interactions between the ICy ligands and the graphene layer explains the lower stability of these Ru NPs against sintering. Altogether, these results highlight the importance of the presence of the pyrene tag in the NHC used as a modifier to control the selectivity and the stability of these functionalized catalysts.
To test the recyclability of Ru@rGO/pyr-IMes0.5 a multi-addition experiment was performed. Specifically, the hydrogenation of acetophenone was carried out during seven consecutive additions of the substrate (every 12 h) using Ru@rGO/pyr-IMes0.5 as a catalyst. Interestingly, we observed that the catalyst not only retained the activity during this multi-addition experiment, but also improved the selectivity to 1-cyclohexyletanol (4) (Fig. 6). This progressive increase in the reactivity during the first addition cycles is explained by the formation of a more active surface under catalytic conditions, due to the gradual reduction of the Ru metal surface (which is partially oxidized in the as-synthesized Ru NPs as was observed by XPS). The TEM analysis of Ru@rGO-pyr-IMes0.5 after the multi-addition experiment revealed Ru NPs similar in size and distribution to the as-prepared ones (Fig. S25†). These results underline the high stability of this functionalized catalyst under catalytic conditions during long reaction times (up to 86 h).
 | ||
| Fig. 6 Multi-addition experiment for the hydrogenation of acetophenone catalyzed by Ru@rGO/pyr-IMes0.5. Conditions: Acetophenone (0.5 mmol), 0.5 mol% cat (0.0025 mmol metal), THF, 50 bar H2, 130 °C, 12 h. Every 12 hours, acetophenone (0.5 mmol) was added to the reaction mixture. Conversions and selectivities were determined by GC using dodecane as the internal standard and confirmed by GC-MS. | ||
Finally, to verify the heterogeneous nature of Ru@rGO/pyr-IMes0.5, a “hot filtration” experiment was performed during the hydrogenation reaction of acetophenone (130 °C, 50 bar H2). After 2 h reaction, the solid Ru@rGO/pyr-IMes0.5 was separated by thermal filtration and the mother liquor solution was transferred to an empty reactor and then pressurized and heated. No change in the conversion was observed after 4 hours under the same catalytic conditions. Specifically, the conversion was maintained at 59% (Table S3, see the ESI†), while 88% conversion was observed in the presence of Ru@rGO/pyr-IMes0.5 (Fig. 5c). In addition, the ICP analysis after the thermal filtration did not show any metal leaching (see the Experimental section), confirming the heterogeneity of the catalyst.
Conclusions
The catalytic properties of graphene-supported Ru NPs were finely tuned by modifying their metal surface with pyrene-tagged NHC ligands. A combined study comprising EA and BET surface area analysis allowed us to determine the precise amount of pyr-IMes incorporated into the different functionalized catalysts. CO chemisorption studies confirmed both the coordination of NHC ligands to the ruthenium surface and the presence of free Ru sites on these functionalized catalysts. Ligand-stabilized Ru NPs were used as model catalysts for surface studies. The results suggested the presence of a second coordination sphere of pyr-IMes ligands on these non-supported Ru NPs, probably formed by π–π stacking interactions between the aromatic groups of the pyrene-tagged NHCs. Despite the high surface coverage of these non-supported Ru NPs, they still have a large number of available active sites, as was confirmed by DRIFT and solid-state NMR analyses using CO as a molecular probe. Interestingly, by XPS we were able to distinguish the different nature and interaction modes of pyr-IMes ligands on the functionalized catalysts. For example, in Ru@rGO/pyr-IMes0.5, the pyrene-tagged NHC can be found in three different ways: (i) as a protonated carbene, (ii) coordinated to the ruthenium surface and (iii) directly interacting with the graphene support. According to the catalytic results, we observed that both the activity and selectivity of these graphene-supported Ru NPs were highly dependent on the surface coverage degree. This clear correlation was investigated using acetophenone hydrogenation as a model reaction. The higher the amount of surface NHC ligands, the lower the activity of the catalyst, but the higher the selectivity towards 1-phenylethanol. Moreover, at higher surface coverages the hydrodeoxygenation side-reaction is practically suppressed. This increase in the selectivity at the expense of the activity is explained by the blockage of metal active sites by surface ligands. The reactivity of Ru@rGO/pyr-IMes0.5, which displays the best balance between activity and selectivity, was also evaluated in the hydrogenation of other substrates of interest, such as 1-methylindole or HMF, among others. Finally, the stability of the modified and non-modified catalysts was studied by TEM analysis after catalysis, observing an evident correlation between the nanoparticle surface ligand coverage and the stability of the supported Ru NPs against sintering. The presence of the pyrene tag on the NHC used as a modifier is crucial to control both the selectivity and stability of these ligand-functionalized supported Ru NPs. In addition, Ru@rGO/pyr-IMes0.5 was employed in multi-addition and “hot filtration” experiments, showing high stability and recyclability. To sum up, these results prove that it is possible to control the activity, selectivity and stability of graphene-supported Ru NPs by modifying their metal surface with organic molecules, making them potential catalysts for industrial applications, since they unify the benefits of homogeneous (high selectivity) and heterogeneous (stability and recyclability) catalysts.Experimental
General methods
Most chemical operations performed in this work were carried out using conventional Fischer–Porter techniques, Schlenk tubes and a glovebox under a nitrogen atmosphere. Solvents were purified before use, THF (Sigma-Aldrich) by distillation under an argon atmosphere through filtration in the column of a solvent purification system (SPS). The organometallic ruthenium precursor, (1,5-cyclooctadiene)(1,3,5-cyclooctatriene) ruthenium(0) [Ru(COD)(COT)], was purchased from Nanomeps (Toulouse), rGO from Graphenea and acetophenone (99%), decanal (98%), nitrobenzene (99%), styrene (99%), 3-methyl-2-cyclohexenone (98%), quinoline (98%), 1-methylindole (>97%), dodecane (99%) and 1,3-dicyclohexylimidazolium chloride from Merck. All reagents were used without prior purification, except for HMF (97%, Carbosynth) which was purified by filtering with an equimolar silica:alumina mixture, and stored in a refrigerator.
:1 solution of nitric and hydrochloric acid over 12 h. The ICP analysis was performed with an Agilent 7500 CX. The metal content of the ligand stabilized Ru NPs (Ru/pyr-IMes0.2 and Ru/pyr-IMes0.5) was measured by Kolbe MikroLab by ICP-AES.
Synthesis of pyr-IMes·HBr
The synthesis of the imidazolium salt (pyr-IMes·HBr) was achieved by following a synthetic method previously described elsewhere.32Synthesis of graphene-supported Ru NPs
Synthesis of ligand-stabilized Ru NPs
Synthesis of pyr-IMes/rGO
Catalytic hydrogenation reactions
The hydrogenation of acetophenone, 1-methylindole, quinoline and HMF was performed in a 25 mL autoclave engineer reactor, equipped with a mechanical stirrer (750 rpm). Hydrogenation reactions were carried out so that the metal–substrate molar ratio was 1:200. In this way, considering that 0.5 mmol of substrate was used and each catalyst has 3 wt% of the Ru metal, 0.5 mol% of the metal-catalyst was added in each reaction, which means 0.0025 mmol of metal in each case. For all the immobilized and functionalized catalysts, the exact mass added during the catalysis was 10 mg.
Thus, 10 mg of the catalyst was dispersed in 9 mL of THF and sonicated for 60 minutes. The suspension was transferred into the reactor, which was purged with H2 three times and finally pressurized with 35 bar H2. The reactor was then heated at 150 °C, reaching a final pressure of 50 bar. The catalyst was kept under these conditions for one hour, to reduce and activate the active ruthenium species. Before the substrate (0.5 mmol) and dodecane (0.3 mmol) diluted in 1 mL of THF were injected with the help of a 250 μL Hamilton syringe, the temperature was reduced to 130 °C. After maintaining the reaction for 20 h, the reactor temperature was lowered to room temperature and the reactor was depressurized. The resulting suspension was collected, and a fraction was filtered to separate the catalyst from the rest of the solution, which was analyzed by GC and GC-MS.
Kinetic experiments
For the kinetic experiments, the reactor was charged, pressurized and heated under the required conditions, and aliquots were taken from the reaction medium every hour. Then, the aliquot was analyzed by GC using dodecane as the internal standard.Multi-addition experiments
For the multi-addition experiment, the autoclave was charged, pressurized and heated under the required conditions for acetophenone hydrogenation (130 °C, 50 bar H2, 10 mL THF). Every 12 hours, an aliquot was taken from the reaction medium and a new starting substrate (i.e. acetophenone) was added to the reactor. The aliquots were analyzed by GC using dodecane as the internal standard and confirmed by GC–MS. The activity and selectivity were retained over 7 catalytic cycles confirming the stability of the Ru-NP catalysts.“Hot filtration” experiment
For the “hot filtration” experiment, the reactor was charged, pressurized and heated under the required conditions for acetophenone hydrogenation (130 °C, 50 bar H2, 10 ml THF). After 2 h reaction, Ru@rGO/pyr-IMes0.5 was removed by thermal filtration, and another clean and empty autoclave was charged with the mother liquor solution, pressurized and heated under the same reaction conditions. Then, the conversion of acetophenone was determined after 4 h and compared to that at 2 h (Table S2, ESI†). No change in the conversion was observed. After the “hot filtration”, the mother liquor solution was analyzed by ICP, with the Ru detected being negligible.Author contributions
C. C.-N. and A. G.-Z. performed the synthesis and structural/spectroscopic characterization and catalytic experiments and participated in writing – review and editing. A. M.-C. performed the synthesis and spectroscopic characterization of the NHC ligands. P. O.-B., A. C. and J. M. supervised the project and participated in writing – review and editing. L. M. M.-P. conceived the idea of the project and wrote the original draft with input from the other authors. All authors contributed to the design of the experiments, analysis of the results and preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Instituto de Tecnología Química (ITQ), Consejo Superior de Investigaciones Científicas (CSIC), Universitat Politècnica de València (UPV) and ‘Servei Central d'Instrumentació Científica (SCIC) de la Universitat Jaume I’ for the facilities and MICIU/AEI/FEDER (RTI2018-098237-B-C22) and Universitat Jaume I (UJI-B2018-23) for the financial support. We also thank the Electron Microscopy Service of the UPV for the TEM facilities, Jose A. Vidal-Moya (ITQ, CSIC-UPV) for the MAS-NMR measurements and J. Gaona for his assistance with the catalytic reactions. A. G.-Z. acknowledges “Programa de Ayudas del Aula Cemex Sostenibilidad” for the completion of the Master's Thesis in Sustainable Chemistry and C. C.-N. gratefully thanks the Generalitat Valenciana predoctoral fellowship (GVA: ACIF/2019/076). A. M.-C. thanks Generalitat Valenciana for a grant (ACIF/2019/100).Notes and references
- (a) D. Astruc, Chem. Rev., 2020, 120, 461 CrossRef CAS PubMed; (b) P. Serp and K. Philippot, Nanomaterials in Catalysis, Wiley-VCH, Weinheim, Germany, 2013 CrossRef; (c) D. Astruc, Nanoparticles and Catalysis, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852 CrossRef CAS PubMed.
- (a) M. R. Axet and K. Philippot, Nanoparticles in Catalysis: Advances in Synthesis and Applications, Wiley-VCH, Weinheim, Germany, 2021 Search PubMed; (b) L. M. Martínez-Prieto and P. W. N. M. van Leeuwen, Ligand effects in ruthenium nanoparticle catalysis, 2020, pp. 407–448 Search PubMed.
- M. R. Axet and K. Philippot, Chem. Rev., 2020, 120, 1085 CrossRef CAS PubMed.
- (a) C. A. Schoenbaum, D. K. Schwartz and J. W. Medlin, Acc. Chem. Res., 2014, 47, 1438 CrossRef CAS; (b) S. Kunz, Top. Catal., 2016, 59, 1671 CrossRef CAS.
- (a) K. R. Kahsar, D. K. Schwartz and J. W. Medlin, J. Am. Chem. Soc., 2014, 136, 520 CrossRef CAS PubMed; (b) I. Schrader, S. Neumann, A. Šulce, F. Schmidt, V. Azov and S. Kunz, ACS Catal., 2017, 7, 3979 CrossRef CAS; (c) P. D. Coan, L. D. Ellis, M. B. Griffin, D. K. Schwartz and J. W. Medlin, J. Phys. Chem. C, 2018, 122, 6637 CrossRef CAS; (d) A. Šulce, J. Backenköhler, I. Schrader, M. D. Piane, C. Müller, A. Wark, L. C. Ciacchi, V. Azov and S. Kunz, Catal. Sci. Technol., 2018, 8, 6062 RSC; (e) P. D. Coan, C. A. Farberow, M. B. Griffin and J. W. Medlin, ACS Catal., 2021, 11, 3730 CrossRef CAS.
- (a) M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072 CrossRef CAS PubMed; (b) A. Corma and H. Garcia, Top. Catal., 2008, 48, 8 CrossRef CAS.
- Q. L. Zhu and Q. Xu, Chem, 2016, 1, 220 CAS.
- (a) Y.-Z. Chen, G. Cai, Y. Wang, Q. Xu, S.-H. Yu and H.-L. Jiang, Green Chem., 2016, 18, 1212 RSC; (b) A. Noujima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2011, 50, 2986 CrossRef CAS PubMed.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Coord. Chem. Rev., 2016, 312, 99 CrossRef CAS.
- J. Lee, K. S. Novoselov and H. S. Shin, ACS Nano, 2011, 5, 608 CrossRef CAS PubMed.
- (a) E. D. Grayfer, L. S. Kibis, A. I. Stadnichenko, O. Y. Vilkov, A. I. Boronin, E. M. Slavinskaya, O. A. Stonkus and V. E. Fedorov, Carbon, 2015, 89, 290 CrossRef CAS; (b) D. E. García-Rodríguez, L. H. Mendoza-Huizar and C. Díaz, Appl. Surf. Sci., 2017, 412, 146 CrossRef; (c) I. Fampiou and A. Ramasubramaniam, J. Phys. Chem. C, 2012, 116, 6543 CrossRef CAS.
- A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef.
- (a) W. A. Herrmann, J. GoobenL and M. Spiegler, J. Organomet. Chem., 1997, 547, 357 CrossRef CAS; (b) M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677 CrossRef CAS; (c) L. A. Schaper, S. J. Hock, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2012, 52, 270 CrossRef PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed.
- (a) M. Koy, P. Bellotti, M. Das and F. Glorius, Nat. Catal., 2021, 4, 352 CrossRef CAS; (b) C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C. H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986 CrossRef CAS; (c) C. Cerezo-Navarrete, P. Lara and L. M. Martínez-Prieto, Catalysts, 2020, 10, 1144 CrossRef CAS.
- E. C. Hurst, K. Wilson, I. J. S. Fairlamb and V. Chechik, New J. Chem., 2009, 33, 1837 RSC.
- U. Hintermair, S. M. Hashmi, M. Elimelech and R. H. Crabtree, J. Am. Chem. Soc., 2012, 134, 9785 CrossRef CAS PubMed.
- (a) P. Lara, O. Rivada-Wheelaghan, S. Conejero, R. Poteau, K. Philippot and B. Chaudret, Angew. Chem., Int. Ed., 2011, 50, 12080 CrossRef CAS PubMed; (b) L. M. Martínez-Prieto, E. A. Baquero, G. Pieters, J. C. Flores, E. De Jesús, C. Nayral, F. Delpech, P. W. N. M. Van Leeuwen, G. Lippens and B. Chaudret, Chem. Commun., 2017, 53, 5850 RSC.
- M. D. de los Bernardos, S. Perez-Rodriguez, A. Gual, C. Claver and C. Godard, Chem. Commun., 2017, 53, 7894 RSC.
- K. V. S. Ranganath, J. Kloesges, A. H. Schäfer and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 7786 CrossRef CAS PubMed.
- (a) E. A. Baquero, S. Tricard, J. C. Flores, E. de Jesús and B. Chaudret, Am. Ethnol., 2014, 126, 13436 CrossRef; (b) E. A. Baquero, S. Tricard, Y. Coppel, J. C. Flores, B. Chaudret and E. De Jesús, Dalton Trans., 2018, 47, 4093 RSC.
- (a) C. Richter, K. Schaepe, F. Glorius and B. J. Ravoo, Chem. Commun., 2014, 50, 3204 RSC; (b) L. M. Martínez-Prieto, A. Ferry, P. Lara, C. Richter, K. Philippot, F. Glorius and B. Chaudret, Chem. – Eur. J., 2015, 21, 17495 CrossRef PubMed; (c) L. M. Martinez-Prieto, A. Ferry, L. Rakers, C. Richter, P. Lecante, K. Philippot, B. Chaudret and F. Glorius, Chem. Commun., 2016, 52, 4768 RSC.
- (a) L. M. Martínez-Prieto, L. Rakers, A. M. López-Vinasco, I. Cano, Y. Coppel, K. Philippot, F. Glorius, B. Chaudret and P. W. N. M. van Leeuwen, Chem. – Eur. J., 2017, 23, 12779 CrossRef PubMed; (b) L. C. Moraes, R. C. Figueiredo, J. P. Espinós, F. Vattier, A. Franconetti, C. Jaime, B. Lacroix, J. Rojo, P. Lara and S. Conejero, Nanoscale, 2020, 12, 6821 RSC.
- (a) C. Taglang, L. M. Martínez-Prieto, I. del Rosal, L. Maron, R. Poteau, K. Philippot, B. Chaudret, S. Perato, A. Sam Lone, C. Puente, C. Dugave, B. Rousseau and G. Pieters, Angew. Chem., 2015, 127, 10620 CrossRef; (b) A. Palazzolo, S. Feuillastre, V. Pfeifer, S. Garcia-Argote, D. Bouzouita, S. Tricard, C. Chollet, E. Marcon, D.-A. Buisson, S. Cholet, F. Fenaille, G. Lippens, B. Chaudret and G. Pieters, Angew. Chem., Int. Ed., 2019, 58, 4891 CrossRef CAS PubMed.
- (a) D. Gonzalez-Galvez, P. Lara, O. Rivada-Wheelaghan, S. Conejero, B. Chaudret, K. Philippot and P. W. N. M. Van Leeuwen, Catal. Sci. Technol., 2013, 3, 99 RSC; (b) P. Lara, A. Suárez, V. Collière, K. Philippot and B. Chaudret, ChemCatChem, 2013, 6, 87 CrossRef; (c) M. Axet, S. Conejero and I. C. Gerber, ACS Appl. Nano Mater., 2018, 1, 5885 CrossRef CAS.
- (a) P. Tegeder, M. Freitag, K. M. Chepiga, S. Muratsugu, N. Möller, S. Lamping, M. Tada, F. Glorius and B. J. Ravoo, Chem. – Eur. J., 2018, 24, 18682 CrossRef CAS PubMed; (b) R. Ye, A. V. Zhukhovitskiy, R. V. Kazantsev, S. C. Fakra, B. B. Wickemeyer, F. D. Toste and G. A. Somorjai, J. Am. Chem. Soc., 2018, 140, 4144 CrossRef CAS PubMed.
- J. B. Ernst, S. Muratsugu, F. Wang, M. Tada and F. Glorius, J. Am. Chem. Soc., 2016, 138, 10718 CrossRef CAS PubMed.
- A. Palazzolo, T. Naret, M. Daniel-Bertrand, D. Buisson, S. Tricard, P. Lesot, Y. Coppel, B. Chaudret, S. Feuillastre and G. Pieters, Angew. Chem., 2020, 132, 21065 CrossRef.
- (a) D. Ventura-Espinosa, S. Martín, H. García and J. A. Mata, J. Catal., 2021, 394, 113 CrossRef CAS; (b) D. Ventura-Espinosa, S. Martín and J. A. Mata, J. Catal., 2019, 375, 419 CrossRef CAS; (c) A. Mollar-Cuni, D. Ventura-Espinosa, S. Martín, Á. Mayoral, P. Borja and J. A. Mata, ACS Omega, 2018, 3, 15217 CrossRef CAS PubMed.
- (a) L. M. Martínez-Prieto, M. Puche, C. Cerezo-Navarrete and B. Chaudret, J. Catal., 2019, 377, 429 CrossRef; (b) C. Cerezo-Navarrete, Y. Mathieu, M. Puche, C. Morales, P. Concepción, L. M. Martínez-Prieto and A. Corma, Catal. Sci. Technol., 2021, 11, 494 RSC.
- D. Ventura-Espinosa, S. Sabater and J. A. Mata, J. Catal., 2017, 352, 498 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. B. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS.
- (a) L. M. Martinez-Prieto, I. Cano, A. Marquez, E. A. Baquero, S. Tricard, L. Cusinato, I. del Rosal, R. Poteau, Y. Coppel, K. Philippot, B. Chaudret, J. Campora and P. W. N. M. van Leeuwen, Chem. Sci., 2017, 8, 2931 RSC.
- L. M. Martinez-Prieto, C. Urbaneja, P. Palma, J. Campora, K. Philippot and B. Chaudret, Chem. Commun., 2015, 51, 4647 RSC.
- F. Novio, K. Philippot and B. Chaudret, Catal. Lett., 2010, 140, 1 CrossRef CAS.
- M. Koy, P. Bellotti, M. Das and F. Glorius, Nat. Catal., 2021, 4, 352 CrossRef CAS.
- F. Martinez-Espinar, P. Blondeau, P. Nolis, B. Chaudret, C. Claver, S. Castillón and C. Godard, J. Catal., 2017, 354, 113 CrossRef CAS.
- A. Rühling, K. Schaepe, L. Rakers, B. Vonhören, P. Tegeder, B. J. Ravoo and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 5856 CrossRef PubMed.
- P. W. N. M. van Leeuwen, Homogeneous Catalysis-Understanding the Art, Kluwer Academic Publishers, Netherlands, 2004 Search PubMed.
- (a) A. Denicourt-Nowicki, B. Leger and A. Roucoux, Phys. Chem. Chem. Phys., 2011, 13, 13510 RSC; (b) D. Duraczynska, A. Drelinkiewicz, E. Bielanska, E. M. Serwicka and L. Litynska-Dobrzynska, Catal. Lett., 2011, 141, 83 CrossRef CAS.
- M. Yang, G. Cheng, D. Xie, T. Zhu, Y. Dong, H. Ke and H. Cheng, Int. J. Hydrogen Energy, 2018, 43, 8868 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy02063c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |