
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Elucidating the role of earth alkaline doping in perovskite-based methane dry reforming catalysts†
Parastoo
Delir Kheyrollahi Nezhad
a,
Maged F.
Bekheet
 b,
Nicolas
Bonmassar
c,
Albert
Gili
d,
Franz
Kamutzki
b,
Aleksander
Gurlo
b,
Andrew
Doran
e,
Sabine
Schwarz
f,
Johannes
Bernardi
f,
Sebastian
Praetz
g,
Aligholi
Niaei
a,
Ali
Farzi
a and
Simon
Penner
*c
b,
Nicolas
Bonmassar
c,
Albert
Gili
d,
Franz
Kamutzki
b,
Aleksander
Gurlo
b,
Andrew
Doran
e,
Sabine
Schwarz
f,
Johannes
Bernardi
f,
Sebastian
Praetz
g,
Aligholi
Niaei
a,
Ali
Farzi
a and
Simon
Penner
*c
aReactor & Catalyst Research Lab, Department of Chemical Engineering, Faculty of Chemical and Petroleum Engineering, University of Tabriz, Tabriz, Iran
bFachgebiet Keramische Werkstoffe/Chair of Advanced Ceramic Materials, Institut für Werkstoffwissenschaften und -technologien, Technische Universität Berlin, Hardenbergstr. 40, 10623 Berlin, Germany
cDepartment of Physical Chemistry, University of Innsbruck, Innrain 52c, A-6020 Innsbruck, Austria. E-mail: simon.penner@uibk.ac.at; Fax: +4351250758199; Tel: +4351250758003
dInstitut für Chemie, Technische Universität Berlin, Sekretariat TC 8, Straße des 17. Juni 124, 10623 Berlin, Germany
eAdvanced Light Source, Lawrence Berkeley National Laboratory Berkeley, California 94720, USA
fUniversity Service Center for Transmission Electron Microscopy, TU Wien, Wiedner Hauptstrasse 8-10, A-1040 Vienna, Austria
gInstitute of Optics and Atomic Physics, Technische Universität Berlin, Hardenbergstraße 36, 10623 Berlin, Germany
First published on 6th January 2022
Abstract
To elucidate the role of earth alkaline doping in perovskite-based dry reforming of methane (DRM) catalysts, we embarked on a comparative and exemplary study of a Ni-based Sm perovskite with and without Sr doping. While the Sr-doped material appears as a structure-pure Sm1.5Sr0.5NiO4 Ruddlesden Popper structure, the undoped material is a NiO/monoclinic Sm2O3 composite. Hydrogen pre-reduction or direct activation in the DRM mixture in all cases yields either active Ni/Sm2O3 or Ni/Sm2O3/SrCO3 materials, with albeit different short-term stability and deactivation behavior. The much smaller Ni particle size after hydrogen reduction of Sm1.5Sr0.5NiO4, and of generally all undoped materials stabilizes the short and long-term DRM activity. Carbon dioxide reactivity manifests itself in the direct formation of SrCO3 in the case of Sm1.5Sr0.5NiO4, which is dominant at high temperatures. For Sm1.5Sr0.5NiO4, the CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 ratio exceeds 1 at these temperatures, which is attributed to faster direct carbon dioxide conversion to SrCO3 without catalytic DRM reactivity. As no Sm2O2CO3 surface or bulk phase as a result of carbon dioxide activation was observed for any material – in contrast to La2O2CO3 – we suggest that oxy-carbonate formation plays only a minor role for DRM reactivity. Rather, we identify surface graphitic carbon as the potentially reactive intermediate. Graphitic carbon has already been shown as a crucial reaction intermediate in metal-oxide DRM catalysts and appears both for Sm1.5Sr0.5NiO4 and NiO/monoclinic Sm2O3 after reaction as crystalline structure. It is significantly more pronounced for the latter due to the higher amount of oxygen-deficient monoclinic Sm2O3 facilitating carbon dioxide activation. Despite the often reported beneficial role of earth alkaline dopants in DRM catalysis, we show that the situation is more complex. In our studies, the detrimental role of earth alkaline doping manifests itself in the exclusive formation of the sole stable carbonated species and a general destabilization of the Ni/monoclinic Sm2O3 interface by favoring Ni particle sintering.
:H2 ratio exceeds 1 at these temperatures, which is attributed to faster direct carbon dioxide conversion to SrCO3 without catalytic DRM reactivity. As no Sm2O2CO3 surface or bulk phase as a result of carbon dioxide activation was observed for any material – in contrast to La2O2CO3 – we suggest that oxy-carbonate formation plays only a minor role for DRM reactivity. Rather, we identify surface graphitic carbon as the potentially reactive intermediate. Graphitic carbon has already been shown as a crucial reaction intermediate in metal-oxide DRM catalysts and appears both for Sm1.5Sr0.5NiO4 and NiO/monoclinic Sm2O3 after reaction as crystalline structure. It is significantly more pronounced for the latter due to the higher amount of oxygen-deficient monoclinic Sm2O3 facilitating carbon dioxide activation. Despite the often reported beneficial role of earth alkaline dopants in DRM catalysis, we show that the situation is more complex. In our studies, the detrimental role of earth alkaline doping manifests itself in the exclusive formation of the sole stable carbonated species and a general destabilization of the Ni/monoclinic Sm2O3 interface by favoring Ni particle sintering.
1. Introduction
The dry reforming of methane (DRM) reaction represents a convenient method for the simultaneous abatement of two harmful greenhouse gases carbon dioxide and methane.1–19 In recent years, several material classes have been screened for the development of promising catalysts and a better understanding of the reaction mechanism on an atomic level.20–25 Materials include intermetallic compounds, oxides, or metal-oxide systems. Mechanistic-wise, there is consensus that a bifunctional operating mechanism for both methane and carbon dioxide activation usually prevails.26–28 While the archetypical metal-based methane dry reforming catalyst nickel is capable of both methane and carbon dioxide activation,29 the serious drawbacks of Ni particle sintering at high temperatures have led to the development of more complex catalytic entities. On the most advanced material level, metal-oxide systems evolving from the in situ decomposition and self-activation of more complex structures during dry reforming of methane represent the state-of-the-art.17,18,23,27,28,30,31 Perovskite-based materials are especially well-suited to prepare metal-oxide systems with superior catalytic properties following in situ activation, as their ABO3 stoichiometry (A and B being tri- and/or bivalent metal ions) through careful selection of the A and B elements allows to trigger decomposition of the perovskite structure usually into a B/A2O3 entity.23,27,28 This concept is best appreciated by exemplifying the behavior of the archetypical LaNiO3 perovskite, whose DRM behavior has been scrutinized in every possible detail. LaNiO3 can be decomposed into a Ni/La2O3 system both by self-activation and a pre-reduction treatment in hydrogen. Ni crystallite sizes and carbonation tendency of La2O3 as a prerequisite of reversible carbon dioxide activation are strongly dependent on the pre-treatment and the doping level on the A- and B sites.17,18,20,23,27,28 Doping generally is a viable way to steer the structural decomposition into prospective catalytic materials not only by influencing the structural instability of the perovskite directly,23,25 but also by exploiting the surface chemical properties of the resulting oxide entity of the metal-oxide systems. The surface basicity and acidity of the oxide directly translate to the carbon dioxide activation properties.32,33 Basic surface sites enhance the carbon dioxide activation usually by reversible oxy-carbonate formation.32,33 In the case of LaNiO3, the corresponding Ni/La2O2CO3 interface is suspected to host the active sites.23,27,28 Even though reversible carbonate formation is imperative, also the presence of very stable spectator carbonates, e.g. BaCO3,20 in contact with Ni are reported to help in structurally stabilizing the active interface. One leading theme in the proper understanding of the catalytic activation of perovskite materials is the necessity to follow the activation by in situ characterization. It has been shown for LaNiO3 and a corresponding perovskite-related Ruddlesden–Popper structure La2NiO4, that before entering the final metal-oxide state, the perovskite structure goes through a sequence of transformations, including the formation of oxygen-deficient structures, transient oxide phases or polymorphic transitions. All of these intermediate steps could be directly related to changes in the catalytic activity.20,27,28 Apart from oxycarbonate formation, also the reactivity of surface-bound graphitic carbon was reported to play a crucial role in enhancing the DRM activity, especially on metal-oxide materials.31In order to search for alternative DRM materials, Sm-containing catalysts have evolved as potentially promising. This is essentially referred to the fact that Sm2O3, like La2O3, is reported to be capable of forming a samarium oxycarbonate species34–36 and that the Ni/Sm2O3 interface showed superior ability in the DRM process.37 A particular important sub-class of Sm-based perovskite catalysts are the associated cobalt-containing materials. Evidence for the existence and use of SmCoO3 (and Sm2CoO4 intermediate) for efficient syngas production via DRM exists, and the potentially beneficial addition of earth alkaline metals to stabilize the active metal (i.e., Co) species was highlighted. For the corresponding Ni-based materials, the situation is more complex from a structural viewpoint. Both the perovskite structure SmNiO3 and its Ruddlesden–Popper related phase Sm2NiO4 cannot be successfully synthesized structure-pure via a simple preparation and annealing process,38,39 since the ionic radius of the Sm3+ ion is already too small (compared to e.g., La3+) to form a stable perovskite structure based on the Goldschmidt tolerance factor. Stabilization via doping is, therefore, necessary. To potentially take advantage of the surface chemical properties and eventually of structural stabilization, we opted for Sr2+ doping.
In the present contribution, we focus on a direct comparison of a Ni–Sm material prepared with and without additional Sr doping to assess the effect of doping on the structural stability and the catalytic DRM properties. Given the mere suspicion of the beneficial action of the added earth alkaline dopant, we are able to address the open questions how i) and if the structural stabilization works, ii) to assess the influence of the Sr doping on the oxy-carbonate formation, iii) to identify possible alternative reaction/carbon dioxide activation pathways in the presence of Sr doping and iv) to comparatively investigate the short-term stability and deactivation behavior.
The starting materials are a Ni(NiO)/Sm2O3 composite material (without Sr doping) and structure- and phase-pure Sm1.5Sr0.5NiO4 (with Sr doping). The resulting Sm1.5Sr0.5NiO4 stoichiometry represents a new class of a perovskite-related Ruddlesden–Popper perovskite, where decomposition of the perovskite and nickel exsolution under hydrogen atmosphere and self-activation under DRM conditions is possible. We show that the final fate of the decomposed Sm1.5Sr0.5NiO4 structure both after pre-treatment with hydrogen and after self-activation are Ni particles in a composite mixture with Sm2O3, SrO (and SrCO3 after self-activation). The transition into that state is structurally complex, and via synchrotron-based in situ XRD reveals several crystalline transient phases and through different Ni particle sizes as a consequence of (pre-)treatment also Ni-oxide interfaces of different extent and reactivity. We are able to establish structure–activity correlations by relating these changes to the catalytic DRM profiles and identifying similarities and differences to the state without Sr doping. Complementing the in situ studies, we characterize important structural points along the experiment axis also by ex situ and in situ X-ray photoelectron spectroscopy, electron microscopy (TEM) and, X-ray absorption (XANES) to correlate surface and bulk structural and morphological properties.
2. Experimental
2.1. Materials synthesis
Ni(O)/Sm2O3 and Sm1.5Sr0.5NiO4 were synthesized using an auto-ignition sol–gel method. Stoichiometric amounts of the required amounts of Sm(NO3)3·6H2O, Sr(NO3)2, and Ni(NO3)2·6H2O were weighted and dissolved in de-ionized water. Afterwards, glycine was added to the solution in a molar ratio of NO3−:NH2− = 1:1 in order to induce the formation of glycine–metal complexes. The resulting solution was stirred at 80 °C for 2 h to evaporate excess water. The final gel-like residue was heated to 250 °C, causing self-ignition at 1200 °C and the formation of stoichiometric amounts of metal oxides. The formed powders were ground slightly in an agate mortar and finally calcined at 900 °C for 12 h. The resulting Sm1.5Sr0.5NiO4 and Ni(O)/Sm2O3 samples were distributed in four batches with each 50 mg to perform the catalytic treatments in different conditions namely under H2 (only hydrogen pre-reduction), DRM (only self-activation), H2-DRM (consecutive hydrogen reduction followed by DRM reaction) and DRM-DRM (two consecutive DRM cycles without hydrogen pre-reduction).
2.2. Structural, morphological and elemental characterization
The specific surface area prior to catalysis was assessed by BET surface quantification via nitrogen adsorption at 77 K. A Quantachrome Nova2000 surface and pore size analyzer was used for all measurements, yielding low BET areas of around 2 m2 g−1.Ex situ powder X-ray diffraction (PXRD) was carried out using a STOE Stadi P powder diffractometer operating in transmission geometry and exploiting monochromatized MoKα1 radiation (λ = 0.7093 Å). A 2θ range of 64° and a step size of 0.015° was used. A focussing Ge(111) primary beam monochromator, as well as a linear position-sensitive detector system, was additionally employed.
In situ synchrotron-based PXRD experiments have been conducted in both DRM mixtures and under pure hydrogen at beamline 12.2.2, Advanced Light Source (ALS) at Lawrence Berkeley National Laboratory, in a cell previously described in ref. 40 and 41. All diffraction patterns were measured in angle-dispersive transmission mode with a focussed 25 keV monochromatic beam (λ = 0.4984 Å/30 μm spot size). The powders were heated in a 0.7 mm outer diameter quartz capillary under quasi-flowing conditions (CH4:CO2 = 1:1, gas flow: 10:10 mL min−1 for DRM; pure hydrogen, gas flow 10 mL min−1, GSHV = 600000 N mL h−1 gcat−1). Heating was performed using a SiC furnace with an infrared light source up to 800 °C at a rate of 10 °C min−1. All gases were injected through a 0.5 mm outer diameter tungsten tube. The patterns were recorded using a Perkin Elmer flat panel detector (XRD 1621 with dark image and strain correction.40,41 Rietveld refinement was performed using the FULLPROF program.42 The profile function 7 (Thompson-Cox-Hastings pseudo-Voigt convoluted with axial divergence asymmetry function)43 was used in all refinements. The resolution function of the diffractometers was obtained from the structure refinement of a LaB6 standard.
Surface characterization was performed using ex situ and in situ X-ray photoelectron spectroscopy (XPS). For ex situ studies, a Thermo Scientific MultiLab 2000 spectrometer, equipped with a monochromatic Al Kα X-ray source (E = 1486 eV) and an Alpha 110 hemispherical analyzer, was used. The base pressure lies in the 10−10 mbar range, and charging of the sample upon measurement is compensated by a flood gun supplying electrons with a kinetic energy of 6 eV. The used pass energy was 20 eV. Relevant high-resolution spectra in the relevant Sm 3d, Ni 2p, Sr 3d, C 1s, and O 1s regions have been collected and used for qualitative and quantitative analysis. For Ni, after background subtraction using a Shirley-type function, deconvolution of the Ni 2p peak into Ni2+ and metallic Ni components was done, obeying the spin-orbit coupling of the individual Ni 2p1/2 and Ni 2p3/2 peaks for each relevant component. The full width at half maximum has been fixed for the individual deconvoluted Ni species as an additional constraint. In contrast, the position of the Ni components was left floating to account for the constant change of the Ni chemical environment during the decomposition of Sm1.5Sr0.5NiO4.
The UHV system used for the in situ near-ambient pressure X-ray photoelectron spectroscopy (NAP XPS) is a customized SPECS setup. Maintaining a base pressure below 10−10 mbar, it comprises an analysis chamber, which can be backfilled with oxidative, reductive and reactive gas atmospheres up to 30 mbar, a PHIOBOS 150 NAP hemispherical energy analyzer with an 1D-DLD detector, a μFOCUS 600 NAP monochromatic small spot X-ray source (Al Kα) and a Flood Gun (FG22/35). A complementary mass spectrometer allows the operando surface characterization. A four-axis manipulator adapted for laser heating and gas-phase reactions is used for all experiments. High-resolution spectra of the Ni 2p, O 1s, C 1s, Sm 3d and Sr 3d regions were collected in selected temperature steps from room temperature to 750 °C. The spectra were fitted with a Shirley-type background. Quantitative analysis was based on the relative sensitivity factors (RSFs), as well as the different inelastic mean free paths. To monitor the appearance of surface-bound Ni and the carbon dioxide activation capabilities, the sample was sequentially heated in 0.2 mbar hydrogen and 0.2 mbar carbon dioxide in 50–100 °C steps from 25 °C to 800 °C, respectively.
Structural and morphological characterization has been further performed by ex situ transmission electron microscopy (TEM) at the University Service Facility for Transmission Electron Microscopy (USTEM) at TU Vienna using a FEI Tecnai F20 S-TWIN analytical high-resolution microscope operated at 200 kV in combination with a windowless Apollo XLTW silicon drift detector for EDX experiments.
X-ray absorption measurements were carried out with a novel self-developed wavelength-dispersive spectrometer in von Hámos geometry.44,45 The spectrometer is equipped with a microfocus X-ray tube, a curved highly annealed pyrolytic graphite mosaic crystal and a hybrid photon counting CMOS detector with 512 × 1030 pixel and a pixel size of 75 μm × 75 μm. The tube was operated with a voltage and current of 15.8 kV and 1870 μA for Ni K-edge, 12.8 kV and 1710 μA for Sm LIII-edge, and 19.8 kV and 14900 μA for Sr K-edge measurements. As the Ni reference, a 2 μm Ni foil was used. All other references and samples were in powder form and prepared as wax-pellets (mixed with Hoechst Wax C), due to their low concentration of Ni. The blending of the sample and reference sample with Hoechst Wax was necessary to achieve pellets with an adequate thickness for stability. After stirring wax and sample material in a mortar to get a homogenous mixture, a pellet with a 13 mm diameter was pressed by using a hydraulic pellet press with force up to 6 tons for not longer than 60 s. As the samples were measured in transmission mode, the absorption spectrum was acquired by measuring once with and once without the sample. The measurement time for each sample varied between 8 to 10 h depending on the thickness of the prepared sample. All references and samples were constantly moved during the measurements to minimize the effects of local thickness inhomogeneity. The beam size on the samples is around 3 mm × 3 mm. The gathered spectral range is covering the Ni K absorption edge at 8332 eV. Normalization of the spectra, as well as linear combination fitting (LCF), was done by using the XAS analyzing and processing software ATHENA which is part of the Demeter software package.46
2.3. Catalytic testing
The performance of the catalysts with respect to DRM activity was tested in a home-built quartz fixed-bed tubular flow reactor (inner diameter = 7 mm, outer diameter = 9 mm, catalyst bed length: 2.5 cm). Each gas required for DRM (CH4:CO2:He = 1:1:3 ratio; GHSV = 60000 N mL gcat h−1) was injected through a corresponding mass flow controller (MKS), where helium acts both as a carrier gas and as a heat conductor. 100 mg catalyst powder was used for each test and homogeneously distributed using thoroughly degassed quartz wool over the entire catalyst bed. Heating was achieved using a Linn High Therm furnace up to 800 °C at a fixed rate of 10 °C min−1. An S-type Pt/PtRh thermocouple placed in close vicinity to the sample ensured accurate temperature reading. The output gas was continuously extracted through a capillary and analyzed by a quadrupole mass spectrometer mounted in cross-beam geometry (Balzers QMA 125). Hydrogen pre-treatments were carried out in 1 bar flowing hydrogen at a rate of 10 mL min−1 up to 800 °C. We have performed several DRM cycles after the selected experimental sequences (cf. section 2.1.). In this respect, the term “first” or “second” cycle is used for DRM measurements, which have been consecutively performed, irrespective of the pre-treatment.
Due to the bifunctional operating mechanism of DRM catalysts on perovskite basis, normalization of the catalytic activity solely to the surface area of the exsolved Ni particles would grossly overestimate the intrinsic, structure-insensitive catalytic role of Ni and is therefore not considered (i.e., calculating metal surface-based turnover frequencies is not meaningful during in situ activation). To derive a qualitative understanding and judgment of structure–activity correlations from temperature-programmed DRM experiments, we rely on onset temperatures of catalytic activity, conversion (%) and H2/CO product ratios. The relative catalytic activity is, therefore, compared on the basis of conversion vs. temperature plots.
3. Results and discussion
3.1. Catalyst with Sr doping: the role of Sm1.5Sr0.5NiO4 in DRM
:methane = 1:1 mixture), with the simultaneous presence of both orthorhombic and rhombohedral SrCO3 after DRM operation. Carbonation of SrO can also be observed after a post-DRM treatment after a hydrogen pre-treatment. The resulting diffractograms are almost identical (green vs. red diffractogram in Fig. 1). A stationary phase composition after multiple consecutive DRM cycles is observed (blue diffractogram), which exhibits only minor differences to the post hydrogen-DRM measurement. The phase compositions of all samples are quantified using Rietveld refinement of XRD data (Table 1). Small amounts of graphitic carbon are observed in the spent catalyst after the DRM treatment, before disappearing during the short-term stability tests for 12 hours (Fig. S1 and S2†). The presence of graphitic carbon is confirmed by using two models for the Rietveld refinement analysis (i.e., with and without including graphitic carbon). As shown in Fig. S1,† the inclusion of graphitic carbon in the analysis leads to a better match between experimental and calculated diffraction data, in particular for the peak at 2θ ∼ 12.05° corresponding to (002) reflection of graphitic carbon. As shown in Table 1, the decomposition of the Sm1.5Sr0.5NiO4 catalyst in H2 results in a smaller crystallite size of Ni compared to decomposition under DRM conditions. Moreover, a higher amount of c-Sm2O3 is formed by decomposing the catalyst under DRM, while a negligible amount of this phase (∼2.4 wt%) is formed in the H2 atmosphere.
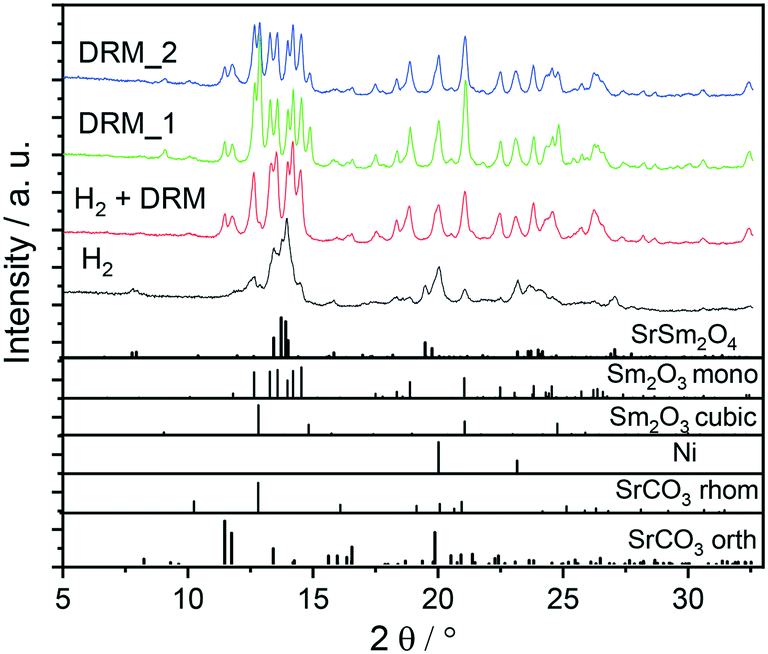 | ||
| Fig. 1 Ex situ XRD patterns of Sm1.5Sr0.5NiO4 after selected pre-treatments and activation experiments. Black trace: hydrogen treatment under flowing pure hydrogen, red trace: post-DRM treatment after pre-reduction in hydrogen, green trace: DRM treatment without hydrogen pre-treatment, blue trace: after two consecutive DRM cycles. Final temperature: 800 °C, heating ramp: 10 °C min−1. The lower panel indicates the phase assignment to the respective reference structures. The unindexed broad shoulder at 2θ ~ 12.05° corresponds to (002) reflection of graphitic carbon. Wavelength: λ = 0.7093 Å. | ||
| Catalytic conditions | Sm1.5Sr0.5NiO4 catalyst | Ni/Sm2O3 catalyst | ||
|---|---|---|---|---|
| Phase composition (wt%) | Ni crystallite size (nm) | Phase composition (wt%) | Ni crystallite size (nm) | |
| Before catalytic experiment | 98.3% Sm1.5Sr0.5NiO4 | — | 68.2% m-Sm2O3 | — |
| 1.7% m-Sm2O3 | 13.6% c-Sm2O3 | |||
| 18.2% NiO | ||||
| Reduction in H2 | 37.7% SrSm2O4 | 9.2(1) | — | — |
| 39.8% m-Sm2O3 | ||||
| 2.4% c-Sm2O3 | ||||
| 20.1% Ni | ||||
| After 1 DRM cycle | 45.8% m-Sm2O3 | 16.0(1) | 70.6% m-Sm2O3 | 21(1) |
| 22.8% c-Sm2O3 | 14.5% c-Sm2O3 | |||
| 15.0% o-SrCO3 | 14.9% Ni | |||
| 1.9% rh-SrCO3 | ||||
| 14.5% Ni | ||||
| After pre-reduction in H2 and 1 DRM cycle | 63.9% m-Sm2O3 | 12.9(1) | 53.2% m-Sm2O3 | 29.4(1) |
| 17.1% o-SrCO3 | 13.3% c-Sm2O3 | |||
| 1.6% rh-SrCO3 | 11.6% Ni | |||
| 14.1% Ni | 21.9% C | |||
| 3.3% C | ||||
| After 2 consecutive DRM cycles without H2 pre-reduction | 39.4% m-Sm2O3 | 19.8(1) | — | — |
| 10.6% c-Sm2O3 | ||||
| 14.3% o-SrCO3 | ||||
| 1.0% rh-SrCO3 | ||||
| 11.1% Ni | ||||
| 23.6% C | ||||
| After 2 consecutive DRM cycles and stability test for 12 h | 36.0% m-Sm2O3 | 29.3(1) | 48.5% m-Sm2O3 | 29.4(1) |
| 31.9% c-Sm2O3 | 10.6% c-Sm2O3 | |||
| 17.3% o-SrCO3 | 10.3% Ni | |||
| 14.8% Ni | 30.6% C | |||
| After pre-reduction in H2, 1 DRM cycle and stability test for 12 h | 66.3% m-Sm2O3 | 16.3(1) | 54.8% m-Sm2O3 | 29.7(1) |
| 18.9% o-SrCO3 | 11.1% c-Sm2O3 | |||
| 14.8% Ni | 11.5% Ni | |||
| 22.6% C | ||||
The complementary structure and morphology in the initial state and after the selected pre-reduction and activation treatments by electron microscopy (Fig. 2) reveals a loose network of rounded, ∼200 nm-sized individual grains, which show a uniform elemental distribution of Sm, Ni, Sr, and O in the as-synthesized calcined state (top-most row, Fig. 2). Corroborating the XRD results, we observe the decomposition of Sm1.5Sr0.5NiO4 after all treatments. While the Sm and O distribution remain almost uniform, the exsolution of small Ni particles (≤30 nm in size) is prevalent both after the treatment in hydrogen and in the DRM mixture. After the latter, Sr is also increasingly randomly distributed. Most striking is the clear difference in Ni particle size after hydrogen treatment compared to after self-activation during DRM operation. Already obvious in the Ni-K EDX maps, the HRTEM images in Fig. S3† indicate a much smaller particle size after a hydrogen treatment (<15 nm, blue framed panel in Fig. S3†) in comparison to after a DRM treatment (>20 nm, red-framed panel in Fig. S3†), which agrees with the XRD results (Table 1).
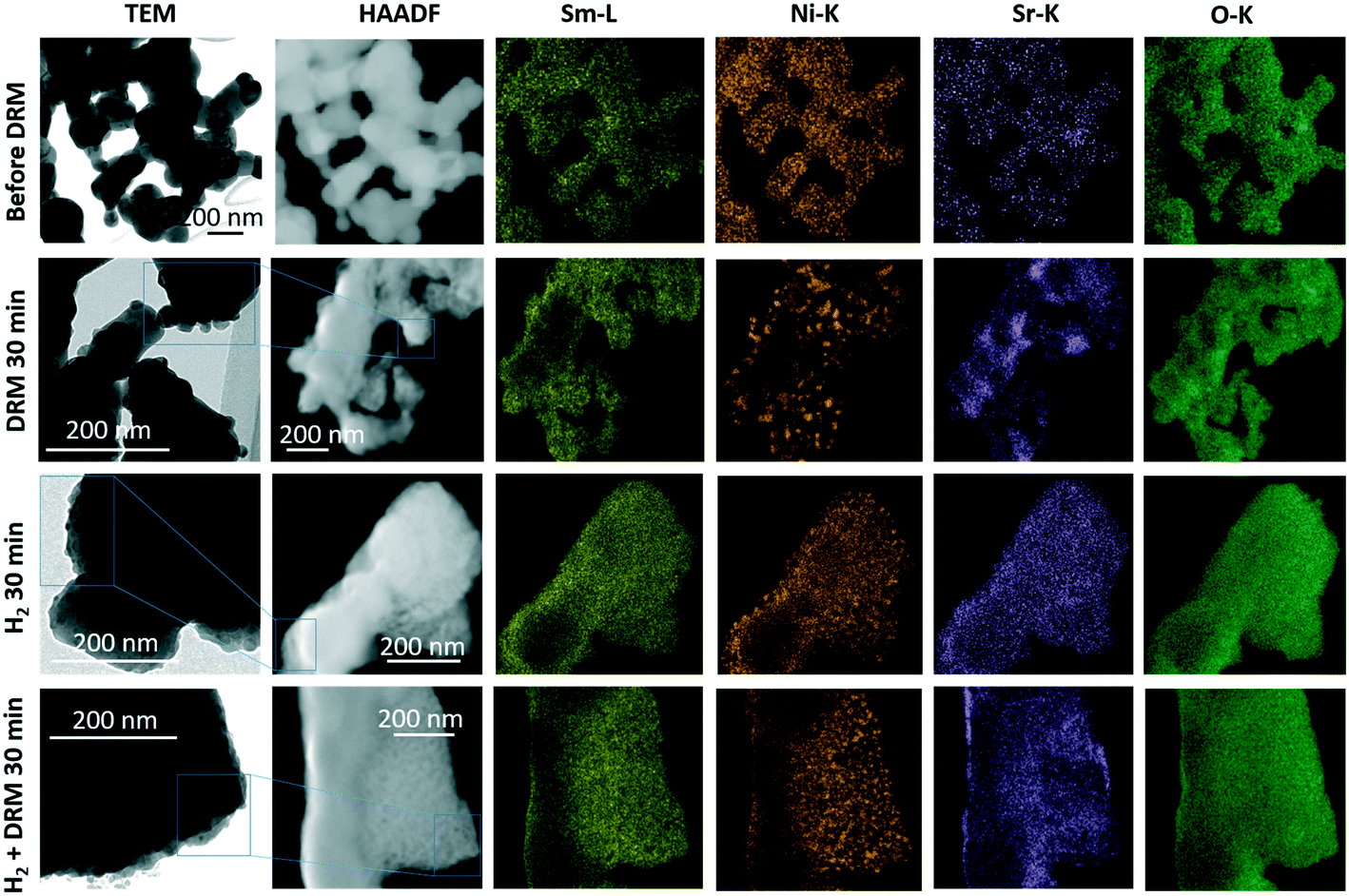 | ||
| Fig. 2 TEM/STEM overview analysis of the Sm1.5Sr0.5NiO4 Ruddlesden–Popper structure in the initial state (top row) and after selected in situ activation treatments in a 1:1 CO2:CH4 dry reforming methane mixture for 30 min (second row from the top), 1 bar flowing hydrogen for 30 min (second row from the bottom) and 1 bar flowing hydrogen for 30 min followed by a treatment in a 1:1 CO2:CH4 dry reforming methane mixture for 30 min (bottom-most row). Final temperatures in all cases 800 °C. The first column depicts overview bright-field TEM images, the second column the respective HAADF images alongside the EDX analysis based on the Sm-L (yellow), Ni-K (orange), Sr-K (magenta) and O-K edges (green). | ||
Of particular relevance for DRM operation is the resistance to coking. Although the presence of especially surface carbon is not always a disadvantage and under special circumstances can act as a reactive intermediate,31 depositions of carbon usually lead to catalyst deactivation. To assess the coking propensity of the Sm1.5Sr0.5NiO4 material, Fig. S4† shows the elemental distribution before and after an in situ activation treatment in an 1:1 CO2:CH4 dry reforming methane mixture for 30 min with a special focus on the carbon distribution. While Sm and O do not feature strong intensity differences before and after the treatment, carbon has only slightly accumulated during the treatment, but no particular agglomeration was observed. In particular, no carbon nanotube- or whisker-like features are observed. This confirms that coking is effectively suppressed during DRM operation on Sm1.5Sr0.5NiO4 – in strong contrast to the archetypical Ruddlesden–Popper structure La2NiO4, where coking is a particular issue.18,20 Although XRD analysis reveals the formation of small amounts of graphitic carbon in the spent catalyst after treatment under DRM with subsequent decomposition in H2 or DRM, all these graphitic carbon disappeared after the short-term stability tests for 12 hours (see Table 1). These results indicate that the formed graphitic carbon is an active intermediate species that reacts with CO2 to form syngas upon a longer reaction time. The formation of active graphitic carbon is also confirmed by the in situ XRD data discussed in section 3.2.
The first sequence of the hydrogen pre-treatment is shown as a set of in situ collected temperature-dependent X-ray diffraction patterns in Fig. S5† panels A–B and as weight-fraction analysis in Fig. 3. Sm1.5Sr0.5NiO4 is quantitatively transformed into oxygen-deficient Sm1.5Sr0.5NiO3 (which remains stable under reductive atmosphere up to 600 °C) following the reaction equation
| Sm1.5Sr0.5NiO4 + H2 → Sm1.5Sr0.5NiO3 + H2O |
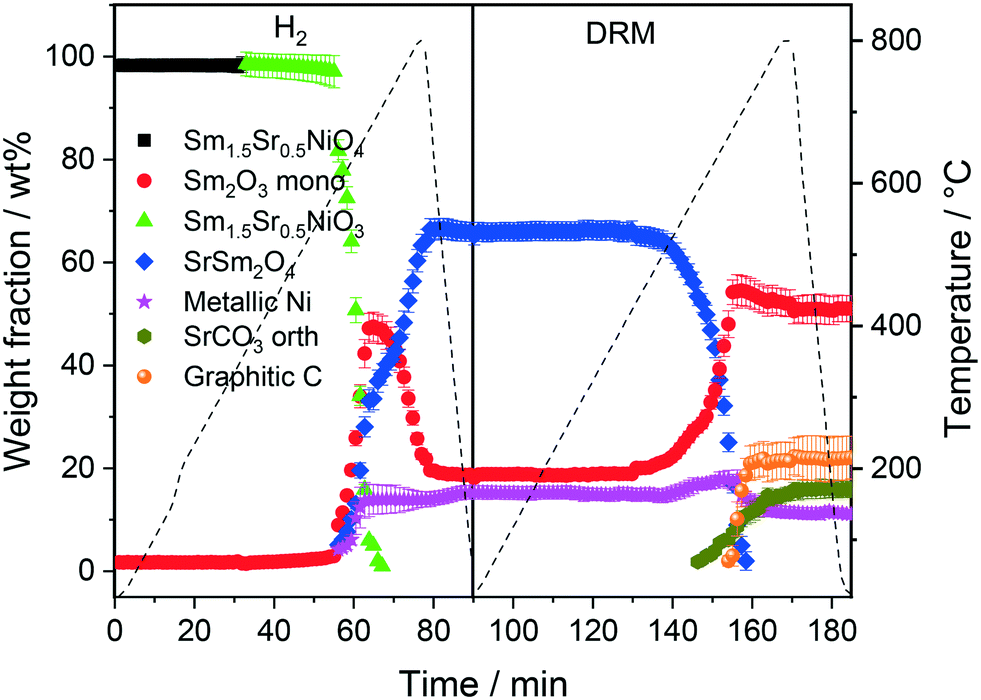 | ||
| Fig. 3 Weight fractions of different crystalline phases formed during the consecutive heating and cooling cycles of Sm1.5Sr0.5NiO4 in H2 and DRM atmospheres as a function of temperature and time obtained by Rietveld refinement of the in situ collected XRD patterns. | ||
At 700 °C, the stability limit of monoclinic Sm2O3 is reached, causing an enhancement in the formation of SrSm2O4. This transformation also proceeds during the cooling stage until a stationary composition of 65 wt% SrSm2O4, 18 wt% monoclinic Sm2O3 and 17 wt% metallic Ni is reached. Closely connected to the structural transformations is the evolution of the Ni crystallite size during hydrogen pre-reduction (Fig. 4). In the early stage of Ni exsolution starting at 600 °C, the Ni crystallite size triples from 4 nm to 12 nm within a narrow temperature window of 50 °C up to 650 °C, above which it essentially stagnates up to 800 °C. The evolution of Ni and monoclinic Sm2O3 is more or less correlated. The further accelerated formation of SrSm2O4 resulting from monoclinic Sm2O3 decomposition has only little effect on the Ni crystallite size.
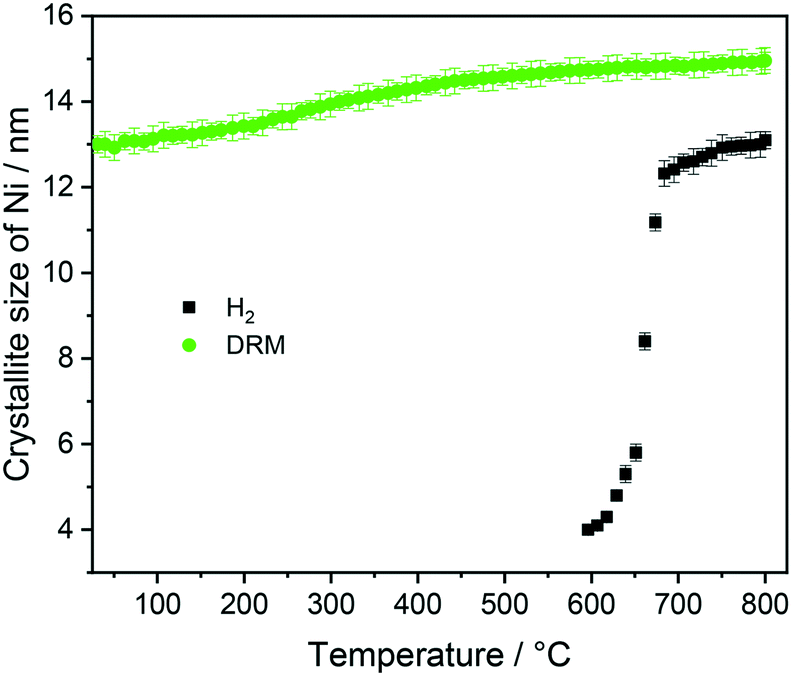 | ||
| Fig. 4 Crystallite size evolution of metallic Ni formed during heating Sm1.5Sr0.5NiO4 up to 800 °C in H2 and DRM (after H2 treatment) atmosphere as a function of temperature obtained by Rietveld refinement of the in situ collected XRD patterns. | ||
In the subsequent DRM experiment (Fig. S3† panels C and D and Fig. 3), the Ni/monoclinic Sm2O3/SrSm2O4 interface is structurally stable up to 550 °C. Intertransformation of SrSm2O4 and monoclinic Sm2O3 proceeds until 700 °C, accompanied by the parallel formation of orthorhombic SrCO3 at and above 650 °C according to the reaction equation
| SrSm2O4 + CO2 → SrCO3 + Sm2O3 |
Graphitic carbon starts to be formed at 660 °C, suggesting the activation of the catalyst by decomposition of CH4. Above 700 °C, a stationary composition – also remaining unaltered during re-cooling – of 51 wt% monoclinic Sm2O3, 16 wt% orthorhombic SrCO3, 22 wt% graphitic carbon and 11 wt% metallic Ni is reached. The final phase composition of the recovered sample at room temperature with respect to Sr-, Sm-, and Ni-containing phases is comparable to that observed in the spent catalyst after H2 pre-treatment and one cycle of DRM (Table 1). A considerable higher stability of SrSm2O4 under hydrogen compared to DRM atmospheres is observed. The amount of metallic Ni remains essentially constant throughout the hydrogen pre-treatment above 700 °C, re-cooling to room temperature and the entire DRM operation. This goes along with only a minor increase in Ni crystallite size during DRM operation from 13 to 15 nm. Important for the understanding of the reaction mechanism and the establishment of structure–activity correlations, we note the apparent missing of a crystalline bulk Sm-oxycarbonate phase Sm2O2CO3 throughout the experiment. Such oxycarbonate phases are usually associated with the reversible CO2 capture/release cycle necessary for high DRM activity and are a recurrent theme for La-based perovskite catalysts.17,18,26 To check for the possibility of the existence of exclusively surface-bound oxy-carbonates, we performed an in situ X-ray photoelectron spectroscopic heating experiment in 0.2 mbar carbon dioxide between 25 °C and 750 °C (Fig. S6†) after a hydrogen pre-treatment to 800 °C to induce the formation of a Ni/Sm2O3/SrO material. The experimental conditions match those of the in situ XRD experiments. Corroborating the in situ XRD results, carbonation of Sr (presumably as SrCO3) is visible at 500 °C in the Sr 3d spectra. On the contrary, no indication of the formation of Sm (oxy) carbonates is observed in the respective Sm 3d spectra. In fact, neither in the bulk, nor on the surface such species are visible.
To determine whether the structural state during and after hydrogen reduction is different from direct self-activation in the dry reforming mixture, Fig. 5 and S7† panels A–B show the corresponding analysis of the in situ X-ray diffraction patterns during two consecutive DRM cycles. Several differences to the hydrogen pre-treatment arise. At first, Sm1.5Sr0.5NiO4 is remarkably stable under DRM operation up to 800 °C. Decomposition of Sm1.5Sr0.5NiO4 during the isothermal period at 800 °C occurs within minutes yielding a phase mixture of metallic Ni, SrO and a mixture of cubic and monoclinic Sm2O3. Within the isothermal period, complex structural transformations involve carbonation of SrO into rhombohedral SrCO3 and the subsequent polymorphic intertransformation of rhombohedral SrCO3 and orthorhombic SrCO3. Upon re-cooling to room temperature and re-heating in the DRM mixture during the second cycle, no substantial structural changes occur, with the exception of the exclusive presence of orthorhombic SrCO3, in contrast to the first cycle where the rhombohedral modification was present. The difference between the first and second consecutive DRM cycle, which might explain this apparent existence of two different SrCO3 modifications, is the direct carbonation of SrO during the first cycle, whereas orthorhombic SrCO3 is the result of a polymorphic intertransformation. We can only speculate that the stabilization of rhombohedral SrCO3 during the first DRM cycle is a result of the unique structural features evolving through the decomposition of Sm1.5Sr0.5NiO4. It is known that orthorhombic SrCO3 (the stable modification under ambient conditions) undergoes a structural transition into rhombohedral SrCO3 upon heating, but the occurrence of rhombohedral SrCO3 at around 800 °C is well below the orthorhombic-to-rhombohedral transformation temperature (∼900 °C).48–51 The decomposition of rhombohedral SrCO3 upon re-cooling is in accordance with the literature, as the transformation is reversible and rhombohedral SrCO3 cannot be quenched to room temperature.49
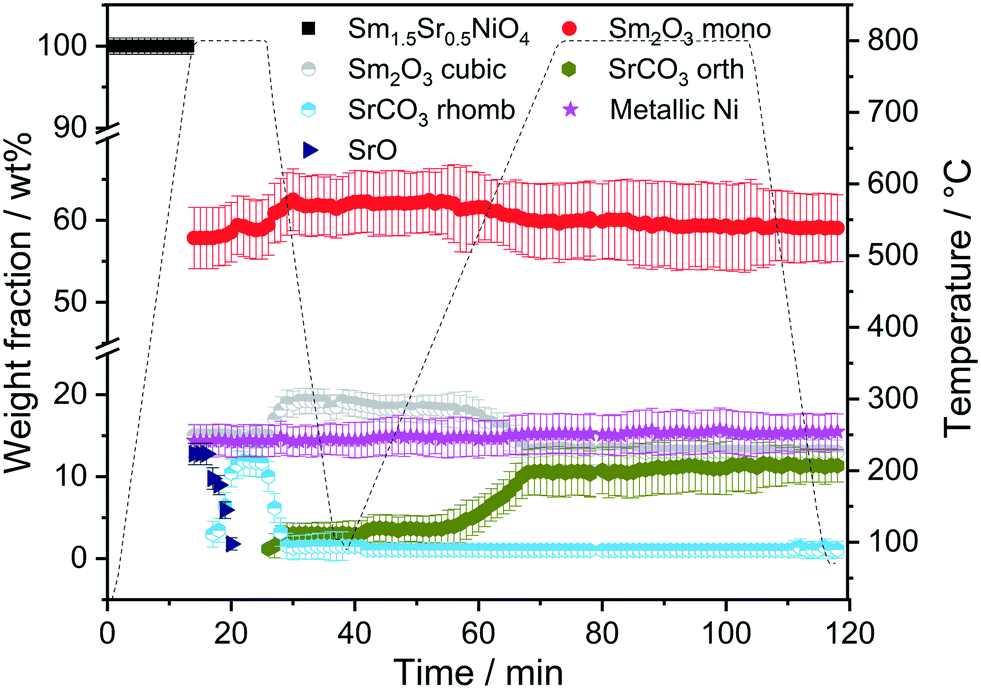 | ||
| Fig. 5 Weight fractions of different crystalline phases formed during heating and cooling Sm1.5Sr0.5NiO4 in DRM atmospheres (CO2:CH4 = 1:1) for 10 and 30 min as a function of temperature and time obtained by Rietveld refinement of the in situ collected XRD patterns. | ||
Five points are striking: the first difference is the missing Sm1.5Sr0.5NiO3 and SrSm2O4 structures that were prominently observed during hydrogen pre-reduction. As Sm1.5Sr0.5NiO3 is apparently stabilized by oxygen vacancies, the appearance of SrSm2O4 is intrinsically connected to Sm1.5Sr0.5NiO3. Under the less-reducing DRM conditions, the absence of Sm1.5Sr0.5NiO3 and SrSm2O4 is therefore logical. Secondly, the absence of a crystalline Sm-oxycarbonate structure is equally striking. The only observed carbonated phase is again SrCO3. Thirdly, Fig. S8† panels A and B reveal that the full-width half-maximum of the Ni (111) reflection of the decomposed sample in the first DRM cycle is smaller than that of Ni formed in the pre-reduction in H2, and does not remarkably change after the second DRM cycle. Rietveld refinement reveals that the crystallite size as a result of Sm1.5Sr0.5NiO4 decomposition in the DRM mixture at 18 nm is much larger compared to pre-reduction in hydrogen already in the first cycle, which does not remarkably change at the end of the isothermal period at 800 °C after the second cycle. The crystallite size values of Ni determined by the in situ XRD experiments match those determined for Ni in the spent catalyst under the same catalytic conditions (see Table 1). Apparently, the stabilization of a smaller Ni crystallite size during hydrogen pre-reduction is a direct consequence of the transformation of Sm1.5Sr0.5NiO4 to Sm1.5Sr0.5NiO3 and further to SrSm2O4, Sm2O3 and Ni. Moreover, a recent study showed that the formation of SrCO3 might increase the crystallite size of metallic Ni in SrNiOx catalysts under DRM conditions.52 Fourthly, although no c-Sm2O3 is formed during hydrogen pre-reduction, the decomposition of the catalyst under DRM results in 19 wt% and 13 wt% of this phase at the end of the first and second DRM cycle, respectively. These results suggest that the transition temperature between cubic and monoclinic Sm2O3 phase strongly depends on the treatment atmosphere, which agrees with previous works.53,54 Finally, as shown in Fig. S8C,† graphitic carbon is formed during DRM with preceding hydrogen reduction, suggesting a higher activity for the catalytic decomposition of CH4. However, as shown in Table 1, graphitic carbon is also observed in the spent catalyst after two consecutive DRM cycles without H2 pre-reduction, which might be explained by the more controlled pressure of gas mixture in the catalytic experiments as compared to the in situ XRD experiments. These results are consistent with previous studies highlighting that the methane and carbon dioxide conversion and the amount of carbon formation are strongly pressure-dependent.55
To corroborate the findings from in situ XRD and to detect eventual local structural and chemical differences after the reduction and activation treatments, Fig. 6 and 7, as well as Fig. S9 and S10 in the ESI,† highlight an in-depth electron microscopy evaluation. After hydrogen reduction at 800 °C (Fig. 6), the formation of Ni particles is observed, but exclusively locally connected to both Sm and Sr. Extra formation of Sm2O3 is also observed, but these areas are devoid of Ni particles. This is clear by comparing the intensities in the blue boxed area in the four individual EDX maps. Correlating these results to the XRD analysis, we note the exclusive presence of the SrSm2NiO4 spinel structure containing both Sm and Sr. Sm2O3, alongside Ni is also present in the composition mixture after hydrogen reduction. From the local phase analysis by TEM, we infer the crucial importance of both Sm and Sr to accelerate the exsolution of the Ni particles.
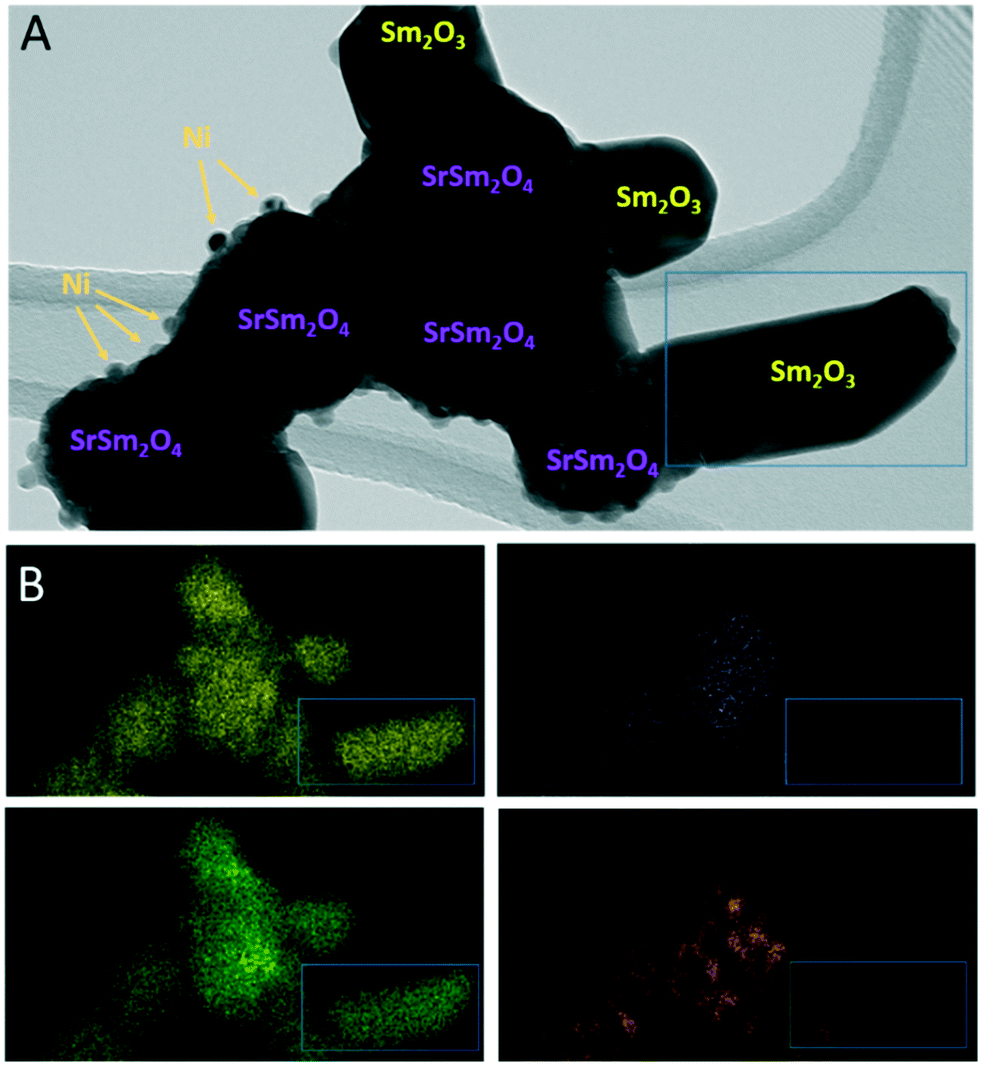 | ||
| Fig. 6 TEM/EDX analysis of Sm1.5Sr0.5NiO4 after hydrogen reduction at 800 °C. Panel A: Bright field overview TEM image. Phase composition of individual grains and particles as inferred from X-ray diffraction (Fig. 1) and EDX analysis is indexed. Panel B: EDX analysis using the Sm-L (yellow), Sr-K (purple), O-K (green) and Ni-K (orange) intensities. | ||
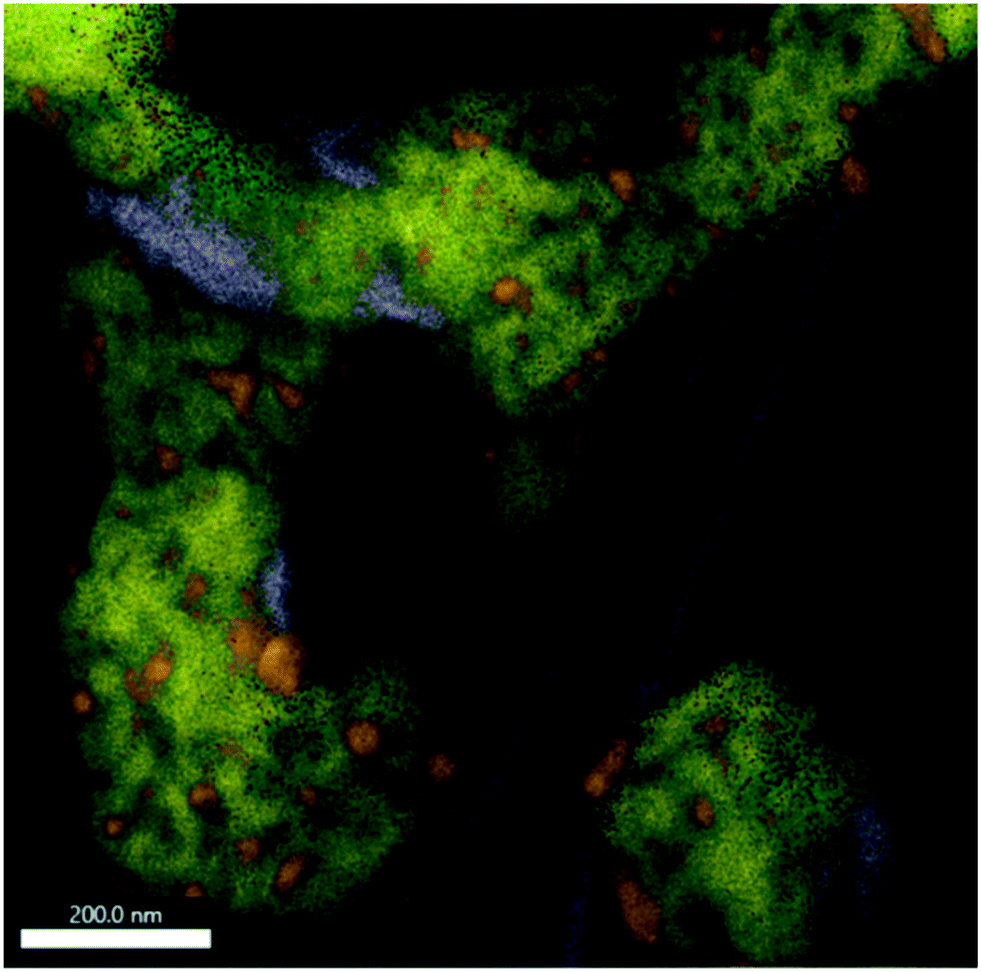 | ||
| Fig. 7 EDX analysis of Sm1.5Sr0.5NiO4 after a DRM reaction at 800 °C followed by an isothermal period for 30 min at 800 °C. The overlay of Sm-L (yellow), Sr-K (purple), O-K (green), C-K (magenta) and Ni-K (orange) intensities is shown. The individual maps are shown in Fig. S10.† | ||
If the Sm1.5Sr0.5NiO4 structure is heated in the DRM mixture up to 800 °C, XRD indicated the extraordinary stability of Sm1.5Sr0.5NiO4 up to 800 °C and the presence of a mixture of SrO, Sm2O3 and exsolved Ni after re-cooling to room temperature (Fig. 5 and S7 and S9†). Fig. S10† shows that Ni is always found next to Sm and Sr, but never in contact with Sm alone. We still observe isolated Sm2O3 patches similar to the one in Fig. 6. In addition, isolated pure Sr-containing areas – in contrast to Sm2O3 – are also present. Carbon is mostly homogeneously distributed and also appears in areas with either pure Sm or Sr. In the lower right corner of Fig. 7 (cf. also Fig. S10†), an elongated Sr-containing grain is seen, with significant carbon intensity in the same local area. We conclude that this grain is associated with SrCO3, confirming the XRD results. XRD did not give hints towards the formation of Sm-oxy-carbonate phases (but only Sm2O3), and it appears that the carbon is mostly associated with graphitic carbon covering Sm2O3 grains.
As indicated in Fig. S11 and Table S1,† surface elemental characterization of the initial state indicate the exclusive presence of Ni (+II), as expected from the stoichiometry of Sm1.5Sr0.5NiO4. Pre-reduction or activation in the DRM mixture introduces a considerable amount of metallic Ni (Ni0), more or less independent of the treatment. On the surface, the ratio of Ni2+:Ni0 is approximately 1:1, which corroborates the TEM and in situ XRD results.
As shown in Fig. 8 panel A, the normalized Ni K-edge X-ray absorption near-edge structure of the initial Sm1.5Sr0.5NiO4 material is best fitted with the corresponding spectrum of the isostructural La2NiO4 Ruddlesden–Popper structure as the oxidation state of Ni in both compounds is +II. Accordingly, after any treatments in either hydrogen or DRM mixture, the Ni K edge structure is best fitted with metallic Ni in the oxidation state 0. As can already be deduced from the TEM experiments and the in situ and ex situ XRD analysis, the near-edge structure of the Sm LIII edge exhibits no distinct changes and is, normalized to Sm2O3, characteristic for Sm in the oxidation state +III (Fig. S12 panel A†). More distinct changes are observed for the Sr K edge (Fig. S12 panel B†). Both before and after DRM, the Sr spectrum of the initial Sm1.5Sr0.5NiO4 sample is essentially characterized by Sr +II, as referenced to potential SrO or SrCO3 phases. A chemical discrimination solely on the basis of the XANES data is difficult, as all Sr-related phases appear in the same oxidation state. It appears, however, that the fingerprints of the Sr K edge in the near-edge region of the Sm1.5Sr0.5NiO4 sample match those of the SrO slightly better in terms of tailing of two main peaks.
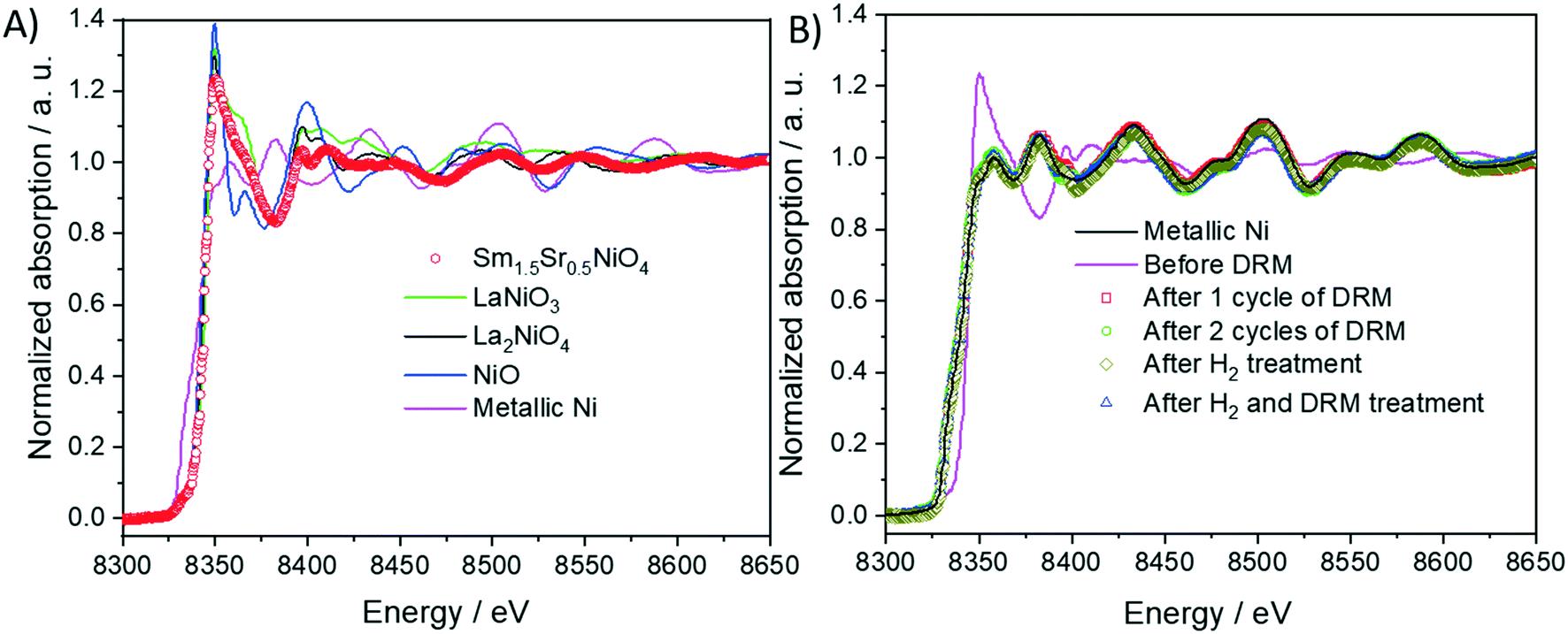 | ||
| Fig. 8 Panel A: Normalized Ni K-edge X-ray absorption near-edge structure (XANES) of Sm1.5Sr0.5NiO4 material compared with those of the reference materials (Ni, NiO, LaNiO3 and La2NiO4) and before and after different DRM experiments (panel B). The spectra are compared to that of metallic Ni as a reference material. | ||
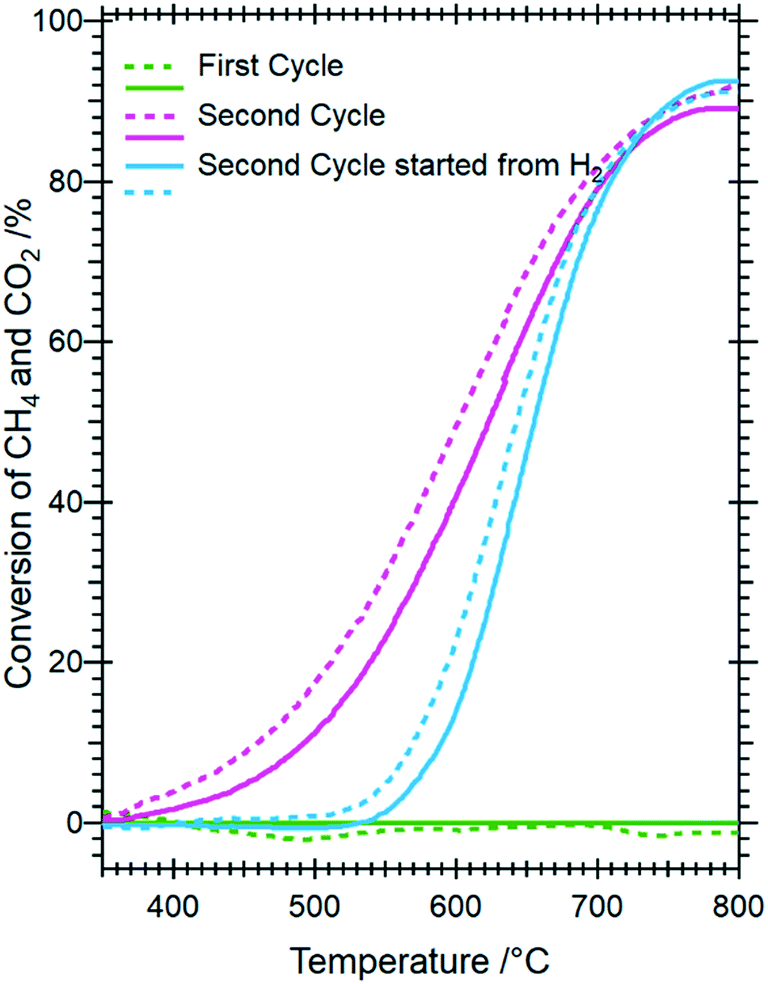 | ||
| Fig. 9 Catalytic methane dry reforming profiles of Sm1.5Sr0.5NiO4 as a function of hydrogen pre-treatment and activation in the DRM mixture. Reaction conditions: 1 bar flowing hydrogen; CO2:CH4 (1:1) mixture. Full lines indicate methane conversion and dashed lines CO2 conversion. | ||
3.2. Ni/Sm2O3 without Sr doping: catalytic DRM properties and structural characterization
To clarify the role of Sr doping in perovskite DRM catalysts, we have synthesized the corresponding catalyst without Sr in a similar fashion. The X-ray diffraction pattern in the calcined state before activation indicates the exclusive presence of NiO (18.2 wt%), cubic Sm2O3 (13.6 wt%) and monoclinic Sm2O3 (68.2%) composite material. As expected, without Sr doping a Sm–Ni perovskite or Ruddlesden–Popper phase could structurally not be stabilized (Fig. 10). Also, the composite material is self-activating in the DRM reaction mixture, manifesting itself as a very sharp activity increase at 720 °C. In a second DRM cycle and after a corresponding hydrogen pre-activation, the catalytic DRM profiles essentially follow the other profiles with Sr doping (Fig. 11). The XRD pattern shows that the initial activity increase is clearly associated with the reduction of NiO to metallic Ni and that the active material is Ni/Sm2O3, where Sm2O3 is present in the cubic and monoclinic polymorphs.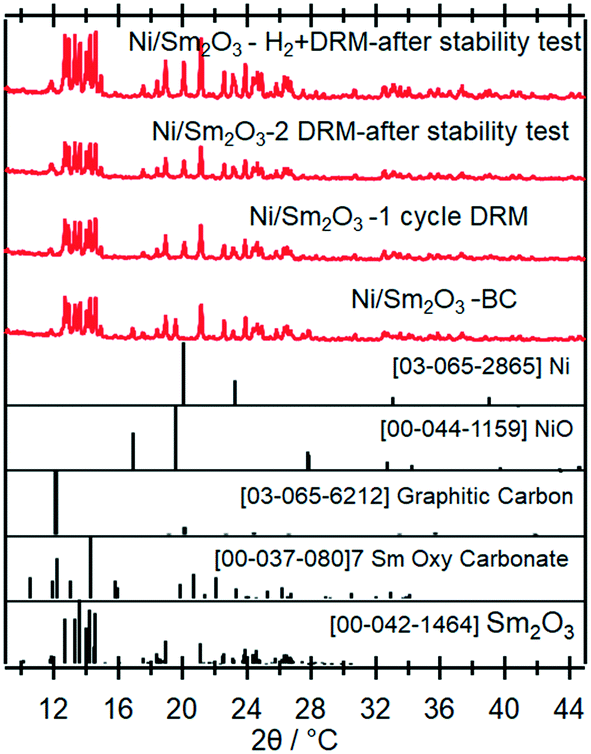 | ||
| Fig. 10 Ex situ X-ray diffraction patterns of the undoped pure Ni/Sm2O3 catalyst in the initial state and after selected activaton treatments as indicated. Wavelength: λ = 0.7093 Å. | ||
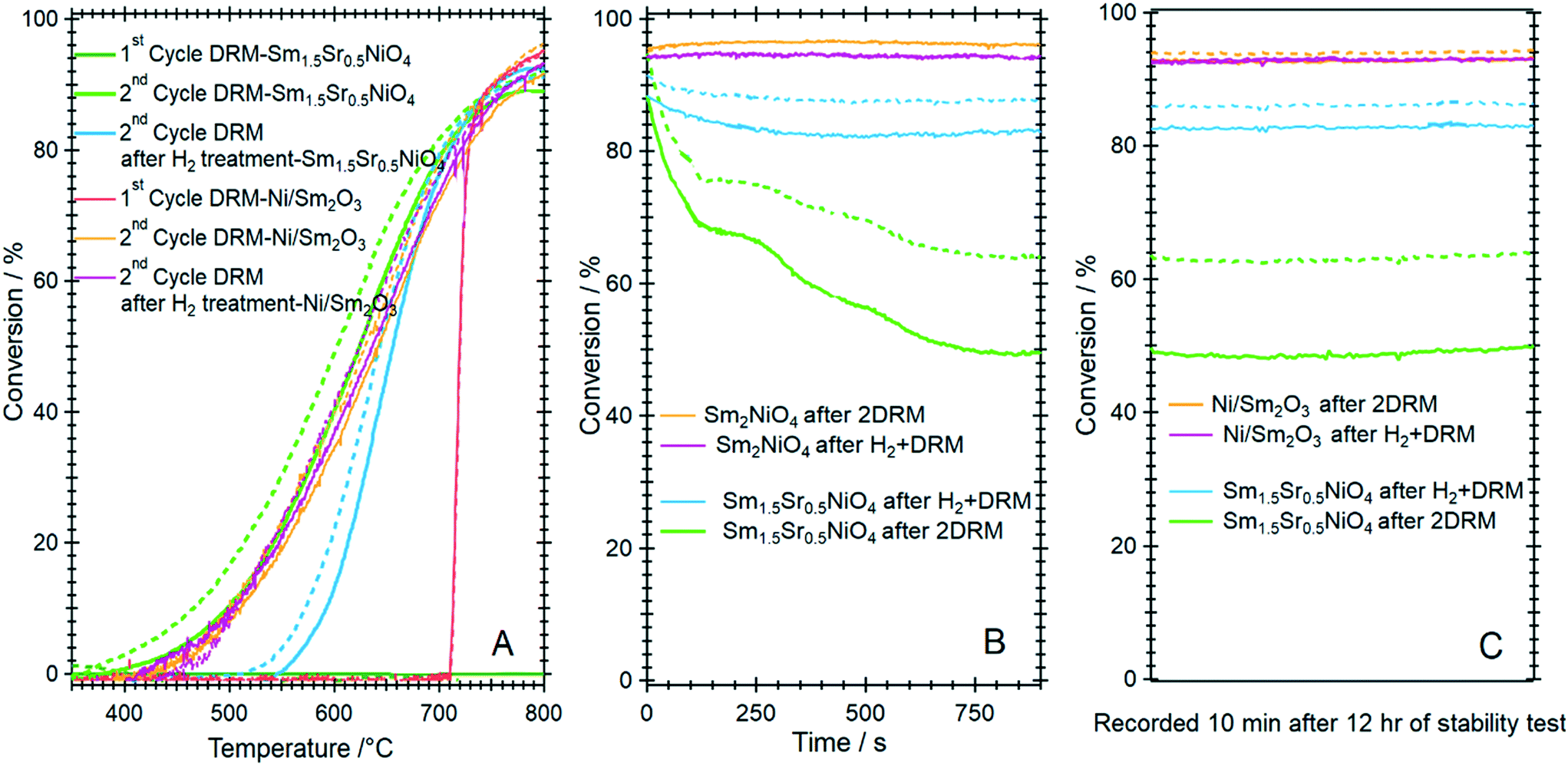 | ||
| Fig. 11 Panel A: Catalytic methane dry reforming profiles of Ni/Sm2O3 in comparison to Sm1.5Sr0.5NiO4 after direct activation in the DRM mixture and after hydrogen pre-activation. Panel B and C: Comparative short-term deactivation studies of Ni/Sm2O3 and Sm1.5Sr0.5NiO4 as a function of hydrogen pre-treatment and activation in the DRM mixture. Reaction conditions: 1 bar flowing hydrogen; CO2:CH4 (1:1) mixture. Full lines indicate methane conversion and dashed lines CO2 conversion. | ||
Striking differences between the Sr-doped and undoped material arise in the short-term deactivation studies (Fig. 11, panels B and C). While the undoped Ni/Sm2O3 catalyst shows a stable DRM performance irrespective of the pre-treatment even after 12 h time-on-stream at 800 °C and also the 1:1 H2:CO product ratio is maintained (as a consequence of catalytic DRM performance), the Sr-doped material deactivates very fast within minutes at 800 °C and the conversion – albeit stable afterwards – drops to almost 50%. Without hydrogen pre-treatment, the deactivation is most pronounced and can be directly related to the formation of a high amount of SrCO3 in the first DRM cycle. This in consequence induces an increase in Ni crystallite size, eventually decreases the number of accessible Ni sites and enhances the propensity towards deactivation by increasing the coverage with carbon species.52 As shown in Table 1, the crystallite size of Ni after DRM without hydrogen pre-reduction is ∼16 nm, which is almost 30% higher in comparison to the one with hydrogen pre-reduction (∼13 nm). Although a much larger Ni crystallite size for the catalyst without Sr doping is observed, this catalyst exhibits better short-term activity. These results suggest that the crystallite size of Ni is not the only explanation for the stable activity. Other factors, such as the presence and relative abundance of SrCO3, monoclinic Sm2O3 and reactive graphitic carbon phases in the catalyst, are of equal importance. As shown in Table 1 and Fig. S13 and S14,† high amounts of graphitic carbon and monoclinic Sm2O3 phases are observed in the spent Ni/Sm2O3 catalysts without Sr doping. One major issue concerns the carbon dioxide activation capability of both the Sr-doped, as well as the undoped material. For the former, we have ample evidence that the activity increase during DRM is not directly connected to the formation of a Sm2O2CO3 oxycarbonate phase. Neither XRD, XPS or TEM give any hint towards such a phase.
3.3. Discussion of the bulk and surface structural transformations and structure–activity correlation: Ni/Sm2O3vs. Sm1.5Sr0.5NiO4
To put the bulk and surface structural transformations into mechanistic and literature perspective and to subsequently correlate the structural changes to catalytic activity, we at first revisit the questions outlined in the introductory section and comparatively discuss the influence of Sr doping. Secondly, we address two peculiar structural issues that set the Sm1.5Sr0.5NiO4 material apart from similar catalysts: the absence of crystalline bulk Sm-oxycarbonate phases and the exclusive presence of SrCO3 as the only bulk-carbonated phase. As ample evidence for the generally beneficial role of adding Sm to DRM catalysts and the possibility of Sm2O3 to enter a reversible CO2 capture and release cycle has been presented,57–64 we first focus on the role of Sm2O3 in our catalysts. Despite the obvious importance of Sm2O3, the exact operating mechanism is far from clear.The role of added earth alkaline dopants to amplify the DRM activity of perovskite and Ni-based catalysts has already been assessed in detail. The introduction of such dopants is done either through a simple impregnation step or via introduction into the perovskite lattice. Either way, the active component finally is an oxide phase forming a phase boundary with metals capable of methane activation, such as Ni or Co. If the dopant is introduced into the lattice, a reduction or in situ activation step is necessary to form this phase boundary. Several earth alkaline metal oxides such as CaO, MgO, BaO, or SrO have been used to improve the DRM activity.21,57,64,65 Mechanistic-wise, the beneficial action usually manifests itself either structurally by stabilizing small metal particle sizes to prevent deactivation by sintering or by directly exploiting the surface chemical properties of the oxides to enhance the CO2 activation capabilities of the catalyst by invoking a bifunctional synergism. Activation of CO2 is significantly enhanced via basic oxide surface sites. In the case of earth alkaline oxides, which feature the formation of very stable carbonate structures usually incapable of entering a reversible CO2 activation cycle, stabilization effects are reported to be more prominent. It has been shown that even the formation of stable BaCO3 or SrCO3 phases can stabilize active Ni and cobalt phases during DRM operation.21,57,64 Specifically, SrCO3 is suspected to be essential for high DRM activity via structurally supporting active cobalt species.64
We have noted the principal capability of formation of a Sm2O2CO3 oxy-carbonate species, but none of our bulk or surface characterization tools provides evidence for such an Sm-oxycarbonate structure during DRM operation. This leaves the presence of an amorphous Sm-oxycarbonate species or other activation pathways, proceeding through the presence of active graphitic carbon and/or monoclinic Sm2O3 as an explanation. As shown in Table 1, high amounts of monoclinic Sm2O3 and graphitic carbon remain in the spent Ni/Sm2O3 catalysts after 12 hours of DRM regardless of the pre-treatment conditions (e.g., in H2 or self-activation during DRM).
Two types of carbon could in principle be formed on the spent nickel-based catalysts under DRM: reactive-surface-type and encapsulating-type carbon. The latter is the most common reason for catalyst deactivation, while reactive surface carbon resulting from CH4 decomposition is a necessary pathway for syngas production.31 No encapsulating-type carbon has been observed in the spent catalysts by TEM. Moreover, the short-term activity of Ni/Sm2O3 is accompanied by the formation of a high amount of graphitic carbon. These results suggest that the graphitic carbon formed under DRM in this work is of reactive-surface type. Due to the fast deactivation of Sm1.5Sr0.5NiO4 during the second DRM cycle, this reactive-surface carbon is successfully oxidized by CO2. Thus, no graphitic carbon has been observed in the spent catalyst after stability tests for 12 hours. In addition, the composition of the catalyst support or the associated interfacial properties might add to this difference in the catalytic properties. It has been reported that the reaction of carbon species with CO2 on Ni/La2O3/Sm2O3 catalysts could take place both via CO2 adsorption on Ni and CO2 adsorption at the La2O3/Sm2O3 interface.34 Intrinsically connected to the interfacial properties is the much higher weight % ratio of monoclinic Sm2O3 to cubic Sm2O3 (>4) after 12 hours of DRM operation. As this ratio drops to ∼1.1 for the Sr-containing catalyst with the lowest conversion rates after 12 hours time-on-stream, this explains the superior catalytic performance of Ni/Sm2O3 catalyst without Sr doping. The presence of monoclinic Sm2O3 as the main phase in the catalyst after the H2 pre-reduction steps (Table 1) additionally suggests a high oxygen vacancy concentration in this structure. The presence of oxygen vacancies in monoclinic Sm2O3 will facilitate CO2 adsorption and dissociation, as reported for other DRM catalysts.56 Moreover, the strong interaction of CO2 with monoclinic Sm2O3 also generates active sites for the adsorption and the reaction of fragmented carbon-containing intermediates, such as CHx, enhancing the CO2/CH4 conversion.34 Thus, hydrogen pre-reduction of the Sr-containing catalyst counteracts the detrimental poperties of the presence of Sr at least to some extent by stabilizing monoclinic Sm2O3 and increasing the ratio of monoclinic Sm2O3-to-cubic Sm2O3 (Fig. 11).
4. Conclusions
Our results on the influence of earth alkaline doping of perovskite-based catalysts using Sr as a representative and exemplary model in methane dry reforming yield a complex picture contrasting the simple previously reported exclusive beneficial role. Although the initial structures are already different as a consequence of the general instability of many Sm-containing perovskite materials without external doping, all catalysts irrespective of doping self-activate either during hydrogen pre-reduction or direct DRM activation. While this activation process for the undoped NiO/monoclinic Sm2O3 catalysts is associated with the simple reduction of NiO to metallic Ni, the doped Sm1.5Sr0.5NiO4 structure goes through a sequence of structural transformations before entering the Ni/Sm2O3/SrO(SrCO3) state. Transient oxygen-deficient Sm1.5Sr0.5NiO3 and SrSm2O4 spinel structures are both observed. The role of Sr is ambivalent: while it serves as a structural stabilizing aid, it acts as a carbon dioxide sink and structurally destabilizes the specific Ni/monoclinic Sm2O3 interface via enhancement of Ni particle sintering, causing catalyst deactivation. In particular, we have no evidence of the presence of a Sm-oxycarbonate structure (neither on the surface nor in the bulk), previously suspected to be responsible for the reversible carbon dioxide capture/release cycle. Rather, we have identified surface graphitic carbon in connection with the particular presence of monoclinic Sm2O3 after DRM treatments on the catalysts with and without Sr doping as a possible reactive intermediate, in line with recent studies on other metal-oxide systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. Penner acknowledges funding from the Austrian Science Fund (FWF) within the SFB project F4503-N16 “Functional Oxide Surfaces and Interfaces” and the DACH project I2877-N34. The work was performed within the framework of the research platform “Materials- and Nanoscience” and the special PhD program “Reactivity and Catalysis” at the University of Innsbruck. The authors further thank the Advanced Light Source (which is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231), where in situ XRD measurements were conducted at beamline 12.2.2 in the framework of the AP program ALS-08865. A. Gili appreciates the support of Unifying Systems in Catalysis (UniSysCat), funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008/1 – 390540038.References
- O. Muraza and A. Galadima, A Review on Coke Management During Dry Reforming of Methane, Int. J. Energy Res., 2015, 39, 1196–1216, DOI:10.1002/er.3295.
- M. Usman, W. M. A. Wan Daud and H. F. Abbas, Dry Reforming of Methane: Influence of Process Parameters - A Review, J. Renewable Sustainable Energy, 2015, 45, 710–744, DOI:10.1016/j.rser.2015.02.026.
- K. Mondal, S. Sasmal, S. Badgandi, D. R. Chowdhury and V. Nair, Dry Reforming of Methane to Syngas: A Potential Alternative Process For Value Added Chemicals - A Techno-Economic Perspective, Environ. Sci. Pollut. Res., 2016, 23, 22267–22273, DOI:10.1007/s11356-016-6310-4.
- H. Er-Rbib, C. Bouallou and F. Werkoff, Dry Reforming of Methane-Review of Feasibility Studies, Chem. Eng. Trans., 2012, 29, 163–168, DOI:10.3303/CET1229028.
- J. H. Edwards and A. M. Maitra, The Chemistry of Methane Reforming With Carbon Dioxide And Its Current And Potential Applications, Fuel Process. Technol., 1995, 42, 269–289, DOI:10.1016/0378-3820(94)00105-3.
- R. Dębek, K. Zubek, M. Motak, P. Da Costa and T. Grzybek, Effect of Nickel Incorporation Into Hydrotalcite-Based Catalyst Systems For Dry Reforming of Methane, Res. Chem. Intermed., 2015, 41, 9485–9495, DOI:10.1007/s11164-015-1973-x.
- A. Al-Fatesh, Suppression of Carbon Formation In CH4 –CO2 Reforming by Addition of Sr into Bimetallic Ni–Co/γ-Al2O3 Catalyst, J. King Saud Univ., Eng. Sci., 2015, 27, 101–107, DOI:10.1016/j.jksues.2013.09.006.
- E. Ruckenstein and Y. H. Hu, Carbon Dioxide Reforming of Methane over Nickel/Alkaline Earth Metal Oxide Catalysts, Appl. Catal., A, 1995, 133, 149–161, DOI:10.1016/0926-860X(95)00201-4.
- Z. Hao, Q. Zhu, Z. Jiang, B. Hou and H. Li, Characterization of Aerogel Ni/Al2O3 Catalysts And Investigation on Their Stability For CH4-CO2 Reforming In A Fluidized Bed, Fuel Process. Technol., 2009, 90, 113–121, DOI:10.1016/j.fuproc.2008.08.004.
- A. F. Lucredio, J. M. Assaf and E. M. Assaf, Methane Conversion Reactions on Ni Catalysts Promoted With Rh: Influence of Support, Appl. Catal., A, 2011, 400, 156–165, DOI:10.1016/j.apcata.2011.04.035.
- T. D. Gould, M. M. Montemore, A. M. Lubers, L. D. Ellis, A. W. Weimer, J. L. Falconer and J. W. Medlin, Enhanced Dry Reforming of Methane on Ni And Ni-Pt Catalysts Synthesized By Atomic Layer Deposition, Appl. Catal., A, 2015, 492, 107–116, DOI:10.1016/j.apcata.2014.11.037.
- K. M. Kang, H. W. Kim, Il. W. Shim and H. Y. Kwak, Catalytic Test of Supported Ni Catalysts With Core/Shell Structure For Dry Reforming of Methane, Fuel Process. Technol., 2011, 92, 1236–1243, DOI:10.1016/j.fuproc.2011.02.007.
- I. G. Crnivec, P. Djinovic, B. Erjavec and A. Pintar, Effect of Synthesis Parameter on Morphology And Activity of Bimetallic Catalysts For CH4-CO2 Reforming, Chem. Eng. J., 2012, 207-208, 297–307, DOI:10.1016/j.cej.2012.06.107.
- X. Zhang, C. S. M. Lee, D. Michel, P. Mingos and D. O. Hayward, Carbon Dioxide Reforming of Methane With Pt Catalyst Using Microwave Electric Heating, Catal. Lett., 2003, 88, 129–139, DOI:10.1023/A:1024049403422.
- K. Nagaoka, M. Okamura and K. I. Aika, Titania Supported Ruthenium As A Coking Resistant Catalyst For High Pressure Dry Reforming of Methane, Catal. Commun., 2001, 2, 255–260, DOI:10.1016/S1566-7367(01)00043-7.
- J. J. Juan, M. C. Roman-Martınez and M. J. Illan-Gomez, Nickel Catalyst Activation In The Carbon Dioxide Reforming of Methane: Effect of Pretreatments, Appl. Catal., A, 2009, 355, 27–32, DOI:10.1016/j.apcata.2008.10.058.
- C. Batiot-Dupeyrat, G. Valderrama, A. Meneses, F. Martinez, J. Barrault and J. Tatibouët, Pulse Study of CO2 Reforming of Methane over LaNiO3, Appl. Catal., A, 2003, 248, 143–151, DOI:10.1016/S0926-860X(03)00155-8.
- G. Sierra Gallego, F. Mondragón, J.-M. Tatibouët, J. Barrault and C. Batiot-Dupeyrat, Carbon Dioxide Reforming of Methane Over La2NiO4 As Catalyst Precursor - Characterization of Carbon Deposition, Catal. Today, 2008, 133-135, 200–209, DOI:10.1016/j.cattod.2007.12.075.
- I. Wender, Reactions Of Synthesis Gas, Fuel Process. Technol., 1996, 48, 189–297, DOI:10.1016/S0378-3820(96)01048-X.
- N. Bonmassar, M. F. Bekheet, L. Schlicker, A. Gili, A. Gurlo, A. Doran, Y. Gao, M. Heggen, J. Bernardi, B. Klötzer and S. Penner, In Situ-Determined Catalytically Active State of LaNiO3 in Methane Dry Reforming, ACS Catal., 2020, 10, 1102–1112, DOI:10.1021/acscatal.9b03687.
- Y. Zhiwei, J. Jun, Z. Yu, L. Fubing, Z. Jiang, S. Yan, G. Haifeng and W. Haiyan, Insights into The Deactivation Mechanism of Metal Carbide Catalysts For Dry Reforming of Methane via Comparison of Nickel-Modified Molybdenum and Tungsten Carbides, RSC Adv., 2016, 6, 19944–19951, 10.1039/C5RA24815A.
- N. Köpfle, T. Götsch, M. Grünbacher, E. Carbonio, M. Hävecker, A. Knop-Gericke, L. Schlicker, A. Doran, D. Kober, A. Gurlo, S. Penner and B. Klötzer, Zirconium-Assisted Activation of Palladium To Boost Syngas Production by Methane Dry Reforming, Angew. Chem., Int. Ed., 2018, 57, 14613–14618, DOI:10.1002/anie.201807463.
- S. Kühl, H. Düdder, F. Girgsdies, K. Kähler, M. Muhler and M. Behrens, Perovskites As Precursors For Ni/La2O3 Catalysts In The Dry Reforming of Methane: Synthesis by Constant pH Co-Precipitation, Reduction Mechanism And Effect of Ru-Doping, Z. Anorg. Allg. Chem., 2017, 643, 1088–1095, DOI:10.1002/zaac.201700141.
- J. Alvarez, G. Valderrama, E. Pietri, M. J. Pérez-Zurita, C. U. de Navarro, E. F. Sousa-Aguiar and M. R. Goldwasser, Ni–Nb-Based Mixed Oxides Precursors For The Dry Reforming of Methane, Top. Catal., 2011, 54, 170–178, DOI:10.1007/s11244-011-9636-7.
- G. R. Moradi, F. Khosravian and M. Rahmanzadeh, Effects of Partial Substitution of Ni By Cu In LaNiO3 Perovskite Catalyst For Dry Methane Reforming, Chin. J. Catal., 2012, 33, 797–801, DOI:10.1016/S1872-2067(11)60378-1.
- M. F. Bekheet, N. P. D. Kheyrollahi, N. Bonmassar, L. Schlicker, A. Gili, S. Praetz, A. Gurlo, A. Doran, Y. Gao, M. Heggen, A. Niaei, A. Farzi, S. Schwarz, J. Bernardi, B. Klötzer and S. Penner, Steering the Methane Dry Reforming Reactivity of Ni/La2O3 Catalysts by Controlled in Situ Decomposition of Doped La2NiO4 Precursor Structures, ACS Catal., 2021, 43–59, DOI:10.1021/acscatal.0c04290.
- C. Papadopoulou, H. Matralis and X. Verykios, Utilization of Biogas as a Renewable Carbon Source: Dry Reforming of Methane, Catalysis for Alternative Energy Generation, 2012, pp. 57–127, DOI:10.1007/978-1-4614-0344-9_3.
- P. Delir Kheyrollahi Nezhad, M. F. Bekheet, N. Bonmassar, L. Schlicker, A. Gili, F. Kamutzki, A. Gurlo, A. Doran, Y. Gao, M. Heggen, S. Schwarz, J. Bernardi, A. Niaei, A. Farzi, B. Klötzer and S. Penner, Mechanistic In Situ Insights Into The Formation, Structural And Catalytic Aspects of The La2NiO4 Intermediate Phase In The Dry Reforming of Methane over Ni-Based Perovskite Catalysts, Appl. Catal., A, 2021, 612, 117984, DOI:10.1016/j.apcata.2020.117984.
- L. Xu, W. Liu, X. Zhang, L. Tao, L. Xia, X. Xu, J. Song, W. Zhou, X. Fang and X. Wang, Ni/La2O3 Catalysts For Dry Reforming of Methane: Insights into The Factors Improving The Catalytic Performance, ChemCatChem, 2019, 11, 2887–2899, DOI:10.1002/cctc.201900331.
- S. Penner and P. Delir Kheyrollahi Nezhad, Steering the Catalytic Properties of Intermetallic Compounds and Alloys in Reforming Reactions by Controlled in Situ Decomposition and Self-Activation, ACS Catal., 2021, 5271–5286, DOI:10.1021/acscatal.1c00718.
- A. Gili, L. Schlicker, M. F. Bekheet, O. Görke, S. Penner, M. Grünbacher, T. Götsch, P. Littlewood, T. J. Marks, P. C. Stair, R. Schomäcker, A. Doran, S. Selve, U. Simon and A. Gurlo, Surface Carbon as a Reactive Intermediate in Dry Reforming of Methane to Syngas on a 5% Ni/MnO Catalyst, ACS Catal., 2018, 8, 8739–8750 CrossRef CAS.
- B. Bakiz, F. Guinneton, M. Arab, A. Benlachemi, S. Villain, P. Satre and J.-R. Gavarri, Carbonatation And Decarbonatation Kinetics In The La2O3-La2O2CO3 System Under CO2 Gas Flows, Adv. Mater. Sci. Eng., 2010, 2010, 360597, DOI:10.1155/2010/360597.
- O. Hirsch, K. O. Kvashnina, L. Luo, M. J. Süess, P. Glatzel and D. Koziej, High-Energy Resolution X-ray Absorption And Emission Spectroscopy Reveals Insight into Unique Selectivity of La-Based Nanoparticles For CO2, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15803–15808, DOI:10.1073/pnas.1516192113.
- W. D. Zhang, B. S. Liu, Y. P. Zhan and Y. L. Tian, Syngas Production Via CO2 Reforming of Methane over Sm2O3– La2O3-Supported Ni Catalyst, Ind. Eng. Chem. Res., 2009, 48, 7498–7504, DOI:10.1021/ie9001298.
- O. U. Osazuwa, H. D. Setiabudi, R. A. Rasid and C. K. Cheng, Syngas Production via Methane Dry Reforming: A Novel Application of SmCoO3 Perovskite Catalyst, J. Nat. Gas Sci. Eng., 2017, 37, 435–448, DOI:10.1016/j.jngse.2016.11.060.
- O. U. Osazuwa and C. K. Cheng, Catalytic Conversion of Methane and Carbon Dioxide (Greenhouse Gases) into Syngas over Samarium-Cobalt-Trioxides Perovskite Catalyst, J. Cleaner Prod., 2017, 148, 202–211, DOI:10.1016/j.jclepro.2017.01.177.
- Z. Taherian, M. Yousefpour, M. Tajally and B. Khoshandam, A Comparative Study of ZrO2, Y2O3 and Sm2O3 Promoted Ni/SBA-15 Catalysts for Evaluation of CO2/Methane Reforming Performance, Int. J. Hydrogen Energy, 2017, 42(26), 16408–16420, DOI:10.1016/j.ijhydene.2017.05.095.
- R. Jaramillo, F. Schoofs, S. D. Ha and S. Ramanathan, High Pressure Synthesis of SmNiO3 Thin Films and Implications for Thermodynamics of the Nickelates, J. Mater. Chem. C, 2013, 1(13), 2455, 10.1039/C3TC00844D.
- G. Amow and S. J. Skinner, Recent Developments In Ruddlesden–Popper Nickelate Systems For Solid Oxide Fuel Cell Cathodes, J. Solid State Electrochem., 2006, 10(8), 538–546, DOI:10.1007/s10008-006-0127-x.
- A. Doran, L. Schlicker, C. M. Beavers, S. Bhat, M. F. Bekheet and A. Gurlo, Compact Low Power Infrared Tube Furnace for in Situ X-Ray Powder Diffraction, Rev. Sci. Instrum., 2017, 88, 13903, DOI:10.1063/1.4973561.
- L. Schlicker, A. Doran, P. Schneppmüller, A. Gili, M. Czasny, S. Penner and A. Gurlo, Transmission in Situ and Operando High Temperature X-Ray Powder Diffraction in Variable Gaseous Environments, Rev. Sci. Instrum., 2018, 89, 33904, DOI:10.1063/1.5001695.
- J. Rodriguez-Carvajal, Recent Developments of The Program FULLPROF, In Commission on Powder Diffraction (IUCr), Newsletter, 2001, vol. 26, pp. 12 –19 Search PubMed.
- L. W. Finger, D. E. Cox and A. P. Jephcoat, A Correction For Powder Diffraction Peak Asymmetry Due to Axial Divergence, J. Appl. Crystallogr., 1994, 27, 892–900, DOI:10.1107/S0021889894004218.
- C. Schlesiger, L. Anklamm, H. Stiel, W. Malzer and B. Kanngießer, XAFS Spectroscopy by An X-Ray Tube Based Spectrometer Using a Novel Type of HOPG Mosaic Crystal and Optimized Image Processing, J. Anal. At. Spectrom., 2015, 30, 1080–1085, 10.1039/C4JA00303A.
- C. Schlesiger, S. Praetz, R. Gnewkow, W. Malzer and B. Kanngießer, Recent Progress In The Performance of HAPG Based Laboratory EXAFS And XANES Spectrometers, J. Anal. At. Spectrom., 2020, 35, 2298–2304, 10.1039/D0JA00208A.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: Data Analysis For X-Ray Absorption Spectroscopy Using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541, DOI:10.1107/S0909049505012719.
- T. Opravil, P. Ptacek, F. Soukal, E. Bartonickova and J. Wasserbauer, Solid State Synthesis of SrY2O4 and SrSm2O4, J. Therm. Anal. Calorim., 2016, 123, 181–194, DOI:10.1007/s10973-015-4950-0.
- S. M. Antao and I. Hassan, The Orthorhombic Structure of CaCO3, SrCO3, PbCO3 and BaCO3: Linear Structural Trends, Can. Mineral., 2009, 47, 1245–1255, DOI:10.3749/canmin.47.5.1245.
- B. Fubini, F. Di. Renzo and F. S. Stone, Strontianite-Aragonite Solid Solutions SrxCa1-xCO3: Effect of Composition on The Orthorhombic-Rhombohedral Phase Transition and The Conversion to Oxide Solid Solutions SrxCa1-xO, J. Solid State Chem., 1988, 77, 281–292, DOI:10.1016/0022-4596(88)90250-2.
- K. Iwafuchi, C. Watanabe and R. Otsuka, On The Transition Temperatures of BaCO3 and SrCO3 as DTA Temperature Reference Materials, Thermochim. Acta, 1983, 64, 381–386, DOI:10.1016/0040-6031(83)85013-8.
- E. Rapopart and C. Pistorius, Orthorhombic-Disordered Rhombohedral Transition in SrCO3 and BaCO3 to 40 Kilobars, J. Geophys. Res., 1967, 72, 6353–6357, DOI:10.1029/JZ072i024p06353.
- B. C. da Silva, P. H. Bastos, R. B. S. Junior, N. R. Checca, R. Frety and S. T. Brandao, Perovskite-type catalysts based on nickel applied in the Oxy-CO2 reforming of CH4: Effect of Catalyst Nature And Operative Conditions, Catal. Today, 2021, 369, 19–30, DOI:10.1016/j.cattod.2020.07.060.
- H. Özdemir, M. A. Öksüzömer and M. A. Gürkayanak, Studies on Oxidative Coupling of Methane Using Sm2O3-Based Catalysts, Chem. Eng. Commun., 2019, 206, 48–60, DOI:10.1080/00986445.2018.1471400.
- A. Ayub, H. Bahruji and A. H. Mahadi, Ni Nanoparticles on Reducible Metal Oxides (Sm2O3, CeO2, ZnO) as Catalysts for CO2 Methanation, BCREC, 2021, vol. 16, pp. 641–650, DOI:10.9767/bcrec.16.3.10948.641-650.
- M. Khoshtinat Nikoo and N. A. S. Amin, Thermodynamic Analysis of Carbon Dioxide Reforming of Methane in View of Solid Carbon Formation, Fuel Process. Technol., 2011, 92, 678–691, DOI:10.1016/j.fuproc.2010.11.027.
- Y. Lu, Y. Guo, P. S. Moyo, Y. Zhao, S. Wang and X. Ma, Enhanced Catalytic Performance of Nix-V@HSS Catalysts For The DRM Reaction: The Study of Interfacial Effects on Ni-VOx Structure With A Unique Yolk-Shell Structure, J. Catal., 2021, 396, 65–80, DOI:10.1016/j.jcat.2021.02.005.
- Z. Taheran, V. Gharahshiran, F. Fazlikhani and M. Yousefpour, Catalytic Performance of Samarium-Modified Ni Catalysts over Al2O3–CaO Support for Dry Reforming of Methane, Int. J. Hydrog. Energy, 2021, 46, 7254–7262, DOI:10.1016/j.ijhydene.2020.11.196.
- A. Jahangiri, H. Pahlavanzandeh and H. Aghabozorg, Synthesis, Characterizationand Catalytic Study of Sm Doped LaNiO3 Nanparticles in Reforming of Methane with CO2 and O2, Int. J. Hydrog. Energy, 2012, 37, 9977–9984, DOI:10.1016/j.ijhydene.2012.03.128.
- O. Osazuwa, H. Setiabudi, A. Abdullah and C. Cheng, Syngas Production From Methane Dry Reforming over SmCoO3 Perovskite Catalyst: Kinetics and Mechanistic Studies, Int. J. Hydrog. Energy, 2017, 23, 1–15, DOI:10.1016/j.ijhydene.2017.03.061.
- A. N. Shirsat, S. R. Bharadwaj and D. Das, Thermochemistry of Decomposition of RE2O2CO3 (RE=Sm, Eu), Thermochim. Acta, 2008, 477, 38–41, DOI:10.1016/j.tca.2008.08.008.
- V. V. Voronov, L. D. Isakhova, V. V. Kashin, S. Yu. Russanov and V. B. Tsvetkov, Microstructure and Propertiesof Single-Crystalline Rare Earth Oxide Fibers, J. Surf. Invest.: X-Ray, Synchrotron Neutron Tech., 2011, 5, 986–991, DOI:10.1134/S1027451011100235.
- K. C. Patil, G. V. Chandrashekar, M. V. George and C. N. R. Rao, Infrared Spectra and Thermal Decompositions of Metal Acetates and Dicarboxylates, Can. J. Chem., 1968, 46, 257, DOI:10.1139/v68-040.
- B. V. Ayodele, M. R. Khan and C. K. Cheng, Greenhouse Gases Abatement by Catalytic Dry Reforming of Methane to Syngas over Samarium Oxide-Supported Cobalt Catalyst, Int. J. Environ. Sci. Technol., 2017, 14, 2769–2782, DOI:10.1007/s13762-017-1359-2.
- K. Omata, N. Nukui, T. Hootai, Y. Showa and M. Yamada, Strontium Carbonate-Supported Cobalt Catalyst For Dry Reforming of Methane Under Pressure, Catal. Commun., 2004, 5, 755–758, DOI:10.1016/j.catcom.2004.09.012.
- Y. Song, E. Ozdemir, S. Ramesh, A. Adishev, S. Subramanian, A. Harale, M. Albuali, B. A. Fadhel, A. Jamal, D. Moon, S. H. Choi and C. T. Yavuz, Dry Reforming of Methane by Stable Ni-Mo Nanocatalysts on Single-Crystalline MgO, Science, 2020, 367, 777–781, DOI:10.1126/science.aav2412.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy02044g |
| This journal is © The Royal Society of Chemistry 2022 |