
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethanol oxidation on Au(332): methyl formate selectivity and surface deactivation under isothermal conditions†
Christoph D.
Feldt
 a,
Thorren
Kirschbaum
ab,
Jian Liang
Low
a,
Wiebke
Riedel
*a and
Thomas
Risse
a
a,
Thorren
Kirschbaum
ab,
Jian Liang
Low
a,
Wiebke
Riedel
*a and
Thomas
Risse
a
aInstitut für Chemie und Biochemie, Freie Universität Berlin, Arnimallee 22, 14195 Berlin, Germany. E-mail: wiebke.riedel@fu-berlin.de; risse@chemie.fu-berlin.de
bHelmholtz-Zentrum Berlin für Materialien und Energie, Hahn-Meitner-Platz 1, 14109 Berlin, Germany
First published on 21st January 2022
Abstract
Methanol oxidation on the stepped Au(332) surface was investigated by pulsed isothermal molecular beam (MB) experiments. The effect of the surface temperature as well as the influence of changes in the methanol and atomic oxygen flux on the partial oxidation to methyl formate was studied. A maximum in methyl formate formation is observed at 250 K under the applied single collision conditions. Increasing the methanol to oxygen ratio was found to increase the selectivity to methyl formate and decrease unwanted overoxidation to surface deactivating formate detected by in situ infrared reflection absorption spectroscopy (IRAS). The results show evidence for the importance of an additional deactivation mechanism for methyl formate formation connected to methanol which is active under oxygen-deficient conditions at low temperatures. Moreover, the measurements suggest a small number of sites to be highly reactive for methyl formate formation which are preferentially blocked under oxygen-deficient conditions.
Introduction
Gold based catalysts have been the subject of numerous investigations since the first publication by Haruta.1 The development of nanoporous gold (np-Au) renewed the interest, as these fully metallic catalysts also exhibit high activity in oxidation reactions2–5 despite the lack of an oxidic support and ligament sizes being about an order of magnitude larger than the size of supported Au nanoparticles of active supported gold catalysts.1,6,7 np-Au catalysts are typically obtained by etching a less noble metal, such as silver or copper, from an alloy with gold. The resulting material has a porous structure and contains mainly gold, but also residuals of the less noble metal. This residual metal plays an important role in the activation of molecular oxygen, which is the rate limiting step for aerobic oxidation catalysis.2,8–11 In the partial oxidation of methanol, np-Au shows high selectivity to methyl formate at high conversion.2 Reduced contact times, however, enhance formaldehyde formation,11 while increased oxygen or Ag contents favor total oxidation.2,11–13 Moreover, the selectivity is affected by the presence of different oxygen species, including oxidic phases.12,14 Under typical reaction conditions, however, the oxygen surface concentration is assumed to be rather low.11,13 Recently, also, carbonaceous deposits, presumably located on residual Ag, were reported for non-steady state conditions at low temperatures.14To gain more insights into the underlying microscopic mechanisms, model studies on single crystal surfaces under well-defined ultra-high vacuum (UHV) conditions as well as theoretical investigations have been conducted. Based on temperature programmed reaction (TPR) studies with Au surfaces pre-covered with activated oxygen, a reaction mechanism has been proposed:15–17 Initially formed methoxy species yield formaldehyde upon hydrogen abstraction, which is the rate limiting step in the formation of methyl formate in case activated oxygen is present. In the presence of oxygen, the (formally) abstracted hydrogen leaves the gold surface predominantly as water or methanol, while formaldehyde may either desorb, react with methoxy and oxygen into methyl formate or become further oxidized finally yielding the total oxidation product CO2. In agreement with studies on np-Au,2,3 an increased oxygen pre-coverage was reported to enhance total oxidation in TPR measurements.15,18,19 Similarly, kinetic modelling suggested reduced methanol to oxygen ratios to favor total oxidation, while lowering the oxygen pressure (at rather high methanol to oxygen pressure ratios) in np-Au catalysts is predicted to enhance formaldehyde formation.20,21
TPR investigations have mainly been conducted on low-index surfaces, while np-Au catalysts contain a significant number of low-coordinated Au sites which presumably alter the reactivity.22–24 Moreover, Au surfaces pre-covered with activated oxygen exhibit, even at low coverage, oxygen islands or oxidic phases next to oxygen atoms and thus, at least, locally high oxygen concentrations.19 Recent pulsed isothermal molecular beam experiments using an atomic oxygen beam provided by a thermal cracker suggested a different reactivity of AuOx phases, i.e. accumulated, residual oxygen, as compared to atomic oxygen provided during the oxygen pulses.25 Formate species formed in the presence of AuOx phases were found to poison the stepped Au(332) surface with respect to the formation of methyl formate.25 Furthermore, methoxy and formaldehyde formation occurs in the presence of AuOx phases, while methyl formate formation as well as (oxidative) decomposition of formate species proceeds preferentially in the presence of atomic oxygen.
In this study, the influence of the surface temperature as well as the oxygen and methanol fluxes on the (partial) oxidation of methanol is investigated by pulsed isothermal MB experiments using the stepped Au(332) surface as a model system for np-Au. In particular, the effects of rather oxygen-deficient conditions and increased surface temperature on the undesired surface deactivation are examined, in addition to their influence on the (initial) selectivity to methyl formate.
Experimental details
The measurements were performed in a UHV setup with two chambers which has been described before.26 One chamber is equipped with a sputter gun (IQE 11/35, SPECS), a low-energy electron diffraction (LEED) system (Omicron MCP LEED), an Auger spectrometer (PHI 11-010, Perkin Elmer), and a quadrupole mass spectrometer (Prisma, Pfeiffer) for temperature programmed desorption (TPD) measurements using a Feulner cup to enhance the sensitivity. In the second chamber, two effusive molecular beams27 and a thermal atomic oxygen source (Dr. Eberl MBE-Komponenten GmbH) being operated as an effusive beam are installed and can be modulated by automated valves and shutters. A stagnation flow monitor with a high precision ion gauge (360 Stabil-Ion, Granville-Phillips) is used to measure the pressure at the sample position. During pulsed isothermal MB experiments, gas phase species are detected using a quadrupole mass spectrometer (MAX-500HT, Extrel), while surface adsorbates are monitored by in situ IRAS measurements in grazing reflection geometry employing an IR spectrometer (IFS 66v, Bruker, 256 scans, nominal resolution of 4 cm−1, zero filling factor of 16). Using Mo clamps, the Au(332) single crystal (10 mm diameter, 2 mm thick, Mateck) is pressed onto a boron nitride heater (HT-01, Momentive) mounted to a home-made Mo-holder which is connected to a liquid nitrogen cooled Cu block, allowing for sample cooling down to approx. 100 K. The crystal temperature is measured by applying a type K thermocouple inserted in a 0.2 mm hole in the Au crystal edge. A commercial PID controller (3508, Eurotherm) is used to monitor the thermocouple voltage and to control the sample temperature in TPD and isothermal experiments.Repeated cycles of Ar+ ion sputtering (1000 V, 7–10 μA, 15 min) and subsequent annealing to 1000 K i. vac. for 10 min were employed to clean the Au(332) surface until a sharp LEED image expected for the (332) surface was observed.25,28 Methanol (Roth, ≥99.98% or 13C-methanol, 99 atom% 13C, Sigma Aldrich; both dried over molecular sieves 3 Å) and methyl formate (Sigma Aldrich, ≥99.8%) were further purified by repeated freeze–pump–thaw cycles and dosed onto the sample surface by an effusive molecular beam. The impurities in 12C-methanol were further investigated by GC-MS and LC-MS measurements (see the ESI†). Oxygen (Air Liquide, 99.998%) was used as received. For removal of carbonyl species from the CO (Linde, 99.997%) feed, a liquid nitrogen cryo-trap was used. Using a beam monitor, the pressure at the sample position was calibrated as a function of the inlet pressure of the beam using Ar gas. A thermal cracker (T = 1615 °C, 12.45 V, 12.60 A and for some experiments shown in the ESI† 1700 °C, 15.8 V, 13.85 A) was employed to provide atomic oxygen. TPD measurements were conducted to calibrate the flux of atomic oxygen comparing the integrated signal intensities for O2 desorption from an Au(332) surface with respect to O2 desorption from a Pt(111) surface after O2 exposure at 300 K resulting in a p(2 × 2) structure with a coverage of 0.25 monolayers (MLs).29,30 The saturation coverage of oxygen on Au(332) achieved by exposure to atomic oxygen from the thermal cracker was 2.1 MLs (1 ML corresponding to 1.4 × 1015 cm−2, and thus, to one O atom per Au surface atom). Calibration measurements with varying fluxes of methyl formate provided by a well-defined effusive molecular beam source were conducted to quantify the QMS signal intensity for methyl formate formation, applying a non-reactive flag at the sample position to ensure comparable scattering conditions. Reference measurements with a well-defined flux of methanol provided by an effusive molecular beam source were conducted after each experiment to calibrate the measured QMS intensity. Moreover, pulsed MB experiments were repeated to ensure the reproducibility of the results.
Results and discussion
Fig. 1 shows the results of pulsed isothermal molecular beam experiments of the partial methanol oxidation on Au(332) as a function of the surface temperature. In these experiments, a continuous flux of methanol (52.7 × 1013 s−1 cm−2, p(MeOH) = 19.6 × 10−7 mbar) was dosed onto the surface, while a flux of atomic oxygen (2.6 × 10−3 ML s−1 ≈ 0.4 × 1013 s−1 cm−2) provided by a thermal cracker was pulsed onto the surface (5 pulses, 200 s on, 600 s off). In Fig. 1a, the initial methyl formate formation rates are displayed relative to the maximum rate observed in this series for temperatures ranging between 220 K and 300 K. While the effective methyl formate formation rate increases when the temperature is raised from 220 K to 250 K, it decreases for higher temperatures. For a surface temperature of 300 K, the methyl formate formation essentially vanishes under the applied conditions. This temperature dependence can be understood considering that an increased surface temperature not only results in a faster coupling reaction to methyl formate, but also accelerates the desorption of molecular species, such as methanol and formaldehyde, essentially removing them from the reaction network under the applied single collision conditions. As formaldehyde needs to couple to surface bound methoxy to yield methyl formate according to the proposed mechanism15–17 schematically depicted in Fig. 2, a reduced residence time at elevated temperature will eventually tip the balance away from the coupling reaction to methyl formate towards formaldehyde desorption. It should be noted that the position of the rate maximum is expected to depend on the applied fluxes, shifting to higher temperatures for a higher flux, and thus pressure, on the surface. | ||
| Fig. 1 Results of pulsed isothermal MB experiments on methanol oxidation to methyl formate on Au(332) for various sample temperatures applying a constant flux of methanol (52.7 × 1013 s−1 cm−2, p(MeOH) = 19.6 × 10−7 mbar) and pulsing (200 s on, 600 s off) atomic oxygen (2.6 × 10−3 ML s−1 ≈ 0.4 × 1013 s−1 cm−2). (a) Initial methyl formate formation rates R normalized to the maximum rate detected in this series as a function of the surface temperature. (b) Relative decrease in methyl formate formation rate ΔR across the pulse sequence for the different surface temperatures given relative to the initial rate obtained for the respective surface temperature: 220 K (black squares), 250 K (red circles), 280 K (blue triangles). | ||
 | ||
| Fig. 2 Schematic depiction of the reaction mechanism for methanol oxidation on gold as previously proposed.15–17 | ||
In a previous study, the methyl formate formation rate was found to decrease across the pulse sequence as formate species accumulate on the surface.25 It can be speculated that an increase in surface temperature may prevent such a surface deactivation process by accelerating the (oxidative) decomposition of the formate species and thereby freeing up the surface sites. However, it can be seen from Fig. 1b that the methyl formate formation rate decreases for all studied surface temperatures across the pulse sequence. The decrease in rate relative to the first pulse is overall similar for temperatures between 220 K and 280 K and clearly does not diminish at higher temperatures. In fact, it appears to be even slightly stronger for the highest surface temperature of 280 K. This is consistent with the enhanced formation of formate at 280 K, as the increased diffusion rate and the reduced transient concentration of molecules is expected to foster the formation of AuOx phases at the expense of the transient concentration of atomic oxygen. The AuOx phases are known to favor the formation of formate, while atomic oxygen is beneficial for methyl formate formation and also shown to be active in total oxidation of formate.25 Thus, it is clear that the deactivation of the Au(332) surface is not lifted under the applied conditions by an increased surface temperature, before methyl formate formation ceases.
The flux dependence of the methanol oxidation to methyl formate on Au(332) was studied by pulsed isothermal MB experiments at 230 K (Fig. 3). In these experiments, atomic oxygen was similarly pulsed (200 s on, 300 s off), while methanol was supplied continuously in excess in the gas phase. Experiments were conducted with a rather high oxygen atom flux (0.4 × 1013 s−1 cm−2, 2.6 × 10−3 ML s−1, approx. 0.5 ML per pulse, Fig. 3a and b) and with a significantly lower oxygen atom flux (0.08 × 1013 s−1 cm−2, 0.6 × 10−3 ML s−1, approx. 0.12 ML per pulse, Fig. 3c and d). For each set of oxygen fluxes, measurements with a high (52.7 × 1013 s−1 cm−2, 19.6 × 10−7 mbar, Fig. 3b and d) and a low methanol flux (4.3 × 1013 s−1 cm−2, 1.6 × 10−7 mbar, Fig. 3a and c) were performed (see also Table 1 for details). For ease of reading, the flux conditions are abbreviated as the following, e.g. hiMloO denoting the experiment applying a high methanol flux and a low oxygen flux (see also Table 1). Upon increasing the oxygen flux, the initial methyl formate formation rate at the beginning of the pulse sequence increases for both methanol fluxes (see also Table 1). This is qualitatively in agreement with the expectation that methyl formate formation is limited by the availability of activated oxygen. However, the increase in the rate of methyl formate formation is considerably lower than the five-fold increase of oxygen flux, a direct consequence of the complex reaction network with competing reaction paths. When increasing the methanol flux, the initial methyl formate formation rate also increases, at least slightly, despite the applied excess of methanol flux (factor of ≥10, see also Table 1). To this end, it is important to note that oxygen does not desorb from Au(332) at 230 K,25 while there is no stable methanol adsorption on gold at 230 K (methanol desorption temperature: 155 K on Au(111),31 200 K on clean Au(110),16 220 K on 0.2 ML O pre-covered Au(111)15). Hence, the methanol to oxygen ratio on the surface is significantly reduced as compared to that in the gas phase. It should be noted that the increase of the initial rate is less pronounced for the high methanol and the high oxygen fluxes as compared to the lower fluxes. In all measurements of this series, the methyl formate formation rate decreases across the pulse sequence. The rate reduction at the end of the pulse sequence relative to the initial rate is overall very similar (approx. 40%) for the different applied conditions, except for the loMloO measurement (Fig. 3c) where the relative rate decrease is smaller (approx. 10%). For the high oxygen fluxes used here, the rate decrease across the pulse sequence was previously found to be correlated to the accumulation of formate species blocking the active surface sites for methyl formate formation.25
 | ||
| Fig. 3 Pulsed isothermal MB experiments of the partial methanol oxidation on Au(332) to methyl formate (molecular peak: m/z = 60) at 230 K under a continuous methanol flux, while applying pulses of atomic oxygen (200 s, delay 300 s). An atomic oxygen flux of 2.6 × 10−3 ML s−1 ≈ 0.4 × 1013 s−1 cm−2 is applied in (a) and (b), while in (c) and (d) a flux of 0.6 × 10−3 ML s−1 ≈ 0.08 × 1013 s−1 cm−2 was used. The methanol flux applied in (a) and (c) was 4.3 × 1013 s−1 cm−2 (p(MeOH) = 1.6 × 10−7 mbar), while in (b) and (d) a methanol flux of 52.7 × 1013 s−1 cm−2 (p(MeOH) = 19.6 × 10−7 mbar) was used. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :f(O) for the pulsed isothermal MB experiments on the methanol oxidation at 230 K on Au(332) (Fig. 3 and 4) and the initial methyl formate formation rate ν0(MF)
:f(O) for the pulsed isothermal MB experiments on the methanol oxidation at 230 K on Au(332) (Fig. 3 and 4) and the initial methyl formate formation rate ν0(MF)
| Name | f(CH3OH) [1013 s−1 cm−2] | f(O) [1013 s−1 cm−2] | Approx. flux ratio f(CH3OH):f(O) |
Approx. ν0(MF) [1011 s−1 cm−2] | |
|---|---|---|---|---|---|
| a | loMhiO | 4.3 | 0.4 | 10 | 4 |
| b | hiMhiO | 52.7 | 0.4 | 130 | 4.8 |
| c | loMloO | 4.3 | 0.08 | 55 | 1.6 |
| d | hiMloO | 52.7 | 0.08 | 660 | 2.8 |
In situ IRAS measurements conducted during the oxygen pulse as well as during the delay times of the pulsed isothermal MB experiments are displayed in Fig. 4. For the loMhiO measurement (Fig. 4a), a strong signal at around 1332 cm−1 and weaker signals at around 2832 cm−1 and 2907 cm−1 are clearly detectable and are assigned to the νs(OCO) stretching, νaCH and a combination of the νs(OCO) and νaCH modes of bidentate formate species based on the results by Senanayake et al. for Au(111).25,32 As previously reported,25 the formate intensity effectively increases mainly during the delay times between the oxygen pulses, demonstrating the accumulation of residual oxygen during the pulse for the loMhiO measurement. The nearly constant intensity during the oxygen pulses was explained by the increased (oxidative) decomposition of formate due to a higher concentration of (atomic) oxygen.25 These formate signals exhibit a lower intensity, when a lower oxygen flux (Fig. 4c and d) or higher methanol flux (Fig. 4b and d) is applied. For the hiMloO experiment (Fig. 4d), the signals are basically absent. These observations agree with the expectation that overoxidation to formate, as compared to methyl formate formation, should be reduced for lower oxygen contents (relative to methanol).
 | ||
| Fig. 4 In situ IRAS measurements conducted during the pulsed isothermal MB experiments of the methanol oxidation on Au(332) at 230 K (see also Fig. 3) under a continuous methanol flow, while applying pulses of atomic oxygen (200 s, delay 300 s). An atomic oxygen flux of 2.6 × 10−3 ML s−1 ≈ 0.4 × 1013 s−1 cm−2 is applied in (a) and (b), while in (c) and (d) a flux of 0.6 × 10−3 ML s−1 ≈ 0.08 × 1013 s−1 cm−2 was used. The methanol flux applied in (a) and (c) was 4.3 × 1013 s−1 cm−2 (p(MeOH) = 1.6 × 10−7 mbar), while in (b) and (d) a methanol flux of 52.7 × 1013 s−1 cm−2 (p(MeOH) = 19.6 × 10−7 mbar) was used. IRAS measurements (duration approx. 3 min) were conducted during the oxygen pulses (blue, start approx. 5–10 s after the beginning of oxygen pulse) and also in the delay times (red, start after approx. 10–20 s after the end of oxygen pulse, when the methyl formate formation rate was roughly decreased to background level) across the pulse sequence (from top to bottom). | ||
In the measurements with a high methanol flux, additional broad signals at around 2948 cm−1, 2918 cm−1, and 2816 cm−1 and a sharper one at around 1015 cm−1 are observed during the oxygen pulse, while being absent in the delay times. The intensity of these species is somewhat lower for the experiment with a low oxygen flux (hiMloO, Fig. 4d) as compared to that in the hiMhiO measurement (Fig. 4b). These results point to the presence of methoxy species, which is corroborated by the good agreement with the value reported for the νC–O mode of methoxy in EELS measurements on Cu(110) with a signal at around 1015 cm−1.33 The signal of the νC–O mode is, however, red-shifted compared to previous HREELS results for Au(111) displaying at 160 K a signal at 1060 cm−1.15 Yet, it should be noted that these HREELS measurements exhibit a signal at 1020 cm−1 for a surface temperature of 225 K which is in fair agreement with the position observed here for Au(332). The signals at around 2948 cm−1, 2918 cm−1 and 2816 cm−1 can be assigned to the C–H stretching modes of methoxy. While the C–H stretching signals differ in shape and are red-shifted as compared to previous reports for HREEL spectra on Au(111),1513C-methanol adsorbed at low temperature on clean Au(332) yielded IRAS signals in the C–H stretching region which are similar in shape, but are shifted (Fig. S1†). A signal shift is expected (at least) due to the use of a different isotopologue of methanol. The spectral shape of the C–H stretching region, however, is expected to be similar for methanol and methoxy supporting the assignment to methoxy. Across the pulse sequence, the intensity of these methoxy signals during the oxygen pulse decreases for the hiMhiO measurement, while the signal intensity of formate related lines increases (Fig. 4b) which is consistent with the poisoning of adsorption sites for methoxy by formate species. For the hiMloO experiment (Fig. 4d), where no clear formate accumulation is detected, in contrast, the intensity of the methoxy signals appears to even increase at the beginning of the pulse sequence, exhibiting only a slight intensity decrease towards the end of the pulse sequence.
Thus, in agreement with expectations, in situ IRAS shows a lower accumulation of formate species for lower oxygen and higher methanol fluxes. Yet, the relative surface deactivation for methyl formate formation does not differ accordingly, especially for the measurements with a low oxygen flux: the loMloO measurement displays only a slight surface deactivation, despite significant formate accumulation (Fig. 3c and 4c), while the hiMloO measurement exhibits a pronounced surface deactivation for methyl formate, even though no clear evidence for formate accumulation is detected (Fig. 3d and 4d). Thus, formate (alone) cannot account for the loss in reactivity towards partial oxidation to methyl formate. Instead, an additional mechanism appears to contribute to the observed surface deactivation for methyl formate which is not clearly detectable by IRAS measurements. Please note that the (formally) abstracted hydrogen atoms were previously reported to predominantly react into water or methanol15 both desorbing rapidly from Au(332) at 230 K. Hydrogen was reported for TPR experiments to desorb in trace amounts in the oxidation of methanol on Au(110) at 250 K,16i.e. above the temperature for associative hydrogen desorption observed at 215 K.34 Thus, also small amounts of potentially formed hydrogen should desorb at 230 K and are not expected to cause surface deactivation. Moreover, it should be emphasized that accumulation of oxygen on the surface is not the origin of this additional deactivation: (i) accumulating residual oxygen was connected to formate formation25 which is not clearly detected for the hiMloO measurement (Fig. 3d and 4d). From the loMloO measurement (Fig. 3c and 4c), it can be estimated that less than 0.25 MLs of residual oxygen are sufficient to clearly observe the formation of formate species (after the third pulse). The (maximum) amount of oxygen which may accumulate on the surface was estimated based on the absolute methyl formate formation rate and the applied oxygen flux, neglecting any oxygen consumption due to formaldehyde desorption or CO2 formation. For the hiMloO experiment (Fig. 3d and 4d), a maximum of 0.3 ML oxygen at the end of the measurement may have accumulated, potentially sufficient for formate formation which is however not detected, suggesting the oxygen accumulation to be negligible under these conditions. (ii) For the loMhiO measurement where formate formation in the delay times attests to the presence of residual oxygen, the transient pulse shape does not show decreasing methyl formate formation rates across the pulse duration, suggesting no significant deactivating effect of accumulating oxygen on the methyl formate formation under these conditions. (iii) The additional deactivation is clearly observed for the hiMloO measurement corresponding to the highest applied methanol excess in the gas phase in this series, suggesting a sufficiently high methanol surface concentration for a fast consumption of rather low amounts of oxygen (an upper bound of 0.04–0.07 MLs of unreacted oxygen per pulse can be estimated based on the methyl formate formation).
To further understand the surface deactivation mechanisms, the relative reduction of the methyl formate formation rate (with respect to the initial rate) is displayed as a function of the oxygen and the methanol exposure (after the first oxygen pulse) in Fig. 5a and b, respectively. For the (three) experiments in which significant formate accumulation on the surface is observed (Fig. 4a–c), the relative decrease of the rate exhibits a similar correlation with the oxygen exposure, further validating the role of formate in the deactivation under these conditions. In contrast, the hiMloO measurement (green symbols, Fig. 5a), for which essentially no formate was detected, exhibits a significantly faster deactivation, indicating the contribution of other deactivation mechanism(s). Additional indication for the importance of an additional mechanism can be found comparing the experiments with a high oxygen flux (red and black symbols in Fig. 5a): the relative rate decrease as a function of the oxygen exposure is the same, but the amount of formate observed by IRAS is significantly different (see Fig. 4a and b) which is inconsistent with formate being the only cause for the observed deactivation.
 | ||
| Fig. 5 Methyl formate rate decrease ΔR relative to the initially observed rate at the beginning of the pulse sequence for the different pulsed isothermal MB experiments conducted at 230 K on Au(332) (see also Fig. 3 and 4) as a function of (a) the oxygen exposure and (b) the methanol exposure (after the first oxygen pulse): low methanol and high oxygen flux (black squares, Fig. 3a), high methanol and high oxygen flux (red circles, Fig. 3b), low methanol and low oxygen flux (blue triangles, Fig. 3c), and high methanol and low oxygen flux (green triangles, Fig. 3d). The connecting line is a guide to the eye. | ||
Considering next the relative rate reduction as a function of the methanol exposure (Fig. 5b), the correlation varies significantly between the different experiments. For the loMhiO experiment, with the lowest applied methanol to oxygen flux ratio (black symbols, Fig. 5b), the relative rate reduction is steeper than those for the other measurements attributed to the highest formate accumulation. In contrast, for the highest methanol to oxygen flux ratio (hiMloO) where no clear formate accumulation was detected, a kind of bimodal behavior is observed, i.e. the initially steep decrease flattens towards the end of the pulse sequence (green symbols, Fig. 5b). The initial steep decrease is similar to that observed for the loMloO measurement (blue symbols, Fig. 5b), while the flatter slope is similar to that observed for the hiMhiO measurement (red symbols, Fig. 5b). While the slightly steeper slope for the loMloO measurement (blue symbols, Fig. 5b), as compared to the initial slope for the highest methanol to oxygen flux ratio (hiMloO, green symbols, Fig. 5b), may be (partially) attributed to formate accumulation blocking active surface sites, the rather shallow initial relative rate decrease for hiMloO (red symbols, Fig. 5b), where also significant formate signals are detected, cannot be explained in a similar manner, but points to the importance of a different deactivation mechanism under oxygen-deficient conditions which causes an even faster, methanol dependent deactivation than the formate accumulation under the investigated conditions.
Additional pulsed isothermal MB experiments (with a different methanol batch) were conducted to test this hypothesis: extending the delay time between the oxygen pulses and thus, increasing the methanol exposure, while keeping the methanol to oxygen ratio during the oxygen pulse constant, resulted in a stronger methyl formate rate reduction for a constant oxygen exposure (Fig. S2†). In addition, prolonging the methanol exposure before the first oxygen pulse resulted in an even reduced initial rate (Fig. S3†). Both results are consistent with the (proposed) blocking of surface sites for methyl formate formation by species related to the exposure to methanol. This deactivation pathway is further justified by the rate decrease during each oxygen pulse, as expected for a decreasing number of available surface sites reactive for methyl formate formation. With this batch of methanol, in situ IRAS across the pulse sequence evidenced the accumulation of species containing C–H bonds; however, there is a lack of clear signals attributable to CO modes (Fig. S2†). These species block the surface for CO adsorption (Fig. S4†) and were found to be oxidizable by atomic oxygen at 230 K, as previously shown for formate species.25 Furthermore, a short annealing i. vac. to 310 K and 450 K results in the reduction and even the disappearance of IRAS signals that characterize these species restoring the ability of the surface to adsorb CO and re-establishing the surface activity for methyl formate formation (Fig. S4 and S5†). These results suggest the C–H bond containing species to be molecular species bound moderately strongly on the Au(332) surface, presumably an impurity present in the methanol. It should be noted that high purity methanol was used also in these experiments which did not display, however, clear evidence for impurities in the QMS measurements implying that even very small concentrations may be detrimental to the surface reactivity towards methyl formate. For the measurements with a high methanol flux (Fig. 3 and 4), a similar effect may have caused the observed deactivation as indicated by a comparable correlation to a high methanol flux and rather oxygen-deficient conditions as well as by decreasing methyl formate formation rates across the pulse duration for methanol-rich conditions. Presumably, a different type of species accumulates which may escape IRAS detection due to very low concentrations or the metal surface selection rule preventing detection of vibrational modes parallel to the surface. Due to the lack of IRAS signals, it is unfortunately impossible to distinguish which of the trace impurities in the methanol is causing the deactivation. The more pronounced deactivation observed at low oxygen flux indicates that these species can possibly be oxidized by atomic oxygen at 230 K on the Au(332) surface, as was shown for the contaminants having an IR spectrum discussed above.
The selectivity towards methyl formate with respect to the number of supplied oxygen atoms is given in Fig. 6a as a function of the methanol to oxygen flux ratio. Significant fragmentation in the QMS of methanol and methyl formate hampers quantification of formaldehyde desorption which is expected to compete effectively with methyl formate formation under the applied single collision conditions. Similarly, the quantification of the CO2 formation rate resulting from the total oxidation of methanol on Au(332) is not possible because of background reactions of the chamber also exposed to atomic oxygen. Please note that methyl formate is exclusively formed on the Au surface. Thus, the selectivity towards methyl formate S(MF) is based on quantitative calibrations of the atomic oxygen flux f(O) and the methyl formate flux f(MF) based on the QMS signal (m/z = 60) considering that two oxygen atoms are required for methyl formate formation from methanol:
 | (1) |
 | ||
| Fig. 6 Selectivity to methyl formate S(MF) with respect to the number of supplied oxygen atoms for the pulsed isothermal MB experiments of the methanol oxidation on Au(332) at 230 K shown in Fig. 3 displayed as a function of (a) the methanol to atomic oxygen flux ratio f(CH3OH):f(O) and (b) the relative IRAS signal intensity as indicated by the amplitude of the formate band at 1332 cm−1 at the end of the pulse sequence relative to the maximum observed in this series. The initial selectivity (pulse 1, solid symbols) and the selectivity at the end of the pulse sequence (pulse 5, open symbols) are shown. | ||
A discussion of the observed selectivity in a simplified kinetic model assuming low coverage, constant availability of reactive sites, etc. is hampered by the complexity of the reaction network, e.g. the presence of deactivation processes discussed above. The effect of the latter is readily seen for the experiments done at low oxygen flux, for which a high methanol pressure (hiMloO) exhibits a significantly higher initial selectivity than that observed for the loMloO experiment, while the selectivity is significantly lower and more importantly similar for both methanol pressures at the end of the pulse series.
Based on the proposed reaction mechanism, a high methanol to oxygen ratio on the surface is expected to be beneficial for methyl formate formation, while the lowering of this ratio should favor overoxidation.2,15,18–21 In addition, the competition of formaldehyde desorption with subsequent coupling to methyl formate is expected to lower the selectivity towards methyl formate for lower surface concentrations, in specific also for lower surface concentrations of methoxy, in the case of single scattering conditions. While these considerations allow reconciling the changes in the initial selectivity found for the two pairs of experiments using high and low oxygen fluxes, respectively, the higher selectivity towards methyl formate found for a methanol to oxygen ratio of 55 (loMloO) as compared to the hiMhiO experiment with a ratio of 130 cannot be understood based on these simplified considerations. The IRAS results presented above clearly show that the hiOhiM conditions lead to a significant transient concentration of methoxy on the surface as well as a sizeable amount of unreacted oxygen at the end of the pulse which gives rise to the formation of formate species in the delay periods.25 In contrast to that, the loMloO IRAS experiments show no indication for a significant transient methoxy concentration and a considerably reduced formation of formate. Hence, the transient concentration of ‘excess’ activated oxygen, i.e. oxygen which does not contribute to methyl formate formation, but tends to form AuOx phases and is known to shift the selectivity towards overoxidation products, has to be considered as one factor determining the selectivity of the surface. This interpretation is further corroborated by the fact that the selectivity drops monotonously with increasing amounts of excess oxygen atoms at the end of the pulse as estimated from the relative IR intensity of formate (Fig. 6b). It is important to note that overoxidation is rather efficient for the hiMhiO experiment even though there is a significant transient concentration of methoxy, whose surface concentration is critical for methyl formate selectivity as it needs to react with transiently formed formaldehyde before the latter becomes overoxidized or desorbs. This implies that the methanol supply from the gas phase is not sufficient to react with the oxygen atoms deposited on the surface predominantly to methyl formate, corroborating the expectation that the ratio of methanol to oxygen on the surface is significantly smaller than expected based on the impinging fluxes. The blocking of surface sites by oxygen as well as methoxy found for the hiMhiO measurement also allows explaining the less pronounced increase of the initial rates for high oxygen and high methanol fluxes as compared to the low fluxes (Fig. 3).
Given the significant role of methoxy species on the reaction network, it is interesting to discuss the intensity of the methoxy species observed for the experiments with high methanol flux in more detail. Based on the previously suggested reaction mechanism15–17 (see also Fig. 2), the steady state concentration of methoxy [H3CO]stst should be obtained by equating the rate of its formation (reaction of methanol with activated oxygen) to the rate(s) of its consumption (reactions to formaldehyde or methyl formate):
 | (2) |
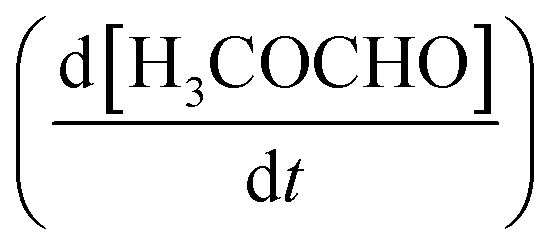 is proportional to the surface concentration of methoxy:
is proportional to the surface concentration of methoxy: | (3) |
For the hiMloO experiment, an increase of the methoxy concentration is observed if comparing the first and the second pulse. This can be understood along the same lines as the discussion above, as this increase in methoxy intensity is associated with a significant drop in methyl formate selectivity (Fig. 5b, green symbols), and hence a reduction in one of the depletion channels for methoxy (eqn (2)). However, this reduction in selectivity between the first two pulses is not associated with significant site blocking for methoxy. In contrast, the methanol related deactivation dominates under oxygen-deficient conditions. This observation can be understood assuming a preferential blocking of a small number of sites which are, however, highly reactive for methyl formate formation. Please note that under these conditions, a fraction of deactivating species clearly below the per mill range is sufficient for blocking of a small number of sites. While the exact nature of these highly reactive sites towards methyl formate formation cannot be determined from these measurements, viable candidates for minority sites on the stepped Au(332) surface which bind the methanol related deactivating species strongly are low-coordinated sites, such as steps or kinks (present due to a miscut with respect to the ideal (332) orientation28).
As compared to np-Au catalysts, the methyl formate formation decreases in the isothermal MB experiments on Au(332) already at rather low temperatures, where methanol conversion with molecular oxygen on np-Au is still small, but increases significantly for higher temperatures.2,3 Similar to the enhanced selectivity on np-Au, this can be attributed to effective re-adsorption and subsequent coupling to methyl formate of desorbing formaldehyde (and methanol) for long contact times in np-Au catalysts11 as compared to the single collision conditions applied in the MB measurements. At lower temperatures, the isothermal MB experiments show under all applied conditions a deactivation of the Au(332) surface for methyl formate formation. The deactivation due to formate accumulation observed for oxygen-rich conditions is expected to be less relevant for np-Au studies with rather low oxygen concentrations under typically applied conditions. However, for increased oxygen or Ag contents, this channel may become more important resulting in unwanted overoxidation products, such as CO2, for higher temperatures.2,11–13 Under oxygen-deficient conditions, a different deactivation related to methanol or an impurity in methanol was found to be important on Au(332). While surface deactivation by impurities may appear to be a trivial issue, this methanol related deactivation is very effective at low temperatures under oxygen-deficient conditions, thereby blocking the active sites on the Au(332) surface for methyl formate formation, even for high purity methanol. Thus, in np-Au studies with molecular oxygen where the surface concentration of activated oxygen is typically rather low,11,13 a similar mechanism may be relevant. Specifically, it may be important for achieving steady state conditions at low temperatures. So far, steady state reactivity in the methanol oxidation to methyl formate on np-Au has been reported for temperatures ≥20 °C,2 while oxygen activation on np-Au catalysts proceeds already at temperatures significantly below 0 °C as evidenced by CO oxidation.4,39 Moreover, carbonaceous deposits were recently reported to be present on np-Au catalysts under non-steady state conditions with decreasing amounts as the temperature increases to 323 K and further to 423 K, which is the steady-state reaction temperature applied in the np-Au study.14 While the authors suggested the carbonaceous deposits to be adsorbed on residual Ag in the np-Au catalyst, our results indicate that even Au sites may be blocked at low temperatures after high exposures to methanol, even when high purity methanol is used.
Conclusion
The influence of surface temperature and variation of the flux on the (partial) methanol oxidation over the stepped Au(332) surface was investigated by pulsed isothermal molecular beam experiments. In these measurements, atomic oxygen provided by a thermal cracker was pulsed, while methanol was provided continuously, at an excess in the gas phase.With increasing surface temperature, the methyl formate formation rate exhibits a maximum at 250 K under the applied (single collision) conditions, as the desorption of methanol and formaldehyde limits the coupling reaction to methyl formate. The surface deactivation observed across the pulse sequence is not lifted by an increased surface temperature in the investigated temperature range, but even slightly increases.
Variation of the methanol and oxygen fluxes demonstrated a reduced surface deactivation across the pulse sequence, when both the methanol flux and the atomic oxygen flux were chosen to be rather small. For oxygen-rich conditions, i.e. low methanol to oxygen flux ratios, in situ IRAS measurements evidence the increased formation of formate species blocking the surface for methoxy and methyl formate formation. At high methanol fluxes and under rather oxygen-deficient conditions, an additional deactivation mechanism is effective in reducing methyl formate formation at low surface temperatures which may also be important for achieving steady state conditions on np-Au catalysts at low temperatures. In contrast to deactivation by formate species, the results indicate that this methanol related deactivation preferentially blocks a small number of highly reactive sites for methyl formate formation, while methoxy formation is (initially) not reduced by this deactivation.
In agreement with expectations, the selectivity to methyl formate increased for higher methanol to oxygen ratios. Deviations from this trend were observed, however, for conditions where the number of available surface sites is expected to be limited. A maximum selectivity to methyl formate of 65% was achieved for the highest applied methanol to atomic oxygen flux ratio of approx. 660.
These results demonstrate the importance of deviations from the idealized methanol oxidation mechanism on gold arising from different deactivation pathways and thus, provide additional insights into the processes under isothermal conditions at low temperatures.
Author contributions
This manuscript was written through contributions of all the authors. All the authors have given approval to the final version of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the German Research Foundation (DFG) within the framework of research unit 2231 “NAGOCAT” project no. RI 1025/3-1(2). We thank S. Eltayeb and K. Tang for technical support. C. D. F. thanks the International Max-Planck Research School “Functional Interfaces in Physics and Chemistry” for support and the IMPRS for Elementary Processes in Physical Chemistry. We would like to acknowledge the assistance of the department SupraMS of the Core Facility BioSupraMol supported by the DFG.References
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Novel Gold Catalysts for the Oxidation of Carbon-Monoxide at a Temperature Far Below 0 °C, Chem. Lett., 1987, 405–408 CrossRef CAS.
- A. Wittstock, V. Zielasek, J. Biener, C. M. Friend and M. Bäumer, Nanoporous Gold Catalysts for Selective Gas-Phase Oxidative Coupling of Methanol at Low Temperature, Science, 2010, 327, 319–322 CrossRef CAS PubMed.
- A. Wittstock, J. Biener and M. Bäumer, Nanoporous Gold: A New Material for Catalytic and Sensor Applications, Phys. Chem. Chem. Phys., 2010, 12, 12919–12930 RSC.
- C. X. Xu, J. X. Su, X. H. Xu, P. P. Liu, H. J. Zhao, F. Tian and Y. Ding, Low Temperature CO Oxidation over Unsupported Nanoporous Gold, J. Am. Chem. Soc., 2007, 129, 42–43 CrossRef CAS PubMed.
- H. M. Yin, C. Q. Zhou, C. X. Xu, P. P. Liu, X. H. Xu and Y. Ding, Aerobic Oxidation of D-Glucose on Support-Free Nanoporous Gold, J. Phys. Chem. C, 2008, 112, 9673–9678 CrossRef CAS.
- R. Meyer, C. Lemire, S. K. Shaikhutdinov and H.-J. Freund, Surface Chemistry of Catalysis by Gold, Gold Bull., 2004, 37, 72–124 CrossRef CAS.
- G. C. Bond, Gold: A Relatively New Catalyst, Catal. Today, 2002, 72, 5–9 CrossRef CAS.
- J. Erlebacher, M. J. Aziz, A. Karma, N. Dimitrov and K. Sieradzki, Evolution of Nanoporosity in Dealloying, Nature, 2001, 410, 450–453 CrossRef CAS PubMed.
- L. C. Wang, Y. Zhong, D. Widmann, J. Weissmüller and R. J. Behm, On the Role of Residual Ag in Nanoporous Au Catalysts for CO Oxidation: A Combined Microreactor and Tap Reactor Study, ChemCatChem, 2012, 4, 251–259 CrossRef CAS.
- L. C. Wang, Y. Zhong, H. J. Jin, D. Widmann, J. Weissmüller and R. J. Behm, Catalytic Activity of Nanostructured Au: Scale Effects Versus Bimetallic/Bifunctional Effects in Low-Temperature CO Oxidation on Nanoporous Au, Beilstein J. Nanotechnol., 2013, 4, 111–128 CrossRef PubMed.
- L. C. Wang, M. L. Personick, S. Karakalos, R. Fushimi, C. M. Friend and R. J. Madix, Active Sites for Methanol Partial Oxidation on Nanoporous Gold Catalysts, J. Catal., 2016, 344, 778–783 CrossRef CAS.
- B. Zugic, L. C. Wang, C. Heine, D. N. Zakharov, B. A. J. Lechner, E. A. Stach, J. Biener, M. Salmeron, R. J. Madix and C. M. Friend, Dynamic Restructuring Drives Catalytic Activity on Nanoporous Gold-Silver Alloy Catalysts, Nat. Mater., 2017, 16, 558–564 CrossRef CAS PubMed.
- L. C. Wang, C. M. Friend, R. Fushimi and R. J. Madix, Active Site Densities, Oxygen Activation and Adsorbed Reactive Oxygen in Alcohol Activation on NpAu Catalysts, Faraday Discuss., 2016, 188, 57–67 RSC.
- B. Zugic, et al., Evolution of Steady-State Material Properties During Catalysis: Oxidative Coupling of Methanol over Nanoporous Ag0.03Au0.97, J. Catal., 2019, 380, 366–374 CrossRef CAS.
- B. J. Xu, X. Y. Liu, J. Haubrich, R. J. Madix and C. M. Friend, Selectivity Control in Gold-Mediated Esterification of Methanol, Angew. Chem., Int. Ed., 2009, 48, 4206–4209 CrossRef CAS PubMed.
- D. A. Outka and R. J. Madix, Bronsted Basicity of Atomic Oxygen on the Au(110) Surface -Reactions with Methanol, Acetylene, Water and Ethylene, J. Am. Chem. Soc., 1987, 109, 1708–1714 CrossRef CAS.
- B. J. Xu, J. Haubrich, T. A. Baker, E. Kaxiras and C. M. Friend, Theoretical Study of O-Assisted Selective Coupling of Methanol on Au(111), J. Phys. Chem. C, 2011, 115, 3703–3708 CrossRef CAS.
- B. J. Xu, C. G. F. Siler, R. J. Madix and C. M. Friend, Ag/Au Mixed Sites Promote Oxidative Coupling of Methanol on the Alloy Surface, Chem. – Eur. J., 2014, 20, 4646–4652 CrossRef CAS PubMed.
- F. Hiebel, S. Karakalos, Y. F. Xu, C. M. Friend and R. J. Madix, Structural Differentiation of the Reactivity of Alcohols with Active Oxygen on Au(110), Top. Catal., 2018, 61, 299–307 CrossRef CAS.
- C. Reece, M. Luneau and R. J. Madix, Dissecting the Performance of Nanoporous Gold Catalysts for Oxygen-Assisted Coupling of Methanol with Fundamental Mechanistic and Kinetic Information, ACS Catal., 2019, 9, 4477–4487 CrossRef CAS.
- C. Reece, M. Luneau, C. M. Friend and R. J. Madix, Predicting a Sharp Decline in Selectivity for Catalytic Esterification of Alcohols from Van Der Waals Interactions, Angew. Chem., Int. Ed., 2020, 59, 10864–10867 CrossRef CAS PubMed.
- W. L. Yim, et al., Universal Phenomena of CO Adsorption on Gold Surfaces with Low-Coordinated Sites, J. Phys. Chem. C, 2007, 111, 445–451 CrossRef CAS.
- G. Tomaschun, W. Dononelli, Y. Li, M. Bäumer, T. Klüner and L. V. Moskaleva, Methanol Oxidation on the Au(310) Surface: A Theoretical Study, J. Catal., 2018, 364, 216–227 CrossRef CAS.
- T. Fujita, et al., Atomic Origins of the High Catalytic Activity of Nanoporous Gold, Nat. Mater., 2012, 11, 775–780 CrossRef CAS PubMed.
- C. D. Feldt, T. Gimm, R. Moreira, W. Riedel and T. Risse, Methanol Oxidation on Au(332): An Isothermal Pulsed Molecular Beam Study, Phys. Chem. Chem. Phys., 2021, 23, 21599–21605 RSC.
- R. Moreira, Setup of a Molecular Beam Apparatus to Study the Reactivity of Single Crystal Surfaces and Its Application to CO Oxidation on Au(332), PhD thesis, Freie Universtität Berlin, Berlin, 2018 Search PubMed.
- J. Libuda, I. Meusel, J. Hartmann and H. J. Freund, A Molecular Beam/Surface Spectroscopy Apparatus for the Study of Reactions on Complex Model Catalysts, Rev. Sci. Instrum., 2000, 71, 4395–4408 CrossRef CAS.
- M. J. Prieto, E. A. Carbonio, R. Landers and A. de Siervo, Structural and Electronic Characterization of Co Nanostructures on Au(332), Surf. Sci., 2013, 617, 87–93 CrossRef CAS.
- K. Mortensen, C. Klink, F. Jensen, F. Besenbacher and I. Stensgaard, Adsorption Position of Oxygen on the Pt(111) Surface, Surf. Sci., 1989, 220, L701–L708 CrossRef CAS.
- P. R. Norton, J. A. Davies and T. E. Jackman, Absolute Coverages of CO and O on Pt(111) - Comparison of Saturation CO Coverages on Pt(100), (110) and (111) Surfaces, Surf. Sci., 1982, 122, L593–L600 CrossRef CAS.
- J. Gong, D. W. Flaherty, R. A. Ojifinni, J. M. White and C. B. Mullins, Surface Chemistry of Methanol on Clean and Atomic Oxygen Pre-Covered Au(111), J. Phys. Chem. C, 2008, 112, 5501–5509 CrossRef CAS.
- S. D. Senanayake, D. Stacchiola, P. Liu, C. B. Mullins, J. Hrbek and J. A. Rodriguez, Interaction of CO with OH on Au(111): HCOO, CO3, and HOCO as Key Intermediates in the Water-Gas Shift Reaction, J. Phys. Chem. C, 2009, 113, 19536–19544 CrossRef CAS.
- B. A. Sexton, A. E. Hughes and N. R. Avery, Surface Intermediates in the Reaction of Methanol, Formaldehyde and Methyl Formate on Cu(110), Appl. Surf. Sci., 1985, 22-3, 404–414 Search PubMed.
- A. G. Sault, R. J. Madix and C. T. Campbell, Adsorption of Oxygen and Hydrogen on Au(110)-(1x2), Surf. Sci., 1986, 169, 347–356 CrossRef CAS.
- D. A. Outka and R. J. Madix, Acid-Base and Nucleophilic Chemistry of Atomic Oxygen on the Au(110) Surface - Reactions with Formic Acid and Formaldehyde, Surf. Sci., 1987, 179, 361–376 CrossRef CAS.
- B. K. Min and C. M. Friend, Heterogeneous Gold-Based Catalysis for Green Chemistry: Low-Temperature Co Oxidation and Propene Oxidation, Chem. Rev., 2007, 107, 2709–2724 CrossRef CAS PubMed.
- M. L. Personick, B. Zugic, M. M. Biener, J. Biener, R. J. Madix and C. M. Friend, Ozone-Activated Nanoporous Gold: A Stable and Storable Material for Catalytic Oxidation, ACS Catal., 2015, 5, 4237–4241 CrossRef CAS.
- Y. Li, W. Dononelli, R. Moreira, T. Risse, M. Bäumer, T. Klüner and L. V. Moskaleva, Oxygen-Driven Surface Evolution of Nanoporous Gold: Insights from Ab Initio Molecular Dynamics and Auger Electron Spectroscopy, J. Phys. Chem. C, 2018, 122, 5349–5357 CrossRef CAS.
- V. Zielasek, B. Jürgens, C. Schulz, J. Biener, M. M. Biener, A. V. Hamza and M. Bäumer, Gold Catalysts: Nanoporous Gold Foams, Angew. Chem., Int. Ed., 2006, 45, 8241–8244 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: IRAS measurements of 13C methanol on Au(332). Pulsed isothermal MB experiments with in situ IRAS measurements on methanol oxidation on Au(332) with 13C methanol for various delay times between oxygen pulses as well as extended methanol exposure prior to the first oxygen pulse. CO IRAS measurements of the Au(332) surface after the pulsed isothermal MB experiment and subsequent annealing. Pulsed isothermal MB experiments with in situ IRAS measurements on methanol oxidation on Au(332) for the annealed surface after a prior pulsed isothermal MB experiment. See DOI: 10.1039/d1cy02034j |
| This journal is © The Royal Society of Chemistry 2022 |