
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Auto-tandem catalytic reductive hydroformylation with continuous multiphase catalyst recycling†
Sebastian
Püschel
 a,
Enes
Hammami
a,
Thorsten
Rösler
a,
Kira R.
Ehmann
a,
Andreas J.
Vorholt
*a and
Walter
Leitner
ab
a,
Enes
Hammami
a,
Thorsten
Rösler
a,
Kira R.
Ehmann
a,
Andreas J.
Vorholt
*a and
Walter
Leitner
ab
aMolecular Catalysis, Max Planck Institute for Chemical Energy Conversion, Stiftstr. 34-36, 45470 Mülheim an der Ruhr, Germany. E-mail: andreas-j.vorholt@cec.mpg.de
bInstitute for Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany
First published on 22nd November 2021
Abstract
The production of alcohols from olefin-enriched Fischer–Tropsch products represents a promising route for CO2-neutral bio-synthetic fuels. Tandem-catalytic systems as alternatives for the conventional two-step production of long-chain alcohols are attractive in terms of energy and resource efficiency. Herein, we present the first rhodium-based catalytic system capable of direct conversion of olefins to alcohols in a biphasic liquid/liquid system. After optimizing the reaction conditions for the biphasic operation, an alcohol selectivity of up to 64% was achieved, while aldehydes and olefin isomers were observed as main by-products. By employing water-soluble alkanolamines, the catalyst is immobilized in a water phase and can be easily separated from the product containing an organic phase with a rhodium loss of as low as 0.1%. After investigation of various reaction parameters, a TON of 128 in batch operation was achieved. Furthermore, the developed catalyst recycling strategy was implemented in a continuously operated miniplant, reaching a TTON for alcohol production of 1236 after 50 hours.
Introduction
Biosynthetic fuels present a chance to reduce carbon dioxide emissions in the current fleet of vehicles without major changes in the current road transport infrastructure or vehicle engines.1–3 One pathway to synthetic fuels is the biogas to olefin-selective Fischer–Tropsch (FT) concept.4 By subsequent functionalization of the FT-derived olefins, tailor-made fuel properties are accessible. The hydroformylation/hydrogenation sequence to alcohols of FT olefins allows for further utilization of bio-derived syngas for the production of alcohols.5–7 Several alcohols, e.g. n-octanol, were proven to reduce emissions and to improve combustion characteristics by introducing oxygen into the fuel mix.2,8–10In industrial hydroformylation, alcohols are usually produced in multi-step processes via aldehyde intermediates.11 These aldehydes are usually isolated and utilized as platform chemicals to form a wide variety of products, e.g. amines, acetals or carboxylic acids as well as alcohols. However, in the context of fuels, the direct conversion from olefins to alcohols without an intermediate step can be beneficial in terms of resource efficiency.12,13 To achieve such a one-step synthesis, tandem catalysis is necessary. Several catalytic systems capable of so called “reductive hydroformylation” are known in the literature: orthogonal systems, using separate catalysts for either reaction step14,15 and assisted systems, where each step is carried out under different reaction conditions with the same or a trigger-modified catalyst.6
In terms of efficiency, auto-tandem catalysis is the most desirable option13 – since both reaction steps are catalysed under the same reaction conditions and only a single catalyst precursor is used. Auto-tandem catalytic reductive hydroformylation was first described for cobalt-based catalysts, which require harsh conditions and lead to rather poor selectivity.16,17 Rhodium is the most active metal in hydroformylation reactions; hence, most recent investigations focus on rhodium-based systems, typically in combination with tertiary alkylamines.18–25 In a recent publication from our group, the role and necessary properties of amines in reductive hydroformylation were characterized.26
However, the mentioned systems inherit the most common drawback of homogeneous catalysis: difficult recycling of the expensive rhodium catalyst. In industrial hydroformylation, the Ruhrchemie–Rhône-Poulenc process represents a milestone by introducing water-soluble phosphine ligands, thus enabling the recycling of the precious catalyst (Fig. 1) in a liquid/liquid multiphase system.27–29 In order to apply this type of catalyst recycling to a system containing tertiary alkylamines, water-soluble equivalents are necessary.
 | ||
| Fig. 1 Monophasic vs. liquid–liquid-biphasic reaction system. | ||
Various alkanolamines, predominantly used as surfactants or in gas purification,30 show high water solubility and therefore represent an interesting, comparably cheap and air-stable group of molecules to transfer this system into a biphasic process. Furthermore, a water-based system is not only ecologically desirable12 but water has also shown promoting effects in rhodium-catalysed reductive hydroformylation.6,31
In this manuscript, we present the first amine-based biphasic catalyst system capable of reductive hydroformylation with high selectivity towards alcohols. After transferring the reaction system into a liquid–liquid biphasic system, various influences on the phase behaviour and catalytic activity and stability in recycling were investigated. The feasibility of the catalyst recycling approach was proven in a continuous flow.
Results and discussion
The reaction network of the reductive hydroformylation reaction is depicted in Scheme 1. To mimic an FT product, a mixture of 1-octene and n-heptane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mass) was used as a model substrate. The substrates α-olefins can react with either linear 1a or branched 2a aldehydes by the addition of carbon monoxide and hydrogen to the C
:1 mass) was used as a model substrate. The substrates α-olefins can react with either linear 1a or branched 2a aldehydes by the addition of carbon monoxide and hydrogen to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. Internal olefins 3 can be formed by isomerization. Additionally, hydrogenation of the olefin takes place, leading to the already present corresponding paraffin 4. Hydroformylation of an internal olefin leads to further branched aldehydes 5a. All formed aldehydes can undergo hydrogenation to alcohols, linear (1b) and branched (2b + 5b). Since alcohols are produced from aldehydes formed in the hydroformylation step, the combined yield of all hydroformylation products (Yhyfo, 1a + 1b + 2a + 2b + 5a + 5b) is used to describe the first reaction step of the tandem-catalytic system. The second step is characterized by the alcohol yield (Yalcohols, 1b + 2b + 5b). Linear and branched alcohols represent the desired reaction products: primary alcohols as fuel additives. Hence, regioselectivity is of minor importance in this system.
C double bond. Internal olefins 3 can be formed by isomerization. Additionally, hydrogenation of the olefin takes place, leading to the already present corresponding paraffin 4. Hydroformylation of an internal olefin leads to further branched aldehydes 5a. All formed aldehydes can undergo hydrogenation to alcohols, linear (1b) and branched (2b + 5b). Since alcohols are produced from aldehydes formed in the hydroformylation step, the combined yield of all hydroformylation products (Yhyfo, 1a + 1b + 2a + 2b + 5a + 5b) is used to describe the first reaction step of the tandem-catalytic system. The second step is characterized by the alcohol yield (Yalcohols, 1b + 2b + 5b). Linear and branched alcohols represent the desired reaction products: primary alcohols as fuel additives. Hence, regioselectivity is of minor importance in this system.
 | ||
| Scheme 1 Hydroformylation and hydrogenation reaction network. | ||
In a previous contribution of our group, necessary properties of amines in reductive hydroformylation have been determined in a monophasic system, in which tertiary alkylamines (e.g. triethylamine) were used.
The largest influence was determined to be the basicity of the amine; only amines with a pKa value of 10 ± 1 led to the formation of alcohols.26 To achieve a green, water-based liquid–liquid biphasic system for catalyst recycling, water-soluble amines are necessary.
Fig. 2 shows the transfer between a mono- and biphasic system. By switching to water as the solvent and water-soluble alkanolamines, a liquid–liquid system can be achieved. Because of the high polarity of water and the use of alkanolamines, products and substrates are expected to separate in an organic phase, thus enabling simple catalyst recycling by phase separation.
 | ||
| Fig. 2 Transfer to a biphasic system by switching to water and polar amines. | ||
Batch experiments
Various alkanolamines show high water solubility, and examples and their respective pKa and logP (water/octanol partition coefficient) values are given in Chart 1. Because DMAE is a promising candidate (pKa value close to 10), it was chosen for the initial experiments. Starting with the adapted reaction conditions (Table 1) optimized for the monophasic system previously published by our group,26 only low hydroformylation rates and alcohol yields are observable (Fig. 3).
 | ||
| Chart 1 Variation of amine. Conditions: ccat = 0.5 mol% (substrate), T = 100 °C, tR = 1.5 h, p = 90 bar, CO/H2 = 1:2, φorg = 0.4, Vliq = 30 mL, water:amine = 1.5, n = 2000 min−1, catalyst precursor = [Rh(acac)(CO)2], Yhyfo = Yaldehydes + Yalcohols. | ||
| Parameter | Monophasic system26 | Adapted reaction conditions |
|---|---|---|
| Catalyst concentration | 0.5 mol% | 0.5 mol% |
| Amine | Diethylmethylamine | Dimethylaminoethanol (DMAE) |
| Amine:rhodium |
400:1 |
400:1 |
| Reaction volume | 3.4 mL | 30 mL |
| Solvent | Acetonitrile (2.4 mL) | Water (10.3 mL) |
| Temperature | 100 °C | 100 °C |
| Syngas pressure (CO:H2) |
30 bar (1:2) |
30 bar (1:2) |
| Reaction time | 1.5 h | 1.5 h |
 | ||
| Fig. 3 Variation of syngas pressure. Conditions: ccat = 0.5 mol% (substrate), Rh:amine = 1:400, T = 100 °C, tR = 1.5 h, CO/H2 = 1:2, φorg = 0.4, Vliq = 30 mL, water:amine = 1.5, n = 2000 min−1, catalyst precursor = [Rh(acac)(CO)2], amine = DMAE. | ||
The low activity in this water-based reaction system compared to our previous approach can be explained by the solubilities of the gaseous substrates. Under standard conditions, the solubility of hydrogen in water is one order of magnitude lower compared to acetonitrile.32 To achieve similar CO and H2 concentrations in the catalyst phase, the syngas pressure was increased (Fig. 3). As expected, higher pressures lead to increased hydroformylation and aldehyde hydrogenation rates.
At 90 bar, a yield of 57% for the alcohols was achieved (E), which is comparable to the results of the monophasic system. However, rhodium leaching is increased at higher syngas pressure.
Two potential pathways for increased catalyst leaching exist here: the formation of non-polar carbonyl complexes and increased amine leaching caused by higher alcohol fractions in the product, drawing rhodium to the organic phase. To investigate this effect further, experiments with a constant H2 partial pressure of 40 bar and varying CO partial pressures were conducted. Increased CO partial pressures are detrimental to catalyst immobilization, while H2 partial pressure predominantly influences aldehyde hydrogenation (Fig. 4). Furthermore, experiments carried out at 30 bar CO with different H2 pressures lead to different alcohol yields (H: 47% and C: 57%) but almost identical leaching (25 ppm) of the rhodium catalyst. In contrast, an experiment with 40 bar of CO partial pressure (I) showed yields in the same range (52%) but showed an increased rhodium leaching (34 ppm).
 | ||
| Fig. 4 Variation of CO partial pressure. Conditions: ccat = 0.5 mol% (substrate), Rh:amine = 1:400, T = 100 °C, tR = 1.5 h, φorg = 0.4, Vliq = 30 mL, water:amine = 1.5, n = 2000 min−1, catalyst precursor = [Rh(acac)(CO)2], amine = DMAE. | ||
This indicates that catalyst leaching is most likely caused by the formation of carbonyl species rather than induced by amine leaching due to high alcohol concentrations in the reaction product.
At CO partial pressures above 20 bar, nearly full conversion of the olefin is reached. Since the formation of isomers and especially the hydrogenation of olefins are undesired, varying the temperature may lead to improved selectivity.
Increasing the temperature predominantly accelerates the hydrogenation of olefins (Fig. 5). Since this is a “dead end” for the reaction, the formation of paraffins is undesired. Furthermore, increased temperatures are detrimental to both steps of reductive hydroformylation and form less aldehydes, and less hydrogenation of the intermediate occurs. However, at 80 °C, the hydrogenation of the olefin substrate is inhibited without a large influence on aldehyde hydrogenation, resulting in increased selectivity. Further reducing the temperature led to a significant decrease of the catalytic activity.
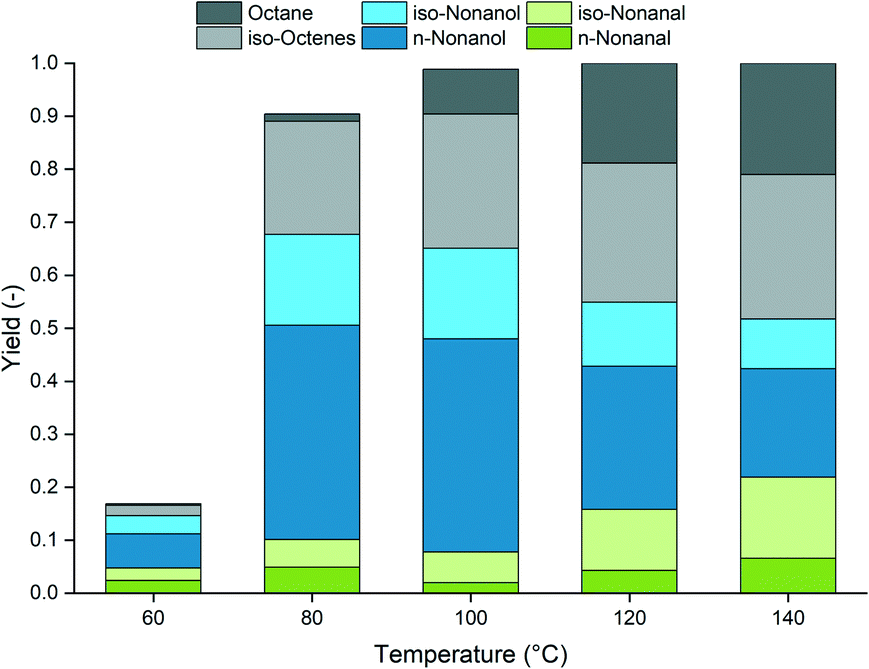 | ||
| Fig. 5 Variation of reaction temperature. Conditions: ccat = 0.5 mol% (substrate), Rh:amine = 1:400, tR = 1.5 h, p = 90 bar, CO/H2 = 1:2, φorg = 0.4, Vliq = 30 mL, water:amine = 1.5, n = 2000 min−1, catalyst precursor = [Rh(acac)(CO)2], amine = DMAE. | ||
After optimizing the most important reaction parameters, a variety of different amines with changing alkyl groups and a number of alcohol groups were investigated. Although modifications of the alkyl groups will predominantly influence the polarity and partition (logP, negative values represent the affinity to the aqueous phase) of the amine, the introduction of additional hydroxy groups increases the polarity but at the same time decreases the pKa value of the amine (Chart 1).
Given the high excess of amine with respect to rhodium of 1 to 400, the amine accounts for 26% of the total mass in the reaction. Hence, the amine influences not only the catalyst but also the phase behaviour of the reaction.
Hence, the mass ratio between water and amine in the aqueous phase was kept at a constant level of 1.5 to 1. While resulting in similar yields compared to DMAE, DEAE leads to significantly increased amine and rhodium leaching (Chart 1). This can be attributed to the less polar structure (logP value) compared to DMAE; the trend continues for DIPAE and DBAE. The loss of alcohol selectivity for DIPAE is probably caused by the steric hindrance introduced by the isopropyl groups.
Substitution of the ethanol group with a 1,2-propanediol moiety (DMAPD) partially inhibits the formation of alcohols while a similar hydroformylation yield is observable. Here, the presence of two hydroxy-groups counteracts the amine leaching into the organic product phase.
When replacing a methyl group with a phenyl group (MPhAE), the reaction leads to an almost quantitative aldehyde yield, and only traces of alcohols were detected. This and the linear-branched ratio of 1 lead to the assumption that the reaction was catalysed by rhodium carbonyl complexes instead of an amine-modified catalyst, as also indicated by high rhodium leaching.
As it would be expected for their basicity influence, amines with additional alcohol groups (MDEA pKa = 8.5, TEA pKa = 7.7) lead to decreased alcohol yields. Since DMAE combines high alcohol yields with low amine leaching, all following experiments were conducted using DMAE.
Additionally, the determined reaction conditions were applied to an actual FT product containing olefins and paraffins in the C5 to C10 range. Fig. 6 shows the successful conversion of all the olefins to their corresponding alcohols in comparable yields.
 | ||
| Fig. 6 FT mixture experiment. Conditions: ccat = 0.5 mol% (substrate, composition in the ESI†), Rh:amine = 1:400, T = 80 °C, tR = 1.5 h, p = 90 bar, CO/H2 = 1:2, φorg = 0.4, Vliq = 30 mL, water:amine = 1.5, n = 2000 min−1, catalyst = Rh(acac)(CO)2, amine = DMAE. C5 isomers could not be detected due to low concentration and volatility. | ||
The developed catalytic system is capable of converting an olefin–paraffin mixture with varying chain lengths to alcohols in a single conversion step, eliminating the intermediate purification steps via auto-tandem catalysis. Furthermore, the results are similar to the results obtained with the model substrate, hence proving the feasibility of the chosen approach.
Design of experiment
In a system where one main component is part of the catalytic system and acts as a solvent at the same time, cross-dependencies of various parameters are expected. A classical one-factor-at-a-time approach is not suitable if one part of the solvent system strongly interacts with the catalyst at the same time. Hence, a design of experiment (DoE) approach was chosen to investigate the influence of the phase fractions and the catalyst concentration. The parameters and investigated ranges are given in Table 2.| Parameter | Unit | Low limit | High limit |
|---|---|---|---|
| Organic phase | Volume fraction | 0.2 | 0.8 |
| Water | Mass fraction (aq. phase) | 0.15 | 0.7 |
| DMAE | Mass fraction (aq. phase) | 0.1 | 0.32 |
| Catalyst | Mol% (olefin) | 0.125% | 1% |
From the initial experiments and a simulation using Aspen Plus (details in the ESI†), it was found that a water–amine mass ratio of >1.5 has to be maintained to ensure biphasic behaviours. Therefore, this was applied as a secondary condition when designing the experiments. By implementing the three mixture components and the catalyst concentration, other parameters can be evaluated as well: water–amine ratio, rhodium–amine ratio and the catalyst concentration in the aqueous phase.
Fig. 7 shows the results obtained for the hydroformylation and aldehyde hydrogenation (alcohol) yields.
 | ||
| Fig. 7
Y
hyfo (top) and Yalcohols (bottom) over the water–amine ratio, organic phase volume fraction and catalyst concentration. Conditions: T = 80 °C, tR = 1.5 h, p = 90 bar, CO/H2 = 1:2, φorg = 0.4, Vliq = 30 mL, n = 2000 min−1, catalyst precursor = [Rh(acac)(CO)2], amine = DMAE, Yhyfo = Yaldehydes + Yalcohols. | ||
Although hydroformylation activity can be observed over a broad range of parameters, alcohols are only formed if a high catalyst concentration and a high amine concentration are combined. Furthermore, an interaction between rhodium and the amine is indicated by the inhibition of the hydroformylation as well as the hydrogenation activity when low catalyst loadings are paired with high amine concentrations.
Catalyst recycling
ICP-MS measurements were used to quantify the amount of catalyst leaching into the organic phase during all the DoE experiments. Fig. 8 shows that a water–amine ratio of 1.5 leads to successful catalyst immobilization for low rhodium concentrations. | ||
| Fig. 8 Rhodium leaching over the catalyst concentration and water–amine ratio. | ||
By increasing the water–amine ratio (low amine content in the aqueous phase), even low catalyst loadings show significant loss of rhodium into the product phase. High catalyst concentrations in the water-amine phase also lead to increased catalyst leaching. A water–amine mass ratio of 1.5 was found to be beneficial for the selectivity towards alcohols (Fig. 7) as well as catalyst immobilization (Fig. 8).
Meanwhile, the observed rhodium leaching does not correlate with the leaching of the stabilizing amine as observed when varying the amine (Fig. 9). The rather low leaching of DMAE (<6%) seems not to induce rhodium leaching. In contrast, high alcohol yields and the corresponding amine leaching occur at a water–amine ratio of 1.5, i.e. high amine concentrations where rhodium leaching is low.
 | ||
| Fig. 9 Amine and rhodium loss over the alcohol yield. | ||
After optimization of the most important reaction parameters, a batch recycling experiment was conducted. After the reaction, the organic product phase was withdrawn and a new substrate was added to the catalyst phase, which remained in the reactor.
As can be seen in Fig. 10, the catalyst recycling after 5 runs was successful; however, it was observed that the alcohol selectivity decreases after each run.
 | ||
| Fig. 10 Batch recycling experiment. Conditions: ccat = 0.5 mol% (substrate), Rh:amine = 1:400, T = 80 °C, tR = 1.5 h, p = 75 bar, CO/H2 = 1:2, φorg = 0.4, Vliq = 120 mL, water:amine = 1.51, n = 2000 min−1, catalyst precursor = Rh(acac)(CO)2, amine = DMAE. | ||
This can be attributed to amine leaching, since the water leaching values are lower because of its high polarity, and the ratio between water and amine increases with each recycling, which was proven to cause rhodium leaching and low alcohol selectivity (Fig. 8). Hence, replenishment of the amine is necessary to achieve stable catalyst recycling.
Continuous flow experiment
After identifying the important reaction parameters, the biphasic catalyst recycling was used in a pilot plant designed for liquid–liquid biphasic catalyst recycling33 (Fig. 11). The details of the setup are included in the ESI.†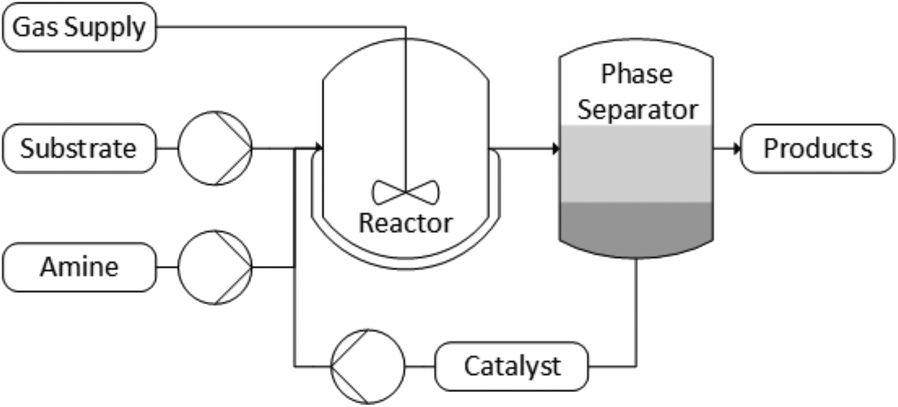 | ||
| Fig. 11 Simplified pilot plant diagram. | ||
For this proof-of-concept experiment, reaction conditions were chosen so that incomplete conversion and only partial alcohol selectivity are achieved. This allows precise observation of changes in catalytic activity or selectivity. Fig. 12 shows the successful application of the reaction system in a continuous flow. After the start-up procedure (first 6 hours), rather stable hydroformylation activity and alcohol yields were observed. After 22 hours of operation, the hydrogenation rate of aldehyde intermediates decreased while hydroformylation activity remained unchanged.
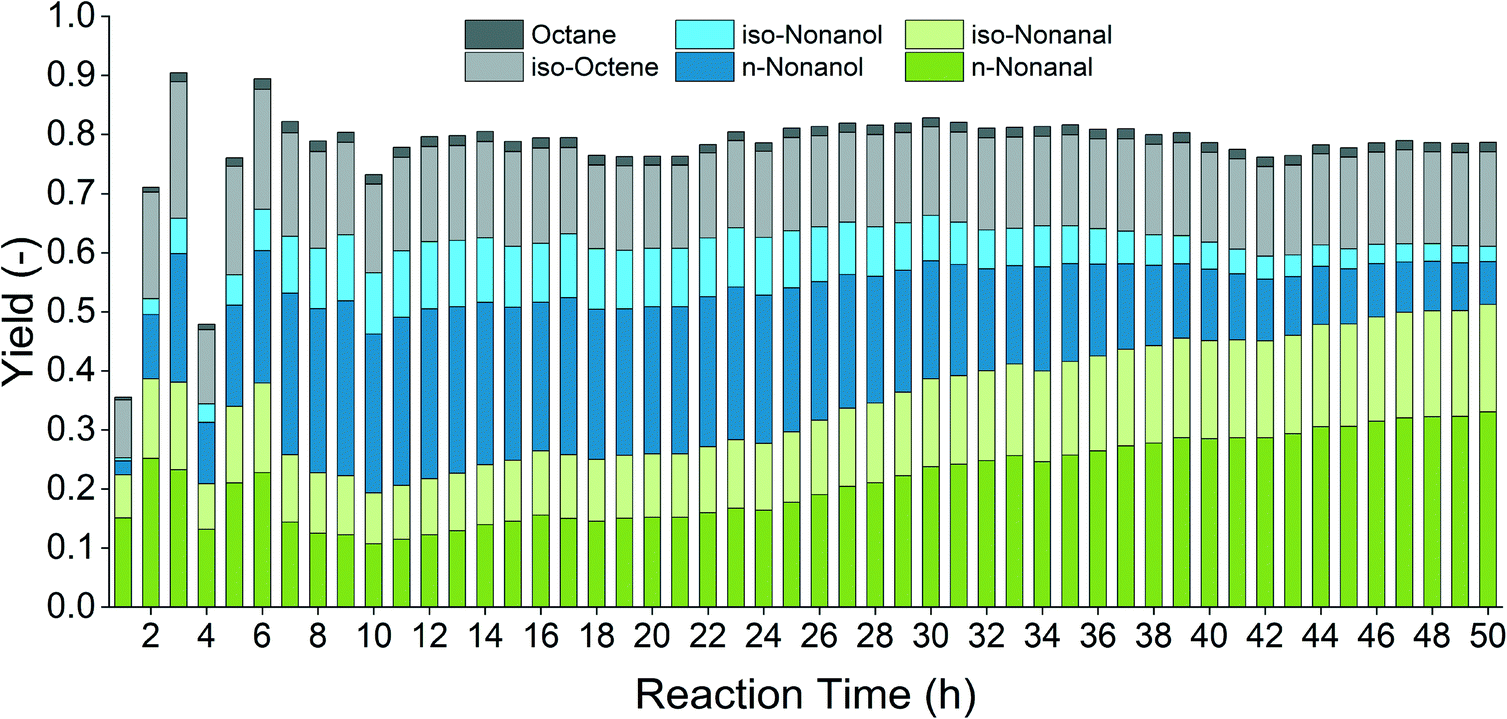 | ||
| Fig. 12 Proof-of-concept in a continuous flow. Conditions: ccat = 0.5 mol% (substrate), Rh:amine = 1:400, T = 80 °C, p = 90 bar, CO/H2 = 1:2, φorg = 0.4, Vliq = 120 mL, water:amine = 1.5, n = 2000 min−1, catalyst = Rh(acac)(CO)2, amine = DMAE, VFeed = 20 mL h−1, Vamine = 1 mL h−1. | ||
According to the behaviour observed in batch experiments, this can be attributed to amine leaching and therefore an increased water–amine ratio (Fig. 13). To counteract the loss of amine, a predetermined feed of 1 ml h−1 DMAE was applied throughout the experiment. The actual amine leaching value was slightly higher, since the used pilot plant is not equipped with online-analytics of amine concentration, and the amine loss was detected with time delay. Counteracting this effect by an additional amine feed after 33 and 40 hours led to the stabilization of alcohol yields and reduced rhodium leaching; however, during the period between 20 and 40 hours, the unfavourably high water–amine ratio has caused irreversible rhodium leaching, reducing the catalyst concentration to levels where alcohol formation is inhibited.
 | ||
| Fig. 13 Water–amine ratio and rhodium leaching observed in the continuous flow experiment. Conditions: see Fig. 12. | ||
For further optimization, advanced process control with the aid of online-analytics is necessary. With precise control of the amine feed, the developed recycling strategy is applicable in a continuous flow. Despite the loss of catalyst due to amine leaching, a total turnover number (TTON) for the hydroformylation step of 3145 was achieved. Because of the selectivity loss caused by amine and rhodium leaching, the alcohol production TTON reached 1236.
Conclusions
In this work, a multiphase catalyst recycling approach for reductive hydroformylation was investigated in batches, batch recycling and continuous operation. This reaction system represents the first rhodium-based catalytic system for the auto-tandem catalytic production of alcohols from C5–C10 olefins in a liquid/liquid multiphase system.The influence of temperature, syngas pressure and structure of the used alkanolamine has been characterized. Non-linear interactions of the reaction components were investigated using design of experiment. In these experiments, rhodium–amine interaction was evident. Although rather high catalyst loadings were used, the developed recycling approach with catalyst immobilization in a water/alkanolamine phase led to a rhodium leaching of less than 1%. After the determination of the suitable reaction conditions for the biphasic operation, alcohol yields of up to 64% were achieved with the main side products being valuable aldehydes and internal olefins.
As a proof of concept, the system has been operated in a continuous flow. A pilot plant was operated for 50 hours, reaching a TTON of 1236 for alcohol production and 3145 for the hydroformylation step. The dropping alcohol selectivity revealed a strong dependency of the catalyst stability on the precise control of the water–amine ratio. Hence, for further investigation of the developed process, in situ measurement of the amine lost into the product stream is necessary.
By switching from acetonitrile as the solvent to water and with the ability to recycle the precious metal catalyst, a potential one-step route for the production of alcohols as renewable, synthetic fuel additives was developed.
Materials and methods
Chemicals
1-Octene (99+%), 1-heptanol (98%) and [Rh(acac)(CO)2] (98.5%) were acquired from Acros Organics. n-Heptane (99+%) and 2-propanol (99+%) were purchased from Carl Roth GmbH & Co. KG. N,N-Diethylaminoethanol (DEAE, >99.5%), N,N-diethanolmethylamine (MDEA, >99%), 2-(methylphenylamino)ethanol (MPhAE, 98%), N,N-diisopropylaminoethanol (DIPAE, >99%) and triethanolamine (TEA, >99%) were obtained from Sigma Aldrich. N,N-Dimethylaminoethanol (DMAE, >99%), N,N-dibutylaminoethanol (DBAE, >99%) and 3-dimethylamino-1,2-propanediol (DMAPD, >98%) were obtained from TCI Chemicals. A Merck Milli-Q purification system was used to prepare ultrapure water (TOC <3 ppb, conductivity <0.055 μS cm−1). Carbon monoxide (99997%) and hydrogen (99999%) were supplied by Westfalen AG. The FT product was produced in the experiments in context of the REDIFUEL project with a cobalt-based catalyst developed by the Spanish Research Council in a process operated by VTT Technical Research Centre of Finland Ltd. The C5–C10 fraction was separated by distillation carried out by Neste Oy.Experimental setup
Batch experiments were conducted using a Parr Instruments 4560 high-pressure stainless-steel autoclave head with a custom-made 250 mL windowed reaction vessel. The reactor was heated with an electrical heating jacket and stirred using a mechanically driven 4-pitched-blade agitator. Continuous experiments were conducted using a pilot plant setup. A detailed description of the pilot plant can be found in the ESI.†Experimental procedure
All the solid and liquid components were handled under standard Schlenk conditions. Liquid components were purged with argon before use to remove any potential oxygen content. The catalyst precursor [Rh(acac)(CO)2] was weighed in a Schlenk tube in an oxygen- and water-free glovebox and then dissolved in the respective alkanolamine and Milli-Q water. The catalyst and substrate solutions were filled into the reactor with a syringe under an argon counterflow. After filling, the reactor was closed and pressurized with carbon monoxide and hydrogen and electrically heated to reaction temperature. A stirring rate of 150 min−1 was applied to facilitate heat transfer during heat-up. When the reaction temperature was reached, the reaction was started by increasing the stirring rate to 2000 min−1. In recycling experiments, the upper organic phase was withdrawn through a draft tube and a fresh substrate was fed to the reactor using an HPLC pump. The pilot plant was filled using the same procedure as for the batch experiments. Additionally, the plant was purged with argon three times before the experiment. In continuous flow experiments, the reactor was run in batch mode for 3 h, followed by a loop operation of the reactor and decanter for another 3 h. After this start-up procedure, the continuous operation was started by the activation of the feed pumps.Analytics
Yields were determined by gas chromatography. A Shimadzu Nexis GC-2030 gas chromatograph with a flame ionization detector (FID) and an additional thermal conductivity detector (TCD) was used. Both lines were equipped with a Restek Corp. RTX-5 polysiloxane column with a 30 m length, 0.25 mm internal diameter and 1 μm film thickness each. Hydrogen was used as a carrier gas. The samples were injected by a Shimadzu AOC-20iPlus injection system connected to a Shimadzu AOC-20sPlus autosampler. The reaction samples (175 mg) were diluted with 2-propanol (800 mg), and 1-heptanol (25 mg) was added as the internal standard. ICP-MS measurements were conducted using a Shimadzu ICPMS-2030, and the digestions were carried out using nitric acid and a CEM Corp. Mars 6 microwave reactor.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The project Robust and Efficient processes and technologies for Drop In renewable FUELs for road transport (REDIFUEL) has received funding from the European Union's Horizon 2020 research and innovation programme under Grant Agreement no. 817612. Open Access funding provided by the Max Planck Society.References
- W. Leitner, J. Klankermayer, S. Pischinger, H. Pitsch and K. Kohse-Höinghaus, Angew. Chem., Int. Ed., 2017, 56, 5412–5452 CrossRef CAS PubMed.
- A. J. Janssen, F. W. Kremer, J. H. Baron, M. Muether, S. Pischinger and J. Klankermayer, Energy Fuels, 2011, 25, 4734–4744 CrossRef CAS.
- A. García, J. Monsalve-Serrano, D. Villalta, M. Zubel and S. Pischinger, Energy Convers. Manage., 2018, 177, 563–571 CrossRef.
- K. Jeske, A. C. Kizilkaya, I. López-Luque, N. Pfänder, M. Bartsch, P. Concepción and G. Prieto, ACS Catal., 2021, 4784–4798 CrossRef CAS PubMed.
- G. M. Torres, R. Frauenlob, R. Franke and A. Börner, Catal. Sci. Technol., 2015, 5, 34–54 RSC.
- O. Diebolt, C. Müller and D. Vogt, Catal. Sci. Technol., 2012, 2, 773–777 RSC.
- S. Tuomi, E. Kurkela, P. Simell and M. Reinikainen, Fuel, 2015, 139, 220–231 CrossRef CAS.
- P. Hellier, M. Talibi, A. Eveleigh and N. Ladommatos, Proc. Inst. Mech. Eng., Part D, 2018, 232, 90–105 CrossRef CAS.
- H. A. Choudhury, S. Intikhab, S. Kalakul, M. Khan, R. Tafreshi, R. Gani and N. O. Elbashir, Energy Fuels, 2017, 31, 11266–11279 CrossRef CAS.
- B. Graziano, S. Schönfeld, B. Heuser and D. Pelerin, ATZheavy duty, 2020, 13, 36–41 CrossRef.
- C. Kohlpaintner, M. Schulte, J. Falbe, P. Lappe, J. Weber and G. D. Frey, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2013 Search PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- D. E. Fogg and E. N. Dos Santos, Coord. Chem. Rev., 2004, 248, 2365–2379 CrossRef CAS.
- F. M. S. Rodrigues, P. K. Kucmierczyk, M. Pineiro, R. Jackstell, R. Franke, M. M. Pereira and M. Beller, ChemSusChem, 2018, 11, 2310–2314 CrossRef CAS PubMed.
- K. Takahashi, M. Yamashita, T. Ichihara, K. Nakano and K. Nozaki, Angew. Chem., Int. Ed., 2010, 49, 4488–4490 CrossRef CAS PubMed.
- J. L. van Winkle, S. Lorenzo, R. C. Morris and R. F. Mason, US Pat. 3420898, 1969 Search PubMed.
- L. H. Slaugh and R. D. Mullineaux, US Pat. 3239569A, 1966 Search PubMed.
- S. Fuchs, D. Lichte, M. Dittmar, G. Meier, H. Strutz, A. Behr and A. J. Vorholt, ChemCatChem, 2017, 9, 1436–1441 CrossRef CAS.
- S. Fuchs, D. Lichte, T. Jolmes, T. Rösler, G. Meier, H. Strutz, A. Behr and A. J. Vorholt, ChemCatChem, 2018, 10, 4126–4133 CrossRef CAS.
- D. Gorbunov, M. Nenasheva, E. Naranov, A. Maximov, E. Rosenberg and E. Karakhanov, Appl. Catal., A, 2021, 623, 118266 CrossRef CAS.
- M. R. L. Furst, V. Korkmaz, T. Gaide, T. Seidensticker, A. Behr and A. J. Vorholt, ChemCatChem, 2017, 9, 4319–4323 CrossRef CAS.
- C. Becquet, F. Berche, H. Bricout, E. Monflier and S. Tilloy, ACS Sustainable Chem. Eng., 2021, 9, 9444–9454 CrossRef CAS.
- T. Vanbésien, E. Monflier and F. Hapiot, Green Chem., 2016, 18, 6687–6694 RSC.
- J. Tonziello and M. Vellini, Energy Procedia, 2011, 4, 637–644 CrossRef CAS.
- L. L. W. Cheung, G. Vasapollo and H. Alper, Adv. Synth. Catal., 2012, 354, 2019–2022 CrossRef CAS.
- T. Rösler, K. R. Ehmann, K. Köhnke, M. Leutzsch, N. Wessel, A. J. Vorholt and W. Leitner, J. Catal., 2021, 400, 234–243 CrossRef.
- B. Cornils and E. G. Kuntz, J. Organomet. Chem., 1995, 502, 177–186 CrossRef CAS.
- C. W. Kohlpaintner, R. W. Fischer and B. Cornils, Appl. Catal., A, 2001, 221, 219–225 CrossRef CAS.
- M. Beller, B. Cornils, C. D. Frohning and C. W. Kohlpaintner, J. Mol. Catal. A: Chem., 1995, 104, 17–85 CrossRef CAS.
- M. Frauenkron, J.-P. Melder, G. Ruider, R. Rossbacher and H. Höke, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, pp. 405–431 Search PubMed.
- J. K. MacDougall, M. C. Simpson, M. J. Green and D. J. Cole-Hamilton, J. Chem. Soc., Dalton Trans., 1996, 1161–1172 RSC.
- Purwanto, R. M. Deshpande, R. V. Chaudhari and H. Delmas, J. Chem. Eng. Data, 1996, 41, 1414–1417 CrossRef CAS.
- S. Püschel, S. Störtte, J. Topphoff, A. J. Vorholt and W. Leitner, ChemSusChem, 2021, 14(23), 5226–5234 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy02000e |
| This journal is © The Royal Society of Chemistry 2022 |