
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical ruthenium-catalysed C–H activation in water through heterogenization of a molecular catalyst†
Jan
Bühler‡
 a,
Jonas
Zurflüh‡
a,
Sebastian
Siol
b,
Olivier
Blacque
a,
Laurent
Sévery
*a and
S. David
Tilley
*a
a,
Jonas
Zurflüh‡
a,
Sebastian
Siol
b,
Olivier
Blacque
a,
Laurent
Sévery
*a and
S. David
Tilley
*a
aDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland. E-mail: david.tilley@chem.uzh.ch
bEmpa – Swiss Federal Laboratories for Materials Science and Technology, Überlandstrasse 129, 8600 Dübendorf, Switzerland
First published on 14th January 2022
Abstract
Efficient catalytic oxidative C–H activation of organic substrates remains an important challenge in synthetic chemistry. Here, we show that the combination of a transition metal catalyst, surface immobilisation and an electrochemical potential provide a promising approach to effecting these transformations in aqueous solution. A ruthenium-based molecular catalyst [Ru(tpy)(pic-PO3H2)(Cl)] (where tpy is 2,2′:6′,2′′-terpyridine, pic-PO3H2 is 4-phosphonopyrid-2-ylcarboxylic acid) was synthesised and fully characterised. Oxidation of benzyl alcohol with the catalyst in aqueous media using ceric ammonium nitrate as terminal oxidant resulted in a rapid deactivation of the catalyst. Immobilisation of the catalyst on a mesoporous indium tin oxide electrode surface through the phosphonate anchoring group was shown to circumvent the issues observed in solution. Using the heterogeneous catalyst system, the oxidation of a variety of organic substrates with varying bond dissociation energies was demonstrated with turnover numbers of up to 346. Finally, surface-analysis of the functionalised electrodes after catalysis revealed that fragmentation of the complex during the reaction was the limiting factor for catalytic performance.
Introduction
Over the past few decades, C–H bond functionalisation has become one of the most important research topics in modern synthetic chemistry.1–9 As C–H bonds are ubiquitous in organic molecules, transforming them directly into desired functional groups is a highly efficient and valuable synthetic strategy.10–17 Transition metal complexes that can mediate transformations of sp2 and sp3 carbon–hydrogen bonds via hydrogen atom abstraction, C–H insertions and C–H oxidations or via the formation of organometallic intermediates provide the basis of this research field.10,18–20 C–H functionalisation remains a highly significant research field as evidenced by the constant output of new strategies to improve selectivity and efficiency and to increase the substrate scope.21In this regard, high valent ruthenium–oxo complexes have been reported to be powerful oxidants towards various organic substrates. Ru(VI)– and Ru(VIII)–oxo complexes are known for their ability to oxidise C–H bonds in hydrocarbons22,23 and alcohols.24,25 Ru(IV)–oxo–polypyridyl complexes display activity in oxidative activation of C–H bonds,26–28 as well as in epoxidation29,30 and alcohol oxidation.31,32 Due to their well-defined and reversible redox behaviour, Ru(IV)–oxo–polypyridyl complexes as well as their corresponding Ru(III) hydroxy- and Ru(II) aqua equivalents have been intensively studied.33,34
In 2011, Kojima and co-workers employed a Ru(IV) ([RuIV(O)(H+TPA)(bpy)]3+) complex to stoichiometrically oxidise C–H bonds in organic substrates at ambient temperatures using acetonitrile as a solvent.35 The oxidant efficiency was found to be inversely proportional to the bond dissociation energy (BDE) of the substrate within this set of substrates. A more recent work from Dhuri and co-workers investigated the complex [RuIV(O)(tpy)(bpm)]2+ where they shed light on mechanistic processes on cyclohexene as organic substrate.36 The results of the combined theoretical and experimental study suggested that the allylic C–H oxidation is the dominant pathway and proceeds via H atom abstraction.
Typically, the active Ru(IV)oxo species is formed through proton-coupled electron transfer from the corresponding ruthenium aqua-complex.33 This can be achieved by using chemical oxidants such as [CeIV(NO3)6](NH4)2 (CAN) or alternatively by electrochemical oxidation. Electrosynthetic approaches recently experienced a considerable renaissance as a tool in synthetic chemistry as they offer advantages such as real time (catalytic) current monitoring and the use of electrical potential instead of chemical oxidants.37,38 However, the disadvantages of this approach include undesired diffusion limitations of active catalyst complexes to and from the electrode and short circuit reactions at the counter electrode. These issues can be addressed by immobilising the catalyst on the electrode surface.39,40
Meyer and co-workers demonstrated that electrochemically prepared, high valent Ru(IV)–oxo complexes readily oxidise benzyl alcohol (BnOH) to benzaldehyde.31,41 A [RuII(tpy-PO3H2)(H2O)3]2+ complex that was immobilised on a nanostructured indium tin oxide (ITO) electrode could be electrochemically regenerated after each cycle to achieve a final turnover of 130 (2e− oxidation) before complete deactivation. Meyer and co-workers found that the rate constant of dissolved and anchored Ru–(poly)pyridyl complexes are comparable for oxidation reactions of a set of organic substrates.42 The use of electrosynthetic techniques with the immobilised ruthenium complex as a catalyst is clearly superior to the use of chemical oxidants, in terms of atom efficiency and minimised waste generation. Despite the advantages of the electrosynthetic approach, the electrocatalytic oxidation of organic substrates in aqueous environment is under-investigated and merits further research. This holds true especially for immobilised molecular catalysts. Here, we show that a homogeneous ruthenium polypyridyl complex, [RuII(tpy)(pic-PO3H2)Cl] (where tpy is 2,2′:6′,2′′-terpyridine, pic-PO3H2 is 4-phosphonopyrid-2-ylcarboxylic acid), can be adapted from using chemical oxidants for C–H activation in solution into a homogeneous electrocatalytic system, and finally into a heterogeneous catalytic system (Fig. 1) to oxidise a variety of alcohol and hydrocarbon substrates with high turnover numbers of up to 346. Further, the catalyst exhibits high selectivity (faradaic efficiency of 69–95%) towards oxidation of organic substrates over water oxidation. In contrast to previous related work, we focus on the use of water as a green solvent, which helps to minimise the generation of toxic solvent waste and produces a highly desirable co-product at the counter electrode in the form of H2.
 | ||
| Fig. 1 Concept of the different oxidation processes explored. a) Generation of the active catalyst in solution using a chemical oxidant. b) Electrochemical oxidation of the catalyst in solution. c) Electrochemical oxidation of the immobilised catalyst. | ||
Results and discussion
Synthesis and characterisation
The design of our catalyst was inspired by the work of Chatterjee and co-workers, where a ruthenium oxo complex, [RuIV(tpy)(pic)(O)]ClO4, was used as a stoichiometric oxidant for the epoxidation of alkenes in acetonitrile.30,43 A phosphonic acid substituent on the picolinic acid ligand was selected for catalyst immobilisation, as this group displays enhanced stability on metal oxide surfaces in acidic environment compared to carboxylic acids.44–47 Metalation of 2,2′:6′,2′′-terpyridine (tpy) with the Ru(III) chloride precursor (RuCl3) in refluxing ethanol resulted in the isolation of [RuIII(tpy)Cl3] (1).48,49 The 4-phosphonopyrid-2-ylcarboxylic acid (2, pic-PO3H2) ligand was prepared by palladium-catalysed Hirao cross-coupling of 4-bromopicolinate with diethyl phosphite and subsequent ester hydrolysis.50 [RuIII(tpy)Cl3] was then reacted with pic-PO3H2 in the presence of NEt3 as reductant to give [RuII(tpy)(pic-PO3H2)Cl] (3, Scheme 1). X-Ray crystallographic studies on single crystals grown through vapor diffusion from MeOH with THF as the antisolvent show a near octahedral geometry around the Ru metal centre, with the chloride ligand and the picolinic nitrogen occupying axial positions, the tpy fully chelating in the equatorial plane and the carboxylate moiety of pic-PO3H2 at the remaining equatorial coordination site. | ||
| Scheme 1 Synthesis of [Ru(tpy)(pic-PO3H2)Cl] (3) from [Ru(tpy)Cl3] (1) and 4-phosphonopyrid-2-ylcarboxylic acid (2). | ||
The complex is essentially isostructural to cis-[RuII(tpy)(pic)Cl] described by Llobet and co-workers.51 The trans-isomer, with the carboxylate moiety in the axial position, was only observed in small amounts and could be separated by preparative HPLC. This is consistent with previous reports of [RuII(tpy)(pic)Cl] synthesised from ethanolic H2O,43 whereas synthesis from EtOH lead to the formation of both isomers.51,52
C–H oxidation under homogeneous conditions
To validate the possibility of utilising aqua-complex [RuII(tpy)(pic-PO3H2)(H2O)]+(Cl)− (4) as an electrochemical catalyst for organic substrate oxidation, stoichiometric experiments with CAN as the oxidising agent and BnOH as the substrate were performed and followed by 1H NMR spectroscopy (Fig. 2). For this purpose, complex 3 was dissolved in D2O leading to the rapid exchange of the chloride ligand and formation of the cationic aqua-complex [RuII(tpy)(pic-PO3H2)(H2O)]+(Cl)− (4, Fig. 2a) which is readily oxidised to [RuIV(tpy)(pic-PO3H2)(O)]+ (5, Fig. 2b) by treatment with two equivalents of CAN. The presence of the paramagnetic complex can be readily observed by the strong broadening of the signals in the recorded 1H NMR spectrum (Fig. 2b). Upon addition of two equivalents of BnOH, the aqua-complex is regenerated and one equivalent of the substrate is oxidised to benzaldehyde (Fig. 2c). In theory, one equivalent of substrate is adequate to fully reduce the oxo-complex, however, under these conditions a substantial amount of benzoic acid is also formed from the oxidation of benzaldehyde. Thus, two equivalents of substrate were added to simplify the analysis of the obtained products. | ||
| Fig. 2 Chemical oxidation of [RuII(tpy)(pic-PO3H2)(H2O)]+(Cl)− (4) to [RuIV(tpy)(pic-PO3H2)(O)]+ (5) and subsequent substrate oxidation. NMR spectra in D2O showing (a) the Ru(II) aqua-complex, (b) addition of two equivalents of ammonium cerium nitrate leading to the formation of a Ru(IV) oxo-complex, and (c) addition of two equivalents of benzyl alcohol leading to oxidation to benzaldehyde and reduction of the Ru(IV) oxo-complex to the Ru(II) aqua-complex. | ||
After successfully showing stoichiometric oxidation from complex 4, it was tested for catalytic conversion in solution using excess substrate and oxidising agent. When 20 equivalents each of CAN and benzyl alcohol were added, 27% of the substrate was converted to benzaldehyde, with only minor amounts of benzoic acid forming. This corresponds to a final turnover number of 5.4 before complete deactivation of the catalyst (Fig. S1†).
Several drawbacks of employing complex 4 as a homogeneous catalyst were apparent from the stoichiometric and homogeneous catalytic experiments. The oxo-complex 5 has a limited solubility in aqueous media and readily precipitates from solution at concentrations above 1 mM. Addition of substrate to precipitated [RuIV(tpy)(pic-PO3H2)(O)]Cl then leads to the formation of a green complex that could no longer be converted into 4. This is assumed to be an oxo-bridged dinuclear ruthenium species, which has been observed for similar ruthenium complexes.44,53 These types of complexes are not catalytically active and their formation is not reversible, which leads to deactivation of the catalyst over time.
Homogeneous electrocatalytic oxidation
Cyclic voltammograms (CVs) of complex 4 in aqueous solution (pH 2.4) show two oxidation features at positive potentials (Fig. 3a): reversible redox peaks at 0.65 V vs. NHE indicating the [RuII(tpy)(pic-PO3H2)(H2O)]+/[RuIII(tpy)(pic-PO3H2)(OH)]+ transition, followed by a peak with an onset at around 1.05 V vs. NHE that is attributed to the [RuIII(tpy)(pic-PO3H2)(OH)]+/[RuIV(tpy)(pic-PO3H2)(O)]+ oxidation. The subsequent current onset at 1.45 V vs. NHE coincides with the oxidation to RuV, which has been shown to be active for water oxidation in the closely related complex [Ru(tpy)(pic)(H2O)]+.52 The addition of BnOH resulted in catalytic current attributed to substrate oxidation, with an onset that immediately follows the oxidation to the active RuIV species. The catalytic current density increases with increasing concentration of the organic substrate (Fig. 3b). We note that catalytic oxidation of BnOH does not occur in the absence of Ru complex (Fig. S2†). | ||
| Fig. 3 (a) Cyclic voltammograms of [Ru(tpy)(pic-PO3H2)(H2O)]+(Cl)− (0.1 mM) in aqueous Na2SO4 (0.1 M, pH 2.4). (b) Cyclic voltammograms scans after addition of varying amounts of benzyl alcohol. Both recorded on an FTO electrode at 100 mV s−1. | ||
Kinetic information for the catalytic oxidation could be approximated from the CVs according to previously reported procedures (Fig. S3†).54,55 The current responses of the ic/ip ratios, where ic is the catalytic plateau current and ip is the peak current without substrate, were measured at different scan rates with varying substrate concentrations at 1.35 V vs. NHE (Fig. S3†). A rate constant of kcat = 41 ± 14 M−1 s−1 was measured for the homogeneous electrocatalytic oxidation of benzyl alcohol with corresponding two-electron transfer from the catalyst. At a substrate concentration of 0.01 M, this corresponds to a turnover frequency (TOF) of 0.41 ± 0.14 s−1.
Catalyst immobilisation
Encouraged by these results, complex 3 was immobilised on a metal-oxide surface for further investigations. Surface-attachment of 3 on optically transparent nanostructured indium tin oxide (meso-ITO) films (Fig. S4†) was achieved by soaking the substrate in a 1 mM methanolic solution of the complex. The extent of loading was determined by UV-visible measurements of the catalyst, desorbed by soaking the functionalised substrates in aqueous KOH (pH 14) (Fig. S5†). The geometric surface loading was found to be 15.4 ± 0.7 nmol cm−2 (Table S1†).CV measurements of immobilised 4, obtained by soaking of the surface anchored 3 in aqueous bisulfate buffer, show the two expected redox features for the transitions of RuII/RuIII and RuIII/RuIV at 0.75 and 1.15 V vs. NHE, respectively, followed by the onset of water oxidation at 1.60 V (Fig. 4). If the potential is cycled between 0.2 and 1.45 V, at less positive potentials than the onset of the water oxidation, the reduction of RuIV to RuIII is more pronounced (Fig. S6†).
 | ||
| Fig. 4 Cyclic voltammogram of [Ru(tpy)(pic-PO3H2)(H2O)]+(Cl)− (4) immobilised on a meso-ITO electrode in aqueous Na2SO4 (0.1 M, pH 2.4) at 100 mV s−1. | ||
To obtain a deeper understanding of the electrochemical properties of immobilised complex 4, the pH dependence of the redox potentials was investigated in the pH range of 2.0–5.0. The Pourbaix diagram of complex 4 on meso-ITO (Fig. 5) shows that the potential for the RuII/RuIII redox couple is pH independent in the range of pH 2.0–3.5 and displays approximately Nernstian behaviour in the range of pH 3.5–5.0. The slope of −66 mV pH−1 indicates the presence of proton coupled electron transfer (PCET), where the transfer of a proton accompanies the one electron oxidation.56 The RuIII/RuIV redox couple exhibits pH dependency with a two proton one electron process from pH 2.0–3.5 and a simple proton coupled electron transfer from pH 3.5–5.0. The pKa value of [RuIII(tpy)(pic-PO3H2)(H2O)]2+ can thus be determined to be 3.5. The bond strength of the oxygen–hydrogen bond in [RuIII(tpy)(pic-PO3H2)(OH)]+ can be calculated to be 93–95 kcal mol−1 using methods developed by Bordwell and co-workers, giving an estimate for the maximal BDE a potential substrate can exhibit to successfully be oxidised by the catalyst.57,58 A third pH dependent redox event with a slope of −62 mV pH−1 can be observed. This is assigned as a ligand-based oxidation with concomitant loss of a proton by comparison with the redox behaviour of the pic-PO3H2 ligand (Fig. S7†) and immobilised [Fe(tpy)(pic-PO3H2)Cl] (Fig. S8†). The catalytically non-active, lower homologue of the ruthenium complex is used as a comparison due to its very similar structure and high potentials needed for the FeIII/FeIV transition, which in contrast to complex 4 does not overlap with the oxidation of pic-PO3H2. This ligand-based PCET redox-event also occurs for a homogeneous sample of 4 but cannot be observed with the phosphonodiethylester analogue [Ru(tpy)(pic-PO3Et2)Cl] (Fig. S9†).
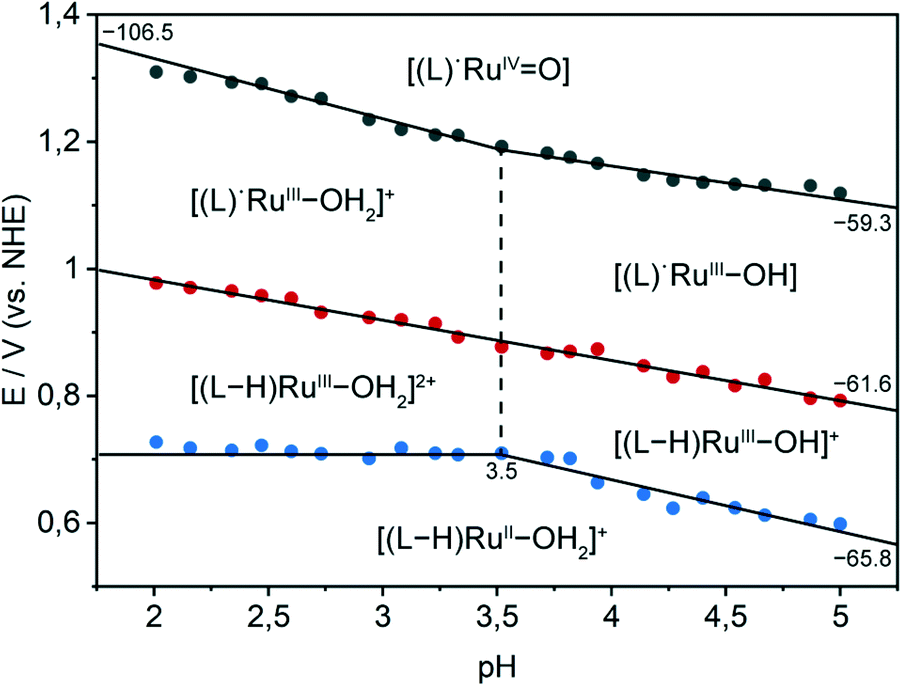 | ||
| Fig. 5 Pourbaix diagram of [Ru(tpy)(pic-PO3H2)(H2O)]+(Cl)− immobilised on a meso-ITO electrode in the pH range of 2.0–5.0 (pKa value is denoted by the vertical dashed line, slopes are given in mV pH−1). | ||
Significant similarities to the Pourbaix diagram of a homogeneous solution of complex 3 (Fig. S10†) can be observed. The two diagrams show essentially equal pKa values and slopes for the immobilised and solution-phase catalyst. The RuII/RuIII redox couple for the immobilised catalyst is shifted to less positive potentials by 150 mV compared to the solution, indicating a considerable facilitation of the catalyst oxidation by the immobilisation on the metal-oxide electrode surface. Further, in the homogeneous case the ligand-based oxidation (shifted by 200 mV) occurs at a lower potential then the RuII/RuIII transition.
Electrocatalysis with the immobilised complex
The electrocatalytic activity of the immobilised complex 4 towards C–H bond activation was explored by chronoamperometry experiments using a set of five different substrates with bond dissociation energies (BDE) ranging from 76.0 (1,4-cyclohexadiene) to 94.9 kcal mol−1 (n-butanol). BDEs, concentrations, conversions, faradaic yields and turnover numbers are summarised in Table 1. All experiments were allowed to proceed for 2 h at a potential of 1.45 V vs. NHE in aqueous Na2SO4 (0.1 M, pH 2.4). The concentration of the substrates was set at 10 mM, except for toluene and 1,4-cyclohexadiene, where the concentration was limited by the solubility in water and saturated solutions were used. The oxidation of 1,4-cyclohexadiene afforded benzene as the only product (Fig. S11†). This result is consistent with previous observations that H atom abstraction is the dominant pathway for allylic C–H bond oxidations by RuIV–oxo species.34,36 A CV with toluene as substrate revealed catalytic current with an onset directly following the RuIV oxidation event (Fig. 6 and S12†). NMR studies revealed a conversion of 4.8% and benzyl alcohol was found to be the only product of this reaction. At prolonged reaction times, further oxidation to benzaldehyde was detected in trace amounts. Chronoamperometry with 9H-fluorene-2,7-disulfonic acid (S2-fluorene) gave the four-electron oxidised ketone product S2-fluorenone (Fig. S13†). Only traces of the alcohol intermediate were found by NMR. We observe that higher BDE tends to lead to slower conversion rates, which is also reflected in the lower turnover values for the latter two substrates. We note that the oxidation of toluene and S2-fluorene likely follows hydrogen atom transfer with subsequent oxygen rebound of the formed radical with the resulting RuIII hydroxo complex.35,59 However, elucidation of the mechanisms at play were not part of this work.| Substrate | Concentration (mM) | BDE (kcal mol−1)60 | Conversion (%, CA) | Conversion (%, NMR) | Faradaic yield (%) | TON |
|---|---|---|---|---|---|---|
| 1,4-Cyclohexadiene | 8.7 | 76.0 | 11.9 | 10.3 | 86 | 346 |
| Benzyl alcohol | 10.0 | 79.0 | 12.7 | 11.1 | 87 | 343 |
| S2-Fluorene | 10.0 | 82.0 | 2.7 | 2.6 | 95 | 79 |
| Toluene | 5.7 | 89.7 | 6.5 | 4.8 | 74 | 117 |
| n-Butanol | 10.0 | 94.9 | 6.2 | 4.2 | 68 | 129 |
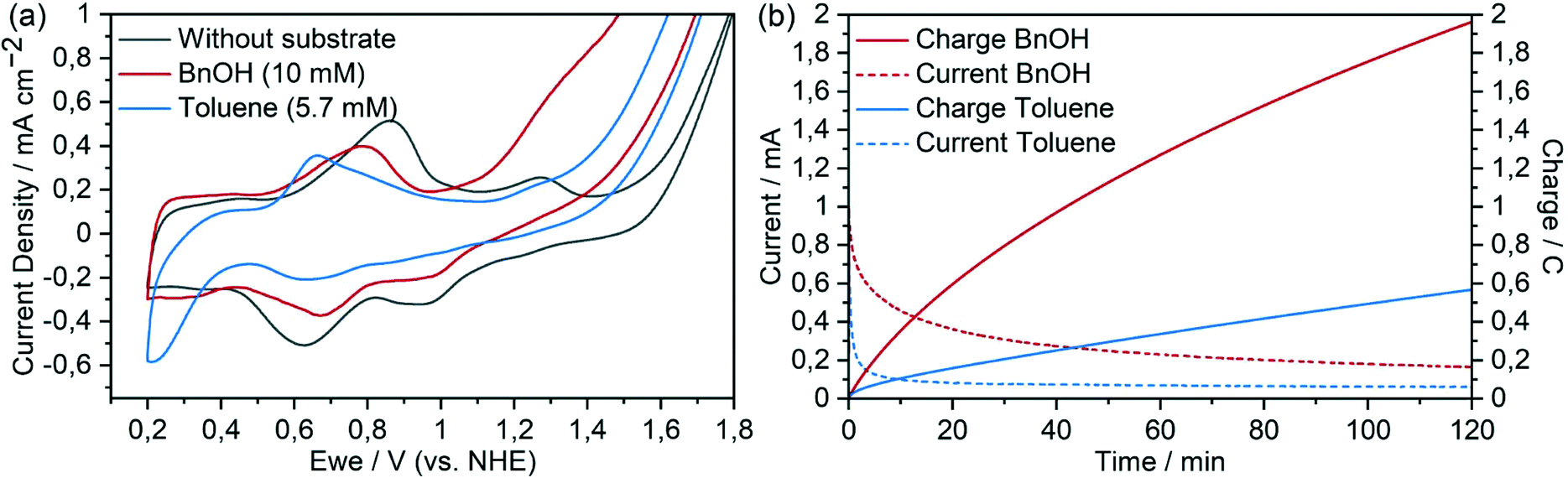 | ||
| Fig. 6 (a) Cyclic voltammograms of [Ru(tpy)(pic-PO3H2)(H2O)]+(Cl)− immobilised on a meso-ITO electrode in aqueous Na2SO4 (0.1 M, pH 2.4) and after addition of benzyl alcohol (10 mM) and toluene (5.7 mM) at 100 mV s−1. (b) Chronoamperometry of benzyl alcohol (10 mM) and toluene (5.7 mM) solutions in aqueous Na2SO4 (0.1 M, pH 2.4) at 1.45 V vs. NHE. | ||
This correlation again became apparent with benzyl alcohol as the organic substrate which – like 1,4-cyclohexadiene – has a BDE below 80 kcal mol−1 (Fig. 6 and S14†) and showed a significantly higher turnover number compared to toluene. The onset of the catalytic current following the RuII/RuIII oxidation indicates that catalyst–substrate interaction is possible even at the RuIII state for molecules with low C–H bond strengths. This is also observed in the previously mentioned oxidation of 1,4-cyclohexadiene and is in agreement with the observation that the BDE of the O–H bond in [RuII(tpy)(pic-PO3H2)(H2O)]+ is 76–78 kcal mol−1. Our intention to transfer the catalyst from a homogeneous system with CAN as a chemical oxidant into an entirely electrocatalytic system under purely aqueous conditions was achieved at this point. The use of the same organic substrate allows a first direct comparison: as anticipated, the problems of precipitation and dimerization could be overcome by the immobilisation of complex 4 onto the meso-ITO working electrode. In contrast to homogeneous experiments with CAN as the oxidant and the same substrate, the complex performed several hundred turnovers. The initial rate of the catalytic oxidation of the immobilized catalyst could be calculated from the current (after 10 s, minimizing contributions from capacitive processes) along with the electrode area and previously determined loading. For the two-electron process, the TOF with 10 mM substrate concentration was determined to be 0.18 ± 0.01 s−1. This value is in the same order of magnitude as is the case for the homogeneous system (0.41 ± 0.14 s−1), suggesting that the activity of the catalyst was roughly retained when immobilized to the electrode surface (in agreement with the work of Meyer and co-workers42). The faradaic efficiency for this experiment was 87% as determined by integration of product peaks of NMR spectra measured after chronoamperometry. Electrocatalytic oxidation of n-butanol, which has significantly stronger C–H bond energy compared to benzyl alcohol, still reached 129 turnovers at a conversion of 4.2% and a faradaic yield of 68% (Fig. S15†). The dehydrogenation reaction with ruthenium oxo species was reported to proceed via either hydrogen atom abstraction or concerted PCET,33,61,62 although the mechanism at play in this case was not investigated. We note that for 1,4-dioxane (BDE = 96.0 kcal mol−1) no catalytic current was observed, which matches well our previously calculated C–H bond strength of 93–95 kcal mol−1 that the catalyst should be able to oxidise.
Although the heterogeneous electrocatalytic system showed the most promising results in terms of faradaic efficiency and turnover numbers for a broad scope of substrates, the limitations become apparent when considering the long-term stability of the catalytic system. The catalytic current decreases steadily over time for all chronoamperometry experiments and generally falls below 100 μA after 2 h. The reasons for this current drop could be desorption of the catalyst from the electrode surface with subsequent deactivation or decomposition of the catalyst on the surface. Comparison of the functionalised electrode before and after the 2 h electrocatalytic experiments by X-ray photoelectron spectroscopy (XPS) revealed that the latter phenomena could play the dominant role in the deactivation process (Fig. 7). Almost no signs of ruthenium were found on the electrode surface after catalysis, while the intensity of the phosphorous signal did not obviously change. The signal for nitrogen was reduced, but a residual amount could be measured on the electrode surface. These results suggest that the pic-PO3H2 ligand remains bound to the surface, but the ruthenium along with the tpy-fragment dissociated from the anchored complex. Alternatively, the deactivation of the catalyst could occur via N-oxidation on the terpyridine ligand, leading to its dissociation.63 We further note that no sign of oxidative cleavage of the phosphorus carbon bond was observed, which had previously been observed for water-oxidation catalysts.64
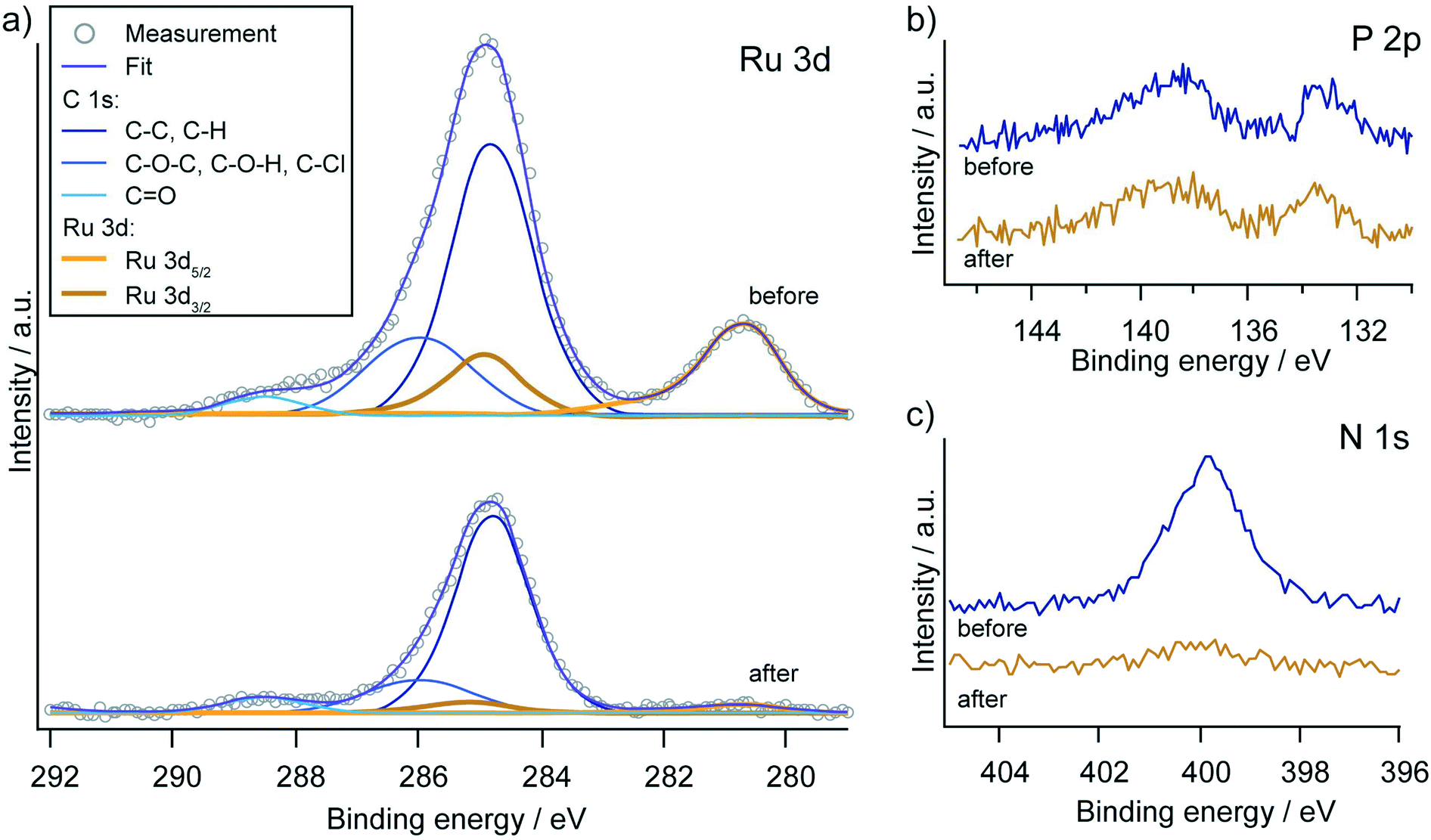 | ||
| Fig. 7 XPS core level spectra of the C 1s/Ru 3d, P 2p and N 1s regions before and after 2 h electrocatalytic synthesis. a) Fits of the C 1s and Ru 3d core level emissions reveal a complete loss of the Ru signal. b) The peak in the P 2p region (134 eV) is retained, indicating stable binding of the 4-phosphonopyrid-2-ylcarboxylic acid moiety. The signal at 138 eV is the Sn 4s core level emission from the ITO substrate. c) The N 1s signal is significantly decreased after catalysis. | ||
Conclusions
In summary, we have synthesised and characterised a ruthenium terpyridine complex with a picolinic acid moiety containing a phosphonate anchoring group for immobilisation on metal-oxide surfaces. The complex was tested and shown to be active in homogenous chemical and electrochemical catalytic experiments before immobilisation on an indium tin oxide electrode. While previous reports with similar complexes have extensively studied water oxidation, we reported on the oxidation of organic substrates in aqueous media. The heterogenized catalyst showed high turnover numbers and selectivity over water oxidation in the electrochemical oxidation of different organic substrates with varying BDEs of their carbon hydrogen bonds, improving upon previously reported immobilised ruthenium catalyst [Ru(tpy-PO3H2)(H2O)3]2+.41 XPS measurements suggest that decomposition of the anchored catalyst on a time scale of hours limits the activity of the heterogeneous catalyst system.Improvements in performance and stability of the system would support the application of immobilised electrocatalysts in synthesis. Some approaches, including potential tuning through substitution and improved design of the ligands to prevent decomposition of the metal complex, could provide a significant step forward to practical uses of this catalytic system. Finally, mechanistic studies will be necessary to provide important information for the development of an overall more efficient and broadly applicable catalyst.
Author contributions
L. S. and S. D. T. designed the project and supervised the research; J. B. and J. Z. performed most of the experimental work and characterisation; S. S. performed XPS measurements and analysis; O. B. performed XRD measurements. The manuscript was written by J. B. and J. Z. and edited by L. S. and S. D. T.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the University of Zurich and the University Research Priority Program LightChEC for funding.References
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed.
- X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790–6791 CrossRef CAS PubMed.
- D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18566–18569 CrossRef CAS PubMed.
- J. Wencel-Delord and F. Colobert, Chem. – Eur. J., 2013, 19, 14010–14017 CrossRef CAS PubMed.
- A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851–863 CrossRef CAS PubMed.
- M. Costas, Coord. Chem. Rev., 2011, 255, 2912–2932 CrossRef CAS.
- D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649–8709 CrossRef CAS PubMed.
- S. Das, C. D. Incarvito, R. H. Crabtree and G. W. Brudvig, Science, 2006, 312, 1941 CrossRef CAS PubMed.
- A. R. Dick and M. S. Sanford, Tetrahedron, 2006, 62, 2439–2463 CrossRef CAS.
- K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS PubMed.
- Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC.
- J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC.
- D.-S. Kim, W.-J. Park and C.-H. Jun, Chem. Rev., 2017, 117, 8977–9015 CrossRef CAS PubMed.
- S. Jin, J. Kim, D. Kim, J.-W. Park and S. Chang, ACS Catal., 2021, 11, 6590–6595 CrossRef CAS.
- P. E. Gormisky and M. C. White, J. Am. Chem. Soc., 2013, 135, 14052–14055 CrossRef CAS PubMed.
- Q. Zhao, G. Meng, S. P. Nolan and M. Szostak, Chem. Rev., 2020, 120, 1981–2048 CrossRef CAS PubMed.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed.
- A. Gunay and K. H. Theopold, Chem. Rev., 2010, 110, 1060–1081 CrossRef CAS PubMed.
- S. Rej, A. Das and N. Chatani, Coord. Chem. Rev., 2021, 431, 213683 CrossRef CAS.
- C.-M. Che, J.-L. Zhang, R. Zhang, J.-S. Huang, T.-S. Lai, W.-M. Tsui, X.-G. Zhou, Z.-Y. Zhou, N. Zhu and C. K. Chang, Chem. – Eur. J., 2005, 11, 7040–7053 CrossRef CAS PubMed.
- V. Piccialli, Molecules, 2014, 19, 6534–6582 CrossRef PubMed.
- E. L. Lebeau and T. J. Meyer, Inorg. Chem., 1999, 38, 2174–2181 CrossRef CAS PubMed.
- G. Green, W. P. Griffith, D. M. Hollinshead, S. V. Ley and M. Schröder, J. Chem. Soc., Perkin Trans. 1, 1984, 681–686 RSC.
- L. K. Stultz, M. H. V. Huynh, R. A. Binstead, M. Curry and T. J. Meyer, J. Am. Chem. Soc., 2000, 122, 5984–5996 CrossRef CAS.
- Y. Hirai, T. Kojima, Y. Mizutani, Y. Shiota, K. Yoshizawa and S. Fukuzumi, Angew. Chem., 2008, 120, 5856–5860 CrossRef.
- C.-M. Che, K.-W. Cheng, M. C. W. Chan, T.-C. Lau and C.-K. Mak, J. Org. Chem., 2000, 65, 7996–8000 CrossRef CAS PubMed.
- K. Jitsukawa, Y. Oka, S. Yamaguchi and H. Masuda, Inorg. Chem., 2004, 43, 8119–8129 CrossRef CAS PubMed.
- D. Chatterjee, Inorg. Chim. Acta, 2008, 361, 2177–2182 CrossRef CAS.
- A. K. Vannucci, J. F. Hull, Z. Chen, R. A. Binstead, J. J. Concepcion and T. J. Meyer, J. Am. Chem. Soc., 2012, 134, 3972–3975 CrossRef CAS PubMed.
- C.-M. Che, W.-T. Tang, W.-O. Lee, K.-Y. Wong and T.-C. Lau, J. Chem. Soc., Dalton Trans., 1992, 1551–1556 RSC.
- T. Ishizuka, H. Kotani and T. Kojima, Dalton Trans., 2016, 45, 16727–16750 RSC.
- J. R. Bryant and J. M. Mayer, J. Am. Chem. Soc., 2003, 125, 10351–10361 CrossRef CAS PubMed.
- T. Kojima, K. Nakayama, K. Ikemura, T. Ogura and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 11692–11700 CrossRef CAS PubMed.
- S. N. Dhuri, K.-B. Cho, Y.-M. Lee, S. Y. Shin, J. H. Kim, D. Mandal, S. Shaik and W. Nam, J. Am. Chem. Soc., 2015, 137, 8623–8632 CrossRef CAS PubMed.
- T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484–2496 CAS.
- D. Pletcher, Electrochem. Commun., 2018, 88, 1–4 CrossRef CAS.
- T. E. Rosser and E. Reisner, ACS Catal., 2017, 7, 3131–3141 CrossRef CAS.
- L. Sévery, J. Szczerbiński, M. Taskin, I. Tuncay, F. Brandalise Nunes, C. Cignarella, G. Tocci, O. Blacque, J. Osterwalder, R. Zenobi, M. Iannuzzi and S. D. Tilley, Nat. Chem., 2021, 13, 523–529 CrossRef PubMed.
- B. J. Hornstein, D. M. Dattelbaum, J. R. Schoonover and T. J. Meyer, Inorg. Chem., 2007, 46, 8139–8145 CrossRef CAS PubMed.
- L. A. Gallagher and T. J. Meyer, J. Am. Chem. Soc., 2001, 123, 5308–5312 CrossRef CAS PubMed.
- D. Chatterjee, A. Sengupta and A. Mitra, Polyhedron, 2007, 26, 178–183 CrossRef CAS.
- Y. Zhu, D. Wang, Q. Huang, J. Du, L. Sun, F. Li and T. J. Meyer, Nat. Commun., 2020, 11, 4610 CrossRef CAS PubMed.
- E. Bae, W. Choi, J. Park, H. S. Shin, S. B. Kim and J. S. Lee, J. Phys. Chem. B, 2004, 108, 14093–14101 CrossRef CAS.
- M. R. Norris, J. J. Concepcion, C. R. K. Glasson, Z. Fang, A. M. Lapides, D. L. Ashford, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2013, 52, 12492–12501 CrossRef CAS PubMed.
- I. Gillaizeau-Gauthier, F. Odobel, M. Alebbi, R. Argazzi, E. Costa, C. A. Bignozzi, P. Qu and G. J. Meyer, Inorg. Chem., 2001, 40, 6073–6079 CrossRef CAS PubMed.
- B. P. Sullivan, J. M. Calvert and T. J. Meyer, Inorg. Chem., 1980, 19, 1404–1407 CrossRef CAS.
- F. P. Dwyer, H. A. Goodwin and E. C. Gyarfas, Aust. J. Chem., 1963, 16, 42–50 CrossRef CAS.
- Z. A. Dziuganowska, K. Ślepokura, J.-N. Volle, D. Virieux, J.-L. Pirat and P. Kafarski, J. Org. Chem., 2016, 81, 4947–4954 CrossRef CAS PubMed.
- M. A. Hoque, A. D. Chowdhury, S. Maji, J. Benet-Buchholz, M. Z. Ertem, C. Gimbert-Suriñach, G. K. Lahiri and A. Llobet, Inorg. Chem., 2021, 60, 5791–5803 CrossRef CAS PubMed.
- A. Llobet, P. Doppelt and T. J. Meyer, Inorg. Chem., 1988, 27, 514–520 CrossRef CAS.
- Q.-Q. Yang, X. Jiang, B. Yang, Y. Wang, C.-H. Tung and L.-Z. Wu, iScience, 2020, 23, 100969 CrossRef CAS PubMed.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS PubMed.
- M. D. Kärkäs, T. Åkermark, H. Chen, J. Sun and B. Åkermark, Angew. Chem., Int. Ed., 2013, 52, 4189–4193 CrossRef PubMed.
- F. G. Bordwell, J. P. Cheng and J. A. Harrelson, J. Am. Chem. Soc., 1988, 110, 1229–1231 CrossRef CAS.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- J. W. Darcy, B. Koronkiewicz, G. A. Parada and J. M. Mayer, Acc. Chem. Res., 2018, 51, 2391–2399 CrossRef CAS PubMed.
- Comprehensive Handbook of Chemical Bond Energies, CRC Press, 2007 Search PubMed.
- S. Ohzu, T. Ishizuka, Y. Hirai, H. Jiang, M. Sakaguchi, T. Ogura, S. Fukuzumi and T. Kojima, Chem. Sci., 2012, 3, 3421–3431 RSC.
- T. Kojima, Y. Hirai, T. Ishizuka, Y. Shiota, K. Yoshizawa, K. Ikemura, T. Ogura and S. Fukuzumi, Angew. Chem., Int. Ed., 2010, 49, 8449–8453 CrossRef CAS PubMed.
- J. B. C. Mack, K. L. Walker, S. G. Robinson, R. N. Zare, M. S. Sigman, R. M. Waymouth and J. Du Bois, J. Am. Chem. Soc., 2019, 141, 972–980 CrossRef CAS PubMed.
- J. Odrobina, J. Scholz, M. Risch, S. Dechert, C. Jooss and F. Meyer, ACS Catal., 2017, 7, 6235–6244 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General information, general procedures, analytical data, XRD, SEM, TOF measurements and catalytic data. CCDC 2094375. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cy01999f |
| ‡ J. B. and J. Z. contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2022 |