
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancing catalytic performance of AuPd catalysts towards the direct synthesis of H2O2 through incorporation of base metals†
Alexandra
Barnes‡
a,
Richard J.
Lewis‡
 *a,
David J.
Morgan
ab,
Thomas E.
Davies
a and
Graham J.
Hutchings
*a
*a,
David J.
Morgan
ab,
Thomas E.
Davies
a and
Graham J.
Hutchings
*a
aMax Planck–Cardiff Centre on the Fundamentals of Heterogeneous Catalysis FUNCAT, Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: LewisR27@cardiff.ac.uk; Hutch@cardiff.ac.uk
bHarwell XPS, Research Complex at Harwell (RCaH) Didcot, OX11 0FA, UK
First published on 12th February 2022
Abstract
The introduction of small quantities of tertiary base metals into supported AuPd nanoparticles results in improved catalytic performance towards the direct synthesis of H2O2, compared to the bi-metallic analogue. This enhanced activity can be attributed to the electronic modification of Pd and the formation of domains of mixed Pd oxidation state. In particular the introduction of Ni is observed to result in initial rates of H2O2 synthesis, where the contribution from competitive degradation reactions is negligible, in excess of three times that achieved over the supported AuPd catalyst.
Introduction
Hydrogen peroxide (H2O2) is a highly effective, environmentally friendly oxidant, with the only by-product of its application being water. Finding application in sectors such as pulp manufacture, where its efficiency as a bleaching agent is sought, or in chemical synthesis, which utilizes its high active oxygen content, H2O2 is rapidly superseding traditional oxidants such as permanganate or perchlorate.1 In recent years global H2O2 production has grown at a rate of 4% per annum, with this increase in demand primarily driven by the chemical synthesis sector. Currently H2O2 production on an industrial scale is met entirely via the anthraquinone oxidation (AO) process, accounting for 95% of global H2O2 supply. While highly efficient there are numerous environmental and economic concerns associated with the AO process, chief amongst these is related to poor atom-efficiency, with the anthraquinone H2-carrier molecule requiring periodic replacement due to its unselective, over-hydrogenation.2 Furthermore, due to economies of scale, H2O2 production via the AO process is typically centralised, necessitating the transport and storage of H2O2 concentrations far in excess of that often required by the end-user, resulting in the dilution of H2O2 prior to use and effectively wasting large quantities of energy utilised in the distillation and concentration process.3 In addition, due to its relative instability, decomposing to water under relatively mild temperatures or basic conditions, H2O2 produced via the AO process is often shipped in the presence of acidic promoters,4,5 which require separation from product streams and can deleteriously effect reactor lifetime.6 These cumulative drawbacks pass on significant costs to the end user and would be reduced or removed altogether via the on-site production of H2O2.The direct synthesis of H2O2 from molecular H2 and O2 (Scheme 1) offers an attractive alternative to the AO process and would alleviate many of the concerns associated with the current means of production, allowing for the synthesis of stabilizer free H2O2 at appropriate concentrations, at site of final use.
 | ||
| Scheme 1 Reaction pathways associated with the direct synthesis of H2O2 from H2 and O2. | ||
Despite significant attention within the academic and patent literature and over 100 years of academic pursuit7 the direct route to H2O2 has yet to overcome issues associated with catalytic performance. Although in recent years great strides have been made in improving catalyst selectivity through the incorporation of numerous secondary metals into supported Pd catalysts. Perhaps most extensively studied has been the enhancement in catalytic efficacy through the alloying of Pd with Au,8–12 however numerous investigations have demonstrated that the addition of a range of abundant metals including Fe,13,14 Sn,15,16 Ni,17,18 In,19 Ag,20 Zn,21,22 Te23 and Co24 can similarly enhance catalytic performance. Further studies have focussed on the incorporation of dopant levels of precious metals, in particular Pt, into supported Pd,25–28 Au29 and AuPd catalysts.30–32 Typically, the improvement in catalytic performance has been ascribed to a combination of isolation of contiguous Pd ensembles,33–35 widely believed to be key in promoting the production of H2O as a result of O–O bond cleavage, in addition to the electronic modification of Pd.36
The sol-immobilisation procedure is a promising method for the production of supported metal nanoparticles, allowing for enhanced control over nanoparticle size and a more uniform particle-to-particle composition in comparison to alternative catalyst preparation techniques, such as impregnation.37–39
With these previous studies in mind, we now investigate the efficacy of tertiary metal incorporation into supported AuPd catalysts towards the direct synthesis of H2O2, with a particular focus on non-precious metals to reduce costs.
Experimental
Catalyst preparation
A series of mono-, bi- and tri-metallic 1% AuPdX/TiO2 (X = Pt, Zn, Ga, Ni, Sn, Co, Cu, In) catalysts have been prepared (on a weight basis) by a sol-immobilisation procedure, based on methodology previously reported in the literature, which has been shown to result in enhanced precious metal dispersion by limiting particle growth and agglomeration.23 The procedure to produce 1% AuPd(0.975)Ni(0.025)/TiO2 (1 g) is outlined below where the total metal loading is 1 wt%, the combined weight loading of Au and Pd is 0.975 wt% and that of Ni is 0.025 wt%, in all cases the Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd ratio is 1:1 (mol mol−1). A similar methodology to that outlined below was utilised for all mono- and bi-metallic catalysts. Table S1† reports the exact quantities of precursors used to synthesise the key catalysts used within this work.
:Pd ratio is 1:1 (mol mol−1). A similar methodology to that outlined below was utilised for all mono- and bi-metallic catalysts. Table S1† reports the exact quantities of precursors used to synthesise the key catalysts used within this work.
Aqueous solutions of HAuCl4·3H2O (0.322 mL, 12.25 mg mL−1, Strem Chemicals), PdCl2 (0.356 mL, 6 mg mL−1, Sigma Aldrich) and Ni(NO3)2 (570 μL, 1.08 mg mL−1, Sigma Aldrich) were added to deionised water (400 mL) under vigorous stirring conditions at room temperature. The resulting solution was allowed to stir for 2 minutes prior to the addition of polyvinylalcohol (PVA) (1.30 mL, 1 wt% MW = 9000–10000 gmol−1, 80% hydrolysed, Sigma Aldrich) such that the weight ratio of metal:PVA was 1:1.3. The resulting solution was stirred for 2 minutes prior to the addition of a freshly prepared solution of NaBH4 (4.015 mL, 0.1 M), such that the molar ratio of NaBH4:(Au + Pd) was 5:1 and the molar ratio of NaBH4:(tertiary metal) was 10:1. Upon the addition of NaBH4 the mixture turned dark brown and was stirred vigorously for an additional 30 min followed by the addition of TiO2 (0.99 g, Degussa P25). The solution was acidified to pH 1 via the addition of H2SO4 (>95%) and stirred for 1 h. Following this, the suspension was filtered under vacuum, washed thoroughly with distilled water, then dried under vacuum (30 °C, 16 h) followed by calcination (static air, 3 h, 400 °C, 10 °C min−1).
Catalyst testing
Direct synthesis of H2O2
Hydrogen peroxide synthesis was evaluated using a Parr Instruments stainless steel autoclave with a nominal volume of 100 mL, equipped with a PTFE liner so that total volume is reduced to 66 mL, and a maximum working pressure of 2000 psi. To test each catalyst for H2O2 synthesis, the autoclave liner was charged with catalyst (0.01 g) and solvent (methanol (5.6 g, HPLC grade, Fischer Scientific) and H2O (2.9 g, HPLC grade, Fischer Scientific)). The charged autoclave was then purged three times with 5% H2/CO2 (100 psi) before filling with 5% H2/CO2 to a pressure of 420 psi, followed by the addition of 25% O2/CO2 (160 psi), with the pressure of 5% H2/CO2 and 25% O2/CO2 given as gauge pressures. The reactor was not continually fed with reactant gas. The reaction was conducted at a temperature of 2 °C for 0.5 h with stirring (1200 rpm). The above reaction parameters are based on optimum conditions we have previously used for the synthesis of H2O2.30 The H2O2 productivity was determined by titrating aliquots of the final solution after reaction with acidified Ce(SO4)2 (0.0085 M) in the presence of ferroin indicator. Catalyst productivities are reported as molH2O2 kgcat−1 h−1. To collect a series of data points, as in the case of Fig. 3, it should be noted that individual experiments were carried out and the reactant mixture was not sampled on-line.The catalytic conversion of H2 and selectivity towards H2O2 were determined using a Varian 3800 GC fitted with TCD and equipped with a Porapak Q column.
H2 conversion (eqn (1)) and H2O2 selectivity (eqn (2)) are defined as follows:
 | (1) |
 | (2) |
Gas replacement experiments for the direct synthesis of H2O2
An identical procedure to that outlined above for the direct synthesis reaction was followed for a reaction time of 0.5 h. After this, stirring was stopped and the reactant gas mixture was vented prior to replacement with the standard pressures of 5% H2/CO2 (420 psi) and 25% O2/CO2 (160 psi). The reaction mixture was then stirred (1200 rpm) for a further 0.5 h. To collect a series of data points, as in the case of Fig. 5, it should be noted that individual experiments were carried out and the reactant mixture was not sampled on-line.Catalyst reusability in the direct synthesis and degradation of H2O2
In order to determine catalyst reusability, a similar procedure to that outlined above for the direct synthesis of H2O2 is followed utilising 0.05 g of catalyst. Following the initial test, the catalyst was recovered by filtration and dried (30 °C, 16 h, under vacuum); from the recovered catalyst sample 0.01 g was used to conduct a standard H2O2 synthesis or degradation test.Degradation of H2O2
Catalytic activity towards H2O2 degradation was determined in a similar manner to the direct synthesis activity of a catalyst. The autoclave liner was charged with solvent (methanol (5.6 g, HPLC grade, Fischer Scientific) and H2O (2.9 g, HPLC grade, Fischer Scientific)) and H2O2 (50 wt% 0.69 g, Sigma Aldrich), with the solvent composition equivalent to a 4 wt% H2O2 solution. From the resulting solution, two 0.05 g aliquots were removed and titrated with acidified Ce(SO4)2 using ferroin as an indicator to determine an accurate concentration of H2O2 at the start of the reaction. Subsequently the catalyst (0.01 g) was added to the reaction media and the autoclave was purged with 5%H2/CO2 (100 psi) prior to being pressurised with 5% H2/CO2 (420 psi). The reaction solution was cooled to a temperature of 2 °C, prior to stirring (1200 rpm) for 0.5 h. After the reaction was complete the catalyst was removed from the reaction mixture and two 0.05 g aliquots were titrated against the acidified Ce(SO4)2 solution using ferroin as an indicator. The degradation activity is reported as molH2O2 kgcat−1 h−1.In all cases reactions were run multiple times, over multiple batches of catalyst, with the data being presented as an average of these experiments. The catalytic activity toward the direct synthesis and subsequent degradation of H2O2 was found to be consistent to within ±3% on the basis of multiple reactions.
Characterisation
The as-prepared aqueous sols, contained in a quartz cuvette, were optically characterised using a UV-vis spectrometer (V-570, JASCO) operating over the 200 to 800 nm wavelength range.Thermo Scientific K-Alpha+ photoelectron spectrometer was used to collect XP spectra utilising a micro-focused monochromatic Al Kα X-ray source operating at 72 W. Samples were pressed into a copper holder and analysed using the 400 μm spot mode at pass energies of 40 and 150 eV for high-resolution and survey spectra respectively. Charge compensation was performed using a combination of low energy electrons and argon ions, which resulted in a C(1s) binding energy of 284.8 eV for the adventitious carbon present on all samples and all samples also showed a constant Ti(2p3/2) of 458.5 eV. All data was processed using CasaXPS v2.3.2442 using a Shirley background, Scofield sensitivity factors43 and an electron energy dependence of −0.6 as recommended by the manufacturer. Peak fits were performed using a combination of Voigt-type functions and models derived from bulk reference samples where appropriate. Analysis of catalytic samples, after use in the direct synthesis of H2O2 was conducted after the sample was dried under vacuum (30 °C, 16 h).
The bulk structure of the catalysts was determined by powder X-ray diffraction using a (θ–θ) PANalytical X'pert Pro powder diffractometer using a Cu Kα radiation source, operating at 40 keV and 40 mA. Standard analysis was carried out using a 40 min run with a back filled sample, between 2θ values of 10–80°. Phase identification was carried out using the International Centre for Diffraction Data (ICDD).
Transmission electron microscopy (TEM) was performed on a JEOL JEM-2100 operating at 200 kV. Samples were prepared by dispersion in ethanol by sonication and deposited on 300 mesh copper grids coated with holey carbon film. Energy dispersive X-ray spectroscopy (XEDS) was performed using an Oxford Instruments X-MaxN 80 detector and the data analysed using Aztec software. Aberration corrected scanning transmission electron microscopy (AC-STEM) was performed using a probe-corrected Hitachi HF5000 S/TEM, operating at 200 kV. The instrument was equipped with bright field (BF), high angle annular dark field (HAADF) and secondary electron (SE) detectors for high spatial resolution STEM imaging experiments. This microscope was also equipped with a secondary electron detector and dual Oxford Instruments XEDS detectors (2 × 100 mm2) having a total collection angle of 2.02 sr.
Total metal leaching from the supported catalysts was quantified via inductively coupled plasma mass spectrometry (ICP-MS). Post-reaction solutions were analysed using an Agilent 7900 ICP-MS equipped with I-AS auto-sampler. All samples were diluted by a factor of 10 using HPLC grade H2O (1%HNO3 and 0.5% HCl matrix). All calibrants were matrix matched and measured against a five-point calibration using certified reference materials purchased from Perkin Elmer and certified internal standards acquired from Agilent.
To allow for quantification of total metal loading catalytic samples were digested via a HF assisted microwave digestion method using a Milestone Connect Ethos UP microwave with an SK15 sample rotor. Digested samples were analysed by inductively coupled plasma-optical emission spectroscopy (ICP-OES). All calibrants were matrix matched and measured against a five-point calibration using certified reference materials purchased from Perkin Elmer and certified internal standards acquired from Agilent. Actual metal loadings of key catalytic samples are provided in Table S2.†
Results and discussion
Prior to preparation of the tertiary metal catalysts the as-synthesised precious metal colloids consisting of Au and a range of secondary metals were analysed by UV/vis spectroscopy (Fig. S2†) with no characteristic plasmon resonance band for Au observed in the bi-metallic colloids, indicative of the formation of alloyed nanoparticles. It should be noted that such analysis alone cannot definitely identify the presence of alloyed species, as the data does not provide any insight into the environment of the other constituent metals. However, the metals chosen as dopants have also been widely reported to readily from bi-metallic alloy with Pd, this in conjunction with our UV/vis analysis supports the formation of tertiary metal alloys. Our initial studies established the ability of a range of metals (Pt, Ni, Co, Cu, In, Sn, Ga, Zn) at dopant concentrations (theoretical loading of 0.025 wt%), to modify the catalytic performance of a 1% AuPd(1.00)/TiO2 catalyst, prepared via a sol-immobilisation methodology,31 towards the direct synthesis and subsequent degradation of H2O2 (Table 1).| Catalyst | Productivity/molH2O2 kgcat−1 h−1 | H2O2 Conc./wt% | Degradation/molH2O2 kgcat−1 h−1 |
|---|---|---|---|
|
H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 2 °C, 1200 rpm. H2O2 degradation reaction conditions: catalyst (0.01 g), H2O2 (50 wt% 0.68 g) H2O (2.22 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 0.5 h, 2 °C, 1200 rpm. Note: values in parentheses refer to metal loading of (Au + Pd) or tertiary metal. In all instances total metal loading is 1 wt% and Au:Pd:X = 1:1 (mol mol−1).
|
|||
| 1% Au(1.00)/TiO2 | 3 | 0.007 | 3 |
| 1% Pd(1.00)/TiO2 | 42 | 0.085 | 41 |
| 1% AuPd(1.00)/TiO2 | 61 | 0.125 | 215 |
| 1% AuPd(0.975)Pt(0.025)/TiO2 | 106 | 0.216 | 158 |
| 1% AuPd(0.975)Ni(0.025)/TiO2 | 107 | 0.215 | 203 |
| 1% AuPd(0.975)Sn(0.025)/TiO2 | 78 | 0.159 | 170 |
| 1% AuPd(0.975)Cu(0.025)/TiO2 | 94 | 0.188 | 169 |
| 1% AuPd(0.975)Co(0.025)/TiO2 | 71 | 0.143 | 228 |
| 1% AuPd(0.975)In(0.025)/TiO2 | 77 | 0.154 | 163 |
| 1% AuPd(0.975)Ga(0.025)/TiO2 | 70 | 0.142 | 159 |
| 1% AuPd(0.975)Zn(0.025)/TiO2 | 100 | 0.197 | 191 |
In keeping with our previous studies,30,31,44,45 the incorporation of Pt into supported AuPd nanoparticles was seen to result in a significant improvement in catalytic activity towards H2O2 synthesis (106 molH2O2 kgcat−1 h−1), compared to the bi-metallic 1% AuPd(1.00)/TiO2 analogue (61 molH2O2 kgcat−1 h−1). Interestingly, the addition of several base metals; Ni, (107 molH2O2 kgcat−1 h−1) Zn, (100 molH2O2 kgcat−1 h−1) and Cu (94 molH2O2 kgcat−1 h−1) was also observed to improve catalytic activity considerably, achieving H2O2 synthesis rates far greater than the bimetallic 1% AuPd(1.00)/TiO2 catalyst, and comparable to that offered by 1% AuPd(0.975)Pt(0.025)/TiO2 analogue. While the incorporation of Ni17,18,46 and Zn21,22 into supported precious metal catalysts has previously been reported to result in an improvement in catalytic performance towards H2O2 production, the addition of Cu, either into AuPd47 or Pd-only48 catalysts has been found to inhibit catalyst activity towards H2O2 synthesis. With DFT studies by Joshi et al. indicating that the formation of the intermediate hydroperoxy (OOH*) species and in turn H2O2, to be thermodynamically unfavourable over Cu-containing supported catalysts.49 Although these prior studies have typically focused on the incorporation of Cu at concentrations far greater than that utilised in this work.
Building on our initial observations and with a focus on the Ni, Zn and Cu containing catalysts, we subsequently investigated the effect of tertiary metal content on catalytic activity towards the direct synthesis and subsequent degradation of H2O2 (Fig. 1a–c). In keeping with our previous studies into the introduction of small quantities of Pt into supported AuPd catalysts,30,31 the addition of Ni, Zn and Cu at 0.025 wt% was found to improve catalytic performance towards H2O2 formation considerably. However, further tertiary metal addition led to a reduction in H2O2 synthesis rates. As expected the addition of Cu at relatively high loadings was observed to have a significant detrimental effect on catalytic activity towards H2O2 formation,47,48 with the activity of the 1% AuPd(0.9)Cu(0.1)/TiO2 catalyst (38 molH2O2 kgcat−1 h−1) significantly lower than the parent 1% AuPd(1.00)/TiO2 material. Analysis of these catalytic series by XPS (Fig. S3†) reveals that the introduction of small concentrations of the tertiary metal dopant (Ni, Zn, Cu) into a AuPd nanoalloy results in a considerable shift in Pd speciation from that observed in the bimetallic catalyst. We further observe that the optimal catalytic compositions (1% AuPd(0.975)Ni(0.025)/TiO2, 1% AuPd(0.975)Cu(0.025)/TiO2 and 1% AuPd(0.975)Zn(0.025)/TiO2) consist of a mixture of Pd0 and Pd2+, with the addition of greater concentrations of the dopant metal generally leading to a shift towards Pd2+, which is known to offer lower rates of H2O2 synthesis than Pd0 species.50 This correlates well with our observations, with catalytic activity towards H2O2 synthesis decreasing with the addition of greater concentrations of the dopant metal.
 | ||
| Fig. 1 The effect of tertiary metal incorporation into 1% AuPd(1.00)/TiO2 on catalytic activity towards the direct synthesis of H2O2. (a) 1% AuPd(X)Ni(1−X)/TiO2, (b) 1% AuPd(X)Cu(1−X)/TiO2, (c) 1% AuPd(X)Zn(1−X)/TiO2. H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 2 °C, 1200 rpm. | ||
With the evident improvement in activity upon introduction of low concentrations of Ni, Zn and Cu into a supported AuPd catalyst, we were subsequently motivated to investigate this sub-set of four catalysts in order to gain further insight into the underlying cause for the observed differences in performance.
An assessment of catalytic selectivity towards H2O2 and H2 conversion of the set of key catalysts is presented in Table 2. Upon incorporation of the three base metals (Ni, Cu and Zn), H2 conversion was observed to increase significantly in comparison to the bimetallic AuPd catalyst, correlating well with the observed improvement in H2O2 synthesis rates. However, unlike in our earlier studies into supported AuPd catalysts that incorporate dopant levels of Pt30,31 the presence of Ni, Cu and Zn did not result in an improvement in H2O2 selectivity, with this metric decreasing somewhat when compared to the 1% AuPd(1.00)/TiO2 catalyst (59%). While this could lead to the inference that the incorporation of the tertiary metals results in a reduction in catalytic selectivity it is important to make such comparisons at near-equivalent rates of H2 conversion. A comparison of the selectivity of the supported catalysts at near iso-conversion is presented in Table S3,† from which it is clear that, while the introduction of the transition metals at dopant concentrations does reduce catalytic selectivity, the extent of such a reduction is not as substantial as may be inferred from the data presented in Table 2. Regardless it is therefore possible to conclude that the enhanced performance of the 1% AuPd(0.975)X(0.025)/TiO2 catalysts is related to the ability of the transition metals to increase H2O2 production, rather than promote catalytic selectivity.
| Catalyst | H2 conversion/% | H2O2 selectivity/% | Productivity/molH2O2 kgcat−1 h−1 | H2O2 Conc./wt% | Reaction rate/mmolH2O2 mmolmetal−1 min−1 | Degradation/molH2O2 kgcat−1 h−1 |
|---|---|---|---|---|---|---|
|
H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 2 °C, 1200 rpm. H2O2 degradation reaction conditions: catalyst (0.01 g), H2O2 (50 wt% 0.68 g) H2O (2.22 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 0.5 h, 2 °C, 1200 rpm. Note 1: reaction rates upon are calculated based on the as determined metal content (see Table S2†). Note 2: values in parentheses refer to metal loading of (Au + Pd) or tertiary metal. In all instances total metal loading is 1 wt% and Au:Pd:X = 1:1 (mol mol−1).
|
||||||
| 1% AuPd(1.00)/TiO2 | 12 | 59 | 61 | 0.125 | 14.06 | 215 |
| 1% AuPd(0.975)Ni(0.025)/TiO2 | 32 | 41 | 107 | 0.215 | 25.16 | 294 |
| 1% AuPd(0.975)Cu(0.025)/TiO2 | 31 | 40 | 94 | 0.188 | 21.39 | 169 |
| 1% AuPd(0.975)Zn(0.025)/TiO2 | 24 | 50 | 100 | 0.197 | 23.30 | 191 |
Evaluation of the as-prepared 1% AuPd(0.975)X(0.025)/TiO2 catalysts by XPS can be seen in Fig. 2. As discussed above, the introduction of low quantities of Ni, Cu and Zn results in a significant shift in Pd oxidation state, towards Pd0, coinciding with an increase in H2O2 synthesis rate and decreased selectivity towards H2O2 (Table 2). While this observation may be surprising given the oxidative heat treatment (400 °C, 3 h, static air) applied to these materials prior to use it is in keeping with our earlier studies into supported AuPdPt catalysts, where the introduction of Pt was found to be a key modifier of Pd oxidation state.31 With the performance of Pd-based catalysts towards H2O2 synthesis well known to be highly dependent on Pd oxidation state, with domains of mixed Pd2+–Pd0 species offering enhanced performance compared to those with a predominance of Pd in either oxidation state,31,51 it is possible to correlate the shift in catalytic activity with changes in Pd speciation.
 | ||
| Fig. 2 Pd(3d)/Au(4d) region for (a) 1% AuPd(1.00)/TiO2, (b) 1% AuPd(0.975)Ni(0.025)/TiO2, (c) 1% AuPd(0.975)Cu(0.025)/TiO2 and (d) 1% AuPd(0.975)Zn(0.025)/TiO2 catalysts key: Au(4d) (red), Pd0 (blue), Pd2+ (magenta). | ||
With our XPS analysis revealing a modification in Pd oxidation state results from the incorporation of dopant concentrations of Ni, Cu and Zn we were subsequently motivated to probe the key catalytic species via CO-DRIFTS (Fig. 3). Perhaps unsurprisingly the DRIFTS spectra of all catalysts is dominated by Pd–CO bands. The peak observed at 1990 cm−1 represents CO bonded linearly to low co-ordination Pd sites (i.e. edge or corner sites, denoted Pd–CO), while the broad feature, which is centred around 1940 cm−1 represents the 2- and 3-fold adsorption of CO on Pd. Upon the introduction of small quantities of dopant metal into the AuPd nanoalloy, a small red-shift in both the band related to the linearly bonded CO on Pd and the bridging CO species. This shift is possibly a result of the charge-transfer to Pd d-orbitals, resulting in enhanced back donation to 2π CO molecular orbitals. In keeping with our observations, Ouyang et al.34 have previously reported a similar transfer of electron density upon the alloying of Au and Pd with an associated suppression of O–O bond scission, which is in keeping the observed loss of catalytic activity towards H2O2 degradation, which results from the incorporation of small quantities of tertiary base metal.
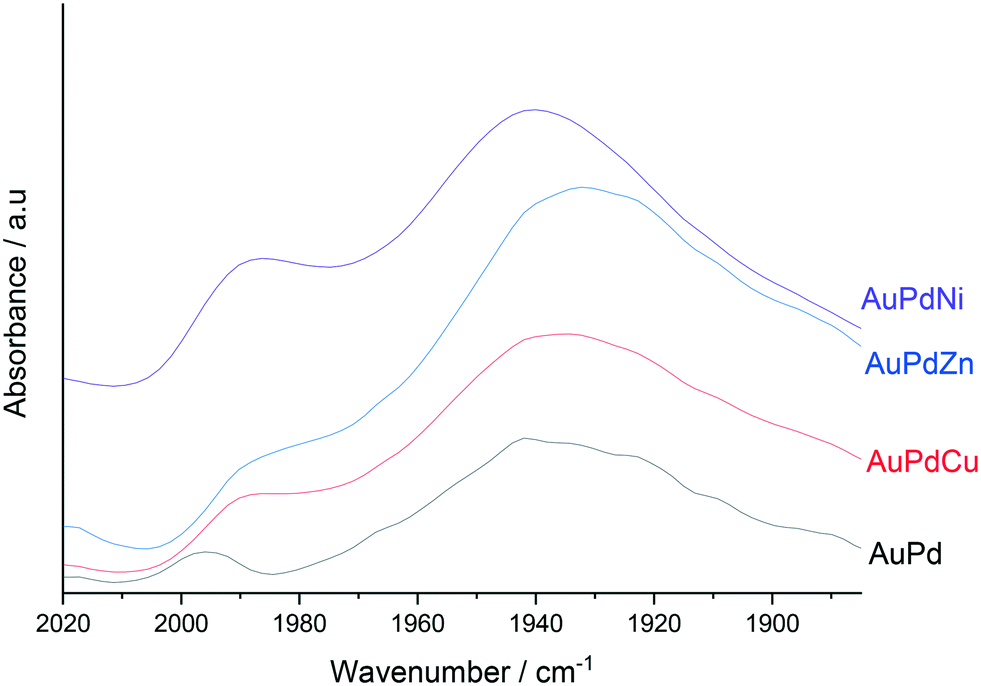 | ||
| Fig. 3 CO-DRIFTS spectra for 1% AuPd(0.975)X(0.025)/TiO2 catalysts in the as-prepared state. | ||
Numerous studies have elucidated the dependence between particle size and catalytic performance towards the direct synthesis of H2O2,52,53 with work by Tian et al. in particular highlighting that an optimal particle size in the sub-nanometre range is desirable for achieving high catalytic performance towards H2O2 production, with larger nanoparticles favouring H2O2 degradation pathways.54,55 Measurements of mean particle size for the various 1% AuPd(0.975)X(0.025)/TiO2 catalysts, as determined from the bright field transmission electron micrographs presented in Fig. S4† are reported in Table 3, with minimal variation observed across the subset of catalysts. As such, it is reasonable to propose that the enhancement in catalytic activity, achieved through incorporation of tertiary base metals cannot be associated with increased nanoparticle dispersion. We consider that these observations, in addition to our analysis via UV/vis-spectroscopy, XPS and CO-DRIFTS strongly support the formation of tri-metallic alloyed nanoparticles.
| Catalyst | Mean particle size/nm (standard deviation) | Productivity/molH2O2 kgcat−1 h−1 (H2O2 wt%) |
|---|---|---|
|
Note: values in parentheses refer to metal loading of (Au + Pd) or tertiary metal. In all instances total metal loading is 1 wt% and Au:Pd:X = 1:1 (mol mol−1).
|
||
| 1% AuPd(1.00)/TiO2 | 4.9 (1.66) | 61 (0.125) |
| 1% AuPd(0.975)Ni(0.025)/TiO2 | 4.1 (1.47) | 107 (0.215) |
| 1% AuPd(0.975)Cu(0.025)/TiO2 | 5.4 (1.35) | 94 (0.188) |
| 1% AuPd(0.975)Zn(0.025)/TiO2 | 5.8 (1.40) | 98 (0.197) |
Time-on-line studies comparing H2O2 synthesis rates over the bi-metallic 1% AuPd(1.00)/TiO2 and tri-metallic 1% AuPd(0.975)X(0.025)/TiO2 catalysts can be seen in Fig. 4, with a stark difference in catalytic activity observed. Indeed, the enhanced activity of the 1% AuPd(0.975)X(0.025)/TiO2 catalysts is clear, with all tertiary metal containing catalysts achieving H2O2 concentrations (0.22–0.26 wt%) far greater than that of the AuPd analogue (0.17 wt%), with the net concentrations of H2O2 achieved over the dopant containing catalysts comparable to that we have previously reported over an optimised 1% AuPdPt/TiO2 catalyst, prepared via a similar methodology, under identical reaction conditions.31 Evaluation of the catalysts by XPS over the course of the H2O2 synthesis reaction indicates a clear shift towards Pd0, which may be expected given the reductive atmosphere used during H2O2 synthesis (Fig. S5†).
 | ||
| Fig. 4 Comparison of the catalytic activity as a function of reaction time. Key: 1% AuPd(1.00)/TiO2 (inverted green triangles), 1% AuPd(0.975)Ni(0.025)/TiO2 (black squares), 1% AuPd(0.975)Cu(0.025)/TiO2 (blue triangles) and 1% AuPd(0.975)Zn(0.025)/TiO2 (red circles). H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 2 °C, 1200 rpm. | ||
The improved performance of the 1% AuPd(0.975)X(0.025)/TiO2 catalysts is further highlighted through comparison of calculated reaction rates (Table S4†) both at reaction times where there is assumed to be no contribution from subsequent degradation reactions and over the course of our standard 0.5 h reaction. Indeed the initial rate of the 1% AuPd(0.975)X(0.025)/TiO2 catalysts is between 2.6 and 3.5 times that of the AuPd analogue. Further evaluation of catalytic activity over multiple sequential H2O2 synthesis tests can be seen in Fig. 5 with a marked enhancement in H2O2 concentration observed for all tertiary metal containing catalysts, when compared to the bimetallic catalyst. After running the reaction five consecutive times, H2O2 concentrations produced over the 1% AuPd(0.975)X(0.025)/TiO2 catalysts (0.47–0.58 wt%) were observed to be between 15 and 33% greater than that observed over the AuPd analogue (0.39 wt%). In particular the 1% AuPd(0.975)Ni(0.025)/TiO2 catalyst was found to achieve concentrations of H2O2 comparable to those previously reported by Freakley et al. using a near 100% selective 3% Pd-2% Sn/TiO2 catalyst,15 this is despite the significantly lower precious metal loading of the catalysts reported within this study.
 | ||
| Fig. 5 Comparison of the catalytic activity over sequential H2O2 synthesis reactions. Key: 1% AuPd(1.00)/TiO2 (inverted green triangles), 1% AuPd(0.975)Ni(0.025)/TiO2 (black squares), 1% AuPd(0.975)Cu(0.025)/TiO2 (blue triangles) and 1% AuPd(0.975)Zn(0.025)/TiO2 (red circles). H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 2 °C, 1200 rpm. | ||
With the requirement to re-use a catalyst successfully at the heart of green chemistry, we next evaluated catalytic activity towards H2O2 synthesis and H2O2 degradation pathways, upon re-use (Table 4). It was found that for all catalysts evaluated H2O2 degradation activity increased significantly upon re-use, coinciding with a reduction in H2O2 synthesis rate. A similar decrease in reaction rate at short reaction times, where the contribution of competitive degradation reactions is assumed to be negligible, was also observed (Table S4†). Notably the increase in H2O2 degradation rates observed over the 1% AuPd(0.975)X(0.025)/TiO2 catalysts was far greater than that observed over the 1% AuPd(1.00)/TiO2 analogue. However, regardless of this loss of selectivity the H2O2 synthesis activity of the 1% AuPd(0.975)X(0.025)/TiO2 catalysts was retained to a far greater extent than that of the bi-metallic analogue. Analysis of the catalysts after use initial use in the direct synthesis reaction, by XPS (Fig. S6,† with elemental quantification shown in Table S5†) revealed a clear shift in Pd-oxidation state, towards Pd0 in all cases, with a further increase in the proportion of Pd0 observed upon second use. With the enhanced activity of Pd0 species towards H2O2 degradation well known56,57 it is therefore possible to, at least in part, attribute the decreased H2O2 selectivity to the in situ reduction of Pd2+ to Pd0 species.
| Catalyst | Productivity/molH2O2 kgcat−1 h−1 (H2O2 wt.%) | Reaction rate/mmolH2O2 mmolmetal−1 min−1 | Degradation/molH2O2 kgcat−1 h−1 | Metal leached/% (ppb) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Use 1 | Use 2 | Use 1 | Use 2 | Use 1 | Use 2 | Au | Pd | X (Ni, Cu, Zn) | |
|
H2O2 direct synthesis reaction conditions: catalyst (0.01 g), H2O (2.9 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 25% O2/CO2 (160 psi), 0.5 h, 2 °C 1200 rpm. H2O2 degradation reaction conditions: catalyst (0.01 g), H2O2 (50 wt% 0.68 g) H2O (2.22 g), MeOH (5.6 g), 5% H2/CO2 (420 psi), 0.5 h, 2 °C 1200 rpm. B.D.L = below detection limits. Note 1: reaction rates upon first use (use 1) are calculated based on the actual metal loading as determined by HF digestion (see Table S2†). Reaction rates upon second use (use 2) account for metal leaching during the initial H2O2 synthesis reaction, as determined by ICP-MS analysis of the post reaction solutions (see Table 4). Note 2: values in parentheses refer to metal loading of (Au + Pd) or tertiary metal. In all instances total metal loading is 1 wt% and Au:Pd:X = 1:1 (mol mol−1).
|
|||||||||
| 1% AuPd(1.00)/TiO2 | 61 (0.125) | 44 | 14.06 | 10.54 | 215 | 268 | 0 | 1.90 (4.8) | — |
| 1% AuPd(0.975)Ni(0.025)/TiO2 | 107 (0.215) | 95 | 25.16 | 22.37 | 203 | 348 | 0 | 2.52 (6.3) | B.D.L |
| 1% AuPd(0.975)Cu(0.025)/TiO2 | 94 (0.188) | 80 | 21.39 | 18.94 | 191 | 391 | 0 | 0.42 (1.1) | B.D.L |
| 1% AuPd(0.975)Zn(0.025)/TiO2 | 100 (0.197) | 95 | 23.30 | 18.40 | 169 | 327 | 0 | 1.74 (4.4) | B.D.L |
For any heterogeneous catalyst operating in a three-phase system the possibility of leaching of active metals and resulting homogeneous contribution to the observed catalytic activity is of great concern. This is particularly true given the ability of homogeneous Pd species to catalyse the direct synthesis of H2O2.58,59 Analysis of the post-reaction solution by ICP-MS (Table 4) revealed the high stability of Au in all cases, however, a degree of Pd (approx. 1–6 ppb) was observed. Notably any potential leaching of the tertiary metal was below the detectable limits of ICP-MS.
Conclusions
The addition of low quantities of earth abundant metals (Ni, Cu, Zn) into supported AuPd nanoparticles results in a significant enhancement in catalytic activity towards the direct synthesis of H2O2, with the activity of the optimal AuPdCu, AuPdNi and AuPdZn catalysts observed to be 1.5–1.8 times greater than that of the bi-metallic analogue. The resulting enhancement is found to be largely associated with increased rates of H2 conversion, rather than through enhancement in catalytic selectivity as in the case of supported AuPdPt catalysts. This improvement is considered to derive from the electronic modification of Pd oxidation state, with the addition of low concentrations of tertiary metals found to promote the formation of Pd2+–Pd0 domains. While both the bimetallic AuPd catalyst and those materials with high tertiary metal content are found to consist predominantly of one Pd oxidation state, although further investigation is needed to fully deconvolute the effect of electronic modification from restructuring of the alloyed nanoparticles. Although catalytic stability may be a concern, primarily resulting from the in situ reduction of Pd2+ the addition of the dopant metal was found to retain Pd speciation of the fresh material to a greater extent than the AuPd analogue. We consider these catalysts represent a promising basis for further exploration for the direct synthesis of H2O2.Author contributions
A. B. and R. J. L. conducted catalytic synthesis, testing and data analysis. A. B., R. J. L., D. J. M. and T. E. D. conducted catalyst characterisation and corresponding data processing. R. J. L. and G. J. H. contributed to the design of the study and provided technical advice and result interpretation. R. J. L. wrote the manuscript and ESI,† with all authors commenting on and amending both documents. All authors discussed and contributed to the work.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors wish to thank the Cardiff University electron microscope facility for the transmission electron microscopy. XPS data collection was performed at the EPSRC National Facility for XPS (‘HarwellXPS’), operated by Cardiff University and UCL, under contract No. PR16195. The Max Planck Centre for Fundamental Heterogeneous Catalysis (FUNCAT) is gratefully acknowledged for financial support.References
- R. J. Lewis and G. J. Hutchings, ChemCatChem, 2019, 11, 298–308 CrossRef CAS.
- J. K. Edwards, S. J. Freakley, R. J. Lewis, J. C. Pritchard and G. J. Hutchings, Catal. Today, 2015, 248, 3–9 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- J. R. Scoville and I. A. Novicova (Cottrell Ltd.), US5900256, 1996.
- P. Wegner, (Wegner Paul C.), US20050065052A1, 2003.
- Y. T. G. Gao, X. Gong, Z. Pan, K. Yong and B. Zong, Chin. J. Catal., 2020, 41, 1039–1047 CrossRef.
- H. Henkel and W. Weber (Henkel AG and Co KGaA), US1108752A, 1914.
- J. K. Edwards, B. Solsona, P. Landon, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2005, 236, 69–79 CrossRef CAS.
- J. K. Edwards, B. Solsona, E. N. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef CAS PubMed.
- A. Staykov, T. Kamachi, T. Ishihara and K. Yoshizawa, J. Phys. Chem. C, 2008, 112, 19501–19505 CrossRef CAS.
- Y. Nomura, T. Ishihara, Y. Hata, K. Kitawaki, K. Kaneko and H. Matsumoto, ChemSusChem, 2008, 1, 619–621 CrossRef CAS PubMed.
- P. K. Sajith, A. Staykov, M. Yoshida, Y. Shiota and K. Yoshizawa, J. Phys. Chem. C, 2020, 124, 13231–13239 CrossRef CAS.
- C. M. Crombie, R. J. Lewis, R. L. Taylor, D. J. Morgan, T. E. Davies, A. Folli, D. M. Murphy, J. K. Edwards, J. Qi, H. Jiang, C. J. Kiely, X. Liu, M. S. Skjøth-Rasmussen and G. J. Hutchings, ACS Catal., 2021, 11, 2701–2714 CrossRef CAS.
- A. Santos, R. J. Lewis, D. J. Morgan, T. E. Davies, E. Hampton, P. Gaskin and G. J. Hutchings, Catal. Sci. Technol., 2021, 11, 7866–7874 RSC.
- S. J. Freakley, Q. He, J. H. Harrhy, L. Lu, D. A. Crole, D. J. Morgan, E. N. Ntainjua, J. K. Edwards, A. F. Carley, A. Y. Borisevich, C. J. Kiely and G. J. Hutchings, Science, 2016, 351, 965–968 CrossRef CAS PubMed.
- F. Li, Q. Shao, M. Hu, Y. Chen and X. Huang, ACS Catal., 2018, 8, 3418–3423 CrossRef CAS.
- D. A. Crole, R. Underhill, J. K. Edwards, G. Shaw, S. J. Freakley, G. J. Hutchings and R. J. Lewis, Philos. Trans. R. Soc., A, 2020, 378, 20200062 CrossRef CAS PubMed.
- S. Maity and M. Eswaramoorthy, J. Mater. Chem. A, 2016, 4, 3233–3237 RSC.
- S. Wang, R. J. Lewis, D. E. Doronkin, D. J. Morgan, J. Grunwaldt, G. J. Hutchings and S. Behrens, Catal. Sci. Technol., 2020, 10, 1925–1932 RSC.
- J. Gu, S. Wang, Z. He, Y. Han and J. Zhang, Catal. Sci. Technol., 2016, 6, 809–817 RSC.
- S. Wang, K. Gao, W. Li and J. Zhang, Appl. Catal., A, 2017, 531, 89–95 CrossRef CAS.
- N. M. Wilson, J. Schröder, P. Priyadarshini, D. T. Bregante, S. Kunz and D. W. Flaherty, J. Catal., 2018, 368, 261–274 CrossRef CAS.
- P. Tian, F. Xuan, D. Ding, Y. Sun, X. Xu, W. Li, R. Si, J. Xu and Y. Han, J. Catal., 2020, 385, 21–29 CrossRef CAS.
- Y. Wang, H. Pan, Q. Lin, Y. Shi and J. Zhang, Catalysts, 2020, 10, 303 CrossRef.
- G. Bernardotto, F. Menegazzo, F. Pinna, M. Signoretto, G. Cruciani and G. Strukul, Appl. Catal., A, 2009, 358, 129–135 CrossRef CAS.
- S. Quon, D. Y. Jo, G. Han, S. S. Han, M. Seo and K. Lee, J. Catal., 2018, 368, 237–247 CrossRef CAS.
- T. Deguchi, H. Yamano, S. Takenouchi and M. Iwamoto, Catal. Sci. Technol., 2018, 8, 1002–1015 RSC.
- M. Kim, G. Han, X. Xiao, J. Song, J. Hong, E. Jung, H. Kim, J. Ahn, S. S. Han, K. Lee and T. Yu, Appl. Surf. Sci., 2021, 562, 150031 CrossRef CAS.
- T. Ricciardulli, J. S. Adams, M. DeRidder, A. P. van Bavel, A. M. Karim and D. W. Flaherty, J. Catal., 2021, 404, 661–678 CrossRef CAS.
- R. J. Lewis, K. Ueura, Y. Fukuta, S. J. Freakley, L. Kang, R. Wang, Q. He, J. K. Edwards, D. J. Morgan, Y. Yamamoto and G. J. Hutchings, ChemCatChem, 2019, 11, 1673–1680 CrossRef CAS.
- X. Gong, R. J. Lewis, S. Zhou, D. J. Morgan, T. E. Davies, X. Liu, C. J. Kiely, B. Zong and G. J. Hutchings, Catal. Sci. Technol., 2020, 10, 4635–4644 RSC.
- H. V. Nguyen, K. Y. Kim, H. Nam, S. Y. Lee, T. Yu and T. S. Seo, Lab Chip, 2020, 20, 3293–3301 RSC.
- H. C. Ham, G. S. Hwang, J. Han, S. W. Nam and T. H. Lim, J. Phys. Chem. C, 2009, 113, 12943–12945 CrossRef CAS.
- L. Ouyang, G. Da, P. Tian, T. Chen, G. Liang, J. Xu and Y. Han, J. Catal., 2014, 311, 129–136 CrossRef CAS.
- H. C. Ham, J. A. Stephens, G. S. Hwang, J. Han, S. W. Nam and T. H. Lim, Catal. Today, 2011, 165, 138–144 CrossRef CAS.
- J. Li, T. Ishihara and K. Yoshizawa, J. Phys. Chem. C, 2011, 115, 25359–25367 CrossRef CAS.
- A. Villa, D. Wang, G. M. Veith, F. Vindigni and L. Prati, Catal. Sci. Technol., 2013, 3, 3036–3041 RSC.
- A. A. Herzing, M. Watanabe, J. K. Edwards, M. Conte, Z. Tang, G. J. Hutchings and C. J. Kiely, Faraday Discuss., 2008, 138, 337 RSC.
- J. K. Edwards, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Faraday Discuss., 2008, 138, 225 RSC.
- A. Santos, R. J. Lewis, G. Malta, A. G. R. Howe, D. J. Morgan, E. Hampton, P. Gaskin and G. J. Hutchings, Ind. Eng. Chem. Res., 2019, 58, 12623–12631 CrossRef CAS.
- M. Piccinini, E. N. Ntainjua, J. K. Edwards, A. F. Carley, J. A. Moulijn and G. J. Hutchings, Phys. Chem. Chem. Phys., 2010, 12, 2488–2492 RSC.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci., 2021, 5, 100112 CrossRef.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- J. K. Edwards, J. Pritchard, P. J. Miedziak, M. Piccinini, A. F. Carley, Q. He, C. J. Kiely and G. J. Hutchings, Catal. Sci. Technol., 2014, 4, 3244–3250 RSC.
- J. K. Edwards, J. Pritchard, L. Lu, M. Piccinini, G. Shaw, A. F. Carley, D. J. Morgan, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2014, 53, 2381–2384 CrossRef CAS PubMed.
- M. J. Banisalman, H. W. Lee, H. Koh and S. S. Han, ACS Appl. Mater. Interfaces, 2021, 13, 17577–17585 CrossRef CAS PubMed.
- M. H. Ab Rahim, R. D. Armstrong, C. Hammond, N. Dimitratos, S. J. Freakley, M. M. Forde, D. J. Morgan, G. Lalev, R. L. Jenkins, J. A. Lopez-Sanchez, S. H. Taylor and G. J. Hutchings, Catal. Sci. Technol., 2016, 6, 3410–3418 RSC.
- F. Alotaibi, S. Al-Mayman, M. Alotaibi, J. K. Edwards, R. J. Lewis, R. Alotaibi and G. J. Hutchings, Catal. Lett., 2019, 149, 998–1006 CrossRef CAS.
- A. M. Joshi, W. N. Delgass and K. T. Thomson, J. Phys. Chem. C, 2007, 111, 7384–7395 CrossRef CAS.
- R. Burch and P. R. Ellis, Appl. Catal., B, 2003, 42, 203–211 CrossRef CAS.
- L. Ouyang, P. Tian, G. Da, X. Xu, C. Ao, T. Chen, R. Si, J. Xu and Y. Han, J. Catal., 2015, 321, 70–80 CrossRef CAS.
- S. Kim, D. Lee, K. Lee and E. A. Cho, Catal. Lett., 2014, 144, 905–911 CrossRef CAS.
- G. Giorgianni, S. Abate, G. Centi and S. Perathoner, ChemCatChem, 2019, 11, 550–559 CrossRef CAS.
- P. Tian, L. Ouyang, X. Xu, C. Ao, X. Xu, R. Si, X. Shen, M. Lin, J. Xu and Y. Han, J. Catal., 2017, 349, 30–40 CrossRef CAS.
- P. Tian, D. Ding, Y. Sun, F. Xuan, X. Xu, J. Xu and Y. Han, J. Catal., 2019, 369, 95–104 CrossRef CAS.
- V. R. Choudhary, A. G. Gaikwad and S. D. Sansare, Catal. Lett., 2002, 83, 235–239 CrossRef CAS.
- A. G. Gaikwad, S. D. Sansare and V. R. Choudhary, J. Mol. Catal. A: Chem., 2002, 181, 143–149 CrossRef CAS.
- S. J. Freakley, N. Agarwal, R. U. McVicker, S. Althahban, R. J. Lewis, D. J. Morgan, N. Dimitratos, C. J. Kiely and G. J. Hutchings, Catal. Sci. Technol., 2020, 10, 5935–5944 RSC.
- D. P. Dissanayake and J. H. Lunsford, J. Catal., 2002, 206, 173–176 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy01962g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |