
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterometallic cooperativity in divalent metal ProPhenol catalysts: combining zinc with magnesium or calcium for cyclic ester ring-opening polymerisation†
Weronika
Gruszka
a,
Haopeng
Sha
a,
Antoine
Buchard
 b and
Jennifer A.
Garden
*a
b and
Jennifer A.
Garden
*a
aEaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: j.garden@ed.ac.uk
bDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
First published on 16th November 2021
Abstract
Homobimetallic zinc complexes have shown extremely high activity in lactide (LA) ring-opening polymerisation (ROP). In contrast, heterobimetallic ROP catalysts combining zinc with other divalent heterometals such as Mg and Ca are unknown, despite heterometallic cooperativity leading to improved catalyst performance with other heterocombinations. Here, we report the first heterobimetallic LA ROP catalysts featuring only divalent metals, by systematically replacing one Zn centre in a bis-Zn ProPhenol complex with Mg or Ca. The Mg/Zn and Ca/Zn complexes are highly active and outperform their homobimetallic counterparts in rac-LA and ε-caprolactone (ε-CL) ROP. The Ca/Zn complex exhibits the highest activity, converting 48 eq. rac-LA in 5 s and 380 eq. ε-CL in 30 s at room temperature. The activity enhancements are credited to Lewis acidic Mg and Ca enhancing monomer coordination as well as altering the catalyst electronics by increasing the polarity of the Zn–Et initiating group. The heterobimetallic catalysts also display unusual two-step kinetic plots in rac-LA ROP, which is attributed to the initial catalyst preference for the coordination and insertion of one of the rac-LA stereoisomers.
Introduction
Cyclic ester ring-opening polymerisation (ROP) is a promising strategy for the production of degradable polymers,1–3 such as poly(lactic acid) (PLA) from bioderived lactide (LA) and poly(ε-caprolactone) (PCL). These aliphatic polyesters have packaging,4 electronic and biomedical applications,5 where the material properties are dictated by the polymer microstructure. While these polymers can be prepared using organocatalysts and enzymes, well-defined organometallic ROP complexes have been especially efficient at combining high catalytic activities with exquisite polymerisation control.1 In particular, homobimetallic complexes (e.g. bis-Al, Hf, In, Mg, Ti, Y, Zn and Zr) have exhibited significant activity enhancements compared to their monometallic counterparts.6 This has been attributed to close proximity between multiple metal sites facilitating monomer activation and nucleophilic attack, which are key steps in ROP.6–10 Nature has long exploited multi- and often hetero-metallic catalysis, where (hetero)metallic enzymes enable and accelerate biological transformations.11,12Inspired by nature, chemists have utilised heterometallic cooperativity in different fields including metal–halogen exchange,13 C–H bond activation,14 asymmetric catalysis15 and olefin polymerisation.16 Some heterometallic catalysts exhibit a cooperative effect, where the metal centres work together to achieve activities and selectivities that are “greater than the sum of their parts”.16–19 However, the concept remains relatively underexplored in cyclic ester ROP, with most catalyst design instead focussed on ligand modification, in spite of the exceptional activity and selectivity enhancements achieved with some heterometallic ROP catalysts. A recent survey revealed emerging structural trends for heterometallic cooperativity in ROP.20 The most active heterometallic catalysts generally comprise medium/large Lewis acidic metals (M1 = group 1, 2 or lanthanide) combined with a more electronegative metal (M2), e.g. Al/Zn,21 La/Mg,22 Li/In,23 Li/Mg and Li/Zn,24 Li/Sm,25 Mg/Al,26 Na/Sm,27 Sm/Al,28 and Ti/Zn.29 Typically supported by M1-O-M2 ligand frameworks, electronic communication between the heterometals can enable the formation of “ate” complexes through partial or complete transfer of an anion from the harder metal to the softer, more carbophilic metal (e.g. Li+ with MgR3−/ZnR3− to form lithium magnesiates or lithium zincates, respectively, Fig. 1).18 We recently reported two heterometallic Na/Zn2 and K/Zn2 catalysts supported by a ProPhenol ligand,30 which outperform the homometallic analogues by combining exceptional activity (Na or K) with good polymerisation control (Zn2). The K/Zn2 analogue is the most active heterometallic catalyst for rac-LA ROP reported to date (kobs = 1.7 × 10−2 s−1). Based on experimental and computational studies, the high activity of the Na/Zn2 and K/Zn2 ProPhenol complexes was attributed to an “ate” activation simultaneously enhancing both the Lewis acidity of the hard metal (M1 = Na or K) and the nucleophilicity of the alkoxide co-ligands surrounding the softer metal (M2 = Zn).18,31,32 These features were proposed to enhance monomer coordination (M1) and nucleophilic attack (from the anionic activation of the co-ligand(s) on M2); both are key mechanistic steps in cyclic ester ROP.
 | ||
| Fig. 1 General structural motifs of “ate” complexes and heterometallic M/Zn ProPhenol complexes (M = Mg or Ca) reported in this work. | ||
Most of the reported heterometallic ROP catalysts feature MI/MII and MI/MIII combinations.20 To the best of our knowledge, no heterometallic catalysts comprising only divalent metals (M1II/M2II) have been reported for LA ROP. This is surprising, considering that some of the most active catalysts for LA ROP are bis-Zn(II) complexes.33–36 Furthermore, heterobimetallic Mg/Zn complexes have outperformed homobimetallic Zn and Mg analogues in carbon dioxide (CO2)/epoxide ring-opening copolymerisation (ROCOP) and asymmetric transformations.19,37 A study by Trost et al. demonstrated that an in situ generated Mg/Zn ProPhenol complex gives enhanced activity and diastereoselectivity (vs. the bis-Zn ProPhenol analogue) in asymmetric Michael reactions.38 Despite this heterometallic activity enhancement, the homometallic bis-Zn ProPhenol complex remains the catalyst of choice for asymmetric transformations, with over 50 applications reported since 2000.39 The excellent catalyst performance is attributed to the two inequivalent Zn centres; one (tricoordinate) Zn acts as a Lewis acid whereas the second (tetracoordinate) Zn acts as the source of a Brønsted base, enabling simultaneous nucleophile and electrophile activation. The different roles of these two metals are likely to be elevated by replacing the tricoordinate Zn with Lewis acidic Mg or Ca but this remains unexplored. Divalent Mg and Ca are particularly attractive metal choices due to their low cost and toxicity, and earth abundance. Herein, we report the first isolated heterobimetallic Zn-based ProPhenol catalysts featuring Mg and Ca heterometals, and describe their propensity to form magnesium and calcium zincates (Fig. 1).13,40 We also employ these ProPhenol complexes in cyclic ester ROP and reveal how the divalent heterometal nature influences the activity compared to the homobimetallic analogues. For the first time within ROP, this is done by performing a systematic study, where one Zn centre in the bis-Zn ProPhenol complex is directly replaced with Mg or Ca without significantly altering the coordination environment of the metal centres or the number of initiating groups.
Results and discussion
Synthesis of homo- and hetero-metallic ProPhenol complexes
Heterobimetallic complexes [LMgZnEt(THF)(HMDSH)] (1) and [LCaZnEt(THF)(HMDSH)] (2) were synthesised via sequential deprotonation of the ProPhenol ligand (LH3) with 1 eq. [M(HMDS)2(THF)x] (HMDS = hexamethyldisilazane, where M = Mg (x = 0) or Ca (x = 2)) followed by 1 eq. ZnEt2, in THF at room temperature (Scheme 1). Firstly, the monometallation of LH3 with Mg(HMDS)2 and Ca(HMDS)2(THF)2 was determined by NMR spectroscopy in THF-d8, which showed that the reaction was complete within 15 minutes at room temperature. Diagnostic phenolic-OH1H NMR signals were observed at 5.61 ppm for [LHMg] and 4.93 ppm for [LHCa] (Fig. S1†), suggesting that LH3 was deprotonated at both benzylic OH positions in spite of the greater acidity of phenolic protons compared to benzylic OH protons (respective pKa values of 9.95 and 13–15). This strong selectivity for benzylic-OH deprotonation was attributed to the steric bulk of the HMDS co-ligands, as metalation of LH3 with 1 eq. of Mg(CH2SiMe3)2 or Mg(nBu)2 gave a mixture of products, which included bis-Mg species as well as unreacted LH3. However, other factors, including metal ionic radius and Brønsted basicity may also play a role. DOSY NMR analysis indicated that [LHMg] displayed a dimeric structure in THF-d8 whilst [LHCa] is monomeric, although both displayed evidence of further aggregation to trimeric and dimeric species, respectively (Fig. S2 and S3†). To allow benchmarking experiments (vide infra), the homobimetallic complex [LZn2Et] (3, Scheme 1) and heterotrimetallic complexes [LNaZn2Et2(THF)2] (4, Fig. 2) and [LKZn2Et2(THF)2] (5, Fig. 2) were synthesised as previously reported,30,36 whereas [LMg2HMDS] (6) and [LCa2HMDS] (7) were synthesised via deprotonation of LH3 with 2 eq. of Mg(HMDS)2 or Ca(HMDS)2(THF)2, respectively, in THF at R.T. (Scheme 1, refer to ESI† for details). | ||
| Scheme 1 Synthesis of heterobimetallic Mg/Zn (1) and Ca/Zn (2) and homobimetallic Zn (3), Mg (6) and Ca (7)41 ProPhenol complexes. a Based on DFT calculations, phenolic OH is proposed to be labile and could be on the pyrrolidine N or on the phenolic O (see ESI†). b For clarity, THF and HMDSH are not shown in the chemical structures of 1 and 2 as the 1H NMR chemical shifts and DOSY analysis suggest these Lewis donors are in coordinative equilibrium with THF-d8 solvent (see Fig. S4 and S8†). Reagents and conditions: (i) 1 eq. Mg(HMDS)2 or Ca(HMDS)2(THF)2, THF, R.T., 15 min; (ii) 1 eq. ZnEt2, THF, R.T., 16 h; (iii) 2 eq. ZnEt2, Mg(HMDS)2 or Ca(HMDS)2(THF)2, THF, R.T., 16 h. | ||
 | ||
| Fig. 2 The Zn–Et 1H NMR shifts and the electronegativity and ionic radii values for complexes 1–5. The complexes are shown in the order of increasing heterometal electronegativity. | ||
Deprotonation of [LHMg] and [LHCa] with 1 eq. ZnEt2 in THF at room temperature generated heterobimetallic complexes 1 and 2 as characterised by NMR spectroscopy, mass spectrometry and elemental analysis (see ESI†). 1H NMR analysis of 1 (Fig. S4†) and 2 (Fig. S8†) in THF-d8 indicated that both complexes are asymmetric, evidenced by the doublet of doublets at 6.58 and 6.64 ppm, and 6.60 and 6.61 ppm, respectively, corresponding to the meta-phenolic protons on the ligand backbone. The coupling of the meta-phenolic protons in 1 and 2 was confirmed by COSY NMR, indicating that the two heterometals are part of the same complex. While the related bis-Zn complex 3 was also asymmetric in the solution-state,36 the more upfield resonances of the meta-phenolic protons in 1–2vs.3 (6.70 and 6.74 ppm, Fig. S19†) suggest increased shielding due to incorporation of the more electropositive Mg and Ca (Fig. 2). Importantly, other 1H NMR signals of 1–2 were also distinct from 3, suggesting that no metal redistribution to homobimetallic species occurs in solution. In line with previous reports of magnesium and calcium zincates,13,40 in both 1 and 2 Zn is proposed to bear the nucleophilic/Brønsted basic ethyl (Et) group, as the 1H NMR resonances of 1 (−0.06 and 0.89 ppm) and 2 (−0.13 and 1.00 ppm) lie significantly closer to the chemical shifts of ZnEt2 (0.03 and 1.12 ppm) compared to MgEt2 (δ = −0.76 and 1.19 ppm). Indicative of “ate” character, the Et moieties in 1–2 were significantly shifted upfield compared to homobimetallic 3 (δ = 0.70 and 1.57 ppm, Fig. 2).36 This trend was also observed with related heterometallic Na/Zn2 and K/Zn2 ProPhenol complexes 4 and 5, where the Zn–Et shifts were observed at −0.48–−0.30 and 0.55 ppm (4) and −0.52–−0.38 and 0.58 ppm (5) (Fig. 2).30 The Zn–Et 1H NMR shifts reflect the electronegativity of the heterometal, with less electronegative metals (from Zn to K) giving a greater upfield shift due to the increased polarity of the Zn–Et bonds (χZn 1.65, δZn–CH2 = 0.70 ppm > Mg χMg 1.31, δZn–CH2 = −0.06 ppm > Ca χCa 1.00, δZn–CH2 = −0.13 ppm > Na χNa 0.93, δZn–CH2 = −0.48–−0.30 ppm > K χK 0.82, δZn–CH2 = −0.52–−0.38 ppm) (Fig. 2).42 These observations suggest that the heterometal increases the polarity (and nucleophilicity) of the Zn–R bond. The Mg and Ca centres in 1 and 2, respectively, are proposed to form strong M-HMDSH and -THF adducts as 1 eq. of each of these Lewis donors were observed by NMR analysis of 1 and 2, with both complexes undergoing full or partial decomposition upon HMDSH removal (by in vacuo solvent removal at ambient temperature and at 130 °C, azeotroping with toluene and/or washing 1–2 with pentane).
DFT optimisation indicated that the lowest energy molecular structures of 1′ and 2′ (′ denotes computationally modelled structures, see ESI†) involve coordination of both THF and HMDSH, with hydrogen bonding between the HMDSH proton and a benzylic O atom of L (Tables S8 and S9,†Fig. 3).
 | ||
| Fig. 3 Molecular structures of 1′ and 2′ with the lowest free enthalpies, as computed by DFT (refer to Tables S8 and S9†). | ||
Furthermore, DFT also suggested that the THF molecule coordinated to Mg or Ca in 1′ and 2′, respectively, faces in the opposite direction to the Zn–Et moiety relative to the phenol ring plane, with retention of the R,R configuration at the N atoms upon metal coordination, as previously demonstrated for 4′–5′;30 a similar Zn-THF/Et orientation was previously shown for a related [LZn2(4-nitrophenol)] complex by X-ray crystallography.43 These results highlight the ability of Mg and Ca to coordinate Lewis donor(s), and DOSY NMR analysis of 1 and 2 in THF-d8 and toluene-d8 (Fig. S5 and S6, S9–S10†) indicated that while both HMDSH and THF are present, they are likely to be in a coordinative equilibrium with the complexes in the solution-state. These observations suggested that 1 and 2 could be suitable candidates for ROP catalysis, as (Lewis basic) monomer coordination is a key step in lactone polymerisation.
Benchmarking heterometallic ProPhenol complexes against homometallic analogues in rac-LA ROP
Complexes 1 and 2 exhibited excellent activity in rac-LA ROP with 1 eq. benzyl alcohol (BnOH) in toluene at 60 °C at 1 mol% catalyst loading, and maintained high activity at lower catalyst loadings of 0.33–0.1 mol%, producing PLA with Mn values up to 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 g mol−1 (Tables 1 and S1†). The presence of BnOH was essential for the high activity of 1–2 (entries 1 and 6, Table 1), which was attributed to the in situ deprotonation of BnOH by Zn–Et bonds of 1–2 (vide infra). Heterobimetallic 1 and 2 were benchmarked against their homobimetallic counterparts to investigate the nature of heterometallic cooperativity with 1–2 in rac-LA ROP (Tables 1 and S1†). Complex 2 was significantly more active than 1 (entries 8 and 2, Table 1) and outperformed both the bis-Zn complex 3 (entry 11) and bis-Ca complex 7 (Scheme 1, entry 15). Interestingly, 1 showed similar activity to the bis-Zn complex 3 (entries 3 and 12, Table 1),36 yet was more active than the bis-Mg complex 6 (Scheme 1, entry 14) under the same reaction conditions. This was somewhat surprising as the increased Lewis acidity of Mg vs. Zn (Fig. 2) was expected to enhance rac-LA coordination and thus the activity of 1 and 6vs.3. The M–OR bond strength of the active M–OBn species formed in situ (vide infra) may play a key role in dictating the activity of 1, 3 and 6, as Zn–O bonds are known to be weaker than Mg–O bonds (bond dissociation energies: Zn–O ΔHf298 = 284 kJ mol−1 and Mg–O ΔHf298 = 394 kJ mol−1);44 cleavage of this bond is crucial during cyclic ester ROP. The overall activity trend with complexes 1–3 and 6–7 in rac-LA ROP was therefore deduced as: Ca/Zn > bis-Ca > Mg/Zn ≈ bis-Zn > bis-Mg. The superior activity of 2 likely stemmed from the increased availability of monomer coordination sites on Ca2+ (ionic radii: Ca2+ = 100 pm vs. Mg2+ = 72 pm and Zn2+ = 74 pm, Fig. 2);45 a similar trend was observed with the related Na/Zn2 and K/Zn2 complexes 4 and 5 which feature large alkali metal centres (Na+ = 102 pm and K+ = 138 pm),30,45 as well as literature reports of other heterometallic ROP catalysts.20
100 g mol−1 (Tables 1 and S1†). The presence of BnOH was essential for the high activity of 1–2 (entries 1 and 6, Table 1), which was attributed to the in situ deprotonation of BnOH by Zn–Et bonds of 1–2 (vide infra). Heterobimetallic 1 and 2 were benchmarked against their homobimetallic counterparts to investigate the nature of heterometallic cooperativity with 1–2 in rac-LA ROP (Tables 1 and S1†). Complex 2 was significantly more active than 1 (entries 8 and 2, Table 1) and outperformed both the bis-Zn complex 3 (entry 11) and bis-Ca complex 7 (Scheme 1, entry 15). Interestingly, 1 showed similar activity to the bis-Zn complex 3 (entries 3 and 12, Table 1),36 yet was more active than the bis-Mg complex 6 (Scheme 1, entry 14) under the same reaction conditions. This was somewhat surprising as the increased Lewis acidity of Mg vs. Zn (Fig. 2) was expected to enhance rac-LA coordination and thus the activity of 1 and 6vs.3. The M–OR bond strength of the active M–OBn species formed in situ (vide infra) may play a key role in dictating the activity of 1, 3 and 6, as Zn–O bonds are known to be weaker than Mg–O bonds (bond dissociation energies: Zn–O ΔHf298 = 284 kJ mol−1 and Mg–O ΔHf298 = 394 kJ mol−1);44 cleavage of this bond is crucial during cyclic ester ROP. The overall activity trend with complexes 1–3 and 6–7 in rac-LA ROP was therefore deduced as: Ca/Zn > bis-Ca > Mg/Zn ≈ bis-Zn > bis-Mg. The superior activity of 2 likely stemmed from the increased availability of monomer coordination sites on Ca2+ (ionic radii: Ca2+ = 100 pm vs. Mg2+ = 72 pm and Zn2+ = 74 pm, Fig. 2);45 a similar trend was observed with the related Na/Zn2 and K/Zn2 complexes 4 and 5 which feature large alkali metal centres (Na+ = 102 pm and K+ = 138 pm),30,45 as well as literature reports of other heterometallic ROP catalysts.20
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Time (min) | Conv.a (%) | M n,obs (g mol−1) | M n,calc (g mol−1) | Đ |
| 100 eq. LA, [LA] = 1 M in toluene. LA and pre-catalyst pre-stirred separately for 3 min in toluene at 60 °C before mixing and initiation with BnOH. a Conversion calculated using 1H NMR spectroscopy. b M n,obs and Đ determined by SEC using polystyrene standards in THF. Values corrected by Mark–Houwink factor (0.58).46 c M n,calc of polymers calculated from the monomer conversion Mn,calc = M0 × ([M]/[I]) × conversion assuming 1 chain per catalyst. d No BnOH used. e 500 eq. LA. f 1000 eq. LA. g Polymerisation performed in THF solvent. | ||||||
| 1d | 1 | 5 | 0 | — | — | — |
| 2 | 1 | 1.25 | 25 | 1900 | 3600 | 1.12 |
| 3 | 1 | 10 | 84 | 5600 | 12100 |
1.13 |
| 4e | 1 | 60 | 95 | 23600 |
68500 |
1.07 |
| 5f | 1 | 90 | 81 | 27800 |
116700 |
1.24 |
| 6d | 2 | 2 | 13 | — | — | — |
| 7 | 2 | 0.08 | 32 | 1300 | 4600 | 1.19 |
| 8 | 2 | 1.25 | 82 | 4700 | 11800 |
1.26 |
| 9e | 2 | 25 | 97 | 20600 |
69900 |
1.55 |
| 10f | 2 | 45 | 85 | 30134 |
122500 |
1.45 |
| 11 | 3 | 1.25 | 27 | 1900 | 3900 | 1.22 |
| 12 | 3 | 10 | 87 | 8100 | 12500 |
1.07 |
| 13 | 6 | 1.25 | 25 | 2000 | 3600 | 1.16 |
| 14 | 6 | 10 | 66 | 4600 | 9500 | 1.21 |
| 15 | 7 | 1.25 | 64 | 4800 | 9200 | 1.51 |
| 16g | 1 | 10 | 16 | — | — | — |
| 17g | 2 | 1.25 | 80 | 5800 | 11500 |
1.55 |
| 18g | 3 | 10 | 7 | — | — | — |

This was supported by the DFT structures of 1′–2′ (Fig. 3), which suggested a more facile coordination of the sterically-hindered HMDSH to Ca in 2′ than Mg in 1′, evidenced by a significantly shorter bond between Ca and HMDSH (Ca–N = 2.785 Å) compared to Mg-HMDSH (Mg–N = 3.661 Å); this could potentially be translated to LA coordination and activation, resulting in the enhanced activity of 2vs.1. The higher activity of 2 (vs.1 and 3) also aligns with the greater polarity (and nucleophilicity) of the Zn–Et (and Zn–OBn) bond in 2 indicated by the NMR analysis (Fig. 2), which is likely to accelerate ring-opening and propagation during rac-LA ROP. In addition, the electronic communication between Zn and Ca centres in 2 may result in “ate”-type activation, further influencing both the monomer coordination (by amplifying the Lewis acidity of Ca+) and M–OR bond nucleophilicity (anionic formulation of ZnR3−), and enhancing the activity compared to the homobimetallic analogues. Complex 2 also displayed comparable activity to the Na/Zn2 complex 4 in rac-LA ROP under the same reaction conditions (32 eq. rac-LA converted in 5 s with 2vs. 35 eq. with 4)30 despite being bimetallic and featuring one less metal site for monomer coordination (vs. trimetallic 4), and comprising a single initiating group with only one PLA chain growing per catalyst (vs. two chains with 4). However, 2 was only half as active as the K/Zn2 analogue 5 (68 eq. rac-LA converted in 5 s with 5).30 This provides further support for the importance of a large and Lewis acidic metal centre, as K+ is larger than Ca2+, as well as the polarity of the Zn–Et initiating units, with 5 including two initiating groups with enhanced polarity (thus nucleophilicity) vs. those of 2 (Fig. 2). Complex 2 also displayed high catalyst activity in THF solvent, both at 60 °C and room temperature, whereas 1 and 3 were inhibited under the same reaction conditions (Tables 1 and S2†). The additional coordination sites on Ca2+ (vs. Zn2+ and Mg2+) are proposed to partially relieve competitive THF/LA coordination to Ca/Mg/Zn. The activity enhancements and improved tolerance of Ca/Zn catalyst 2 towards Lewis donor solvents highlights the benefits of designing polymerisation catalysts with carefully selected heterometallic combinations.
Alcoholysis of heterometallic ethyl complexes with BnOH
To further understand the origins of heterometallic cooperativity with 1 and 2 in LA ROP, we investigated the nature of the active catalytic species formed upon alcoholysis of 1 and 2 with 1 eq. BnOH (Fig. 4). NMR-monitoring of these reactions in THF-d8 showed deprotonation of BnOH to form [LMgZnOBn] (8) and [LCaZnOBn] (9) is rapid at room temperature (<15 min), evidenced by the disappearance of Zn–Et and BnOH 1H NMR signals and appearance of an ethane signal at 0.84 ppm (Fig. S34 and S36†). Unlike homobimetallic [LZn2OBn] (10),36[LMg2OBn] (11, Fig. S15†) and [LCa2OBn] (12, Fig. S17†), all of which are symmetric, benzoxide complexes 8 and 9 are asymmetric in the solution-state as indicated by the broad doublet of doublets at 6.58 and 6.60 ppm (8) and 6.54 and 6.64 ppm (9) (Fig. 4), corresponding to the meta- and meta’-phenolic protons on the ligand backbone; COSY NMR analysis indicated coupling between these protons in both 8 and 9. The asymmetry of 8–9 observed by NMR spectroscopy was therefore attributed to the presence of two heterometals. The 1H NMR spectra of 8 and 9 also include doublets of doublets at 5.29 and 5.63 ppm (8) and 5.38 and 5.73 ppm (9), corresponding to the benzylic PhCH2–O(complex) protons from the OBn moiety. Alkoxide ligands are well known to bridge between two proximal metal centres,6,20 and DFT calculations suggested that bridging OBn co-ligands (between Mg and Zn in 8, and Ca and Zn in 9) are more favourable than coordination to the individual metal centres (Tables S11 and S13, Fig. S74 and S75†). Lewis donor coordination of THF and HMDSH to 8–9 was probed by DFT (Tables S12 and S14†), which suggested that the lowest energy structure of 8′ includes the coordination of 1 eq. of THF (ΔG = −9.1 kcal mol−1), albeit coordination of 1 eq. of THF and HMDSH was also favoured (ΔG = −8.8 kcal mol−1, Table S12†). In the case of 9′, coordination of 2 eq. of THF was more favoured (ΔG = −8.6 kcal mol−1) than coordination of 1 eq. of THF and HMDSH (ΔG = −6.3 kcal mol−1, Table S14†). | ||
| Fig. 4 Alcoholysis of heterobimetallic 1 and 2 with 1 eq. BnOH and the meta-phenolic 1H NMR region of the product mixtures formed overlaid with the homobimetallic analogues (THF-d8 at R.T; bis-Zn, 10; bis-Mg, 11; bis-Ca, 12). 1a) complex 1 + 1 eq. BnOH, 1b) complex 10 only, 1c) complex 11 only; 2a) complex 2 + 1 eq. BnOH, 2b) complex 10 only, 2c) complex 12 only. | ||
While NMR analysis showed that heterobimetallic 8 and 9 were the major products (approx. 76 and 80%, respectively) formed upon the reaction of 1 and 2 with 1 eq. BnOH in THF solvent at room temperature (Fig. S34 and S36†), some in situ formation of homobimetallic 10 (12%) and 11 (12%) was observed upon alcoholysis of 1, and formation of 10 (10%) and 12 (10%) along with 9 was detected by NMR spectroscopy and MALDI-ToF spectrometry (Fig. 4 and S34–S37†). The meta-phenolic proton resonances were particularly diagnostic, as a doublet of doublets was observed with asymmetric 8 and 9 whereas a singlet was observed for symmetric homobimetallic species 10–12 (Fig. 4). The hetero-/homo-bimetallic product distribution remained unchanged with variable temperature 1H NMR in THF-d8 at 5–55 °C and when 1 and 2 were reacted with 1 eq. BnOH under different reactions, such as in toluene or THF at 60 or 0 °C and under more dilute conditions; we were therefore unable to isolate purely heterobimetallic 8 and 9. Interestingly, the 1:1 combination of homobimetallic 10 and 11 in THF-d8 gave partial rearrangement to heterobimetallic 8 after heating the mixture to 60 °C (44% after 7 h; 51% after 39 h; Fig. S38†). Notably, the heterobimetallic product was accompanied by an unbalanced stoichiometry of homobimetallic 10 and 11 (36% and 20%, respectively), which was attributed to the presence of trace by- and/or decomposition products in the 1H NMR spectra due to the pro-longed heating of the reaction. While heat was required to generate 8 from a mixture of 10 and 11, the 1:1 mixture of 10 and 12 partially rearranged after 16 h to form 9 even at room temperature (44% vs.10 (18%) and 12 (38%), Fig. S39†), with 68% of 9 observed at 60 °C after 7 h, albeit with some product decomposition observed.
Kinetic studies of rac-, L- and D-LA ROP
Interestingly, kinetic studies showed that both 1/BnOH and 2/BnOH display slower polymerisation rates while converting the first 50 eq. of rac-LA (kobs = 1.8 × 10−3 and 7.2 × 10−3 s−1, respectively, Fig. 5), followed by a more rapid propagation of the remaining 50 eq. (kobs = 3.2 × 10−3 and 3.4 × 10−2 s−1, respectively) in toluene at 60 °C. The difference in rate was less pronounced with 1; similarly, bis-Zn complex 3 also displayed a subtle two-step rac-LA ROP kinetic plot (kobs = 2.2 × 10−3 and 3.9 × 10−3 s−1, Fig. S33†). The unusual kinetic profiles of 1–3 in rac-LA ROP were attributed to alcoholysis occurring in the early stages of the polymerisation, as well as catalyst preference for the coordination and insertion of one of the rac-LA stereoisomers, which could stem from 1–3 being arranged to favour D- or L-LA coordination. Indeed, kinetic plots of L-LA and D-LA ROP with 1–2 did not show the same two-step profile as with rac-LA ROP (Fig. 5). Complex 1 displayed higher activity for L-LA ROP (kobs = 3.7 × 10−3 s−1) than D-LA ROP (kobs = 1.5 × 10−3 s−1; Table S3†), which reflected the kobs values observed for the two-step rac-LA ROP with 1 (Fig. 5, left). Complex 2 gave more pronounced rate differences between D-LA and L-LA (Fig. 5 right, Table S4†), and exhibited the highest polymerisation rate for L-LA ROP (kobs = 8.4 × 10−2 s−1; vs.D-LA ROP, kobs = 2.2 × 10−2 s−1). Despite the distinct rate differences in L-LA and D-LA ROP, 1–2 displayed only a slight isotactic bias (max. Pi = 0.64, Table S1†) during the conversion of the first ∼30 eq. rac-LA in toluene at 60 °C. Nearly atactic PLA was produced at higher monomer conversions (>50%, Table S1†), which was tentatively attributed to the high propagation rates and transesterification (vide infra). Unfortunately, neither lowering the polymerisation temperature to −36 °C in THF nor increasing the dilution of 2 improved the stereocontrol (Table S2†). On the basis of the kinetic studies, it is plausible that the slower propagation and slight stereocontrol observed when converting the first 50 eq. of rac-LA with both 1–2 in toluene at 60 °C is due to polymerisation of the “slower” D-LA stereoisomer first (Fig. 5). This may potentially be followed by in situ ligand rearrangement and/or decoordination of a Lewis donor (e.g. HMDSH or THF) to facilitate the coordination and insertion of the second “faster” L-LA stereoisomer. The proposed complex rearrangement is further supported by the induction periods observed in L-LA ROP (Fig. 5) with 1 (approx. 2.6 min) and 2 (approx. 20 s), whereas no induction periods were observed for D-LA ROP (Fig. 5). In addition, PLA produced from L-LA and D-LA with 1 showed epimerisation, which was more pronounced for L-LA (Pi decreasing from 0.99 to 0.48 between 40 s and 2.5 min, Table S3†) than D-LA (minimum Pi = 0.70, Table S3†); this was attributed to the induction period during L-LA ROP. No epimerisation of LA/PLA was observed with 2, which was attributed to the significantly faster propagation rate (Table S4,†Fig. 5). DFT calculations of L-LA/D-LA coordination to 8′–9′ and the transition states of the first nucleophilic attack on L-LA/D-LA supported the experimental results. In both cases, the transition states found were lower for L-LA ring-opening than for D-LA (with 8′: ΔΔGTS(L-LA) = 17.1 kcal mol−1 and ΔΔGTS(D-LA) = 22.4 kcal mol−1; with 9′: ΔΔGTS(L-LA) = 18.3 kcal mol−1 and ΔΔGTS(D-LA) = 26.4 kcal mol−1, Tables S15 and S17†). | ||
| Fig. 5 Plots of ln([LA]0/[LA]t) vs. time (min) for rac-LA, L-LA and D-LA ROP with 1–2 in the presence of 1 eq. BnOH (100 eq. LA, [LA] = 1 M, toluene, 60 °C). | ||
However, for both 8′ and 9′, the initial coordination of D-LA to Mg and Ca in 8′ (ΔG = −7.2 kcal mol−1) and 9′ (ΔG = −8.8 kcal mol−1), respectively, was more favoured than L-LA coordination (with 8′ ΔG = −2.5 kcal mol−1; with 9′ ΔG = −2.8 kcal mol−1; Tables S16 and S18†). It is tentatively proposed that the lower energy transition state for nucleophilic attack on L-LA, but less favourable L-LA coordination, stems from less sterically hindered coordination of D-LA to Mg and Ca in 8′ and 9′ than of L-LA (Fig. S76–S79† for 8′ and Fig. 6 and S80 and S81† for 9′). However, once coordinated, the increased steric congestion around Ca and Mg in 8′ and 9′ reduces the distance between the M–OBn bond and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety of L-LA with both 8′ (2.889 Å vs. 3.161 Å with D-LA) and 9′ (3.179 Å vs. 3.464 Å with D-LA). This feature likely accelerates the nucleophilic attack and propagation of L-LA with 8 and 9, thus resulting in a lower transition state energy (vs.D-LA). Notably, coordination of sterically hindered HMDSH (1 eq.) to Mg and Ca in 8′ and 9′ was also more favoured in the presence of D-LA (with 8′ ΔG = −7.0 kcal mol−1; with 9′ ΔG = −8.6 kcal mol−1) than of L-LA (with 8′ ΔG = −3.1 kcal mol−1; with 9′ ΔG = −3.4 kcal mol−1, Tables S16 and S18†).
O moiety of L-LA with both 8′ (2.889 Å vs. 3.161 Å with D-LA) and 9′ (3.179 Å vs. 3.464 Å with D-LA). This feature likely accelerates the nucleophilic attack and propagation of L-LA with 8 and 9, thus resulting in a lower transition state energy (vs.D-LA). Notably, coordination of sterically hindered HMDSH (1 eq.) to Mg and Ca in 8′ and 9′ was also more favoured in the presence of D-LA (with 8′ ΔG = −7.0 kcal mol−1; with 9′ ΔG = −8.6 kcal mol−1) than of L-LA (with 8′ ΔG = −3.1 kcal mol−1; with 9′ ΔG = −3.4 kcal mol−1, Tables S16 and S18†).
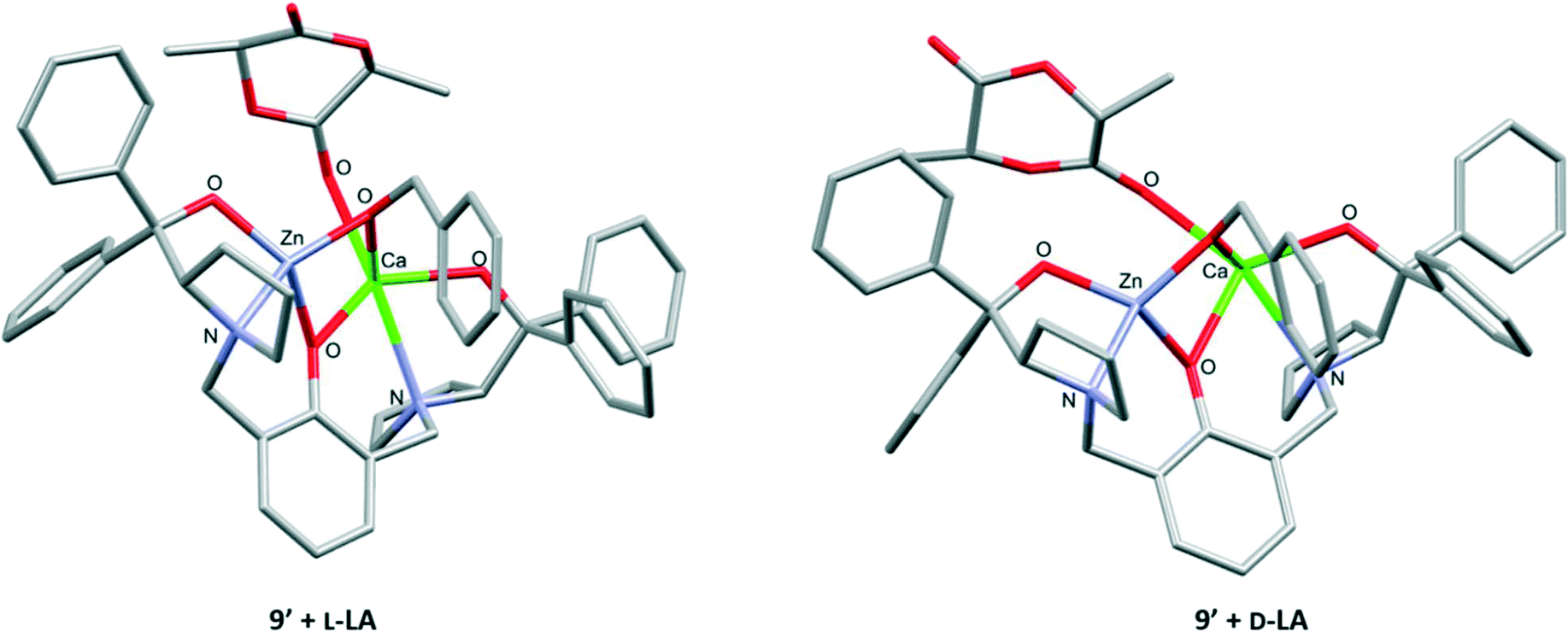 | ||
| Fig. 6 DFT computational structures of 9′ with coordinated L-LA or D-LA (refer to Table S18†). | ||
Reactivity insights: molecular weight control and the effect of HMDSH
In spite of the two-step kinetic profiles, complexes 1–2 were relatively well-controlled rac-LA ROP initiators in the presence of 1 eq. BnOH, indicated by a generally linear relationship between Mn and monomer conversion and narrow dispersity values in toluene at 60 °C (Table 1 and S1, Fig. S20 and S22†). Complex 1 demonstrated slightly improved polymerisation control compared to 2 (Đ ≤ 1.14 and Đ ≤ 1.27, respectively), and both complexes displayed similar dispersities to the bis-Zn analogue 3 (Đ = 1.07–1.22).36 While 1 gave comparable dispersities to the bis-Mg complex 6 (Đ = 1.12–1.21, Table S1†), complex 2 was more controlled than the bis-Ca counterpart 7 (Đ = 1.37–2.01, Table S1†). Together with the enhanced activity of heterobimetallic 2vs. homobimetallic analogues 3 and 7, these observations highlight the benefit of incorporating both Ca and Zn within one structure to combine the high activity of Ca with the good control of Zn. Good to moderate dispersities were also observed with 2 in THF solvent at 60 °C (Đ ≤ 1.55, Table S2, Fig. S29†) and at room temperature (Đ ≤ 1.49, Fig. S31†). The discrepancy between the observed and calculated Mn values observed with both 1–2 was attributed to transesterification reactions as MALDI-ToF analysis showed the expected α-benzoxy, ω-hydroxy (major series) and α-hydroxy, ω-hydroxy (minor series) end-capped PLA (Fig. S40–S47†). Notably, the molecular weight agreement was slightly improved with 2 in THF at 60 °C (Fig. S29†), which could be caused by reduced transesterification in THF solvent stemming from increased steric hindrance in the metal pocket due to THF coordination to Ca/Zn, as demonstrated by DFT calculations with 9′ (Table S14†).Importantly, despite the presence of 1 eq. HMDSH with 1 and 2, no HMDS-capped PLA was detected by MALDI-ToF analysis. Control reactions with 1 eq. BnOH and HMDSH in rac-LA ROP under the conditions employed with 1–2 showed no rac-LA conversion. These results suggest that in the presence of 1 or 2 with 1 eq. BnOH, the polymerisation proceeds via a coordination–insertion mechanism mediated by the in situ generated M–OBn co-ligand (vide supra).
The effect of HMDSH on 1 and 2 in rac-LA ROP was probed with kinetic studies in the presence of an additional 1 eq. of exogenous HMDSH (2 eq. in total) in toluene at 60 °C (Table S5, Fig. S48 and S49†). While the polymerisations also proceeded in two stages, including 1 eq. of exogenous HMDSH gave different effects on the polymerisation rates with 1 and 2. With 1 and 1 eq. of exogenous HMDSH, both the initial polymerisation rate (conversion of <50 eq. rac-LA, kobs = 1.9 × 10−3 s−1, Fig. S48†) and the latter rate (conversion of >50 eq. rac-LA, kobs = 3.2 × 10−3 s−1, Fig. S48†) were essentially the same as in the absence of exogenous HMDSH (1st kobs = 1.8 × 10−3 s−1 and 2nd kobs = 3.2 × 10−3 s−1, Fig. 5). The addition of further exogenous HMDSH (3–5 eq. in total) gave only a slight drop in rac-LA conversion (from 79% to 70%, Table S5†). The small influence of further equivalents of HMDSH on the activity of 1 was supported by the DFT calculations, as the lowest energy structure of 8′ in the presence of LA does not involve HMDSH coordination and instead either involves coordination of 1 eq. of THF (with L-LA) or no Lewis donors (with D-LA, Table S16†). Furthermore, despite numerous attempts, DFT modelling of the coordination of 2 eq. HMDSH to 8′ was unsuccessful, with a systematic decoordination of the 2nd eq. of HMDSH occurring. Conversely, with 2, the initial stage (conversion of approx. 50 eq. rac-LA) was accelerated when 1 eq. of exogenous HMDSH was added (kobs = 2.9 × 10−2 s−1, Fig. S49†vs. kobs = 7.2 × 10−3 s−1 with no exogenous HMDSH, Fig. 5), whereas the latter rate was reduced (kobs = 6.7 × 10−3 s−1, Fig. S49†vs. kobs = 3.4 × 10−2 s−1 with no exogenous HMDSH, Fig. 5). DFT calculations with 9′ also suggested that the coordination of 2 eq. HMDSH is unlikely and that the lowest energy molecular structure of 9′ includes 1 eq. of THF in the presence of both L-LA and D-LA (Table S18†). The presence of 2 eq. HMDSH improved the agreement between observed and calculated Mn for both 1 and 2 (+1 eq. BnOH, Fig. S50 and S51†), similar to the observations with 2 in THF solvent (vs. toluene, vide supra). While the exact role of the exogenous HMDSH remains unclear, it is plausible that it has a greater effect on the activity of 2vs.1 (+ 1 eq. BnOH) due to the larger ionic radius and higher Lewis acidity of Ca2+vs. Mg2+ enhancing the coordination of HMDSH.
Reactivity insights: using isolated heterometallic benzoxide mixtures in rac-LA ROP
Previous studies on complex 10 (bis-Zn) showed that the isolated complex gave improved activity (ten-fold) and molecular weight control in rac-LA ROP compared to the in situ generated analogue (from alcoholysis of 3).36 The multi-component mixtures of 8/10/11 and 9/10/12, attained by reaction of 1 and 2 with 1 eq. BnOH in toluene at 60 °C for 1 h, respectively, were therefore tested in rac-LA ROP in toluene at 60 °C (Table S6†). These mixtures of 8/10/11 and 9/10/12 were isolated by removing toluene in vacuo, and were used without purification as redistribution to the homobimetallic complexes was observed in solution (vide supra). Pre-forming the active metal-benzoxide species improved the initiation with the Mg/Zn 8/10/11 mixture, with a four-fold increase in the initial rate (8/10/11, kobs = 7.9 × 10−3 s−1, Fig. S52†) vs. the 1/BnOH system (Fig. 5), yet lowered the second rate slightly (from kobs = 3.2 × 10−3 s−1 to kobs = 2.4 × 10−3 s−1, Fig. S52†). The isolated Ca/Zn 9/10/12 mixture displayed a linear kinetic plot with a reduced rac-LA ROP rate (kobs = 2.5 × 10−3 s−1, Fig. S53†) compared to the 2/BnOH system (kobs = 7.2 × 10−3 s−1 and 3.4 × 10−2 s−1, Fig. 5). This result and the more modest rate enhancement observed between the “isolated” and in situ generated Mg/Zn benzoxide species compared to 10 (bis-Zn) were tentatively attributed to nucleophilic ring-opening of LA requiring cleavage of both the Zn- and Mg/Ca–OBn bonds of the bridging OBn moiety when the isolated 8/10/11 and 9/10/12 mixtures were used. Compared to Zn, both Ca and Mg have higher oxophilicity and metal–O bond dissociation energies (⊖(Mg) = 0.6, ΔHf298 = 394 kJ mol−1; ⊖(Ca) = 0.3, ΔHf298 = 464 kJ mol−1; ⊖(Zn) = 0.2, ΔHf298 = 284 kJ mol−1).44,47 Similarly to the trends observed with 3 ([LZn2Et]) and 6 ([LMg2HMDS]) + 1 eq. BnOH (vide supra), the Mg > Ca > Zn oxophilicity and metal–O bond dissociation energy trends also reflect the activities of isolated homobimetallic benzoxide complexes 10–12 in rac-LA ROP. Indeed, 10 (bis-Zn) displayed the highest activity polymerising 99 eq. rac-LA in 2 min, which was attributed to the lower Zn–O bond strength (facilitating nucleophilic attack). The bis-Ca (12) and bis-Mg (11) complexes were significantly slower, with 12 requiring 5 min to convert 83 eq. rac-LA and 11 polymerising only 12 eq. rac-LA in 10 min in toluene at 60 °C (Table S6†). It is tentatively proposed that the reduced M–OR bond strength in 10 might outweigh the potentially enhanced monomer coordination to the more oxophillic Mg and Ca in 11 and 12, respectively. However, it is difficult to entirely decouple the influence of the metal–alkoxide nucleophilicity from metal Lewis acidity and ionic radius as all of these are key catalytic features in cyclic ester ROP. The agreement between observed and calculated Mn values was improved with both “isolated” multi-component 8/10/11 and 9/10/12 mixtures (Table S6, Fig. S54 and S55†) compared to the in situ1/BnOH and 2/BnOH systems (Fig. S20 and S22,† respectively), albeit with the Mn values slightly higher than expected. This is tentatively attributed to not all of the catalyst being active due to aggregation in toluene,36 as DOSY NMR analysis of the 8/10/11 and 9/10/12 mixtures suggests the presence of monomer/dimer equilibria in toluene-d8 (Fig. S56 and 57†) but monomeric structures in THF-d8 (Fig. S58 and S59†).Catalyst scope
Both 1 and 2 (with 1 eq. BnOH) were also extremely active in ε-caprolactone (ε-CL) and δ-valerolactone (δ-VL) ROP (Table S7†), with 1 converting 86 eq. ε-CL and 99 eq. δ-VL and 2 converting 99 eq. of both monomers in just 5 s in toluene at R.T. Complexes 1 and 2 also maintained high activity in ε-CL ROP at 0.5 mol% catalyst loading, converting 405 and 380 eq. ε-CL in 10 min and 30 s at R.T., respectively, producing PCL with Mn up to 54200 g mol−1 (Table S7†). Both 1 and 2 significantly outperform the analogous bis-Zn complex 3, which required 4.5 min to convert 90 eq. ε-CL at 60 °C.36 Despite a single initiating group and fewer metal centres in 1–2, the heterobimetallic complexes (+ 1 eq. BnOH) also showed superior activity compared to 4 ([LNaZn2Et2] + 2 eq. BnOH), which polymerised 53 eq. ε-CL in 5 s at R.T.30 To the best of our knowledge, the outstanding activities of 1–2 in ε-CL ROP make these complexes not only more active than 3 and 4–5, but also rival the multi- hetero-metallic La/Mg allyl complex reported by Bochmann and co-workers,22 which previously displayed the highest activity among heterometallic ε-CL ROP catalysts, converting 198 eq. ε-CL in 20 s at R.T. Notably, the reactivity enhancements with 1–2vs.3–5 in ε-CL ROP are more significant than those in rac-LA ROP, which highlights the importance of tailoring the heterometal combination and optimisation towards the ROP of different monomers.
Conclusions
In summary, this systematic study shows that the direct replacement of one Zn centre from a bis-Zn ProPhenol complex with Mg or Ca boosts the activity in cyclic ester ROP. To the best of our knowledge, these Mg/Zn and Ca/Zn complexes are the first heterometallic LA ROP catalysts where both metals are divalent. Within rac-LA ROP, the Ca/Zn and Mg/Zn complexes outperform their bis-Ca and bis-Mg analogues, with the overall activity trend deduced as: Ca/Zn > bis-Ca > Mg/Zn ≈ bis-Zn > bis-Mg. Since only divalent metals are employed, these studies enable the influence of the heterometal to be directly studied in cyclic ester ROP. In contrast, with previously reported heterometallic ROP catalysts, the activity comparisons between hetero- and homo-metallic complexes have been limited by differences in the number of initiating groups and the coordination environments of the metal centres. While NMR and DFT studies suggest that the initiating Et moiety remains on Zn, incorporation of Mg or Ca results in “ate”-type activation, which may concurrently enhance the polarity (and nucleophilicity) of the Zn–Et/OBn bonds (Ca/Zn > Mg/Zn) and the Lewis acidity of Mg and Ca (Ca > Mg). These features are likely to accelerate nucleophilic attack and monomer coordination, respectively, in ROP, as well as in organic transformations. Whilst the more electronegative Zn likely acts as the nucleophile source, monomer coordination is proposed to primarily occur on Lewis acidic Mg and Ca, as suggested by the coordination of THF and HMDSH to these metals, with a greater availability of coordination sites on the larger Ca2+vs. Mg2+. These amplified heterometallic catalytic features may also elevate the activity of ProPhenol complexes in asymmetric addition reactions (e.g. aldol and Mannich) by increasing the deprotonation power of the Brønsted basic Zn–Et group to generate known and new organic nucleophiles, which may subsequently add to an electrophilic partner activated via coordination to the Lewis acidic heterometal (Mg, Ca).39 Furthermore, detailed kinetic experiments suggest that the Mg/Zn and Ca/Zn complexes display unusual two-step kinetic plots, which, to the best of our knowledge, have not yet been reported in rac-LA ROP. Based on experimental and DFT results, the two-step kinetic plots are tentatively attributed to the catalyst preference for the coordination and insertion of the “slower” D-LA stereoisomer first, followed by the “faster” L-LA stereoisomer. In addition to the activity enhancements, these results highlight another possible benefit of heterometallic catalysts. While no polymer stereocontrol was exhibited here (possibly due to high polymerisation rates and/or epimerisation), these findings indicate that the heterometallic ProPhenol complexes could exhibit a higher degree of stereocontrol than the bis-Zn analogue, which could potentially be exploited in other ring-opening (co)polymerisations as well as an extensive range of asymmetric addition reactions that have been efficiently catalysed by the bis-Zn ProPhenol complex.39 Indeed, a Mg/Zn ProPhenol catalyst system has shown promise in asymmetric Michael reactions and outperformed the bis-Zn ProPhenol complex, however this complex was generated in situ and was not characterised.38 Our work therefore reports the first examples of isolated and characterised Mg/Zn and Ca/Zn ProPhenol catalysts, which can not only elevate polymerisation activities but also potentially improve and lead to new asymmetric transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the CRITICAT Centre for Doctoral Training and EPSRC (W.G., EP\L016419\1), Royal Society (J.A.G., RSG\R1\180101; A.B., UF\160021 fellowship), British Ramsay Memorial Trust (J.A.G.), L'Oréal-UNESCO For Women in Science (J.A.G.) and UKRI Future Leaders Fellowship (Grant MR/T042710/1) for funding. We would also like to thank Dr Faye Cruickshank at the Scottish Instrumentation and Resource Centre for Advanced Mass Spectrometry (The University of Edinburgh) for performing the APPI-MS analysis.Notes and references
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC.
- C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed.
- C.-S. Ha and J. A. Gardella, Chem. Rev., 2005, 105, 4205–4232 CrossRef CAS.
- A. B. Kremer and P. Mehrkhodavandi, Coord. Chem. Rev., 2019, 380, 35–57 CrossRef CAS.
- D. M. Lyubov, A. O. Tolpygin and A. A. Trifonov, Coord. Chem. Rev., 2019, 392, 83–145 CrossRef CAS.
- A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007–9023 RSC.
- L. Chen, W. Li, D. Yuan, Y. Zhang, Q. Shen and Y. Yao, Inorg. Chem., 2015, 54, 4699–4708 CrossRef CAS.
- F. Isnard, M. Lamberti, L. Lettieri, I. D'Auria, K. Press, R. Troiano and M. Mazzeo, Dalton Trans., 2016, 45, 16001–16010 RSC.
- K. C. MacLeod and P. L. Holland, Nat. Chem., 2013, 5, 559–565 CrossRef CAS.
- C. Wombwell and E. Reisner, Dalton Trans., 2014, 43, 4483–4493 RSC.
- T. D. Bluemke, W. Clegg, P. García-Alvarez, A. R. Kennedy, K. Koszinowski, M. D. McCall, L. Russo and E. Hevia, Chem. Sci., 2014, 5, 3552–3562 RSC.
- A. J. Martinez-Martinez, A. R. Kennedy, R. E. Mulvey and C. T. O'Hara, Science, 2014, 346, 834–837 CrossRef CAS PubMed.
- M. Shibasaki, H. Sasai and T. Arai, Angew. Chem., Int. Ed. Engl., 1997, 36, 1236–1256 CrossRef.
- S. K. Mandal and H. W. Roesky, Acc. Chem. Res., 2010, 43, 248–259 CrossRef CAS PubMed.
- P. Buchwalter, J. Rose and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed.
- S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332–8405 CrossRef CAS PubMed.
- J. A. Garden, P. K. Saini and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 15078–15081 CrossRef CAS.
- W. Gruszka and J. A. Garden, Nat. Commun., 2021, 12, 3252–3265 CrossRef CAS PubMed.
- W. Yao, Y. Mu, A. Gao, W. Gao and L. Ye, Dalton Trans., 2008, 3199–3206 RSC.
- L. F. Sánchez-Barba, D. L. Hughes, S. M. Humphrey and M. Bochmann, Organometallics, 2006, 25, 1012–1020 CrossRef.
- M. Normand, E. Kirillov, T. Roisnel and J.-F. Carpentier, Organometallics, 2012, 31, 1448–1457 CrossRef CAS.
- J. Char, E. Brulé, P. C. Gros, M. N. Rager, V. Guérineau and C. M. Thomas, J. Organomet. Chem., 2015, 796, 47–52 CrossRef CAS.
- W. Li, Z. Zhang, Y. Yao, Y. Zhang and Q. Shen, Organometallics, 2012, 31, 3499–3511 CrossRef CAS.
- A. J. Gaston, Z. Greindl, C. A. Morrison and J. A. Garden, Inorg. Chem., 2021, 60, 2294–2303 CrossRef CAS PubMed.
- H.-T. Sheng, J.-M. Li, Y. Zhang, Y.-M. Yao and Q. Shen, Polyhedron, 2008, 27, 1665–1672 CrossRef CAS.
- J. Hao, J. Li, C. Cui and H. W. Roesky, Inorg. Chem., 2011, 50, 7453–7459 CrossRef CAS PubMed.
- H.-Y. Chen, M.-Y. Liu, A. K. Sutar and C.-C. Lin, Inorg. Chem., 2010, 49, 665–674 CrossRef CAS PubMed.
- W. Gruszka, A. Lykkeberg, G. S. Nichol, M. P. Shaver, A. Buchard and J. A. Garden, Chem. Sci., 2020, 11, 11785–11790 RSC.
- R. E. Mulvey, Acc. Chem. Res., 2009, 42, 743–755 CrossRef CAS.
- G. Wittig, F. J. Meyer and G. Lange, Ann. Chem., 1951, 571, 167–201 CAS.
- A. Thevenon, C. Romain, M. S. Bennington, A. J. P. White, H. J. Davidson, S. Brooker and C. K. Williams, Angew. Chem., Int. Ed., 2016, 55, 8680–8685 CrossRef CAS PubMed.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed.
- C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350–11359 CrossRef CAS PubMed.
- W. Gruszka, L. C. Walker, M. P. Shaver and J. A. Garden, Macromolecules, 2020, 53, 4294–4302 CrossRef CAS.
- G. Trott, J. A. Garden and C. K. Williams, Chem. Sci., 2019, 10, 4618–4627 RSC.
- B. M. Trost and S. Hisaindee, Org. Lett., 2006, 8, 6003–6005 CrossRef CAS PubMed.
- B. M. Trost, C.-I. J. Hung and G. Mata, Angew. Chem., Int. Ed., 2020, 59, 4240–4261 CrossRef CAS PubMed.
- M. Westerhausen, Coord. Chem. Rev., 2008, 252, 1516–1531 CrossRef CAS.
- F. Buch, PhD thesis, Universität Duisburg-Essen, 2008 Search PubMed.
- A. L. Allred, J. Inorg. Nucl. Chem., 1961, 17, 215–221 CrossRef CAS.
- Y. Xiao, Z. Wang and K. Ding, Chem. – Eur. J., 2005, 11, 3668–3678 CrossRef CAS.
- J. A. Dean, Lange's Handbook of Chemistry, McGraw-Hill Handbooks, New York, 1999 Search PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 1998, 31, 2114–2122 CrossRef CAS.
- K. P. Kepp, Inorg. Chem., 2016, 55, 9461–9470 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, EA, MS characterisation data, polymer MALDI-ToF, SEC, kinetic and DOSY studies and DFT calculations. See DOI: 10.1039/d1cy01914g |
| This journal is © The Royal Society of Chemistry 2022 |