Formic acid generating in situ H2 and CO2 for nitrite reduction in the aqueous phase†
Received
11th August 2021
, Accepted 9th February 2022
First published on 9th February 2022
Abstract
The aim of this work is to explore and to understand the effect of pH, concentrations and presence of oxygen traces on the reduction of nitrite in drinking water with Pd/γ-Al2O3, using formic acid as an in situ hydrogen supplier. Formic acid can reduce nitrite in the pH range between 4.5 and 8, producing negligible amounts of ammonium. By investigating the effect of pH, traces of oxygen and formic acid concentration on the conversion rates of both formic acid and nitrite, it is found that both the rate of conversion on nitrite with formic acid and the rate of formic acid decomposition are controlled by competitive adsorption of nitrite and formic acid on Pd, forming chemisorbed NO and chemisorbed H, respectively. The adsorbed species are studied with ATR-IR spectroscopy. Formic acid decomposition requires an ensemble of empty sites, favored by a low surface coverage of NO. The NO surface coverage, on the other hand, decreases with increasing hydrogen coverage, by converting NO to N2. The H-coverage in turn depends on the rate of formic acid decomposition. This causes an apparent order for the rate of formic acid decomposition of 1.4 in formic acid. In short, the surface coverage of NO should not be too high in order to have sufficient empty sites available for formic acid decomposition. When the pH of the solution is below 4.5, homogeneous disproportionation of nitric acid occurs forming nitrate and NO, resulting in catalyst poisoning with NO. The catalyst shows no activity at pH above 8, as formate ions are not reactive under such conditions.
1. Introduction
Pollution of water with nitrate and nitrite is increasing globally, caused by emissions from agriculture and industry,1 which threatens human health, as they are carcinogenic and cause blue-baby-syndrome, diabetes, and liver damage.2–5 Therefore, the World Health Organization (WHO) set their maximum acceptable concentrations in drinking water, i.e. 50 mg L−1 for nitrate, 3 mg L−1 for nitrite, and 1.5 mg L−1 for ammonia, respectively.6
Nitrate and nitrite ions can be removed from water by biological denitrification, ion exchange and catalytic reduction. Biological denitrification is not possible for drinking water, as nutrients to sustain the growth of bacteria are absent.7 Ion exchange technology produces concentrated brines that would need further treatment.7 Catalytic reduction of nitrate is one of the most promising methods, converting nitrate to harmless molecular nitrogen with high efficiency.8–23 For practical applications, further improvement of the selectivity to N2 is necessary for purification of drinking water, preventing any formation of ammonia.
Since the first paper on catalytic reduction of nitrate by Vorlop and Tacke,3 numerous studies7,24–32 have been reported, mostly using hydrogen as the reducing agent. It is well known that reduction of nitrate proceeds in two steps requiring a bimetallic catalyst.33–38 First, nitrate is reduced to nitrite, requiring a non-noble promotor such as e.g. Cu, which is generally rate determining. Furthermore, conversion of nitrite is much faster and determines the selectivity to ammonium and nitrogen. The reactions are shown below. Therefore, in this paper nitrite reduction is studied as a model reaction.
| |  | (1) |
| |  | (2) |
| |  | (3) |
Using hydrogen as the reducing agent has disadvantages in terms of hydrogen transportation and storage at high pressure. In addition, it is well known that the selectivity to nitrogen can be improved by decreasing the pH,
39–42e.g. by co-feeding CO
2 in many lab studies. For practical application, this method is less suitable. Note that the reaction consumes protons and the pH will increase during the reaction, which can be buffered with
e.g. CO
2. Formic acid, however, can be used as an alternative reductant, decomposing not only to H
2 but also to CO
2, buffering the pH of the solution during the reaction. A few studies on catalytic reduction of nitrate with formic acid as the reducing agent
35,43–45 indeed report low selectivity to ammonium.
IR spectroscopy used with an attenuated total reflection in situ cell (ATR-IR), depositing a thin catalyst layer on the internal reflection crystal, is an ideal technique for studying adsorbed species at the solid–liquid interface. In our group, ATR-IR spectroscopy has been used for both CO oxidation and nitrite reduction with H2 in the aqueous phase.46–50 NO adsorbed on Pd was observed as an important intermediate species during nitrite reduction, which readily converts to N2.
The goal of this work is to study the performance of a Pd catalyst in the reduction of nitrite with formic acid under well controlled conditions. Our recent work demonstrated that formic acid51 decomposes over Pd catalysts in the absence of nitrite and that deactivation caused by chemisorbed CO can be prevented by adding trace amounts of oxygen. CO2 and H2 are formed, resulting in the generation of both the reductant for nitrite hydrogenation and in situ buffer for the solution. The influence of the pH, initial formic acid concentration and effect of trace amounts of oxygen are therefore studied. ATR-IR spectroscopy is used for the first time to identify intermediate species on the Pd surface for nitrite reduction with formic acid. The interplay between formic acid decomposition and nitrite reduction is discussed.
2. Experimental
2.1. Materials
Commercial γ-Al2O3 powder (BASF) with a surface area of 195 m2 g−1 was used as the catalyst support. Tetra-ammine-palladium(II) nitrate solution (10 wt% in H2O, purity 99.99%, Sigma-Aldrich) was used as the palladium precursor. Sodium nitrite (99.99%) which is purchased from Sigma-Aldrich was used to prepare nitrite (NO2−) solutions. The pH during catalyst preparation was adjusted with an aqueous ammonium solution (50% v/v water, Sigma-Aldrich). Formic acid (≥98%), sodium formate and sodium hydroxide were purchased from Sigma-Aldrich. All aqueous solutions were prepared using ultra purified water (Millipore, Synergy).
2.2. Catalyst preparation
The preparation method is described in detail elsewhere.52 In brief, the Pd/γ-Al2O3 catalyst (1 wt% Pd) was prepared by wet impregnation. Typically, 10 g of support particles (sieved to smaller than 38 μm, mean particle size 22 μm) was calcined at 600 °C for 4 hours, removing any organic contamination. The support was suspended in 100 mL MilliQ water and the pH of the solution was adjusted to 9 by adding ammonia solution. Next, 3 grams of the palladium precursor solution (Pd(NO3)2·4NH3) was slowly added to the suspension and stirred at room temperature for at least 1 hour. The water was evaporated in a rotary evaporator for 2 hours, followed by calcination in air at 400 °C for 3 hours (heating rate 5 K min−1) and reduction in hydrogen (33% in nitrogen, total flow rate 90 mL min−1) at the same temperature for 3 hours. Catalyst with 3 wt% Pd loading was prepared following the same procedure, except for adding more Pd precursor. This catalyst was used for ATR-IR experiments in order to obtain sufficient intensity in the IR spectra.
2.3. Catalyst characterization
The surface area of the prepared catalyst was determined based on the BET N2-adsorption isotherms obtained at 77 K (Micromeritics Tristar). The Pd loading was determined with X-ray fluorescence spectroscopy, XRF (Philips PW 1480). The accessible metal surface area was determined with CO chemisorption at room temperature (Chemisorb 2750, Micromeritics). The sample was reduced at room temperature in hydrogen for 1 hour and flushed with He at the same temperature for 0.5 hours, followed by pulsing with CO, recording the response using a TCD detector. We assumed that every accessible Pd surface atom adsorbed one CO molecule.
2.4. Catalytic tests and analysis
The activity and selectivity of the catalysts were measured in a 1 L batch reactor at 20 °C and atmospheric pressure. The glass reactor with 10 cm inner diameter and 12.7 cm height has four connections on the reactor lid for gas-in, gas-out, sampling and stirring shaft, respectively.52 The possible reactions involved in formic acid decomposition and nitrite reduction are given in the following equations:| |  | (2) |
| |  | (3) |
| |  | (4) |
| |  | (5) |
Typically, in a standard experiment 0.10 g catalyst was suspended in 0.3 L MilliQ water and stirred at 625 rpm under 1 bar helium for at least 1 h, removing dissolved oxygen. The reaction is initialized by introducing 3 mL NaNO2 solution (100 mmol L−1) and 3 mL formic acid (1 M) at the same time in the glass reactor. Note that these volumes are not precisely controlled, resulting in some variation in the initial concentrations. The pH of the solution was varied between 2.8 and 9 by adding appropriate amounts of the sodium hydroxide solution (1 M) to formic acid solution (1 M). The initial pH of the solution was measured after injecting the concentrated formic acid and sodium hydroxide solution to the bulk solution. The formic acid concentration was varied between 5 mM and 40 mM. The formic acid concentration is defined as the sum of the concentrations of formic acid and formate ions and is termed as “formic acid” throughout the paper. During the experiment, the reactor is flushed with 50 ml min−1 He. This type of experiment is termed as “inert-flow-through”, in contrast to “ambient-batch” meaning that the reactor is not flushed with inert gas both before and during the reaction, implying batch operation regarding the gas phase. Table 1 summarizes all the experimental conditions. All the experiments were performed with small catalyst particle size and low Pd loading to minimize the effects of any concentration gradients in the porous catalyst particles.
Table 1 Range of operating conditions for the reduction of nitrite with formic acid in a slurry reactor
| Reaction temperature, °C |
20 |
| Reaction volume, L |
0.3 |
| Stirring speed, rpm |
625 |
| Catalyst particle size, μm |
≤38 |
| Total operating pressure, bar |
1 |
| Amount of the catalyst, g |
0.05–0.1 |
| Initial nitrite concentration, mmol L−1 |
1 |
| Initial formic acid concentration, mmol L−1 |
5–40 |
| pH of the solution |
2.5–9 |
| Mole of oxygen present at ambient atmosphere, without degassing (dissolved in liquid and in gas cap), mmol (ref. 53) |
6.6 |
Samples were taken using a 2.5 mL syringe (BD Plastipak), filtering through a syringe filter (PTFE, 0.2 μm, Whatman), removing the catalyst particles. Formic acid, nitrite, nitrate and ammonium concentrations in the reactants and products were measured with an ion-chromatograph (DIONEX, ICS 3000) equipped with an UltiMate autosampler. It is well known that exclusively ammonia and nitrogen form during the reduction of nitrite,13,40,46,54,55 including nitrite reduction with formic acid,45 and nitrogen was calculated based on the mass balance. All the experiments on the reduction of nitrite with formic acid were performed while flowing He through the reactor as described, resulting in concentrations of gas products below the detection limit of the GC.
The formic acid conversion, nitrite conversion, ammonium yield, nitrate yield and nitrogen yield are calculated using the following equations:
| |  | (6) |
| |  | (7) |
| |  | (8) |
| |  | (9) |
| |  | (10) |
where [HCOOH]
t0 is the initial concentration of formic acid, [NO
2−]
t0 is the initial nitrite concentration, and [HCOOH]
t1, [NO
2−]
t1, [NH
4+]
t1 and [NO
3−]
t1 are the concentration of each compound at
t1.
2.5. ATR-IR
The preparation of a catalyst layer on the ATR crystal is described in detail elsewhere.42,46–48,56 The catalyst with a relatively high Pd loading, i.e. 3 wt% Pd/γ-Al2O3, was used to improve the quality of the IR spectra. Briefly and similar to that described in ref. 42, a suspension containing 0.1 g 3 wt% Pd/γ-Al2O3 or γ-Al2O3 dispersed in 20 mL 2-propanol was prepared. In order to prevent the cracking of the catalyst layer, the suspension was sonicated with an ultrasonic processor (Fisher Scientific-705) for 1 hour. Subsequently, the suspension was spray-coated on a trapezoidal ZnSe crystal (52.5 mm × 20 mm × 2 mm, facet angle 45°, Anadis instruments BV), which was placed on a hot plate at 150 °C, resulting in about 5 mg catalyst on the crystal. Then, the coated crystal was calcined at 300 °C (1 °C min−1) for 1 h in N2 atmosphere (20 mL min−1). It was mounted in a home-build in situ attenuated total reflection infrared spectroscopy (ATR-IR) cell which has been described in detail elsewhere.46,56Fig. 1a shows a SEM micrograph of the catalyst layer in top view, showing a reasonable homogeneous layer coated on the surface. Fig. 1b shows that the thickness of the layer is about 5 μm and uniform. The experimental setup is shown in Scheme 1. The cell was mounted in the sample compartment of an infrared spectrometer (Tensor 27, Bruker) equipped with a liquid nitrogen cooled MCT detector. All the liquid flows were pumped by a peristaltic pump (Verderflex) downstream of the ATR-IR cell, preventing the formation of gas bubbles in the cell (Scheme 1).
 |
| | Fig. 1 SEM micrographs of (a) top view and (b) cross section view of Pd/γ-Al2O3 on a glass plate with identical dimensions to the ZnSe crystal. | |
 |
| | Scheme 1 Scheme of the ATR-IR setup. | |
Once the cell was assembled in the IR spectrometer, it was flushed with Ar/H2O with a flow rate of 0.5 mL min−1 until a stable water spectrum was obtained. Subsequently, the cell with the catalyst layer was flushed with different solutions, including nitrite (10 mM) both at pH 4 and 7 or a mixture of formic acid (10 mM) and nitrite solutions (10 mM), at both pH 3 and pH 5. All the solutions were degassed with Ar before the experiment for at least 3 h. ATR-IR spectra were recorded at room temperature (20 ± 1 °C) and each spectrum was acquired by averaging of 128 scans taken with a resolution of 4 cm−1. The interval between the start of two subsequent spectra was 120 s. Once the water spectrum was stable, the background spectrum was collected. The catalyst layers were re-used a few times and comparable results were obtained, indicating that the catalyst layer is stable during the experiments.
3. Results
3.1. Characterization
Table 2 reports the Pd loading of the prepared catalysts, both close to the targeted values of 1 wt% and 3 wt%, respectively. The 1 wt% catalyst was used in the kinetic batch experiments, whereas the 3 wt% catalyst was used for the ATR-IR experiments, increasing the intensity of IR adsorption bands of species adsorbed on the Pd surface. The specific surface area of the support and 1 wt% Pd/γ-Al2O3 catalyst is similar, indicating that the structure and porosity of the support remained unchanged. In contrast, 3 wt% Pd/γ-Al2O3 shows a minor decrease in surface area, indicating that some pores might be blocked. The metal dispersion of the catalyst with 3 wt% loading is lower than that with 1 wt% metal loading, as expected.
Table 2 Characterization of the two catalysts
| Catalyst materials |
Pd loading (%) |
Specific surface area (m2 g) |
Dispersion (%) |
| γ-Al2O3 |
N.A. |
198 |
N.A. |
| 1 wt% Pd/γ-Al2O3 |
0.9 |
195 |
56 |
| 3 wt% Pd/γ-Al2O3 |
3.1 |
180 |
35 |
3.2. Nitrite reduction with formic acid: effect of pH
Fig. 2 presents the result of two typical nitrite reduction experiments with formic acid in a slurry reactor, at pH 2.8 and pH 4.8, respectively. We conducted the same experiment at different pH values in the window between 2.8 and 9. Fig. 2a shows insignificant conversion of formic acid at pH 2.8 over three hours reaction time. In contrast, nitrite is significantly converted while nitrate is being formed. Since the Pd catalyst was exposed to ambient atmosphere and was not reduced before the reaction, oxidation of nitrite by PdO might contribute to the conversion. Based on the dispersion of Pd and assuming monolayer coverage of O, this can contribute a maximum of 3% of the nitrate amount detected in Fig. 2a. Therefore, the majority of nitrate is formed via another reaction, to be discussed later. Ammonium, the common side product of nitrite reduction, was not detected under pH 2.8 condition.
 |
| | Fig. 2 Concentration profile of formic acid, nitrite, nitrate and ammonium at a) pH 2.8 and b) pH 4.8, respectively (100 mg 0.9 wt% catalyst, 20 mM formic acid solution, and 1 mM nitrite, inert-flow-through, 50 mL min−1 He). | |
Fig. 2b shows significant yet small conversion of formic acid at pH 4.8, instead of no conversion at pH 2.8. In contrast, nitrite conversion is much lower at pH 4.8, while no nitrate is formed. Formation of ammonium as a side product is not detected in both experiments and the ammonium concentration remains below the detection limit in all experiments in this study (0.1%). Thus, nitrogen is the only significant product of reduction of nitrite with formic acid, according to the overall redox reaction:45
| | | 3HCOOH + 2NO2− → 3CO2 + N2 + 2H2O + 2OH− | (11) |
This remarkable selectivity is ascribed to the relatively slow decomposition of formic acid, keeping the effective concentration of H
2 and chemisorbed H low, which is favourable for preventing ammonia formation.
3,57 In addition, CO
2 formed during formic acid decomposition acts as a local pH buffer, keeping the pH low which is also known to suppress the formation of ammonium.
58,59
Fig. 3 presents the influence of pH on the formic acid conversion, nitrite conversion and product distribution after 3 hours reaction time, including the final observations in Fig. 2. Consistent with the data in Fig. 2, no significant conversion of formic acid is detected when the pH of the solution is below 4, whereas formation of nitrate is observed. In contrast, formic acid is consumed and nitrite is hydrogenated to N2 at pH 4.8 or higher, without any formation of NO3−. The catalyst showed maximal nitrite conversion without formation of nitrate at pH 4.8 and the activity dropped with further increasing pH. No conversion whatsoever of formic acid and nitrite is observed at pH 9.
 |
| | Fig. 3 Nitrite conversion, formic acid conversion and nitrate concentration under different pH values and inert-flow-through mode after 3 hours (100 mg 0.9 wt% catalyst, 20 mM formic acid, 1 mM nitrite, 50 mL min−1 He). Please note that the formic acid conversion is multiplied with a factor of four. | |
3.3. ATR-IR
3.3.1. Nitrite adsorption.
Fig. 4 presents the spectra obtained on exposure to nitrite solution for at least 10 minutes. Only one peak is observed at 1235 cm−1 on both ZnSe (dark yellow line) and the γ-Al2O3 layer (blue line), which is assigned to free nitrite in solution. This is in good agreement with previous studies by Ebbesen et al.48,50,56 and Zhao et al.42 The red line displays the spectrum of Pd/γ-Al2O3 exposed to nitrite at pH 7. In addition to the peak at 1235 cm−1, also observed on bare ZnSe and γ-Al2O3, two additional peaks are observed at 1330 and 1425 cm−1, assigned to adsorbed NOx− (x = 2, 3) species.56 Note that the catalyst was stored in ambient atmosphere and was not reduced before the experiment, which could result in the formation of nitrate by the reaction of adsorbed oxygen on Pd with nitrite. The noise between 1600 and 1680 cm−1 is an artifact caused by the subtraction of the water signal.60,61 The black line represents the spectrum on Pd/γ-Al2O3 exposed to nitrite at pH 4. The three peaks at 1235, 1330 and 1425 cm−1 are identical with the experiment at pH 7. However, an extra peak is observed at 1723 cm−1, assigned to NO adsorbed on Pd, based on our previous ATR-IR studies on nitrite reduction,42,46,48,50,56i.e. in experiments where NO is formed via reduction of nitrite with H2.46,50 Apparently, adsorbed NO (NOads) can also form in the absence of H2 at pH 4 but not at pH 7.
 |
| | Fig. 4 ATR-IR spectra obtained after exposure to nitrite solutions: the dark yellow line represents the nitrite spectrum on ZnSe at pH 7, the blue line represents the nitrite spectrum of the γ-Al2O3 layer under pH 7, the red and black lines represent the nitrite spectrum of the Pd/γ-Al2O3 layer under pH 7 and 4, respectively (5 mg catalyst, 3 wt% Pd/γ-Al2O3, 10 mM nitrite, 0.5 mL min−1 flow rate). | |
3.3.2. Reaction of formic acid and nitrite.
Fig. 5a shows ATR-IR spectra obtained by flowing a solution containing both formic acid and nitrite at pH 3 over the catalyst layer for 38 minutes. The intensities of the peaks increase with time. The strong peak at 1581 cm−1 is assigned to free formic acid51 and the peak at 1235 cm−1 is assigned to free nitrite as discussed above. The peak at 1716 cm−1 is assigned to NO adsorbed on Pd as discussed above as well42,46,48,50,56 and the small red-shift is likely due to differences in the surface coverage, influencing the extent of dipole–dipole coupling. The complex overlapping peaks between 1300 and 1500 cm−1 are assigned to free formic acid in water51 and NOx− (x = 2, 3) species (Fig. 4, 1425 and 1330 cm−1).
 |
| | Fig. 5 a) Reduction of nitrite with formic acid at pH 3 for 38 min, b) after stopping the flow to mimic the batch reaction for another 38 min, and c) zoom in the range between 1700 and 2200 cm−1 in figure b (5 mg 3 wt% catalyst on ZnSe, 10 mM formic acid, 10 mM nitrite, 0.5 mL min−1 flow rate). | |
After 38 min, the liquid flow was stopped, leaving the solution in the reactor in contact with the catalyst layer, mimicking a batch reaction experiment. The important observation in Fig. 5b is that the spectrum does not change at all with time, indicating that there is no detectable conversion of formic acid, in agreement with observations in batch experiments at low pH, as shown in Fig. 2a. Fig. 2a also shows typically 20% conversion of nitrite in 38 minutes, which is apparently not detectable in the ATR-IR experiment. Fig. 5c shows a zoom-in of Fig. 5b in the range between 1700 and 2200 cm−1, demonstrating the absence of chemisorbed CO on Pd metal.
Fig. 6a shows the time-evolved ATR-IR spectra during exposure to formic acid and nitrite solution at higher pH, i.e. pH 5. Clearly, the intensities of all the peaks increase again with time. It is noticeable that a new shoulder peak is observed at 1510 cm−1 with increasing intensity in time. Based on our previous study on nitrite hydrogenation with ATR-IR, the peak at 1510 cm−1 was assigned to NH2,ads on the Pd surface (1510 cm−1) by Ebbesen et al.46 as an intermediate species during nitrite reduction. It should be noted though that a peak at the same wavelength (1510 cm−1) is also observed during decomposition of formic acid.51 The other peaks observed in the window between 1300 and 1500 cm−1 are again assigned to free formic acid and adsorbed NOx− (x = 2, 3). Further carbonates, generated during the reaction, also contribute to additional IR peaks62,63 in the 1300–1500 cm−1 range, complicating any detailed interpretation in this range.
 |
| | Fig. 6 a) ATR-IR spectra of the reduction of nitrite with formic acid at pH 5 for 38 min, b) after stopping the flow, mimicking a batch reaction for another 38 min, and c) zoom in figure b in the range of 1700 and 2200 cm−1 (5 mg 3 wt% catalyst on ZnSe, 10 mM formic acid solution, 10 mM nitrite, 0.5 mL min−1 flow rate). | |
After flowing formic acid and nitrite for 38 min, the flow was again stopped to mimic a batch experiment. Fig. 6b shows that peak intensities change significantly with reaction time, in contrast to Fig. 5b. Both peaks assigned to formic acid (1581 cm−1) and bulk nitrite (1235 cm−1) gradually decreased in intensity, in qualitative agreement with the conversion of formic acid via reaction with nitrite as observed in Fig. 2b. The peaks assigned to adsorbed NO (1716 cm−1) and the intermediate species (1510 cm−1) appear stable during the experiment. The zoom-in Fig. 6c shows minor but significant formation of chemisorbed CO during the batch experiment.
3.4. Effect of oxygen on nitrite reduction
Table 3 reports the nitrite conversion and formic acid conversion achieved after 2 hours of reaction when starting the experiment with air present in the reactor, as described as ambient-batch mode. The presence of oxygen slows down the conversion of both formic acid and nitrite, although the effect on formic acid conversion is smaller. The efficiency of formic acid to reduce nitrite is defined as:| |  | (12) |
The presence of traces of oxygen clearly decreases the efficiency and a larger fraction of formic acid apparently decomposes or reacts with oxygen, instead of reacting with nitrite.
Table 3 Influence of the presence of oxygen on nitrite reduction with formic acid (pH = 5, 100 mg 0.9 wt% catalyst, 1 mM nitrite, 20 mM formic acid)
| Atmosphere |
Initial nitrite (mM) |
Initial formic acid (mM) |
Formic acid conversion at 2 h (%) |
Nitrite conversion at 2 h (%) |
Formic acid efficiency (%) |
| Inert-flow-through |
1 |
20 |
5.1 ± 0.3 |
33.3 ± 1 |
49 ± 4 |
| Ambient-batch |
1 |
20 |
3.1 ± 0.1 |
8.9 ± 0.1 |
22 ± 1 |
3.5. Formic acid concentration
The influence of the concentration of formic acid on the rate of nitrite reduction was measured between 5 and 40 mM under inert-flow-through at pH 5. Fig. S1–S4† present the concentration profiles and Table 4 summarizes the results, reporting the conversion of formic acid and nitrite after 2 hours of reaction. Nitrite conversion increases with increasing formic acid concentration as expected. Surprisingly, the formic acid conversion also increases with increasing initial concentration.
Table 4 Formic acid and nitrite conversion with different formic acid concentrations at pH 5 and under inert-flow-through conditions (100 mg 0.9 wt% catalyst, 1 mM nitrite, 2 h)
| Initial formic acid (mM) |
Initial nitrite (mM) |
Formic acid conversion at 2 h (%) |
Nitrite conversion at 2 h (%) |
Formic acid efficiency (%) |
| 40 |
1 |
12.4 ± 0.1 |
66.1 ± 0.1 |
20 ± 1 |
| 20 |
1 |
5.1 ± 0.3 |
33.3 ± 1 |
49 ± 4 |
| 10 |
1 |
4.6 ± 1 |
13 ± 3 |
42 ± 19 |
| 5 |
1 |
2.4 ± 0.1 |
0.94 ± 0.05 |
12 ± 10 |
3.6. Effect of H2 addition
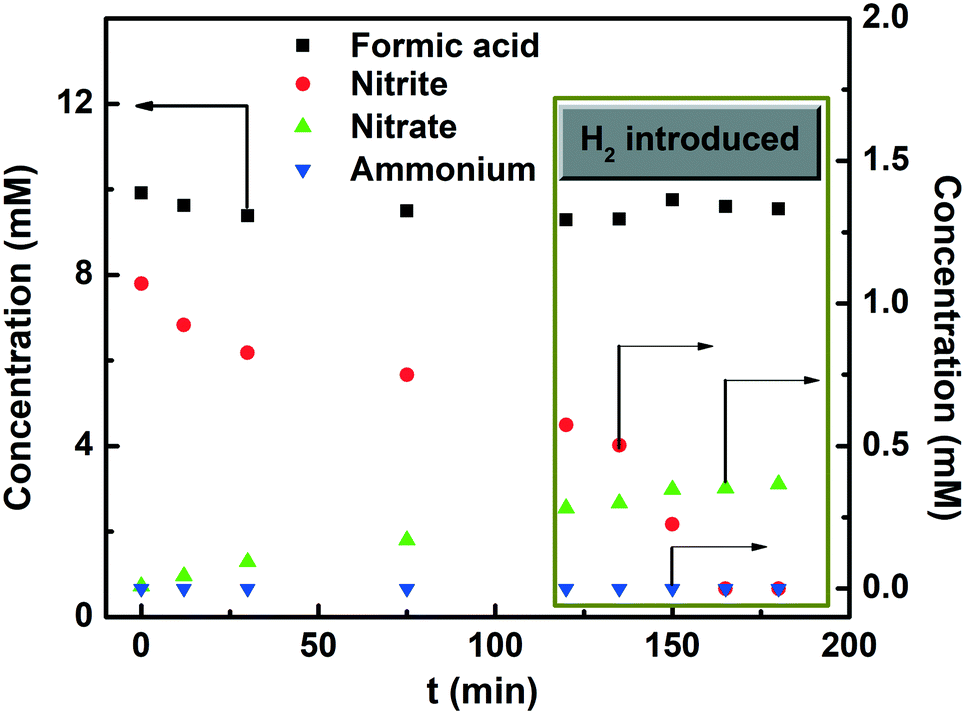
Fig. 7 presents the concentration profiles of formic acid, nitrite, ammonium and nitrate during the reduction of nitrite with formic acid at pH 3. Pure H2 gas was introduced to the reactor after two hours, resulting in fast conversion of nitrite without producing any detectable ammonium, i.e. the concentration remained below the detection limit of 1 μM. This result demonstrates that the catalyst remains active for nitrite reduction with H2 gas.
 |
| | Fig. 7 Nitrite reduction with formic acid (pH = 3) performed in ambient-batch mode (100 mg 0.9 wt% catalyst, 10 mM formic acid, 1 mM nitrite); hydrogen is introduced (1 bar, 100 mL min−1 flow rate) in the solution after 2 h of reaction. | |
4. Discussion
The results obtained at low pH will be discussed first, followed by a detailed discussion for the case of intermediate pH values resulting in the reaction of nitrite and formic acid. Finally, the absence of any activity at high pH will be discussed.
4.1.a. Nitrite conversion at low pH
As shown in Fig. 2a and 3, formic acid is not converted at low pH, while nitrite was consumed and nitrate was formed at the same time. Garron et al.45 reported a similar phenomenon, i.e. formation of nitrate at low pH.
Nitrite is converted to nitrate in a stoichiometric ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. 2a) according to homogeneous disproportionation of nitrous acid forming NO and nitric acid (eqn (14)). This reaction proceeds exclusively at sufficiently low pH, shifting the equilibrium in eqn (13) to the right.45,64 The reaction in eqn (15) closes the catalytic cycle with formation of acid, in this case formic acid, acting as a catalyst.64
:1 (Fig. 2a) according to homogeneous disproportionation of nitrous acid forming NO and nitric acid (eqn (14)). This reaction proceeds exclusively at sufficiently low pH, shifting the equilibrium in eqn (13) to the right.45,64 The reaction in eqn (15) closes the catalytic cycle with formation of acid, in this case formic acid, acting as a catalyst.64
| | | HCOOH + NO2− ↔ HNO2 + HCOO− | (13) |
| | | 3HNO2 → 2NO + HNO3 + H2O | (14) |
| | | HNO3 + HCOO− → NO3− + HCOOH | (15) |
As shown in
Fig. 4, adsorbed NO is observed with ATR-IR at pH 3 based on the peak at 1723 cm
−1, confirming that the homogeneous disproportionation reaction is significant at low pH. Note that formic acid was absent in the ATR-IR experiment in
Fig. 4, illustrating that any acid catalyses the homogeneous disproportionation reaction. This is confirmed with two control experiments in the absence of any catalyst in ambient atmosphere in the presence of formic acid and HCl, respectively (Fig. S5
†). In both cases conversion of nitrite to nitrate is indeed observed.
Conversion of nitrite at low pH is apparently not detectable in the ATR-IR experiment (Fig. 5b), in contrast to the batch experiment (Fig. 2a). This is likely caused by the fact that any gaseous products formed, i.e. NO, remain in the ATR-IR reactor, instead of being flushed out of the reactor in the batch experiment. The presence of NO probably delays the reversible reaction when not flushed out. It should be noted that it is assumed that the qualitative observations on the presence of adsorbed NO, obtained with the catalyst with 3 wt% Pd loading used for the ATR-IR experiment, is also valid for the catalyst with 1 wt% Pd loading, used for the kinetic experiments.
4.1.b. Formic acid conversion at low pH
Fig. 2a, 3 and 5b demonstrate the absence of any conversion of formic acid at low pH. However, decomposition of formic acid would be expected to proceed over Pd, also in the absence of nitrite. In our recent work,51 we reported that Pd catalysts partially convert formic acid, although deactivating during the experiment caused by CO poisoning. Fig. 5c shows the absence of any peaks in ATR-IR to be assigned to chemisorbed CO, implying that the Pd sites are not occupied with CO. Clearly, the lack of activity for formic acid decomposition is not caused by CO poisoning.
The inactivity for formic acid decomposition is attributed to the formation of chemisorbed NO. Noticeably, adsorbed NO is detected in ATR-IR (Fig. 5), according to the peak at 1716 cm−1, maintaining constant intensity during the experiment mimicking a batch experiment (Fig. 5b). Fig. 7 shows that introduction of hydrogen results in rapid conversion of nitrite, without forming any nitrate. This indicates that the species adsorbed on Pd, responsible for hindering the formic acid decomposition, does not deactivate the catalyst for nitrite hydrogenation with H2. This agrees well with the proposition that adsorbed NO is responsible, as previous ATR-IR studies showed that adsorbed NO on Pd converts readily with H2 to N2.46 In fact, NO is a key surface-intermediate in reduction of nitrite with H2 over Pd catalysts. In short, adsorbed NO impedes the decomposition of formic acid on the catalyst, without impacting the dissociative adsorption of H2 on the catalyst surface.
An alternative hypothesis that the catalyst is deactivated for formic acid decomposition by nitrate can be rejected. Addition of nitrate in a formic acid decomposition experiment has no effect on the rate of decomposition of formic acid (Fig. S6†). Nitrate is not converted in this experiment because nitrate reduction requires a bimetallic catalyst, for instance the Cu–Pd catalyst,28,35,43–45 instead of a monometallic Pd catalyst.
Garron et al.45 reported, besides the formation of nitrate at low pH, nitrite reduction by formic acid, which is not in agreement with our results. The difference might be caused by differences in concentration and the support used, i.e., silica instead of alumina in this study.
4.2. Nitrite reduction with formic acid at intermediate pH
The catalytic reaction of nitrite and formic acid at pH above 4.5 results in simultaneous conversion of formic acid and nitrite (Fig. 2b and 3). Nitrate was not formed, implying that the homogeneous disproportionation reaction forming NO does not proceed and any catalyst deactivation by NO was prevented. This is qualitatively in agreement with the ATR-IR results in Fig. 6b, demonstrating decreasing concentrations of both nitrite (1235 cm−1) and formic acid (1581 cm−1) when operating in batch mode at pH 5. The surprisingly high conversion of nitrite suggested by Fig. 6b, compared to the low conversion in batch experiments (Fig. 2b), despite a much higher formic acid/nitrite molar ratio (2069:2069:1) in the ATR-IR experiment compared to 719:36:1 in the batch experiment. Remarkably, reaction rates normalized to the number of Pd surface atoms are quite similar, as calculated in ESI† Section S1. Possibly, the high conversion of nitrite in ATR-IR is caused the fact that gasses are not flushed out of the reactor, in contrast to the batch experiment as discussed before. H2 accumulates in the ATR-IR reactor as the formic acid efficiency is far below 100% (Table 4), enhancing nitrite conversion in the ATR-IR cell.
It should also be noted that the conversion of formic acid cannot be estimated as deconvolution of the complex set of peaks between 1300 and 1700 cm−1 would be required, which is not possible with the data available.
Interestingly, adsorbed NO is observed both at pH 3 (Fig. 5b) and pH 5 (Fig. 6b) but the origin is completely different. The NOads peak at pH 3 in Fig. 5b is due to the homogeneous disproportionation reaction, as discussed above. NO formed homogeneously chemisorbs on the Pd surface, preventing dissociation of formic acid to form H2 and chemisorbed H. In the absence of chemisorbed H, chemisorbed NO cannot convert to N2 or ammonia.
In contrast, adsorbed NO formed in the presence of formic acid and nitrite at pH 5 in Fig. 6b is a reaction intermediate of nitrite reduction. Scheme 2 presents a reaction scheme with the most important pathways, occurring during nitrite reduction with formic acid. Note that most of these steps contain multiple lumped elementary reactions. In step 1, formic acid decomposes to adsorbed Hads and CO2, requiring at least two active sites. This step may require even a larger ensemble of Pd sites to allow formic acid to decompose, as Navlani-García et al.65 reported that formic acid decomposition is structure sensitive. Once Hads is produced via step 1, Hads can be consumed via steps 2, 3, 4 and 5. In step 2, two Hads atoms desorb associatively to H2 leaving the Pd surface, lowering the efficiency of formic acid. Step 3 and step 4 are derived from the nitrite reduction mechanism discussed in our previous study.52
 |
| | Scheme 2 Reaction scheme of nitrite reduction with formic acid. | |
When oxygen is present, step 5 influences the performance. Table 3 shows that nitrite conversion was much higher in inert atmosphere than in the presence of traces of oxygen, also decreasing the formic acid efficiency. This is expected as oxygen reacts readily with chemisorbed H, competing with steps 3 and 4 and resulting in decreased nitrite conversion and decreased formic acid efficiency in the presence of oxygen traces.
It is surprising that the presence of traces of oxygen decreases formic acid conversion (Table 3), opposite to the observation in our recent work on formic acid decomposition.51 It was shown that formic acid decomposition is enhanced by oxygen in two ways: first, low oxygen concentrations prevent poisoning of the catalyst by oxidizing and removing chemisorbed CO and second, higher oxygen concentrations enhance the conversion of formic acid via oxidation.51 It might be that the enhancing effect of removal of chemisorbed CO by oxygen is not significant under the conditions in this study as the intensity of the IR peak attributed to chemisorbed CO is rather weak (Fig. 6c). Nevertheless, the decrease in formic acid conversion remains surprising and another effect apparently dominates.
Earlier work by Ebbesen showed that high coverage of NOads forms on Pd exposed to nitrite at low Hads surface coverage, based on ATR-IR spectroscopy data.46 This was explained by reasoning that the rate of step 4 depends more strongly on the H-coverage as many hydrogen atoms are required to convert NOads to products. The rate of reaction (3) is less sensitive to the H-coverage, requiring only one H atom and a proton. The presence of oxygen consumes hydrogen according to reaction (5), decreasing the H coverage. This will delay reaction (4) more strongly than reaction (3), decreasing the rate of consumption of NOads more strongly than decreasing the rate of formation and therefore the concentration of NOads increases. Consequently, fewer empty active sites are available for formic acid decomposition, lowering the rate of step 1 and thus the rate of formic acid conversion in the presence of oxygen. Obviously, decreasing the formic acid concentration also causes a decrease in H coverage by decreasing the rate of reaction (1). Also in that case, the concentration of NOads increases and the concentration of free Pd sites decreases, decreasing further the rate of reaction (1). Therefore, the apparent reaction order in formic acid is larger than one, i.e. 1.4 (Table 4 and Fig. S7†). In short, the availability of Hads on the surface needs to be in balance with NOads so that sufficient open sites remain available for formic acid to adsorb dissociatively.
From a practical point of view, high formic acid efficiency is preferred. The best efficiencies are obtained at intermediate formic acid concentration (Table 4) and in the absence of any air (Table 3), but values below 50% are not sufficient. A high rate at the expense of low efficiency is expected at high formic acid concentration as H2 generation is too fast compared to hydrogen consumption. In semi-batch operation this results in H2 flushing out of the reactor. Therefore, the reaction in the batch mode without gas flowing through the reactor during the reaction is preferred, preventing H2 gas from escaping the reactor. On the other hand, continuous operation for water would be much more practical, obviously. An advantage is that formation of ammonium is negligible because formic acid releases adsorbed H very slowly via decomposition compared with H2 gas flowing continuously through the reactor.66 The reason for low efficiency at very low formic acid concentration is not clear and would require additional research to understand.
4.3. Nitrite reduction with formic acid at high pH
At very high pH (9) no reaction is observed because formate ions cannot decompose to provide H2 under these conditions,51 in good agreement with the literature.67–69 Therefore, no conversion of both formic acid and nitrite is expected. Also, adsorbed NO is not detected at higher pH, in agreement with the fact that the disproportionation reaction (eqn (14)) proceeds exclusively at low pH.
5. Conclusions
This work demonstrates that nitrite reduction with formic acid is possible in the pH range between 4.5 and 8 compatible with drinking water, producing negligible amounts of ammonium. The rate of conversion of nitrite with formic acid and the rate of decomposition of formic acid are controlled by competitive adsorption of nitrite and formic acid on the Pd sites, forming chemisorbed NO and chemisorbed H, respectively. Dissociative adsorption of formic acid requires empty Pd sites, possibly an ensemble of empty sites, which is favoured by a low coverage of chemisorbed NO. The coverage of NO is low if sufficient adsorbed H is available to convert NO rapidly to N2, which in turn depends on the rate of dissociative adsorption of formic acid. This causes the formic acid decomposition to exhibit an apparent order of 1.4 in formic acid. The homogeneous disproportionation reaction of nitric acid proceeds when the pH is below 4.5, resulting in catalyst poisoning with NO generated by the homogeneous disproportionation reaction, independent of the catalyst. The catalyst shows no activity at pH above 8 due to the fact that formate ions are not reactive under such conditions.
Author contributions
P. Xu: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft. S. Agarwal: supervision, visualization, writing – review & editing. L. Lefferts: funding acquisition, project administration, supervision resources, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge financial support from China Scholarship Council. We are grateful to K. Altena-Schildkamp and T. M. L. Velthuizen for chemical analysis. We acknowledge B. Geerdink for technical support.
References
- G. Gulis, M. Czompolyova and J. R. Cerhan, An Ecologic Study of Nitrate in Municipal Drinking Water and Cancer Incidence in Trnava District, Slovakia, Environ. Res., 2002, 88, 182–187, DOI:10.1006/enrs.2002.4331.
- C. S. Bruning-Fann and J. B. Kaneene, The effects of nitrate, nitrite and N-nitroso compounds on human health: a review, Vet. Hum. Toxicol., 1993, 35, 521–538 CAS , http://www.ncbi.nlm.nih.gov/pubmed/8303822 (accessed September 20, 2017).
- S. Hörold, K.-D. Vorlop, T. Tacke and M. Sell, Development of catalysts for a selective nitrate and nitrite removal from drinking water, Catal. Today, 1993, 17, 21–30, DOI:10.1016/0920-5861(93)80004-K.
- V. Matějů, S. Čižinská, J. Krejčí and T. Janoch, Biological water denitrification-A review, Enzyme Microb. Technol., 1992, 14, 170–183, DOI:10.1016/0141-0229(92)90062-S.
- J. Schullehner, B. Hansen, M. Thygesen, C. B. Pedersen and T. Sigsgaard, Nitrate in drinking water and colorectal cancer risk: A nationwide population-based cohort study, Int. J. Cancer, 2018, 143, 73–79, DOI:10.1002/ijc.31306.
- W. H. Organization, Guidelines for Drinking-water Quality, 4th edn, 2011 Search PubMed.
- A. Kapoor and T. Viraraghavan, Nitrate Removal From Drinking Water—Review, J. Environ. Eng., 1997, 123, 371–380, DOI:10.1061/(ASCE)0733-9372(1997)123:4(371).
- A. Bothner-By and L. Friedman, The reaction of nitrous acid with hydroxylamine, J. Chem. Phys., 1952, 20, 459–462, DOI:10.1063/1.1700442.
- J. J. F. Scholten, Metal Surface Area and Metal Dispersion in Catalysts, Stud. Surf. Sci. Catal., 1979, 685–714, DOI:10.1016/S0167-2991(09)60244-5.
- A. Obuchi, S. Naito, T. Onishi and K. Tamaru, Mechanism of catalytic reduction of NO by H2 or CO on a Pd foil; Role of chemisorbed nitrogen on Pd, Surf. Sci., 1982, 122, 235–255, DOI:10.1016/0039-6028(82)90076-0.
- S. B. Oblath, S. S. Markowitz, T. Novakov and S. G. Chang, Reaction of Nitrite Ion with Hydroxylamine-N-sulfonate in Aqueous Solution, Inorg. Chem., 1983, 22, 579–583, DOI:10.1021/ic00146a002.
-
T. Tacke, PhD Dissertation, Tech. Univ. Carolo-Wilhelmina, 1991, DOI:10.1016/0039-6028(91)90317-L.
- J. Wärn, I. Turunen, T. Salmi and T. Maunula, Kinetics of nitrate reduction in monolith reactor, Chem. Eng. Sci., 1994, 49, 5763–5773, DOI:10.1016/0009-2509(94)00331-9.
- E. E. A. (EEA), https://www.eea.europa.eu/data-and-maps/figures/nitrate-concentration-in-groundwater, 2002, DOI:10.1016/0039-6028(91)90317-L.
- M. Vospernik, A. Pintar, G. Berčič and J. Levec, Experimental verification of ceramic membrane potentials for supporting three-phase catalytic reactions, J. Membr. Sci., 2003, 223, 157–169, DOI:10.1016/S0376-7388(03)00320-X.
- K. T. Ranjit and B. Viswanathan, Photocatalytic reduction of nitrite and nitrate ions to ammonia on M/TiO2 catalysts, J. Photochem. Photobiol., A, 1997, 108, 73–78, DOI:10.1016/S1010-6030(96)04505-4.
- A. Pintar, G. Berčič and J. Levec, Catalytic liquid-phase nitrite reduction: Kinetics and catalyst deactivation, AIChE J., 1998, 44, 2280–2292, DOI:10.1002/aic.690441017.
- J. K. Chinthaginjala and L. Lefferts, Support effect on selectivity of nitrite reduction in water, Appl. Catal., B, 2010, 101, 144–149, DOI:10.1016/j.apcatb.2010.09.023.
- J. K. Chinthaginjala, J. H. Bitter and L. Lefferts, Thin layer of carbon-nano-fibers (CNFs) as catalyst support for fast mass transfer in hydrogenation of nitrite, Appl. Catal., A, 2010, 383, 24–32, DOI:10.1016/j.apcata.2010.05.013.
- H. C. Aran, J. K. Chinthaginjala, R. Groote, T. Roelofs, L. Lefferts, M. Wessling and R. G. H. H. Lammertink, Porous ceramic mesoreactors: A new approach for gas–liquid contacting in multiphase microreaction technology, Chem. Eng. J., 2011, 169, 239–246, DOI:10.1016/j.cej.2010.11.005.
- Y. Zhao, J. A. Baeza, N. Koteswara Rao, L. Calvo, M. A. Gilarranz, Y. D. Li and L. Lefferts, Unsupported PVA- and PVP-stabilized Pd nanoparticles as catalyst for nitrite hydrogenation in aqueous phase, J. Catal., 2014, 318, 162–169, DOI:10.1016/j.jcat.2014.07.011.
- R. Brunet Espinosa and L. Lefferts, Ni in CNFs: Highly Active for Nitrite Hydrogenation, ACS Catal., 2016, 6, 5432–5440, DOI:10.1021/acscatal.6b01375.
- A. J. Lecloux, Chemical, biological and physical constrains in catalytic reduction processes for purification of drinking water, Catal. Today, 1999, 53, 23–34, DOI:10.1016/S0920-5861(99)00100-5.
- G. Strukul, R. Gavagnin, F. Pinna, E. Modaferri, S. Perathoner, G. Centi, M. Marella and M. Tomaselli, Use of palladium based catalysts in the hydrogenation of nitrates in drinking water: from powders to membranes, Catal. Today, 2000, 55, 139–149, DOI:10.1016/S0920-5861(99)00233-3.
- O. M. Ilinitch, L. V. Nosova, V. V. Gorodetskii, V. P. Ivanov, S. N. Trukhan, E. N. Gribov, S. V. Bogdanov and F. P. Cuperus, Catalytic reduction of nitrate and nitrite ions by hydrogen: Investigation of the reaction mechanism over Pd and Pd-Cu catalysts, J. Mol. Catal. A: Chem., 2000, 158, 237–249, DOI:10.1016/S1381-1169(00)00070-4.
- Y. Sakamoto, K. Nakamura, R. Kushibiki, Y. Kamiya and T. Okuhara, A Two-stage Catalytic Process with Cu–Pd Cluster/Active Carbon and Pd/β-Zeolite for Removal of Nitrate in Water, Chem. Lett., 2005, 34, 1510–1511, DOI:10.1246/cl.2005.1510.
- D. Gašparovičová, M. Králik, M. Hronec, Z. Vallušová, H. Vinek and B. Corain, Supported Pd-Cu catalysts in the water phase reduction of nitrates: Functional resin versus alumina, J. Mol. Catal. A: Chem., 2007, 264, 93–102, DOI:10.1016/j.molcata.2006.08.081.
- U. Prüsse, M. Hähnlein, J. Daum and K. D. Vorlop, Improving the catalytic nitrate reduction, Catal. Today, 2000, 55, 79–90, DOI:10.1016/S0920-5861(99)00228-X.
- A. Pintar, J. Batista, J. Levec and T. Kajiuchi, Kinetics of the catalytic liquid-phase hydrogenation of aqueous nitrate solutions, Appl. Catal., B, 1996, 11, 81–98, DOI:10.1016/S0926-3373(96)00036-7.
- J. Sá, J. Montero, E. Duncan and J. A. Anderson, Bi modified Pd/SnO2 catalysts for water denitration, Appl. Catal., B, 2007, 73, 98–105, DOI:10.1016/j.apcatb.2006.06.012.
- H. O. N. Tugaoen, S. Garcia-Segura, K. Hristovski and P. Westerhoff, Challenges in photocatalytic reduction of nitrate as a water treatment technology, Sci. Total Environ., 2017, 599–600, 1524–1551, DOI:10.1016/j.scitotenv.2017.04.238.
- S. Tyagi, D. Rawtani, N. Khatri and M. Tharmavaram, Strategies for Nitrate removal from aqueous environment using Nanotechnology: A Review, J. Water Process. Eng., 2018, 21, 84–95, DOI:10.1016/j.jwpe.2017.12.005.
- G. Mendow, N. S. Veizaga, C. A. Querini and B. S. Sánchez, A continuous process for the catalytic reduction of water nitrate, J. Environ. Chem. Eng., 2019, 7, 102808, DOI:10.1016/j.jece.2018.11.052.
- S. Guo, K. Heck, S. Kasiraju, H. Qian, Z. Zhao, L. C. Grabow, J. T. Miller and M. S. Wong, Insights into Nitrate Reduction over Indium-Decorated Palladium Nanoparticle Catalysts, ACS Catal., 2018, 8, 503–515, DOI:10.1021/acscatal.7b01371.
- Y. Ding, W. Sun, W. Yang and Q. Li, Formic acid as the in-situ hydrogen source for catalytic reduction of nitrate in water by PdAg alloy nanoparticles supported on amine-functionalized SiO2, Appl. Catal., B, 2017, 203, 372–380, DOI:10.1016/j.apcatb.2016.10.048.
- O. S. G. P. Soares, M. F. R. Pereira, J. J. M. Órfão, J. L. Faria and C. G. Silva, Photocatalytic nitrate reduction over Pd-Cu/TiO2, Chem. Eng. J., 2014, 251, 123–130, DOI:10.1016/j.cej.2014.04.030.
- S. Hamid, S. Bae, W. Lee, M. T. Amin and A. A. Alazba, Catalytic Nitrate Removal in Continuous Bimetallic Cu–Pd/Nanoscale Zerovalent Iron System, Ind. Eng. Chem. Res., 2015, 54, 6247–6257, DOI:10.1021/acs.iecr.5b01127.
- S. Jung, S. Bae and W. Lee, Development of Pd-Cu/hematite catalyst for selective nitrate reduction, Environ. Sci. Technol., 2014, 48, 9651–9658, DOI:10.1021/es502263p.
- K. N. Heck, S. Garcia-Segura, P. Westerhoff and M. S. Wong, Catalytic Converters for Water Treatment, Acc. Chem. Res., 2019, 52, 906–915, DOI:10.1021/acs.accounts.8b00642.
- R. Brunet Espinosa, D. Rafieian, R. S. Postma, R. G. H. Lammertink and L. Lefferts, Egg-shell membrane reactors for nitrite hydrogenation: Manipulating kinetics and selectivity, Appl. Catal., B, 2018, 224, 276–282, DOI:10.1016/j.apcatb.2017.10.058.
- Y. Zhao, W. Liang, Y. Li and L. Lefferts, Effect of chlorine on performance of Pd catalysts prepared via colloidal immobilization, Catal. Today, 2017, 297, 308–315, DOI:10.1016/j.cattod.2017.01.028.
- Y. Zhao, N. Koteswara Rao and L. Lefferts, Adsorbed species on Pd catalyst during nitrite hydrogenation approaching complete conversion, J. Catal., 2016, 337, 102–110, DOI:10.1016/j.jcat.2016.02.007.
- E. K. Choi, K. H. Park, H. B. Lee, M. Cho and S. Ahn, Formic acid as an alternative reducing agent for the catalytic nitrate readuction in aqueous media, J. Environ. Sci., 2013, 25, 1696–1702, DOI:10.1016/S1001-0742(12)60226-5.
-
Y. Wu, Y. Hu, J. Cheng and Y. Guo, Catalytic reduction of nitrate by Pd/SnO2 catalyst using formic acid as reducing agent, 2010 4th Int. Conf. Bioinforma. Biomed. Eng. ICBBE 2010, 2010, pp. 2–5, DOI:10.1109/ICBBE.2010.5515602.
- A. Garron and F. Epron, Use of formic acid as reducing agent for application in catalytic reduction of nitrate in water, Water Res., 2005, 39, 3073–3081, DOI:10.1016/j.watres.2005.05.012.
- S. D. Ebbesen, B. L. Mojet and L. Lefferts, In situ ATR-IR study of nitrite hydrogenation over Pd/Al2O3, J. Catal., 2008, 256, 15–23, DOI:10.1016/j.jcat.2008.02.013.
- S. D. Ebbesen, B. L. Mojet and L. Lefferts, The influence of water and pH on adsorption and oxidation of CO on Pd/Al2O3-an investigation by attenuated total reflection infrared spectroscopy, Phys. Chem. Chem. Phys., 2009, 11, 641–649, 10.1039/B814605E.
- S. D. Ebbesen, B. L. Mojet and L. Lefferts, Mechanistic Investigation of the Heterogeneous Hydrogenation of Nitrite over Pt/Al2O3 by Attenuated Total Reflection Infrared Spectroscopy, J. Phys. Chem. C, 2009, 113, 2503–2511, DOI:10.1021/jp8081886.
- B. L. Mojet, S. D. Ebbesen and L. Lefferts, Light at the interface: the potential of attenuated total reflection infrared spectroscopy for understanding heterogeneous catalysis in water, Chem. Soc. Rev., 2010, 39, 4643–4655, 10.1039/C0CS00014K.
- S. D. Ebbesen, B. L. Mojet and L. Lefferts, Effect of pH on the Nitrite Hydrogenation Mechanism over Pd/Al2O3 and Pt/Al2O3: Details Obtained with ATR-IR Spectroscopy, J. Phys. Chem. C, 2011, 115, 1186–1194, DOI:10.1021/jp106521t.
- P. Xu, F. D. Bernal-juan and L. Lefferts, Effect of oxygen on formic acid decomposition over Pd catalyst, J. Catal., 2021, 394, 342–352 CrossRef CAS.
- P. Xu, S. Agarwal and L. Lefferts, Mechanism of nitrite hydrogenation over Pd/γ-Al2O3 according a rigorous kinetic study, J. Catal., 2020, 383, 124–134, DOI:10.1016/j.jcat.2020.01.003.
-
Fondriest Environmental Inc., Fundamentals of Environmental Measurements: Dissolved Oxygen, 2016, https://www.fondriest.com/environmental-measurements/parameters/water-quality/dissolved-oxygen/.
- R. Zhang, D. Shuai, K. A. Guy, J. R. Shapley, T. J. Strathmann and C. J. Werth, Elucidation of Nitrate Reduction Mechanisms on a Pd-In Bimetallic Catalyst using Isotope Labeled Nitrogen Species, ChemCatChem, 2013, 5, 313–321, DOI:10.1002/cctc.201200457.
- I. Mikami, Y. Sakamoto, Y. Yoshinaga and T. Okuhara, Kinetic and adsorption studies on the hydrogenation of nitrate and nitrite in water using Pd-Cu on active carbon support, Appl. Catal., B, 2003, 44, 79–86, DOI:10.1016/S0926-3373(03)00021-3.
- S. D. Ebbesen, B. L. Mojet and L. Lefferts, In Situ attenuated total reflection infrared (ATR-IR) study of the adsorption of NO2-, NH2OH, and NH4+ on Pd/Al2O3 and Pt/Al2O3, Langmuir, 2008, 24, 869–879, DOI:10.1021/la7027725.
- R. S. Postma, R. Brunet Espinosa and L. Lefferts, Competitive Adsorption of Nitrite and Hydrogen on Palladium during Nitrite Hydrogenation, ChemCatChem, 2018, 10, 3770–3776, DOI:10.1002/cctc.201800523.
- C. Franch, R. G. H. Lammertink and L. Lefferts, Partially hydrophobized catalyst particles for aqueous nitrite hydrogenation, Appl. Catal., B, 2014, 156–157, 166–172, DOI:10.1016/j.apcatb.2014.03.020.
- F. Deganello, L. F. Liotta, A. Macaluso, A. M. Venezia and G. Deganello, Catalytic reduction of nitrates and nitrites in water solution on pumice-supported Pd-Cu catalysts, Appl. Catal., B, 2000, 24, 265–273, DOI:10.1016/S0926-3373(99)00109-5.
- M. J. Weaver, S. Zou and C. Tang, A concerted assessment of potential-dependent vibrational frequencies for nitric oxide and carbon monoxide adlayers on low-index platinum-group surfaces in electrochemical compared with ultrahigh vacuum environments: Structural and electrostatic implicati, J. Chem. Phys., 1999, 111, 368–381, DOI:10.1063/1.479279.
- R. Gómez, A. Rodes, J. M. Orts, J. M. Feliu and J. M. Pérez, FTIRS and electrochemical characterization of NO adlayers on Pt(hkl) generated upon immersion in an acidic solution of nitrite, Surf. Sci., 1995, 342(1–3), L1104–L1110, DOI:10.1016/0039-6028(95)00890-X.
- J. E. Pander, M. F. Baruch and A. B. Bocarsly, Probing the Mechanism of Aqueous CO2Reduction on Post-Transition-Metal Electrodes using ATR-IR Spectroelectrochemistry, ACS Catal., 2016, 6, 7824–7833, DOI:10.1021/acscatal.6b01879.
- M. F. Baruch, J. E. Pander, J. L. White and A. B. Bocarsly, Mechanistic Insights into the Reduction of CO2 on Tin Electrodes using in Situ ATR-IR Spectroscopy, ACS Catal., 2015, 5, 3148–3156, DOI:10.1021/acscatal.5b00402.
- R. B. King, N. K. Bhattacharyya and K. D. Wiemers, Noble metal catalyzed hydrogen generation from formic acid in nitrite-containing simulated nuclear waste media, Environ. Sci. Technol., 1996, 30, 1292–1299, DOI:10.1021/es950524c.
- M. Navlani-García, K. Mori, A. Nozaki, Y. Kuwahara and H. Yamashita, Investigation of Size Sensitivity in the Hydrogen Production from Formic Acid over Carbon-Supported Pd Nanoparticles, ChemistrySelect, 2016, 1, 1879–1886, DOI:10.1002/slct.201600559.
- P. Xu, S. Agarwal, J. F. Albanese and L. Lefferts, Enhanced transport in Gas-Liquid-Solid catalytic reaction by structured wetting properties: Nitrite hydrogenation, Chem. Eng. Process., 2020, 148, 107802, DOI:10.1016/j.cep.2020.107802.
- L. Yang, X. Hua, J. Su, W. Luo, S. Chen and G. Cheng, Highly efficient hydrogen generation from formic acid-sodium formate over monodisperse AgPd nanoparticles at room temperature, Appl. Catal., B, 2015, 168–169, 423–428, DOI:10.1016/j.apcatb.2015.01.003.
- J. Li, W. Chen, H. Zhao, X. Zheng, L. Wu, H. Pan, J. Zhu, Y. Chen and J. Lu, Size-dependent catalytic activity over carbon-supported palladium nanoparticles in dehydrogenation of formic acid, J. Catal., 2017, 352, 371–381, DOI:10.1016/j.jcat.2017.06.007.
- Q. F. Deng, Z. F. Zhang, F. J. Cui and L. H. Jia, Highly dispersed Pd-MnOx nanoparticles supported on graphitic carbon nitride for hydrogen generation from formic acid-formate mixtures, Int. J. Hydrogen Energy, 2017, 42, 14865–14871, DOI:10.1016/j.ijhydene.2017.04.294.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cy01448j |
| ‡ Present address: Infineum UK Ltd, Milton Hill Business & Technology Centre, PO Box 1, Abingdon, Oxfordshire, UK, OX13 6BB. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*