
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRare-earth based tetrapyrrolic sandwiches: chemistry, materials and applications
Alexander G.
Martynov
 a,
Yoji
Horii
b,
Keiichi
Katoh
c,
Yongzhong
Bian
de,
Jianzhuang
Jiang
*de,
Masahiro
Yamashita
*f and
Yulia G.
Gorbunova
*ag
a,
Yoji
Horii
b,
Keiichi
Katoh
c,
Yongzhong
Bian
de,
Jianzhuang
Jiang
*de,
Masahiro
Yamashita
*f and
Yulia G.
Gorbunova
*ag
aA.N. Frumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences, 119071, Leninskiy pr., 31, bldg.4, Moscow, Russia
bDepartment of Chemistry, Faculty of Science, Nara Women's University, Nara 630-8506, Japan
cDepartment of Chemistry, Graduate School of Science, Josai University, 1-1 Keyakidai, Sakado, Saitama 350-0295, Japan
dBeijing Key Laboratory for Science and Application of Functional Molecular and Crystalline Materials, Department of Chemistry, School of Chemistry and Biological Engineering, University of Science and Technology Beijing, Beijing, China
eDaxing Research Institute, and Beijing Advanced Innovation Center for Materials Genome Engineering, University of Science and Technology Beijing, Beijing, China. E-mail: jianzhuang@ustb.edu.cn
fDepartment of Chemistry, Graduate School of Science, Tohoku University, 6-3 Aramaki-Aza-Aoba, Aoba-Ku, Sendai 980-8578, Japan. E-mail: yamasita@agnus.chem.tohoku.ac.jp
gN.S. Kurnakov Institute of General and Inorganic Chemistry, Russian Academy of Sciences, 119991, Leninskiy pr., 31, Moscow, Russia. E-mail: yulia@igic.ras.ru
First published on 31st October 2022
Abstract
The unique properties of natural tetrapyrrolic compounds have inspired the rapid growth of research interest in the design and synthesis of artificial porphyrinoids and their metal complexes as a basis of modern functional materials. A special role in the design of such materials is played by sandwich complexes formed by tetrapyrrolic macrocycles with rare earth elements, especially lanthanides. The development of synthetic approaches to the functionalization of tetrapyrrolic compounds and their rare earth complexes has facilitated the intensive development of new applications over the last decade. As a way of expanding the functionalities of rare earth complexes, sophisticated examples have been obtained, including mixed-ligand complexes, π-extended analogues, covalently linked and fused sandwiches, complexes with less-common tetrapyrrols, sandwiches with non-tetrapyrrolic macrocycles and even complexes containing up to six stacked ligands. This review intends to offer a general overview of the preparation of such sophisticated REE tetrapyrrolic sandwiches over the last decade as well as emphasizes the current challenges and perspectives of their application in areas such as single-molecule magnetism (SMM), organic field-effect transistors (OFET), conductive materials and nonlinear optics (NLO).
 Alexander G. Martynov | Alexander G. Martynov graduated from the Higher Chemical College of the Russian Academy of Sciences in 2006. He received his PhD and Dr Hab. degrees in 2008 and 2020. In 2022 he was awarded the Moscow government prize for young scientists. In 2022 he became a Professor of the Russian Academy of Sciences. Currently he is a leading researcher at the Frumkin Institute of Physical Chemistry and Electrochemistry RAS. His research interests are in the areas of spectroscopy of tetrapyrrolic macrocycles and tetrapyrrol-based materials with tuneable properties including REE sandwich complexes for magnetic applications, agents for photodynamic therapy and carbene transfer catalysts. |
 Yoji Horii | Yoji Horii was born in Gumma, Japan in 1989. He received his Bachelor's (2012), Master's (2014) and PhD (2017) degrees at Tohoku University under the supervision of Professor Masahiro Yamashita. He worked on magnetism and electronic states of phthalocyaninato-lanthanide single-molecule magnets. He moved to Osaka University as the postdoctoral fellow in April 2017. In 2019, he moved to Tohoku University as a postdoctoral fellow. Since April 2020, he has been working as an assistant professor at Nara Women's University. His current research interests include molecular magnetism, two dimensional nanomaterials and soft materials. |
 Keiichi Katoh | Keiichi Katoh is currently an associate professor in the Department of Chemistry of Josai University where he is leading research activities in inorganic chemistry and magnetism. He received his PhD in 2003 from the Graduate University for Advanced Studies (Institute for Molecular Science). After being a post-doctoral fellow at Hokkaido University (Research Institute for Electronic Science) until 2004, he worked as a post-doctoral fellow at Kyoto University (Institute for Chemical Research) until 2008, and he joined the Prof. Yamashita group as a post-doctoral fellow at Tohoku University in 2008. From 2009 to 2020, he was appointed to an assistant professor in the Department of Chemistry of Tohoku University. He works on the development of solid state spin systems based on molecular magnetic materials. |
 Yongzhong Bian | Yongzhong Bian was born in Shanxi, China. He received his BSc (1998) and PhD (2005) (with Prof. Jiangzhuang Jiang) from Shandong University. He carried out a JSPS postdoctoral fellowship (2005–2007) at the University of Tokyo with Prof. Takuzo Aida. He became an associate professor at Shandong University in 2007, and moved to the University of Science and Technology Beijing as a full professor in 2009. His current research centers on porphyrin chemistry, encompasses applications in Fluorescent chemosensing and bioimaging, photodynamic therapy, photochemical catalysis and molecular recognition. |
 Jianzhuang Jiang | Jianzhuang Jiang was born in Heilongjiang, China. He received his BSc (1985), MSc (1988), and PhD (1993) (with Tsinglien Chang) from the Peking University. During his doctoral study (1990–1992), he obtained a Fellowship from the Ministry of Culture, Science, and Sport of Japan and carried out his PhD work at the Osaka University under the guidance of Kenichi Machida and Ginya Adachi. He became a Postdoctoral Fellow at Peking University with Tsinglien Chang (1993–1994), a Visiting Scholar at The Chinese University of Hong Kong with Dennis K. P. Ng and Thomas C. W. Mak (1995–1996), and a Postdoctoral Fellow at the Queensland University of Technology with Dennis P. Arnold (1998–2000). He joined Shandong University and the University of Science and Technology Beijing in 1996 and 2008, respectively. He has been working on porphyrin- and phthalocyanine-based materials for more than thirty years to develop new single-molecule magnets, organic field-effect transistors, optical materials, energy materials, and porous materials. |
 Masahiro Yamashita | Masahiro Yamashita received his BSc degree in 1977, MSc in 1979, and DSc in 1982 from Kyushu University. After his graduation, he joined the Institute for Molecular Science (IMS). In 1983, he was appointed as an Assistant Professor at Kyushu University. In 1988, he was appointed as an Associate Professor at Nagoya University, and he was promoted to a full Professor at the same university in 1998. He was a full Professor at Tokyo Metropolitan University from 2000 to 2004. He moved to Tohoku University as a full professor in 2005. He has been honored with the Inoue Scientific Award (2002), the Chemical Society of Japan Award for Creative Work (2005), the Award of Japan Society of Coordination Chemistry (2014), Mukai Award (2019), and the Chemical Society of Japan Award (2020). He is now an Associate Member of the Science Council of Japan. He is a Fellow of the Royal Society of Chemistry (FRSC). |
 Yulia G. Gorbunova | Yulia G. Gorbunova is a Full member of Russian Academy of Sciences (2022) and main researcher at the Russian Academy of Sciences (Kurnakov Institute of General and Inorganic Chemistry RAS (IGIC RAS) and Frumkin Institute of Physical Chemistry and Electrochemistry RAS). She graduated in Chemistry from the Lomonosov Moscow State University and received her PhD (1995) and habilitation (2006) from IGIC RAS. Her current research interests are related to the chemistry of tetrapyrrolic compounds as the basis of functional materials, including molecular switchers, nonlinear optics, electrochromic materials, SMM, sensors and hybrid organo-inorganic nanomaterials. She was a recipient of the Award of European Academy of Science for young researcher (2001), the Russian Government prize in the field of science and techniques (2002) and the Chugaev Award in coordination chemistry (2010). In 2016 Prof. Gorbunova was awarded by order “Chevalier dans l’Ordre national des Palmes Academiques” by the Government of France. |
1. Introduction
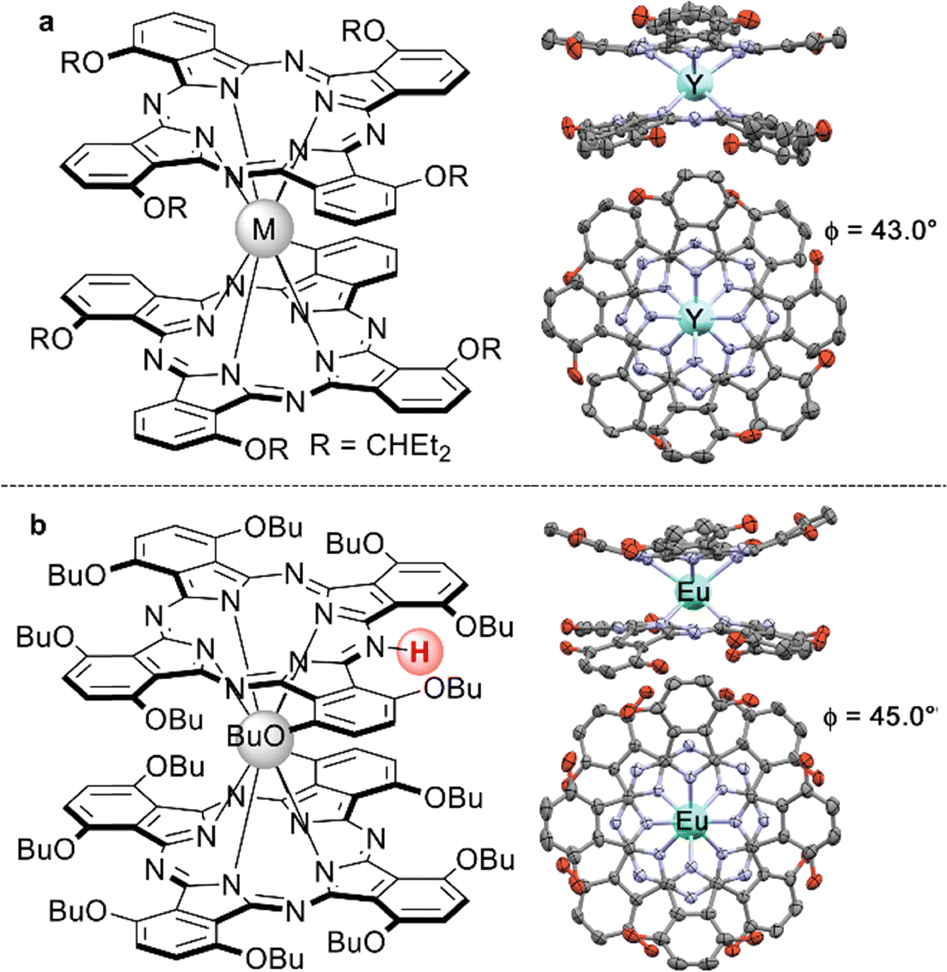
Just as it is difficult to overestimate the role of natural porphyrins in the functioning of living beings, it is also difficult to disagree with the fact that synthetic porphyrinoids have greatly contributed to the development of modern materials science. In particular, this has been made possible by a variety of unique photophysical, electrochemical, catalytic, magnetic, and other properties provided by both the tetrapyrroles themselves and the metal ions placed in their cavities.Among various coordination compounds formed by tetrapyrrole macrocycles, the complexes with rare earth elements (REE) attract special attention.1–6 Due to large ionic radii and high coordination numbers, these elements form sandwich double- and triple-decker complexes with porphyrins (Por), phthalocyanines (Pc) and related macrocycles (Fig. 1), which, with some exceptions, are not observed for the other metals of the periodic table. Tightly coupled π-systems of stacked aromatic macrocycles provide these sandwiches with unique optical and electrochemical properties, and the presence of 4f-electron shells in paramagnetic lanthanide (Ln) metal centres expands the area of their application to the actively developing and promising fields of molecular magnetism and spintronics.7–11
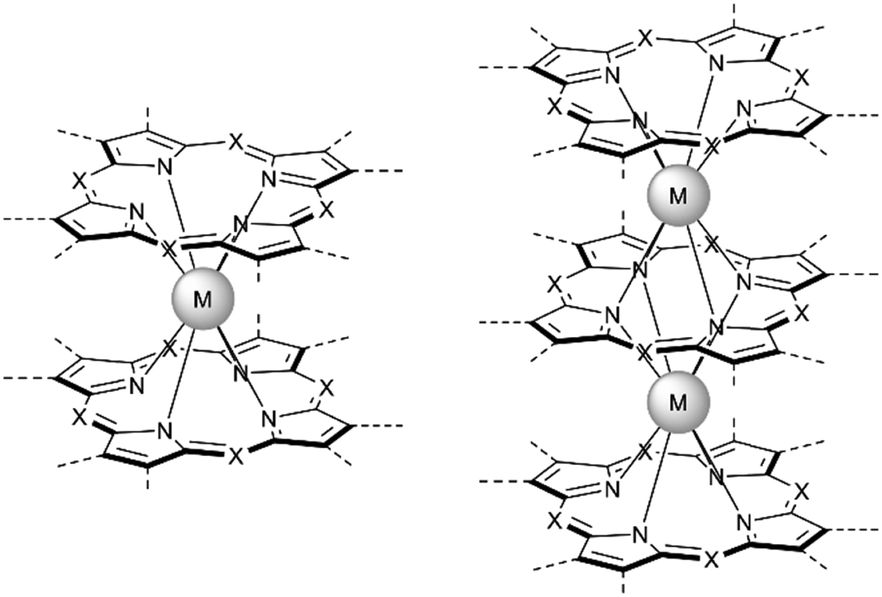 | ||
| Fig. 1 Double- and triple-decker sandwich complexes formed by REE ions with tetrapyrrolic macrocycles, X = C-Ar, C-Alk, and N. | ||
Interestingly, the somewhat detective chemistry of sandwich complexes with tetrapyrrolic ligands started almost simultaneously with rise of phthalocyanines, although the first element to form double-decker complexes – tin(IV) – did not belong to rare earth elements. The synthesis of its bisphthalocyaninate, Sn(Pc)2, was reported in 1936 by Linstead et al.12 The authors used the reaction of (Pc)SnCl2 and Na2(Pc), and this raise-by-one-story approach much later became the main way to various heteroleptic and mixed ligand sandwiches.
Much later, in 1965 Kirin and Moskalev et al. reported seminal work on the template reaction between REE acetates and melted phthalonitrile.13 The authors isolated surprisingly stable green and blue complexes, where metal ions were sandwiched between two Pc ligands – M(Pc)2; however, at that time the details of their electronic structure and the nature of the intriguing redox behaviour remained unclear. In particular, thin films formed by bisphthalocyaninates were able to reversibly change colour when a potential was applied or when these films were treated with volatile oxidants and reducing agents. The electrochromic behaviour of bisphthalocyaninates was explained in 1979 by Corker et al.14 who used UV-Vis and EPR spectra to show that M(Pc)2 undergoes ligand-centred redox-triggered interconversion between three forms of trivalent metal complexes – anionic [M3+(Pc2−)2]−, neutral [(Pc−˙)M3+(Pc2−)]0 and cationic [M3+(Pc−)2]+ forms. Due to the pronounced difference in spectral signatures these three forms possessed different colours, and for many years they were known as blue, green and red forms, respectively. Apart from changes in UV-Vis spectra, switching between redox states caused modulation of NIR absorbance, and the nature of NIR bands of neutral green complexes, has been discussed in the works of Simon et al.,15 Aroca et al.,16 Tomilova and Lukyanets et al.17
Importantly, under harsh conditions of template condensation, apart from double-decker complexes, REE ions can also form blue triple-decker sandwiches M2(Pc)3,18,19 where Pc ligands are isoelectronic with those in blue forms of bisphthalocyaninates. Because of the similarity of their optical properties and lack of available mass-spectral characterization these complexes were sometimes mistaken for each other. These contradictions have been eliminated by single-crystal X-ray assignment performed in 1980 for neodymium bisphthalocyaninate, Nd(Pc)2 by Kasuga et al.,20 and in 1999 for dilutetium tris-(tetra-15-crown-5-phthalocyaninate), Lu2[(15C5)4Pc]3 by Troyanov et al.21
The survey for milder conditions where double- and triple-deckers could be selectively obtained, afforded several convenient protocols, which are commonly used in synthetic practice nowadays. These protocols include template condensation of phthalonitriles and REE salts in high boiling alcohols in the presence of DBU as a phthalonitrile activator as suggested by De Cian et al.,22 and also direct reactions of pre-synthesized phthalocyanine ligands with metal salts as suggested by Takahashi et al.23,24 While the former method yields double-decker sandwiches, the latter method furnishes both double- and triple-decker complexes, and later studies showed that the selectivity of complex formation can be governed either by the presence or absence of DBU, respectively.25,26
In the case of phthalocyanines both template and direct methods are equally applicable for the synthesis of REE sandwiches, however, in the case of porphyrins only direct method can be used to synthesize double- and triple-decker complexes. First example of homoleptic REE bisporphyrinates was reported in 1977 by Buchler et al. with the example of quadrivalent cerium complex Ce(TTP)2 formed upon the reaction of Ce(acac)3 with tetra-p-tolylporphyrin, H2(TTP) in refluxing 1,2,4-trichlorobenzene (TClB).27 The authors noticed a remarkable analogy between the structure and the redox-properties of this complex and the “special pair” formed by chlorophyl molecules in the photosynthetic systems of green plants.28 Later more double- and triple-decker porphyrinates were isolated and characterized,29–31 and it was found that porphyrin ligands tended to form sandwiches only with early and middle lanthanides, while Pc ligands could form sandwiches with all REE ions, including the relatively small Sc3+ ions.32,33
Finally, REE complexes provided the marriage of porphyrins and phthalocyanines in mixed ligand sandwiches, where both types of ligands are combined and strongly coupled due to intramolecular orbital interactions providing them with peculiar optical properties of panchromatic absorbers. Synthesis and crystallographic characterization of the first example of such complexes were reported in 1986 by Moussavi et al. with the example of mixed ligand triple-decker complexes with tetra-p-anisylporphyrin (Pc)M(Pc)M(TAnP), where M = Nd, Eu, and Gd.34
The rapid development of the chemistry of sandwich REE complexes was driven both by advances in spectroscopy and physicochemical characterization of these challenging objects and breakthrough discoveries which affected many areas of modern chemistry. Among them we should inevitably mention the first organic semiconductor – lutetium bisphthalocyaninate, Lu(Pc)2, described for the first time in 1987 by Turek et al.,35 and especially the first example of a 4f single molecule magnet – terbium(III) bisphthalocyaninate, Tb(Pc)2, reported in 2003 by Ishikawa et al.36
Because of this, the largely fundamental interest in these compounds was gradually replaced by a focus on applied research where archetypal double- and triple-decker complexes M(Pc)2 and M2(Pc)3 coexist with more sophisticated compounds, including mixed-ligand complexes, π-extended analogues, covalently linked and fused sandwiches, complexes with less-common tetrapyrrols, sandwiches with non-tetrapyrrolic macrocycles and even complexes containing up to six stacked ligands obtained by coordination or supramolecular bonding. Moreover, these complexes have been recognized as molecular switches whose properties can be altered by the reversible self-assembly processes, protonation, redox reactions, etc., opening the way to the creation of “smart” materials.
These sophisticated complexes began to attract the most attention over the last decade; thus, the aim of the present review is mainly to summarize the advances in the chemistry of REE tetrapyrrolic sandwiches over this period to supplement the previously published comprehensive reviews1–6 with new data. Another goal of this review is to emphasize the current challenges and perspectives of sandwich complex application in various fields such as single-molecule magnetism (SMM), organic field-effect transistors (OFET) and conductive materials, and nonlinear optics (NLO).
2. Advances in the synthesis of functionalized multiple-decker sandwiches
2.1 New homo- and heteroleptic double- and triple-decker complexes bearing symmetrically substituted tetrapyrolic ligands
From the synthetic viewpoint, sandwich complexes with symmetrically substituted tetrapyrrolic ligands are the most available objects, particularly due to facile synthesis and separation procedures. The following section will include various classes of homo- and heteroleptic sandwiches with symmetrical ligands. In the latter case, ligands belong to the same classes of tetrapyrrolic macrocycles.For example, Kong et al. used template condensation of 4,5-di(α-naphthoxy)phthalonitrile with Eu(acac)3 and DBU in refluxing n-PentOH to make a double-decker complex Eu[(NaphO)8Pc]2 (Scheme 1) which was used as a component of a flexible ambipolar OFET,38 and it also revealed sensing properties towards NO2 and NH3 (cf. Section 4).39 To synthesize a triple-decker complex Eu2[(NaphO)8Pc]3, raise-by-one-story approach was applied, namely octa-naphthyloxy-phthalocyanine was condensed with the double-decker complex (Scheme 1).40 The resulting trisphthalocyaninate could form self-assembled microstructures whose morphology and n-/p-mobilities could be controlled by solvent–vapour annealing procedure (cf. Section 4.2). Similar approaches were used to synthesize various examples of other aryloxy-substituted sandwiches, including heteroleptic complexes, which were designed as high-performance air-stable ambipolar organic thin film transistors,41–44 optical limiters45–48 and single molecule magnets.49
 | ||
| Scheme 1 Synthesis of α-naphthyloxy-substituted Eu(III) bis- and trisphthalocyaninates.38,40 | ||
Phenyl-substituted phthalo- and naphthalocyanines were used by Dubinina et al. for the synthesis of REE complexes.50,51 The selectivity of complex formation upon direct metalation of these ligands depended on conditions affording either monophthalo- and naphthalocyaninates or sandwich complexes (Scheme 2). While double-decker complexes were obtained for both types of macrocycles, triple-decker complexes could be obtained by only using phthalocyanine ligands.
 | ||
| Scheme 2 Synthesis of lanthanide complexes with phenyl-substituted phthalo- and naphthalocyanines.50,51 | ||
Single-crystal XRD studies of Lu(Ph8Pc)2 revealed intramolecular edge-to-face π–π stacking interactions between peripheral phenyl groups, reinforcing the overall intramolecular interactions between Pc ligands (Fig. 2).50 In turn, it resulted in a strong hypsochromic shift of NIR bands in UV-Vis-NIR spectra of bisphthalocyaninates – for example the intervalence band shifted from 1575 nm in the spectrum of Tb(tBu4Pc)2 to 1433 nm in the spectrum of Tb(Ph8Pc)2. In contrast, benzo-annelation going from phthalo- to naphthalocyaninates caused bathochromic shifts of intervalence bands by ca. 200 nm.
 | ||
| Fig. 2 Top view of the X-ray structure of Lu(Ph8Pc)2 (CCDC PIXGUA). Dark blue lines show edge-to-face π-stacking interactions.50 | ||
Measurements of conductivity of thin drop-casting films formed by phenyl-substituted terbium(III) phthalocyaninates showed that in the series of mono-, bis- and trisphthalocyaninates the complex Tb(Ph8Pc)2 had the largest conductivity of 5 × 10−10 S cm−1, which is however much smaller than the values obtained for unsubstituted double-deckers.51 Optical microscopy data showed that such a low conductivity value was partially caused by film inhomogeneity. AFM studies of films, formed by triple-decker complex Tb2(Ph8Pc)3 showed that it forms ring-shaped nanostructures – nano-coffee rings, which appear due to an increase in the surface tension of the solutions of this complex in comparison with mono- and bisphthalocyaninates. This feature can be used for nano-patterning of various substrates.
Attention to crown-substituted sandwiches was attracted mainly in the context of fabrication of electronic devices due to their ability to form thin films with well-defined morphology.58,60 While investigating the formation of ultrathin Langmuir films by crown-substituted bisphthalocyaninates, Selektor et al. discovered the phenomenon of orientation-induced redox isomerism in planar supramolecular systems with Ce[(15C5)4Pc]2 as the example.61 Depending on the surface pressure at the water/air interface the complex underwent valence tautomeric interconversion: Ce4+[L2−]2 ↔ [L2−]Ce3+[L˙−]; moreover, films with different redox states of metal centres could be transferred from water to solid surfaces to characterize the valence state by various spectroscopic techniques. The phenomenon was also found to be general for most of bisphthalocyaninates, which are formed by lanthanides exhibiting di- and trivalent states – Sm, Eu, Er, Tm, and Yb.62,63
Homonuclear trisphthalocyaninates of strongly paramagnetic lanthanides – Ln2[(15C5)4Pc]3, Ln = Tb, Dy, Ho, and Tm were studied by variable-temperature NMR showing the possibility of their application as thermosensors in the physiological temperature range (303–323 K)64,65 due to the sensitivity of lanthanide-induced shifts to temperature. The Tb(III) complex was found to have the highest sensitivity of this shift among the reported paramagnetic complexes of 4f and d elements with polydentate ligands in organic media.
Several types of crown-substituted complexes were synthesized to study single-molecule magnetism,66,67 which is particularly interesting for this type of REE sandwiches since it can be tuned by the interaction of crown-ether macrocycles with alkali metal cations. Such tuning was demonstrated for the first time by Horii et al.67 with heteroleptic terbium(III) complex [(15C5)4Pc]Tb(Pc) synthesized from (Pc)Tb(acac), 4′,5′-dicyanobenzo-15-crown-5 and DBU in hexanol as the example (cf. Section 3.3.1 for more details on SMM studies).
Alternative one-pot strategy towards this complex was proposed by Martynov et al.68 It was based on the generation of (Pc)Tb(acac) by transmetalation of a lanthanum(III) complex La(Pc)2 with Tb(acac)3·nH2O and the reaction of this intermediate with tetra-15-crown-5-phthalocyanine, H2[(15C5)4Pc], in the presence of DBU in refluxing 1-chloronaphthalene (ClN) and OctOH mixture (Scheme 3a). The yield of the complex was 24%, and the presence of DBU was shown to be crucial for the formation of the double-decker complex – in the absence of DBU the mixtures of homonuclear triple-decker complexes (Pc)Tb[(15C5)4Pc]Tb(Pc) and [(15C5)4Pc]Tb[(15C5)4Pc]Tb(Pc) were obtained.69,70
 | ||
| Scheme 3 Transmetalation of La(Pc)2 in the synthesis of heteroleptic crown-substituted bisphthalocyaninates (a) and heteronuclear trisphthalocyaninates (b).66,68,71,72 | ||
Original variant of the raise-by-one-story approach was proposed to synthesize heteronuclear triple-decker complexes, [(15C5)4Pc]M*[(15C5)4Pc]M(Pc), using La(Pc)2 as a donor of unsubstituted phthalocyaninate-dianion (Scheme 3b).71,72 Thus, its reaction with crown-substituted bisphthalocyaninates, M*[(15C5)4Pc]2 and M(acac)3·nH2O, in refluxing ClN resulted in remarkably rapid – 10–15 min – formation of the target heteronuclear complexes with a wide range of pairs of dia- and paramagnetic metal centers (M ≠ M* = Nd, Sm, Eu, Gd–Yb, Y) in high yields. SMM properties of homonuclear di-terbium(III) complex and heteronuclear terbium(III)/yttrium(III) complexes were studied and the effects of intramolecular ferromagnetic interactions together with the impact of coordinating surrounding were revealed.66
A heteroleptic octopus-type Tb(III) bisphthalocyaninate bearing a 15-crown-substituted ligand and a ligand with eight thioacetate-terminated flexible tentacles was synthesized by Shokurov et al. via the raise-by-one-story approach starting from tetrahydropyranyl-protected phthalocyanine and half-sandwich Tb(III) crown-phthalocyaninate followed by the cleavage of protective groups, the introduction of iodine atoms and their nucleophilic substitution with thioacetate residues (Scheme 4).73 The resulting octopus formed a self-assembled monolayer (SAM) on gold with a face-to-face orientation of the molecules, serving as an anchor for further surface modification, namely the binding of additional crown phthalocyanine molecules via potassium ion bridges. The enhanced redox-switching ability of the bilayer thus formed was demonstrated (cf. Section 4.3).
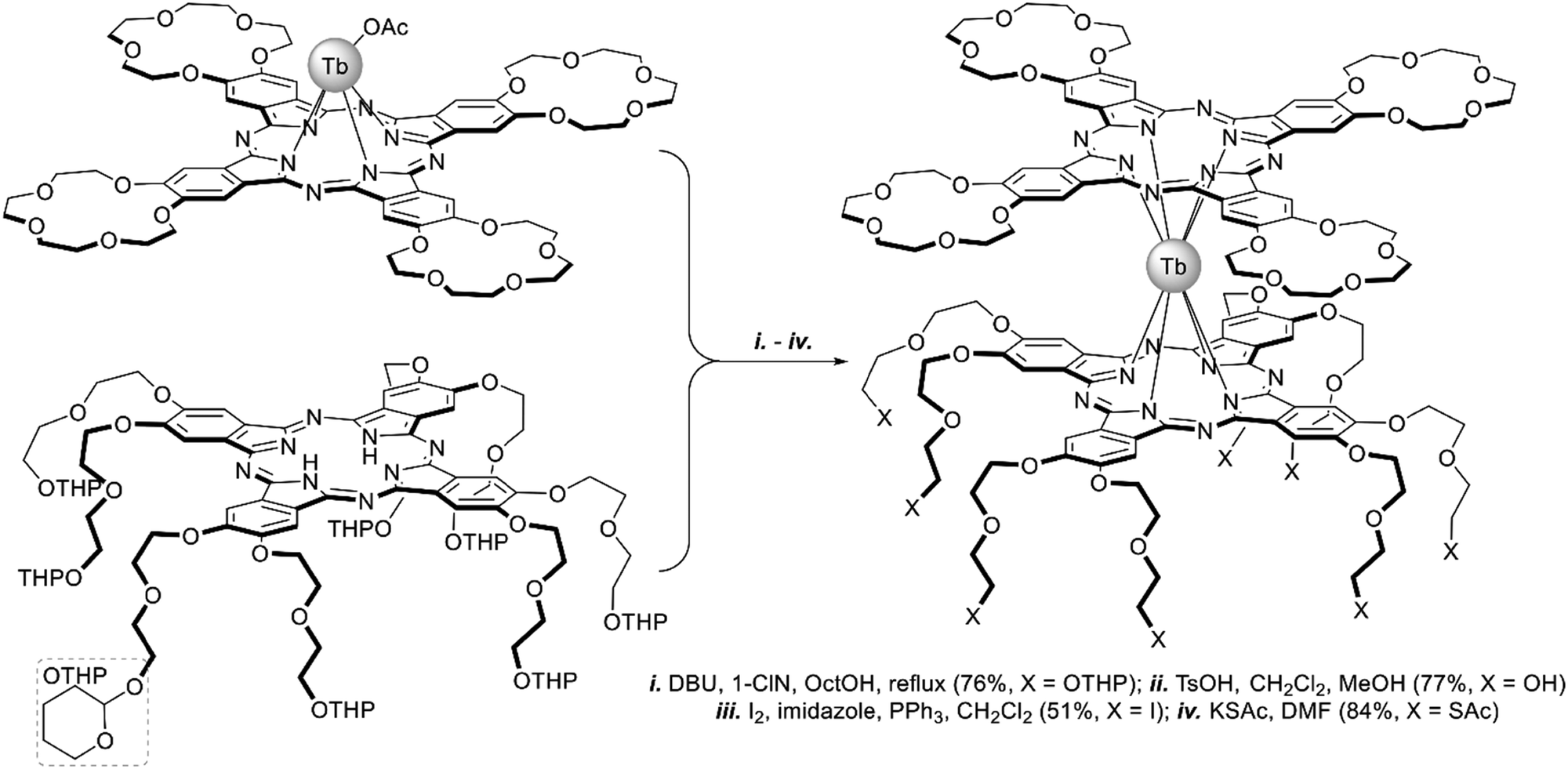 | ||
| Scheme 4 Synthesis of heteroleptic octopus-type Tb(III) bisphthalocyaninate.73 | ||
 | ||
| Scheme 5 Synthesis of chiral bisphthalocyaninates on the examples of Y[(BNPO)4Pc]2 (a, only S-enantiomer is shown),74 and menthyl-substituted bisphthalocyaninates M[(MenthO)4Pc]2 (b).75 | ||
Another naturally occurring chiral terpene, (−)-myrtenal, was used by Zheng et al. to synthesize the dicyanobenzo-annelated pinene derivative which underwent template condensation to furnish double-decker complexes M[(pinene)4Pc]2, M = Eu, Er, and Lu (Scheme 6).76 Introduction of bulky conformationally rigid hydrocarbon substituents was made with the aim to decrease both inter- and intramolecular interactions between Pc ligands. Such tuning of intermolecular interactions decreased the aggregation of complexes and enhanced their film-forming ability. Decrease of intramolecular interactions resulted in a significant red shift of the intervalence band in the NIR region in comparison with other double-decker complexes, improving the electrochromic properties of compounds thus modified. Finally, chirality of complexes was manifested in their CD-spectra.
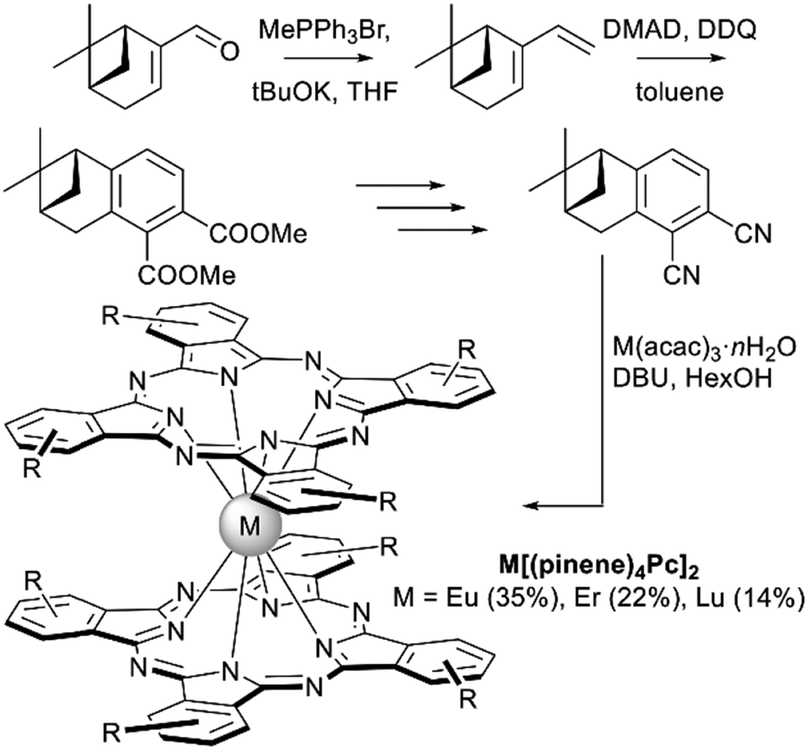 | ||
| Scheme 6 Key steps leading to pinene-annelated phthalonitrile and bisphthalocyaninates (DMAD – dimethyl acetylene dicarboxylate). Only one isomer of double-decker complexes is shown.76 | ||
Terbium(III) bisphthalocyaninate substituted with chiral (S)-2-(dodecyloxy)propoxy groups was synthesized by Gonidec et al. as a liquid crystalline material with tuneable magnetic properties.77 This double-decker complex was synthesized by the direct metalation of the corresponding phthalocyanine (Scheme 7) with lithium bis(trimethylsilyl)amide as a basic reagent promoting complex formation. Similar to mentyl-substitued complexes, the metal-free ligand was CD-silent, while the double-decker complex showed a pronounced CD signal in the Q-band region. Also, the complex showed mesogenic behaviour and both crystalline and disordered states of the same specimen could be kinetically trapped upon measurements of SMM behaviour at low temperatures. Both structural states showed markedly different magnetization dynamics revealing important influence of the structural environment of a single-molecule magnet taking advantage of its phase behaviour.
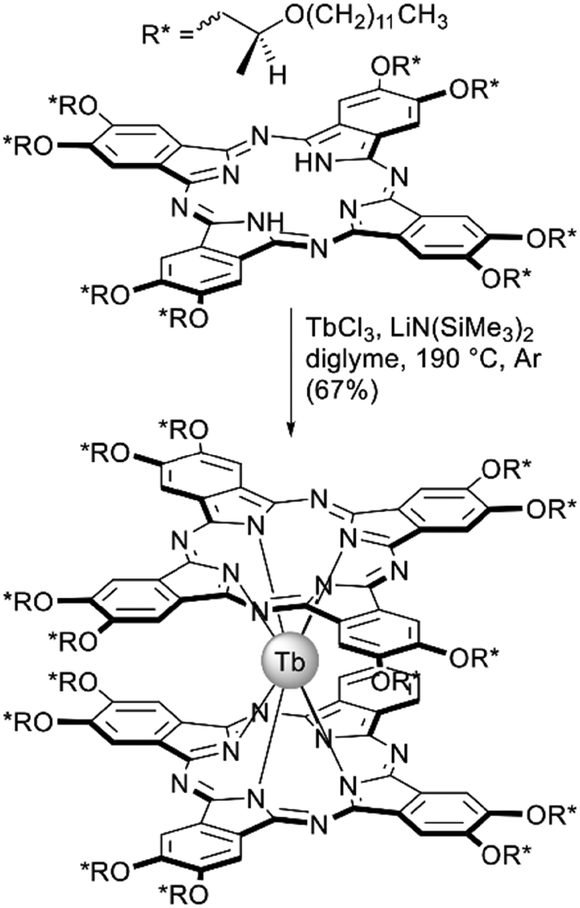 | ||
| Scheme 7 Synthesis of mesogenic terbium(III) bisphthalocyaninate, bearing (S)-2-(dodecyloxy)propoxy-substituents.77 | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) mixture yielded double-decker complexes in mediocre yields.80
:1 v/v) mixture yielded double-decker complexes in mediocre yields.80
 | ||
| Scheme 8 Synthesis of bisphthalocyaninates based on octa-(di-n-butylamino)phthalocyanine.81,82 | ||
The complex Tb[(Bu2N)8Pc]2 revealed excellent SMM behaviour which could be further improved by the synthesis of a heteroleptic complex [(Bu2N)8Pc]Tb(Pc) (cf. Section 3.2.2).81 This complex was synthesized by the raise-by-one-story approach involving the metalation of H2[(Bu2N)8Pc] with Tb(acac)·nH2O in refluxing OctOH and in situ reaction of the generated Tb(III) monophthalocyaninate with Li2(Pc). In the same manner, erbium(III) complex [(Bu2N)8Pc]Er(Pc) was also synthesized and its anionic form was a first sandwich-type phthalocyanine-based erbium(III) compound with field-induced slow relaxation.82
Chiral heteroleptic complex was synthesized in a similar manner by the reaction of [(Bu2N)8Pc]Tb(acac) with H2[(BNPO)4Pc],83 and it also revealed excellent SMM characteristics. Such superior SMM behaviour of amino-substituted complexes was explained using DFT calculations in terms of the electrostatic effects of electron-rich ligands.
 | ||
| Scheme 9 Synthesis of the imide substituted double-decker complexes.86 | ||
Another extremely electron-deficient Tb(III) bis(octa-isopropyl)phthalocyaninate where all hydrogen atoms were replaced with fluorine atoms was studied by Gonidec et al.87 It could be electrochemically reduced to di- and trianionic forms, and magnetic characteristics of these forms were studied by low-temperature MCD (cf. Section 3.4).
Neodymium(III) bis(octachlorophthalocyaninate), Nd(Cl8Pc)2, was synthesized by Kuzmina et al. via two alternative strategies. Template condensation of 4,5-dichlorophthalonitrile with Nd(OAc)3·H2O and DBU in refluxing PentOH yielded only 10% of the target complex; however, its yield could be improved to 87% using the raise-by-one-story reaction between H2(Cl8Pc) and (Cl8Pc)Nd(OAc) and MeOLi in a refluxing mixture of trichlorobenzene and cetyl alcohol. This sandwich complex revealed improved optical limiting behaviour in comparison with nonhalogenated derivatives due to the heavy atom effect, although halogenated monophthalocyaninates turned out to be better optical limiters in comparison with sandwich complexes.90 Interestingly, 4,5-dibromophthalonitrile did not yield any traces of double-decker complexes under the conditions of template synthesis.91
Sandwiches based on tetra-α-substituted Pc ligands were reported for the first time by Wang et al. in 2005 with tetra-[α-(3-pentyloxy)]phthalocyaninates, M[(α-Et2CHO)4Pc]2, M = Eu, Lu, and Y as examples (Fig. 3a).93 These complexes were synthesized in a straightforward manner by the reaction of metal-free phthalocyanine with the corresponding M(acac)3·nH2O in refluxing OctOH, and the major C4h isomer of europium(III) complex was characterized using XRD, which showed its elegant pinwheel-like structure. SMM behaviour of terbium(III) complex was also studied, showing the clear dependence of the magnetic properties of the complex on its redox state (cf. Section 3.4).94
 | ||
| Fig. 3 Homoleptic nonperipherally-substituted bisphthalocyaninates: (a) C4h isomer of M[(α-C5H11O)4Pc]2 and X-ray structure of the Y(III) complex (CCDC NAZTEO),93 and (b) meso-protonated complex HM[(α-BuO)8Pc]2 with the X-ray structure of the Eu(III) complex (CCDC XUXCOJ01).95 Substituents in the X-ray structures are truncated to oxygen atoms; hydrogen atoms are omitted. | ||
The synthesis of complexes with octa-α-substituted ligands (Fig. 3b) turned out to be more challenging. In 2010 Gao et al. showed that the reaction of H2[(α-BuO)8Pc] with M(acac)3·nH2O and DBU in OctOH did not yield any traces of sandwich complexes; however, it was found that crown-ethers catalyse this reaction, and the best yield of the complex HM[(α-BuO)8Pc]2 – 36.5% – was obtained in the presence of dibenzo-18-crown-6.95 Crystallographic characterization of this complex revealed severe distortion of Pc ligands because of the sterical hindrance, caused by the eight proximate butoxy-groups as well as the presence of a proton at one of meso-nitrogen atoms. Localization of this proton in solution was shown by Damjanović et al. using the 1H-NMR study of the paramagnetic terbium(III) complex HTb[(α-BuO)8Pc]2,96 and the impact of protonation on the SMM behaviour of this complex was also demonstrated by Horii et al. (cf. Section 3.5).97
2.2 Sandwich complexes with low-symmetry phthalocyanine ligands
Lowering the symmetry of phthalocyanines is a powerful tool to tune their physical-chemical properties,98 which is particularly important in the field of nonlinear optics as well as in the fabrication of hybrid materials, where it allows the introduction of anchor groups which afford immobilization of complexes on various supports.This approach was optimized by Alpugan et al. with the example of europium(III)-templated cross-condensation of 4,5-di(n-hexylthio)phthalonitrile as component A with phthalonitriles, bearing one or two 5-hydroxypentylthio-groups as component B (Scheme 10).100 Using the stoichiometric ratio of precursors yielded a mixture of six products with laborious separation issues. However, due to similar reactivities of both components, the distribution of products is expected to be close to statistical, thus increasing the ratio of A:B the yield of products, containing more than one B unit can be decreased, and the optimal ratio of 10:1 was selected to obtain two main products – complexes Eu(A4)2 and (A4)Eu(A3B), whose separation was facilitated by increased polarity of the asymmetric compound. Reproducible 10–12% yields were obtained for A7B-type complexes based on hydroxylated phthalonitrile. Further functionalization of complexes was achieved by their mesylation and substitution of mesyl groups with azide as a possible substrate for click-chemistry.
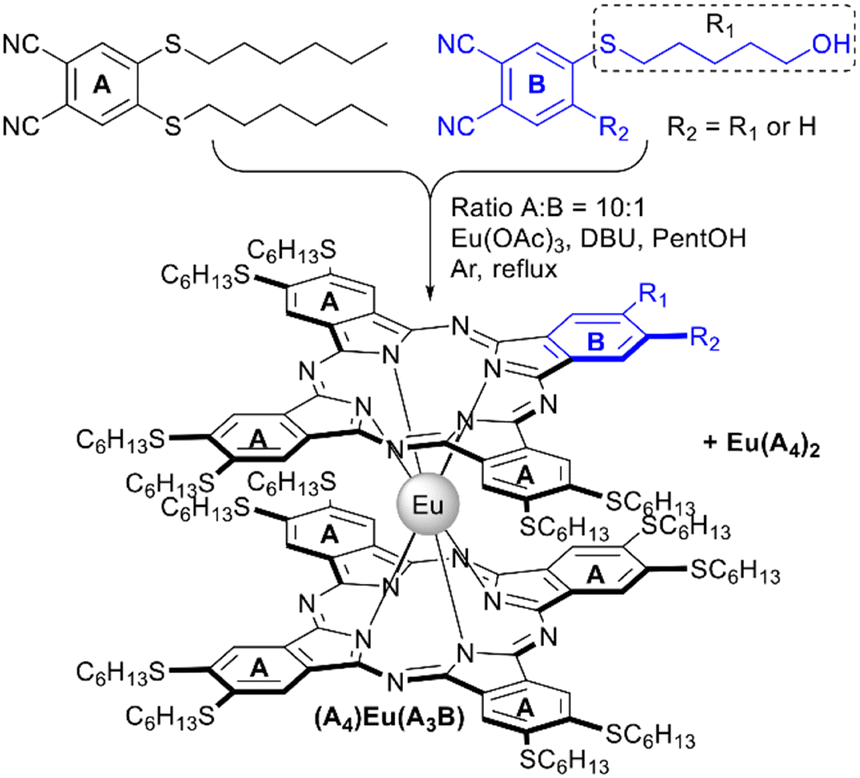 | ||
| Scheme 10 Selective synthesis of a functionalized A7B-type europium(III) bisphthalocyaninate.100 | ||
Direct metalation of A3B-type ligand was attempted by Oluwole et al. for the synthesis of homoleptic europium(III) bis- and trisphthalocyaninates bearing diethyleneglycol chains with terminal OH groups (Scheme 11).101 Unexpectedly, direct metalation of this ligand with Eu(OAc)3 in refluxing OctOH neither in the absence nor in the presence of DBU failed to produce target sandwiches, likely due to side processes associated with the interaction of diethyleneglycol groups with Eu3+ ions. However, protection of OH-groups with dihydropyran in starting phthalocyanine prior to complex formation allowed smooth conversion of the protected derivative into double-decker and triple-decker complexes, and the selectivity was controlled by the addition of DBU. Deprotection of THPO-groups yielded OH-substituted sandwiches, which were immobilized on the surface of quantum dots to give the first examples of hybrid optical limiters based on sandwich REE complexes.
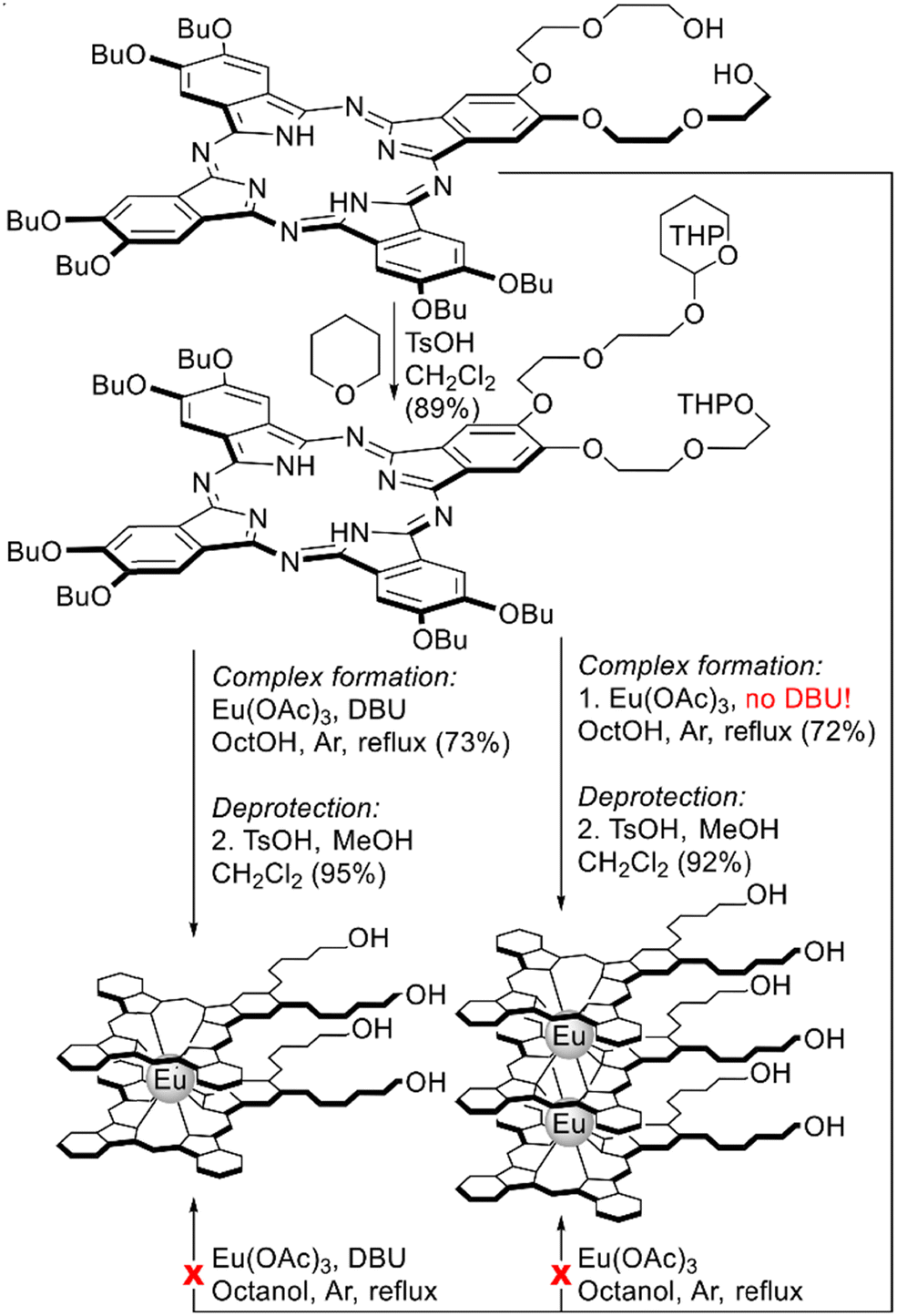 | ||
| Scheme 11 Synthesis of sandwich europium(III) complexes bearing diethyleneglycol substituents with terminal OH-groups.101 | ||
A similar necessity to introduce protective groups was observed by Pushkarev et al. with the example of metalation of the phthalocyanine ligand bearing one phenolic OH group conjugated with the Pc system (Scheme 12a).102 Degradation of the unprotected ligand was observed upon metalation with europium and lutetium acetylacetonates in high boiling solvents; however, the benzylated derivative smoothly produced double-decker complexes where benzyl protections were removed by treatment with sulphuric acid.
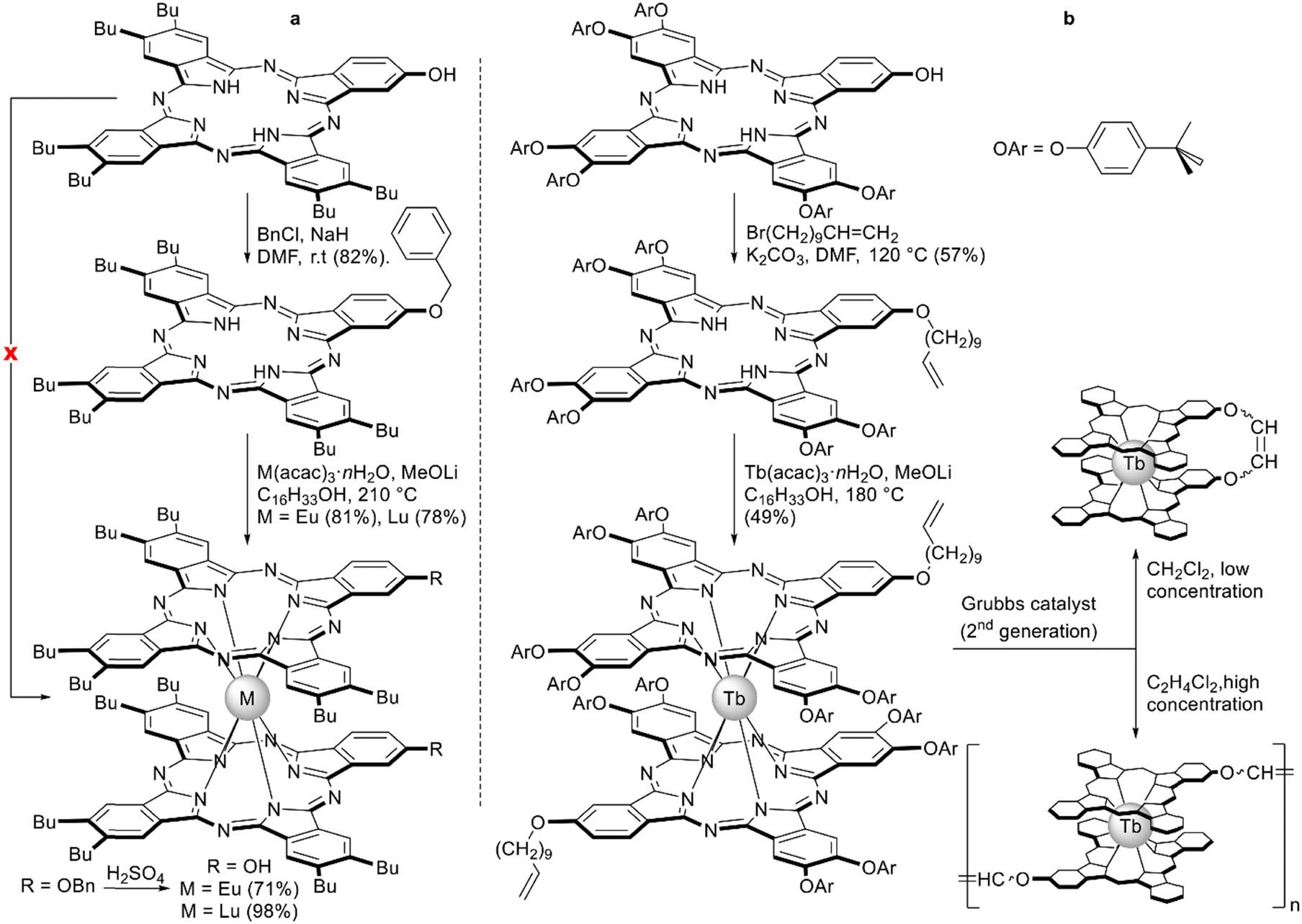 | ||
| Scheme 12 Synthesis of sandwich complexes starting from hydroxy-substituted phthalocyanine ligands.102,103 | ||
This approach allowed Pedrini et al. to synthesize homoleptic terbium(III) bisphthalocyaninate with A3B-type ligands bearing undec-10-en-1-yloxy groups, which were used to obtain covalent 1D architectures using diene metathesis (Scheme 12b).103 This reaction turned out to be concentration-dependent – the product of intramolecular cyclisation of two ω-alkenyl chains was obtained when metathesis was performed in dichloromethane solution at 2.4 mM concentration. Increasing the concentration by an order of magnitude afforded the mixture of oligomers containing species from dimer to pentamer according to MALDI TOF mass-spectrometry. Importantly, the magnetic properties of this mixture were only slightly modified after the formation of the bond between molecules, highlighting the potential of this reaction scheme to form multinuclear magnetic assemblies.
Studies of metalation of covalently bonded dimeric phthalocyanines in the synthesis of sandwich complexes revealed that this reaction can proceed either in the intra- or intermolecular mode leading to mononuclear complexes or oligomeric assemblies (Scheme 13).
 | ||
| Scheme 13 Examples of sandwich complexes formed with dimeric tetrapyrrols: (a) intra-104vs. intermolecular105,106 binding of lanthanides with clamshell-phthalocyanine H4[c-Pc2]; (b) 24-crown-8-interlocked bisphthalocyaninates M[(24C8)Pc2];107 and (c) binaphthol-strapped chiral bis(porphyrinato)cerium(IV) complexes.108 | ||
For example, clamshell-type diphthalocyanine H4[c-Pc2] with an o-xylyl bridge studied by Pushkarev et al. underwent mainly intracavity metalation upon interaction with lanthanide acetylacetonates (Scheme 13a), although some oligomers were detected by MALDI TOF mass-spectrometry.104 To form well-defined dimeric bisphthalocyaninates the starting clamshell ligand can be treated with lanthanide monophthalocyaninates.105,106 With a diterbium complex (tBu4Pc)Tb[c-Pc2]Tb(tBu4Pc) as the example ferromagnetic intermetallic interactions were observed leading to elongation of magnetic relaxation times in comparison with the monomeric terbium complex Tb(tBu4Pc)2 (cf. Section 3.3.1).105
The balance of intra- vs. intermolecular complexation was studied by Birin et al. for butoxy-substituted diphthalocyanine with 24-crown-8 bridge H4[(24C8)Pc2].107 Its interaction with M(acac)3, M = La, Ce, and Eu, and DBU in ClN yielded mixtures of mononuclear complexes and olygomers, and the yields of monomeric intercavity complexes (Scheme 13b) increased with the decrease of the metal size, gaining 40% for the Ce(IV) complex. The latter complex revealed unusual solvatochromic behaviour when going from chloroform to toluene, which was not observed in the case of homoleptic symmetrical Ce[(15C5)4Pc]2. Such behaviour was also studied by NMR spectroscopy which revealed solvent-induced changing of sandwich conformation.
Intracavity complexes of cerium(IV) with diporphyrins with chiral BINOL bridges Ce[(BINOL-n)Por2], n = 1 or 2 (Scheme 13c), were synthesized by Lu et al. using metalation of the corresponding ligands with cerium(III) acetylacetonate in trichlorobenzene in the presence of metal lithium, which was added to activate the porphyrin.108 CD measurements evidenced that (R)- and (S)-BINOL-strapped complexes formed perfect mirror spectra with negative and positive signs in the Soret band regions respectively. This observation suggests that the C2-chirality of a BINOL strap is transcribed to the double-decker core as a defined chiral twist in the inter-porphyrin arrangement. The intensity of signals also depended on the length of the aliphatic bridge – (CH2CH2)n between BINOL and CePor2 units, indicating the possibility to tune the efficiency of the intramolecular chirality transfer.
Homoleptic complexes with low-symmetric ABAB-type Pc ligands have been designed as NLO materials, bearing in mind the concept of octupolar molecules, which can be approximated by a cube with alternating donor and acceptor groups in the corners (Scheme 14). With this aim, double-deckers were synthesized by direct metalation of the corresponding Pcs bearing either four hexylthio-groups – H2[(SHex)4Pc],109,110 or alternating pairs of α-PentO and β-Cl substituents H2[(α-PentO)4Cl4Pc].111,112
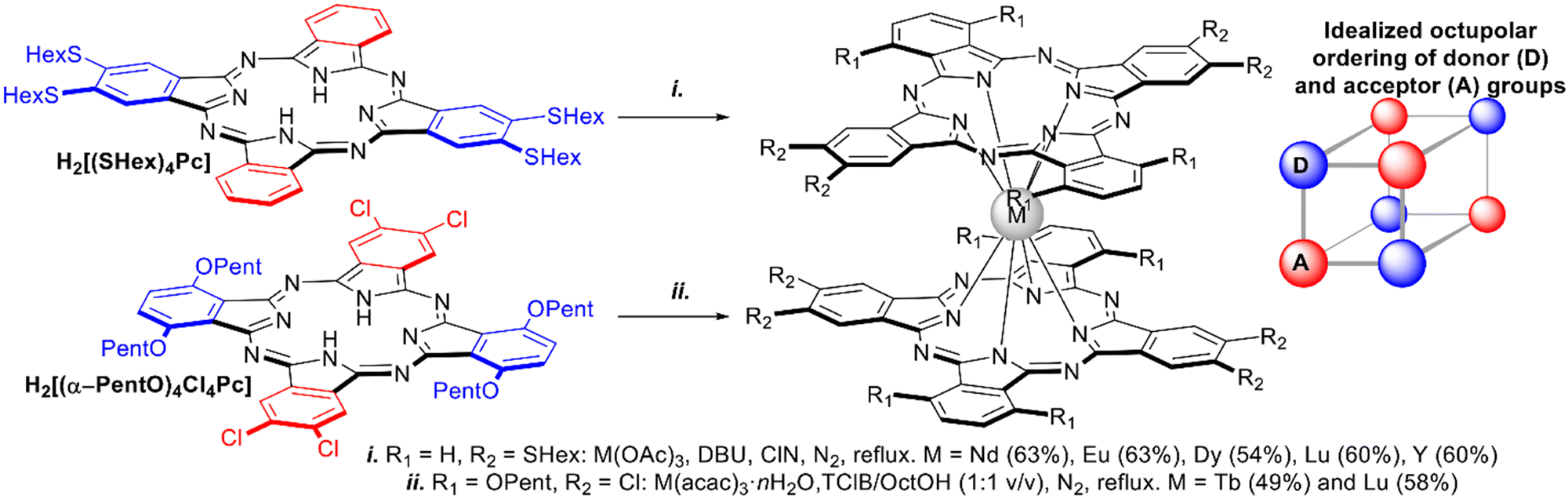 | ||
| Scheme 14 Synthesis of octupolar bisphthalocyaninates based on ABAB-type ligands.109–112 | ||
Homoleptic terbium(III) bis-porphyrinate HTb(DADAPor)2 was synthesized by the reaction of metal-free tetra-meso-substituted porphyrin bearing alternating donor- and acceptor p-diphenylamino-phenyl and p-carboxymethylphenyl-ethynyl groups (Scheme 15).113 Although X-ray structure of this complex was not obtained, DFT calculations suggested that intramolecular interactions between donor and acceptor fragments should stabilize distorted cuboid conformation of this sandwich with a twist angle near 17°. This structural feature provided the studied complex with the largest off-resonant first hyperpolarizability as evaluated by harmonic light scattering measurements. Details on NLO studies of synthesized octupolar sandwiches are given in Section 5.1.
 | ||
| Scheme 15 Synthesis of octupolar bisporphyrinate with ABAB-type ligands.113 | ||
For example, bisphthalocyaninates bearing one pyrene-containing A3B ligand were synthesized by Klyatskaya et al. starting from Li2(Pc) which was treated with M(acac)3 in ClN generating (Pc)M(acac) species, M = Tb, Dy, and Ho.114,115 Their further interaction with dilithium complex of pyrenyl-substituted phthalocyanine Li2[(Pyrene)Pc] yielded 20–24% of the target heteroleptic complexes [(Pyrene)Pc]M(Pc), which were separated from homoleptic complexes by column chromatography (Scheme 16). Isolation of the heteroleptic triple-decker terbium(III) complex with one terminal unsubstituted ligand and two adjacent pyrenyl-substituted ligands was also reported; the order of ligands was determined by collision induced dissociation using Fourier transform ion cyclotron resonance and an Orbitrap mass spectrometer.116
Due to the presence of pyrenyl group in the double-decker terbium(III) complex, it could be anchored to the surface of carbon nanotubes via π–π interactions. The anisotropy energy barrier and the magnetic relaxation time of the resulting conjugate are both increased compared with those of pure crystalline [(Pyrene)Pc]Tb(Pc).114 Also this conjugate behaved as an original spin-valve device, where localized magnetic moments lead to a magnetic field dependence of the electronic transport through single-walled carbon nanotubes, resulting in magnetoresistance ratios up to 300% at temperatures lower than 1 K (cf. Section 3.8).117
 | ||
| Scheme 16 Synthesis of pyrenyl-substituted bisphthalocyaninates [(Pyrene)Pc]M(Pc).114,115 | ||
Interestingly, the ability of the pyrenyl group to participate in stacking interactions governed the crystal packing of the [(Pyrene)Pc]Tb(Pc) complex – its molecules formed head-to-tail dimers with the shortest intermolecular distances of 3.345–3.369 Å which is in good agreement with the general values observed for π–π stacking (Fig. 4).115
 | ||
| Fig. 4 Structure of a head-to-tail supramolecular dimer, formed in crystal packing by molecules of [(Pyrene)Pc]Tb(Pc) (CCDC LOGBEQ).115 Hexyl substituents, hydrogen atoms and solvate molecules are omitted for clarity. The shortest intermolecular distances are shown with blue dotted lines. | ||
Heteroleptic bisphthalocyaninates, bearing one fullerene-appended ligand, were synthesized by Ballesteros et al. starting from a A3B-type ligand with one benzylic alcohol group H2[(BnOH)Pc] whose reaction with acetylacetonates in o-dichlorobenzene yielded the corresponding monophthalocyaninates [(BnOH)Pc]M(acac), M = Sm, Eu and Lu (Scheme 17).118 These complexes were used as templates in reactions with phthalonitrile to construct unsubstituted Pc ligands yielding heteroleptic sandwiches which were esterified with fulleropyrrolidine carboxylic acid C60-COOH. Photophysical characterization of the resulting complexes [(BnOCO-C60)Pc]M(Pc) showed that their components totally lack electronic interactions in the ground state; however, photoexcitation of the C60 fragment at 387 nm lead to a fast and long-lived charge transfer from M(Pc)2 to C60 π-systems. In contrast, the short-lived nature of the bisphthalocyaninate excited states fails to trigger analogous charge-transfer reactions upon photoexcitation of this moiety.
 | ||
| Scheme 17 Synthesis of fullerene-substituted bisphthalocyaninates [(BnOCO-C60)Pc]M(Pc).118 | ||
Monophthalocyaninates can act as templates in cross-condensation of pairs of different phthalonitriles A and B paving the way to heteroleptic complexes where one of the ligands is represented by various combinations AnB4−n, n = 0–4. This one-pot approach was used by Pan et al. to synthesize a series of tetrathiafulvalene(TTF)-fused heteroleptic europium(III) double-deckers [(TTF)nPc]Eu(Pc).119 With this aim (Pc)Eu(acac) reacted with the mixture of unsubstituted and TTF-fused phthalonitriles (Scheme 18) and the mixture of the resulting complexes could be successfully separated by size-exclusion chromatography together with chromatography on silica to separate the cis- and trans-isomers of [(TTF)2Pc]Eu(Pc). Introduction of the TTF units onto double-decker complexes had strong effects on their UV-Vis spectra and electrochemical properties, revealing the significant contribution of these units to the electronic structure of double-deckers.
 | ||
| Scheme 18 One-pot synthesis of a series of tetrathiafulvalene (TTF)-fused bisphthalocyaninates [(TTF)nPc]Eu(Pc), n = 0–4.118 | ||
Unprecedent heteroleptic complexes with open-chain triisoindole-1-one ligands (R-TIO)Lu(Pc), R = Me or iPr, were unexpectedly isolated by Wang et al. from the conventional raise-by-one-story synthesis of heteroleptic bisphthalocyaninates via template condensation of bulky diaryloxy-phthalonitriles with lutetium(III) monophthalocyaninate and DBU in refluxing pentanol (Scheme 19).120 It was found that the target [(R2PhO)8Pc]Lu(Pc) complexes were sole products of template condensation only when it was performed under strictly anaerobic conditions. However, the presence of even 5 ppm of oxygen in inert gas media terminated the reaction at the stage of uncyclized isoindole oligomeric derivatives rather than the phthalocyanine chromophores. The yields of TIO-containing sandwiches could be increased up to 25.8% when the reactions were performed in a pure oxygen atmosphere. Experiments with labeled 18O2 showed that the carbonyl group in the TIO ligand contains 16O atom, suggesting that it comes from the solvent. TIO ligands were stabilized both by bulky aryloxy-groups as well as by half-sandwich part of the molecule, and demetalation of these complexes resulted in complete degradation of triisoindole-1-ones.
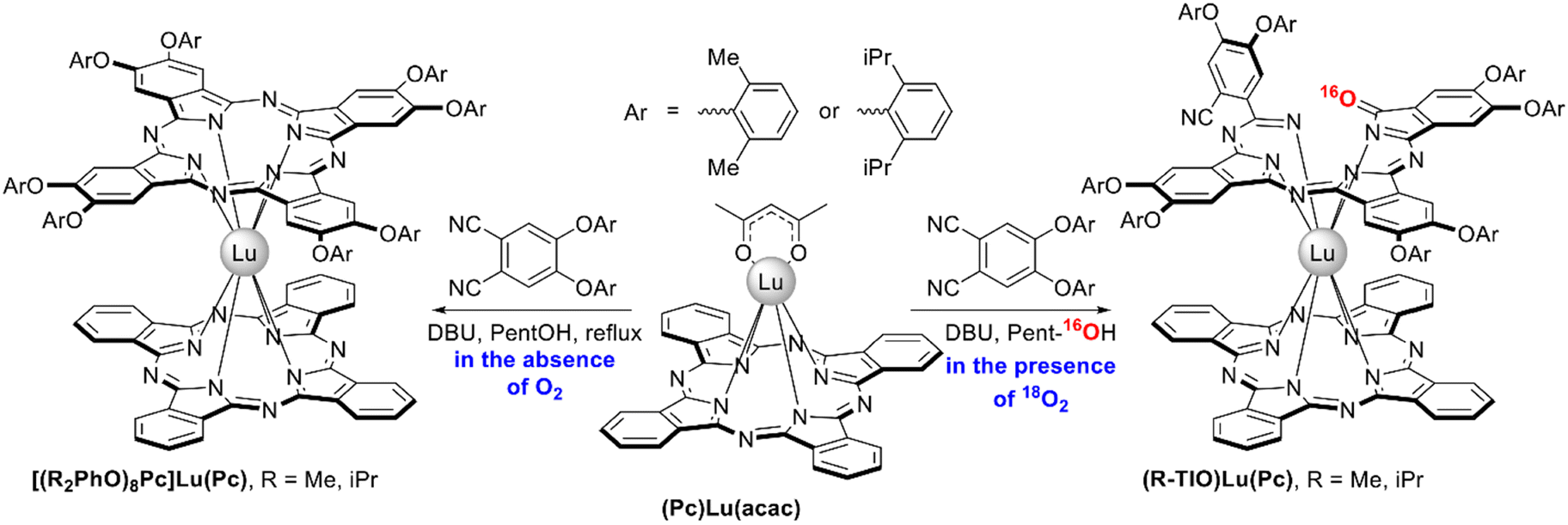 | ||
| Scheme 19 Synthesis of heteroleptic double-deckers with triisoindole-1-one ligands.120 | ||
2.3 Mixed ligand sandwich complexes
In hierarchy of sandwich complexes mixed ligand double- and triple-deckers combining tetrapyrrolic macrocycles from different families (Por, Pc, Nc, etc.) attract special attention due to wider possibilities of tuning of both structural and functional characteristics in comparison with homoligand complexes. Among the most important features in the context of the present review we should mention specific acid–base equilibria in double-decker Por-Pc complexes (cf. Section 3.5). Panchromatic absorbance appearing from orbital interactions between stacked ligands is particularly important for the elaboration of broadband light harvesting materials.121 Combination of ligands with different sterical demands in sandwich complexes also affords precise control of twist angles between these ligands which in turn rules the shape of the coordination polyhedron of the metal centre bound to these ligands and affects its SMM behaviour (cf. Section 3.2.1). | ||
| Scheme 20 Synthesis of homo- and heteronuclear mixed ligand complexes (TAnP)M*(Pc)M(Pc), M* = M = Tb or M* ≠ M = Tb/Y.122 | ||
Interest in studies of mixed ligand sandwiches requires efficient synthetic pathways with improved yields, which can be realized either by optimization of experimental protocols, or by application of auxiliary functionalities in macrocycles, governing the regioselectivity of complex formation.
With the aim to synthesize early lanthanide complexes based on tetra-meso-aryl-porphyrins and crown-phthalocyanine Birin et al. proposed one-pot procedures implying reactions between corresponding porphyrins, M(acac)3 and 15-crown-5-phthalonitrile, the latter undergoes template condensation with simultaneous formation of sandwich complexes (Scheme 21).123–126 The balance of concurrent stages was studied127 and the optimal conditions were found leading either to mixtures of double- and triple-decker complexes or selectively to triple-decker complexes with a Por–Pc–Por ligand arrangement. The crucial role of electron-donating crown-ether groups in the synthesis of such triple-deckers was demonstrated, when unsubstituted phthalonitrile was used instead of crown-substituted precursor; the synthesis terminated at the step of mixed-ligand double-decker complex.128 Going from early to late lanthanides drastically decreased the yield of triple-deckers, thus, to synthesize a terbium(III) complex it was necessary to use the raise-by-one procedure with the addition of terbium(III) porphyrinates to pre-synthesized double-decker complexes.129
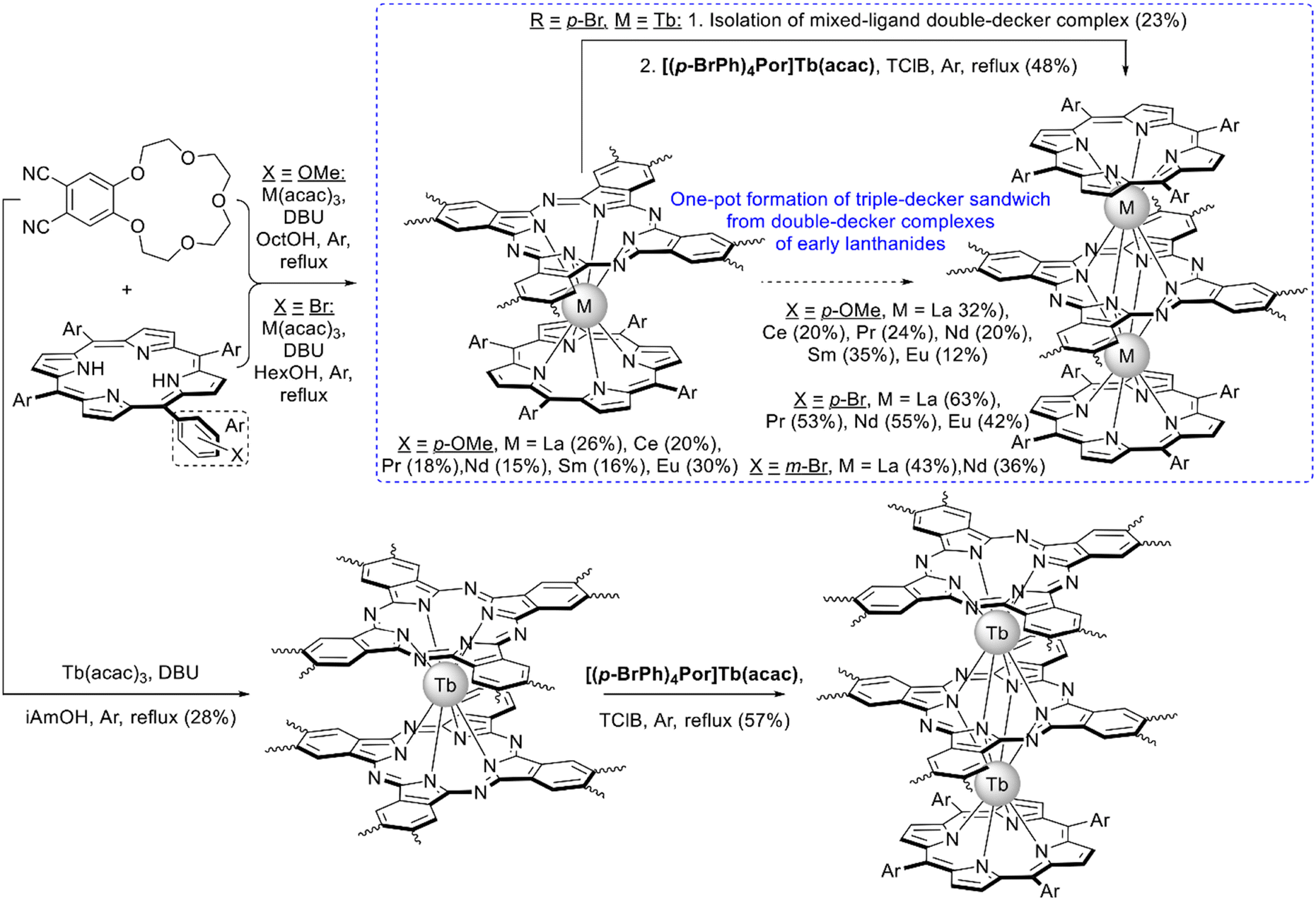 | ||
| Scheme 21 Synthesis of crown-substituted mixed ligand sandwich complexes.123–129 | ||
Classical conditions of the raise-by-one-story approach to Por–Pc–Pc mixed ligand complexes via the interaction of monoporphyrinates with bisphthalocyaninates in high-boiling solvents turned out to be inapplicable in the synthesis of complexes with phthalocyanines bearing bulky substituents. To circumvent this drawback Jin et al. used microwave activation, the thus optimized conditions allowed the synthesis of series of complexes in high yields (Scheme 22).130 Importantly, MW activation allowed the speeding-up of the synthetic procedures, thereby minimizing possible thermolysis of labile compounds. This benefit allowed the synthesis of complex with free meso-positions in porphyrin ligands, while under classical solvothermal synthesis of similar complexes starting from 5,15-diphenylporphyrin its degradation was observed under prolonged refluxing of reaction mixtures.128
 | ||
| Scheme 22 Microwave-mediated synthesis of lanthanide porphyrin/phthalocyanine triple-deckers bearing bulky substituents.130 | ||
Straightforward synthesis of mixed ligand triple-deckers leading to the regioselective Por–Pc–Por arrangement of ligands was achieved by González-Lucas et al.131 who applied a diporphyrin with a flexible decamethylene spacer (Scheme 23). This pincer ligand sequentially captures lanthanide ions and phthalocyanines to efficiently form desired triple-decker complexes in high yield when it reacts with stoichiometric amounts of La(acac)3 and H2(Pc). In a similar manner, complexes with Pr(III), Nd(III), Sm(III) and Eu(III) were synthesized; however, attempts to synthesize complexes with a smaller metal centre – Dy(III) resulted in the formation of a double-decker complex linked with a free-base porphyrin residue. Reaction of this complex with La(acac)3 resulted in the formation of an intermediate heteronuclear La(III)/Dy(III) complex which however underwent complete transmetalation with the formation of a homonuclear lanthanum(III) complex.
 | ||
| Scheme 23 Synthesis of sandwich mixed ligand complexes with spacered diporphyrin and X-ray structure of dilanthanum(III) complex (CCDC LUVWUX). Only one of the symmetry-imposed disorder components is shown. Hydrogen atoms and solvent molecules have been omitted for clarity.131 | ||
To broaden the range of useful properties of mixed ligand complexes, they can be functionalized with redox-active groups or hydrogen-bond donors which can form supramolecular aggregates of various architectures via self-assembling processes.
A series of ferrocene-decorated mixed ligand sandwiches were synthesized by Zhu et al. using the raise-by-one-story approach starting from 5,15-diferrocenylporphyrin in 20–5% yields for double-decker complexes and 70% for a triple-decker sandwich (Fig. 5).132 Electrochemical studies of these complexes revealed two consecutive Fc-based one-electron oxidation waves, suggesting the effective electronic coupling between these units which could be finely tuned by structural modifications of the sandwich framework. Thus, the separation between these waves increased with the decrease of the REE ionic radius, but it decreased when going from a double- to triple-decker complex.
 | ||
| Fig. 5 Ferrocene-decorated double- and triple-decker mixed ligand complexes.132 | ||
Mixed ligand europium(III) double-deckers bearing from one to four n-octylamino-groups (Fig. 6) were synthesized and used as tectons for self-assembled nanostructures, formed upon injection of concentrated solutions of complexes in toluene into excess of methanol.133,134 The morphology of the resulting aggregates was studied by various methods, and it was demonstrated that it governed their functional characteristics. For example, in the case of (A1-TPP)Eu(Pc) and (A4-TPP)Eu(Pc) this procedure resulted in the fabrication of uniform well-defined nano-rods and nano-sheets with conductivities of 3.85 × 10−5 and 2.48 × 10−5 S m−1, respectively. This difference was attributed mainly to the more effective intermolecular π-electron delocalization in nano-sheets which was concluded from the analysis of X-ray diffraction patterns of the nanostructures.134
 | ||
| Fig. 6 Amino-substituted mixed ligand complexes (A1-TPP)Eu(Pc): R1 = NHOct, R2 = R3 = H; (A2-TPP)Eu(Pc): R1 = R2 = NHOct, R3 = H; (A4-TPP)Eu(Pc): R1 = R2 = R3 = NHOct.133,134 | ||
Mixed-ligand Pc–Nc complexes can be synthesized using the raise-by-one-story approach. Thus, neutral complex (Nc)Tb(Pc) was synthesized in a mediocre yield by template condensation of 2,3-naphthalonitrile in the coordination sphere of terbium(III) monophthalocyaninate in the presence of DBU and ammonium molybdate (Scheme 24i).136 High quality crystals of its cationic complex were obtained by the electrochemical oxidation of neutral (Nc)Tb(Pc)0 in dichloromethane in the presence of NBu4PF6 electrolyte.
 | ||
| Scheme 24 Template (i) vs. direct (ii) synthesis of complexes containing phthalo- and naphthalocyanine ligands.43,136,137 | ||
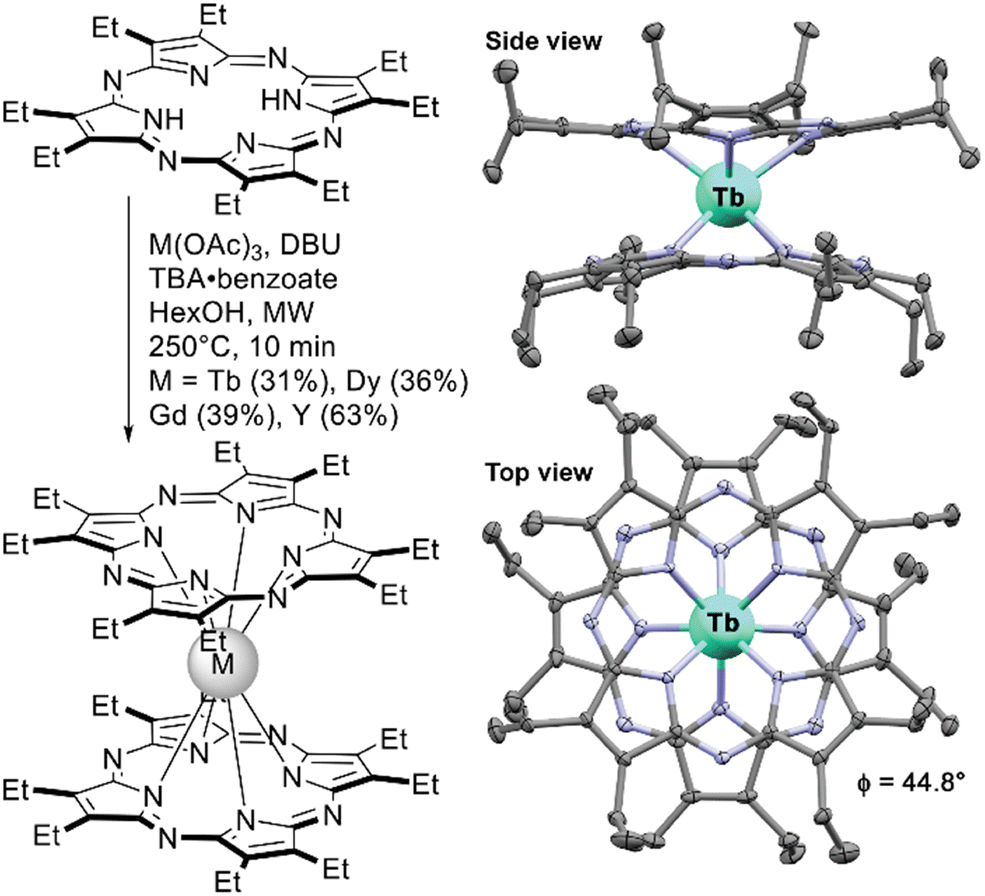 | ||
| Scheme 25 Synthesis of double-decker complexes with octaethylporphyrazine and the X-ray structure of the complex Tb(OEPz)2 (CCDC QOQPIX).140 Hydrogen atoms and solvate molecules are not shown for clarity. | ||
The interaction of naphthalocyanine ligands with monophthalocyaninates using lithium methoxide in the mixture of TClB and cetyl alcohol allowed Dubinina et al. to optimize the synthesis of mixed ligand phthalo-/naphthalocyaninates (Scheme 24ii).43,137 It afforded a series of complexes containing various substituents in both Pc and Nc rings, and the yields of sandwiches were high even for the late lanthanides. AFM studies of thin films formed by lutetium(III) Pc–Nc complexes evidenced that their tendency to aggregate depended on the substitution patterns, gaining maximum for (Ph8Nc)Lu(Ph8Pc), and in general the series of complex behaved as semiconductors with small activation energy.43
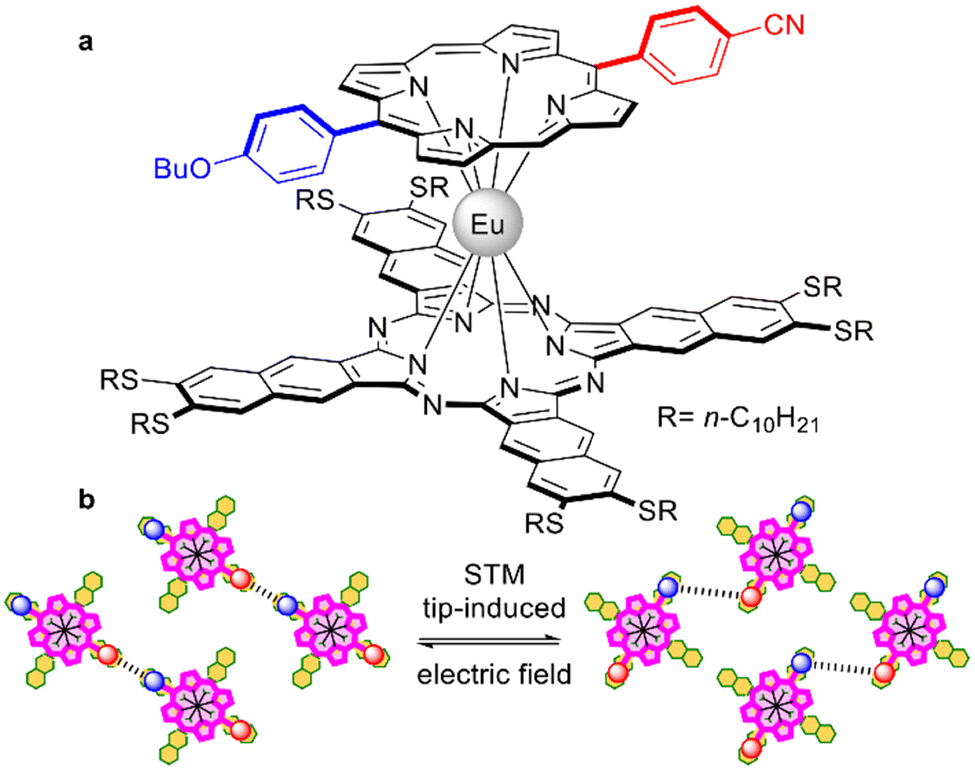
A mixed ligand europium(III) complex with decylthio-substituted naphthalocyanine and push-pull porphyrin (Fig. 7a) was synthesized by Stefak et al., who used the template condensation of 6,7-di(n-decylthio)-2,3-naphthalonitrile in the presence of the corresponding monoporphyrinate.138 This complex was suggested as a rotor which can be absorbed on the Cu(111) surface in a porphyrin-up fashion, forming a network where molecules undergo simultaneous and coordinated rotational switching under the influence of an STM tip-induced electric field by applying biases above 1 V at 80 K (Fig. 7b).139
2.4 Sandwiches with less-common tetrapyrrolic ligands
Advances in the synthesis of novel tetrapyrrolic ligands were often followed by the introduction of these ligands into the chemistry of sandwich REE complexes. The following section will include examples of double- and triple-deckers with porphyrazines including hetero-annulated derivatives, tetrabenzoporphyrins, and core-modified macrocycles – tetrabensotriazaporphyrins (TBTAP), corroles and N-confused porphyrins. Most of these complexes are yet to be comprehensively explored in terms of applicability, thus this section can give a summary inspiring their further study.A series of neutral double-decker complexes with an octaethylporpyrazine ligand (OEPz) with Tb(III), Dy(III), Gd(III) and Y(III) metal centres was reported in 2014 by Giménez-Agulló et al. Sandwiches were synthesized starting from H2(OEPz) using microwave activation.140 Interestingly, the synthetic protocol included the necessity to add tetrabutylammonium benzoate as an auxiliary reagent – in its absence double-decker complexes could not be obtained. The authors claimed that this salt was needed to stabilize the intermediate – anionic form [M3+(OEPz2−)2]−TBA+, which underwent oxidation to the neutral radical form during reaction workup.
X-ray structure of the complex showed that the molecule adapts staggered conformation with SAP surrounding of the metal centre which is a prerequisite of SMM behaviour of Tb(III) and Dy(III) complexes. Indeed, AC magnetometry showed that Tb(OEPz)2 is characterized by high value of magnetization relaxation barrier comparable with the value of Tb(Pc)2.
Sandwich REE complexes with heterocycle-annelated macrocycles were reported for the first time by Tarakanova et al. in 2014141 with complexes with tetradiazepinoporphyrazine (DZPz) ligands as examples. High CH-acidity of the peripheral diazepine rings made them efficient hydrogen bond donors thus the macrocycles themselves were found to form highly stable dimers in solutions.142 Staggered conformations of these dimers reminded the arrangement of tetrapyrrols in REE sandwiches; therefore, additional stabilization of a double-decker scaffold could be expected.143 And indeed, it was shown that direct metalation of H2[(tBuPh)8DZPz] with M(acac)3 and DBU (Scheme 26a) smoothly yielded double-decker complexes at the temperature of refluxing o-dichlorobenzene (180 °C), where classical phthalocyanines selectively form only monophthalocyaninates.141,143–145 Importantly, diazepine rings were susceptible to hydrolysis in alkaline media, which made template synthesis of REE complexes with DZPz ligands less efficient than direct metalation.141 DFT modelling of the resulting double-deckers suggested the presence of intramolecular hydrogen bonds between endocyclic CH-groups of diazepine rings and meso-nitrogen atoms of opposite ligands (Scheme 26b).
 | ||
| Scheme 26 (a) Synthesis of double-decker complexes with tetradiazepinoporphyrazine ligands. (b) Optimized structure of Lu(Ph8DZPz)2− in top and side views, showing hydrogen bonds between CH2 groups of diazepine rings and meso-N atoms. Other hydrogen atoms are omitted for clarity. Model was built using Cartesian coordinates, provided in the ESI in ref. 143. | ||
Among other differences in the reactivity of DZPz and Pc ligands the stabilization of anionic forms of double-decker complexes should be emphasized. In contrast to M(Pc)2− which readily undergoes aerobic oxidation to neutral forms, the synthesized complexes M[(tBuPh)8DZPz]2− remained in this form during chromatographic purification, and the addition of strong oxidants like bromine was needed to generate neutral forms which revealed characteristic intervalence bands in the NIR region.143 The cerium complex fell out of this trend and its double-decker complex was stabilized in the neutral form with a quadrivalent metal centre.145 Its electrochemical reduction to trivalent state was characterized by slow electron transfer kinetics which was explained by strong structural rearrangement of the sandwich molecule following the electron transfer step as well as shielding of the metal center by the bulky macrocycles.
Interestingly, solutions of anionic double-deckers with DZPz ligands were stable for several months showing no evidence of aggregation or decomposition; however, in the case of neutral forms evolution of their UV-Vis spectra was observed, depending on the nature of the metal centre. First of all, upon storage of solutions of M[(tBuPh)8DZPz]20˙ in CH2Cl2 the hypsochromic shift and broadening of Q-bands were observed, and DLS studies of aged solutions revealed the formation of aggregates with a hydrodynamic radius of 20 nm. The tendency of neutral forms to aggregate was explained by the possibility of inversion of diazepine moieties leading to the reorientation of the intramolecular hydrogen bonds in the double-decker followed by the formation of intermolecular hydrogen bonds. In turn, it weakened the stabilizing effect of intramolecular H-bonding, and it resulted in lowering of stability in early lanthanide complexes – slow demetalation of La[(tBuPh)8DZPz]20˙ was observed even after 1 day of solution storage, while lutetium(III) complex was stable towards demetalation ever after five days.143
In the case of mixed ligand complexes [(tBuPh)8DZPz]M(Pc), M = La, Eu and Lu, spontaneous aggregation due to inversion of diazepine ring was observed even for anionic forms.146 In contrast to homoleptic complexes these double-deckers could form only dimeric species, which were observed by NMR spectroscopy. Tendency to aggregation followed the growth of the ionic radius of lanthanide ion due to an increase of interligand distance, weakening of intramolecular CH…N interactions and facilitation of diazepine ring inversion. The same trend was observed in the case of neutral forms generated during spectroelectrochemical studies. Thus, the intermolecular self-assembly of sandwich complexes containing DZPz decks can be controlled by changing the nature of both peripheral substituents and REE ions.
Soluble tetrathieno[2,3-b]porphyrazine annelated with cyclohexane rings was used by Dubinina et al. to synthesize lutetium(III) complexes expecting that the presence of sulphur-containing heterocycles can provide the resulting complexes with improved NLO properties.147 Starting metal-free ligand was synthesized from tiophenedicarbonitrile by Zn-templated condensation and demetalation. Reactivity of the resulting ligand was quite unusual – upon its reaction with Lu(OAc)3·4H2O and MeOLi in refluxing mixture of TClB and cetyl alcohol only mono- and triple-decker complexes were obtained, which were separated by preparative TLC (Scheme 27). Although the formation of double-decker was detected by mass-spectrometry, it could not be isolated because of its low stability associated with the presence of π-radical.
 | ||
| Scheme 27 Synthesis of lutetium(III) complexes with substituted tetrathieno[2,3-b] porphyrazine.147 | ||
 | ||
| Scheme 28 Synthetic pathways to sandwich complexes with benzoporphyrin and corrin ligands. | ||
Thus, interaction of tetra(bicyclo[2.2.2])octadienoporphyrin with Gd(OAc)3 in refluxing TClB was studied by Xu and Liu et al.149 Under these conditions metalation of tetrapyrrolic macrocycles was accompanied by retro Diels-Alder aromatization producing TBP ligands. Heating of the reaction mixture for 15–17 h resulted in the formation of a double-decker complex Gd(TBP)2, and increase of the reaction time to 45–48 h resulted in the predominant formation of a triple-decker complex Gd2(TBP)3.150,151 Magnetic properties of complexes were studied revealing antiferromagnetic interactions between π- and f-electrons in Gd(TBP)2 and between two Gd3+ ions in the case of Gd2(TBP)3.
Interaction of tetrabenzoporphyrin H2(TBP) itself with M(acac)3, M = La, Nd, Gd, Dy, and Lu was used by Galanin et al. to generate half-sandwich complexes (TBP)M(acac) which reacted with Li2(Pc) to form mixed ligand double-deckers (TBP)M(Pc) in 48–74% yields.152 A similar strategy was applied to synthesize complexes starting from tert-butyl-substituted benzoporphyrin H2[(tBu)4TBP].153
Original strategy leading to homoleptic complexes M(TBP)2, M = Gd, Er, and Lu, was proposed using a dipyrrolic precursor (PhIm)2 prepared from readily available phthalimide PhIm.154 This procedure compares favourably with the time-consuming protocol using tetra(bicyclo[2.2.2])octadienoporphyrin due to the availability of starting compounds.
Application of acyclic olygopyrrolic precursors affords more options in the synthesis of various porphyrinoid derivatives. Condensation of 2-methylquinoline with a tripyrrolic compound (PhIm)3 prepared from phthalimide afforded meso-(quinolin-5-yl)tetrabenzoporphyrin H2(QnlTBP) which was also used to synthesize mixed ligand complexes.155 The aforementioned dipyrrolic compound (PhIm)2 can be dimerized in the presence of lanthanide acetates and hydrazine with the formation of complexes with the 1-tethyltetrabenzo-octadehydrocorrin ligand.156 Depending on the ratio of reagents, either half-sandwiches or homoleptic double-decker complexes could be obtained. The reaction of the former complex with phthalonitrile can be used to synthesize phthalocyanine-corrin mixed ligand double- and triple-decker complexes.
 | ||
| Scheme 29 (a) Synthesis of homo- and heteroleptic complexes with meso-phenyl-tetrabenzotriazaporphyrin, H2(Ph-TBTAP).157,158 (b) Side products of dephenylation. (c) X-ray structure of the complex Eu(Ph-TBTAP)2 (CCDC ZUNJID).158 Hydrogen atoms are not shown for clarity, meso-phenyl groups are highlighted with blue colour. (d) Anti- vs. syn-conformers of M(Ph-TBTAP)2 complexes. | ||
Comparison of UV-Vis spectra of M(Pc)2, (Ph-TBTAP)M(Pc) and M(Ph-TBTAP)2 revealed that replacement of one or two meso-nitrogen atoms with C-Ph groups resulted in gradual perturbation of electronic systems, leading to complicated spectral patterns with split Q- and intervalence bands, which is not typical for symmetrical complexes. Electrochemical studies of new complexes evidenced that the introduction of two bulky phenyl rings has a profound effect on frontier orbital levels due to weakening of the intramolecular interaction between macrocyclic ligands.
Single-crystal X-ray characterization of Eu(Ph-TBTAP)2 showed that the molecule adapt syn-conformation (Scheme 29c and d), although conformational analysis performed by DFT calculations suggested that anti-form should be more stable and this form is observed as a sole state in solution according to NMR studies. Thus, a less stable conformer is formed in the solid state due to the advantage in energy of the molecular packing.
First examples of REE complexes with corroles were synthesized only in 2013 by Buckley et al. either by metalation of tris-meso-aryl-corrole with M[N(SiMe3)2]3, M = La and Tb or by the reaction of lithium corrolate with GdCl3.161 In both the cases half-sandwiches were isolated, where the coordination sphere of lanthanide ions was saturated with neutral ligands – dimethoxyethane or trimethyltriazanonane. A similar procedure was used to synthesize cerium complexes with corrole ligands and axial ligand-dependent self-assembling of the resulting half-sandwiches was studied.162
Mixed ligand complexes containing corroles and phthalocyanine ligands were synthesized by the reaction of pre-synthesized monophthalocyaninates with corroles in the presence of DBU in octanol (Scheme 30).163–168 Several substituted macrocycles were introduced into this reaction, but invariantly on substituents and REE ions in all cases mixed ligand triple-deckers were isolated with inner Cor and terminal Pc ligands. Moreover, reaction of half-sandwich corrole complex with metal-free phthalocyanine and one-pot reaction of corrole, metal-free Pc and metal salts both yielded the identical triple-deckers and their yields were systematically larger than that when the first method was used.166
 | ||
| Scheme 30 (a) Synthesis of mixed ligand complexes with phthalocyanine and corrole macrocycles. (b) X-ray structure of the complex Eu2[(OctO)8Pc]2[(p-ClPh)3Cor] (CCDC HOKQOP).163 Hydrogen atoms and substituents in top views are not shown for clarity. | ||
Some of the synthesized complexes were characterized by XRD revealing that in contrast to other symmetrical triple-deckers two outer phthalocyanine ligands were not staggered, and coordination polyhedra of two metal centres were slightly different, although in all cases they were close to distorted square prisms.163,164,167 This distortion of coordination surrounding manifested in fast relaxation of magnetization because of QTM (the quantum tunnelling of the magnetisation; cf. Section 3.1), as evidenced from AC magnetic susceptibility measurements performed for dysprosium(III) complexes Dy2[(PentO)8Pc]2[(p-XPh)3Cor], X = F or Cl.164
The presence of three protons in corroles suggests that the closed-shell form of triple-decker complexes should be anionic in nature, which makes them different from trisphthalocyaninates, where the charge of two trivalent REE ions is balanced by the charge of three dianionic ligands. Thus, to stabilize the negative charge of the (M3+)2(Cor3−)(Pc2−)2 species a counterion is needed. NMR studies of the europium(III) complex suggested that the charge is balanced by a proton, which is likely to be delocalized over the isoindoline nitrogen atoms of one of the phthalocyanine ligands.163 Air oxidation of the protonated forms with the formation of paramagnetic complexes with delocalized π-electron was observed, which was followed by disappearance of NMR spectra and appearance of intervalence bands in UV-Vis-NIR spectra at ca. 2000 nm.167
Rich electrochemistry of studied complexes was demonstrated, and up to five oxidized states and three reduced states were observed rendering them as potential molecular materials for information storage devices.163,166–168 The potentials of redox transitions could be finely tuned both by variation of metal centres and substituents in the para-positions of meso-phenyl groups, due to linear correlations between the sums of Hammett constants σ of these substituents and electrochemical characteristics of complexes.168
Comparative studies of self-assembling of two europium(III) complexes Eu2(R8Pc)2[(p-ClPh)3Cor], where R = H or octyl revealed that substituents in Pc ligands affected the morphology of aggregates formed by the phase transfer method upon mixing of solutions of complexes in chloroform with excess of methanol.165 TEM and SEM studies of the resulting aggregates showed that the complex with unsubstituted Pc ligands forms nanobelts of 1.5–2.0 μm length and 100 nm width, while the octyloxy-substituted complex forms large scale sheet-like nanostructures with an average length of ca. 500 nm and a width of 300 nm. NLO studies of these complexes showed significant nonlinear reverse saturation absorption and self-defocusing behaviour.
Altogether, these results suggest that these corrole-containing mixed-ligand complexes can be considered as promising candidates for future nano-electronic and optical device applications.
First REE sandwiches with N-confused porphyrins were synthesized in 2012 by Cao et al. on the examples of mixed ligand double-deckers of iPor-Pc type using the typical raise-by-one-story approach with monophthalocyaninate and metal-free N-confused porphyrin (Scheme 31a).171 For the comparison of spectral properties, N-methylated iPor complex was also synthesized.
 | ||
| Scheme 31 (a and b) Synthesis of heteroligand double-decker complexes with N-confused ligands. (c) X-ray structures of [(tBuPh)4iPorH]Lu(Pc) (CCDC YEFZIU), [(tBuPh)4iPorH]Eu(Pc)·MeOH (CCDC YEFZAM)171 and [(p-ClPh)4iPorH]Pr[(tBuPh)4Por] (CCDC BODFIL).172 Solvate molecules and most of hydrogen atoms are not shown for clarity. | ||
In 2014 cross-condensation of common and N-confused porphyrins with La(III) and Pr(III) acetylacetonates was reported, leading to heteroleptic double-decker complexes of Por-iPor type (Scheme 31b).172 Homoleptic bisporphyrinates were isolated as side products, while formation of homoleptic complexes with N-confused ligands was not observed. Interestingly, heteroleptic complexes exhibited NIR band at ca. 850 nm, which was absent in homoleptic bisporphyrinates.
Another type of porphyrin core modification, namely replacement of one of pyrrolic nitrogen atoms with oxygen or sulphur was used for spectacular tuning of magnetic properties of mixed ligand double-deckers. This was demonstrated on the examples of dysprosium(III) complexes with phthalocyanine and oxa- or thiaporphyrin ligands [(tBuPh)4XPor]Dy(Pc), X = O or S (Scheme 32).173 It was demonstrated that although Dy–X bonds are longer than Dy–N bonds, the symmetries of coordination polyhedra did not change upon going from classical porphyrin to its oxa- and thia-analogues. However, the replacement of one of heteroatoms significantly enhanced the anisotropy of the Dy(III) ion providing the complex with one of the largest barriers ever reported for dysprosium(III) complexes with tetrapyrrolic ligands (cf. Section 3.2.2).
 | ||
| Scheme 32 Synthesis of mixed ligand double-decker complexes with oxa- and thiaporphyrin ligands and X-ray structure of [(tBuPh)4SPor]Dy(Pc) (CCDC AHOPUK).173 Hydrogen atoms and solvent molecules are not shown for clarity. | ||
2.5 Complexes based on half-sandwiches with axially coordinated non-tetrapyrrolic ligands
Due to large coordination numbers of REE ions, their half-sandwich complexes tend to have labile coordination spheres. This phenomenon is at the heart of the raise-by-one-story approach leading to the aforementioned heteroleptic and mixed ligand sandwiches, but it is possible to combine ligands of completely different natures in a molecule as part of this approach.Thus, the present section will include sandwich REE complexes, where porphyrin or phthalocyanine ligands coexist with ligands of non-tetrapyrrolic nature. Although they only resemble sandwich structures of archetypal double- and triple-decker complexes, we included them in this section due to the increasing interest in such type of objects providing pathways to novel fluorescent materials, optical limiters, molecular magnets, etc. Finally, some of the structures are curious by themselves, broadening the rich coordination chemistry of both REE and tetrapyrrolic macrocycles.
 | ||
| Scheme 33 Synthesis of mixed-ligand complexes based on half-sandwich phthalocyaninates and Schiff base H2(SB) with X-ray structures of the corresponding complexes – from the left to the right: (Pc)2Dy2(SB)2Ca(MeOH)2 (CCDC FEVDOB),174(Pc)Dy(SB)K(MeOH) (CCDC FEVDUH),174 (Pc)Tb(SB)Zn(Py)2NO3 (CCDC YIMMUE)175 and (Pc)2Dy2(CB)(H2O) (CCDC XILTOD).176 | ||
First examples of such complexes were prepared by Wang et al. from (Pc)M(acac), M = Dy and Y and H2(SB).174 Depending on the base used for deprotonation of H2(SB) complexes of different nuclearities were obtained. Performing this reaction with Ca(OH)2 afforded a quadruple-decker complex where the [Ca(MeOH)2]2+ group bridged two (Pc)M(SB)− units (Scheme 33a), while dinuclear double-deckers (Pc)M[(SB)K(MeOH)] was isolated in the presence of KOH (Scheme 33b). The latter complex dissociated with the formation of double-decker species upon treatment with K2CO3 (Scheme 33c). Fast QTM relaxation was observed in zero-field AC magnetic susceptibility measurements for dysprosium(III) complexes of both types, although long-distance magnetic interactions between two paramagnetic centres in quadruple-decker complex was detected (cf. Section 3.2.2).
Later, Ma et al. synthesized terbium(III) complexes with all alkali metal cations [(SB)M(MeOH)]Tb(Pc), M = Li+ to Cs+ and the possibility to exchange these cations with the Zn2+ ion was demonstrated (Scheme 33d), providing an interesting example of polyfunctional complex with tuneable properties.175
Slow diffusion of the acetonitrile solution of H2(SB) into the solution of (Pc)M(acac), M = Gd and Dy, in chloroform afforded another type of triple-decker complex with bridging (SB)2− unit and chemically non-equivalent REE ions – M2(Pc)2(SB)(H2O) (Scheme 33e).176 One of metal centres was in octahedral surrounding and another one was hepta-coordinated by four isoindole nitrogen atoms of (Pc)2−, two oxygen atoms of a Schiff-base ligand, and one oxygen atom of water molecule. Homoleptic binuclear dysprosium(III) complex with the H2(SB) ligand had the same binding mode; however, incorporation of the phthalocyanine ligand had a profound effect on SMM properties of the mixed-ligand sandwich in comparison with Dy2(SB)3(H2O) in terms of ferromagnetic coupling between paramagnetic ions and energy barrier, Ueff.
Analogous binuclear complexes were synthesized by Li et al. based on a family of chiral ligand prepared by the condensation of 1,2-diphenylethylenediamine and 5-chlorosalicylic aldehyde. Photophysical characterization of such complexes showed their potential in future nonlinear optical applications such as wavelength conversion and optical switching.177,178
By varying the ratio of half-sandwich phthalocyaninate and Schiff base, complexes with different stoichiometries of ligands can be obtained. This was shown by Gao et al. on the example of the reaction between one equivalent of dysprosium(III) phthalocyaninate and two equivalents of tetrathiafulvalene-fused Schiff base H2(SB-TTF) yielding a binuclear complex Dy2(Pc)(SB-TTF)2(MeOH) (Scheme 34a).179 Due to the intrinsic dipole moment, this complex could be absorbed on the surface of HOPG either in Pc-up or S-TTF-up fashion depending on the applied potential, which was demonstrated by scanning tunneling microscopy at the single molecule level, rendering this compound as a component of functional materials fabricated by self-assembly.
 | ||
| Scheme 34 (a) Synthesis of a mixed-ligand complex with tetrathiafulvalene-fused Schiff base with the X-ray structure of complex Dy2(Pc)(SB-TTF)2(MeOH) (CCDC WONKIV).179 (b) Synthesis of sandwiches based on closed-macrocyclic Schiff base and REE monophthalocyaninates with the X-ray structure of complex Dy3(Pc)2(mSB)(OMe)2 (CCDC IKAQAO, only one of two crystallographically independent molecules is shown).180 Hydrogen atoms are omitted for clarity. | ||
Gao et al. used dysprosium(III) or erbium(III) monophthalocyaninates generated in situ as templates for condensation of 2,6-diformyl-4-methylphenol and 1,3-diaminopropane to form unusual trinuclear triple-decker sandwiches with a macrocyclic Schiff ligand and bridging acetate and methoxide counterions M3(Pc)2(mS)(OAc)(OMe)2 (Scheme 34b).180 Interestingly, in the crystal lattice these complexes existed as pairs of symmetrically unrelated molecules with slightly different symmetries of coordination polyhedra. This feature resulted in multiple magnetic relaxation modes found by AC magnetic susceptibility measurements of the dysprosium(III) complex. Pronounced intramolecular interactions between proximal metal centres was observed – ferromagnetic in the case of dysprosium and antiferromagnetic in the case of erbium (cf. Section 3.2.2).
 | ||
| Scheme 35 Synthesis of REE porphyrinates and phthalocyaninates with axially-coordinated cyclen ligands with X-ray structures of [(TPP)Tb(Cyclen)]+Cl− (CCDC LEBMUD) and [(Pc)Tb(Cyclen)]+Cl− (CCDC WENBID).181,182 Hydrogen atoms are omitted for clarity. | ||
Ground multiplet states of a series of porphyrin complexes [(TPP)M(Cyclen)]+Cl− were determined by NMR and magnetic susceptibility data together with CASSCF calculations, giving insight into their electronic structures.183 Intramolecular magnetic interactions between localized 4f-electronic systems and photoexcited Por or Pc π-systems in terbium(III) Por/cyclen and Pc/cyclen complexes was studied by variable-temperature variable-field magnetic circular dichroism spectroscopy providing experimental data to validate the theoretical models describing such interactions.184,185
Interesting sandwich complexes with η5-Cp and η5-Me5Cp ligands sitting-atop scandium(III) phthalocyaninate were synthesized from (Pc)ScCl by Platel et al. (Scheme 36).186 As a first step of this work, template condensation of phthalonitrile in the presence of ScCl3 in ClN was performed, yielding inseparable mixture of target (Pc)ScCl with ca. 15% of unreacted ScCl3. Treatment of this mixture with LiCH(TMS)2 in THF afforded the formation of a soluble binuclear complex (Pc)ScCl2Li(THF)2 which could be crystallized as an individual compound. Its reaction with NaCp or LiCp* in THF provided (Pc)Sc(Cp) and (Pc)Sc(Cp*), respectively, as first representatives of metallophthalocyanine-ocenes.
 | ||
| Scheme 36 Synthesis of scandium phthalocyaninates – (Pc)ScCl2Li(THF)2 (CCDC DUDTED) and (Pc)Sc(Cp) (CCDC DUDTIH).186 Hydrogen atoms are omitted for clarity. | ||
 | ||
| Scheme 37 Synthesis of complexes with hemiporphyrazine ligand188,189 and X-ray structures of (HHp)Eu(Pc) (CCDC GAWCAL) and (HHp)Eu(Hp) (CCDC GAVXUZ).188 Hydrogen atoms are omitted for clarity. | ||
Another class of macrocyclic ligands which was recently introduced into sandwich REE chemistry is hemiporphyrazin, Hp. Synthesis of this 20π-electron antiaromatic relative of azaporphyrins was reported for the first time in 1952 by Elvidge and Linstead.187 However, REE complexes with Hp were reported for the first time only in 2017.188 The complexes (HHp)M(Pc), M = Eu, Lu, were obtained by Liu et al. using reactions of the corresponding (Pc)M(acac) with H2(Hp) and DBU in refluxing pentanol. Metalation of hemiporpyrazine with M(acac)3·nH2O under the same conditions yielded double-decker complex (HHp)M(Hp). ESR and NMR studies of the resulting sandwiches evidenced that they were diamagnetic in contrast to most bisphthalocyaninates existing as free radicals. The proton needed for such state was localized by NMR spectroscopy, XRD and quantum-chemical calculations – all methods evidenced that the proton is bound to the pyridine nitrogen atom. Quantum-chemical calculations of shielding surfaces of both types of complexes revealed that in homoleptic complexes hemiporphyrazine ligands retained their antiaromatic nature; however, combination of antiaromatic Hp and aromatic Pc species provided molecules with overall aromatic behaviour.
XRD characterization of both types of Hp complexes revealed nearly ideal SAP surrounding of metal centres, although somewhat distorted because of the protonation of one of the pyridine groups. Nevertheless, measurements of magnetic properties of (HHp)Dy(Pc) and (HHp)Dy(Hp) revealed excellent SMM properties (cf. Section 3.2.2).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)(OEt)2}3].190 These reactions yielded seven-coordinate complexes with axial tripodal ligands (Scheme 38a and b). Tb(III) and Dy(III) complexes showed field-induced slow relaxation of magnetization; these complexes were convenient models demonstrating the correlation between the magnetic relaxation properties and subtle distortions of the coordination geometry of the paramagnetic lanthanide ions (cf. Section 3.2.2).
O)(OEt)2}3].190 These reactions yielded seven-coordinate complexes with axial tripodal ligands (Scheme 38a and b). Tb(III) and Dy(III) complexes showed field-induced slow relaxation of magnetization; these complexes were convenient models demonstrating the correlation between the magnetic relaxation properties and subtle distortions of the coordination geometry of the paramagnetic lanthanide ions (cf. Section 3.2.2).
 | ||
| Scheme 38 Synthesis of heteronuclear complexes based on REE half-sandwiches. (a and b) Synthesis of Dy(III) porphyrinate (CCDC LIMDOC) and phthalocyaninate (CCDC LIMCUH) with a Kläui ligand. (c) Synthesis of Tb(III) phthalocyaninate bearing a polyoxotungstate ligand (Pc)M(PW11O39)6−(NBu4+)6 (CCDC GUZFOZ), and the X-ray structure of anionic part of the complex (Pc)Tb[PW11O39]6−. Solvate molecules, hydrogen atoms and counterions are omitted for clarity. | ||
Later, half-sandwich Yb(III) with porphyrin derivatives and axial Kläui's ligands attracted great interest as highly near-IR emissive complexes with unprecedented quantum yields up to 63%,191 which contributes to the design of molecular probes for near-IR imaging.192
In 2020 Sarwar et al. reported the synthesis of Tb(III), Dy(III) and Y(III) phthalocyaninates with a monolacunary α-Keggin polyoxotungstate ligand [PVWVI11O39]7−.193 With this aim, the corresponding monophthalocyaninates (Pc)M(OAc) reacted with (NBu4)4H3[PW11O39] in the presence of triethylamine and NBu4Br under mild conditions, yielding anionic complexes with tetrabutylammonium counterions (Pc)M(PW11O39)6−(NBu4+)6 (Scheme 38c). Their characterisation included high resolution mass spectrometry, synchrotron-based single-crystal X-ray diffraction and magnetic studies revealing slow relaxation of the magnetisation for the Dy(III) derivate (cf. Section 3.2.2).
Thus, these complexes were reported for the first time in 1983 in a short communication by Sugimoto et al., describing the reaction between Li2(Pc) and M(dpm)3 in refluxing THF under anaerobic conditions.194 This reaction afforded a series of (μ-Pc)[M(dpm)2]2, where M = Sm to Yb and Y. The X-ray structure of the samarium(III) complex with benzene solvate molecules was determined (Fig. 8), showing the extremely rare representative of binuclear complexes formed by a single tetrapyrrolic macrocycle. A bit later magnetic properties of these unique complexes were studied by Maeda et al., showing the first evidence of intramolecular spin–spin exchange interaction between trivalent REE ions.195,196 However, for many years that followed, these complexes attracted no attention.
 | ||
| Fig. 8 X-ray structure of (μ-Pc)[Sm(dpm)2]2 (CCDC CALWIV).194 Solvate molecules and hydrogen atoms are not shown. | ||
Only in 2014 detailed characterization of magnetic properties of (μ-Pc)[M(dpm)2]2, M = Tb and Dy, was performed by Tsuchiya et al., showing their striking difference from common binuclear triple-deckers.197 Unexpectedly, antiferromagnetic interaction between metal centres was found in contrast to ferromagnetic coupling in the corresponding trisphthalocyaninates. NMR studies showed that aromatic proton resonances were shifted down-field – again, it was in sharp contrast with M2(Pc)3 with large up-field lanthanide-induced shifts. These intriguing results were explained assuming the ligand field potential of the inverted-sandwich complexes gave strong easy-plain-type magnetic anisotropy, where the Jz value of the lowest sublevel has a small value, while trisphthalocyaninates have a strong easy-axis-type magnetic anisotropy. The benzene solvates (μ-Pc)[M(dpm)2]2·2C6H6 showed interesting behaviour with respect to desolvation and exchange of solvation molecules to dichloromethane, affording the pathway for fine tuning of magnetic properties (cf. Section 3.6).198–200
Interesting properties of inverted sandwiches could encourage synthetic studies to broaden this class of REE complexes; however, to date, only dpm ligands could stabilize their unusual architecture. To the best of our knowledge, the sole reported attempt to exchange these anions was made by Ge et al. when the complex (μ-Pc)[Dy(dpm)2]2 was treated with 3-methoxy- or 3-ethoxysalicyllic aldehydes H(LR), R = OMe, OEt.201 However, it resulted in the deterioration of the inverted sandwich scaffold with the formation of a new type of triple-deckers (μ-LR)2Dy2(Pc)2(H2O) where two (DyPc)+ units were bridged by a pair of coplanar deprotonated LR− ligands (Scheme 39a). Although at the first glance both complexes had similar structures invariant to substituents in bridging ligands, their magnetic properties were markedly different. Magnetic studies indicated that the MeO-substituted complex behaved as a zero-field single-molecule magnet with a higher energy barrier, while EtO-substituted complex exhibited a fast tunneling relaxation process (cf. Section 3.2.2). Detailed analysis of the XRD data revealed that one of MeO-groups coordinated to dysprosium(III) ion, while oxygen atoms of both ethoxy-groups remained in the non-coordination mode (Scheme 39b). This difference in the substitution of ligands in the middle layer leads to perturbation of the main magnetic axes, consequently changing the coupling interaction and modulating the magnetic behaviour for this Dy2 system.
 | ||
| Scheme 39 (a) Transformation of inverted sandwich (μ-Pc)[Dy(dpm)2]2 into a triple-decker mixed ligand complex (μ-LR)2Dy2(Pc)2(H2O) upon interaction with 3-alkoxysalicylic aldehydes, H(LR), R = OMe, OEr. (b) Structure of the middle layers formed by deprotonated ligands LR−, coordinated to dysprosium ions (CCDC MESMIJ, R = Me and MESMOP, R = Et).201 | ||
2.6 Sandwich complexes that go beyond triple-deckers
Simple electrostatic considerations suggest that triple-deckers formed by doubly-deprotonated porphyrins and phthalocyanines with a pair of trivalent REE cations are the largest possible electroneutral species. However, there is interest in larger sandwiches, which can fill in the gap between discrete double-/triple-decker complexes and infinite lattices formed by these complexes in solid functional materials. The following section will describe some approaches which were developed over the last decade to obtain discrete multideckers, avoiding electrostatic limitations. | ||
| Scheme 40 (a–c) Synthesis of discrete quadruple-decker complexes Lu2Cd(Pc)4,202Dy2Cd[(BuO)8Pc]4,205 and Eu2Cd[(HexS)8Pc]4206via connection of two double-decker molecules with Cd(II) ions. (c) Structure of complex Dy2Cd[(BuO)8Pc]4 (CCDC IYOCIJ),205 the structure of only one of two crystallographically independent molecules is shown, hydrogen atoms and solvent molecules are omitted. | ||
Both stability and yields of quadriphthalocyaninates could be improved when electron-rich BuO-substituted ligands were used instead of unsubstituted phthalocyaninates. Thus, Wang et al. showed that the reaction of neutral Dy[(BuO)8Pc]2 with Cd(OAc)2·2H2O in refluxing TClB afforded target substituted quadruple-decker complex Dy2Cd[(BuO)8Pc]4 in yield as high as 43% (Scheme 40b).205 In a similar manner, Eu(III) complex with n-hexylthio-substituents Eu2Cd[(HexS)8Pc]4 was synthesized starting from the corresponding double-decker in 84% yield (Scheme 40c).206 Both the OFET and gas sensing properties were studied on the self-assembled film of the latter complex (cf. Section 4).
Single crystals of the Dy(III) complex were obtained by Wang et al., and XRD provided a first ultimate proof of a quadruple-decker structure (Scheme 40d).205 Separation between Dy(III) ions – ca. 6.65 Å was expectedly much larger than in triple-decker complexes; however, it was still enough to detect notable intramolecular interaction between paramagnetic metal centres, which was responsible for the fast quantum tunnelling of magnetization in a zero DC field (cf. Section 3.2.4). Interestingly, introduction of alkoxy and alkylthio groups provided complexes with different conformations. In the case of the BuO-substituted complex both Dy(III) and Cd(II) metal centres were in distorted prismatic surrounding with a twist angle of ca. 20°,205 while in HexS-substituted complex Eu(III) and Cd(II) had lightly deviated SAP coordination geometries with twist angles of ca. 43 and 39°.206
The capability of cadmium bridge to connect pairs of different double-deckers was tested by Wang et al. on the example of homoleptic bisphthalocyaninates M(Pc)2 and mixed-ligand complexes (TClPP)M(Pc), M = Eu and Y.207 In principle, this reaction could lead to isomeric quadruple-decker complexes with different sequences of tetrapyrrols – Pc–Pc–Pc–Por or Pc–Pc–Por–Pc, but both NMR and X-ray crystallography showed that only one type of complex with terminal porphyrin ligand was formed. The complexes were isolated in 9–14% yields. Similarly to homoleptic quadriphthalocyaninates, the synthesized mixed-ligand complex had rich electrochemistry associated with four ligand-centred oxidation and three reduction processes, whose potentials showed dependence on the metal centre.
No surprise, extended sandwiches became attractive objects to study long-distance interactions between paramagnetic metal ions in the context of elaboration of SMM materials and understand mechanisms or magnetization relaxation. Thus, the approach to quadriple-deckers was extended to even higher sandwiches. Wang et al. showed that reactions of M[(BuO)8Pc]2 with one or two equivalents of H2[(BuO)8Pc] and Cd(OAc)2·2H2O allowed the introduction of one or two additional phthalocyanine layers between REE-containing moieties, affording therefore quintuple- and sextuple-decker sandwiches (Scheme 41).208,209 Moreover, this strategy allowed the synthesis of heteroleptic quintuple-decker sandwiches with precise sequences of unsubstituted and octa-pentyloxy-substituted ligands – either Pc–Pc–Pc*–Pc–Pc or Pc–Pc*–Pc*–Pc*–Pc.210 Single-crystal XRD studies of complexes evidenced of smooth elongation of distances between REE ions going from triple- to sextuple-deckers, providing more objects for studies of long-range magnetic interactions (cf. Section 3.2.4).211–214
 | ||
| Scheme 41 Synthesis of discrete quintuple-decker complexes M2Cd2(Pc)5208 and sextuple-decker complexes M2Cd3[(BuO)8Pc]6.209 Butoxy-groups in resulting multi-deckers are not shown. | ||
In principle, this approach can be used to obtain multi-decker complexes with any number of tetrapyrrolic ligands, since further sequential addition of “CdPc” fragments does not violate the electroneutrality requirement in the resulting extended sandwiches. However, the problems with isolation of required multi-decker from the mixture of complexes are highly likely to arise.
Thus, in 2013 Wang et al. showed that Y(III)- or Dy(III)-templated cross-condensation of bis(diiminoisoindoline) with 4,5-dibutoxyphthalonitrile in refluxing pentanol in the presence of DBU furnished target fused bisphthalocyaninates (Pc)M[f-Pc2]M(Pc)OBu in 2.0–4.3% yields (Scheme 42a).216 In the same manner Tb(III) complex was synthesized by Morita et al. in 2.5% yield using hexanol as a solvent for template condensation.217 Using 4,5-di(ethylthio)phthalonitrile in this procedure allowed Wei et al. to increase the yield of the fused bisphthalocyaninate (Pc)M[f-Pc2]M(Pc)SEt to 24.8% (Scheme 42b).218
 | ||
| Scheme 42 Synthesis of fused sandwich phthalocyaninates. (a and b) Binuclear complexes (Pc)M[f-Pc2]M(Pc)XR, XR = OBu or SEt. (c) Tetranuclear complexes (Pc)2M2[f-Pc2]M2(Pc)2OBu. (d) Tri- and tetranuclear complexes (Pc)2M2[f-Pc2]M(Pc)SEt and (Pc)2M2[f-Pc2]M2(Pc)2SEt. Superscripts OBu and SEt indicate that all accessible peripheral positions of the benzene rings contain the corresponding substituents. | ||
Fused complexes with higher nuclearity could be synthesized using variants of the raise-by-one-story approach. Thus, Motita and Katoh et al. treated metal-free BuO-substituted fused phthalocyanine H4[f-Pc2]OBu with double-deckers and Tb(III) or Dy(III) acetylacetonates in TClB to synthesize tetranuclear Tb(III)219 and Dy(III)220 complexes (Pc)2M2[f-Pc2]M2(Pc)2OBu (Scheme 42c). Alternatively, Wei et al. treated SEt-substituted fused Eu(III) bisphthalocyaninate with H2[(SEt)8Pc] and Eu(acac)3 to synthesize tri- and tetranuclear complexes (Pc)M[f-Pc2]M2(Pc)2SEt and (Pc)2M2[f-Pc2]M2(Pc)2SEt which could be separated by chromatography (Scheme 42d).218 In all cases the yields of isolated complexes were relatively small, yet, it was enough to characterize their properties and demonstrate their applicability.
Fused bi-, tri- and tetranuclear substituted complexes were used as redox-active components in sensors to NO2 and NH3 revealing excellent sensitivity to these gasses.218,221 The film formed by the tetranuclear SEt-substituted complex was one of the best gas sensors obtained so far for all solution-processed organic semiconductor films in dry air at room temperature.218 Binuclear SEt-substituted complex was also used as a component of an electrochemical sensor for simultaneous detection of acetaminophen and ascorbic acid.222 SHex-substituted tetranuclear complex was found to form well-defined single crystal microsheets upon self-assembling in solution, which were used to fabricate OFET with high hole and electron mobilities of 18 and 0.3 cm2 V−1 s−1, respectively, representing the best result for phthalocyanine-based OFETs at that time (cf. Section 4).223
A unique example of fused bisporphyrinate synthesized from β-β/meso–meso/β-β triply-linked diporphyrin H4[f-Por2] was reported by Lee et al. in 2019.224 Starting ligand was treated with LiHMDS to generate a tetralithium intermediate which reacted with half-sandwiches (TPP)M(acac) to furnish fused sandwiches (TPP)M[f-Por2]M(TPP), M = Tb and Y in small yields (Scheme 43). Complexes were isolated in neutral states exhibiting an open shell biradical character, as determined from magnetic susceptibility measurements and theoretical calculations. The Tb(III) complex exhibited rich electrochemistry, having more than eight clear peaks in the voltammetry curves; moreover, it has a very small HOMO–LUMO energy gap as evidenced from optical (3700 nm, 0.33 eV) and electrochemical measurements (3400 nm, 0.36 eV). These features together with high stability render these biradicals as promising candidates for use in molecular electronics, optics, and spintronics.
 | ||
| Scheme 43 Synthesis of fused sandwich bisporphyrinates (TPP)M[f-Por2]M(TPP).224 | ||
3. Development of the functionalities of sandwich tetrapyrrole rare-earth single-molecule magnets
In this section, we will provide a comprehensive survey of several types of sandwich tetrapyrrole rare-earth (RE) single-molecule magnets (SMMs) that use a supramolecular approach, combined with a discussion of their functionalities. Importantly, the properties of these molecules are changed significantly by the formation of associations among such molecules. The chemical and physical properties of molecular assemblies composed of a finite number of molecules are also affected by various intermolecular interactions. Currently, researchers are also conducting studies on applying self-assembly strategies that actively apply coordination bonds to molecular magnetic materials. In 2002, Wernsdorfer et al. reported that the introduction of hydrogen bonds and van der Waals forces into a tetranuclear Mn complex SMM (abbreviated as [Mn4O3Cl4(O2CEt)3(py)3] = Mn4) resulted in dimers of the type Mn4⋯Mn4, which exhibit superexchange interactions and suppress the quantum tunnelling of magnetisation (QTM; cf. Section 3.1).225 This phenomenon was achieved via the superexchange interaction between the SMMs, which act as an exchange bias (Hbias ≈ ±M2J/gμB) on the QTM that occurs at a zero magnetic field, resulting in a spin-sublevel energy mismatch. Thus, the magnetic interaction acts as a perturbation between the SMMs that affects the QTM that occurs at a near-zero magnetic field. In other words, it is also possible to control the properties of SMMs using intra/intermolecular magnetic interactions between SMMs. Moreover, to control the magnetic interactions between SMMs, it is possible to use not only hydrogen bonds but also π–π interactions, CH–π interactions, and inclusion (host–guest) interactions associated with ion and molecular recognition. Controlling the spin relaxation time (τ) by utilizing the interactions between the infinite number of intramolecular vibrational modes of SMMs and spin excitation is essential for spintronics. SMMs (molecular crystal) have weak intermolecular interaction and low phonon energy in the acoustic mode. Therefore, the spin-acoustic mode interaction is restricted. On the other hand, by introducing a strong intermolecular interaction into the SMMs, the phonon energy in the acoustic mode can be increased, and interaction with the spin can be expected. In other words, from the viewpoint of controlling magnetic relaxation, the approach of introducing a molecular assembly structure into SMMs is one method (vide infra).Furthermore, ligand modifications, protonation–deprotonation reactions, and redox reactions also cause dramatic changes in SMM characteristics by inducing substantial changes in their coordination environments. Sandwich tetrapyrrole rare-earth-type SMMs have attracted attention for potential use in the fields of spintronic devices and/or quantum technology (QT). However, the performance of these devices depends on the nature of the ground-state electronic structures of the SMM units. By using supramolecular approaches to prepare sandwich tetrapyrrole rare-earth-type SMMs, their ground-state electronic structures and functionality can potentially be controlled (Fig. 9).
 | ||
| Fig. 9 Brief overview of rare-earth sandwich tetrapyrrole SMMs. Double-decker complexes with various kinds of porphyrinoids act as SMMs. The magnetic properties of the double-decker SMMs can be reversibly tuned by redox reactions and protonation–deprotonation reactions. Various approaches for connecting/arranging SMMs have been used to introduce Ln–Ln interactions and tune the coordination environment. | ||
3.1 Brief outline of single-molecule magnets
In 1993, Sessoli, Gatteschi, and co-workers reported that the cluster [Mn12O12(AcO)16(H2O)4]·2AcOH with S = 10 (henceforth abbreviated as Mn12; Fig. 10a), which contains twelve Mn ions (eight Mn3+ with S = 2 and four Mn4+ with S = 3/2), exhibited superparamagnetic properties; they referred to Mn12 as an SMM because it exhibited both single molecule properties and ferromagnetic properties (Fig. 10c).226,227 However, REE-type SMMs are fundamentally different from transition-metal (TM) SMMs, because their SMM behaviour originates from the f electrons (Fig. 10b). Since the orbital angular momentum L does not disappear for f electrons, ground-state multiplets exhibit a total angular momentum J due to spin–orbit coupling (SOC). When the ground-state multiplet is placed in a ligand field (LF) environment, the degeneracy splits into the 2J + 1 sublevels (Fig. 10d). As a result, double-well potentials form, and SMM behaviour emerges (vide infra).36,226 In general, the sublevel splitting in REE-type SMMs (102–103 cm−1)5 is much larger than that of TM-type SMMs (ca. 70 cm−1),228 indicating that REE-type SMMs are more useful in terms of their SMM characteristics, such as large magnetic anisotropy and Ueff (Fig. 10c and d).229,230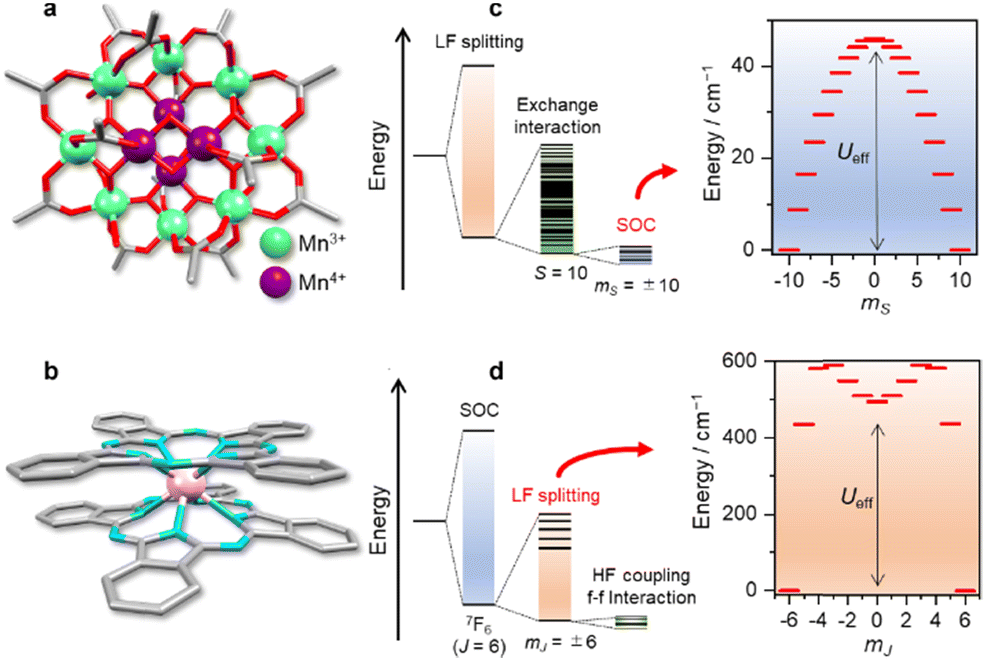 | ||
| Fig. 10 Molecular structures of (a) Mn12 and (b) Tb(Pc)2 based on the cif files from ref. 231 and 232. Schematic energy diagrams of (c) Mn12 and (d) Tb(Pc)2. In the case of Mn12, eight Mn3+ and four Mn4+ ions with d-orbital splitting (LF splitting) couple with each other (exchange interactions) to form an S = 10 state. The L of Mn3+ and Mn4+ is quenched due to the large LF splitting. The zero-field splitting (D = –0.46 cm−1) of S = 10, which mainly originates from second-order SOC, results in a Ueff of ca. 40 cm−1 for spin reversal between mS = +10 and mS = −10. In the case of Tb(Pc)2, S = 3 and L = 3 couple with each other (first-order SOC) to form 7F6. LF splitting of the Tb3+ centre results in a Ueff of ca. 400 cm−1 for spin reversal between mJ = +6 and mJ = −6. mJvs. energy plots were constructed from ref. 233. | ||
In the last 20 years, SMMs with high operating temperature have been synthesized by controlling the LF and/or crystal field (CF) around the metal ion, even in single-ion magnets (SIMs).234 Since the Mn12 cluster complex has been shown to exhibit the SMM phenomena, 3d to 5f metal complexes have been investigated as SMMs (henceforth, the term SMM will be used to refer to SIMs as well).228,230,235–244 SMMs have a fixed number of metal ions and an ordered magnetic ion structure, and exhibit uniaxial magnetic anisotropy due to the introduction of an LF and SOC.226,227,229,245 A brief description of magnetic anisotropy is given here. When a magnetic material is magnetised in various directions, there are directions in which it is easy to magnetise (easy axis of magnetisation) and difficult to magnetise (hard axis of magnetisation). When the direction is in one direction (uniaxial), it is called “uniaxial magnetic anisotropy”. This magnetic anisotropy is caused by the orientation of magnetic moments of the atoms which have the CF and SOC that make up the magnetic material. That is, the stable direction of orbital angular momentum in the crystal affects the direction of spin, which is the source of magnetism. As described later, the 4f orbital shape of rare earth metal ions has a very strong directivity, so it is possible to control the magnetic anisotropy by controlling the appropriate CF (cf. Section 3.2). A characteristic of Mn12 is that the Jahn-Teller axis of Mn3+ ions with magnetic anisotropy is almost parallel to the S4 axis, showing large uniaxial magnetic anisotropy as a single molecule.226 It is possible to control the magnetic properties of metal complexes, such as their electronic state, LF, and magnetic anisotropy, by changing the metal ions and coordinating ligands. In particular, it has been shown that the LF directly affects the magnetic properties. Classical magnets exhibit large magnetisation via three-dimensional interactions among the electron spins of the metal ions, but SMMs involve the uniaxial magnetic anisotropy of one molecule. It is thought that SMMs are also “single-domain magnets”, as intermolecular magnetic interactions are ignored.
Using the effective spin Hamiltonian, we discuss the uniaxial magnetic anisotropy and Ueff in SMMs. The concept of effective spin Hamiltonian  was introduced by M. H. L. Pryce which has only the spin operator being considered. When incorporating the CF and SO into the effective spin Hamiltonian, it can be expressed by D (uniaxial crystal field constant) and E (biaxial crystal field constant). Here, the spin Hamiltonian is given by
was introduced by M. H. L. Pryce which has only the spin operator being considered. When incorporating the CF and SO into the effective spin Hamiltonian, it can be expressed by D (uniaxial crystal field constant) and E (biaxial crystal field constant). Here, the spin Hamiltonian is given by  with the D. In the absence of an external DC magnetic field (H = 0; H and Hdc are used interchangeably), the Zeeman term goes to zero, so DmS2 (mS is the z-component of S) is the splitting energy at zero magnetic field (zero-field splitting, ZFS). For the reasons mentioned above, the splitting width of the ground and highest energy levels is defined as Ueff = |D|S2 for TM-type SMMs, where the D value is the “zero-field splitting constant” and the S is the spin quantum number of the ground state, which mainly originates from second-order SOC (the Ueff of REE-type SMMs is explained in the next section). For SMMs, the degeneracy of the total spin state Stotal is split into 2S + 1 by LF and SOC, a quantized double-well potential is formed by the uniaxial magnetic anisotropy, and the spin inversion between spin-up and spin-downstate caused by the spin sublevel shows Arrhenius-like relaxation (τ = τ0 exp(Ueff/T), Fig. 10c). Here, τ represents the magnetic relaxation time, τ0 is the frequency factor, Ueff is the energy barrier associated with spin inversion, and T is the temperature. At temperatures higher than Ueff associated with spin inversion, the SMM behaves as a paramagnetic material, but when the temperature decreases and the Ueff cannot be overcome, the magnetic moment (spin) is frozen. The temperature at which the τ reaches 100 s is defined as the blocking temperature (TB). SMMs behave like a magnet which shows magnetic hysteresis with coercivity (HC) below TB due to long τ values. In addition, the field cooled (FC) and zero-field cooled (ZFC) measurements of the SMM diverge at TB. The magnitude of magnetic anisotropy is possible to estimate from the M versus HT−1 plot at different T values. If SMMs have large uniaxial magnetic anisotropies and/or the population of low-lying excited states, M versus HT−1 shows non-superposed magnetisation curves.
with the D. In the absence of an external DC magnetic field (H = 0; H and Hdc are used interchangeably), the Zeeman term goes to zero, so DmS2 (mS is the z-component of S) is the splitting energy at zero magnetic field (zero-field splitting, ZFS). For the reasons mentioned above, the splitting width of the ground and highest energy levels is defined as Ueff = |D|S2 for TM-type SMMs, where the D value is the “zero-field splitting constant” and the S is the spin quantum number of the ground state, which mainly originates from second-order SOC (the Ueff of REE-type SMMs is explained in the next section). For SMMs, the degeneracy of the total spin state Stotal is split into 2S + 1 by LF and SOC, a quantized double-well potential is formed by the uniaxial magnetic anisotropy, and the spin inversion between spin-up and spin-downstate caused by the spin sublevel shows Arrhenius-like relaxation (τ = τ0 exp(Ueff/T), Fig. 10c). Here, τ represents the magnetic relaxation time, τ0 is the frequency factor, Ueff is the energy barrier associated with spin inversion, and T is the temperature. At temperatures higher than Ueff associated with spin inversion, the SMM behaves as a paramagnetic material, but when the temperature decreases and the Ueff cannot be overcome, the magnetic moment (spin) is frozen. The temperature at which the τ reaches 100 s is defined as the blocking temperature (TB). SMMs behave like a magnet which shows magnetic hysteresis with coercivity (HC) below TB due to long τ values. In addition, the field cooled (FC) and zero-field cooled (ZFC) measurements of the SMM diverge at TB. The magnitude of magnetic anisotropy is possible to estimate from the M versus HT−1 plot at different T values. If SMMs have large uniaxial magnetic anisotropies and/or the population of low-lying excited states, M versus HT−1 shows non-superposed magnetisation curves.
The temperature change in the spin system reaches thermal equilibrium through heat exchange between the spin and the lattice, and finally moves to the heat bath. Therefore, the information for slow magnetic relaxation can be obtained from alternating current (AC) magnetic measurements. The measurement data are analyzed by using a generalized Debye model containing the dispersion coefficient α. It is possible to estimate values of τ. The magnetic relaxation undergoes a single relaxation process at α = 0, a Cole–Cole plot (χ′′ versus χ′ plot) is semicircular.246 In addition, magnetic relaxation is a phenomenon associated with spin–phonon coupling, in which the lattice vibration of the molecular crystal is mainly dominated by intramolecular vibration. SMMs undergo three types of magnetic relaxation processes due to spin–lattice interactions, i.e., (1) direct, (2) Raman, and (3) Orbach processes.247 The direct process is caused by the absorption or emission of a phonon (ħω), while the Raman process is due to phonon scattering between the ground state and the virtual transition to higher energy levels.248–250 Numerical simulation of the T dependence of τ is carry out using a combination of the magnetic relaxation processes.247,251,252
Furthermore, at extremely low temperatures (e.g. 40 mK), spin inversion via QTM occurs due to the quantum tunneling effect when the energies of the spin sublevels are equal, and the steps in the magnetic hysteresis curves (M versus H) are observed using a micro-SQUID (Fig. 12c).227,245,253 The origin of QTM in SMMs can be understood via an energy level diagram for a system with a tunnelling gap (ΔEtunnel) by the hyperfine (hf) interaction between the electronic spin and the nuclear spin I. QTM is complicated due to the interactions of the ground state with the I, which cause energy level crossing (Fig. 12b).253–255 It is possible to explain QTM using the Landau–Zener–Stückelberg (LZS) model to the ground state ±mS levels.245 Quantum phenomena, such as the QTM observed in SMMs, have been studied in relation to the field of quantum logic (e.g., quantum spin coherence,256 quantum phase transition,257 Berry phase interference,258 and quantum computers259). Interest in the use of molecules as spin qubits is growing rapidly, and several perspective articles and reviews focusing on such molecules have been published recently.260–263 Using SMMs in information storage devices would allow memory capacity to be expanded quite dramatically. Thus, the development of multi-functional SMMs can be expected to continue for several years to come.264 In addition, single-crystal X-ray diffraction analysis and/or theoretical calculations (DFT and ab initio; e.g. CASSCF/RASSI-SO/SINGLE_ANISO calculations)265 for the structure optimisation and the ground state are the most effective methods for investigating the coordination environment of Ln3+ ions in SMMs. Additionally, electron spin resonance (ESR), nuclear magnetic resonance (NMR), inelastic neutron scattering, and muon spin relaxation (μSR) are useful in the investigation of the SMM phenomena.245 It is possible to evaluate the SMM characteristics by using a variety of measurements together with magnetic measurements. Thus, the field of molecular magnetism, which includes SMMs, has been extensively studied recently in physics and nanotechnology contexts, particularly in the areas of molecular spintronics and quantum technology (QT) (Fig. 11).266
 | ||
| Fig. 11 Quantum technology actively using SMMs. Rare-earth sandwich tetrapyrrole SMMs (Tb(Pc)2) have excellent chemical and physical stability, which make it possible to use them in quantum devices. Reprinted with permission from (a) ref. 267. Copyright 2010, American Chemical Society. Reprinted from (b) ref. 268. Copyright 2016, Royal Society of Chemistry. Reprinted from (c) ref. 269. Copyright 2011, MDPI. Reprinted with permission from (d) ref. 270. Copyright 2014, American Association for the Advancement of Science. | ||
For more information, several books and reviews suggested by the authors are included in the reference. In particular, D. Gatteschi, R. Sessoli and J. Villain have published a groundbreaking book on SMMs in 2006, which covers everything from basic principles of physical phenomena to measurement techniques.245
3.2 Brief overview of rare-earth sandwich tetrapyrrole single-molecule magnets
Phthalocyanines feature prominently in diverse research fields, including molecular conductors, solar cells, liquid crystals, pharmaceuticals, and biological chemistry,271,272 as they are highly soluble in several organic solvents, chemically stable and robust, as well as commercially available.273–275 In addition, in recent years, tetrapyrrole has attracted attention as a ligand for SMMs.9,36In order to identify an underlying cause for SMM properties, we review the coordination environment and SMM properties of Ln(Pc)2 with a D4d symmetry. The LF potential around a Tb3+ ion (7F6) with a total angular momentum (J) of 6 splits the ground-state multiplet so that the lowest sublevel has the largest z-component of J (mJ = ±6) and a large energy gap (ca. 400 cm−1) to the next-closest sublevel (mJ = ±5), which is correlated with Ueff (Fig. 10d).36 In 2003, Ishikawa et al. first reported the SMM properties of (NBu4+)[Tb(Pc)2]−, which contains a single rare-earth 4f ion (Ueff = 230 cm−1; τ0 = 6.25 × 10−8 s; HC ≈ 0.01 T@0.04 K).36,276 For the neutral (one-electron-oxidized) species  with an unpaired π-radical (S = 1/2) on the Pc ligands, the Ueff is 410 cm−1.277 In comparison to (NBu4+)[Tb(Pc)2]− and
with an unpaired π-radical (S = 1/2) on the Pc ligands, the Ueff is 410 cm−1.277 In comparison to (NBu4+)[Tb(Pc)2]− and  , the cationic species Tb[(EtO)8Pc]2+ has a much larger Ueff (550 cm−1).84 The cationic species has a shorter interligand distance than the neutral and anionic ones because the electrons are removed from antibonding molecular orbitals.84 In addition, structural changes in Tb[(BuO)8Pc]2+ are induced by oxidation from its neutral/anionic species, whereby the interligand distance decreases upon oxidation (cf. Section 3.4).278 Thus, LF contractions cause strong LF splitting, which directly affects the Ueff value. This result quantified LF contractions in the cationic species. Later, the group of complexes that exhibits SMM characteristics with a mononuclear d/f metal ion also became known as single-ion magnets (SIMs). In particular, Ln(Pc)2-based SMMs are attractive complexes that can be developed using various approaches, because it is possible to control the distance between the Ln3+ ions, the coordination environment, and the electronic state by chemically modifying the Pc ligands.3,4,279 It should be mentioned here that the relationship between the magnitude of the LF splittings and the Ueff is not straightforward. In case of Tb(Pc)2, the Ueff coincides with the energy difference between ground mJ = ±6 and 1st excited mJ = ±5 states (Fig. 10d). The reason why the magnetic relaxations via 2nd or higher LF level as seen in Mn12 do not occur in the series of Tb(Pc)2 remains unclear to date. However, recent research studies on the SMMs indicate the importance of the vibrations of atoms and molecules in the crystalline lattice.248,280,281 For instance, SMMs with a high Ueff (1541 cm−1) and record TB value (80 K) were reported by Guo et al. in 2018, which was achieved by modifying the vibrational modes upon ligand modification of the [Dy(Cp)2]+ (Cp = cyclopentadienyl) complex.234,282,283 The high Ueff of this complex originates from the Orbach process via 4th excited LF levels, meaning that those via lower excited states are effectively suppressed. The control of the coupling between spin and vibrations is therefore extremely important for achieving high-performance SMMs.
, the cationic species Tb[(EtO)8Pc]2+ has a much larger Ueff (550 cm−1).84 The cationic species has a shorter interligand distance than the neutral and anionic ones because the electrons are removed from antibonding molecular orbitals.84 In addition, structural changes in Tb[(BuO)8Pc]2+ are induced by oxidation from its neutral/anionic species, whereby the interligand distance decreases upon oxidation (cf. Section 3.4).278 Thus, LF contractions cause strong LF splitting, which directly affects the Ueff value. This result quantified LF contractions in the cationic species. Later, the group of complexes that exhibits SMM characteristics with a mononuclear d/f metal ion also became known as single-ion magnets (SIMs). In particular, Ln(Pc)2-based SMMs are attractive complexes that can be developed using various approaches, because it is possible to control the distance between the Ln3+ ions, the coordination environment, and the electronic state by chemically modifying the Pc ligands.3,4,279 It should be mentioned here that the relationship between the magnitude of the LF splittings and the Ueff is not straightforward. In case of Tb(Pc)2, the Ueff coincides with the energy difference between ground mJ = ±6 and 1st excited mJ = ±5 states (Fig. 10d). The reason why the magnetic relaxations via 2nd or higher LF level as seen in Mn12 do not occur in the series of Tb(Pc)2 remains unclear to date. However, recent research studies on the SMMs indicate the importance of the vibrations of atoms and molecules in the crystalline lattice.248,280,281 For instance, SMMs with a high Ueff (1541 cm−1) and record TB value (80 K) were reported by Guo et al. in 2018, which was achieved by modifying the vibrational modes upon ligand modification of the [Dy(Cp)2]+ (Cp = cyclopentadienyl) complex.234,282,283 The high Ueff of this complex originates from the Orbach process via 4th excited LF levels, meaning that those via lower excited states are effectively suppressed. The control of the coupling between spin and vibrations is therefore extremely important for achieving high-performance SMMs.
There have been many attempts, both experimental and theoretical, to elucidate the relationship between coordination geometry and LF parameters, which have a great influence on the ground-state multiplet structure and magnetic anisotropy (cf. Sections 3.2.1 and 3.2.2).229 Contractions of square antiprismatic (SAP) coordination environments affect the LF of Ln3+ ions, thus bringing about highly significant changes in the SMM properties.36,84,233,242,252,276,284–287 In general, the LF (or CF) parameter in the Hamiltonian is defined as follows:

Ln(Pc)2 exhibits excellent thermal stability and is attracting attention as an SMM to which the vacuum deposition method can be applied; they can be deposited on various substrates in an ultra-high vacuum (UHV) environment. Ln(Pc)2-based SMMs have excellent physical and chemical stability, and many groups, including ours, are investigating these SMMs as molecular-memory and spintronic device materials (cf. Section 3.8).117,135,232,267,299–306 The unpaired π-radical (S = 1/2) on the Pc ligands in Tb(Pc)2 interacts with the conduction electrons, and Kondo resonance has been observed using scanning tunnelling spectroscopy (STS).302 Furthermore, the Kondo resonance largely depends on the twist angle (θ) of the tetrapyrrole ligand and the electronic structure due to the delocalized π spin state, demonstrating the manipulation of a single spin with a conduction electron.135,302,307 In addition, Ln-Pc-based SMMs are attractive materials for quantum mechanical devices that can be developed based on the separation between the ground-state doublet mJ and the excited states, as well as the hyperfine (hf) interaction between the electronic spin and the nuclear spin I, because it is possible to control the hyperfine QTM (hf-QTM).269,270,303,308–315 The important point here is that each spin (S, J, I) in Tb(Pc)2 shows a cascade phenomenon through spin interactions. Here, Tb(Pc)2 incorporated in a transistor structure is in contact with the source, drain, and gate electrode. The various spins (S, J, I) interact with the injected conduction electrons via the delocalized π electrons, and the conduction electrons recognize changes in the hf-QTM state during magnetic field sweeps.270 This represents a readout mechanism for the nuclear spin state, in which the transport properties of Tb(Pc)2 are controlled by the conduction electrons. Thus, Tb(Pc)2 SMMs have all the properties necessary to serve as a functional component of quantum-mechanical devices (Fig. 12).
 | ||
| Fig. 12 Molecular qubit based on a Tb(Pc)2 SMM. (a) Schematic diagram of the qubit detection. Tb(Pc)2 is placed between Au electrodes. (b) In the Zeeman diagram of Tb(Pc)2, the strong hyperfine coupling between the mJ = ±6 and the nuclear spin (I = 3/2) forms four avoided level crossings with a tunnel gap (ΔE ≈ 1 μK) and four different qubit states (|–3/2〉, |–1/2〉, |+1/2〉, and |+3/2〉). (c) Quantum tunnelling phenomenon due to hyperfine coupling in which the nuclear spin is observed in the magnetisation curve of Tb(Pc)2. Reprinted from ref. 308 with permission from the Royal Society of Chemistry. | ||
| ĤLF = B02Ô02 + B04Ô04 + B44Ô44 + B06Ô06 + B46Ô46 |
 :
:| ĤLF = B02Ô02 + B04Ô04 + B06Ô06. |
Therefore, there is no mixing between |mJ〉 and 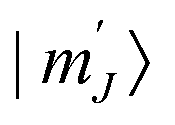 in the ideal SAP geometry, suppressing QTM. It is not easy to construct the SP geometry in double-decker complexes without replacing the Ln3+ ions.316 Instead, SP and SAP geometries around Ln3+ ions have been realized by using porphyrinato–phthalocyaninato triple-decker complexes (Fig. 13).
in the ideal SAP geometry, suppressing QTM. It is not easy to construct the SP geometry in double-decker complexes without replacing the Ln3+ ions.316 Instead, SP and SAP geometries around Ln3+ ions have been realized by using porphyrinato–phthalocyaninato triple-decker complexes (Fig. 13).
 | ||
| Fig. 13 X-ray structures of mixed ligand complexes, showing coordination surrounding symmetries for different metal centres: (a) (Pc)Tb(Pc)Tb(TAnP) (CCDC SAZLIQ);122 (b) (TPP)Tb(Pc)Tb(Pc) (CCDC IJOSUX)317 and (c) (TClPP)Dy[(PhO)8Pc]Dy[(PhO)8Pc] (CCDC MIKSEG).318 Hydrogen atoms and solvate molecules are omitted for clarity. | ||
In 2012, Sakaue et al. reported the crystal structure and SMM properties of the heteroligand triple-decker complex (TAnP)Ln(Pc)Ln*(Pc), Ln* = Ln = Tb, or Ln* ≠ Ln = Tb, Y (Scheme 20 and Fig. 13a).122 The metal center sandwiched by two Pc ligands (Ln*) shows SAP geometry, whereas that sandwiched by Pc and TAnP, namely, Ln, shows SP geometry. It is possible to synthesize heterometal complexes (TAnP)Y(Pc)Tb(Pc) and (TAnP)Tb(Pc)Y(Pc), in which one of the metal ions is replaced with a diamagnetic and isostructural Y3+ ion. The Tb3+ ion of (TAnP)Y(Pc)Tb(Pc) adopts SAP geometry, whereas that of (TAnP)Tb(Pc)Y(Pc) adopts SP geometry. The maxima of the imaginary part of the AC magnetic susceptibility  for (TAnP)Y(Pc)Tb(Pc) occur at much lower frequencies than those of (TAnP)Tb(Pc)Y(Pc) (Fig. 14b). In other words, (TAnP)Y(Pc)Tb(Pc) shows much longer τ values than (TAnP)Tb(Pc)Y(Pc). These differences have been ascribed to the differences in the coordination geometry around the Tb3+ ions. As the Tb3+ ion of (TAnP)Y(Pc)Tb(Pc) adopts SAP geometry, mixing between LF sublevels and the accompanying QTM is suppressed, which results in long τ values.
for (TAnP)Y(Pc)Tb(Pc) occur at much lower frequencies than those of (TAnP)Tb(Pc)Y(Pc) (Fig. 14b). In other words, (TAnP)Y(Pc)Tb(Pc) shows much longer τ values than (TAnP)Tb(Pc)Y(Pc). These differences have been ascribed to the differences in the coordination geometry around the Tb3+ ions. As the Tb3+ ion of (TAnP)Y(Pc)Tb(Pc) adopts SAP geometry, mixing between LF sublevels and the accompanying QTM is suppressed, which results in long τ values.
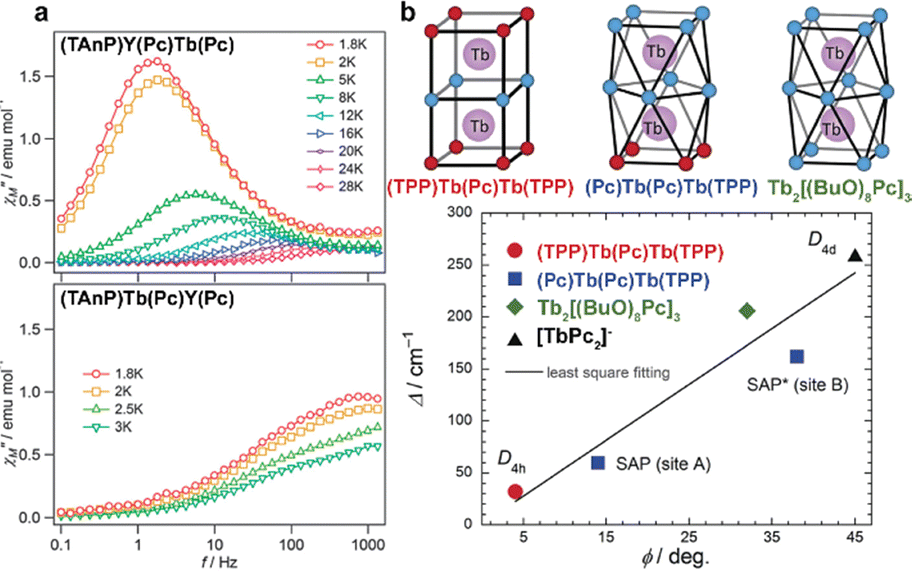 | ||
Fig. 14 (a)  vs. frequency plots for (TAnP)Y(Pc)Tb(Pc) (upper) and (TAnP)Tb(Pc)Y(Pc) (lower). (b) Coordination geometry and θ vs. Ueff plots for the series of Tb3+ triple-decker complexes. Reproduced from (a) ref. 122 and (b) ref. 317 with permission from the Royal Society of Chemistry. vs. frequency plots for (TAnP)Y(Pc)Tb(Pc) (upper) and (TAnP)Tb(Pc)Y(Pc) (lower). (b) Coordination geometry and θ vs. Ueff plots for the series of Tb3+ triple-decker complexes. Reproduced from (a) ref. 122 and (b) ref. 317 with permission from the Royal Society of Chemistry. | ||
In 2016, Katoh et al. reported a detailed study on the crystal structures and the SMM properties of triple-decker complexes composed of two Tb3+ ions, TPP ligands, and Pc.317 In case of the triple-decker complex (TPP)Tb(Pc)Tb(TPP), the coordination geometry of the two Tb3+ ions is close to the SP geometry (θ = 4°), and the Ueff is ca. 50 cm−1. Another triple-decker complex, (Pc)Tb(Pc)Tb(TPP), showed two different types of coordination geometry around the Tb3+ ions (Fig. 14b), and thus two peaks in the  vs. T plots. The coordination geometry around the Tb3+ (Fig. 13b) sandwiched by two Pc ligands is similar to the SAP geometry (θ = 38°), whereas that of the Tb3+ sandwiched by a TPP and a Pc ligand is similar to the SP geometry (θ = 14°). The θ value of the phthalocyaninato triple-decker complex Tb2[(BuO)8Pc]3 is 32°.319 By comparing the magnetic properties of this series of triple-decker complexes, Katoh et al. demonstrated the existence of a linear relationship between the Ueff and θ values (Fig. 14b). This result is consistent with the degree of mixing of the off-diagonal LF terms (B44Ô44 and B46Ô46) due to the deviation from the SAP geometry.
vs. T plots. The coordination geometry around the Tb3+ (Fig. 13b) sandwiched by two Pc ligands is similar to the SAP geometry (θ = 38°), whereas that of the Tb3+ sandwiched by a TPP and a Pc ligand is similar to the SP geometry (θ = 14°). The θ value of the phthalocyaninato triple-decker complex Tb2[(BuO)8Pc]3 is 32°.319 By comparing the magnetic properties of this series of triple-decker complexes, Katoh et al. demonstrated the existence of a linear relationship between the Ueff and θ values (Fig. 14b). This result is consistent with the degree of mixing of the off-diagonal LF terms (B44Ô44 and B46Ô46) due to the deviation from the SAP geometry.
The trend for Dy3+ complexes is similar to that for Tb3+ complexes. In 2013, Kan et al. reported a series of Dy3+ triple-decker complexes composed of phthalocyaninato and porphyrinato ligands (Fig. 13c).318 The θ value of the Ln3+ between a phthalocyaninato and a porphyrinato ligand is narrow (10°), whereas the θ value of Ln3+ surrounded by two phthalocyaninato ligands is wide (25°). The mono-Dy3+ complex with a wide θ value, (TClPP)Y[(PhO)8Pc]Dy[(PhO)8Pc], shows slow magnetic relaxation without a bias DC magnetic field. In contrast, (TClPP)Dy[(PhO)8Pc]Y[(PhO)8Pc] does not show slow magnetic relaxation even in a DC field. The homonuclear Dy3+ complex (TClPP)Dy[(PhO)8Pc]Dy[(PhO)8Pc] shows field-induced SMM properties. These results demonstrate that wider θ values improve the τ values.
In 2012, Wang et al. reported the structural and magnetic properties of the Dy3+ double-decker complexes (TClPP)Dy(Pc) and (TClPP)Dy[(α-Et2CHO)4Pc] (Fig. 15).320 The θ value of (TClPP)Dy(Pc) is 44°, whereas that of (TClPP)Dy[(α-Et2CHO)4Pc] is 38°. (TClPP)Dy(Pc) shows clear peaks in the  vs. T plots in comparison with (TClPP)Dy[(α-Et2CHO)4Pc], indicating that QTM is suppressed in (TClPP)Dy(Pc) due to the SAP coordination geometry.
vs. T plots in comparison with (TClPP)Dy[(α-Et2CHO)4Pc], indicating that QTM is suppressed in (TClPP)Dy(Pc) due to the SAP coordination geometry.

In 2013, Ganivet et al. reported a series of Tb3+ double-decker complexes composed of octa(p-tert-butyl phenoxy)phthalocyanine H2[(tBuPhO)8Pc], tetra(tert-butyl)phthalocyanine H2(tBu4Pc) and unsubstituted phthalocyanine H2(Pc).49 The heteroleptic ligand complexes in which one of the Pc ligands is unsubstituted show higher Ueff values (642–652 cm−1) than the homoleptic ones (394–504 cm−1). Ganivet et al. proposed that electron-donating substituents increase the Tb–N distances, thus pushing the Tb3+ toward the unsubstituted phthalocyanine and hence increasing the Ueff. This concept was subsequently extended by Chen et al. in 2017. These authors reported a high Ueff value (652 cm−1) and a magnetic hysteresis of up to 30 K at a sweep rate of 200 Oe s−1 for the double-decker complex [(Bu2N)8Pc]Tb(Pc), in which one of the Pc ligands is substituted with alkylamino groups (Scheme 8).81 DFT calculations of the optimized structures for [(Bu2N)8Pc]Tb(Pc) and the corresponding homoligand complex Tb[(Bu2N)8Pc]2 revealed that the electrostatic potential of (Bu2N)8Pc is greater in the heteroligand Pc/(Bu2N)8Pc complex than in the homoligand (Bu2N)8Pc complex (Fig. 16a). This result supports the enhancement of Ueff in heteroligand complexes.
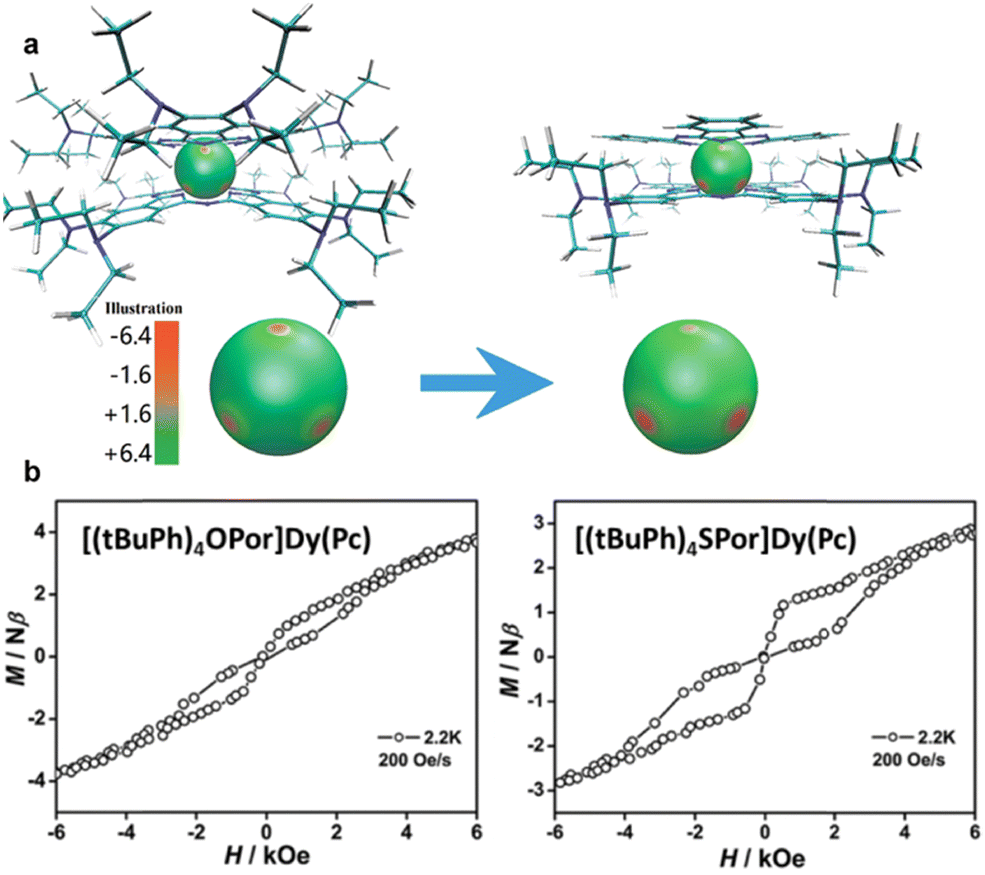 | ||
| Fig. 16 (a) Electrostatic potential around Tb3+ in the Tb[(Bu2N)8Pc]2 and [(Bu2N)8Pc]Tb(Pc) double-decker complexes. (b) Magnetic hysteresis curves for atom-replaced double-decker complexes. [(tBuPh)4SPor]Dy(Pc) show larger magnetic hysteresis (right) than [(tBuPh)4OPor]Dy(Pc) (left) at a sweep rate of 200 Oe s−1. Adapted with permission from (a) ref. 81. Copyright 2017, American Chemical Society. Reproduced from (b) ref. 173 with permission from the Royal Society of Chemistry. | ||
The SMM properties of the porphyrazine (Pz)-based double-decker complexes Tb(OEPz)2 and Dy(OEPz)2 were reported by Gimnez et al. (Scheme 25).140 The Ueff values of Tb(OEPz)2 (266 cm−1) and Dy(OEPz)2 (22 cm−1) are similar to those of their Pc analogues. However, Pz complex benefits from its Pc counterpart due to easier processing. They have a higher solubility of up to 50 mM in CH2Cl2 and they can be readily sublimed under relatively mild conditions, that is, 360 °C and 10 mbar. Tb(OEPz)2 and Dy(OEPz)2 were evaporated using organic molecular beam epitaxy at 550 K and absorbed intact on the surfaces of Au(111), Ag(111) and Cu(111) in a flat-on fashion, forming hexagonal close-packed layers.322 Systematic STM studies with multi- and monolayer XPS and complementary DFT calculations were performed showing the preservation of the electronic properties of both ligands and lanthanide centres upon adsorption. Persistence of slow dynamics in Tb(OEPz)2 single molecule magnets embedded in conducting polymers was confirmed by muon spin relaxation on the example of thin films of PEDOT:PSS mixed with the complex, suggesting that the resulting hybrid material can be interesting for organic spintronics.323
Replacing the coordinating N atoms of the porphyrinoid with another element modulates the LF splitting of the Ln3+ ions. In 2015, Cao et al. reported the magnetic properties of [(tBuPh)4OPor]Dy(Pc) and [(tBuPh)4SPor]Dy(Pc), in which one of the coordinating N atoms of the porphyrin unit is replaced with an O or an S atom, respectively (Scheme 32 and Fig. 16b).173 Importantly, the Ueff values of [(tBuPh)4OPor]Dy(Pc) (95 cm−1) and [(tBuPh)4SPor]Dy(Pc) (135 cm−1) are larger than those of the non-replaced analogue [(tBuPh)4Por]Dy(Pc) (Ueff = 28 cm−1). In addition, the Ueff of [(tBuPh)4SPor]Dy(Pc) is the largest among the Dy3+-porphyrinoid sandwich complexes. The difference in the Ueff values between [(tBuPh)4OPor]Dy(Pc) and [(tBuPh)4SPor]Dy(Pc) are also evidenced by the magnetic hysteresis curve, in which the latter complex exhibits larger magnetic hysteresis than former complex does (Fig. 16c). Ab initio calculations revealed that the ground-state doublet of [(tBuPh)4Por]Dy(Pc) has the second largest mJ value of ±13/2, whereas those of [(tBuPh)4OPor]Dy(Pc) and [(tBuPh)4SPor]Dy(Pc) have an mJ value of ±15/2. The coordination of the O and S atoms to Dy3+ is weaker than that of the N atoms, as evident from the experimental bond lengths and calculated charge distributions. The decrease in the LF along the Dy3+–X (X = O, S) direction stabilizes the mJ = ±15/2 states and thus enhances the SMM properties.
In 2018, the SMM properties of hemiporphyrazinato double-decker complexes (Scheme 37) were reported by Liu et al.189 The homoleptic bis(hemiporphyrazinato) complex (HHp)Dy(Hp) and heteroleptic complex (HHp)Dy(Pc) show Ueff values of 56 cm−1 and 40 cm−1, respectively. These Ueff values are larger than those of Pc-based Dy3+ double-decker complexes. The lone electron pair on N in the pyridine in Hp is more localized in comparison with that of the isoindole N atom of Pc. As a result of the stronger LF of Hp compared to that of Pc, the SMM properties of the Hp-based complexes are superior to those of the Pc-based complexes.
SMMs that combine porphyrinoid and non-porphyrinoid ligands are rare. In 2013, Wang et al. reported triple-decker SMMs composed of Pc and the Schiff-base ligand SB. Reaction of the half-sandwich complex [(Pc)Dy(acac)] with H2(SB) affords the triple-decker complex (Pc)2Dy2(SB)(H2O) (Scheme 33), which shows a Ueff value of 19.8 cm−1.176 The two Dy3+ ions of (Pc)2Dy2(SB)(H2O) are crystallographically independent, whereby one adopts a SAP geometry that comprises four N atoms of Pc and two N and two O atoms of SB, while the other adopts a seven-coordinated geometry constructed by four N atoms of Pc, two O atoms of SB, and one oxygen atom of a water molecule. In 2014, Gao et al. reported a similar triple-decker SMM, Dy2(Pc)(SB-TTF)2(MeOH) (Scheme 34a), with a Ueff value of 20 cm−1, which forms assembled structures on highly oriented pyrolytic graphite (HOPG).179 The same author reported the SMM properties of the trinuclear Dy3+ complex Dy3(Pc)2(mS)(OAc)(OMe)2 (Scheme 34b) composed of two Pc ligands and macrocyclic Schiff base liagnd(mS), which exhibits zero-field SMM behavior with a Ueff value of 42 cm−1 and ferromagnetic interactions. In contrast, its Er3+ analogue Er3(Pc)2(mS)(OAc)(OMe)2 exhibts neither SMM properties nor ferromgnetic interactions.180 A similar analogue of the dinuclear Dy3+ complexes (μ-LR)2Dy2(Pc)2(H2O) composed of salicyclic aldehydes H(LR) (R = OMe and OEt) and Pc reported by Ge et al. (Scheme 39) exhibits zero-field SMM behavior with the Ueff values ranging from 27 (R = OEt in the presence of a DC magnetic field) to 47 cm−1 (R = OMe).201 The MeO-substituted complex exhibits better SMM properties than the EtO-subsituted one does because coordination geometry of the latter one deviates from the SAP geometry, which is caused by a long bond length between the ethocy O atom and Dy3+ ion. As the result of difference in coordination geometry and LF, methoxy-substituted complex exhibits intramolecular ferromagnetic interactions between two Dy3+ ions. Reaction of the half-sandwich complex [Dy(Pc)(OAc)] with the polyoxotungstate [PW11O39]6− affords (Pc)Dy[PW11O39]6−(NBu4+)6, which shows a Ueff value of 34 cm−1 at a bias DC field of 500 Oe (Scheme 38c).193 SMMs composed of Kläui's tripodal ligand and a porphyrinoid, (LCo)Ln(Pc) and (LCo)Ln(TPP) (Ln = Tb and Dy) (Scheme 38a and b), were reported by Gao et al.,190 and their Ueff values ranged from 6 to 17 cm−1.
 | ||
| Fig. 17 Field dependence of the magnetisation at 0.03 K with the field applied parallel to the easy axis of magnetisation for (a) (Et4N+)[163Dy0.05Y0.95(Pc)2]− (I = 5/2) and (b) (Et4N+)[164Dy0.05Y0.95(Pc)2]− (I = 0). Insets show magnifications in the ±30 mT region. (c) Crystal structure of triple-decker complex (Pc)Tb(Pc)Tb[(Hex)8Pc]. (d) Hysteresis loop of (Pc)Tb(Pc)Tb[(Hex)8Pc] recorded with a field sweep of 0.140 T s−1. (e) First field derivative for a field sweep from −1 to +1 T of the data in (d), and (f) Zeeman diagram with the field parallel to the magnetic easy axes. Adapted with permission from (a and b) ref. 311. Copyright 2017, Wiley-VCH. Adapted with permission from (c–f) ref. 314. Copyright 2018, American Chemical Society. | ||
Nuclear-spin-driven QTM has also been observed in the Tb3+ triple-decker complex (Pc)Tb(Pc)Tb[(Hex)8Pc], which contains two Tb3+ ions (Tb(1) and Tb(2)) (Fig. 17c–f).314 As will be discussed later, magnetic interactions exist between the Tb3+ ions in the triple-decker complex (cf. Section 3.2.4). Therefore, QTM occurs among |m1J, m1I, m2J, m2I〉. The number of possible quantum states d for the triple-decker complex (d = (2I + 1)2 = 16) is larger than that for [Tb(Pc)2]− (d = (2I + 1)1 = 4). The strategy of using Ln3+–Ln3+ interactions is promising for the construction of multiple qubits, which is required for quantum computers.
where

 ,
,  and
and 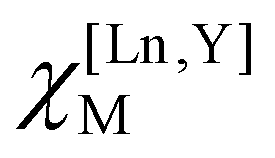 represent the χM values for [Ln,Ln′], [Y,Ln′], and [Ln,Y], respectively. Ishikawa et al. revealed that the behaviour of the ΔχMT values can be explained solely by the magnetic dipolar term, indicating that the magnetic f–f interactions originate from magnetic dipole–dipole interactions. Increases in the ΔχMT in Tb3+, Dy3+, and Ho3+ complexes with decreasing temperature indicate the existence of ferromagnetic interactions. On the other hand, the Er3+ and Tm3+ complexes show antiferromagnetic interactions, which were observed as decreases in ΔχMT. The ΔχMT values of the Yb3+ complex showed only negligible temperature dependence. This complicated behaviour can be interpreted in terms of dipole–dipole interactions. As discussed in the section on double-decker complexes (cf. Section 3.2), Tb3+, Dy3+, and Ho3+ ions show easy-axis magnetic anisotropy when sandwiched by Pc ligands. In other words, the magnetic dipoles of these ions in the [Ln,Ln′] complexes tend to be aligned along the C4 axis (perpendicular to the Pc plane). Therefore, head-to-tail ferromagnetic arrangement of the magnetic dipoles is favored in Tb3+, Dy3+, and Ho3+ complexes. In contrast, Er3+ and Tm3+ sandwiched by Pc ligands show easy-plane magnetic anisotropy. As a result, parallel and antiferromagnetic arrangement of the magnetic dipoles is favored in Er3+ and Tm3+ complexes. The ΔχMT of the Yb3+ complex is less temperature dependent than those of other [Ln,Ln′] complexes because of the weak magnetic anisotropy of Yb3+. The use of diamagnetic and isostructural Y3+ ions demonstrated by Ishikawa et al. is a smart method to accurately determine the effect of magnetic Ln3+–Ln3+ interactions that has been used for a wide variety of Ln3+-based SMMs.
represent the χM values for [Ln,Ln′], [Y,Ln′], and [Ln,Y], respectively. Ishikawa et al. revealed that the behaviour of the ΔχMT values can be explained solely by the magnetic dipolar term, indicating that the magnetic f–f interactions originate from magnetic dipole–dipole interactions. Increases in the ΔχMT in Tb3+, Dy3+, and Ho3+ complexes with decreasing temperature indicate the existence of ferromagnetic interactions. On the other hand, the Er3+ and Tm3+ complexes show antiferromagnetic interactions, which were observed as decreases in ΔχMT. The ΔχMT values of the Yb3+ complex showed only negligible temperature dependence. This complicated behaviour can be interpreted in terms of dipole–dipole interactions. As discussed in the section on double-decker complexes (cf. Section 3.2), Tb3+, Dy3+, and Ho3+ ions show easy-axis magnetic anisotropy when sandwiched by Pc ligands. In other words, the magnetic dipoles of these ions in the [Ln,Ln′] complexes tend to be aligned along the C4 axis (perpendicular to the Pc plane). Therefore, head-to-tail ferromagnetic arrangement of the magnetic dipoles is favored in Tb3+, Dy3+, and Ho3+ complexes. In contrast, Er3+ and Tm3+ sandwiched by Pc ligands show easy-plane magnetic anisotropy. As a result, parallel and antiferromagnetic arrangement of the magnetic dipoles is favored in Er3+ and Tm3+ complexes. The ΔχMT of the Yb3+ complex is less temperature dependent than those of other [Ln,Ln′] complexes because of the weak magnetic anisotropy of Yb3+. The use of diamagnetic and isostructural Y3+ ions demonstrated by Ishikawa et al. is a smart method to accurately determine the effect of magnetic Ln3+–Ln3+ interactions that has been used for a wide variety of Ln3+-based SMMs.
 | ||
| Fig. 18 Molecular structures and ΔχMT values of (Pc)Ln(Pc)Ln′[(BuO)8Pc] denoted as [Ln,Ln′]. The red curves show theoretical data. Adapted with permission from ref. 298. Copyright 2003, American Chemical Society. | ||
The next question is how long the Ln3+–Ln3+ distance must be in order to induce f–f interactions. The Ln3+–Ln3+ distance of the Pc triple-decker complex estimated using DFT calculations is 3.57 Å (this value is consistent with the crystal structure analyses for triple-decker analogues).298 Increasing the Ln3+–Ln3+ distance should affect the magnitude of the f–f interactions.
In 2010, Fukuda et al. reported the first example of a discrete quadruple-decker complex composed of four Pc ligands.202 The reaction of the anionic Lu3+ double-decker complex [Ln(Pc)2]− with Cd2+ acetate afforded (Pc)Lu(Pc)Cd(Pc)Lu(Pc), which is referred to as [LnCdLn] (Fig. 19a). As the two Ln3+ ions are separated by a diamagnetic Cd2+ ion, the Ln3+–Ln3+ distance is longer than those in triple-decker complexes. Fukuda et al. investigated the Ln3+–Ln3+ interactions in [LnCdLn] (Ln = Tb, Dy, Er)325 using the same method as for the series of [Ln,Ln′] complexes298 and revealed the existence of f–f interactions, although their magnitude was low compared to those observed in [Ln,Ln′]. In the case of [TbCdTb] and [DyCdDy], an increase in ΔχMT is observed, indicating intramolecular ferromagnetic interactions between the Ln3+ ions (Fig. 19b). As discussed in the section on [Ln,Ln′], the head-to-tail arrangement of the magnetic dipoles is stabilized when axially anisotropic Ln3+ ions (Tb3+ and Dy3+) are involved. On the other hand, [ErCdEr] shows a decrease in ΔχMT due to the easy-plane magnetic anisotropy of the Er3+ ions.
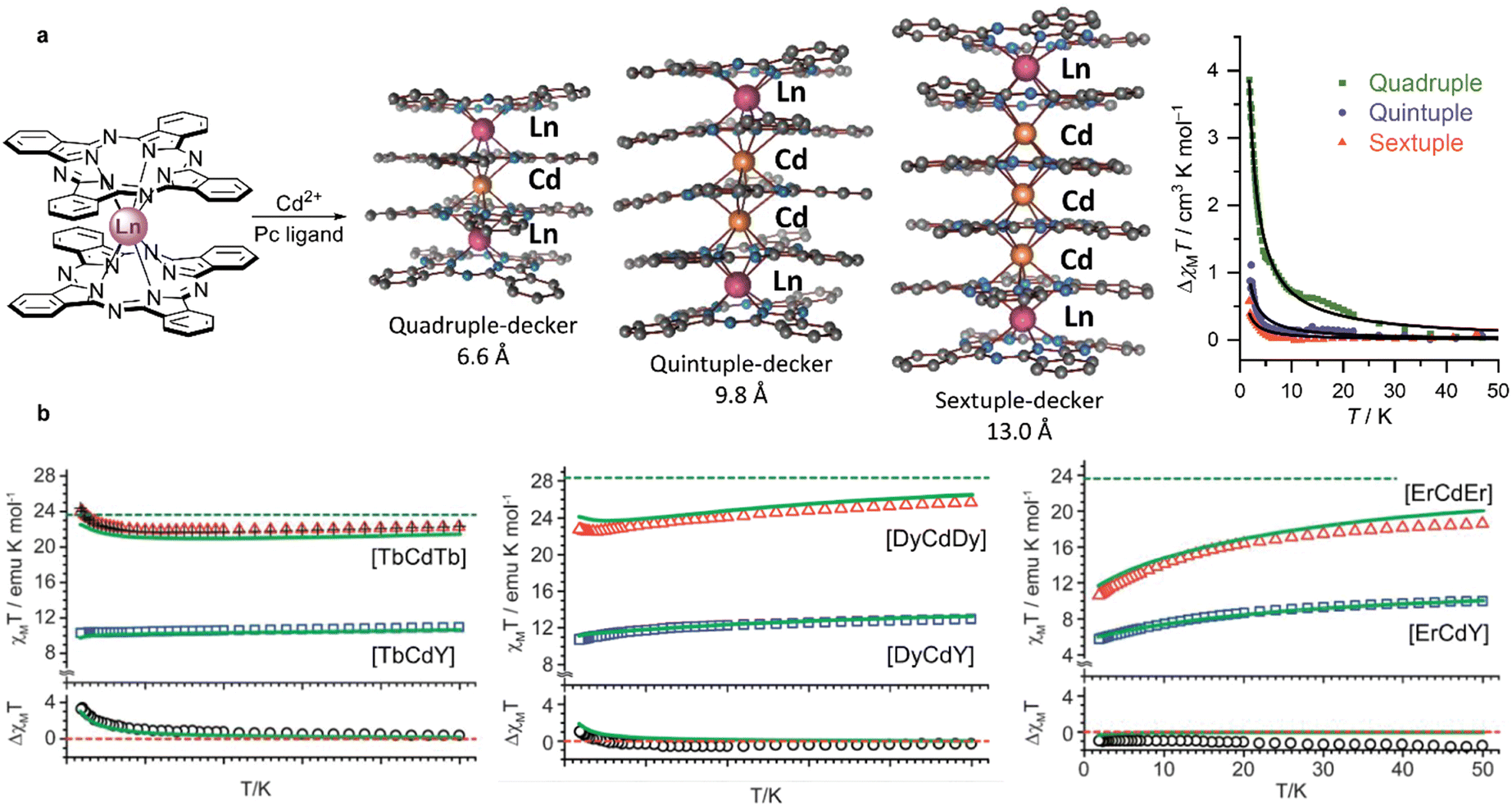 | ||
| Fig. 19 (a) Synthesis and crystal structures of a series of phthalocyaninato multiple-decker complexes; representative molecular crystal structures of the Dy3+ complexes Dy2Cd[(BuO)8Pc]4, Dy2Cd2[(BuO)8Pc]5 and Dy2Cd3[(BuO)8Pc]6; n-butoxy groups in the crystal structures are omitted for clarity. ΔχMT vs. T plots for the Dy3+ multiple-decker complexes were constructed using data from ref. 211, 213 and 326. (b) DC magnetic susceptibilities of quadruple-decker complexes that contain Tb3+, Dy3+ and Er3+ ions. Reproduced from (b) ref. 325 with permission from the Royal Society of Chemistry. | ||
Wang et al. and Katoh et al. have reported the crystal structures of Dy3+ and Tb3+ quadruple-decker complexes composed of (BuO)8Pc ligands, Tb2Cd[(BuO)8Pc]4 and Dy2Cd[(BuO)8Pc]4.205,211 On account of the low solubility of the quadruple-decker complexes composed of unsubstituted Pc ligands, peripheral n-butoxy substitution is mandatory to obtain single crystals suitable for X-ray diffraction analysis. The strategy of using Cd2+ ions to elongate the stacked π-system was further extended by Wang et al. Their group succeeded in obtaining a series of stacked π-systems: the quintuple-decker complexes Ln2Cd2[(BuO)8Pc]5208 and sextuple-decker complexes Ln2Cd3[(BuO)8Pc]6.209 Horii et al. reported the crystal structures of the Dy3+ multiple-decker complexes composed of [(BuO)8Pc] ligands (Fig. 19a) and confirmed that the Dy3+–Dy3+ distances are 9.8 Å for the quintuple-decker complex Dy2Cd2[(BuO)8Pc]5 and 13.0 Å for the sextuple-decker complex Dy2Cd3[(BuO)8Pc]6.213,326 Very weak f–f interactions exist in both complexes, as demonstrated by the increase in their ΔχMT values (Fig. 19a right). These f–f interactions can be used to alter their SMM properties, as discussed in the following section.
3.3 Using f–f interactions to improve SMM properties
In 2002, Wernsdorfer et al. reported the magnetic properties of a SMM dimer composed of the TM-based cluster Mn4.225 The step at zero DC magnetic field in the magnetic hysteresis curve of the monomeric SMM Mn4 disappears in the dimerized SMMs Mn4⋯Mn4 due to the suppression of QTM. The mechanism of the suppression of QTM can be explained as follows. If the probability of QTM between the up (|↑〉) and down spin (|↓〉) states of one SMM is PQTM, the QTM probability between the ferromagnetically coupled states (|↑,↑〉 and |↓,↓〉) in the SMM dimer is expressed as PQTM2, because QTM between these states requires simultaneous spin reversal. Since PQTM2 is much smaller than PQTM, QTM between the coupled spin states is a forbidden transition. Therefore, the coupling of two SMMs suppresses the QTM at zero bias DC magnetic field. This strategy (exchange bias effect) is important when considering SMMs as a possible candidate for information storage, because it can improve data stabilisation at zero DC field. The f–f interactions in multiple-decker complexes can also be used as a source of exchange bias. The effect of f–f interactions on the dynamic magnetic properties of rare-earth SMMs was reported by Ishikawa et al.327 They prepared the homodinuclear Tb3+ triple-decker complex (Pc)Tb(Pc)Tb[(BuO)8Pc] (denoted as [Tb,Tb]) and the two heterodinuclear complexes (Pc)Y(Pc)Tb[(BuO)8Pc] (denoted as [Y,Tb]) and (Pc)Tb(Pc)Y[(BuO)8Pc] (denoted as [Tb,Y]), as summarized in Fig. 18. The mono-Tb3+ complexes [Y,Tb] and [Tb,Y] show clear DC magnetic field (Hdc) dependences in their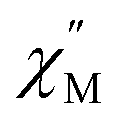 vs. T plots; the peak maxima shift to a higher temperature with increasing Hdc magnitude (Fig. 20a). In contrast, the
vs. T plots; the peak maxima shift to a higher temperature with increasing Hdc magnitude (Fig. 20a). In contrast, the  vs. T plots for [Tb,Tb] are Hdc-independent in the field range of 0 to 3000 Oe (the right side of Fig. 20a). The two peaks in the plots of [Tb,Tb] are due to the two inequivalent Tb3+ ions. The increase in the temperatures of the maxima of [Y,Tb] and [Tb,Y] corresponds to the suppression of QTM by applying an Hdc. The QTM occurs among the energetically degenerate states. However, applying an Hdc resolves the degeneracy of the ground-state doublet |±6〉 due to Zeeman splitting. Therefore, the QTM of [Y,Tb] and [Tb,Y] is suppressed with increasing Hdc, causing the remarkable Hdc dependences of
vs. T plots for [Tb,Tb] are Hdc-independent in the field range of 0 to 3000 Oe (the right side of Fig. 20a). The two peaks in the plots of [Tb,Tb] are due to the two inequivalent Tb3+ ions. The increase in the temperatures of the maxima of [Y,Tb] and [Tb,Y] corresponds to the suppression of QTM by applying an Hdc. The QTM occurs among the energetically degenerate states. However, applying an Hdc resolves the degeneracy of the ground-state doublet |±6〉 due to Zeeman splitting. Therefore, the QTM of [Y,Tb] and [Tb,Y] is suppressed with increasing Hdc, causing the remarkable Hdc dependences of  . In contrast, the Tb3+ ions of [Tb,Tb] are affected by the magnetic dipolar field from the other Tb3+ ion. Therefore, the Tb3+ ions of [Tb,Tb] act as if a DC magnetic field is applied even in the absence of an external Hdc. Therefore, the QTM in [Tb,Tb] is suppressed without an Hdc, resulting in weak Hdc dependence in its
. In contrast, the Tb3+ ions of [Tb,Tb] are affected by the magnetic dipolar field from the other Tb3+ ion. Therefore, the Tb3+ ions of [Tb,Tb] act as if a DC magnetic field is applied even in the absence of an external Hdc. Therefore, the QTM in [Tb,Tb] is suppressed without an Hdc, resulting in weak Hdc dependence in its  vs. T plots.
vs. T plots.
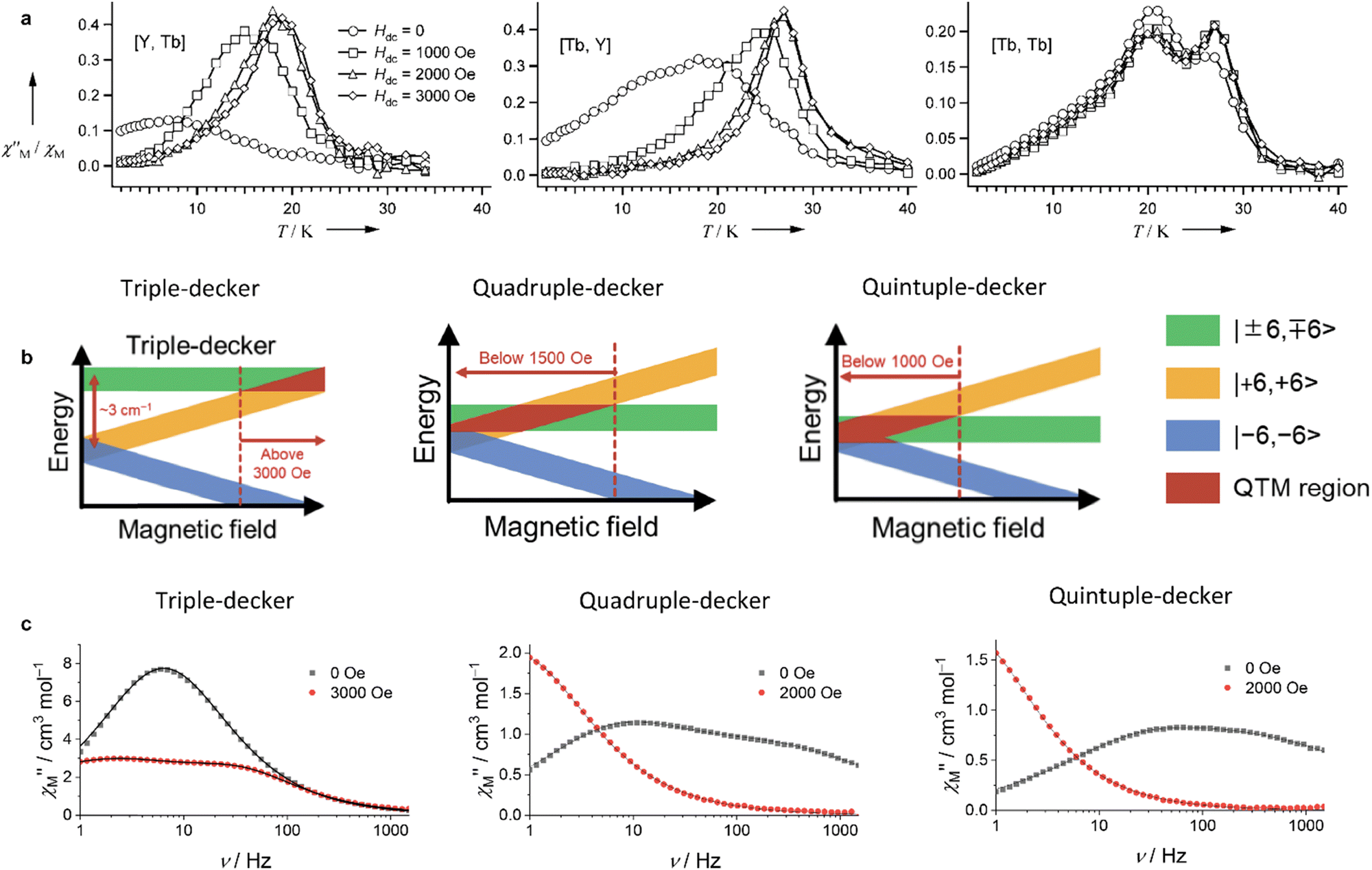 | ||
Fig. 20 (a) DC magnetic field (Hdc) dependences of the 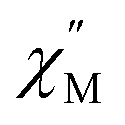 vs. T plots at an AC frequency of 997 Hz for the triple-decker complexes [Y,Tb], [Tb,Y] and [Tb,Tb]. (b) Schematic illustration of the Zeeman diagrams of Tb3+ multiple-decker complexes. (c) Hdc dependences of the vs. T plots at an AC frequency of 997 Hz for the triple-decker complexes [Y,Tb], [Tb,Y] and [Tb,Tb]. (b) Schematic illustration of the Zeeman diagrams of Tb3+ multiple-decker complexes. (c) Hdc dependences of the  vs. ν plots for the series of butoxy-substituted Tb3+ multiple-decker complexes at 4 K based on the data from ref. 319, 328 and 211. Adapted with permission from (a) ref. 327. Copyright 2005, Wiley-VCH. vs. ν plots for the series of butoxy-substituted Tb3+ multiple-decker complexes at 4 K based on the data from ref. 319, 328 and 211. Adapted with permission from (a) ref. 327. Copyright 2005, Wiley-VCH. | ||
The Zeeman diagrams of the triple-decker complexes reported by Sakaue et al. aid in the understanding of the relaxation mechanism of [Tb,Tb].122Fig. 20b is a schematic illustration of the Zeeman diagrams of various multiple-decker Tb3+ complexes.212 Due to the ferromagnetic dipole–dipole interactions between the Tb3+ ions in the triple-decker complex [Tb,Tb], the lowest energy states are |mJ, mJ〉 = |±6, ±6〉 at zero Hdc. The antiferromagnetically coupled doublet |±6, ∓6〉 is located at ca. 3 cm−1. Due to the hyperfine coupling between J and I, the Zeeman diagram of |±6, ±6〉 and |±6, ∓6〉 shows multiple lines, which are depicted as bands in Fig. 20b. At zero DC magnetic field, |+6, +6〉 and |−6, −6〉 are degenerate. However, QTM between |+6, +6〉 and |−6, −6〉 is prohibited because this transition requires simultaneous spin reversal. The Zeeman diagram also predicts energy crossing between |±6, ∓6〉 and |+6, +6〉 at Hdc = ±3000 Oe, indicating that the QTM between |±6, ∓6〉 and |+6, +6〉 is enhanced at 3000 Oe.
In the case of the quadruple-decker complexes, the magnitude of the f–f interactions decreases and the Hdc dependences of τ are changed. Fukuda et al. reported the Hdc dependences of the τ values for homonuclear ([TbCdTb]) and heteronuclear ([TbCdY]) quadruple-decker complexes.329[TbCdTb] has f–f interactions, whereas [TbCdY] does not. At zero Hdc, [TbCdTb] shows longer τ values than [TbCdY]. However, at Hdc = 1000 Oe, the τ values of [TbCdTb] become faster than those of [TbCdY]. Further increasing the Hdc causes the τ of both complexes to become similar. This complicated modulation of τ can again be explained using a Zeeman diagram. Since the magnetic easy axis of the Tb3+ ion in the multiple-decker complex is parallel to the C4 axis, the ferromagnetic head-to-tail arrangement of the magnetic dipoles is stabilized in [TbCdTb]. Therefore, the ground and first excited states of [TbCdTb] are |±6, ±6〉 and |±6, ∓6〉, respectively. Compared to the triple-decker complex, the energy difference between |±6, ±6〉 and |±6, ∓6〉 is smaller in [TbCdTb] due to the weaker f–f interactions. The ground-state doublet |±6, ±6〉 shows Zeeman splitting, whereas |±6, ∓6〉 does not. Due to the hyperfine interactions, |±6, ±6〉 and |±6, ∓6〉 are shown as crowded lines. At zero Hdc, the QTM in [TbCdTb] is suppressed due to the exchange bias effect, which prolongs the τ values. However, |+6, +6〉 and |±6, ∓6〉 cross at around 1000 Oe (middle plot in Fig. 20b). Therefore, the QTM between these is enhanced, decreasing the τ values of [TbCdTb]. This is the reason for the magnetic field dependences of the τ values in [TbCdTb] and [TbCdY]. A similar trend has also been observed in the Dy3+ analogues. Katoh et al. reported that the homonuclear Tb3+ triple-decker complex Tb2[(BuO)8Pc]3 shows two peaks in the imaginary part of the AC magnetic susceptibility in the presence of a non-zero Hdc (3000 Oe) as summarized in the left side of Fig. 20c.319 In contrast, quadruple-decker Tb2Cd[(BuO)8Pc]4328 and quintuple-decker Tb2Cd2[(BuO)8Pc]5211 complexes that contain two Tb3+ ions show dual magnetic relaxations at zero Hdc. Importantly, the dual magnetic relaxations become a single one in the presence of a non-zero Hdc (2000 Oe). The complicated magnetic behaviour in these complexes originates from the differences in the magnitude of the f–f interactions. The dual magnetic relaxations of Tb2[(BuO)8Pc]3 have been observed at 3000 Oe, where level-crossing between |+6, +6〉 and |±6, ∓6〉 occurs. Therefore, one of the dual magnetic relaxations in Tb2[(BuO)8Pc]3 is attributed to the QTM and spin-lattice relaxations among |+6, +6〉 and |±6, ∓6〉. In the case of Tb2Cd[(BuO)8Pc]4 and Tb2Cd2[(BuO)8Pc]5, |±6, ±6〉 and |±6, ∓6〉 show crossing at zero Hdc because of the weaker f–f interactions (small energy difference between |±6, ±6〉 and |±6, ∓6〉), as shown in Fig. 20b. Therefore, magnetic relaxations among these states occur and are observed as the dual magnetic relaxations at zero Hdc. Increasing Hdc resolves the degeneracy of |±6, ±6〉 and |±6, ∓6〉, resulting in the observation of single magnetic relaxation.
The effect of the f–f interactions on the dynamic magnetic properties has been reported in Dy3+ complexes. Katoh et al. reported the SMM properties of the triple-decker complexes Dy2[(BuO)8Pc]3 and DyY[(BuO)8Pc]3.330 Homonuclear Dy2[(BuO)8Pc]3 has intramolecular f–f interactions, whereas DyY[(BuO)8Pc]3 does not. The magnetic relaxation time τ of Dy2[(BuO)8Pc]3 is 1.30 × 10−4 s at 1.82 K. This value is one order of magnitude greater than that for DyY[(BuO)8Pc]3 (3.20 × 10−5 s at 1.9 K), indicating that f–f interactions act as an exchange bias to suppress the QTM at zero Hdc.
Horii et al. reported the exchange bias effect in Dy3+ quadruple-decker (Dy2Cd[(BuO)8Pc]4), quintuple-decker (Dy2Cd2[(BuO)8Pc]5), and sextuple-decker (Dy2Cd3[(BuO)8Pc]6) complexes.213,326 Similar to the triple-decker complexes composed of two Dy3+ ions, the series of multiple-decker complexes shows longer τ values than the mono-Dy3+ complex due to the f–f interactions in the former complex. The exchange bias effect is observed in Dy2Cd3[(BuO)8Pc]6, in which the intramolecular Dy3+–Dy3+ distance is ca. 13 Å.
The use of the clamshell-type phthalocyanine331 reported by Tolbin et al. enables the construction of a discrete stacked π-system without using metal coordination.104 Horii et al. reported a clamshell-type quadruple-decker complex (Pc)Tb[c-Pc2]Tb(Pc) in which the two Tb3+ double-decker units (tBu4Pc)Tb(Pc) are connected by a benzyloxy linker (Scheme 13 and Fig. 21a).105 Ferromagnetic f–f interactions are present between the two double-decker units, as evidenced by the increase in the χMT value at low temperatures because the head-to-tail arrangements of the magnetic dipoles are stabilized. The magnetic hysteresis of (Pc)Tb[c-Pc2]Tb(Pc) at a zero DC magnetic field is wider than that of the monomer double-decker complex (tBu4Pc)Tb(Pc), indicating that the exchange bias is effective in prolonging the τ values. The reaction of (Pc)Tb[c-Pc2]Tb(Pc) with Cd2+ acetate affords the Cd2+-containing complex (Pc)Tb[c-Pc2Cd]Tb(Pc). Although ferromagnetic interactions between the Tb3+ ions exist in (Pc)Tb[c-Pc2Cd]Tb(Pc), it shows a butterfly-shaped magnetic hysteresis curve in which the curve is closed at zero Hdc. These results indicate that the QTM is enhanced (increased PQTM value) by the coordination of a Cd2+ ion.
 | ||
Fig. 21 (a) Molecular structures and magnetic hysteresis curves of the clamshell-type quadruple-decker complexes. (b) Supramolecular dimerisation of the crown-ether-substituted SMM [(15C5)4Pc]Tb(Pc). The maxima in the  vs. ν plots shift toward the low ν region upon dimerisation. (c) Dimerisation of the double-decker SMM H[(α-BuO)8Pc]Tb(Pc) (CCDC FIJBEI) by Na+ ions yielding dimer Na2{[(α-BuO)8Pc]Tb(Pc)}2 (CCDC FIJBIM). Reproduced from (a) ref. 105 with permission from the Royal Society of Chemistry. Adapted with permission from (b) ref. 67. Copyright 2018, Wiley-VCH. vs. ν plots shift toward the low ν region upon dimerisation. (c) Dimerisation of the double-decker SMM H[(α-BuO)8Pc]Tb(Pc) (CCDC FIJBEI) by Na+ ions yielding dimer Na2{[(α-BuO)8Pc]Tb(Pc)}2 (CCDC FIJBIM). Reproduced from (a) ref. 105 with permission from the Royal Society of Chemistry. Adapted with permission from (b) ref. 67. Copyright 2018, Wiley-VCH. | ||
Horii et al. used supramolecular assembly reported by Ishikawa et al.332 to introduce f–f interactions.67 The crown-ether-substituted double-decker complex [(15C5)4Pc]Tb(Pc) forms a supramolecular quadruple-decker complex [K4{[(15C5)4Pc]Tb(Pc)}2]4+ in the presence of excess potassium acetate (Fig. 21b). Quantitative conversion from [(15C5)4Pc]Tb(Pc) to [K4{[(15C5)4Pc]Tb(Pc)}2]4+ was confirmed by paramagnetic 1H NMR analysis and absorption spectra. In addition, ferromagnetic Tb3+–Tb3+ interactions exist in [K4{[(15C5)4Pc]Tb(Pc)}2]4+. At 1.8 K, the τ value of [K4{[(15C5)4Pc]Tb(Pc)}2]4+ is 1000 times longer than that of [(15C5)4Pc]Tb(Pc), indicating that the exchange bias suppresses QTM.
Recently, the crystal structures of the triple-decker complexes dimerized via analogous supramolecular associations using crown ethers and K+ ions were elucidated by Martynov et al.333 The triple-decker complex [(15C5)4Pc]Tb(Pc)Tb(Pc) forms a supramolecular dimer in the presence of K+ ions (Fig. 22a). Such dimerization did not alter the symmetry of coordination surrounding of metal centres, and the Ln⋯Ln distances ranging from 3.5 to 13.2 Å renders these complexes as convenient models to study long-range interactions between paramagnetic metal centres aligned in a collinear fashion. In the case of [(15C5)4Pc]Tb[(15C5)4Pc]Tb(Pc), the reaction with four K+ ions does not induce dimerisation but instead changes the coordination geometry of Tb* from SAP to SP which was demonstrated by crystallographic characterization of isostructural Y3+ complex and its supramolecular inclusion compound (Fig. 22b).334 Therefore, the approach of using (15C5)4Pc is promising not only for introducing f–f interactions, but also for directly changing the LF. Although the SMM properties of [(15C5)4Pc]Tb(Pc)Tb(Pc) and [(15C5)4Pc]Tb[(15C5)4Pc]Tb(Pc) have not yet been reported, modulation of the magnetic anisotropy, which may be correlated with the LF, was demonstrated via NMR analyses.
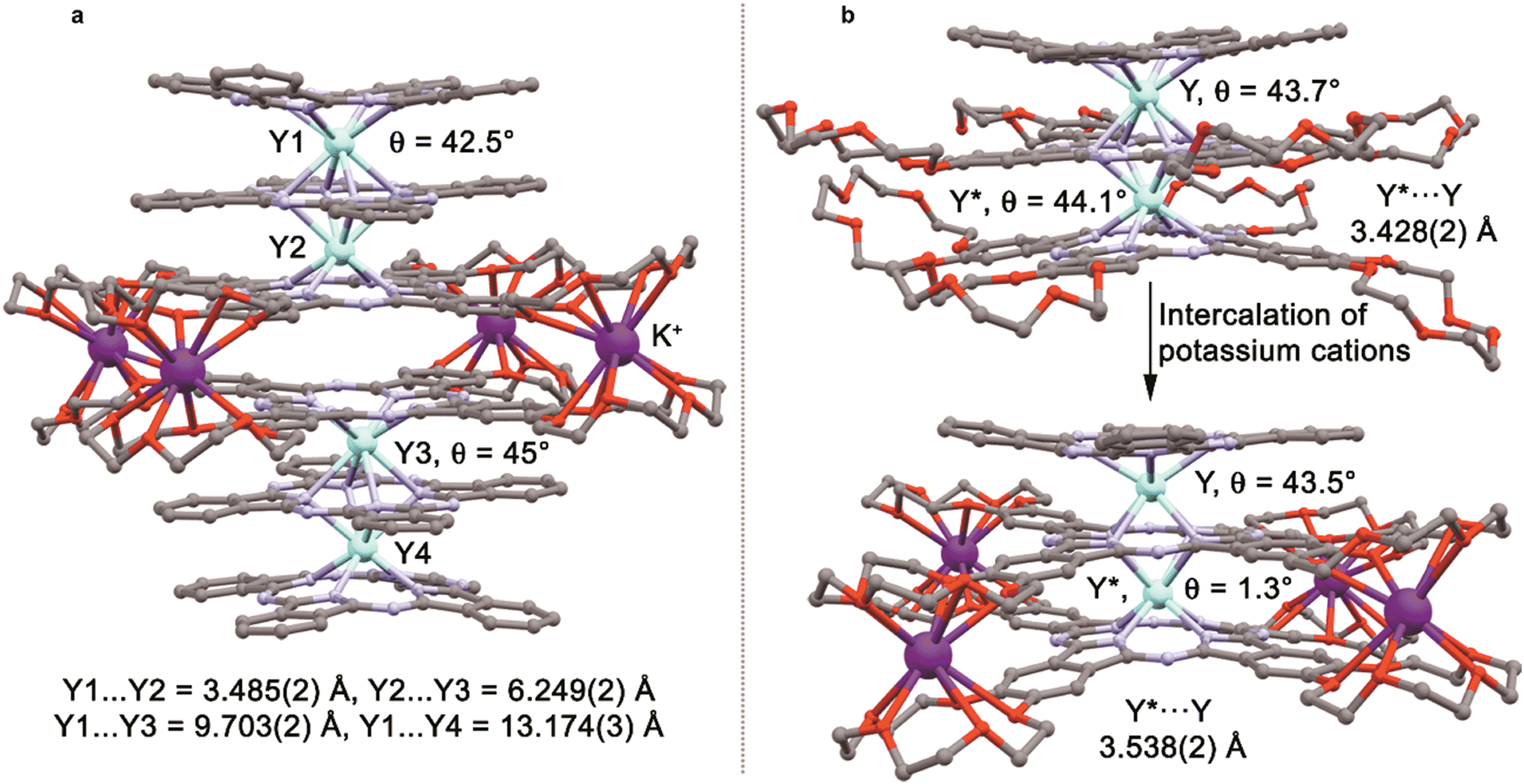 | ||
| Fig. 22 X-ray structure of the supramolecular assemblies formed by heteroleptic Y3+ crown-phthalocyaninates. (a) Dimer formed by [(15C5)4Pc]Y(Pc)Y(Pc) (CCDC SULZIL) in the presence of KBPh4. (b) Intercalation of potassium cations between crown-substituted ligands of [(15C5)4Pc]Y[(15C5)4Pc]Y(Pc) (CCDC ITUJEP and ITUGUC)333,334 without the formation of supramolecular dimers. Tetraphenylborate ions, hydrogen atoms and solvent molecules are not shown for clarity. | ||
The connection of the double-decker SMM H[(α-BuO)8Pc]Tb(Pc) by Na+ was reported by Chen et al. (Fig. 21c).335,336 Compared to the monomeric complex H[(α-BuO)8Pc]Tb(Pc) (Ueff = 125 cm−1), the corresponding dimer complex Na2{[(α-BuO)8Pc]Tb(Pc)}2 shows a larger Ueff value (367 cm−1). The DC magnetic measurements imply that the Tb3+–Tb3+ magnetic interactions in Na2{[(α-BuO)8Pc]Tb(Pc)}2 are very weak. Therefore, the change in the stacking angle from θ = 28° to θ = 34° upon Na+ coordination is the main reason for the increase in Ueff.
In 2013, Wang et al. prepared the first biradical dinuclear Dy3+ triple-decker SMM with a fused phthalocyanine ligand (Pc)Dy(f-Pc2)Dy(Pc)OBu (Scheme 42 and Fig. 23a).328 The distance between the Dy3+ ions coordinated to the fused ring Pc4− is 10 Å or more and π–f interactions are observed. The π-bridge delocalized biradical antiferromagnetic interactions effectively suppress the QTM of the Dy3+-ion-based SMMs, and at temperatures of up to 5 K, butterfly-type magnetic hysteresis with HC was observed.
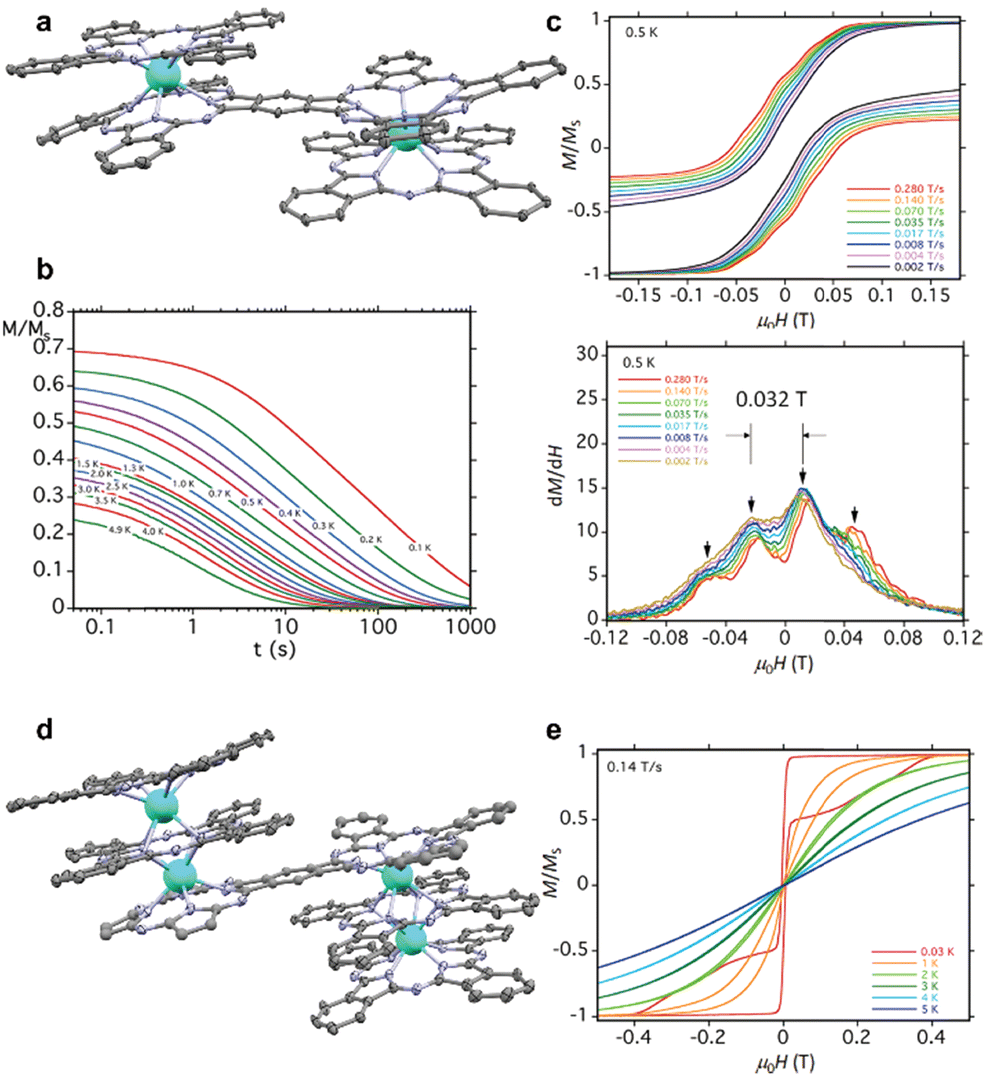 | ||
| Fig. 23 Crystal structure of (a) (Pc)Ln(f-Pc2)Ln(Pc)OBu (Ln3+ = Dy and Tb) and (d) (Pc)2Ln2(f-Pc2)Ln2(Pc)2OBu (Ln3+ = Tb and Dy) and the micro-SQUID-measurement magnetization decay curves (b) and hysteresis loops for oriented crystalline samples of (c) (Pc)Tb(f-Pc2)Tb(Pc)OBu and (e) (Pc)2Dy2(f-Pc2)Dy2(Pc)2OBu. Molecular structures of (a) and (d) based on the cif files from ref. 217 and 220. Protons and n-butoxy groups in (Pc)Ln(f-Pc2)Ln(Pc)OBu and (Pc)2Ln2(f-Pc2)Ln2(Pc)2OBu are omitted for clarity. Adapted with permission from (a and b) ref. 217. Copyright 2018, American Chemical Society. Adapted with permission from (d and e) ref. 220. Copyright 2018, Wiley-VCH. | ||
In 2018, Morita et al. prepared the first dinuclear Tb3+ fused-Pc4− triple-decker SMM (abbreviated as (Pc)Tb(f-Pc2)Tb(Pc)OBu).217 The complex shows an open hysteresis loop with HC = 180 Oe at 1.8 K (these values were obtained from the magnetic data for a magnetically diluted sample). From the crystal structure, the twist angle (θ) between the Pc ligands was determined to be 45°, which indicates an SAP coordination environment. The strict SAP coordination caused the absence of the off-diagonal LF terms (Bqk; q ≠ 0), which eliminates the tunneling gap (ΔE) via level-crossings in the ground state, meaning that QTM processes are suppressed. In addition, solution 1H NMR measurements of (Pc)Tb(f-Pc2)Tb(Pc)OBu show that the coordination environment of the Tb3+ ions affects the anisotropic magnetic susceptibility (χax). Incorporating radicals improves the magnetic properties, as radical-bridged metal complexes exhibit slow magnetic relaxation dynamics due to suppressed QTM, exhibiting high TB values due to strong exchange coupling Jex in comparison with non-radical systems.239,337–339 However, it is not necessary to incorporate strong magnetic interactions between the Ln3+ ions and radical ligands, as shown by (Pc)Tb(f-Pc2)Tb(Pc)OBu. Although the induced intramolecular magnetic interactions using the molecular design of (Pc)Tb(f-Pc2)Tb(Pc)OBu are weak, these interactions play a role in suppressing QTM processes. This prediction was supported by DFT calculations, which were used to analyse the exchange couplings in the complex. The obtained results indicated the presence of magnetic interactions among the four spin centres, including indirect Tb3+–Tb3+ interactions through π-radicals. On the other hand, hyperfine couplings (0.015 cm−1) were observed for an oriented crystalline sample in micro-SQUID measurements (Fig. 23b and c), indicating that the inter- and intramolecular magnetic interactions were very weak, with smaller couplings than that for Tb(Pc)2− (0.017 cm−1).276 Magnetic hysteresis loops with Hc = 860 Oe at 1.8 K were observed for a crystalline sample of (Pc)Tb(f-Pc2)Tb(Pc)OBu (Fig. 23). Considering the relationship between the molecular arrangement and Hc, the weak intermolecular magnetic interactions are more effective in suppressing QTM. These results indicate that the collinearity of magnetic dipole–dipole interactions strongly affects the suppression of QTM.
In 2013, Morita et al. prepared the first tetranuclear Tb3+ fused-phthalocyaninato quintuple-decker SMM (Fig. 23c) by using a fused phthalocyaninato ligand to control the spatial arrangement of the Tb3+ ions in the SMM (Pc)2Tb2(f-Pc2)Tb2(Pc)2OBu.219 In DC magnetic susceptibility measurements, ferromagnetic interactions among the four Tb3+ ions in (Pc)2Tb2(f-Pc2)Tb2(Pc)2OBu were observed, with two kinds of magnetic dipole–dipole interactions. (Pc)2Tb2(f-Pc2)Tb2(Pc)2OBu can be described as a weakly ferromagnetically coupled dimer of the SMM Tb2[(BuO)8Pc]3319 with strong dipole–dipole interactions in the triple-decker moieties and weak dipole–dipole interactions through the fused-Pc4− linking the two Tb2[(BuO)8Pc]3 units. Dual magnetic relaxation processes were observed in (Pc)2Tb2(f-Pc2)Tb2(Pc)2OBu, similar to those in other dinuclear Tb3+–Pc complexes.211,317,340 This provides clear evidence that the magnetic relaxation phenomena depend heavily on the magnetic dipole interactions between the Tb3+ ions in (Pc)2Tb2(f-Pc2)Tb2(Pc)2OBu.
In 2018, Katoh et al. reported the magnetic properties and spin-relaxation processes of the tetranuclear Dy3+ fused-phthalocyaninato quintuple-decker SMM (Pc)2Dy2(f-Pc2)Dy2(Pc)2OBu and its crystal structure (Fig. 23d and e).220 The structure of (Pc)2Dy2(f-Pc2)Dy2(Pc)2OBu can be regarded as a dimer of Dy2[(BuO)8Pc]3 SMMs341 with different magnetic relaxation behaviour corresponding to the octacoordination-geometry sites Dy1 with a C4 symmetry (θ1 = 23°) and Dy2 with a D4d symmetry (θ2 = 45°). The lowest Kramers doublet (KD) of (Pc)2Dy2(f-Pc2)Dy2(Pc)2OBu is thought to be a mixed state of mJ = ±13/2 and ±11/2. At an Hdc of 1750 Oe and T range of 1.8–3.75 K, the QTM is suppressed, and the direct process is enhanced; these depend on the different coordination environments. Consequently, the coordination geometry plays a more significant role in the spin-relaxation phenomena of (Pc)2Dy2(f-Pc2)Dy2(Pc)2OBu than the dipolar bias between Dy3+ ions.341,342 The different conclusions for the Tb3+ and Dy3+ systems are attributable to the ground mJ state for their coordination geometries.36,84,233,242,252,276,284–287
These studies show that the SMM properties can be fine-tuned by introducing magnetic dipole–dipole interactions in a controlled SMM spatial arrangement. In addition, we can expect to obtain nuclear spin molecular qubits using the spin cascade |S = 1/2〉‖J = 6〉‖I = 3/2〉 of the Tb(Pc)2 system by taking advantage of the more complex intramolecular magnetic interactions in (Pc)Tb(f-Pc2)Tb(Pc)OBu.270,303,308
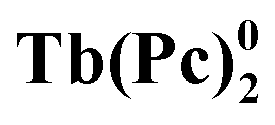 can be used in molecular spintronics due to its SMM characteristics and its physical and chemical stability.344 Investigating the influence of the direction of the magnetic dipole interactions acting among the
can be used in molecular spintronics due to its SMM characteristics and its physical and chemical stability.344 Investigating the influence of the direction of the magnetic dipole interactions acting among the  SMMs in the bulk solid on the magnetic relaxation behaviour is important for controlling its magnetic properties, which depend on the molecular arrangement.345,346 In 2017, Yamabayashi et al. showed that the arrangement of the
SMMs in the bulk solid on the magnetic relaxation behaviour is important for controlling its magnetic properties, which depend on the molecular arrangement.345,346 In 2017, Yamabayashi et al. showed that the arrangement of the  SMMs in crystals affects the ground state, and that QTM is suppressed at low temperatures.347 As a result, it has been suggested that QTM depends on the orientation angle (σ) of the uniaxial magnetic anisotropy with respect to the vector connecting the Tb3+ ions. In addition, QTM is suppressed in the Tb(Pc)2 dimer when the Tb(Pc)2 units are connected with a spacer acting as a Hbiasvia through-space magnetic dipole interactions (dipolar bias).105 Moreover, it is possible to eliminate the influence of the transverse field by arranging the Tb3+ ions in the direction of the C4 axis.97 The probability of QTM occurring in one Tb(Pc)2 unit is PQTM, and the probability of QTM occurring simultaneously in a Tb(Pc)2 dimer is PQTM2, indicating that QTM is effectively suppressed.
SMMs in crystals affects the ground state, and that QTM is suppressed at low temperatures.347 As a result, it has been suggested that QTM depends on the orientation angle (σ) of the uniaxial magnetic anisotropy with respect to the vector connecting the Tb3+ ions. In addition, QTM is suppressed in the Tb(Pc)2 dimer when the Tb(Pc)2 units are connected with a spacer acting as a Hbiasvia through-space magnetic dipole interactions (dipolar bias).105 Moreover, it is possible to eliminate the influence of the transverse field by arranging the Tb3+ ions in the direction of the C4 axis.97 The probability of QTM occurring in one Tb(Pc)2 unit is PQTM, and the probability of QTM occurring simultaneously in a Tb(Pc)2 dimer is PQTM2, indicating that QTM is effectively suppressed.
Magnetic dipole interactions are dominant in quasi-1D molecular magnetic materials, in which (Nc)Tb(Pc) SMM units adopt a structure similar to that of Tb(Pc)2 SMMs (Fig. 24b). In 2018, Katoh et al. reported that the magnetic properties of (Nc)Tb(Pc)0/+ (neutral (Nc)Tb(Pc)0: Ueff = 652 cm−1 with τ0 = 2.5 × 10−12 s; cationic (Nc)Tb(Pc)+: Ueff = 584 cm−1 with τ0 = 5.5 × 10−12 s) with 1D structures are significantly different from those of a magnetically diluted sample ((Nc)Tb0.1Y0.9(Pc)0: Ueff = 342 cm−1 with τ0 = 1.1 × 10−8 s).136 In particular, the τ of (Nc)Tb(Pc)+ (22 s at 1.8 K) in the low-temperature region is five orders of magnitude slower than that of (Nc)Tb0.1Y0.9(Pc)0 (1.2 ms at 1.8 K). Furthermore, the HC of (Nc)Tb(Pc)+ was retained at temperatures of up to ca. 20 K, which is the TB. The single-ion anisotropy of the Tb3+ ions in a 1D structure and the magnetic dipole interactions acting among the molecules determine the direction of the magnetic properties. These results show that the spin dynamics can be improved by manipulating the arrangement of the SMMs in the solid state. There are clear advantages to using quasi-1D SMM systems with magnetic dipole interactions such as (Nc)Tb(Pc)0/+. This strategy could lead to dramatic changes in the spin-blocking phenomena, because τ is slower at a zero magnetic field. In other words, since the probability of QTM occurring in (Nc)Tb(Pc)+ is PQTMn (n > 2), the QTM is effectively suppressed in a 1D system. Therefore, this relatively simple molecular-design method can be used to construct a variety of Ln(Pc)2-SMM-based functional molecular magnetic materials, and new functionalities, such as electronic and ionic conduction, gas adsorption, and various switching characteristics, can be incorporated by controlling the pore size.
 | ||
| Fig. 24 (a) Crystal structure of [(Nc)Tb(Pc)]+PF6− (CCDC YICQIN). Hydrogen atoms and the PF6− anion are omitted for clarity. (b) 1D structure of [(Nc)Tb(Pc)]+PF6− along the c-axis. (c) MH measurement hysteresis loop with a coercivity (HC) of 150 mT (1500 Oe) for a single crystal of [(Nc)Tb(Pc)]+PF6− at 1.8 K (H//c-axis). The data were normalized to the value in a field of 7 T. (d) 1D structure of [Dy(Pc)2]Ix along the c-axis. Hydrogen atoms are omitted for clarity. (e) MH curve of [Dy(Pc)2]Ix for a polycrystalline sample at 2.2 K (top). Magnetoresistance (MR) measurement of [Dy(Pc)2]Ix for a single crystal at 2.2 K. Adapted with permission from (a–c) ref. 136 and (d and e) ref. 348. Copyright 2018 and Copyright 2021, Wiley-VHC. | ||
These Ln(Pc)2-SMM-based materials are expected to exhibit galvanomagnetic effects due to the correlation between the localized f electrons and conduction electrons. There have been several reports of partially oxidized 1D [Ln(Pc)2]Ix complexes (Ln = Ce, Pr, Sm, Ho, and Yb).349–353 In 2021, a 1D arrangement of a partially oxidized Dy3+ double-decker SMM, [Dy(Pc)2]Ix (1.95 < x < 2.26), was reported by Sato et al. (Fig. 24d).348 Comparison of the Arrhenius plots with those of a magnetically diluted sample [Dy0.02Y0.98(Pc)2]Ix revealed that the τ for [Dy(Pc)2]Ix is four times slower at 1.8 K ([Dy(Pc)2]Ix: 0.0050 s; [Dy0.02Y0.98(Pc)2]Ix: 0.0013 s), and that its QTM at low temperatures is suppressed by the dipolar bias in the 1D array. In [Dy(Pc)2]Ix, the Pc ligands were partially oxidized, and the I3− ion was present as a counter ion. Based on the polarised reflection spectra of [Dy(Pc)2]Ix, the π-radicals were delocalized in a 1D column and behaved as a conductor even at low temperatures. Given that conductivity was observed at temperatures lower than TB, the negative magnetoresistance (MR) effects of ca. 1% corresponding to the magnetic hysteresis of the SMM were observed when the galvanomagnetic characteristics were investigated at 2.2 K. This indicated that the galvanomagnetic properties between the SMMs and the electrical conduction characteristics due to the π–f interactions could be observed. Our groups are continuing to investigate the galvanomagnetism due to π–f interactions in partially oxidized [Ln(Pc)2]Ix 1D complexes. This strategy may pave the way for the construction of electro-conductive SMMs as molecular spintronic devices via a spin-cascade system.
In 2014, Wang et al. reported cocrystals of C60 with the SMM (Pc)Dy(TCIPP), along with the relationship among the coordination environment, intermolecular interactions, and the SMM behaviour.360 Focusing on the twist angle θ between the ligands, it has been reported that the initial angle (43.60° for (Pc)Dy(TCIPP)) does not change dramatically after co-crystallisation (41.22°/44.45° for (Pc)Dy(TCIPP)·0.5C60, 44.41° for (Pc)Dy(TCIPP)·C60, and 44.35° for (Pc)Dy(TCIPP)·2C60). However, the frequency (ν) dependences of the alternating current (AC) measurements changed due to the different molecular arrangements, depending on the number of incorporated C60 molecules.
Combining a triple-decker SMM with C60 can be expected to reduce the overall symmetry and thus reveal the effects of the LF term. In 2020, Katoh et al. reported the dinuclear Tb3+ triple-decker complex (TPP)Tb(Pc)Tb(TPP), which contains porphyrin units on the outside to bind with C60 (henceforth abbreviated as [(TPP)Tb(Pc)Tb(TPP)]·C60).321 The supramolecular complex [(TPP)Tb(Pc)Tb(TPP)]·C60 (Fig. 25a and b) was prepared by assembling C60 with the dinuclear Tb3+ triple-decker complex (TPP)Tb(Pc)Tb(TPP) with quasi-D4h symmetry to investigate the relationship between the coordination symmetry and SMM properties (Fig. 25e and f). The two Tb3+ sites of (TPP)Tb(Pc)Tb(TPP) are equivalent, and θ was determined to be 3.62° (Fig. 25d). On the other hand, the two Tb3+ sites of [(TPP)Tb(Pc)Tb(TPP)]·C60 are not equivalent. The θ values for sites Tb1 and Tb2 were determined to be 3.67° and 33.8°, respectively, due to a change in the coordination symmetry of (TPP)Tb(Pc)Tb(TPP) upon association with C60 (Fig. 25c). At 1.8 K, (TPP)Tb(Pc)Tb(TPP) and [(TPP)Tb(Pc)Tb(TPP)]·C60 undergo different magnetic relaxations, and the changes in the ground state affect the spin dynamics. (TPP)Tb(Pc)Tb(TPP) and [(TPP)Tb(Pc)Tb(TPP)]·C60 relax via QTM at zero Hdc, and the H dependences of their τ values are similar at H > 1500 Oe. On the other hand, at H < 1500 Oe, their τ values show different behaviour, since the off-diagonal terms (Bqk; q ≠ 0) affect the magnetic relaxation mechanism. Based on the temperature and H dependences of τ, the spin dynamics can be explained by spin–phonon interactions together with the direct and Raman mechanisms. Thus, a supramolecular approach can be used to control the magnetic anisotropy along the C4 rotation axis and the spin-dynamic properties in Ln3+–Pc multiple-decker complexes.
 | ||
| Fig. 25 Cocrystals of rare-earth phthalocyaninato sandwich complexes and fullerene derivatives. (a–c) Crystal structure of [(TPP)Tb(Pc)Tb(TPP)]·C60. The inclusion of C60 changes the coordination environment of the Tb3+ ion and affects the SMM characteristics. (d) Coordination environment of (TPP)Tb(Pc)Tb(TPP) before the subsumption of C60. The micro-SQUID-measurement hysteresis loops (e) and alternating current (AC) magnetic susceptibility data (f) of [(TPP)Tb(Pc)Tb(TPP)]·C60. (g) The cocrystal [(Pc)Tb(OEP)]3(Li+@C60·PF6−)2 forms a quasi-kagome lattice due to the two kinds of isosceles triangles (A and B). Protons and crystal solvents of [(TPP)Tb(Pc)Tb(TPP)]·C60 and [(Pc)Tb(OEP)]3(Li+@C60·PF6−)2 are omitted for clarity. Adapted with permission from (a–f) ref. 321. Copyright 2020, Wiley-VHC. Reproduced from (g) ref. 361 with permission from the Royal Society of Chemistry. | ||
In 2020, Iwami et al. reported the cocrystallisation of a lithium-ion-encapsulating fullerene (Li+@C60), C60 and C70 with a (Pc)Tb(OEP) SMM (Fig. 25g).361 The OEP unit interacted with the fullerene derivatives to form five different cocrystals: [(Pc)Tb(OEP)]3(Li+@C60·PF6−)2, [(Pc)Tb(OEP)](Li+@C60·BF4−), [(Pc)Tb(OEP)]C70·C6H14, [(Pc)Tb(OEP)]C60·C6H14, and [(Pc)Tb(OEP)]C60·Et2O. For the cocrystals containing Li+@C60 ([(Pc)Tb(OEP)]3(Li+@C60·PF6−)2 and [(Pc)Tb(OEP)](Li+@C60·BF4−)), the use of different counter anions resulted in different crystal packings. In particular, [(Pc)Tb(OEP)]3(Li+@C60·PF6−)2 was found to have a unique quasi-kagome lattice packing that induces intermolecular ferromagnetic interactions. Depending on the crystallisation conditions, different packings, i.e., 1D column packings for [(Pc)Tb(OEP)](Li+@C60·BF4−) and [(Pc)Tb(OEP)]C70·C6H14 and AABB-type arrays of (Pc)Tb(OEP) and C60 dimers for [(Pc)Tb(OEP)]C60·C6H14 and [(Pc)Tb(OEP)]C60·Et2O, were formed. In addition, the magnetic properties of the (Pc)Tb(OEP) units were modulated via cocrystallisation.
The relationship between the changes in the coordination geometry of the same molecule and the SMM properties induced via a supramolecular approach was also investigated. The corresponding results showed that the magnetic anisotropy along the C4 rotation axis of the metal sites in the dinuclear Ln3+–Pc multiple-decker complexes could be controlled by forming a supramolecular complex, which could lead to switchable SMMs.
3.4 Correlation between redox states and SMM properties
Porphyrinoids are redox-active ligands due to their extended π-systems and high redox stability. Therefore, sandwich complexes composed of porphyrinoids are also redox active. In this section, we will explain the relationship between redox states and SMM properties. The Ln3+ ions (except for Ce3+) are in essence redox inert. Therefore, the redox reactions of the sandwich complexes presented here occur on the porphyrinoid ligands.Ishikawa and Takamatsu et al. reported that the Ueff values of the anionic forms of the double-decker complexes (NBu4+){Tb[(EtO)8Pc]2}− and (NBu4+){Dy[(EtO)8Pc]2}− are enhanced in their corresponding cationic forms, {Tb[(EtO)8Pc]2}+(SbCl6−)84 and {Dy[(EtO)8Pc]2}+(SbCl6−).362 The mechanism of the structural changes due to the redox reactions can be explained using molecular orbital theory363 (Fig. 26a and b). As was briefly introduced in Section 3.2, the oxidized species of Tb3+–Pc and Dy3+–Pc double-decker complexes tend to show improved SMM properties compared to the reduced ones due to the short ligand-to-metal distance in the oxidized form, which result in greater LF splitting and Ueff values. The molecular orbitals of the sandwich complexes are constructed from the combination of the Pc ligands. The stacked dimerisation shown in the double-decker complexes constructs the antibonding HOMO orbital and the bonding HOMO−1 orbital (Fig. 26a). The removal of electrons (oxidation of the complex) increases the bond order between the ligands, causing axial compression and a decrease in the interligand distance. Although this mechanism is widely accepted, recent ab initio calculations based on the CASSCF/RASSI-SO method have indicated that the magnitude of the LF splitting does not strongly depend on the structural changes induced by the redox reactions, and that the calculated energy levels of the first excited doublets (|mJ〉 = |±5〉) for Tb3+ double-decker complexes are ca. 300 cm−1.293,364,365 Therefore, the 8% increase in the Ueff of (NBu4+){Tb[(EtO)8Pc]2}− due to oxidation may arise from thermally assisted QTM via higher excited LF sublevels.
 | ||
Fig. 26 (a) Molecular orbitals of [Ln(Pc)2]−. Removal of two electrons from the antibonding HOMO affords the cationic form [Ln(Pc)2]+ with a shorter Ln3+–Pc distance. (b)  vs. T plots for anionic ((NBu4+){Tb[(EtO)8Pc]2}−) and cationic ({Tb[(EtO)8Pc]2}+(SbCl6−)) Tb3+ double-decker complexes. (c) Magnetic hysteresis behaviour of a Tb3+ double-decker complex in a series of redox states ([Tb(iprPc)2]−/0/+) as captured using MCD spectroscopy. (d) Structure of the perfluoro double-decker complexes [Tb(F64Pc)2]−/2−/3−. Adapted with permission from (a) ref. 363. Copyright 2006, Elsevier Ltd. Adapted with permission from (b) ref. 84 (c) ref. 366 and (d) ref. 87. Copyright 2007, Copyright 2010 and Copyright 2013, American Chemical Society. vs. T plots for anionic ((NBu4+){Tb[(EtO)8Pc]2}−) and cationic ({Tb[(EtO)8Pc]2}+(SbCl6−)) Tb3+ double-decker complexes. (c) Magnetic hysteresis behaviour of a Tb3+ double-decker complex in a series of redox states ([Tb(iprPc)2]−/0/+) as captured using MCD spectroscopy. (d) Structure of the perfluoro double-decker complexes [Tb(F64Pc)2]−/2−/3−. Adapted with permission from (a) ref. 363. Copyright 2006, Elsevier Ltd. Adapted with permission from (b) ref. 84 (c) ref. 366 and (d) ref. 87. Copyright 2007, Copyright 2010 and Copyright 2013, American Chemical Society. | ||
In 2010, Gonidec et al. reported the magnetic properties of the various redox states of the double-decker complex [Tb(iprPc)2]−/0/+ (iprPc = 2,3,9,10,16,17,23,24-octa(isopropylidenedioxy)phthalocyaninato) as detected using magnetic circular dichroism (MCD) spectroscopy (Fig. 26c).366 The advantage of MCD is the measurement of the magnetic properties of the specimen in frozen solution states. Therefore, the effects of the intermolecular magnetic interactions and crystal packing, which could potentially affect the magnetic properties, can be minimized. The MCD intensity vs. magnetic field plots at 1.5 K show magnetic hysteresis for all redox states due to magnetisation blocking. [Tb(iprPc)2]− and [Tb(iprPc)2]+ show butterfly-shaped hysteresis curves, and the opening of the curve is larger in [Tb(iprPc)2]+. Importantly, [Tb(iprPc)2]0 (a ligand π-radical complex) shows clear opening of the hysteresis curve at zero DC magnetic field. These results clearly indicate that the ligand π-radical suppresses QTM.277
In 2010, the same group reported the magnetic properties (MCD spectra) of the highly reduced double-decker complex [Tb(F64Pc)2]−/2−/3− (F64Pc = 1,4,8,11,15,18,22,25-octakis-fluoro-2,3,9,10,16,17,23,24-octakis-perfluoro(isopropyl)phthalocyaninato) stabilized by electron-withdrawing fluoro groups (Fig. 26d).87 Magnetic hysteresis behaviour was observed in all the reduced states. Among these, [Tb(F64Pc)2]2− showed the largest magnetic hysteresis. The non-π-radical complexes ([Tb(F64Pc)2]− and [Tb(F64Pc)2]3−) showed narrowing of the hysteresis curve at zero DC magnetic field, whereas the π-radical species [Tb(F64Pc)2]2− did not show such behaviour. Accordingly, the results of this study corroborate suppression of the QTM at the zero DC field due to a ligand π radical.
The first crystal structure characterisation of the dianionic double-decker complex [Cryptand(Na+)]2[Tb(Pc)2]2− was reported by Konarev et al. in 2019.85 The existence of the ligand π-radical was confirmed based on near-IR absorption and EPR analyses. The interligand distance in [Cryptand(Na+)]2[Tb(Pc)2]2− (3.258 Å) is shorter than that (3.294 Å) in the monoanionic form (PPN+)[Tb(Pc)2]− (PPN+ = bis(triphenylphosphoranylidene)ammonium). Interestingly, [Cryptand(Na+)]2[Tb(Pc)2]2− does not show slow magnetic relaxation or magnetic hysteresis at 1.9 K, which may be related to the relatively strong intermolecular antiferromagnetic interactions confirmed by the DC magnetic susceptibility measurements.
In 2020, Horii et al. reported a comprehensive study on the structural and magnetic properties of multiple-decker Tb3+-(BuO)8Pc complexes (triple- (Tb2[(BuO)8Pc]3), quadruple- (Tb2Cd[(BuO)8Pc]4), and quintuple-decker (Tb2Cd2[(BuO)8Pc]5) complexes) in highly oxidized states.214 Crystal structure analyses and DFT calculations revealed that the multiple-decker complexes show decreasing interligand distances with successive oxidations. More importantly, paramagnetic NMR analyses in the solution state revealed that the axial component of the magnetic susceptibility (χax) also decreases with successive oxidation (Fig. 27a). This behaviour stands in clear contrast to that of the double-decker complexes, in which χax increases with oxidation.278 In other words, the Tb3+ ions in the multiple-decker complexes show decreased magnetic anisotropy upon oxidation. A similar trend was confirmed in the AC magnetic measurements of the oxidized species characterized by X-ray analyses (Fig. 27b). In the case of the triple-decker complex Tb2[(BuO)8Pc]3, the oxidation from the neutral to the two-electron-oxidized state {Tb2[(BuO)8Pc]3}2+(SbCl6−)2 shifts the maxima of the 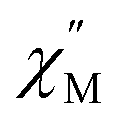 vs. AC frequency (ν) plots to higher ν. In addition, the Ueff decreases from 233 cm−1 (Tb2[(BuO)8Pc]3) to 167 cm−1 ({Tb2[(BuO)8Pc]3}2+(SbCl6−)2) upon oxidation. In the case of the multiple-decker complexes, oxidation causes a decrease in the interligand distances as well as a decrease in the intermetal distances (Fig. 27c). Due to the approach of the positively charged metal ions along the axial direction, the axial LF is decreased. This is the reason for the decrease in Ueff upon oxidation of the multiple-decker complexes.
vs. AC frequency (ν) plots to higher ν. In addition, the Ueff decreases from 233 cm−1 (Tb2[(BuO)8Pc]3) to 167 cm−1 ({Tb2[(BuO)8Pc]3}2+(SbCl6−)2) upon oxidation. In the case of the multiple-decker complexes, oxidation causes a decrease in the interligand distances as well as a decrease in the intermetal distances (Fig. 27c). Due to the approach of the positively charged metal ions along the axial direction, the axial LF is decreased. This is the reason for the decrease in Ueff upon oxidation of the multiple-decker complexes.
 | ||
Fig. 27 (a) χax values for the oxidation states of a (BuO)8Pc multiple-decker complex. (b)  vs. frequency (ν) plots for the redox series of double and triple-decker complexes (Tb2[(BuO)8Pc]3 and {Tb2[(BuO)8Pc]3}2+(SbCl6−)2). (c) Crystal structures of Tb2[(BuO)8Pc]3 and {Tb2[(BuO)8Pc]3}2+(SbCl6−)2 from the cifs in ref. 319 and 214. n-Butoxy groups are omitted for clarity. The triple-decker complex shows a decrease in axial magnetic anisotropy upon oxidation due to the decrease in the LF with the approach of a positively charged Ln3+ ion along the axial direction. Adapted with permission from (a) ref. 214. Copyright 2020, Wiley-VHC. vs. frequency (ν) plots for the redox series of double and triple-decker complexes (Tb2[(BuO)8Pc]3 and {Tb2[(BuO)8Pc]3}2+(SbCl6−)2). (c) Crystal structures of Tb2[(BuO)8Pc]3 and {Tb2[(BuO)8Pc]3}2+(SbCl6−)2 from the cifs in ref. 319 and 214. n-Butoxy groups are omitted for clarity. The triple-decker complex shows a decrease in axial magnetic anisotropy upon oxidation due to the decrease in the LF with the approach of a positively charged Ln3+ ion along the axial direction. Adapted with permission from (a) ref. 214. Copyright 2020, Wiley-VHC. | ||
3.5 Proton-induced switching of the SMM properties
Switching of the SMM properties via external stimuli has great importance for the use of SMMs as information-storage devices due to the requirement to eventually erase stored information.Tanaka and Inose et al.297,367 reported the switching of SMM properties via the protonation/deprotonation of the Tb3+-porphyrinato double-decker complex HTb(TPP)2. In the protonated form of HTb(TPP)2, H+ coordinates to one pyrrole N atom (Fig. 28a). The distance between Tb3+ and the protonated N atom is 2.840 Å; this value is significantly longer than the bond lengths between Tb3+ and the non-protonated N atoms (2.430–2.627 Å). In other words, the Tb3+ ion in HTb(TPP)2 adopts a heptacoordinated structure. The significant deviation from the SAP geometry in HTb(TPP)2 results in the absence of SMM behaviour. When the coordinated proton is removed by DBU (1,8-diazabicyclo[5.4.0]undec-7-ene), the deprotonated form (HDBU+)[Tb(TPP)2]− is obtained. (HDBU+)[Tb(TPP)2]− crystallizes in the space group I4/mmm, and the eight N atoms coordinating to the Tb3+ ion are crystallographically equivalent. Therefore, the Tb3+ of (HDBU+)[Tb(TPP)2]− adopts an octacoordinated SAP geometry, which is one of the most ideal symmetries for SMMs. As a result, (HDBU+)[Tb(TPP)2]− shows slow magnetic relaxations and butterfly-shaped hysteresis, which are characteristics of SMMs. The conversion from (HDBU+)[Tb(TPP)2]− to HTb(TPP)2 is achieved by the addition of acetic acid. Therefore, the protonation/deprotonation process is reversible, enabling the reversible switching of the SMM properties by a single proton.
 | ||
| Fig. 28 (a) Proton-induced on/off switching of SMM properties (HTb(TPP)2 and (HDBU+)[Tb(TPP)2]−). (b) Protonation/deprotonation of the porphyrinato–phthalocyaninato Tb3+ double-decker complex H(Pc)Tb(TPP) and (c) phthalocyaninato–phthalocyaninato double-decker complex HTb[(α-BuO)8Pc]2. (d) Magnetic hysteresis curves of the protonated (HTb[(α-BuO)8Pc]2) and deprotonated form ((HDBU+){Tb[(α-BuO)8Pc]2}−) of the double-decker complex. Reproduced from (a) ref. 297 with permission from the Royal Society of Chemistry. Adapted with permission from (d) ref. 97. Copyright 2018, American Chemical Society. | ||
The protonation/deprotonation of the phthalocyaninato-porphyrinato heteroligand double-decker complex H(Pc)Tb(TPP) was reported by Tanaka et al. (Fig. 28b).368 This complex is obtained as the protonated form H(Pc)Tb(TPP), in which the proton coordinates to the meso-N of the Pc ligand. Since the protonation/deprotonation occurs on the non-coordinating N atom, the changes in the SMM properties induced by deprotonation are not significant compared to those of the homo-porphyrinato complexes. However, the protonated form shows additional magnetic relaxations at low temperatures.
The protonated forms of homo-phthalocyaninato Ln3+ double-decker complexes (Fig. 3) were obtained using the [(α-BuO)8Pc] ligand,95 which has eight n-butoxy groups at the nonperipheral positions of Pc, causing a saddle-shaped distortion of the π-structure (Fig. 3). This distortion causes the meso-N atoms of [(α-BuO)8Pc] to be more basic than those in [(β-BuO)8Pc]. In 2018, Horii et al. reported the modulation of the SMM properties of HTb[(α-BuO)8Pc]2via a protonation/deprotonation reaction (Fig. 28c).97 The protonation of the meso-N atoms in HTb[(α-BuO)8Pc]2 causes asymmetric distortion as well as a low-symmetry structure, whereas deprotonation using DBU affords (HDBU+){Tb[(α-BuO)8Pc]2}−, which is distorted in a highly symmetric windmill-like fashion. The deprotonated form shows larger magnetic hysteresis than the protonated one does. Similar trends were observed in magnetically diluted samples, indicating that the differences between the magnetic properties of the protonated and deprotonated forms originate from the molecular structures rather than the intermolecular magnetic interactions.
The following two examples also show changes in their SMM properties as a result of deprotonation. However, oxidation from the anionic (and protonated) form to the neutral (and deprotonated) form is the driving force for the deprotonation. In 2019, Chen et al. reported the magnetic properties of HTb[(α-C5H11O)4Pc]2 with 3-pentyloxy-substituents (Fig. 3a).94 The Ueff value for HTb[(α-C5H11O)4Pc]2 is 300 cm−1. On the other hand, the Ueff value for the neutral and deprotonated form Tb[(α-C5H11O)4Pc]2 obtained using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) is 324 cm−1, indicating that the SMM properties are enhanced by deprotonation. In contrast to the previous examples, the presence of the ligand π-radical in Tb[(α-C5H11O)4Pc]2 was confirmed by absorption spectroscopy. Therefore, Chen et al. concluded that the enhancement of the SMM properties stems from the radical–f interactions and enhanced LF due to the shorter ligand-to-metal distances in the neutral form.
In 2019, Sun et al. reported the negative effect of deprotonation on SMM properties by comparison of the chiral (phthalocyaninato)(porphyrinato) Dy3+ double-decker complexes (R)/(S)-H[(BNPO)2Pc]Dy(TClPP) and the corresponding neutral unprotonated species (R)/(S)-[(BNPO)2Pc]Dy(TClPP) (Fig. 29).369 The AC magnetic susceptibilities of the protonated form (R)/(S)-[(BNPO)2Pc]Dy(TClPP) show clear frequency dependences, whereas (R)/(S)-[(BNPO)2Pc]Dy(TClPP) shows no frequency dependence even under a bias DC field (Hdc = 2000 Oe). DFT calculations of (R)/(S)-H[(BNPO)2Pc]Dy(TClPP) and (R)/(S)-[(BNPO)2Pc]Dy(TClPP) revealed that the ionic part of the N–Dy bond decreases due to oxidation (deprotonation). This decrease in the electrostatic interactions between the ligand and Dy3+ decreases the uniaxial magnetic anisotropy of (R)/(S)-[(BNPO)2Pc]Dy(TClPP).
 | ||
| Fig. 29 Molecular structures of Dy[(BNPO)2Pc](TClPP). | ||
3.6 Influence of solvation effects in the crystal lattice on SMM properties
The benzene solvates (μ-Pc)[Ln(dpm)2]2·2C6H6 showed interesting behaviour with respect to desolvation and exchange of solvation molecules. Thus, heating of solvates with Ln = Sm, Tb, Dy198 and Er199 at 100 °C was shown to cause desolvation without disintegration of crystals. This procedure had dramatic influences on the structural and magnetic properties of complexes which were demonstrated on a dysprosium analogue. The solvated complex contains one crystallographically independent metal center, which exhibits a single magnetic relaxation process with an energy barrier (Ueff) of 39 cm−1. When desolvated, the complex had two different types of metal centres with slightly different surrounding, and it exhibited field-induced SMM behaviour with two thermally activated magnetic relaxation processes with Ueff which reach 44 cm−1 and 76 cm−1, respectively. Soaking of desolvated crystals in benzene recovered (μ-Pc)[Ln(dpm)2]2·2C6H6 which was confirmed by X-ray powder diffractions and elemental analyses.This work confirmed that the solvated molecules can finely tune the magnetic relaxation mechanisms. Further development of this approach by Ge et al. showed that benzene can be replaced with dichloromethane in the course of “crystal to crystal” transformation.200 Again, the change of solvation state had influence on the magnetic properties, which was demonstrated on the example of (μ-Pc)[Er(dpm)2]2·2C6H6 undergoing transformation to (μ-Pc)[Er(dpm)2]2·2CH2Cl2.200 Magnetic susceptibility measurements showed that the two solvates exhibited dramatically different relaxation behaviour (Fig. 30). While benzene solvate exhibited fast relaxation and an estimated energy barrier of 2.6 cm−1 under 600 Oe DC field, the dichloromethane solvate behaved as a field-induced SMM with a higher energy barrier (Ueff) of 34.3 cm−1. The source of these magnetic differences was tentatively assigned to subtle structural differences caused by the change of the crystal lattice, as confirmed by ab initio calculations.
 | ||
| Fig. 30 Interconversion between solvates (μ-Pc)[Er(dpm)2]2·2C6H6 (CCDC 1494900) and (μ-Pc)[Er(dpm)2]2·2CH2Cl2 (CCDC 1494901) causing drastic change of magnetic relaxation as seen from the corresponding out-of-phase (χ′′) AC susceptibility plots under 600 Oe DC field. Adapted with permission from ref. 200. Copyright 2017, American Chemical Society. | ||
Ruan et al. showed that crystallization of Tb(Pc)2 from CHCl3/MeOH or CH2Cl2/MeOH mixtures afforded orthorhombic crystals Tb(Pc)2 (space group P212121) and Tb(Pc)2·CH2Cl2 (Pnma) solvates respectively.370 Although the structural differences in magnetically active sandwich molecules were only subtle, absorption of CH2Cl2 in the crystal lattice had a profound effect on magnetic relaxation, resulting in a butterfly-shaped hysteresis loop in the case of solvate up to 10 K, while Tb(Pc)2 showed almost no opening even at 2 K. These results are consistent with the conclusions of Yamabayashi et al., shown in Section 3.3.3.347
In the context of this result, an important study by Dailey and Besson on the selective crystallization of four possible polymorphs of M(Pc)2 complexes should be mentioned.371 The authors proposed reproducible and selective crystallization techniques affording pure α-, γ- and δ-phases together with the CH2Cl2 solvate of complexes depending on the solvent, applied for crystallization. Systematic crystallographic and UV-Vis characterization of complexes allowed some of the confusion over chemical composition in previously published literature associated with interconversion between neutral and ionic forms of bisphthalocyaninates to be explained.
3.7 Magnetic studies on surfaces
The properties of molecules significantly change not only through aggregation but also through the interface of organic–inorganic hybrid materials. One of the goals of the research involving sandwich-type tetrapyrrole rare-earth metal–inorganic hybrid materials is their application in spintronic devices.344 In the case of , the magnetic species are composed of the f electron of the central Tb3+ ion and the π radical on the Pc ligands. The Pc ligands are almost flat, and the direction of the magnetic moment (axis of easy magnetisation) of the Tb3+ ion is perpendicular to the Pc ligand with uniaxially anisotropy, indicating that they are single-molecule magnets.36 Furthermore, since they are thermally stable, they can be deposited on various substrates in an ultra-high vacuum (UHV). Several groups have reported on the structures and physical properties of sandwich-type tetrapyrrole rare-earth complexes on various substrates. In this section, we give an overview of the morphologies and magnetic properties of double-decker and triple-decker complexes on different substrates. Since 2008, many examples of Ln(Pc)2 and/or Ln2(Pc)3 complexes on various substrates, including non-magnetic metals (Cu(100),267 Cu(111),372,373 Cu(001),374 Au(111),232,302 Ag(111),375 Ir(111),376 MgO/Ag(100),377 CuO/Cu(110)378), magnetic metals (Co/Ir(111),379 Co/Cu(100),380 Co/Au(111),299 Ni/Cu(100),381 Ni/Ag(100),381 Fe/Cu(100),382 FeMn/Cu(100),382 CoO/Ag(100),383 Mn/Ag(100),383 Ni(111)384), carbon-based substrates (HOPG,385–387 graphite(0001),388 graphene/SiC(0001),389 graphene/Ni(111)390), silicon-based substrates (SiO2(100)391), perovskite (LaSrMnO3/SrTiO3,380 SrVO3(001)392), and composite substrates,393,394 have been reported. Moreover, materials of sandwich-type tetrapyrrole rare-earth metal complexes combined with carbon nanotubes (CNT)/fullerene are shown in Chapter 3.8. In general, eight lobes for both Ln(Pc)2 and/or Ln2(Pc)3 are observed in topographic STM images,301 which reflects the electronic state of the Pc ligand on the vacuum side. On the other hand, for Ln(Pc)x/Au(111), four lobes are observed in the topographic image.232 In addition, the STM height profiles for Ln(Pc)x, Ln(Pc)2, and Ln2(Pc)3 on substrates reflect the molecular structure (crystal structure).301
, the magnetic species are composed of the f electron of the central Tb3+ ion and the π radical on the Pc ligands. The Pc ligands are almost flat, and the direction of the magnetic moment (axis of easy magnetisation) of the Tb3+ ion is perpendicular to the Pc ligand with uniaxially anisotropy, indicating that they are single-molecule magnets.36 Furthermore, since they are thermally stable, they can be deposited on various substrates in an ultra-high vacuum (UHV). Several groups have reported on the structures and physical properties of sandwich-type tetrapyrrole rare-earth complexes on various substrates. In this section, we give an overview of the morphologies and magnetic properties of double-decker and triple-decker complexes on different substrates. Since 2008, many examples of Ln(Pc)2 and/or Ln2(Pc)3 complexes on various substrates, including non-magnetic metals (Cu(100),267 Cu(111),372,373 Cu(001),374 Au(111),232,302 Ag(111),375 Ir(111),376 MgO/Ag(100),377 CuO/Cu(110)378), magnetic metals (Co/Ir(111),379 Co/Cu(100),380 Co/Au(111),299 Ni/Cu(100),381 Ni/Ag(100),381 Fe/Cu(100),382 FeMn/Cu(100),382 CoO/Ag(100),383 Mn/Ag(100),383 Ni(111)384), carbon-based substrates (HOPG,385–387 graphite(0001),388 graphene/SiC(0001),389 graphene/Ni(111)390), silicon-based substrates (SiO2(100)391), perovskite (LaSrMnO3/SrTiO3,380 SrVO3(001)392), and composite substrates,393,394 have been reported. Moreover, materials of sandwich-type tetrapyrrole rare-earth metal complexes combined with carbon nanotubes (CNT)/fullerene are shown in Chapter 3.8. In general, eight lobes for both Ln(Pc)2 and/or Ln2(Pc)3 are observed in topographic STM images,301 which reflects the electronic state of the Pc ligand on the vacuum side. On the other hand, for Ln(Pc)x/Au(111), four lobes are observed in the topographic image.232 In addition, the STM height profiles for Ln(Pc)x, Ln(Pc)2, and Ln2(Pc)3 on substrates reflect the molecular structure (crystal structure).301
In recent years, physical properties of surfaces have been actively studied to advance the research on applications, and together with the progress of nanotechnology, the academic and social importance have increased. Precise control is possible at the atomic level, and organic–inorganic hybrid compounds can be prepared with the surfaces acting as reaction fields. Atoms and molecules adsorbed on surfaces can arrange into various dimensional structures (zero dimensional (0D), one dimensional (1D), two dimensional (2D), and three dimensional (3D)) which exhibit different properties. For example, 2D substances have multifunctional properties. Due to the electronic state of graphene, spin-polarised electron bands form for non-magnetic materials. In addition, graphene is used for molecular spintronics because it utilizes spin–orbit interactions which increases when the π–d orbitals mix at the interface with magnetic metal electrodes.395 At the interface, charge transfer bring about the alteration of the electronic structure, thereby affecting or even quenching the magnetic moment of the molecules. Recently, such spin current control at the interface at the molecular level has become known as spinterface.396 In principle, it is possible to use the spin for quantum information processing (QIP), and spin detection and evaluation are important for realising a quantum computer (QC) and/or spintronic devices.258 Electron spin resonance (ESR), reported by E. Zaboisky in 1945, is one such detection method.
At the same time, scanning tunnelling microscopy (STM) and scanning tunnelling spectroscopy (STS) can be used to identify one magnetic atom adsorbed on a substrate surface and directly observe the density of states (DOS). Thus, they have been attracting attention as a method for single spin detection. STM and STS play a major role in directly investigating the relationship between morphology and electronic structure. For example, for CoPc adsorbed on an Au surface, a symmetric Kondo resonance is observed because only the direct tunnel process from the probe to the substrate is suppressed by the Pc ligand.397 It is possible to study the interactions between magnetic atoms and non-magnetic/magnetic metals or those between magnetic atoms on an atomic scale.398 The Kondo effect, which is the principle of Kondo resonance, was proposed by Jun Kondo in 1964. As the temperature of a metal decreases, its electrical resistance decreases. However, the electrical resistance increases when the temperature is lowered below a certain temperature due to trace amounts of magnetic impurities contained in the metal. This is a phenomenon in which the spins of metallic conduction electrons and the spins of magnetic impurities finally form a Kondo–Yoshida singlet, which has a higher resistance than the metallic state and the electrical resistance remains constant. In this process, as a result of the spin exchange process between the conduction electrons and the magnetic impurities, a resonance state is observed as a sharp peak at the Fermi level (0 V). The temperature at which the electrical conductivity becomes constant at low temperatures is defined as the Kondo temperature (TK), and in the case of STS, it is proportional to the peak width at half maximum of the Kondo resonance obtained from tunnelling current spectroscopy. By applying the Kondo effect to the conduction electrons, it is possible that the conductance changes, and this phenomenon has been attracting attention again recently because of its high utility in spintronics. As a result, the Kondo resonance should be observed even for M(Pc)2, which has a spin source.
A Kondo resonance for a delocalized π radical on the ligand due to the electronic state of Tb(Pc)2 adsorbed on a substrate has been reported, and it is clear that the Kondo resonance observed on Au(111) depends on the molecular conformation (Fig. 31a–c and 32a, b).302,399 In other words, the Kondo resonance can be switched off by changing θ by <8°. In addition, when Cs is deposited on Tb(Pc)2/Au(111), the electronic structure of the molecule changes.400 This is probably because charge transfer occurs when Cs is added, switching on the Kondo resonance. On the other hand, for Tb(Pc)2/Ag(111), Kondo resonance is silent in the first layer of Tb(Pc)2 but active in that of the second layer.375 The π radical recovers by decoupling at the interface. Since the Kondo resonance is silent even for Tb(Pc)2/Cu(111), the interactions between the substrate and  are important for understanding the Kondo resonance.372 In other words, it is possible to control the spin state of
are important for understanding the Kondo resonance.372 In other words, it is possible to control the spin state of  via the interface. In addition, since Kondo resonances depend on the electronic state of ligands, the TK values of delocalized π radicals of Nc2− (naphthalocyaninato) and Pc2− ligands of (Nc)Tb(Pc)/Au(111) differ (Fig. 31d–h).135 Furthermore, for Tb(OEP)2 (OEP2− = 2,3,9,10,16,17,23,24-octaethylporphyrinato)/Au(111), the electronic structure changes significantly due to the addition and desorption of proton at the isoindole nitrogen (Niso) position, meaning that the Kondo resonance could be switched on and off (Fig. 31i–n).307 Moreover, Kondo resonances have been detected for Tb(Pc)x/Au(111) and Tb2(Pc)3/Ag(111) (Fig. 32d).232,401 However, a Kondo resonance for Y2(Pc)3/Au(111) has not been observed under the same conditions as those used for Y(Pc)2/Au(111) (Fig. 32c).302 It has been shown that interactions with a Cu(111) substrate quench the unpaired π electron of
via the interface. In addition, since Kondo resonances depend on the electronic state of ligands, the TK values of delocalized π radicals of Nc2− (naphthalocyaninato) and Pc2− ligands of (Nc)Tb(Pc)/Au(111) differ (Fig. 31d–h).135 Furthermore, for Tb(OEP)2 (OEP2− = 2,3,9,10,16,17,23,24-octaethylporphyrinato)/Au(111), the electronic structure changes significantly due to the addition and desorption of proton at the isoindole nitrogen (Niso) position, meaning that the Kondo resonance could be switched on and off (Fig. 31i–n).307 Moreover, Kondo resonances have been detected for Tb(Pc)x/Au(111) and Tb2(Pc)3/Ag(111) (Fig. 32d).232,401 However, a Kondo resonance for Y2(Pc)3/Au(111) has not been observed under the same conditions as those used for Y(Pc)2/Au(111) (Fig. 32c).302 It has been shown that interactions with a Cu(111) substrate quench the unpaired π electron of  , explaining the lack of a Kondo resonance. In addition, it has been shown that the unpaired π electron expected for an isolated molecule of
, explaining the lack of a Kondo resonance. In addition, it has been shown that the unpaired π electron expected for an isolated molecule of  is quenched upon adsorption onto Cu(001). In the case of Nd(Pc)2/Cu(100), quenching occurs due to charge rearrangement at the interface.402 On the other hand, a 4f metal centre of Dy(Pc)2/Cu(001) contributes to the Kondo resonance.374 From density functional theory (DFT) calculations, the Kondo resonance occurs due to the coupling between the Dy3+ ion and the Cu electron bath which is mediated by strong hybridization between Dy d and f orbitals.
is quenched upon adsorption onto Cu(001). In the case of Nd(Pc)2/Cu(100), quenching occurs due to charge rearrangement at the interface.402 On the other hand, a 4f metal centre of Dy(Pc)2/Cu(001) contributes to the Kondo resonance.374 From density functional theory (DFT) calculations, the Kondo resonance occurs due to the coupling between the Dy3+ ion and the Cu electron bath which is mediated by strong hybridization between Dy d and f orbitals.
 | ||
| Fig. 31 STM image of the sandwich-type tetrapyrrole rare-earth metal complexes on the Au(111) surface. (a) STM image of an Isolated Tb(Pc)2 molecule. (b) Molecular structure of Tb(Pc)2 which is shown in the upper (lower) Pc ligand is coloured in blue (silver). (c) A simulated STM image of Tb(Pc)2 obtained by using DFT calculations. (d) Molecular structure of (Nc)Tb(Pc). (e–h) STM and DFT simulated STM images of isolated Pc-up and Nc-up ligands of (Nc)Tb(Pc). White bars in (a), (e), and (g) are 1 nm. (i and j) A schematic model of the operation caused by the injection of tunnelling electrons into a (HOEP)Tb(OEP). (i) Proton (H) atom hopping and (j) H atom desorption. (k–n) STM and DFT simulated STM images of isolated (k and m) Tb(OEP)2 and (l and n) (HOEP)Tb(OEP). Topographic images, which is similar to those in (k) and (l), observed by the operations in (i) and (j). Reproduced from (a–c) ref. 302. Copyright 2011, Nature Research. Adapted with permission from (d–h) ref. 135. Copyright 2013, American Chemical Society. Reproduced from (i–n) ref. 307. Copyright 2018, Royal Society of Chemistry. | ||
 | ||
| Fig. 32 STS measurement of the sandwich-type tetrapyrrole rare-earth metal complexes on a metal substrate near the Fermi level. (a) Change in the Kondo peak height of Tb(Pc)2/Au(111) when the tip position is moved along the blue line (bottom) in the STM topography panel. The different colours correspond to the dI/dV intensity, described above the figure. (b) dI/dV spectra measured at the upper Pc ligand of Tb(Pc)2/Au(111) at positions I (centre) and II (ligand) of the molecule. (c) dI/dV spectra and STM topography of Y(Pc)2/Au(111) (I) and Y2(Pc)3/Au(111) (II). (d) STM topography (i) and topography with staggered three Pc ligands (iii) of Tb2(Pc)3/Ag(111). (ii) Map of the fitted Kondo temperature (TK) on a single Tb2(Pc)3/Ag(111). (iv) Averaged point spectra showing the Kondo peak (red x's on ii) on a single Tb2(Pc)3/Ag(111). Reproduced from (a–c) ref. 302. Copyright 2011, Nature Research. Reproduced from (d) ref. 401. Copyright 2018, Royal Society of Chemistry. | ||
Research on the Kondo resonance induced at the interface between a light rare-earth tetrapyrrole sandwich compounds has been reported. For (Pc)Ce(TPP)0 and 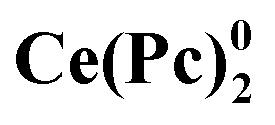 , the formal charge of Ce4+ resulting from the donation of four negative charges to both Pc2− and TPP2− anions causes a 4f0 electronic configuration.403,404 In this case, no Kondo resonance is expected. However, (Pc)Ce(TPP)/Ag(111) exhibits a Kondo resonance but (Pc)Ce(TPP)/Cu(111) does not.403 Thus, a formal 3 +/4+ oxidation state would result in one electron in the 4f orbital and one unpaired π electron in a radical state (˙−)/2− in the ligand for a valence fluctuating system, such as
, the formal charge of Ce4+ resulting from the donation of four negative charges to both Pc2− and TPP2− anions causes a 4f0 electronic configuration.403,404 In this case, no Kondo resonance is expected. However, (Pc)Ce(TPP)/Ag(111) exhibits a Kondo resonance but (Pc)Ce(TPP)/Cu(111) does not.403 Thus, a formal 3 +/4+ oxidation state would result in one electron in the 4f orbital and one unpaired π electron in a radical state (˙−)/2− in the ligand for a valence fluctuating system, such as 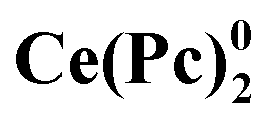 , in which the Ce ion has a valence charge of about 3.57 estimated from Ce LIII-edge X-ray absorption near edge structure spectrum (XANES).403 These systems with fluctuating valences could lead to a ligand-spin induced Kondo resonance similar to those observed for Cu(Pc)/Ag(100) and Tb(Pc)x/Au(111).405 Intermediate valence states make it difficult to explain the presence of a Kondo resonance. Strong hybridization of the molecular orbitals and substrate could result in the quenching of the spin at the interface, eliminating the Kondo resonance. In addition, ligand–ligand interactions in Pc based molecules have been shown to reduce molecule–substrate coupling via the modification of the molecule/substrate distance or charge transfer and therefore to reduce or even quench TK.135,405 For example, the competition between the Kondo effect and Ruderman–Kittel–Kasuya–Yosida (RKKY) coupling in 1D and 2D lattices has been reported.135,406 Thus, theoretical and experimental results are needed to fully understand the spin states at the interface involved in the Kondo process.
, in which the Ce ion has a valence charge of about 3.57 estimated from Ce LIII-edge X-ray absorption near edge structure spectrum (XANES).403 These systems with fluctuating valences could lead to a ligand-spin induced Kondo resonance similar to those observed for Cu(Pc)/Ag(100) and Tb(Pc)x/Au(111).405 Intermediate valence states make it difficult to explain the presence of a Kondo resonance. Strong hybridization of the molecular orbitals and substrate could result in the quenching of the spin at the interface, eliminating the Kondo resonance. In addition, ligand–ligand interactions in Pc based molecules have been shown to reduce molecule–substrate coupling via the modification of the molecule/substrate distance or charge transfer and therefore to reduce or even quench TK.135,405 For example, the competition between the Kondo effect and Ruderman–Kittel–Kasuya–Yosida (RKKY) coupling in 1D and 2D lattices has been reported.135,406 Thus, theoretical and experimental results are needed to fully understand the spin states at the interface involved in the Kondo process.
The magnetic anisotropy and coercivity can be investigated using soft X-ray magnetic circular dichroism (XMCD) measurements on Ln(Pc)2 (Ln3+ = Tb and Dy) on a substrate (Fig. 33).267 Moreover, the magnetic properties of rare-earth metal ions three-dimensionally surrounded by organic ligands in an element-selective manner can be analysed, and the origin of the magnetic anisotropy of SMMs can be determined from the viewpoint of their electronic states. Soft XMCD can be used on a single atomic layer when synchrotron radiation is used. The uniaxial magnetic anisotropy of the Tb3+ ion of Tb(Pc)2 can be determined by measuring the change in the XMCD spectrum when the magnetic field is applied in different directions (vertical and in-plane directions to the substrate). By performing sum-rule analysis on the obtained spectrum, magnetic hysteresis can be obtained from the quantitative evaluation of the Tb 4f magnetic moment and the magnetisation curve for each element. In addition, by applying the sum rule to the XMCD (and/or XAS) spectra, the orbital magnetic moment Lz and spin magnetic moment 2Sz of the Tb3+ ions can be calculated separately. Studies on the magnetic properties of Ln(Pc)2 (Ln3+ = Tb and Dy) on various substrates using XMCD have been reported.345,380,387,391 The ferromagnetic Ni layer forms out-of-plane (OP)/Cu(111) and in-plane (IP)/Ag(100), depending on the type of substrate (Fig. 33d–g).381 When Tb(Pc)2 is adsorbed on these Ni layers, the elemental decomposition hysteresis loop changes according to the magnetisation direction of the ferromagnetic Ni layer due to the magnetic interaction between Tb(Pc)2 and Ni layer. Tb(Pc)2/Fe/Cu(111) has the same antiferromagnetic relationship as the Ni system.382 When Li is deposited, the ferromagnetic exchange interactions change probably because charge transfer occurs when Li is added. Furthermore, it has been reported that the SMM characteristics of Tb(Pc)2 and Dy(Pc)2 adsorbed on the MgO substrate are better (Fig. 33c).377,407 The improvement in the SMM properties is believed to be due to the effects of spin-phonon coupling on the magnetic stabilities of surface-adsorbed SMMs. This suggests that the use of substrates with low phonon density of states is beneficial for the construction of molecular-based spintronics devices and for further increases in the blocking temperature (TB) of surface-adsorbed Ln(Pc)2 (Ln3+ = Tb and Dy).
 | ||
| Fig. 33 XMCD hysteresis curves for Tb(Pc)2 on a metal substrate. (a) Schematic illustration of a Tb(Pc)2 on an ultrathin MgO film on Ag(100). (b) STM image of a 2D film of Tb(Pc)2 on a double-layer film of MgO on Ag(100). (c) Hysteresis loop obtained with XMCD at 3 K and the best fit calculations of magnetisation dynamics for Tb(Pc)2/MgO/Ag(100) and Tb(Pc)2/Ag(100). (d) Schematic illustration of the magnetic anisotropy axis of Tb(Pc)2/Ni(OP)/Cu(100) and (f) elemental Ni and Tb resolved hysteresis loops from XMCD at 8 K. (e) Schematic illustration of the magnetic anisotropy axis of Tb(Pc)2/Ni(IP)/Ag(100) and (g) elemental Ni and Tb resolved hysteresis loops from XMCD at 8 K. Adapted with permission from (a and b) ref. 377. Copyright 2016, Wiley-VCH. Reproduced from (c) ref. 407. Copyright 2019, Wiley-VCH. Reproduced from (d–g) ref. 305. Copyright 2016, Royal Society of Chemistry. | ||
Spintronics of SMMs, like Tb(Pc)2 on a substrate, has been prepared using the spin state control. With respect to Tb(Pc)2 adsorbed on a ferromagnetic Co layer grown on an Ir(111), the spin-polarised tunnel current changes according to the application direction (parallel/antiparallel) of the external magnetic field (Bext) (Fig. 34a).379 It depends on the magnetisation direction of the chip and the sample, and the pattern of spin polarisation of the molecular orbital is observed (Fig. 34b–d). It has been shown that the spin of Tb(Pc)2 is antiferromagnetically coupled to the magnetisation of Co-islands grown on Au(111) regardless of the adsorption site of the Tb(Pc)2.299 Both edge and flat-lying molecules exhibit a stable spin direction opposite to the magnetisation direction of the Co-islands to which they are attached (Fig. 34e). More importantly, the spin state of Tb(Pc)2 can be switched independently of the Co island by applying Bext (Fig. 34f). The magnetic asymmetries of edge and flat molecules are reversed due to the difference in the DOS due to the Tb(Pc)2-Co-island interactions between the two different adsorption configurations. Stable spin polarisation is inferred from the substantial hysteresis observed in the magnetisation curve of the Tb(Pc)2 SMM even with the frozen magnetization of the Co island (Fig. 34g). The observation of significant openings of the hysteresis in the magnetisation curves suggests suppression of the quantum tunnelling of the magnetisation (QTM) process.
 | ||
| Fig. 34 Spin state control of Tb(Pc)2 on a ferromagnetic Co layer. (a) Schematic illustration of the SP-STM experimental setting of a Tb(Pc)2/Co/Ir(111) by applying an external magnetic field (Bext) which is parallel (right) and antiparallel to the magnetisation direction (left). (b) Schematic illustration of the spin-split on the LUMO of Tb(Pc)2 on the ferromagnetic Co layer. (c) Spin-resolved point-mode spectroscopy. The parallel (blue line) and the antiparallel magnetisation directions (red line) (above). Difference in the spectroscopic curves (below). (d) The spin polarisation of the molecular orbitals which have opposite spin polarisations: red (U = +1.0 V) and blue (U = +1.3 V). (e) STM topographic image of Tb(Pc)2/Co/Au(111), which is grouped in islands Co 1 and Co 2. (f) Conductance mapping for Tb(Pc)2/Co/Au(111) obtained with Vs = −600 mV and Bext = +1 T (i) and −1 T (ii). (iii) Difference mapping ΔG (+1 T, −1 T). The scale of dI/dV (top) and the ΔG (bottom) are shown in the colour bar scales. (g) dI/dV signal measured as a function of external magnetic field (Bext) for Tb(Pc)2/Co/Au(111), which are shown in (f). The hysteresis loops of dI/dV shown in both edge and flat-lying molecular positions. Adapted with permission from (a–d) ref. 379. Copyright 2012, Nature Research. Adapted with permission from (e–g) ref. 299. Copyright 2019, American Institute of Physics. | ||
SMMs and/or molecular nano magnets (MNM) have potential for technological applications in magnetic storage and for implementing qubits in the quantum computing (QC).266 The application in magnetic storage requires high anisotropic energy barriers for spin reversal (Ueff) and QTM suppression, whereas QC requires a QTM process, such as a spin cascade |S = 1/2〉‖J = 6〉‖I = 3/2〉 in the  system, for qubit reading and manipulation (the spin cascade mechanism for qubit is described in Section 3.2).270 Therefore, both applications can be achieved using organic–inorganic hybrid materials by developing viable means for coordinating the QTM process in a controlled manner. An approach that uses the hybrid materials to control and enhance the properties of sandwich-type tetrapyrrole rare-earth metal SMMs will accelerate further research towards the realisation of spintronic devices made of functional sandwich-type tetrapyrrole rare-earth metal complexes.
system, for qubit reading and manipulation (the spin cascade mechanism for qubit is described in Section 3.2).270 Therefore, both applications can be achieved using organic–inorganic hybrid materials by developing viable means for coordinating the QTM process in a controlled manner. An approach that uses the hybrid materials to control and enhance the properties of sandwich-type tetrapyrrole rare-earth metal SMMs will accelerate further research towards the realisation of spintronic devices made of functional sandwich-type tetrapyrrole rare-earth metal complexes.
3.8 Rare-earth phthalocyaninato sandwich complex/carbon nanotube hybrid materials
The internal nanospace of carbon nanotubes (CNTs) can be regarded as a reaction vessel that is 1/1000 smaller than a microreactor.408 Therefore, these chemical reaction fields, which have a diameter range of 1–3 nm (100 nm to 0.1 mm in length), are expected to have a great influence on reaction chemistry and/or nanoelectronics research in the 21st century.409–412 It has been shown that atoms and molecules can be contained in the nanospace of CNTs.413–418 Focusing on the use of the stable one-dimensional nanospace of a CNT as a host for host–guest structures, various SMM–CNT hybrid materials (i.e., with single-walled CNTs (SWCNTs): internal diameter of ca. 3 nm and multi-walled CNTs (MWCNTs): internal diameters of (5–50 nm)) have been reported for nanoelectronics and spintronics research.344,419–421In 2009, Kyatskaya et al. first reported a [(Pyrene)Pc]Tb(Pc)–SWCNT hybrid material, in which a [(Pyrene)Pc]Tb(Pc) (one Pc ligand is substituted with three hexyl groups and one 4-(4-pyren-1-ylbutoxy) group) was attached to the outside of a SWCNT, along with its magnetic properties.114[(Pyrene)Pc]Tb(Pc)–SWCNT shows a Coulomb blockade in which a Tb(Pc)2 derivative and a SWCNT act as a quantum dot in a field-effect-transistor (FET) device. In the spin valves of [(Pyrene)Pc]Tb(Pc)–SWCNT, the maximum magnetic resistance ratio between the parallel state and the antiparallel state is approximately 300% at submillikelvin temperatures.117,312 In 2011, in a related study, Urdampilleta et al. reported hybrid devices that consist of chemical vapor deposition (CVD)-grown CNT transistors decorated with Tb(Pc)2 SMMs.269,309 The devices were achieved by tailoring the supramolecular π–π interactions between the Tb(Pc)2 SMMs and CNTs (Tb(Pc)2–CNT) (Fig. 35a). Magnetoresistance measurements revealed steep steps, which were related to the magnetisation reversal of the individual Tb(Pc)2 SMMs. The electron-transport properties of these devices are strongly dependent on the relative magnetisation orientation of the grafted SMMs. In this material, the Tb(Pc)2 SMMs act as a local spin polarizer and analyser on the CNT electron-conduction channel. As a result, reluctance ratios of up to several hundred percent were observed at submillikelvin temperatures. These results suggest that a spin valve with a larger magnetoresistance ratio can be prepared by using SMMs as the spin source instead of ferromagnets. These systems were demonstrated to detect differential conductance (dI/dV) by taking advantage of the switching of the QTM steps by the corresponding magnetic field (μ0Hz < ±50 mT). In other words, readout of the nuclear spin state was realized using Tb(Pc)2 SMMs.
 | ||
| Fig. 35 Supramolecular spin valves based on the Tb(Pc)2–SWCNT hybrid device. (a) Schematic illustration of the device. (b) Conductance hysteresis loops of Tb(Pc)2–SWCNT. dI/dV measured at 40 mK with an in-plane magnetic field applied along the easy axial direction (0°). Bias and temperature dependences of the conductance hysteresis loops of Tb(Pc)2–SWCNT. (c) Scale map of the dI/dV hysteresis as a function of the in-plane magnetic field and source drain voltage (Vds). (d) Temperature dependence of the conductance hysteresis loops. (e) Supramolecular structure of the Tb(Pc)2@SWCNT hybrid material. TEM image and schematic illustration of Tb(Pc)2@SWCNT. (f) STM image showing empty (red dot) and filled (black dot) regions. Comparative STS spectra measured at the red and black dotted points in STM image. The STS of Tb(Pc)2 adsorbed on an Au(111) surface is illustrated as plot III (blue) in (f) as a reference. Reprinted from (a–d) ref. 269. Copyright 2011, MDPI. Reproduced from (e and f) ref. 422. Copyright 2021, Royal Society of Chemistry. | ||
In 2019, Abd El-Mageed et al. reported a supramolecular structure formed by a Tb(BDP)2–SWCNT hybrid material, in which Tb3+-5,15-bisdodecylporphyrin (BDP) double-decker SMMs were attached to the outside of an SWCNT.423 Scanning tunneling microscopy (STM) confirmed an ordered, self-organized spiral array on the SWCNT surface stabilized by non-sharing (π-stacking) interactions. In addition, atomic force microscopy (AFM) images exhibited a distinctive debundling effect due to the adsorption of the  SMMs on the SWCNT surface with some helical array structures. The SMM properties of Tb(BDP)2–SWCNT were investigated, and the hybrid materials interestingly showed larger butterfly-shaped hysteresis loops than the
SMMs on the SWCNT surface with some helical array structures. The SMM properties of Tb(BDP)2–SWCNT were investigated, and the hybrid materials interestingly showed larger butterfly-shaped hysteresis loops than the  SMMs alone, indicating a slow τ for Tb(BDP)2–SWCNT. So far, electron-transport properties have not yet been reported for Tb(BDP)2–SWCNT.
SMMs alone, indicating a slow τ for Tb(BDP)2–SWCNT. So far, electron-transport properties have not yet been reported for Tb(BDP)2–SWCNT.
In 2021, Katoh et al. first reported the encapsulation of Tb(Pc)2 SMMs in the internal nanospace of SWCNTs (Tb(Pc)2SMM@SWCNT).422 The magnetic and electronic properties of the Tb(Pc)2SMM@SWCNT hybrids (Fig. 35e) were investigated in detail using DC and AC magnetic susceptibility measurements, transition electron microscopy (TEM), scanning electron microscopy (SEM), STM, and STS. By arranging the Tb(Pc)2 SMMs in the 1D internal nanospaces of the SWCNTs, it is possible to investigate the essential SMM characteristics of Tb(Pc)2 without considering LF effects. Moreover, it appears that the electron correlation between Tb(Pc)2 and the SWCNT can affect the electron transport and/or electromagnetic properties. Since bandgap modulation was observed due to the encapsulation of Tb(Pc)2 in SWCNTs, these materials should be useful in FETs. Furthermore, as the stable internal nanospace of SWCNTs is used, it is thought that the density of the SMMs in the SMM–SWCNT hybrid material can be controlled, and the hybrids should be usable as spin valves. This strategy may pave the way for the construction of SMM–SWCNT hybrid materials for operable molecular spintronic devices using the quantum effects of individual SMMs.
4. Semiconducting materials based on REE sandwich complexes
Discovery of semiconducting properties of phthalocyanines reported by the group of J. Simon in 1987 on the examples of radical complexes LiPc and Lu(Pc)2 is definitely one of important milestones not only for tetrapyrrolic compounds, but for all organic electronics in general.35 This discovery nearly coincided with the first report on organic field effect transistors (OFETs) made in 1986 by Tsumura et al. with polytiophene conducting layer,424 thus no surprise that the same year Lu(Pc)2 was introduced into OFET technology.OFETs attract increasing research interest since they were first reported due to their potential for application in flexible displays, integrated circuits and low-cost electronic devices. A typical OFET device consists of an organic semiconducting layer, a dielectric layer, and three electrodes (source, drain, and gate electrodes). Three key parameters define the performance of an OFET device, namely charge carrier mobility (μ), current on/off ratio, and threshold voltage (VT). The organic semiconducting layer is the most important part to determining the OFET performance, though other factors, such as the device configuration, the interfaces, and the materials for electrodes and insulators, also can affect the performance.425,426
π-Conjugated tetrapyrrole derivatives, including porphyrins (Pors) and phthalocyanines (Pcs), are important organic semiconducting materials for OFETs because of their unique molecular and electronic structures.427,428 For rare-earth based tetrapyrrolic sandwiches, the molecular energy levels are highly disturbed by the intramolecular π–π interactions between tetrapyrrolic rings, typically possessing destabilized HOMOs and stabilized LUMOs, thus having decreased molecular energy gaps in comparison to the constituent monomeric tetrapyrroles. This is desirable for improving the semiconducting properties of tetrapyrrolic materials, as demonstrated by Guillaud et al. for Lu(Pc)2 and Tm(Pc)2 based OFETs.429 The molecular energy levels of tetrapyrrolic sandwiches can be further tuned by choosing different ligands and rare-earth metal ions, adjusting the number of deckers, and incorporating various substitutes on tetrapyrrolic ligands.1,7 The intermolecular interactions and solubility of sandwiches are also adjustable by such chemical modifications, which can provide further opportunity for the fabrication of solution-processable and high quality films for OFET devices.
Moreover, specific substitution of sandwich complexes may allow for the formation of supramolecular structures that provide intrinsic dielectric insulation at the molecular level. For example, ultrathin solid films of structurally similar octa-butoxy and tetra-crown-ether substituted Eu(III) bisphthalocyaninates, Eu[(BuO)8Pc]2 and Eu[(15C5)4Pc]2, were formed and their transversal conductivity and, consequently, electrochemical activity were studied in an aqueous electrolyte.430 Excellent transfer of charge through monomolecular ultrathin films of crown-substituted complex was observed enabling its redox-multistability, which can be utilized in molecular logic and information storage devices. However, transversal charge transfer was inhibited in the films formed by octa-butoxy-substituted complex, which might be promising for OFET applications.
Depending on the correlation between the molecular energy levels of the semiconducting layer and the work function of source–drain electrodes, holes or electrons can be preferentially injected from the source electrode to the semiconducting layer, and transported to the drain electrode, leading to p- or n-type transporting modes, respectively. If both types of charge carriers can be injected and transported, an ambipolar OFET device is achieved, which, however, is of great importance in complementary metal oxide semiconductor (CMOS) logic circuits.431 Most tetrapyrrolic sandwiches can exhibit p-type OFET performance in air with usual source–drain electrodes (such as Au, with a work function of 5.1 eV), because of the suitable HOMO energy levels of sandwiches for hole injection (Table 1). However, unipolar n-type operations are often detected under an inert (or reducing) gas atmosphere, because of the relatively high LUMO energy levels of sandwiches, which cannot resist the air-derived electron traps. On the other hand, due to the versatility of tuning the HOMO and LUMO energy levels of tetrapyrrolic sandwiches by molecular design,432 single-component air-stable ambipolar OFET devices based on tetrapyrrolic sandwiches have become the research focus in recent years (Table 2).
| Compounds/deposition method | Hole mobility [cm2 V−1 s−1] | V T [V] | I on/Ioff | L and W [μm] | Source–drain electrode | Gate electrode | Insulator | Ref. |
|---|---|---|---|---|---|---|---|---|
| [(OctO)8Pc]Tb(Pc)/LB | 6.4 × 10−4 | 5.5 × 104 | 240 and 28600 |
Au | Si | SiO2 | 433 | |
| [(OctO)8Pc]Lu(Pc)/LB | 1.7 × 10−3 | 102 | ||||||
| Lu[(α-Oct)8Pc]2/SC | 1.5 × 10−3 | −25 | 103 | 10 and 2000 | Au/Cr | n-Type Si | SiO2/OTS | 434 |
| Lu[(α-Oct)8Pc]/SC, TA | 8.0 × 10−3 | −12.5 | 105 | 10 and 2000 | Au/Cr | n-Type Si | SiO2/OTS | |
| Gd[(α-C8H17)8Pc]2/SC | 7.47 × 10−5 | −28.9 | 103 | 5 and 20000 |
Au | Si | SiO2/OTS | 435 |
| Gd[Pc(α-C8H17)8]2/SC | 1.96 × 10−5 | −27.6 | 2 × 102 | 20 and 20000 |
Au | Si | SiO2/OTS | |
| [(OctO)8Pc]Eu[(15C5)4Pc]Eu[(15C5)4Pc]/LB | 0.60 | — | 1.4 × 105 | 240 and 28600 |
Au | Si | SiO2 | 436 |
| [(OctO)8Pc]Ho[(15C5)4Pc]Ho[(15C5)4Pc]/LB | 0.40 | — | — | Si | ||||
| [(OctO)8Pc]Lu[(15C5)4Pc]Lu[(15C5)4Pc]/LB | 0.24 | — | — | Si | ||||
| [(BuO)8Pc]Eu[(15C5)4Pc]Eu[(15C5)4Pc]/LB | 0.0032 | — | 9.3 × 10 | 240 and 28600 |
Au | Si | SiO2 | 437 |
| [(HexO)8Pc]Eu[(15C5)4Pc]Eu[(15C5)4Pc]/LB | 0.014 | — | 8.2 × 103 | |||||
| [(DecO)8Pc]Eu[(15C5)4Pc]Eu[(15C5)4Pc]/LB | 0.21 | — | 9.7 × 104 | |||||
| [(C12H25O)8Pc]Eu[(15C5)4Pc]Eu[(15C5)4Pc]/LB | 0.053 | — | 3.9 × 10 | |||||
| [(HexO)8Pc]Eu[(DEGOMe)8Pc]Eu[(DEGOMe)8Pc]/LB | 0.46 | — | 1.01 × 102 | 240 and 28600 |
Au | Si | SiO2 | 438 |
| [(OctO)8Pc]Eu[(DEGOMe)8Pc]Eu[(DEGOMe)8Pc]/LB | 0.17 | — | 1.02 × 103 | |||||
| [(DecO)8Pc]Eu[(DEGOMe)8Pc]Eu[(DEGOMe)8Pc]/LB | 0.096 | — | 3.85 × 104 | |||||
| [(C12H25O)8Pc]Eu[(DEGOMe)8Pc]Eu[(DEGOMe)8Pc]/LB | 0.014 | — | 1.15 × 102 | |||||
| [(OctO)8Pc]Eu[(12C4)4Pc]Eu[(12C4)4Pc]/LB | 0.03 | — | 2.32 × 103 | 240 and 28600 |
Au | Si | SiO2 | 439 |
| 0.31 | — | 8.59 × 104 | SiO2/HMDS | |||||
| 0.33 | — | 7.91 × 105 | SiO2/OTS | |||||
| [(OctO)8Pc]Eu[(18C6)4Pc]Eu[(18C6)4Pc]/LB | 0.02 | — | 5.5 × 103 | 240 and 28600 |
Au | Si | SiO2 | |
| 0.28 | — | 1.73 × 105 | SiO2/HMDS | |||||
| [(Dec)4Por]Eu[(15C5)4Pc]Eu[(15C5)4Pc]/LB | 0.78 | −3.84 | 2.20 × 104 | 240 and 28600 |
Au | Si | SiO2/HMDS | 440 |
| 0.04 | −4.02 | 4.94 × 103 | SiO2/OTS | |||||
| [(p-PentOPh)4Por]Eu[(15C5)4Pc]Eu[(15C5)4Pc]/LB | 0.05 | −1.19 | 1.48 × 104 | SiO2/HMDS | ||||
| 0.03 | −4.34 | 1.03 × 104 | SiO2/OTS | |||||
| Tb2[(BuO)8Pc]3/DC | 1–2 × 10−5 | — | — | 20 and 150 | — | Si | SiO2 | 441 |
| Compounds/deposition method | Hole mobility [cm2 V−1 s−1] | I on/Ioff | Electron mobility [cm2 V−1 s−1] | I on/Ioff | −VT [V] | L and W [μm] | Source–drain electrode | Gate electrode | Insulator | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Y(Pc)2/DFT | 0.034 | 0.031 | — | — | — | Au | — | — | 432 | |
| La(Pc)2/DFT | 0.17 | 0.088 | — | — | — | Au | — | — | ||
| [(PhO)8Pc]Ho(Pc)/QLS film | 1.7 × 10−4 in air | — | 103 | −21 | 240 and 28600 |
Au | Si | SiO2 | 442 | |
| Ho[(PhO)8Pc]2/QLS film | — | 0.54 in N2 | 104 | 12 | 240 and 28600 |
Au | Si | SiO2 | ||
| Eu[(NaphO)8Pc]2/QLS film | 0.10 in air | 104 | 0.01 in N2 | 102 | 240 and 28600 |
Au | Au | PMMA | 38 | |
| Eu[(SPh)8Pc]2/QLS film | 1.1 × 10−4 in air | — | 0.11 in N2 | — | — | — | Au | Si | SiO2/HMDS | 443 |
| Ho[Pc(SPh)8]2/QLS film | 4.9 × 10−7 in air | 3.0 × 10−3 in N2 | ||||||||
| [(PhS)8Pc]Eu(Pc)/QLS film | 4.4 × 10−3 | |||||||||
| [(PhS)8Pc]Ho(Pc)/QLS film | 2.6 × 10−3 | |||||||||
| (Pc)Eu[f-Pc2]Eu(Pc)OBu/QLS film | 0.8 in air | — | 2.3 in N2 | — | −3 + 12 | 240 and 28600 |
Au | Si | SiO2/HMDS | 221 |
| (TPP)Eu[(PhO)8Pc]Eu[(PhO)8Pc]/QLS film | 0.04 in air | 105 | 0.08 in N2 | 105 | — | 240 and 28600 |
Au | Si | SiO2/HMDS | 444 |
| Eu2[(p-FPhO)8Pc]3/QLS film | 0.24 | — | 0.042 | — | −1.1 + 13 | W = 28.6 mm, L = 240 μm, W/L = 119 | Au | Si | SiO2/HMDS | 44 |
| [(PhO)8Pc]Eu[(PhO)8Pc]Eu(Pc)/QLS film | 0.68 | — | 0.014 | — | — | 240 and 28600 |
Au | Si | SiO2/HMDS | 42 |
| Eu2[(PhO)8Pc]3/QLS film | 0.041 | — | 0.0026 | — | — | |||||
| Eu2[(NaphO)8Pc]3/QLS film | 2.5 × 10−6 | 102 | 2.3 × 10−6 | 102 | −8 + 10 | 240 and 28600 |
Au | Si | SiO2/HMDS | 40 |
| Solvent-vapor annealing 60 °C | 2.6 × 10−7 | 102 | 2.3 × 10−8 | 102 | −1 + 3 | |||||
| Solvent-vapor annealing 80 °C | 4.9 × 10−4 | 103 | 7.1 × 10−5 | 102 | −20 + 18 | |||||
| Solvent-vapor annealing 100 °C | 0.11 | 105 | 0.06 | 105 | −3 + 9 | |||||
| Solvent-vapor annealing 120 °C | 1.1 × 10−2 | 104 | 9.6 × 10−3 | 104 | −4 + 5 | |||||
[(PhO)8Pc]Eu[(PhO)8Pc]Eu[(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCOOH)Por]/QLS film CCOOH)Por]/QLS film |
7.0 × 10−7 | 102 | 7.5 × 10−7 | 102 | −8 + 8 | 240 and 28600 |
Au | Si | SiO2/HMDS | 445 |
|
[(PhO)8Pc]Eu[(PhO)8Pc]Eu[(CCCOOH)Por]/nanoribbon |
0.11 | 106 | 4 × 10−4 | 104 | −3 + 17 | |||||
| CdEu2[(HexS)8Pc]4/QLS film | 5.6 × 10−3 | 10 | 1.2 × 10−3 | 10 | — | — | — | — | — | 206 |
| (Pc)2Eu2[f-Pc2]Eu2(Pc)2SHex/single crystal microsheets | 18 | 103–104 | 0.3 | 103–104 | — | — | Au | Si | SiO2/HMDS | 223 |
| [(Py4Por)Eu[(PhO)8Pc]Eu[(PhO)8Pc]]/QLS film | 6.9 × 10−7 | 102 | 3.07 × 10−5 | 102 | — | — | Au | Si | SiO2/HMDS | 446 |
| [(Py4Por)Eu[(PhO)8Pc]Eu[(PhO)8Pc]]/CdS hybrid QLS film | 0.09 | 104 | 0.01 | 103 | — | |||||
| (TClPP)Y(Pc) and fullerene C60 cocrystals | 3.72 | 102 | 2.22 | 102 | — | 240 and 28600 |
Au | Si | SiO2 | 447 |
| (TFPP)Eu[(PhO)8Pc]Eu[(PhO)8Pc]/QLS film | 6.0 × 10−5 | — | 1.4 × 10−4 | — | −20 + 38 | 240 and 28600 |
Au | Si | SiO2 | 448 |
| (TFPP)Eu[(PhO)8Pc]Eu[(PhO)8Pc]/QLS film/CuPc VCD film: | 0.16 | — | 0.30 | — | −13 + 25 | |||||
| [(p-HOPh)4Por]Eu(Pc)Eu(Pc)/QLS film | 4.85 × 10−8 | 102–103 | 2.33 × 10−8 | 102–103 | — | 240 and 28600 |
Au | Si | SiO2/HMDS | 449 |
| [(p-HOPh)4Por]Eu(Pc)Eu(Pc)/rGO/QLS film | 25.53 | 103–104 | 0.77 | 103–104 | — | |||||
| [(p-HOPh)4Por]Eu[(PhO)8Pc]Eu[(PhO)8Pc]/QLS film | 2.14 × 10−6 | 102–103 | 2.33 × 10−8 | 102–103 | — | |||||
| [(p-HOPh)4Por]Eu[(PhO)8Pc]Eu[(PhO)8Pc]/rGO/QLS film | 30.9 | 103–104 | 39.6 | 103–104 | — |
4.1 p-Type OFETs
In 2005, Su et al. reported LB film-based OFETs by using heteroleptic bis(phthalocyaninato) rare earth complexes [(OctO)8Pc]M(Pc), M = Tb and Lu as the electroactive components, which showed hole mobilities of 6.4 × 10−4 and 1.7 × 10−3 cm2 V−1 s−1, respectively, on a SiO2 dielectric layer with top contacted Au source–drain electrodes.433 Chen et al. designed amphiphilic tris(phthalocyaninato) rare earth complexes [(OctO)8Pc]M[(15C5)4Pc]M[(15C5)4Pc], M = Eu, Ho and Lu, and fabricated LB film-based OFETs with SiO2/Si substrates and top contacted Au source–drain electrodes.436 The devices exhibited highly improved hole mobilities in the range of 0.24–0.60 cm2 V−1 s−1 with an on/off current ratio of 105, which was ascribed to the intramolecular π–π stacking among Pc rings and the J-aggregation in the LB films.437In order to study the substituent effect on the assembling properties and OFET performance of tris(phthalocyaninato) triple-deckers [(CnH2n+1O)8Pc]Eu[(15C5)4Pc]Eu[(15C5)4Pc], n = 4, 6, and 8, different lengths of alkoxy chains were connected to one of the outer phthalocyanine ligands.437 The results indicated that the molecular packing and intermolecular interaction in LB films are closely related to the length of hydrophobic side chains, thus leading to the change of hole mobilities within 0.0032–0.60 cm2 V−1 s−1.
A similar trend was observed for another series of amphiphilic tris(phthalocyaninato) rare earth complexes [(CnH2n+1O)8Pc]Eu[(DEGOMe)8Pc]Eu[(DEGOMe)8Pc], DEGOMe = (OC2H4)2OCH3, n = 6, 8, 10 and 12.438 In the OFET devices with bottom contacted Au source–drain electrodes, the LB films of those triple-deckers also displayed hole mobilities in the range of 0.014–0.46 cm2 V−1 s−1, depending on the length of hydrophobic alkoxy chains.
The hydrophilic sides of tris(phthalocyaninato) rare earth complexes were also modified with different crown ether substituents.439 Replacing the 15-crown-5 rings of [(OctO)8Pc]M[(15C5)4Pc]M[(15C5)4Pc] (Fig. 36a) with 12-crown-4 or 18-crown-6 moieties resulted in different packing modes in LB films. Thus, 12-crown-4 and 15-crown5-substituted complexes formed edge-to-edge connected J-aggregates, providing a charge transfer channel parallel to the Pc rings. In contrast, 18-crown-6-substituted complex formed face-to-face connected H-aggregates in LB films, providing a channel along the long axis of the assemblies (Fig. 36b and c). In addition, the authors found that the surface treatment of the SiO2/Si substrate by hexamethyldisilazane (HMDS) or octadecyltrichlorosilane (OTS) can significantly improve the quality of the LB film, thus leading to an increased carrier mobility.
 | ||
| Fig. 36 (a) Amphiphilic Eu(III) crown-phthalocyaninates [(OctO)Pc]Eu[(crown)4Pc]Eu[(crown)4Pc] and schematic arrangement of complexes [(OctO)8Pc]Eu[(12C4)4Pc]Eu[(12C4)4Pc] – (b) and [(OC8H17)8Pc]Eu[(18C6)4Pc]Eu[(18C6)4Pc] – (c) in the LB film-based OFET devices. Adapted with permission from ref. 439. Copyright 2009, American Chemical Society. | ||
Mixed-ligand (phthalocyaninato)(porphyrinato) Eu(III) triple-deckers (Por*)Eu[(15C5)4Pc]Eu[(15C5)4Pc] where Por* = tetra-meso-decylporphyrinate or tetra-meso-(p-pentyloxyphenyl)porphyrinate were synthesized and tested in LB film-based OFET devices.440 These triple-deckers adopt a “face-to-face” connected “edge-on” orientation on HMDS- or OTS-treated SiO2/Si surfaces, leading to the effective intramolecular π–π stacking in films. As a result, the hole mobility of OFET devices can reach 0.78 cm2 V−1 s−1, with an on/off ratio of 2.2 × 104 and a threshold voltage of −3.84 V.
In addition to LB films, drop-casted and spin-coated films of tetrapyrrolic sandwiches have also been applied for OFET devices. By using spin-coated films of a liquid crystalline bis(phthalocyaninato) lutetium complex Lu[(α-Oct)8Pc]2, Ray et al. fabricated bottom gated OFET devices, which showed a hole mobility of 1.5 × 10−3 cm2 V−1 s−1 with an on/off current ratio of 103 at a threshold voltage of −25 V.434 After thermal annealing, the hole mobility was improved to 8.0 × 10−3 cm2 V−1 s−1, together with an increased on/off ratio by two orders of magnitude and a reduced threshold voltage to half of that of the as-deposited films. Recently, through the study of spin-coated films of the gadolinium complex Gd[(α-Oct)8Pc]2,435 Ray et al. revealed that the OFET performance decreases along with scaling down the channel length of devices. The electronic transport properties of drop-casted films of a triple-decker complex Tb2[(BuO)8Pc]3 were reported by Yamashita et al., which also show p-type mobilities in the range of 1–2 × 10−5 cm2 V−1 s−1.441
4.2 Ambipolar OFETs
The ambipolar semiconducting properties of unsubstituted bis(phthalocyaninato) rare earth complexes were initially reported by Simon and co-workers in 1990s.450 The high intrinsic conductivity of Lu(Pc)2 and Tm(Pc)2 was ascribed to the narrow molecular energy gaps associated with their radical nature.429 The ambipolar charge transfer properties of M(Pc)2, M = Y or La were also studied by density functional theory (DFT) calculations, which suggest that the delocalized radical can induce a high HOMO level and a low LUMO level, leading to a small ionization potential and a large electronic affinity. As a result, small injection barriers for both hole and electron from source–drain electrodes (Au) can be expected for these neutral double-deckers.432 The DFT study also revealed small charge reorganization energy and large transfer integral values in the crystal of M(Pc)2, thus predicted promising ambipolar charge transfer properties. However, in experimental studies, single-component bis(phthalocyaninato) rare earth complex-based OFET devices usually show p-type transporting properties in air, the corresponding n-type operations only can be detected under an inert or reducing atmosphere.By connecting electron withdrawing phenoxyl groups onto one or both Pc rings of rare earth double-deckers, heteroleptic [(PhO)8Pc]Ho(Pc) and homoleptic Ho[(PhO)8Pc]2 were obtained, which exhibit p- and n-type transporting properties, respectively, in the quasi-Langmuir–Shäfer (QLS) film-based OFET devices.442 Introducing other electron withdrawing groups, such as naphthoxyl38 and phenylthiol,443 to the Pc rings of rare earth double-deckers can lead to transforming their semiconducting nature from p-type to ambipolar type by tuning their HOMO/LUMO energy levels.37 A dimeric phthalocyanine ligand was incorporated into rare earth sandwiches, leading to the formation of a yttrium(III) double-decker dimer (Pc)Y[f-Pc2]Y(Pc)OBu (cf. Section 2.6.2),216 which exhibits a unique biradical state and can be fabricated into high quality QLS films, thus displaying very high carrier mobilities of 2.3 and 0.8 cm2 V−1 s−1 for electrons and holes in air and N2, respectively (Table 2).221
Molecular orbital (MO) levels of sandwiches can be adjusted by the addition of another tetrapyrrolic ring to double-deckers. In this way, a mixed (phthalocyaninato)(porphyrinato) europium(III) triple-decker complex (TPP)Eu[(OPh)8Pc]Eu[(OPh)8Pc] was prepared and fabricated into QLS film-based OFET devices, which displayed balanced hole and electron mobilities in air and N2, respectively.444
Tetrapyrrolic sandwich-based air-stable ambipolar OFET devices were first achieved by using a partially fluorinated tris(phthalocyaninato) europium complex Eu2[(p-FPhO)8Pc]3.44 The introduction of electron-withdrawing 4-fluoro-phenoxy groups on the peripheral sites resulted in finely controlled HOMO (−5.27 eV) and LUMO (−4.17 eV) levels; both facilitate charge carrier injection from Au electrodes. In particular, the LUMO level is suitable for electron transport under ambient conditions. The triple-decker molecules also form H-aggregates and adopt an “edge-on” conformation in the QLS films, which can provide intense intermolecular interaction and π-electron delocalization. As a result, the QLS film-based single-component OFET devices display an average hole mobility of 0.24 cm2 V−1 s−1 and an average electron mobility of 0.042 cm2 V−1 s−1 in air, with relatively low threshold voltages of −1.1 and +13 V for holes and electrons, respectively. By taking a similar optimizing strategy, tris(phthalocyaninato) europium complexes [(PhO)8Pc]Eu[(PhO)8Pc]Eu(Pc), Eu2[(PhO)8Pc]3 and Eu2[(NaphO)8Pc]3 were obtained. The HOMO and LUMO levels of these triple-deckers were also successfully tuned into the range for air-stable ambipolar semiconductors by the introduction of electron-withdrawing phenoxy42 or naphthoxy subsititutes.40
Triple-decker complex Eu2[(NaphO)8Pc]3 was used to fabricate ambipolar OFET device by QLS technique, and nanoparticles of the pristine QLS film could be converted into well-defined microstructures using solvent vapour annealing with o-dichlorobenzene (Fig. 37).40 Fragment-like irregular polygons, quasi-four-corner sheets, 2D squares, and clovers were obtained at different temperatures (60, 80, 100 and 120 °C, respectively), providing access to the tuning of OFET device performance due the carrier mobilities depending on morphologies and crystallinities. The best result was achieved for the most crystalline 2D squares-based devices with the carrier mobilities of 0.11 ± 0.03 cm2 V−1 s−1 for holes and 0.06 ± 0.02 cm2 V−1 s−1 for electrons.
 | ||
| Fig. 37 The carrier mobilities for holes and electrons in pristine QLS film of Eu2[(NaphO)8Pc]3 and microstructures obtained by solvent vapour annealing at different temperatures together with SEM images of the corresponding films. Adapted from ref. 40, Copyright (2018) with permission Elsevier. | ||
A further modification was performed by replacing one of the outer Pc ligands with an amphiphilic low-symmetry porphyrin ring (Fig. 38a).445 The resulting triple-decker can form 1-D nanoribbons via a “phase-transfer” process (Fig. 38b), which show improved air-stable ambipolar semiconducting properties with mobilities of 0.11 and 4 × 10−4 cm2 V−1 s−1 for holes and electrons, respectively, 3 to 6 orders higher than those of non-crystalline QLS film (Fig. 38c).
 | ||
| Fig. 38 (a) Amphiphilic mixed-ligand Eu(III) complex and SEM images of films showing crystallinity of films formed by the “phase transfer” process – (b) and non-crystalline films obtained by the quasi-Langmuir–Shäfer process – (c) Adapted with permission from ref. 445. Copyright 2016, American Chemical Society. | ||
Further extension of sandwiches was beneficial for their OFET behaviour. Thus a quadruple-decker complex Eu2Cd[(HexS)8Pc]4 exhibited air-stable ambipolar semiconducting characters due to the extended π-system and narrowed bandgap206 The best result for phthalocyanine-based OFETs was achieved with a dimeric phthalocyanine ligand involved sandwich-type triple-decker complex (Pc)2Eu2[f-Pc2]Eu2(Pc)2SHex.223 This special sandwich can be fabricated into single crystal microsheets, which display high hole and electron mobilities of 18 and 0.3 cm2 V−1 s−1, respectively, with on/off ratios of 103–104.
Another approach for ambipolar OFETs is the fabrication of bicomponent films and/or cocrystals with triple-decker complexes as one of the components. For example, bicomponent thin film transistors of (TPyP)Eu[(OPh)8Pc]Eu[(OPh)8Pc]/CdS show highly improved charge mobilities for holes (0.09 cm2 V−1 s−1) and electrons (0.01 cm2 V−1 s−1) relative to those of single-component thin film transistors of (TPyP)Eu[(PhO)8Pc]Eu[(PhO)8Pc] for holes (6.9 × 10−7 cm2 V−1 s−1) and electrons (3.07 × 10−5 cm2 V−1 s−1).446 OFET devices based on the cocrystals of (TClPP)Y(Pc)Y(Pc) and C60 also display very high mobilities of 3.72 and 2.22 cm2 V−1 s−1 for holes and electrons, respectively, which is proposed to be due to similar LUMO energy levels of the two components and the low reorganization energy values for holes and electrons at the interface.447 On the other hand, ambipolar OFETs have been achieved with a bilayer heterojunction configuration by using triple-decker complexes as air-stable ambipolar semiconductors. The QLS films of (TFPP)Eu[(PhO)8Pc]Eu[(PhO)8Pc] were obtained on vacuum deposited (VCD) CuPc templates, and fabricated into two-component bilayer thin film transistors.448 The devices possess charge mobilities of 0.16 and 0.30 cm2 V−1 s−1 for holes and electrons, respectively, which are 103–105 times of those of the single-component based devices. The improvement of the performance can be attributed to the heterojunction effect and the H-aggregation of triple-deckers on CuPc films. Bilayer heterojunction FETs were also fabricated by QLS films of [TP(OH)P]Eu(Pc)Eu(Pc) and [TP(OH)P]Eu[(PhO)8Pc]Eu[(PhO)8Pc], as the top layers, and reduced graphene oxide (rGO) film as the sublayers. The obtained devices exhibit very high and balanced carrier mobilities of 30.9 and 39.6 cm2 V−1 s−1 for holes and electrons, respectively, again due to the heterojunction effect.449
4.3 Supramolecular assembling of semiconductive materials based on sandwich crown-substituted complexes
The relationship between morphology and conductivity of layers formed by tetrapyrrole sandwiches in electronic devices makes it possible to apply supramolecular tools to control these factors. For this purpose, molecular blocks capable of exhibiting various types of intermolecular interactions, such as hydrophobic and stacking interactions, as well as hydrogen bonds, were synthesized and studied. However, the assembly of such molecules is a trial-and-error method, since the morphology and conductivity of the film cannot be predicted a priori. To some extent, this problem can be solved by applying cation-induced assembly of crown-substituted phthalocyanines, where well-established cooperative effects govern the formation of well-defined supramolecular architectures (cf. Section 3.3.1).Thus, Zvyagina et al. used homoleptic crown-substituted bisphthalocyaninates M[(15C5)4Pc]2, M = Ce, Tb, and Lu as potential molecular blocks for cation-induced formation of 1D supramolecular polymers {M[(15C5)4Pc]2·4+}n where enhanced conductivity was expected (Fig. 39a).451 Noteworthy, this possibility was envisaged in 1994 by Toupance et al. in one of keystone papers on the construction of ionoelectronic materials based on crown-substituted phthalocyanines; however, experimental verification of this hypothesis was reported only in 2021.
 | ||
| Fig. 39 (a) Modes of interaction of M[(15C5)4Pc]2 with potassium cations – intramolecular for M = Tb and Ce vs. intermolecular for M = Lu. (b) The current−voltage characteristic of the drop-cast films of the assemblies formed by M[(15C5)4Pc]2 and KBPh4. Inset shows SEM image of nanowires formed by Lu(III) complex. (c) Schematic illustration of the mechanism of alignment of the {Lu[(15C5)4Pc]2·4KBPh4}n nanowires. Adapted with permission from ref. 451. Copyright 2021, American Chemical Society. | ||
Indeed, interaction of Lu(III) complex with KBPh4 in chloroform–acetonitrile mixture resulted in the intermolecular binding of potassium cations which connect neighbouring double-deckers in a cofacial manner, yielding nanowires with a thickness of 10–50 nm and a length of up to 50 μm. The four-probe technique in a four-stripe layout revealed the superior conductivity of these wires up to 11.4 S cm−1, the highest among them being reported for phthalocyanine assemblies (Fig. 39b). The electric properties of these 1D assemblies made it possible to use technologically feasible methods for their controlled alignment on surfaces under an applied electric field (Fig. 39c).
In striking contrast, cerium- and terbium-based complexes underwent mainly intramolecular cation binding without the formation of conductive nanowires (Fig. 39a and b). Apparently, this is associated with a larger interligand distance in these complexes allowing potassium cations to intercalate between crown-substituted ligands.452 The films, formed by the resulting assemblies had 8 orders of magnitude lower conductivity due to the lack of intermolecular interactions.
The way to controllable immobilization of supramolecular assemblies onto the electroactive surfaces was paved by Shokurov et al., who used the octopus-like double-decker anchor, octopus-Pc (Scheme 4) which can form self-assembled monolayer (SAM) on the surface of Au electrodes (Fig. 40).73 Face-on orientation of the anchors allowed for the subsequent binding of crown-phthalocyanine molecules H2[(15C5)4Pc]via potassium ion bridges. This chemistry was utilized to form a heterogeneous bilayer, in which a single molecule thick adlayer brought an additional redox-state to the system, thus expanding the multistability of the system as a whole. All four redox states available to this system exhibited characteristic absorbance in the visible range, allowing for the switching to be easy read out using optical density measurements. It is expected that the proposed approach can be used in a wide range of switchible materials – single-molecule magnets, conductive and optical devices, etc.
 | ||
| Fig. 40 Formation of a bilayer composed of H2[(15C5)4Pc] molecules appended to octopus-Pc SAM on gold via supramolecular assembly. Adapted with permission from ref. 73. Copyright 2022, Wiley-VCH. | ||
5. Non-linear optical materials based on REE sandwich complexes
Metalloporphyrins (MPor) and metallophthalocyanines (MPcs) are important organic nonlinear optical (NLO) materials due to their two-dimensional π-system.453,454 Among practically relevant properties optical limiting (OL) is particularly interesting as it can be used to protect eyes from damage caused by laser irradiation.Upon the formation of sandwich complexes with rare earth metals, the π-conjugation systems can be further extended along the axial direction of tetrapyrrol plane, leading to three-dimensionally conjugated molecules, which is desirable for improving the NLO response by regulating the intramolecular charge transfer transitions and/or constructing dipolar and multipolar systems.3
5.1 Second-order nonlinear optical materials
For materials having second-order NLO responses at the molecular level, noncentrosymmetric molecules with strong intramolecular dipolar charge distribution are required. Heteroleptic double-decker tetrapyrrolic complexes generally meet this requirement. However, reports in this field are still very rare. The sole example of heteroleptic double-decker complexes studied for the second-order NLO properties is a mixed porphyrin and tetraazaporphyrin zirconium(IV) complex (OEP)Zr(OETAP).455 A negative hyperpolarizability β [(−83 ± 43) × 10−30 esu] was obtained at 1907 nm by the electric field-induced second harmonic (EFISH) measurements.An interesting trial for second-order NLO materials has been extended to homoleptic double-decker complexes with “cubic” octupolar architectures (cf. Section 2.2.2 for structures of complexes). In this regard, Ayhan et al. developed ABAB homoleptic bis(phthalocyaninato) rare earth complexes M[(SHex)4Pc]2, M = Nd, Eu, Dy, Y and Lu,109,110 which represent a new class of cubic molecules with an octupolar charge distribution. Their second-order NLO properties were studied by Hyper–Rayleigh Scattering (HRS) measurements at 1907 nm in chloroform solution. The first molecular hyperpolarizabilities (βHLS) were evaluated to be in the range of 3010 × 10−30 esu to 5760 × 10−30 esu for Y(III) and Lu(III) complexes, respectively, which are among the largest values ever reported for nondipolar molecules. Furthermore, it was found that the βHLS values of these complexes are depended on the number of 4f electrons and ionic radii of the sandwiched lanthanide(III) ions, thus providing opportunity to regulate the second-order NLO responses. Another ABAB homoleptic bis(phthalocyaninato) rare earth complex Lu[(α-PentO)4Cl4Pc]2, containing alternating pentyloxy and chloro substituents on phthalocyanine ligands, was reported by Cao et al.,111 which also showed a high molecular hyperpolarizability (βHLS = 1240 × 10−30 esu at 1907 nm, in chloroform).
By applying a porphyrin ligand with di(phenylamino)phenyl and carboxymethyl–phenylethynyl meso-substituents as typical electron-donating and electron-accepting groups, respectively, Sun et al. synthesized the first octupolar bis(porphyrinato) rare earth complex [HTb(DADAPor)2] (Scheme 15).113 Because of the reduced protonated nature, this compound does not have any absorption in the near-IR range beyond 800 nm. Consequently, an off-resonant hyperpolarizability (βHLS = 1700 × 10−30 esu at 1907 nm, in chloroform) was obtained for this compound, which is the largest off-resonant hyperpolarizability for octupolar molecular materials.
Density functional theory (DFT) studies were also conducted to clarify the nature of the second-order NLO properties of octupolar homoleptic bis(phthalocyaninato) rare earth complexes.456 A series of cubic molecules based on the bis(phthalocyaninato) yttrium Y(Pc)2 skeleton, containing two couples of electron-donating and electron-withdrawing groups in an alternate manner (Fig. 41a), were investigated. According to the calculation results: (1) the static hyperpolarizability (βHLS, 103–104 a.u.) increases along with increasing electronegativity of the peripheral substituents; (2) a dynamic perturbation (excitation) will induce a resonance effect with a much higher dynamic βHLS (104–106 a.u.), which is also positively correlated with the electronegativity of peripheral substituents; (3) the molecular conformation affects the second-order NLO properties. The maximal static βHLS appears at a rotation angle θ of 70°, but not 90°, because Y(Pc)2 molecule employs a cuboid instead of a standard cube (Fig. 41b and c). These findings are useful for the design of new octupolar NLO materials, given that the theoretically predicted maximal βHLS values can reach 105 × 10−30 esu for such octupolar molecules.
 | ||
| Fig. 41 (a) Molecular structures of the series of bis(phthalocyaninato) yttrium double-decker skeletons used for DFT modelling. (b) Plot of the calculated static βHRS as a function of the rotation angle θ for the complex Y(Pc4)2. (c) Cuboid structure of the bis(phthalocyaninato) yttrium double-decker skeleton with a rotation angle of 90°. Adapted with permission from ref. 456. Copyright 2015, Wiley-VCH. | ||
5.2 Third-order nonlinear optical materials
The tetrapyrrol macrocycles are very suitable for the third-order NLO materials due to three special advantages, including (1) the large number of free π electrons playing the role of oscillators driven by an external periodically vibrating electromagnetic field; (2) the extended delocalized π-system acting as a broad oscillating space; and (3) the huge polarizing/hyperpolarizing moments due to the long distance between the peripheral electron-donating and withdrawing substituents. The double-decker and multi-decker tetrapyrrols further improves the total three advantages in comparison with the single-decker ones.The study of third-order NLO materials based on double-decker phthalocyanines started from 1990s. Shirk and coworkers investigated the third-order NLO performance of double-deckers M(Pc)2 (M = Sc, Y, Eu, Gd, Lu) using the degenerate four-wave mixing (DFWM) method.457 The second-order hyper-polarizabilities (γ) and third-order susceptibility χ(3) values of M(Pc)2 are about an order of magnitude higher than those of typical monomeric metallophthalocyanines. Among these double-deckers, the third-order NLO performance of Sc(Pc)2, Lu(Pc)2 and Yb(Pc)2 are better than others. This therefore suggests that the large increase in the hyperpolarizabilities of bis(phthalocyaninato) complexes over those of the monophthalocyaninato compounds could not be simply attributed to the extended delocalization length from monomers to double-deckers.
In the past 20 years, the Z-scan method was widely used in most third-order NLO experiments. Sheng and coworkers studied a series of homoleptic bis(phthalocyaninato) rare earth complexes M[(OctO)8Pc]2, M = Pr, Ce, Sm, and Lu.458–460 The NLO performance of these compounds are tested using the exciting laser wavelength of 800 nm (Z-scan, 170 ns). The two-photon absorption cross-section σ2PA values of this series are in the range of (3–254) × 103 GM, showing their strong nonlinear optical response. More importantly, the authors measured the real part of the second-order molecular hyperpolarizability (γR) of the neutral complex Pr[(OctO)8Pc]20˙ together with its anion Pr[(OctO)8Pc]2− and cation Pr[(OctO)8Pc]2+. They found that the γR values of neutral, anionic and cationic forms are 440 × 10−32, 180 × 10−32, and 220 × 10−32 esu, respectively.460 This is a key experimental phenomenon which shows the free single electron in the neutral radical molecule can be used as a good oscillator in the NLO response. When this free single electron is paired in the anionic form or removed in the cationic form, the NLO response will be significantly weakened. Following a similar mentality, Plekhanov et al. examined another complex Lu[(SHex)8Pc]2.461 Using a similar Z-scan test (300 ns) at an exciting laser wavelength of 1550 nm, the complex achieved a remarkable nonlinear refractive index (n2) value of (300 ± 3) × 10−5 cm2 GW−1. They found that the n2 for unsubstituted complex Lu(Pc)2 is only (228 ± 3) × 10−5 cm2 GW−1, which is a quarter lower than that of Lu[(SHex)8Pc]2. This comparative investigation revealed that the peripheral electron-withdrawing substituents are beneficial to the NLO response because they could provide more free electrons to the π conjugated system.
Karpo et al. studied the NLO properties of a series of butyl-substituted complexes M(Bu8Pc)2, M = Eu, Dy, Er, and Lu in THF solution using the open aperture z-scan technique with 350 ps laser pulses at 532 nm.462 All the compounds exhibited reverse saturable absorption (RSA), and application or rate equations formalism evidenced that RSA is mainly contributed by the triplet state absorption. Absorption cross-sections from the ground state (σ0), excited singlet and triplet (σ1 and σ2) were deduced, evidencing that σ2 ≫ σ0 > σ1, the largest ratio σ2/σ0 was found for erbium complex.
Sekhosana et al. conducted a series of comparative studies on the NLO properties of double-decker phthalocyanines with electron-withdrawing pyridin-4-yloxy substituents (PyO).47,463–466 They found that the imaginary part of the third-order susceptibility χ(3)I value is 106 × 10−12 esu for Yb[(α-OPy)4Pc]2 and 338 × 10−12 esu for Yb[(β-OPy)4Pc]2 (Y20) under the Z-scan test (10 ns, in DMSO) at the wavelength of 532 nm. When the –OR substituents are linked onto the macrocycle α-sites, there will be a p–π-super-conjugation between the pz(O) and π(Pc) orbitals. When the –OR substituents are connected on the β-sites instead of the α-sites, the oscillation space of π electrons will be more effectively extended, which is positive to the periodic oscillation in the nonlinear forced vibration.
Another interesting discovery is that the χ(3)I value for the neutral complex Nd[(tBuPhO)4Pc]0˙ is 377 × 10−12 esu, but it gains spectacular increase to 1280 × 10−12 esu for the ionic complex Nd[(tBuPhO)4Pc]−Nd(OAc)2+.47 Although the latter complex does not have unpaired π-electrons, a huge positive/negative ion-pair is constructed by the bisphthalocyaninate anion and its counter-ion, rendering it as a (δ+)–(δ−) pair with a very strong dipole moment, which shows an improved NLO property.
Throughout the investigations on various RE(Pc)2 molecules, it is easy to find out two rules to get the double-decker tetrapyrrols with high NLO performance: (1) the neutral radical M(Pc)20˙ is a good NLO material due to the additional free single π electron. When this radical complex is oxidized/reduced and then this free single π electron is removed/paired, it lowers the overall NLO response. However, it still can be improved by ion pair formation.47 (2) The peripheral electron-donating substituents, especially the –OR groups, are beneficial to NLO response because they could provide more free electrons to the π conjugated system. If the –OR groups are linking at the β-sites, they will provide an extended oscillating space, and thus an improved NLO response. In comparison with these positions, the efficiency of substitution at α-sites is not obvious.
Sheng and coworkers also tested a series of heteroleptic double-deckers [(OctO)8Pc]M(Pc), M = Y, Ho, Er, Yb, and found that their σ2PA is in the range of (126–183) × 103 GM,467 which is still at the same level of M[(OctO)8Pc]2, M = Pr, Ce, Sm, and Lu series.458–460
Jiang and coworkers explored binuclear phthalocyanine-containing sandwich-type rare-earth complexes.468 They revealed that the χ(3)I value of (Pc)M[f-Pc2]M(Pc)SHex (18.6 × 10−12 esu) is higher than that of Pr[(SHex)8Pc]2 (15.6 × 10−12 esu), due to the enlarged π-conjugation by incorporating a binuclear phthalocyanine ligand.
Chen et al. conducted another comparative study on a series of porphyrin-appended europium(III) bis(phthalocyaninato) complexes (Fig. 42a).469 The χ(3)I values for [(α-ZnPor)Pc]Eu(Pc), [(β-ZnPor)Pc]Eu(Pc) and [(β-ZnPor)2Pc]Eu(Pc) are 111 × 10−22, 74 × 10−22, and 133 × 10−22 m2 V−2 which are in cases smaller than χ(3)I for the unsubstituted Eu(Pc)2 complex (194 × 10−22 m2 V−2). This result shows that the using of larger ligands is not always beneficial to NLO properties. In the porphyrin-appended Pc ligands, there is no unified conjugating system between the Por and Pc macrocycles, which cannot provide a channel for electron oscillation. So the effective electron oscillating space is still located at the Eu(Pc)2 part.
 | ||
| Fig. 42 Examples of π-extended sandwich complexes used for NLO studies: (a) Eu(III) bisphthalocyaninates with appended porphyrin units;469 (b and c) double- and triple-decker complexes with meso-pyrenyl-substituted porphyrin ligands.31 | ||
In order to improve the third-order NLO responses of rare-earth based tetrapyrrolic sandwiches, multi-deckers were constructed to extend the delocalized π-system and increase the oscillating space. However, the χ(3)I value of a homoleptic triple-decker Nd2[(β-tBuPhO)4Pc]3 is found to be 350 × 10−12 esu,470 which is slightly lower than that of the corresponding double-decker Yb[(β-tBuPhO)4Pc]2 (χ(3)I = 420 × 10−12 esu) in the same Z-scan test (10 ns, 0.3 mmol L−1 in DMF).465 Similar results were also obtained in the comparison of mixed (phthalocyaninato)(porphyrinato) europium triple- and double-decker complexes, in which γR for [(Pyr)4Por]Eu(Pc)Eu(Pc) is also lower than γR for [(Pyr)4Por]Eu(Pc) (Fig. 42b and c).31
However, Oluwole et al. showed that symmetrical Eu2[(BuO)8Pc]3 has a higher χ(3)I value in comparison with Eu[(BuO)8Pc]2.101 NLO response and OL properties can be improved by lowering the molecular symmetry via the introduction of diethyleneglycol substituents (Scheme 11), which in turn can be used to graft complexes to quantum dot surfaces gaining even more spectacular improvement of OL characteristics.
The third-order NLO properties of multi-decker sandwiches with more than a three layered structure were also examined by using Z-scan technique.48,209,471–473 Along with expanding the π–π interaction system in the order of triple-, quadruple-, quintuple-, and sextuple-deckers, as expected, the NLO responses are gradually improved due to the extended oscillating space. Remarkable χ(3)I and γR values of 4910 × 10−12 and 12700 × 10−30 esu were recorded for a sextuple-decker Nd3Cd2[(tBuPhO)4Pc]6 in the Z-scan test (10 ns, 532 nm, 3.0 × 10−4 M in CH2Cl2),48 suggesting the great potential for application.
While solution measurements provide fundamental NLO characteristics of synthesized complexes, their practical application requires immobilization of molecules on surfaces or incorporation into polymeric matrices for coating fabrication. The latter approach was used by Liu et al. to form optical limiters based on heteroleptic sextuple-decker complex (Pc)Sm(Pc)Cd(Pc*)Cd(Pc*)Cd(Pc)Sm(Pc), where Pc* = (BuO)8Pc.474 Due to RSA mechanism this complex showed excellent OL characteristics in chloroform solution (Fig. 43a), which inspired the authors to probe its practical application. Thus, this complex was dispersed in gel glass formed by methyltriethoxysilane polycondensation (Fig. 43b), affording materials capable of efficient optical limiting – while linear transmittance of a glass sample containing 1 mass% of complex was 84% (Fig. 43c), the minimum normalized transmittances decreased to 10% (Fig. 43d).
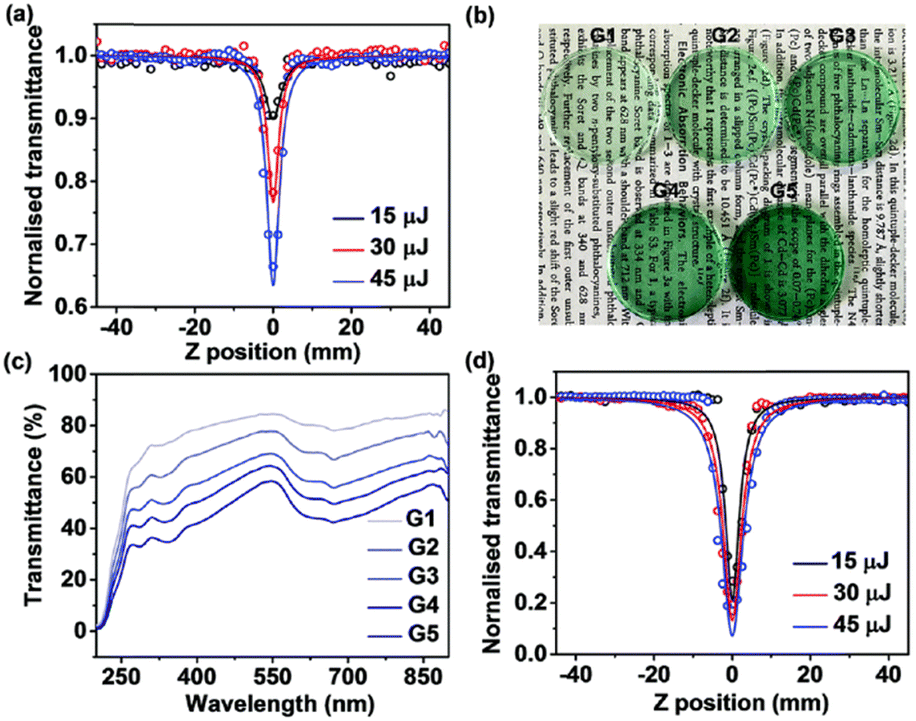 | ||
| Fig. 43 (a) NLO properties of (Pc)Sm(Pc)Cd(Pc*)Cd(Pc*)Cd(Pc)Sm(Pc) in CHCl3 at a concentration of 2.0 × 10−5 mol L−1 at different incident intensities. (b) Photographs of the gel glass of the complex with different concentrations, G1–G5 where the number corresponds to weight percentage of the complex. (c) Transmittance spectra of glasses G1–G5. (d) NLO properties of gel glass G1 at different incident intensities. Reproduced from ref. 474 with permission from the Royal Society of Chemistry. | ||
6. Conclusions
The data presented in this review showed that tetrapyrrole sandwich complexes based on rare earth elements are a unique molecular platform for targeted design of functional materials. Over the last 20 years, the field of REE sandwich complexes has made great progress in both basic and applied research. The variety of architectures and properties of sandwiches presented in this review shows fantastic opportunities for their application in advanced technologies, particularly SMM-based quantum information storage and processing, where spins are used as information carriages and quantum effects play an important role.We can expect that the prospects for the use of REE tetrapyrrolic sandwiches will greatly expand in the coming years both with the further development of their synthetic chemistry and further elaboration of approaches to hybrid materials and thin film-based devices. The latter aspect is particularly important in terms of application, as the requirements in small amounts of complexes needed for the fabrication of such devices partially alleviate the need in large scale synthesis.
Thus, despite the rather large number of papers already published and reviewed, this area of research still has many avenues for further development, so the authors are confident that this review will encourage scientists from various fields of chemistry and materials science to tackle this exciting and promising topic.
List of abbreviations
| 3nCn, n = 4, 5, 6 | 3n-crown-n-ether |
| acac− | Acetylacetonate |
| ClN | Chloronaphthalene |
| c-Pc2 | Clamshell-bisphthalocyanine |
| DClB | Dichlorobenzene |
| dpm− | Dipivaloylmethanate |
| DZPc | Diazepinoporphyrazine |
| f-Pc2 | Fused bisphthalocyanine |
| Hp | Hemiporphyrazine |
| LF | Ligand-field |
| Ln | Lanthanide |
| Menth | Menthyl |
| Naph | Naphthyl |
| Nc | Naphthalocyanine |
| NLO | Nonlinear optics |
| OFET | Organic field-effect transistor |
| P or Por | Porphyrin |
| Pc | Phthalocyanine |
| Pz | Porphyrazine |
| QT | Quantum technology |
| QTM | Quantum tunneling of magnetization |
| REE | Rare earth element |
| SB | Schiff base |
| SMM | Single-molecule magnet |
| SOC | Spin–orbit coupling |
| SWNT | Single-wall nanotube |
| TAnP | Tetra-p-anisylpoprhyrin |
| TBP | Tetrabenzoporphyrin |
| TBTAP | Tetrabenzotriazporphyrin |
| TClB | 1,2,4-Trichlorobenzene |
| TClPP | Tetra-p-chlorophenylpoprhyrin |
| TM | Transition metal |
| TTP | Tetra-p-tolylporphyrin |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. G. Martynov (Chapter 2) thanks Russian Science Foundation (18-73-10174-P). Y. Horii thanks the JSPS KAKENHI Grant Number JP21K14645. K. Katoh thanks the JSPS KAKENHI Grant Number JP24750119, JP15K05467. Y. Bian thanks the Financial support from the National Natural Science Foundation of China (22271012) and Beijing Natural Science Foundation (2202028). J. Jiang thanks the Financial support from the National Natural Science Foundation of China (22235001, 22175020), M. Yamashita thanks the JSPS KAKENHI Grant Number JP19H05631, JP20225003, CREST (JPMJCR12L3) from JST, the National Natural Science Foundation of China (NSFC, 22150710513), and the 111 project (B18030) from China.Notes and references
- D. K. P. Ng and J. Jiang, Chem. Soc. Rev., 1997, 26, 433–442 RSC.
- J. W. Buchler and D. K. P. Ng, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, 2000, vol. 3, pp. 245–294 Search PubMed.
- J. Jiang, K. Kasuga and D. P. Arnold, in Supramolecular Photosensitive and Electroactive Materials, ed. H. S. Nalwa, Elsevier, 2001, pp. 113–210 Search PubMed.
- R. Weiss and J. Fischer, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, 2003, vol. 16, pp. 171–246 Search PubMed.
- V. E. Pushkarev, L. G. Tomilova and V. N. Nemykin, Coord. Chem. Rev., 2016, 319, 110–179 CrossRef CAS.
- V. E. Pushkarev, L. G. Tomilova and Y. V. Tomilov, Russ. Chem. Rev., 2008, 77, 875–907 CrossRef CAS.
- J. Jiang and D. K. P. Ng, Acc. Chem. Res., 2009, 42, 79–88 CrossRef CAS PubMed.
- M. Bouvet, P. Gaudillat and J.-M. M. Suisse, J. Porphyrins Phthalocyanines, 2013, 17, 628–635 CrossRef CAS.
- H. Wang, B. W. Wang, Y. Bian, S. Gao and J. Jiang, Coord. Chem. Rev., 2016, 306, 195–216 CrossRef CAS.
- W. L. Chan, C. Xie, W. S. Lo, J. C. G. Bünzli, W. K. Wong and K. L. Wong, Chem. Soc. Rev., 2021, 50, 12189–12257 RSC.
- X. Tang, Q. Liu, C. Wei, X. Lv, Z. Jin, Y. Chen and J. Jiang, J. Rare Earths, 2021, 39, 113–120 CrossRef CAS.
- P. A. Barrett, C. E. Dent and R. P. Linstead, J. Chem. Soc., 1936, 1719 RSC.
- I. S. Kirin, P. N. Moskalev and Y. A. Makashev, Russ. J. Inorg. Chem., 1965, 10, 1065–1066 Search PubMed.
- G. A. Corker, B. Grant and N. J. Clecak, J. Electrochem. Soc., 1979, 126, 1339–1343 CrossRef CAS.
- D. Markovitsi, T.-H. Tran-Thi, R. Even and J. Simon, Chem. Phys. Lett., 1987, 137, 107–112 CrossRef CAS.
- R. Rousseau, R. Aroca and M. L. Rodríguez-Méndez, J. Mol. Struct., 1995, 356, 49–62 CrossRef CAS.
- L. G. Tomilova, E. V. Chernykh, N. A. Ovchinnikova, E. V. Bezlepko, V. M. Mizin and E. A. Lukyanets, Opt. Spectrosc., 1991, 70, 775–778 CAS.
- K. Kasuga, M. Ando, H. Morimoto and M. Isa, Chem. Lett., 1986, 1095–1098 CrossRef CAS.
- M. M’Sadak, J. Roncali and F. Garnier, J. Chim. Phys., 1986, 83, 211–216 CrossRef.
- K. Kasuga, M. Tsutsui, R. C. Petterson, K. Tatsumi, N. Van Opdenbosch, G. Pepe and E. F. Meyer, J. Am. Chem. Soc., 1980, 102, 4835–4836 CrossRef CAS.
- S. I. Troyanov, L. A. Lapkina, V. E. Larchenko and A. Y. Tsivadze, Dokl. Chem., 1999, 367, 192–196 Search PubMed.
- A. De Cian, M. Moussavi, J. Fischer and R. Weiss, Inorg. Chem., 1985, 24, 3162–3167 CrossRef CAS.
- K. Takahashi, Y. Tomita, Y. Hada, K. Tsubota, M. Handa, K. Kasuga, K. Sogabe and T. Tokii, Chem. Lett., 1992, 759–762 CrossRef CAS.
- K. Takahashi, M. Itoh, Y. Tomita, K. Nojima, K. Kasuga and K. Isa, Chem. Lett., 1993, 1915–1918 CrossRef CAS.
- L. A. Lapkina, E. Niskanen, H. Rönkkömäki, V. E. Larchenko, K. I. Popov and A. Y. Tsivadze, J. Porphyrins Phthalocyanines, 2000, 4, 588–590 CrossRef.
- I. V. Nefedova, Y. G. Gorbunova, S. G. Sakharov and A. Y. Tsivadze, Russ. J. Inorg. Chem., 2005, 50, 165–173 Search PubMed.
- J. W. Buchler, H.-G. Kapellmann, M. Knoff, K.-L. Lay and S. Pfeifer, Zeitschrift für Naturforsch. B, 1983, 38, 1339–1345 CrossRef.
- J. W. Buchler, K. Elsässer, M. Kihn-Botulinski and B. Scharbert, Angew. Chem., Int. Ed. Engl., 1986, 25, 286–287 CrossRef.
- J. W. Buchler, A. De Cian, J. Fischer, M. Kihn-Botulinski, H. Paulus and R. Weiss, J. Am. Chem. Soc., 1986, 108, 3652–3659 CrossRef CAS.
- J. W. Buchler, A. De Cian, J. Fischer, M. Kihn-Botulinski and R. Weiss, Inorg. Chem., 1988, 27, 339–345 CrossRef CAS.
- J. K. Duchowski and D. F. Bocian, J. Am. Chem. Soc., 1990, 112, 8807–8811 CrossRef CAS.
- H. Heiner, A. Tutaß, M. Göldner, U. Cornelissen and H. Homborg, Z. Anorg. Allg. Chem., 2001, 627, 485–497 CrossRef.
- L. A. Lapkina, S. G. Sakharov, N. Y. Konstantinov, V. E. Larchenko, Y. G. Gorbunova and A. Y. Tsivadze, Russ. J. Inorg. Chem., 2007, 52, 1758–1768 CrossRef.
- M. Moussavi, A. De Cian, J. Fischer and R. Weiss, Inorg. Chem., 1986, 25, 2107–2108 CrossRef CAS.
- P. Turek, P. Petit, J. J. Andre, J. Simon, R. Even, B. Boudjema, G. Guillaud and M. Maitrot, J. Am. Chem. Soc., 1987, 109, 5119–5122 CrossRef CAS.
- N. Ishikawa, M. Sugita, T. Ishikawa, S.-Y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- G. Lu, M. Bai, R. Li, X. Zhang, C. Ma, P.-C. Lo, D. K. P. Ng and J. Jiang, Eur. J. Inorg. Chem., 2006, 3703–3709 CrossRef CAS.
- X. Kong, Q. Jia, F. Wu and Y. Chen, Dyes Pigm., 2015, 115, 67–72 CrossRef CAS.
- X. Kong, Z. Dong, Y. Wu, X. Li, Y. Chen and J. Jiang, Chin. J. Chem., 2016, 34, 975–982 CrossRef CAS.
- X. Kong, G. Lu, X. Zhang, X. Li, Y. Chen and J. Jiang, Dyes Pigm., 2017, 143, 203–210 CrossRef CAS.
- X. Kong, X. Zhang, D. Gao, D. Qi, Y. Chen and J. Jiang, Chem. Sci., 2015, 6, 1967–1972 RSC.
- D. Li, H. Wang, J. Kan, W. Lu, Y. Chen and J. Jiang, Org. Electron., 2013, 14, 2582–2589 CrossRef CAS.
- T. V. Dubinina, A. D. Kosov, E. F. Petrusevich, S. S. Maklakov, N. E. Borisova, L. G. Tomilova and N. S. Zefirov, Dalton Trans., 2015, 44, 7973–7981 RSC.
- J. Kan, Y. Chen, D. Qi, Y. Liu and J. Jiang, Adv. Mater., 2012, 24, 1755–1758 CrossRef CAS PubMed.
- K. E. Sekhosana, E. Amuhaya, J. Mack and T. Nyokong, J. Mater. Chem. C, 2014, 2, 5431–5437 RSC.
- K. E. Sekhosana, M. H. Manyeruke and T. Nyokong, J. Mol. Struct., 2016, 1121, 111–118 CrossRef CAS.
- K. E. Sekhosana, R. Nkhahle and T. Nyokong, ChemistrySelect, 2018, 3, 6671–6682 CrossRef CAS.
- K. E. Sekhosana and T. Nyokong, Dyes Pigm., 2020, 172, 107836 CrossRef CAS.
- C. R. Ganivet, B. Ballesteros, G. de la Torre, J. M. Clemente-Juan, E. Coronado and T. Torres, Chem. – Eur. J., 2013, 19, 1457–1465 CrossRef CAS PubMed.
- T. V. Dubinina, K. V. Paramonova, S. A. Trashin, N. E. Borisova, L. G. Tomilova and N. S. Zefirov, Dalton Trans., 2014, 43, 2799–2809 RSC.
- T. V. Dubinina, M. S. Belousov, S. S. Maklakov, V. I. Chernichkin, M. V. Sedova, V. A. Tafeenko, N. E. Borisova and L. G. Tomilova, Dyes Pigm., 2019, 170, 107655 CrossRef CAS.
- N. Kobayashi and A. B. P. Lever, J. Am. Chem. Soc., 1987, 109, 7433–7441 CrossRef CAS.
- O. E. Sielcken, M. M. Van Tilborg, M. F. M. Roks, R. Hendriks, W. Drenth and R. J. M. Nolte, J. Am. Chem. Soc., 1987, 109, 4261–4265 CrossRef CAS.
- T. Toupance, V. Ahsen and J. Simon, J. Am. Chem. Soc., 1994, 116, 5352–5361 CrossRef CAS.
- T. Toupance, H. Benoit, D. Sarazin and J. Simon, J. Am. Chem. Soc., 1997, 119, 9191–9197 CrossRef CAS.
- Y. G. Gorbunova, L. A. Lapkina, A. G. Martynov, I. V. Biryukova and A. Y. Tsivadze, Russ. J. Coord. Chem., 2004, 30, 245–251 CrossRef CAS.
- A. May, P. Majumdar, A. G. Martynov, L. A. Lapkina, S. I. Troyanov, Y. G. Gorbunova, A. Y. Tsivadze, J. Mack and T. Nyokong, J. Porphyrins Phthalocyanines, 2020, 24, 589–601 CrossRef CAS.
- Y. G. Gorbunova, A. G. Martynov and A. Y. Tsivadze, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing, 2012, vol. 24, pp. 271–388 Search PubMed.
- K. P. Birin, Y. G. Gorbunova and A. Y. Tsivadze, J. Porphyrins Phthalocyanines, 2006, 10, 931–936 CrossRef CAS.
- A. G. Martynov, E. A. Safonova, A. Y. Tsivadze and Y. G. Gorbunova, Coord. Chem. Rev., 2019, 387, 325–347 CrossRef CAS.
- S. L. Selektor, A. V. Shokurov, V. V. Arslanov, Y. G. Gorbunova, K. P. Birin, O. A. Raitman, F. Morote, T. Cohen-Bouhacina, C. Grauby-Heywang and A. Y. Tsivadze, J. Phys. Chem. C, 2014, 118, 4250–4258 CrossRef CAS.
- A. V. Shokurov, D. S. Kutsybala, A. G. Martynov, A. V. Bakirov, M. A. Shcherbina, S. N. Chvalun, Y. G. Gorbunova, A. Y. Tsivadze, A. V. Zaytseva, D. Novikov, V. V. Arslanov and S. L. Selektor, Langmuir, 2020, 36, 1423–1429 CrossRef CAS PubMed.
- D. S. Kutsybala, A. V. Shokurov, A. G. Martynov, A. V. Yagodin, V. V. Arslanov, Y. G. Gorbunova and S. L. Selektor, Symmetry, 2022, 14, 340 CrossRef CAS.
- S. P. Babailov, M. A. Polovkova, G. A. Kirakosyan, A. G. Martynov, E. N. Zapolotsky and Y. G. Gorbunova, Sens. Actuators, A, 2021, 331, 112933 CrossRef CAS.
- S. P. Babailov, M. A. Polovkova, E. N. Zapolotsky, G. A. Kirakosyan, A. G. Martynov and Y. G. Gorbunova, J. Porphyrins Phthalocyanines, 2022, 26, 334–339 CrossRef CAS.
- R. J. Holmberg, M. A. Polovkova, A. G. Martynov, Y. G. Gorbunova and M. Murugesu, Dalton Trans., 2016, 45, 9320–9327 RSC.
- Y. Horii, S. Kishiue, M. Damjanović, K. Katoh, B. K. Breedlove, M. Enders and M. Yamashita, Chem. – Eur. J., 2018, 24, 4320–4327 CrossRef CAS PubMed.
- A. G. Martynov, A. V. Bykov, Y. G. Gorbunova and A. Y. Tsivadze, Russ. Chem. Bull., 2018, 67, 2195–2200 CrossRef CAS.
- A. G. Martynov, O. V. Zubareva, Y. G. Gorbunova, S. G. Sakharov, S. E. Nefedov, F. M. Dolgushin and A. Y. Tsivadze, Eur. J. Inorg. Chem., 2007, 4800–4807 CrossRef CAS.
- A. G. Martynov, E. A. Safonova, Y. G. Gorbunova and A. Y. Tsivadze, Russ. J. Inorg. Chem., 2010, 55, 347–354 CrossRef CAS.
- A. Y. Tsivadze, A. G. Martynov, M. A. Polovkova and Y. G. Gorbunova, Russ. Chem. Bull., 2011, 60, 2258–2262 CrossRef CAS.
- M. A. Polovkova, A. G. Martynov, K. P. Birin, S. E. Nefedov, Y. G. Gorbunova and A. Y. Tsivadze, Inorg. Chem., 2016, 55, 9258–9269 CrossRef CAS PubMed.
- A. V. Shokurov, A. V. Yagodin, A. G. Martynov, Y. G. Gorbunova, A. Y. Tsivadze and S. L. Selektor, Small, 2022, 18, 2104306 CrossRef CAS PubMed.
- H. Zhou, K. Wang, D. Qi and J. Jiang, Dalton Trans., 2014, 43, 1699–1705 RSC.
- W. Lv, P. Zhu, Y. Bian, C. Ma, X. Zhang and J. Jiang, Inorg. Chem., 2010, 49, 6628–6635 CrossRef CAS PubMed.
- W. Zheng, B.-B. Wang, J.-C. Lai, C.-Z. Wan, X.-R. Lu, C.-H. Li and X.-Z. You, J. Mater. Chem. C, 2015, 3, 3072–3080 RSC.
- M. Gonidec, F. Luis, À. Vílchez, J. Esquena, D. B. Amabilino and J. Veciana, Angew. Chem., Int. Ed., 2010, 49, 1623–1626 CrossRef CAS PubMed.
- E. A. Kuzmina, T. V. Dubinina and L. G. Tomilova, New J. Chem., 2019, 43, 9314–9327 RSC.
- Y. Chen, W. Cao, K. Wang and J. Jiang, Inorg. Chem., 2015, 54, 9962–9967 CrossRef CAS PubMed.
- Y. Chen, F. Ma, X. Chen, B. Dong, K. Wang, S. Jiang, C. Wang, X. Chen, D. Qi, H. Sun, B. Wang, S. Gao and J. Jiang, Inorg. Chem. Front., 2017, 4, 1465–1471 RSC.
- Y. Chen, F. Ma, X. Chen, B. Dong, K. Wang, S. Jiang, C. Wang, X. Chen, D. Qi, H. Sun, B. Wang, S. Gao and J. Jiang, Inorg. Chem., 2017, 56, 13889–13896 CrossRef CAS PubMed.
- C. Liu, M. Li, Y. Zhang, H. Tian, Y. Chen, H. Wang, J. Dou and J. Jiang, Eur. J. Inorg. Chem., 2019, 2940–2946 CrossRef CAS.
- Y. Chen, F. Ma, Y. Zhang, L. Zhao, K. Wang, D. Qi, H. Sun and J. Jiang, Inorg. Chem. Front., 2018, 5, 2006–2012 RSC.
- S. Takamatsu, T. Ishikawa, S. Koshihara and N. Ishikawa, Inorg. Chem., 2007, 46, 7250–7252 CrossRef CAS PubMed.
- D. V. Konarev, S. S. Khasanov, M. S. Batov, A. G. Martynov, I. V. Nefedova, Y. G. Gorbunova, A. Otsuka, H. Yamochi, H. Kitagawa and R. N. Lyubovskaya, Inorg. Chem., 2019, 58, 5058–5068 CrossRef CAS PubMed.
- M. Gonidec, D. B. Amabilino and J. Veciana, Dalton Trans., 2012, 41, 13632 RSC.
- M. Gonidec, I. Krivokapic, J. Vidal-Gancedo, E. S. Davies, J. McMaster, S. M. Gorun and J. Veciana, Inorg. Chem., 2013, 52, 4464–4471 CrossRef CAS PubMed.
- A. Şenocak, B. Köksoy, E. Demirbaş and M. Durmuş, J. Phys. Org. Chem., 2019, 32, e3907 CrossRef.
- F. Bertani, N. Cristiani, M. Mannini, R. Pinalli, R. Sessoli and E. Dalcanale, Eur. J. Org. Chem., 2015, 7036–7042 CrossRef CAS.
- E. A. Kuzmina, T. V. Dubinina, N. E. Borisova, B. N. Tarasevich, V. I. Krasovskii, I. N. Feofanov, A. V. Dzuban and L. G. Tomilova, Dyes Pigm., 2020, 174, 108075 CrossRef CAS.
- E. A. Kuzmina, T. V. Dubinina, P. N. Vasilevsky, M. S. Saveliev, A. Y. Gerasimenko, N. E. Borisova and L. G. Tomilova, Dyes Pigm., 2021, 185, 108871 CrossRef CAS.
- E. A. Safonova, A. G. Martynov, S. E. Nefedov, G. A. Kirakosyan, Y. G. Gorbunova and A. Y. Tsivadze, Inorg. Chem., 2016, 55, 2450–2459 CrossRef CAS PubMed.
- R. Wang, R. Li, Y. Bian, C.-F. Choi, D. K. P. Ng, J. Dou, D. Wang, P. Zhu, C. Ma, R. D. Hartnell, D. P. Arnold and J. Jiang, Chem. – Eur. J., 2005, 11, 7351–7357 CrossRef CAS PubMed.
- Y. Chen, F. Ma, X. Chen, Y. Zhang, H. Wang, K. Wang, D. Qi, H.-L. Sun and J. Jiang, Inorg. Chem., 2019, 58, 2422–2429 CrossRef CAS PubMed.
- Y. Gao, R. Li, S. Dong, Y. Bian and J. Jiang, Dalton Trans., 2010, 39, 1321–1327 RSC.
- M. Damjanović, Y. Horie, T. Morita, Y. Horii, K. Katoh, M. Yamashita and M. Enders, Inorg. Chem., 2015, 54, 11986–11992 CrossRef PubMed.
- Y. Horii, Y. Horie, K. Katoh, B. K. Breedlove and M. Yamashita, Inorg. Chem., 2018, 57, 565–574 CrossRef CAS PubMed.
- J. Mack and N. Kobayashi, Chem. Rev., 2011, 111, 281–321 CrossRef CAS PubMed.
- N. Ishikawa and Y. Kaizu, Chem. Lett., 1998, 183–184 CrossRef CAS.
- S. Alpugan, Ü. İşci, F. Albrieux, C. Hirel, A. G. Gürek, Y. Bretonnière, V. Ahsen and F. Dumoulin, Chem. Commun., 2014, 50, 7466 RSC.
- D. O. Oluwole, A. V. Yagodin, N. C. Mkhize, K. E. Sekhosana, A. G. Martynov, Y. G. Gorbunova, A. Y. Tsivadze and T. Nyokong, Chem. – Eur. J., 2017, 23, 2820–2830 CrossRef CAS PubMed.
- V. E. Pushkarev, A. Y. Tolbin, N. E. Borisova, S. A. Trashin and L. G. Tomilova, Eur. J. Inorg. Chem., 2010, 5254–5262 CrossRef CAS.
- A. Pedrini, M. Perfetti, M. Mannini and E. Dalcanale, ACS Omega, 2017, 2, 517–521 CrossRef CAS PubMed.
- V. E. Pushkarev, A. Y. Tolbin, F. E. Zhurkin, N. E. Borisova, S. A. Trashin, L. G. Tomilova and N. S. Zefirov, Chem. – Eur. J., 2012, 18, 9046–9055 CrossRef CAS PubMed.
- Y. Horii, K. Katoh, B. K. Breedlove and M. Yamashita, Chem. Commun., 2017, 53, 8561–8564 RSC.
- Y. S. Korostei, V. G. Tarasova, V. E. Pushkarev, N. E. Borisova, A. K. Vorobiev and L. G. Tomilova, Dyes Pigm., 2018, 159, 573–575 CrossRef CAS.
- K. P. Birin, A. I. Poddubnaya, E. V. Isanbaeva, Y. G. Gorbunova and A. Y. Tsivadze, J. Porphyrins Phthalocyanines, 2017, 21, 406–415 CrossRef CAS.
- W. Lu, C. Wang, X. Li, X. Zhan, D. Qi and Y. Bian, Inorg. Chem. Commun., 2017, 81, 18–21 CrossRef CAS.
- M. M. Ayhan, A. Singh, C. Hirel, A. G. Gürek, V. Ahsen, E. Jeanneau, I. Ledoux-Rak, J. Zyss, C. Andraud and Y. Bretonnière, J. Am. Chem. Soc., 2012, 134, 3655–3658 CrossRef CAS PubMed.
- M. M. Ayhan, A. Singh, E. Jeanneau, V. Ahsen, J. Zyss, I. Ledoux-Rak, A. G. Gürek, C. Hirel, Y. Bretonnière and C. Andraud, Inorg. Chem., 2014, 53, 4359–4370 CrossRef CAS PubMed.
- W. Cao, K. Wang, I. Ledoux-Rak and J. Jiang, Inorg. Chem. Front, 2016, 3, 1146–1151 RSC.
- Q. Zhi, F. Ma, C. Wang, Y. Chen, H. Wang, H. Sun and J. Jiang, Eur. J. Inorg. Chem., 2019, 1329–1334 CrossRef CAS.
- J. Sun and Y. Han, RSC Adv., 2017, 7, 22855–22859 RSC.
- S. Kyatskaya, J. R. Galán Mascarós, L. Bogani, F. Hennrich, M. Kappes, W. Wernsdorfer and M. Ruben, J. Am. Chem. Soc., 2009, 131, 15143–15151 CrossRef CAS PubMed.
- S. Klyatskaya, A. Eichhöfer and W. Wernsdorfer, Eur. J. Inorg. Chem., 2014, 4179–4185 CrossRef CAS.
- O. Hampe, S. Klyatskaya, T. Karpuschkin, M. Vonderach, P. Weis, M. Ruben and M. M. Kappes, Int. J. Mass Spectrom., 2012, 325–327, 183–188 CrossRef CAS.
- M. Urdampilleta, S. Klyatskaya, J.-P. Cleuziou, M. Ruben and W. Wernsdorfer, Nat. Mater., 2011, 10, 502–506 CrossRef CAS PubMed.
- B. Ballesteros, G. de la Torre, A. Shearer, A. Hausmann, M. Â. Ã. Herranz, D. M. Guldi and T. Torres, Chem. – Eur. J., 2010, 16, 114–125 CrossRef CAS PubMed.
- H. Pan, L. Gong, W. Liu, C. Lin, Q. Ma, G. Lu, D. Qi, K. Wang and J. Jiang, Dyes Pigm., 2018, 156, 167–174 CrossRef CAS.
- K. Wang, H. Pan and J. Jiang, Chem. – Eur. J., 2015, 21, 18461–18465 CrossRef CAS PubMed.
- Y. Li, Y. Bian, M. Yan, P. S. Thapaliya, D. Johns, X. Yan, D. Galipeau and J. Jiang, J. Mater. Chem., 2011, 21, 11131 RSC.
- S. Sakaue, A. Fuyuhiro, T. Fukuda and N. Ishikawa, Chem. Commun., 2012, 48, 5337 RSC.
- K. P. Birin, Y. G. Gorbunova, A. Y. Tsivadze and A. Y. Tsivadze, J. Porphyrins Phthalocyanines, 2009, 13, 283 CrossRef CAS.
- K. P. Birin, Y. G. Gorbunova and A. Y. Tsivadze, Magn. Reson. Chem., 2010, 48, 505–515 CrossRef CAS PubMed.
- K. P. Birin, Y. G. Gorbunova and A. Y. Tsivadze, Macroheterocycles, 2010, 3, 210–217 CrossRef CAS.
- K. P. Birin, K. a Kamarova, Y. G. Gorbunova and A. Y. Tsivadze, J. Porphyrins Phthalocyanines, 2013, 17, 1027–1034 CrossRef CAS.
- K. P. Birin, Y. G. Gorbunova and A. Y. Tsivadze, Dalton Trans., 2011, 40, 11539–11549 RSC.
- K. P. Birin, A. I. Poddubnaya, Y. G. Gorbunova and A. Y. Tsivadze, Macroheterocycles, 2017, 10, 514–519 CrossRef CAS.
- K. P. Birin, Y. G. Gorbunova and A. Y. Tsivadze, Dalton Trans., 2012, 41, 23–28 RSC.
- H.-G. Jin, X. Jiang, I. A. Kühne, S. Clair, V. Monnier, C. Chendo, G. Novitchi, A. K. Powell, K. M. Kadish and T. S. Balaban, Inorg. Chem., 2017, 56, 4864–4873 CrossRef CAS PubMed.
- D. González-Lucas, S. C. Soobrattee, D. L. Hughes, G. J. Tizzard, S. J. Coles and A. N. Cammidge, Chem. – Eur. J., 2020, 26, 10724–10728 CrossRef PubMed.
- P. Zhu, X. Zhang, H. Wang, Y. Zhang, Y. Bian and J. Jiang, Inorg. Chem., 2012, 51, 5651–5659 CrossRef CAS PubMed.
- X. Wu, W. Lv, Q. Wang, H. Wang, X. Zhang and J. Jiang, Dalton Trans., 2011, 40, 107–113 RSC.
- J. Lu, L. Zhou, Q. Meng, H. Sui, Y. Li and X. Tai, Dyes Pigm., 2015, 113, 138–144 CrossRef CAS.
- T. Komeda, H. Isshiki, J. Liu, K. Katoh, M. Shirakata, B. K. Breedlove and M. Yamashita, ACS Nano, 2013, 7, 1092–1099 CrossRef CAS PubMed.
- K. Katoh, S. Yamashita, N. Yasuda, Y. Kitagawa, B. K. Breedlove, Y. Nakazawa and M. Yamashita, Angew. Chem., Int. Ed., 2018, 57, 9262–9267 CrossRef CAS PubMed.
- T. V. Dubinina, A. D. Kosov, E. F. Petrusevich, N. E. Borisova, A. L. Trigub, G. V. Mamin, I. F. Gilmutdinov, A. A. Masitov, S. V. Tokarev, V. E. Pushkarev and L. G. Tomilova, Dalton Trans., 2019, 48, 13413–13422 RSC.
- R. Stefak, N. Ratel-Ramond and G. Rapenne, Inorg. Chim. Acta, 2012, 380, 181–186 CrossRef CAS.
- Y. Zhang, H. Kersell, R. Stefak, J. Echeverria, V. Iancu, U. G. E. Perera, Y. Li, A. Deshpande, K.-F. Braun, C. Joachim, G. Rapenne and S.-W. Hla, Nat. Nanotechnol., 2016, 11, 706–712 CrossRef CAS PubMed.
- N. Giménez-Agulló, C. S. de Pipaón, L. Adriaenssens, M. Filibian, M. Martínez-Belmonte, E. C. Escudero-Adán, P. Carretta, P. Ballester and J. R. Galán-Mascarós, Chem. – Eur. J., 2014, 20, 12817–12825 CrossRef PubMed.
- E. N. Tarakanova, P. A. Tarakanov, V. E. Pushkarev and L. G. Tomilova, J. Porphyrins Phthalocyanines, 2014, 18, 149–154 CrossRef CAS.
- P. A. Tarakanov, E. N. Tarakanova, P. V. Dorovatovskii, Y. V. Zubavichus, V. N. Khrustalev, S. A. Trashin, K. De Wael, M. E. Neganova, D. V. Mischenko, J. L. Sessler, P. A. Stuzhin, V. E. Pushkarev and L. G. Tomilova, Dalton Trans., 2018, 47, 14169–14173 RSC.
- E. N. Tarakanova, S. A. Trashin, A. O. Simakov, T. Furuyama, A. V. Dzuban, L. N. Inasaridze, P. A. Tarakanov, P. A. Troshin, V. E. Pushkarev, N. Kobayashi and L. G. Tomilova, Dalton Trans., 2016, 45, 12041–12052 RSC.
- E. N. Tarakanova, S. A. Trashin, P. A. Tarakanov, V. E. Pushkarev and L. G. Tomilova, Dyes Pigm., 2015, 117, 61–63 CrossRef CAS.
- E. N. Tarakanova, O. A. Levitskiy, T. V. Magdesieva, P. A. Tarakanov, V. E. Pushkarev and L. G. Tomilova, New J. Chem., 2015, 39, 5797–5804 RSC.
- E. N. Tarakanova, P. A. Tarakanov, A. O. Simakov, T. Furuyama, N. Kobayashi, D. V. Konev, O. A. Goncharova, S. A. Trashin, K. De Wael, I. V. Sulimenkov, V. V. Filatov, V. I. Kozlovskiy, L. G. Tomilova, P. A. Stuzhin and V. E. Pushkarev, Dalton Trans., 2021, 50, 6245–6255 RSC.
- T. V. Dubinina, D. V. Dyumaeva, S. A. Trashin, M. V. Sedova, A. S. Dudnik, N. E. Borisova, L. G. Tomilova and N. S. Zefirov, Dyes Pigm., 2013, 96, 699–704 CrossRef CAS.
- J. H. Helberger, A. von Rebay and D. B. Hevér, Justus Liebigs Ann. Chem., 1938, 533, 197–215 CrossRef CAS.
- H. Y. Xu, C. H. Hu, Q. F. Liu, W. X. Zhao and Y. Liu, Appl. Mech. Mater., 2012, 190–191, 571–574 CAS.
- Q. F. Liu, Adv. Mater. Res., 2013, 750–752, 1816–1821 Search PubMed.
- Q. F. Liu, Adv. Mater. Res., 2013, 690–693, 573–576 Search PubMed.
- N. E. Galanin and G. P. Shaposhnikov, Russ. J. Org. Chem., 2012, 48, 851–857 CrossRef CAS.
- A. I. Koptyaev, N. E. Galanin and G. P. Shaposhnikov, Russ. J. Org. Chem., 2019, 55, 944–950 CrossRef CAS.
- A. I. Koptyaev, N. E. Galanin, V. V. Travkin and G. L. Pakhomov, Dyes Pigm., 2021, 186, 108984 CrossRef CAS.
- A. I. Koptyaev, M. I. Bazanov and N. E. Galanin, Russ. J. Org. Chem., 2020, 56, 788–796 CrossRef CAS.
- N. E. Galanin, L. A. Yakubov and G. P. Shaposhnikov, Russ. J. Org. Chem., 2011, 47, 941–947 CrossRef CAS.
- V. E. Pushkarev, V. V. Kalashnikov, S. A. Trashin, N. E. Borisova, L. G. Tomilova and N. S. Zefirov, Dalton Trans., 2013, 42, 12083 RSC.
- V. E. Pushkarev, V. V. Kalashnikov, A. Y. Tolbin, S. A. Trashin, N. E. Borisova, S. V. Simonov, V. B. Rybakov, L. G. Tomilova and N. S. Zefirov, Dalton Trans., 2015, 44, 16553–16564 RSC.
- Z. Gross, N. Galili and I. Saltsman, Angew. Chem., Int. Ed., 1999, 38, 1427–1429 CrossRef CAS PubMed.
- S. Nardis, F. Mandoj, M. Stefanelli and R. Paolesse, Coord. Chem. Rev., 2019, 388, 360–405 CrossRef CAS.
- H. L. Buckley, M. R. Anstey, D. T. Gryko and J. Arnold, Chem. Commun., 2013, 49, 3104 RSC.
- K. C. Armstrong, S. Hohloch, T. D. Lohrey, R. A. Zarkesh, J. Arnold and M. R. Anstey, Dalton Trans., 2016, 45, 18653–18660 RSC.
- G. Lu, S. Yan, M. Shi, W. Yu, J. Li, W. Zhu, Z. Ou and K. M. Kadish, Chem. Commun., 2015, 51, 2411–2413 RSC.
- G. Lu, C. He, K. Wang, J. Sun, D. Qi, L. Gong, C. Wang, Z. Ou, S. Yan, S. Zeng and W. Zhu, Inorg. Chem., 2017, 56, 11503–11512 CrossRef CAS PubMed.
- G. Lu, J. Li, S. Yan, C. He, M. Shi, W. Zhu, Z. Ou and K. M. Kadish, Dyes Pigm., 2015, 121, 38–45 CrossRef CAS.
- G. Lu, J. Li, X. Jiang, Z. Ou and K. M. Kadish, Inorg. Chem., 2015, 54, 9211–9222 CrossRef CAS PubMed.
- G. Lu, J. Li, S. Yan, W. Zhu, Z. Ou and K. M. Kadish, Inorg. Chem., 2015, 54, 5795–5805 CrossRef CAS PubMed.
- G. Lu, C. He, Y. Fang, L. Wang and W. Zhu, New J. Chem., 2018, 42, 2498–2503 RSC.
- H. Furuta, T. Asano and T. Ogawa, J. Am. Chem. Soc., 1994, 116, 767–768 CrossRef CAS.
- H. Furuta, H. Maeda and A. Osuka, Chem. Commun., 2002, 1795–1804 RSC.
- W. Cao, H. Wang, X. Wang, H. K. Lee, D. K. P. Ng and J. Jiang, Inorg. Chem., 2012, 51, 9265–9272 CrossRef CAS PubMed.
- Y. Zhang, W. Cao, K. Wang and J. Jiang, Dalton Trans., 2014, 43, 9152 RSC.
- W. Cao, C. Gao, Y.-Q. Zhang, D. Qi, T. Liu, K. Wang, C. Duan, S. Gao and J. Jiang, Chem. Sci., 2015, 6, 5947–5954 RSC.
- H. Wang, W. Cao, T. Liu, C. Duan and J. Jiang, Chem. – Eur. J., 2013, 19, 2266–2270 CrossRef CAS PubMed.
- Q. Ma, X. Feng, W. Cao, H. Wang and J. Jiang, CrystEngComm, 2013, 15, 10383 RSC.
- H. Wang, C. Liu, T. Liu, S. Zeng, W. Cao, Q. Ma, C. Duan, J. Dou and J. Jiang, Dalton Trans., 2013, 42, 15355 RSC.
- Z. Li, F. Gao, Z. Xiao, G. Ao, X. Wu, Y. Fang, Z. Nie, T. H. Wei, J. Yang, Y. Wang, X. Zhang, J. Zuo and Y. Song, Dyes Pigm., 2015, 119, 70–74 CrossRef CAS.
- Z. Li, F. Gao, Z. Xiao, X. Wu, J. Zuo and Y. Song, Opt. Laser Technol., 2018, 103, 42–47 CrossRef CAS.
- F. Gao, X.-M. Zhang, L. Cui, K. Deng, Q.-D. Zeng and J.-L. Zuo, Sci. Rep., 2015, 4, 5928 CrossRef PubMed.
- F. Gao, X. Feng, L. Yang and X. Chen, Dalton Trans., 2016, 45, 7476–7482 RSC.
- A. Santria, A. Fuyuhiro, T. Fukuda and N. Ishikawa, Inorg. Chem., 2017, 56, 10625–10632 CrossRef CAS PubMed.
- K. Kizaki, M. Uehara, A. Fuyuhiro, T. Fukuda and N. Ishikawa, Inorg. Chem., 2018, 57, 668–675 CrossRef CAS PubMed.
- A. Santria, A. Fuyuhiro, T. Fukuda and N. Ishikawa, Dalton Trans., 2019, 48, 7685–7692 RSC.
- A. Santria and N. Ishikawa, Inorg. Chem., 2020, 59, 14326–14336 CrossRef CAS PubMed.
- K. Kizaki, A. Santria and N. Ishikawa, Inorg. Chem., 2021, 60, 2037–2044 CrossRef CAS PubMed.
- R. H. Platel, T. Teixeira Tasso, W. Zhou, T. Furuyama, N. Kobayashi and D. B. Leznoff, Chem. Commun., 2015, 51, 5986–5989 RSC.
- J. A. Elvidge and R. P. Linstead, J. Chem. Soc., 1952, 5008 RSC.
- W. Liu, H. Pan, Z. Wang, K. Wang, D. Qi and J. Jiang, Chem. Commun., 2017, 53, 3765–3768 RSC.
- W. Liu, S. Zeng, X. Chen, H. Pan, D. Qi, K. Wang, J. Dou and J. Jiang, Inorg. Chem., 2018, 57, 12347–12353 CrossRef CAS PubMed.
- F. Gao, M.-X. Yao, Y.-Y. Li, Y.-Z. Li, Y. Song and J.-L. Zuo, Inorg. Chem., 2013, 52, 6407–6416 CrossRef CAS PubMed.
- J. Y. Hu, Y. Ning, Y. S. Meng, J. Zhang, Z. Y. Wu, S. Gao and J. L. Zhang, Chem. Sci., 2017, 8, 2702–2709 RSC.
- G. Q. Jin, Y. Ning, J. X. Geng, Z. F. Jiang, Y. Wang and J. L. Zhang, Inorg. Chem. Front., 2020, 7, 289–299 RSC.
- S. Sarwar, S. Sanz, J. van Leusen, G. S. Nichol, E. K. Brechin and P. Kögerler, Dalton Trans., 2020, 49, 16638–16642 RSC.
- H. Sugimoto, T. Higashi, A. Maeda, M. Mori, H. Masuda and T. Taga, J. Chem. Soc., Chem. Commun., 1983, 1234–1235 RSC.
- H. Sugimoto, T. Higashi, A. Maeda, Y. Hirai, J. Teraoka, M. Mori, H. Masuda and T. Taga, J. Less Common Met., 1985, 112, 387–392 CrossRef CAS.
- A. Maeda and H. Sugimoto, J. Chem. Soc., Faraday Trans. 2, 1986, 82, 2019 RSC.
- S. Tsuchiya, A. Fuyuhiro, T. Fukuda and N. Ishikawa, J. Porphyrins Phthalocyanines, 2014, 18, 933–936 CrossRef CAS.
- J.-Y. Ge, H.-Y. Wang, J. Li, J.-Z. Xie, Y. Song and J.-L. Zuo, Dalton Trans., 2017, 46, 3353–3362 RSC.
- J. Ge, Y. Qiu, H. Wang, J. Su, P. Wang and Z. Chen, Chem. – Asian J., 2020, 15, 3013–3019 CrossRef CAS PubMed.
- J.-Y. Ge, L. Cui, J. Li, F. Yu, Y. Song, Y.-Q. Zhang, J.-L. Zuo and M. Kurmoo, Inorg. Chem., 2017, 56, 336–343 CrossRef CAS PubMed.
- J.-Y. Ge, H.-Y. Wang, J. Su, J. Li, B.-L. Wang, Y.-Q. Zhang and J.-L. Zuo, Inorg. Chem., 2018, 57, 1408–1416 CrossRef CAS PubMed.
- T. Fukuda, T. Biyajima and N. Kobayashi, J. Am. Chem. Soc., 2010, 132, 6278–6279 CrossRef CAS PubMed.
- T. Fukuda, K. Hata and N. Ishikawa, J. Am. Chem. Soc., 2012, 134, 14698–14701 CrossRef CAS PubMed.
- T. Fukuda and N. Ishikawa, J. Porphyrins Phthalocyanines, 2014, 18, 615–629 CrossRef CAS.
- H. Wang, K. Qian, K. Wang, Y. Bian, J. Jiang and S. Gao, Chem. Commun., 2011, 47, 9624–9626 RSC.
- G. Lu, X. Kong, H. Wang, Y. Y. Chen, C. Wei, Y. Y. Chen and J. Jiang, New J. Chem., 2019, 43, 15763–15767 RSC.
- H. Wang, K. Wang, Y. Bian, J. Jiang and N. Kobayashi, Chem. Commun., 2011, 47, 6879 RSC.
- H. Wang, N. Kobayashi and J. Jiang, Chem. – Eur. J., 2012, 18, 1047–1049 CrossRef CAS PubMed.
- H. Wang, D. Qi, Z. Xie, W. Cao, K. Wang, H. Shang and J. Jiang, Chem. Commun., 2013, 49, 889–891 RSC.
- C. Liu, W. Yang, Y. Zhang and J. Jiang, Inorg. Chem., 2020, 59, 17591–17599 CrossRef CAS PubMed.
- K. Katoh, Y. Horii, N. Yasuda, W. Wernsdorfer, K. Toriumi, B. K. Breedlove and M. Yamashita, Dalton Trans., 2012, 41, 13582–13600 RSC.
- Y. Horii, K. Katoh, N. Yasuda, B. K. Breedlove and M. Yamashita, Inorg. Chem., 2015, 54, 3297–3305 CrossRef CAS PubMed.
- Y. Horii, K. Katoh, G. Cosquer, B. K. Breedlove and M. Yamashita, Inorg. Chem., 2016, 55, 11782–11790 CrossRef CAS PubMed.
- Y. Horii, M. Damjanović, M. R. Ajayakumar, K. Katoh, Y. Kitagawa, L. Chibotaru, L. Ungur, M. Mas-Torrent, W. Wernsdorfer, B. K. Breedlove, M. Enders, J. Veciana and M. Yamashita, Chem. – Eur. J., 2020, 26, 8621–8630 CrossRef CAS PubMed.
- N. Kobayashi, H. Lam, W. A. Nevin, P. Janda, C. C. Leznoff, T. Koyama, A. Monden and H. Shirai, J. Am. Chem. Soc., 1994, 116, 879–890 CrossRef CAS.
- K. Wang, D. Qi, H. Wang, W. Cao, W. Li, T. Liu, C. Duan and J. Jiang, Chem. – Eur. J., 2013, 19, 11162–11166 CrossRef CAS PubMed.
- T. Morita, M. Damjanović, K. Katoh, Y. Kitagawa, N. Yasuda, Y. Lan, W. Wernsdorfer, B. K. Breedlove, M. Enders and M. Yamashita, J. Am. Chem. Soc., 2018, 140, 2995–3007 CrossRef CAS PubMed.
- C. Wei, G. Lu, C. Guo, X. Lv, X. Tang, Q. Liu, X. Cai, Y. Chen and J. Jiang, Org. Electron., 2021, 93, 106151 CrossRef CAS.
- T. Morita, K. Katoh, B. K. Breedlove and M. Yamashita, Inorg. Chem., 2013, 52, 13555–13561 CrossRef CAS PubMed.
- K. Katoh, T. Morita, N. Yasuda, W. Wernsdorfer, Y. Kitagawa, B. K. Breedlove and M. Yamashita, Chem. – Eur. J., 2018, 24, 15522–15528 CrossRef CAS PubMed.
- G. Lu, K. Wang, X. Kong, H. Pan, J. Zhang, Y. Chen and J. Jiang, ChemElectroChem, 2018, 5, 605–609 CrossRef CAS.
- X. Cai, C. Wei, J. Dong, Q. Liu, Y. Wu, G. Lu, Y. Chen and J. Jiang, J. Mater. Sci.: Mater. Electron., 2019, 30, 1976–1983 CrossRef CAS.
- G. Lu, X. Kong, J. Sun, L. Zhang, Y. Chen and J. Jiang, Chem. Commun., 2017, 53, 12754–12757 RSC.
- S. Lee, K. ichi Yamashita, N. Sakata, Y. Hirao, K. Ogawa and T. Ogawa, Chem. – Eur. J., 2019, 25, 3240–3243 CAS.
- W. Wernsdorfer, N. Aliaga-Alcalde, D. N. Hendrickson and G. Christou, Nature, 2002, 416, 406–409 CrossRef PubMed.
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 268–297 CrossRef CAS PubMed.
- ed. Molecular Nanomagnets and Related Phenomena, S. Gao, Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, vol. 164 Search PubMed.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC.
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed.
- A. R. Farrell, J. A. Coome, M. R. Probert, A. E. Goeta, J. A. K. Howard, M.-H. Lemée-Cailleau, S. Parsons and M. Murrie, CrystEngComm, 2013, 15, 3423–3429 RSC.
- K. Katoh, Y. Yoshida, M. Yamashita, H. Miyasaka, B. K. Breedlove, T. Kajiwara, S. Takaishi, N. Ishikawa, H. Isshiki, Y. F. Zhang, T. Komeda, M. Yamagishi and J. Takeya, J. Am. Chem. Soc., 2009, 131, 9967–9976 CrossRef CAS PubMed.
- N. Ishikawa, M. Sugita, T. Okubo, N. Tanaka, T. Iino and Y. Kaizu, Inorg. Chem., 2003, 42, 2440–2446 CrossRef CAS PubMed.
- F. S. Guo, B. M. Day, Y. C. Chen, M. L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400–1403 CrossRef CAS PubMed.
- S. G. McAdams, A.-M. Ariciu, A. K. Kostopoulos, J. P. S. Walsh and F. Tuna, Coord. Chem. Rev., 2017, 346, 216–239 CrossRef CAS.
- M. Feng and M.-L. Tong, Chem. – Eur. J., 2018, 24, 7574–7594 CrossRef CAS PubMed.
- F. Habib and M. Murugesu, Chem. Soc. Rev., 2013, 42, 3278–3288 RSC.
- H. L. C. Feltham and S. Brooker, Coord. Chem. Rev., 2014, 276, 1–33 CrossRef CAS.
- S. Demir, I. R. Jeon, J. R. Long and T. D. Harris, Coord. Chem. Rev., 2015, 289–290, 149–176 CrossRef CAS.
- P. Zhang, Y. N. Guo and J. Tang, Coord. Chem. Rev., 2013, 257, 1728–1763 CrossRef CAS.
- X.-Y. Wang, C. Avendaño and K. R. Dunbar, Chem. Soc. Rev., 2011, 40, 3213–3238 RSC.
- C. Benelli and D. Gatteschi, Introduction to Molecular Magnetism. From Transition Metals to Lanthanides, 2015 Search PubMed.
- Lanthanides and Actinides in Molecular Magnetism, ed. R. A. Layfield and M. Murugesu, 2015 Search PubMed.
- M. Yamashita and K. Katoh, Molecular Magnetic Materials, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2016, pp. 79–101 Search PubMed.
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, 2006 Search PubMed.
- K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341–351 CrossRef CAS.
- A. Abragam and B. Bleaney, Electron paramagnetic resonance of transition ions, Oxford University Press, 2012 Search PubMed.
- A. Lunghi, F. Totti, R. Sessoli and S. Sanvito, Nat. Commun., 2017, 8, 14620 CrossRef PubMed.
- L. Gu and R. Wu, Phys. Rev. Lett., 2020, 125, 117203 CrossRef CAS PubMed.
- M. Briganti, F. Santanni, L. Tesi, F. Totti, R. Sessoli and A. Lunghi, J. Am. Chem. Soc., 2021, 143, 13633–13645 CrossRef CAS PubMed.
- J. M. Zadrozny, M. Atanasov, A. M. Bryan, C.-Y. Lin, B. D. Rekken, P. P. Power, F. Neese and J. R. Long, Chem. Sci., 2013, 4, 125–138 RSC.
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Y. Koshihara and Y. Kaizu, J. Phys. Chem. B, 2004, 108, 11265–11271 CrossRef CAS.
- W. Wernsdorfer, M. Murugesu and G. Christou, Phys. Rev. Lett., 2006, 96, 3–6 CrossRef PubMed.
- L. Gunther, Quantum Tunneling of Magnetization—QTM ’94, Springer Netherlands, Dordrecht, 1995, pp. 413–434 Search PubMed.
- J. R. Friedman, M. P. Sarachik, J. Tejada and R. Ziolo, Phys. Rev. Lett., 1996, 76, 3830–3833 CrossRef CAS PubMed.
- S. Bertaina, S. Gambarelli, T. Mitra, B. Tsukerblat, A. Müller and B. Barbara, Nature, 2008, 453, 203–206 CrossRef CAS PubMed.
- E. Burzurí, F. Luis, B. Barbara, R. Ballou, E. Ressouche, O. Montero, J. Campo and S. Maegawa, Phys. Rev. Lett., 2011, 107, 097203 CrossRef PubMed.
- W. Wernsdorfer and R. Sessoli, Science, 1999, 284, 133–135 CrossRef CAS PubMed.
- M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789–793 CrossRef CAS PubMed.
- S. Sproules, Electron Paramagnetic Resonance, 2017, vol. 25, pp. 61–97 Search PubMed.
- M. J. Graham, J. M. Zadrozny, M. S. Fataftah and D. E. Freedman, Chem. Mater., 2017, 29, 1885–1897 CrossRef CAS.
- A. Gaita-Ariño, F. Luis, S. Hill and E. Coronado, Nat. Chem., 2019, 11, 301–309 CrossRef PubMed.
- M. Atzori and R. Sessoli, J. Am. Chem. Soc., 2019, 141, 11339–11352 CrossRef CAS PubMed.
- J. Bartolomé, F. Luis and J. F. Fernández, Molecular magnets: physics and applications, Springer-Verlag/Sci-Tech/Trade, 2013 Search PubMed.
- F. Aquilante, J. Autschbach, R. K. Carlson, L. F. Chibotaru, M. G. Delcey, L. De Vico, I. F. Galván, N. Ferré, L. M. Frutos, L. Gagliardi, M. Garavelli, A. Giussani, C. E. Hoyer, G. Li Manni, H. Lischka, D. Ma, P. Å. Malmqvist, T. Müller, A. Nenov, M. Olivucci, T. B. Pedersen, D. Peng, F. Plasser, B. Pritchard, M. Reiher, I. Rivalta, I. Schapiro, J. Segarra-Martí, M. Stenrup, D. G. Truhlar, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, V. P. Vysotskiy, O. Weingart, F. Zapata and R. Lindh, J. Comput. Chem., 2016, 37, 506–541 CrossRef CAS PubMed.
- E. Coronado, Nat. Rev. Mater., 2020, 5, 87–104 CrossRef.
- S. Stepanow, J. Honolka, P. Gambardella, L. Vitali, N. Abdurakhmanova, T. C. Tseng, S. Rauschenbach, S. L. Tait, V. Sessi, S. Klyatskaya, M. Ruben and K. Kern, J. Am. Chem. Soc., 2010, 132, 11900–11901 CrossRef CAS PubMed.
- S. Lumetti, A. Candini, C. Godfrin, F. Balestro, W. Wernsdorfer, S. Klyatskaya, M. Ruben and M. Affronte, Dalton Trans., 2016, 45, 16570–16574 RSC.
- M. Urdampilleta, N. V. Nguyen, J. P. Cleuziou, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Int. J. Mol. Sci., 2011, 12, 6656–6667 CrossRef CAS PubMed.
- S. Thiele, F. Balestro, R. Ballou, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Science, 2014, 344, 1135–1138 CrossRef CAS PubMed.
- A. L. Thomas, Phthalocyanine Research and Applications, Taylor & Francis Inc, 1990 Search PubMed.
- N. Ishikawa, J. Jiang, O. Bekaroğlu, J. Liu, P. Lo and D. K. P. Ng, Functional Phthalocyanine Molecular Materials, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, vol. 135 Search PubMed.
- J. W. Perry, K. Mansour, I. S. Lee, X. Wu, P. V. Bedworth, C. Chen, D. Ng, S. R. Marder, P. Miles, T. Wada, M. Tian and H. Sasabe, Science, 1996, 273, 1533–1536 CrossRef CAS.
- E. Palomares, M. V. Martı and J. R. Durrant, Chem. Commun., 2004, 2112–2113 RSC.
- B. Lim, G. Y. Margulis, J.-H. Yum, E. L. Unger, B. E. Hardin, M. Grätzel, M. D. McGehee and A. Sellinger, Org. Lett., 2013, 15, 784–787 CrossRef CAS PubMed.
- N. Ishikawa, M. Sugita and W. Wernsdorfer, Angew. Chem., Int. Ed., 2005, 44, 2931–2935 CrossRef CAS PubMed.
- N. Ishikawa, M. Sugita, N. Tanaka, T. Ishikawa, S. Koshihara and Y. Kaizu, Inorg. Chem., 2004, 43, 5498–5500 CrossRef CAS PubMed.
- M. Damjanovic, T. Morita, K. Katoh, M. Yamashita and M. Enders, Chem. – Eur. J., 2015, 21, 14421–14432 CrossRef CAS PubMed.
- K. M. Kadish, K. M. Smith and R. Guilard, The porphyrin handbook, Phthalocyanines: spectroscopic and electrochemical characterization, Academic Press, 2003, vol. 16 Search PubMed.
- R. Sessoli, Nature, 2017, 548, 400–401 CrossRef CAS PubMed.
- L. Escalera-Moreno, J. J. Baldoví, A. Gaita-Ariño and E. Coronado, Chem. Sci., 2018, 9, 3265–3275 RSC.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445–11449 CrossRef CAS PubMed.
- C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS PubMed.
- N. Ishikawa, T. Iino and Y. Kaizu, J. Phys. Chem. A, 2002, 106, 9543–9550 CrossRef CAS.
- J. L. Liu, Y. C. Chen, Y. Z. Zheng, W. Q. Lin, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M. L. Tong, Chem. Sci., 2013, 4, 3310–3316 RSC.
- J. L. Liu, Y. C. Chen and M. L. Tong, Chem. Soc. Rev., 2018, 47, 2431–2453 RSC.
- L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092–3104 RSC.
- K. W. H. Stevens, Proc. Phys. Soc., London, Sect. A, 1952, 65, 209–215 CrossRef.
- J. Sievers, Z. Phys. Chem., 1982, 45, 289–296 CAS.
- D. Schmitt, J. Phys., 1986, 47, 677–681 CrossRef.
- C. Görller-Walran and K. Binnemans, Handbook on the physics and chemistry of rare earths. Volume 23, Elsevier, North-Holland, Amsterdam, 1996 Search PubMed.
- L. F. Chibotaru, in Molecular Nanomagnets and Related Phenomena, ed. S. Gao, Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, pp. 185–229 Search PubMed.
- L. Ungur and L. F. Chibotaru, Chem. – Eur. J., 2017, 23, 3708–3718 CrossRef CAS PubMed.
- A. L. Wysocki and K. Park, Inorg. Chem., 2020, 59, 2771–2780 CrossRef CAS PubMed.
- G. Taran, E. Bonet and W. Wernsdorfer, J. Appl. Phys., 2019, 125, 142903 CrossRef.
- G. Taran, E. Bonet and W. Wernsdorfer, Phys. Rev. B, 2019, 99, 180408 CrossRef CAS.
- D. Tanaka, T. Inose, H. Tanaka, S. Lee, N. Ishikawa and T. Ogawa, Chem. Commun., 2012, 48, 7796 RSC.
- N. Ishikawa, T. Iino and Y. Kaizu, J. Am. Chem. Soc., 2002, 124, 11440–11447 CrossRef CAS PubMed.
- F. Ara, H. Oka, Y. Sainoo, K. Katoh, M. Yamashita and T. Komeda, J. Appl. Phys., 2019, 125, 183901 CrossRef.
- K. Katoh, H. Isshiki, T. Komeda and M. Yamashita, Coord. Chem. Rev., 2011, 255, 2124–2148 CrossRef CAS.
- K. Katoh, H. Isshiki, T. Komeda and M. Yamashita, Chem. – Asian J., 2012, 7, 1154–1169 CrossRef CAS PubMed.
- T. Komeda, H. Isshiki, J. Liu, Y.-F. Zhang, N. Lorente, K. Katoh, B. K. Breedlove and M. Yamashita, Nat. Commun., 2011, 2, 217 CrossRef PubMed.
- R. Vincent, S. Klyatskaya, M. Ruben, W. Wernsdorfer and F. Balestro, Nature, 2012, 488, 357–360 CrossRef CAS PubMed.
- A. Candini, S. Klyatskaya, M. Ruben, W. Wernsdorfer and M. Affronte, Nano Lett., 2011, 11, 2634–2639 CrossRef CAS PubMed.
- E. Moreno-Pineda, T. Komeda, K. Katoh, M. Yamashita and M. Ruben, Dalton Trans., 2016, 45, 18417–18433 RSC.
- T. Komeda, K. Katoh and M. Yamashita, Single molecule magnet for quantum information process, 2019 Search PubMed.
- T. Inose, D. Tanaka, J. Liu, M. Kajihara, P. Mishra, T. Ogawa and T. Komeda, Nanoscale, 2018, 10, 19409–19417 RSC.
- E. Moreno-Pineda, C. Godfrin, F. Balestro, W. Wernsdorfer and M. Ruben, Chem. Soc. Rev., 2018, 47, 501–513 RSC.
- M. Ganzhorn, S. Klyatskaya, M. Ruben and W. Wernsdorfer, ACS Nano, 2013, 7, 6225–6236 CrossRef CAS PubMed.
- M. Urdampilleta, S. Klayatskaya, M. Ruben and W. Wernsdorfer, ACS Nano, 2015, 9, 4458–4464 CrossRef CAS PubMed.
- E. Moreno-Pineda, M. Damjanović, O. Fuhr, W. Wernsdorfer and M. Ruben, Angew. Chem., Int. Ed., 2017, 56, 9915–9919 CrossRef CAS PubMed.
- I. V. Krainov, J. Klier, A. P. Dmitriev, S. Klyatskaya, M. Ruben, W. Wernsdorfer and I. V. Gornyi, ACS Nano, 2017, 11, 6868–6880 CrossRef CAS PubMed.
- C. Godfrin, S. Thiele, A. Ferhat, S. Klyatskaya, M. Ruben, W. Wernsdorfer and F. Balestro, ACS Nano, 2017, 11, 3984–3989 CrossRef CAS PubMed.
- E. Moreno-Pineda, S. Klyatskaya, P. Du, M. Damjanović, G. Taran, W. Wernsdorfer and M. Ruben, Inorg. Chem., 2018, 57, 9873–9879 CrossRef CAS PubMed.
- C. Godfrin, R. Ballou, E. Bonet, M. Ruben, S. Klyatskaya, W. Wernsdorfer and F. Balestro, npj Quantum Inf., 2018, 4, 53 CrossRef.
- N. Koike, H. Uekusa, Y. Ohashi, C. Harnoode, F. Kitamura, T. Ohsaka and K. Tokuda, Inorg. Chem., 1996, 35, 5798–5804 CrossRef CAS.
- K. Katoh, B. K. Breedlove and M. Yamashita, Chem. Sci., 2016, 7, 4329–4340 RSC.
- J. Kan, H. Wang, W. Sun, W. Cao, J. Tao and J. Jiang, Inorg. Chem., 2013, 52, 8505–8510 CrossRef CAS PubMed.
- K. Katoh, T. Kajiwara, M. Nakano, Y. Nakazawa, W. Wernsdorfer, N. Ishikawa, B. K. Breedlove and M. Yamashita, Chem. – Eur. J., 2011, 17, 117–122 CrossRef CAS PubMed.
- H. Wang, K. Wang, J. Tao and J. Jiang, Chem. Commun., 2012, 48, 2973 RSC.
- K. Katoh, N. Yasuda, M. Damjanović, W. Wernsdorfer, B. K. Breedlove and M. Yamashita, Chem. – Eur. J., 2020, 26, 4805–4815 CrossRef CAS PubMed.
- B. Cirera, J. Matarrubia, T. Kaposi, N. Giménez-Agulló, M. Paszkiewicz, F. Klappenberger, R. Otero, J. M. Gallego, P. Ballester, J. V. Barth, R. Miranda, J. R. Galán-Mascarós, W. Auwärter and D. Ecija, Phys. Chem. Chem. Phys., 2017, 19, 8282–8287 RSC.
- T. Orlando, M. Filibian, S. Sanna, N. Giménez-Agullo, C. S. De Pipaón, P. Ballester, J. R. Galán-Mascarós and P. Carretta, J. Phys.: Condens. Matter, 2016, 28, 386002 CrossRef CAS PubMed.
- N. Ishikawa, M. Sugita and W. Wernsdorfer, J. Am. Chem. Soc., 2005, 127, 3650–3651 CrossRef CAS PubMed.
- T. Fukuda, W. Kuroda and N. Ishikawa, Chem. Commun., 2011, 47, 11686–11688 RSC.
- Y. Horii, K. Katoh, K. Sugimoto, R. Nakanishi, B. K. Breedlove and M. Yamashita, Chem. – Eur. J., 2019, 25, 3098–3104 CrossRef CAS PubMed.
- N. Ishikawa, S. Otsuka and Y. Kaizu, Angew. Chem., Int. Ed., 2005, 44, 731–733 CrossRef CAS PubMed.
- H. Wang, T. Liu, K. Wang, C. Duan and J. Jiang, Chem. – Eur. J., 2012, 18, 7691–7694 CrossRef CAS PubMed.
- T. Fukuda, K. Matsumura and N. Ishikawa, J. Phys. Chem. A, 2013, 117, 10447–10454 CrossRef CAS PubMed.
- K. Katoh, R. Asano, A. Miura, Y. Horii, T. Morita, B. K. Breedlove and M. Yamashita, Dalton Trans., 2014, 43, 7716–7725 RSC.
- A. Y. Tolbin, V. E. Pushkarev, L. G. Tomilova and N. S. Zefirov, Mendeleev Commun., 2009, 19, 78–80 CrossRef CAS.
- N. Ishikawa and Y. Kaizu, Chem. Phys. Lett., 1993, 203, 472–476 CrossRef CAS.
- A. G. Martynov, M. A. Polovkova, G. S. Berezhnoy, A. A. Sinelshchikova, F. M. Dolgushin, K. P. Birin, G. A. Kirakosyan, Y. G. Gorbunova and A. Y. Tsivadze, Inorg. Chem., 2020, 59, 9424–9433 CrossRef CAS PubMed.
- A. G. Martynov, M. A. Polovkova, G. S. Berezhnoy, A. A. Sinelshchikova, V. N. Khrustalev, K. P. Birin, G. A. Kirakosyan, Y. G. Gorbunova and A. Y. Tsivadze, Inorg. Chem., 2021, 60, 9110–9121 CrossRef CAS PubMed.
- Y. Chen, C. Liu, F. Ma, D. Qi, Q. Liu, H.-L. Sun and J. Jiang, Chem. – Eur. J., 2018, 24, 8066–8070 CrossRef CAS PubMed.
- H. Zhang, R. Wang, P. Zhu, Z. Lai, J. Han, C.-F. Choi, D. K. P. Ng, X. Cui, C. Ma and J. Jiang, Inorg. Chem., 2004, 43, 4740–4742 CrossRef CAS PubMed.
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14236–14239 CrossRef CAS PubMed.
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 3, 538–542 CrossRef CAS PubMed.
- K. Katoh, K. Kagesawa and M. Yamashita, Materials and Energy, World Scientific Publishing, 2018, vol. 10, pp. 271–344 Search PubMed.
- K. Katoh, T. Komeda and M. Yamashita, Chem. Rec., 2016, 16, 987–1016 CrossRef CAS PubMed.
- K. Katoh, Y. Aizawa, T. Morita, B. K. Breedlove and M. Yamashita, Chem. – Eur. J., 2017, 23, 15377–15386 CrossRef CAS PubMed.
- T. Sato, S. Matsuzawa, K. Katoh, B. K. Breedlove and M. Yamashita, Magnetochemistry, 2019, 5, 65 CrossRef CAS.
- D. Pinkowicz, R. Podgajny and B. Sieklucka, Molecular Magnetic Materials, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2016, pp. 279–300 Search PubMed.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed.
- L. Margheriti, D. Chiappe, M. Mannini, P. E. Car, P. Sainctavit, M. A. Arrio, F. B. De Mongeot, J. C. Cezar, F. M. Piras, A. Magnani, E. Otero, A. Caneschi and R. Sessoli, Adv. Mater., 2010, 22, 5488–5493 CrossRef CAS PubMed.
- A. Hofmann and Z. Salman, J. Phys.: Conf. Ser., 2014, 551, 012055 CrossRef.
- T. Yamabayashi, K. Katoh, B. K. Breedlove and M. Yamashita, Molecules, 2017, 22, 1–11 CrossRef PubMed.
- T. Sato, B. K. Breedlove, M. Yamashita and K. Katoh, Angew. Chem., Int. Ed., 2021, 60, 21179–21183 CrossRef CAS PubMed.
- M. S. Haghighi, M. Rath, H. W. Rotter and H. Humborg, Z. Anorg. Allg. Chem., 1993, 619, 1887–1896 CrossRef CAS.
- G. Ostendorp, H. W. Rotter and H. Homborg, Zeitschrift für Naturforsch. B, 1996, 51, 567–573 CrossRef CAS.
- G. Ostendorp, H. W. Rotter and H. Homborg, Z. Anorg. Allg. Chem., 1996, 622, 235–244 CrossRef CAS.
- J. Janczak, R. Kubiak and A. Jezierski, Inorg. Chem., 1999, 38, 2043–2049 CrossRef CAS PubMed.
- R. Kubiak and J. Janczak, Inorg. Chim. Acta, 2010, 363, 2461–2466 CrossRef CAS.
- M. A. Lebedeva, T. W. Chamberlain and A. N. Khlobystov, Chem. Rev., 2015, 115, 11301–11351 CrossRef CAS PubMed.
- P. D. W. Boyd and C. A. Reed, Acc. Chem. Res., 2005, 38, 235–242 CrossRef CAS PubMed.
- D. Sun, F. S. Tham, C. A. Reed, L. Chaker and P. D. W. Boyd, J. Am. Chem. Soc., 2002, 124, 6604–6612 CrossRef CAS PubMed.
- A. L. Litvinov, D. V. Konarev, A. Y. Kovalevsky, I. S. Neretin, P. Coppens and R. N. Lyubovskaya, Cryst. Growth Des., 2005, 5, 1807–1819 CrossRef CAS.
- M. Suzuki, Z. Slanina, N. Mizorogi, X. Lu, S. Nagase, M. M. Olmstead, A. L. Balch and T. Akasaka, J. Am. Chem. Soc., 2012, 134, 18772–18778 CrossRef CAS PubMed.
- T. Ishii, N. Aizawa, R. Kanehama, M. Yamashita, K. ichi Sugiura and H. Miyasaka, Coord. Chem. Rev., 2002, 226, 113–124 CrossRef CAS.
- H. Wang, K. Qian, D. Qi, W. Cao, K. Wang, S. Gao and J. Jiang, Chem. Sci., 2014, 5, 3214–3220 RSC.
- H. Iwami, J. Xing, R. Nakanishi, Y. Horii, K. Katoh, B. K. Breedlove, K. Kawachi, Y. Kasama, E. Kwon and M. Yamashita, Chem. Commun., 2020, 56, 12785–12788 RSC.
- N. Ishikawa, Y. Mizuno, S. Takamatsu, T. Ishikawa and S. Koshihara, Inorg. Chem., 2008, 47, 10217–10219 CrossRef CAS PubMed.
- S. Takamatsu and N. Ishikawa, Polyhedron, 2007, 26, 1859–1862 CrossRef CAS.
- R. Pederson, A. L. Wysocki, N. Mayhall and K. Park, J. Phys. Chem. A, 2019, 123, 6996–7006 CrossRef CAS PubMed.
- Y. Horii, M. Damjanović, K. Katoh and M. Yamashita, Dalton Trans., 2021, 50, 9719–9724 RSC.
- M. Gonidec, E. S. Davies, J. McMaster, D. B. Amabilino and J. Veciana, J. Am. Chem. Soc., 2010, 132, 1756–1757 CrossRef CAS PubMed.
- T. Inose, D. Tanaka, H. Tanaka, O. Ivasenko, T. Nagata, Y. Ohta, S. De Feyter, N. Ishikawa and T. Ogawa, Chem. – Eur. J., 2014, 20, 11362–11369 CrossRef CAS PubMed.
- D. Tanaka, N. Sumitani, T. Inose, H. Tanaka, N. Ishikawa and T. Ogawa, Chem. Lett., 2015, 44, 668–670 CrossRef CAS.
- N. Sun, H. Wang, T. Liu, D. Qi and J. Jiang, Dalton Trans., 2019, 48, 1586–1590 RSC.
- L. Ruan, J. Tong, F. Luo, G. Qin, F. Tian, X. Jiao and X. Zhang, Inorg. Chem., 2021, 125, 10165–10172 CAS.
- M. Dailey and C. Besson, CrystEngComm, 2021, 23, 7151–7161 RSC.
- L. Vitali, S. Fabris, A. M. Conte, S. Brink, M. Ruben, S. Baroni and K. Kern, Nano Lett., 2008, 8, 3364–3368 CrossRef CAS PubMed.
- Z. Deng, S. Rauschenbach, S. Stepanow, S. Klyatskaya, M. Ruben and K. Kern, Phys. Scr., 2015, 90, 98003 CrossRef.
- B. Warner, F. El Hallak, N. Atodiresei, P. Seibt, H. Prüser, V. Caciuc, M. Waters, A. J. Fisher, S. Blügel, J. van Slageren and C. F. Hirjibehedin, Nat. Commun., 2016, 7, 12785 CrossRef CAS PubMed.
- F. Ara, Z. K. Qi, J. Hou, T. Komeda, K. Katoh and M. Yamashita, Dalton Trans., 2016, 45, 16644–16652 RSC.
- Y. S. Fu, J. Schwöbel, S. W. Hla, A. Dilullo, G. Hoffmann, S. Klyatskaya, M. Ruben and R. Wiesendanger, Nano Lett., 2012, 12, 3931–3935 CrossRef CAS PubMed.
- C. Wäckerlin, F. Donati, A. Singha, R. Baltic, S. Rusponi, K. Diller, F. Patthey, M. Pivetta, Y. Lan, S. Klyatskaya, M. Ruben, H. Brune and J. Dreiser, Adv. Mater., 2016, 28, 5195–5199 CrossRef PubMed.
- Y. Zhang, Y. Wang, P. Liao, K. Wang, Z. Huang, J. Liu, Q. Chen, J. Jiang and K. Wu, ACS Nano, 2018, 12, 2991–2997 CrossRef CAS PubMed.
- J. Schwöbel, Y. Fu, J. Brede, A. Dilullo, G. Hoffmann, S. Klyatskaya, M. Ruben and R. Wiesendanger, Nat. Commun., 2012, 3, 953 CrossRef PubMed.
- L. Malavolti, L. Poggini, L. Margheriti, L. Chiappe, P. Graziosi, B. Cortigiani, V. Lanzilotto, F. Buatier de Mongeot, P. Ohresser, E. Otero, F. Choueikani, P. Sainctavit, I. Bergenti, V. A. Dediu, M. Mannini and R. Sessoli, Chem. Commun., 2013, 49, 11506–11508 RSC.
- A. Lodi Rizzini, C. Krull, T. Balashov, J. J. Kavich, A. Mugarza, P. S. Miedema, P. K. Thakur, V. Sessi, S. Klyatskaya, M. Ruben, S. Stepanow and P. Gambardella, Phys. Rev. Lett., 2011, 107, 177205 CrossRef CAS PubMed.
- C. Nistor, C. Krull, A. Mugarza, S. Stepanow, C. Stamm, M. Soares, S. Klyatskaya, M. Ruben and P. Gambardella, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 184402 CrossRef.
- A. Lodi Rizzini, C. Krull, T. Balashov, A. Mugarza, C. Nistor, F. Yakhou, V. Sessi, S. Klyatskaya, M. Ruben, S. Stepanow and P. Gambardella, Nano Lett., 2012, 12, 5703–5707 CrossRef CAS PubMed.
- A. Candini, D. Klar, S. Marocchi, V. Corradini, R. Biagi, V. De Renzi, U. Del Pennino, F. Troiani, V. Bellini, S. Klyatskaya, M. Ruben, K. Kummer, N. B. Brookes, H. Huang, A. Soncini, H. Wende and M. Affronte, Sci. Rep., 2016, 6, 1–8 CrossRef PubMed.
- J. Gómez-Segura, I. Díez-Pérez, N. Ishikawa, M. Nakano, J. Veciana and D. Ruiz-Molina, Chem. Commun., 2006, 2866–2868 RSC.
- M. Gonidec, R. Biagi, V. Corradini, F. Moro, V. De Renzi, U. Del Pennino, D. Summa, L. Muccioli, C. Zannoni, D. B. Amabilino and J. Veciana, J. Am. Chem. Soc., 2011, 133, 6603–6612 CrossRef CAS PubMed.
- D. Klar, A. Candini, L. Joly, S. Klyatskaya, B. Krumme, P. Ohresser, J.-P. Kappler, M. Ruben and H. Wende, Dalton Trans., 2014, 43, 10686–10689 RSC.
- L. Smykalla, P. Shukrynau and M. Hietschold, J. Phys. Chem. C, 2012, 116, 8008–8013 CrossRef CAS.
- G. Serrano, E. Velez-Fort, I. Cimatti, B. Cortigiani, L. Malavolti, D. Betto, A. Ouerghi, N. B. Brookes, M. Mannini and R. Sessoli, Nanoscale, 2018, 10, 2715–2720 RSC.
- S. Marocchi, A. Candini, D. Klar, W. Van Den Heuvel, H. Huang, F. Troiani, V. Corradini, R. Biagi, V. De Renzi, S. Klyatskaya, K. Kummer, N. B. Brookes, M. Ruben, H. Wende, U. Del Pennino, A. Soncini, M. Affronte and V. Bellini, ACS Nano, 2016, 10, 9353–9360 CrossRef CAS PubMed.
- M. Mannini, F. Bertani, C. Tudisco, L. Malavolti, L. Poggini, K. Misztal, D. Menozzi, A. Motta, E. Otero, P. Ohresser, P. Sainctavit, G. G. Condorelli, E. Dalcanale and R. Sessoli, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- H. Oka, K. Katoh, Y. Okada, D. Oka, T. Hitosugi, M. Yamashita and T. Fukumura, Chem. Lett., 2021, 50, 1489–1492 CrossRef CAS.
- M. Serri, M. Mannini, L. Poggini, E. Vélez-Fort, B. Cortigiani, P. Sainctavit, D. Rovai, A. Caneschi and R. Sessoli, Nano Lett., 2017, 17, 1899–1905 CrossRef CAS PubMed.
- F. Pineider, E. Pedrueza-Villalmanzo, M. Serri, A. M. Adamu, E. Smetanina, V. Bonanni, G. Campo, L. Poggini, M. Mannini, C. De Julián Fernández, C. Sangregorio, M. Gurioli, A. Dmitriev and R. Sessoli, Mater. Horiz., 2019, 6, 1148–1155 RSC.
- A. Dahal and M. Batzill, Nanoscale, 2014, 6, 2548–2562 RSC.
- M. Cinchetti, V. A. Dediu and L. E. Hueso, Nat. Mater., 2017, 16, 507–515 CrossRef CAS PubMed.
- A. Zhao, Q. Li, L. Chen, H. Xiang, W. Wang, S. Pan, B. Wang, X. Xiao, J. Yang, J. G. Hou and Q. Zhu, Science, 2005, 309, 1542–1544 CrossRef CAS PubMed.
- V. Madhavan, W. Chen, T. Jamneala, M. F. Crommie and N. S. Wingreen, Science, 1998, 280, 567–569 CrossRef CAS PubMed.
- T. Komeda, H. Isshiki, J. Liu, K. Katoh and M. Yamashita, ACS Nano, 2014, 8, 4866–4875 CrossRef CAS PubMed.
- R. Robles, N. Lorente, H. Isshiki, J. Liu, K. Katoh, B. K. Breedlove, M. Yamashita and T. Komeda, Nano Lett., 2012, 12, 3609–3612 CrossRef CAS PubMed.
- J. Hellerstedt, A. Cahlík, M. Švec, B. De La Torre, M. Moro-Lagares, T. Chutora, B. Papoušková, G. Zoppellaro, P. Mutombo, M. Ruben, R. Zbořil and P. Jelinek, Nanoscale, 2018, 10, 15553–15563 RSC.
- S. Fahrendorf, N. Atodiresei, C. Besson, V. Caciuc, F. Matthes, S. Blügel, P. Kögerler, D. E. Bürgler and C. M. Schneider, Nat. Commun., 2013, 4, 2425 CrossRef PubMed.
- J. Granet, M. Sicot, B. Kierren, S. Lamare, F. Chérioux, F. Baudelet, Y. Fagot-Revurat, L. Moreau and D. Malterre, Nanoscale, 2018, 10, 9123–9132 RSC.
- H. Isago and M. Shimoda, Chem. Lett., 1992, 147–150 CrossRef CAS.
- A. Mugarza, C. Krull, R. Robles, S. Stepanow, G. Ceballos and P. Gambardella, Nat. Commun., 2011, 2, 490 CrossRef PubMed.
- N. Tsukahara, S. Shiraki, S. Itou, N. Ohta, N. Takagi and M. Kawai, Phys. Rev. Lett., 2011, 106, 187201 CrossRef PubMed.
- M. Studniarek, C. Wäckerlin, A. Singha, R. Baltic, K. Diller, F. Donati, S. Rusponi, H. Brune, Y. Lan, S. Klyatskaya, M. Ruben, A. P. Seitsonen and J. Dreiser, Adv. Sci., 2019, 6, 1901736 CrossRef CAS PubMed.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- M. Menon and D. Srivastava, Phys. Rev. Lett., 1997, 79, 4453–4456 CrossRef CAS.
- S. J. Tans, A. R. M. Verschueren and C. Dekker, Nature, 1998, 393, 49–52 CrossRef CAS.
- V. I. Preprint, S. W. Emmons and A. M. Leroi, Nature, 1999, 402, 253–254 Search PubMed.
- W. Lu and C. M. Lieber, Nat. Mater., 2007, 6, 841–850 CrossRef CAS PubMed.
- P. M. Ajayan, T. W. Ebbesen, T. Ichihashi, S. Iijima, K. Tanigaki and H. Hiura, Nature, 1993, 362, 522–525 CrossRef CAS.
- E. Dujardin, T. W. Ebbesen, H. Hiura and K. Tanigaki, Science, 1994, 265, 1850–1852 CrossRef CAS PubMed.
- B. W. Smith, M. Monthioux and D. E. Luzzi, Nature, 1998, 396, 323–324 CrossRef CAS.
- T. Pichler, H. Kuzmany, H. Kataura and Y. Achiba, Phys. Rev. Lett., 2001, 87, 267401 CrossRef CAS PubMed.
- S. Bandow, M. Takizawa, K. Hirahara, M. Yudasaka and S. Iijima, Chem. Phys. Lett., 2001, 337, 48–54 CrossRef CAS.
- R. Kitaura, N. Imazu, K. Kobayashi and H. Shinohara, Nano Lett., 2008, 8, 693–699 CrossRef CAS PubMed.
- M. del Carmen Giménez-López, F. Moro, A. La Torre, C. J. Gómez-García, P. D. Brown, J. van Slageren and A. N. Khlobystov, Nat. Commun., 2011, 2, 407 CrossRef PubMed.
- R. Nakanishi, M. Yatoo, K. Katoh, B. Breedlove and M. Yamashita, Materials, 2016, 10, 7 CrossRef PubMed.
- R. Nakanishi, J. Satoh, K. Katoh, H. Zhang, B. K. Breedlove, M. Nishijima, Y. Nakanishi, H. Omachi, H. Shinohara and M. Yamashita, J. Am. Chem. Soc., 2018, 140, 10955–10959 CrossRef CAS PubMed.
- K. Katoh, J. Sato, R. Nakanishi, F. Ara, T. Komeda, Y. Kuwahara, T. Saito, B. K. Breedlove and M. Yamashita, J. Mater. Chem. C, 2021, 9, 10697–10704 RSC.
- A. I. A. Abd El-Mageed and T. Ogawa, RSC Adv., 2019, 9, 28135–28145 RSC.
- A. Tsumura, H. Koezuka and T. Ando, Appl. Phys. Lett., 1986, 49, 1210–1212 CrossRef CAS.
- C. A. Di, G. Yu, Y. Liu and D. Zhu, J. Phys. Chem. B, 2007, 111, 14083–14096 CrossRef CAS PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- M. H. Hoang, Y. Kim, M. Kim, K. H. Kim, T. W. Lee, D. N. Nguyen, S. J. Kim, K. Lee, S. J. Lee and D. H. Choi, Adv. Mater., 2012, 24, 5363–5367 CrossRef CAS PubMed.
- Y. Zhang, X. Cai, Y. Bian and J. Jiang, in Functional Phthalocyanine Molecular Materials, ed. J. Jiang, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, vol. 135, pp. 275–322 Search PubMed.
- G. Guillaud, M. Al Sadoun, M. Maitrot, J. Simon and M. Bouvet, Chem. Phys. Lett., 1990, 167, 503–506 CrossRef CAS.
- A. V. Shokurov, D. S. Kutsybala, A. G. Martynov, O. A. A. Raitman, V. V. V. Arslanov, Y. G. G. Gorbunova, A. Y. Tsivadze and S. L. Selektor, Thin Solid Films, 2019, 692, 137591 CrossRef CAS.
- F. S. Kim, E. Ahmed, S. Subramaniyan and S. A. Jenekhe, ACS Appl. Mater. Interfaces, 2010, 2, 2974–2977 CrossRef CAS PubMed.
- Y. Zhang, X. Cai, D. Qi, Y. Bian and J. Jiang, J. Phys. Chem. C, 2008, 112, 14579–14588 CrossRef CAS.
- W. Su, J. Jiang, K. Xiao, Y. Chen, Q. Zhao, G. Yu and Y. Liu, Langmuir, 2005, 21, 6527–6531 CrossRef CAS PubMed.
- N. B. Chaure, J. L. Sosa-Sanchez, A. N. Cammidge, M. J. Cook and A. K. Ray, Org. Electron., 2010, 11, 434–438 CrossRef CAS.
- S. Barard, D. Mukherjee, S. Sarkar, T. Kreouzis, I. Chambrier, A. N. Cammidge and A. K. Ray, J. Mater. Sci.: Mater. Electron., 2020, 31, 265–273 CrossRef CAS.
- Y. Chen, W. Su, M. Bai, J. Jiang, X. Li, Y. Liu, L. Wang and S. Wang, J. Am. Chem. Soc., 2005, 127, 15700–15701 CrossRef CAS PubMed.
- Y. Chen, R. Li, R. Wang, P. Ma, S. Dong, Y. Gao, X. Li and J. Jiang, Langmuir, 2007, 23, 12549–12554 CrossRef CAS PubMed.
- R. Li, P. Ma, S. Dong, X. Zhang, Y. Chen, X. Li and J. Jiang, Inorg. Chem., 2007, 46, 11397–11404 CrossRef CAS PubMed.
- Y. Gao, P. Ma, Y. Chen, Y. Zhang, Y. Bian, X. Li, J. Jiang and C. Ma, Inorg. Chem., 2009, 48, 45–54 CrossRef CAS PubMed.
- P. Ma, Y. Chen, N. Sheng, Y. Bian and J. Jiang, Eur. J. Inorg. Chem., 2009, 954–960 CrossRef CAS.
- K. Katoh, K. Yamamoto, T. Kajiwara, J. Takeya, B. K. Breedlove and M. Yamashita, J. Phys.: Conf. Ser., 2011, 303, 012035 CrossRef.
- Y. Chen, D. Li, N. Yuan, J. Gao, R. Gu, G. Lu and M. Bouvet, J. Mater. Chem., 2012, 22, 22142–22149 RSC.
- Y. Chen, X. Kong, G. Lu, D. Qi, Y. Wu, X. Li, M. Bouvet, D. Sun and J. Jiang, Mater. Chem. Front., 2018, 2, 1009–1016 RSC.
- X. Zhang and Y. Chen, Inorg. Chem. Commun., 2014, 39, 79–82 CrossRef.
- G. Lu, X. Kong, P. Ma, K. Wang, Y. Chen and J. Jiang, ACS Appl. Mater. Interfaces, 2016, 8, 6174–6182 CrossRef CAS PubMed.
- X. Zhang, L. Liu, J. Xiao, Z. Sun and P. Li, J. Mater. Res. Technol., 2020, 9, 13682–13691 CrossRef CAS.
- C. Wang, D. Qi, G. Lu, H. Wang, Y. Chen and J. Jiang, Inorg. Chem. Front., 2019, 6, 3345–3349 RSC.
- D. Gao, X. Zhang, X. Kong, Y. Chen and J. Jiang, ACS Appl. Mater. Interfaces, 2015, 7, 2486–2493 CrossRef CAS PubMed.
- X. Kong, G. Lu, X. Wang, S. Zhao, D. Sun, X. Li, Y. Chen and J. Jiang, J. Mater. Sci.: Mater. Electron., 2019, 30, 12437–12446 CrossRef CAS.
- R. Madru, G. Guillaud, M. Al Sadoun, M. Maitrot, J. J. André, J. Simon and R. Even, Chem. Phys. Lett., 1988, 145, 343–346 CrossRef CAS.
- A. I. Zvyagina, A. E. Aleksandrov, A. G. Martynov, A. R. Tameev, A. E. Baranchikov, A. A. Ezhov, Y. G. Gorbunova and M. A. Kalinina, Inorg. Chem., 2021, 60, 15509–15518 CrossRef CAS PubMed.
- A. G. Martynov, Y. G. Gorbunova and A. Y. Tsivadze, Prot. Met. Phys. Chem. Surfaces, 2011, 47, 465–470 CrossRef CAS.
- M. O. Senge, M. Fazekas, E. G. A. Notaras, W. J. Blau, M. Zawadzka, O. B. Locos and E. M. N. Mhuircheartaigh, Adv. Mater., 2007, 19, 2737–2774 CrossRef CAS.
- G. de la Torre, P. Vazquez, F. Agullo-Lopez and T. Torres, Chem. Rev., 2004, 104, 3723–3750 CrossRef CAS PubMed.
- J. P. Collman, J. L. Kendall, J. L. Chen, T. A. Eberspacher and C. R. Moylan, Inorg. Chem., 1997, 36, 5603–5608 CrossRef CAS.
- D. Qi and J. Jiang, ChemPhysChem, 2015, 16, 1889–1897 CrossRef CAS PubMed.
- J. S. Shirk, J. R. Lindle, F. J. Bartoli, Z. H. Kafafi, A. W. Snow and M. E. Boyle, J. Nonlinear Opt. Phys. Mater., 1992, 01, 699–726 CrossRef CAS.
- J. L. Wu, B. Gu, N. Sheng, D. Liu and Y. Cui, Appl. Phys. Lett., 2014, 105, 171113 CrossRef.
- N. Sheng, D. Liu, B. Gu, J. He and Y. Cui, Dyes Pigm., 2015, 122, 346–350 CrossRef CAS.
- B. Ren, N. Sheng, B. Gu, Y. Wan, G. Rui, C. Lv and Y. Cui, Dyes Pigm., 2017, 139, 788–794 CrossRef CAS.
- A. I. Plekhanov, T. V. Basova, R. G. Parkhomenko and A. G. Gürek, Opt. Mater., 2017, 64, 13–17 CrossRef CAS.
- A. B. Karpo, V. E. Pushkarev, V. I. Krasovskii and L. G. Tomilova, Chem. Phys. Lett., 2012, 554, 155–158 CrossRef CAS.
- K. E. Sekhosana and T. Nyokong, Opt. Mater., 2014, 37, 139–146 CrossRef CAS.
- K. E. Sekhosana and T. Nyokong, Opt. Mater., 2015, 47, 211–218 CrossRef CAS.
- K. E. Sekhosana and T. Nyokong, Inorg. Chim. Acta, 2016, 450, 87–91 CrossRef CAS.
- K. E. Sekhosana, M. Shumba and T. Nyokong, Polyhedron, 2017, 138, 154–160 CrossRef CAS.
- N. Sheng, B. Gu, B. Ren, Y. Wang, L. Han, J. Wang, H. Cao, M. Guan, X. Zhai and J. Sha, Dyes Pigm., 2017, 136, 553–558 CrossRef CAS.
- C. Huang, K. Wang, J. Sun and J. Jiang, Eur. J. Inorg. Chem., 2014, 1546–1551 CrossRef CAS.
- L. Chen, R. Hu, J. Xu, S. Wang, X. Li, S. Li and G. Yang, Spectrochim. Acta, Part A, 2013, 105, 577–581 CrossRef CAS PubMed.
- K. E. Sekhosana, E. Amuhaya, S. Khene and T. Nyokong, Inorg. Chim. Acta, 2015, 426, 221–226 CrossRef CAS.
- H. Shang, H. Wang, K. Wang, J. Kan, W. Cao and J. Jiang, Dalton Trans., 2013, 42, 1109–1115 RSC.
- K. E. Sekhosana and T. Nyokong, RSC Adv., 2019, 9, 16223–16234 RSC.
- X. Chen, D. Qi, C. Liu, H. Wang, Z. Xie, T. W. Chen, S. M. Chen, T. W. Tseng and J. Jiang, RSC Adv., 2019, 10, 317–322 RSC.
- C. Liu, W. Yang, J. Wang, X. Ding, H. Ren, Y. Chen, Z. Xie, T. Sun and J. Jiang, Dalton Trans., 2021, 50, 13661–13665 RSC.
| This journal is © The Royal Society of Chemistry 2022 |