
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-internalising antibody–drug conjugates
Nicola
Ashman
 a,
Jonathan D.
Bargh
b and
David R.
Spring
*a
a,
Jonathan D.
Bargh
b and
David R.
Spring
*a
aYusuf Hamied Department of Chemistry University of Cambridge Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: spring@ch.cam.ac.uk
bMedicinal Chemistry, Research and Early Development Cardiovascular, Renal and Metabolism, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
First published on 2nd November 2022
Abstract
Antibody–drug conjugates (ADCs) typically require internalisation into cancer cells to release their cytotoxic payload. However, this places stringent constraints on therapeutic development, requiring cancer targets that have high expression of internalising antigens and efficient intracellular processing. An alternative approach is emerging whereby the payloads can be released extracellularly from cleavable linkers upon binding to poorly-internalising antigens or other tumoral components. This removes the reliance on high antigen expression, avoids potentially inefficient internalisation, and can greatly expand the range of cancer targets to components of the extracellular tumour matrix. This review gives an overview of recent developments towards non-internalising ADCs, including emerging cancer-associated cell surface and extracellular proteins, cancer stromal targeting and the linking chemistry that enables extracellular payload release.
 Nicola Ashman | Nicola Ashman recieved her MChem degree from the University of York in 2019 which included an industrial placement year in Early Chemical Development at AstraZeneca, Macclesfield. Nicola started her PhD with Professor David Spring at the University of Cambridge in 2019, where her research has focused on the generation of novel cleavable linkers for ADCs. |
 Jonathan D. Bargh | Jonathan Bargh obtained his PhD in 2020 from the Department of Chemistry, University of Cambridge under the supervision of Professor David Spring. His research involved the development of enzyme cleavable linkers for antibody–drug conjugates. In 2021 he joined AstraZeneca, Gothenburg, Sweden as a Senior Scientist in Medicinal Chemistry, Research and Early Development Cardiovascular, Renal and Metabolism. |
 David R. Spring | David Spring is currently Professor of Chemistry and Chemical Biology at the University of Cambridge within the Chemistry Department. He received his DPhil (1998) at Oxford University under Sir Jack Baldwin. He then worked as a Wellcome Trust Postdoctoral Fellow at Harvard University with Stuart Schreiber (1999– 2001), after which he joined the faculty at the University of Cambridge. His research programme is focused on the use of chemistry to explore biology. |
Introduction
Antibody–drug conjugates (ADCs) are a class of oncology therapeutics that provide targeted drug delivery by conjugation to a highly specific monoclonal antibody (mAb – synthetic antibodies derived from identical cells) (Fig. 1). This approach combines the targeting ability of antibodies with the cytotoxicity of small-molecule drugs. Its success is reflected in the FDA approval of 12 ADCs to date, with over 80 ADCs in active clinical trials.1 This review focuses on ADCs that do not require internalisation for efficacy, unlike the typical mechanism of action of ADCs.Key learning points(1) Internalising antibody–drug conjugates are limited by poor solid tumour penetration, lack of suitable internalising antigen targets and acquired resistance mechanisms.(2) Non-internalising antibody–drug conjugates can release their payload extracellularly due to extracellular linker cleavage, overcoming issues with inefficient internalisation or internalisation related resistance mechanisms. (3) The chemistry of the ADC linker is essential in imparting stability and selectivity of payload release, determining whether a non-internalising mechanism of action is possible. (4) Many ADC linkers thought to require internalisation for cleavage and payload release can be cleaved extracellularly. (5) There are a multitude of extracellular markers of cancer, and non-internalising cancer associated antigens which can be targeted by non-internalising ADCs. Another emerging approach is to target the tumour stroma or vasculature. |
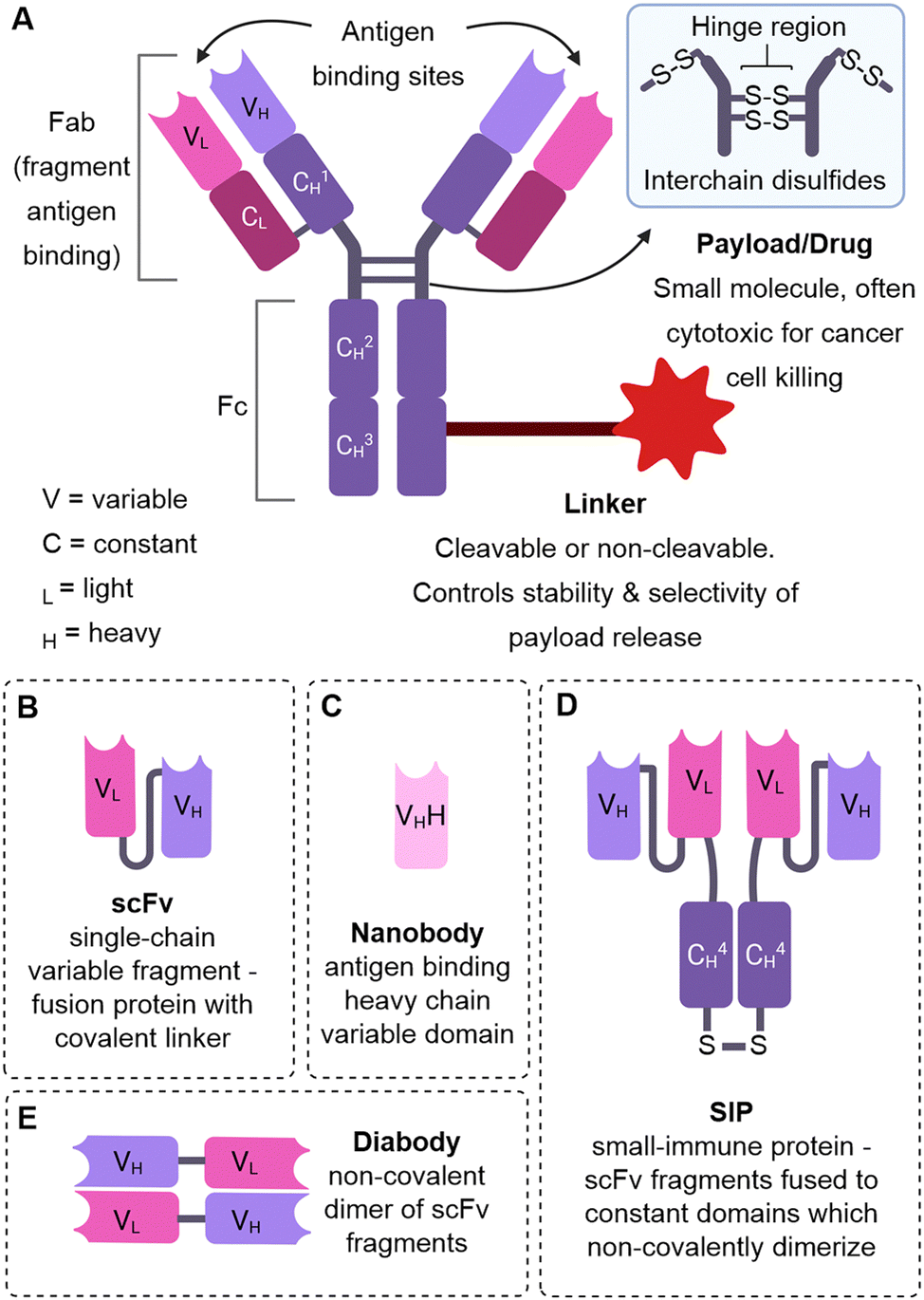 | ||
| Fig. 1 The structure of an ADC and alternative antibody formats. (A) The structure of an ADC (B) single-chain variable fragment (C) nanobody (D) small-immune protein (E) diabody. | ||
Mechanism of action
ADCs are usually designed to function via an internalising mechanism of action: after antibody binding to the target antigen on the cancer cell, the ADC is internalised by endocytosis and then degraded in the lysosome to release the cytotoxic payload (Fig. 2A). If the payload is suitably membrane-permeable, it can diffuse out of the cell in which it was released and into surrounding “bystander” cancer cells, which may or may not express the target antigen (termed the bystander effect). Whilst this approach has been successful, (indeed all currently FDA approved ADCs utilise internalising antibodies), it does suffer from limitations such as the reliance on the overexpression of internalising antigens, which is not a feature of many tumours. Additionally, antibodies are large proteins and thus have limited tumour penetration due to slow diffusion. Another obstacle to tumour penetration is the “antigen barrier”, in which antibodies continually bind to cancer cells in the outer perimeter of the tumour close to blood vessels, preventing their diffusion and binding to cells deeper within the tumour. Cancers can also acquire resistance to internalising ADCs as a result of changes to several processes such as internalisation, trafficking, antigen recycling or lysosomal degradation.2,3 | ||
| Fig. 2 (A) The mechanism of an internalising ADC (B) the mechanisms of non-internalising ADCs. | ||
Whilst it was previously thought that internalisation was essential for the therapeutic efficacy of ADCs, it is increasingly recognised that an alternative mechanism of action can be efficacious. Instead of requiring lysosomal degradation to release the payload intracellularly, the antibody–drug linkage can be designed to be labile in the extracellular tumour environment (Fig. 2B). This exploits the distinct chemical or enzymatic environment of cancer compared to healthy tissue. Payloads released extracellularly can then diffuse into surrounding cancer cells to exert their cytotoxic effect.
A non-internalising mechanism of action may expand the scope of cancer targets since it precludes the stringent requirements of internalising ADCs, such as high internalising antigen expression. Indeed, some non-internalising ADCs target physiological tumoral features common to most aggressive cancers and therefore may be a promising broad-spectrum anticancer approach. Release of the payload extracellularly may also enable deeper penetration of the anticancer agent into the tumour, due to its facile diffusion compared to antibodies, and may maximise the potential bystander effect.4
Internalising ADCs
All 12 currently FDA approved ADCs utilise internalising antibodies (Fig. 3), with the majority (10/12) incorporating cleavable linkers.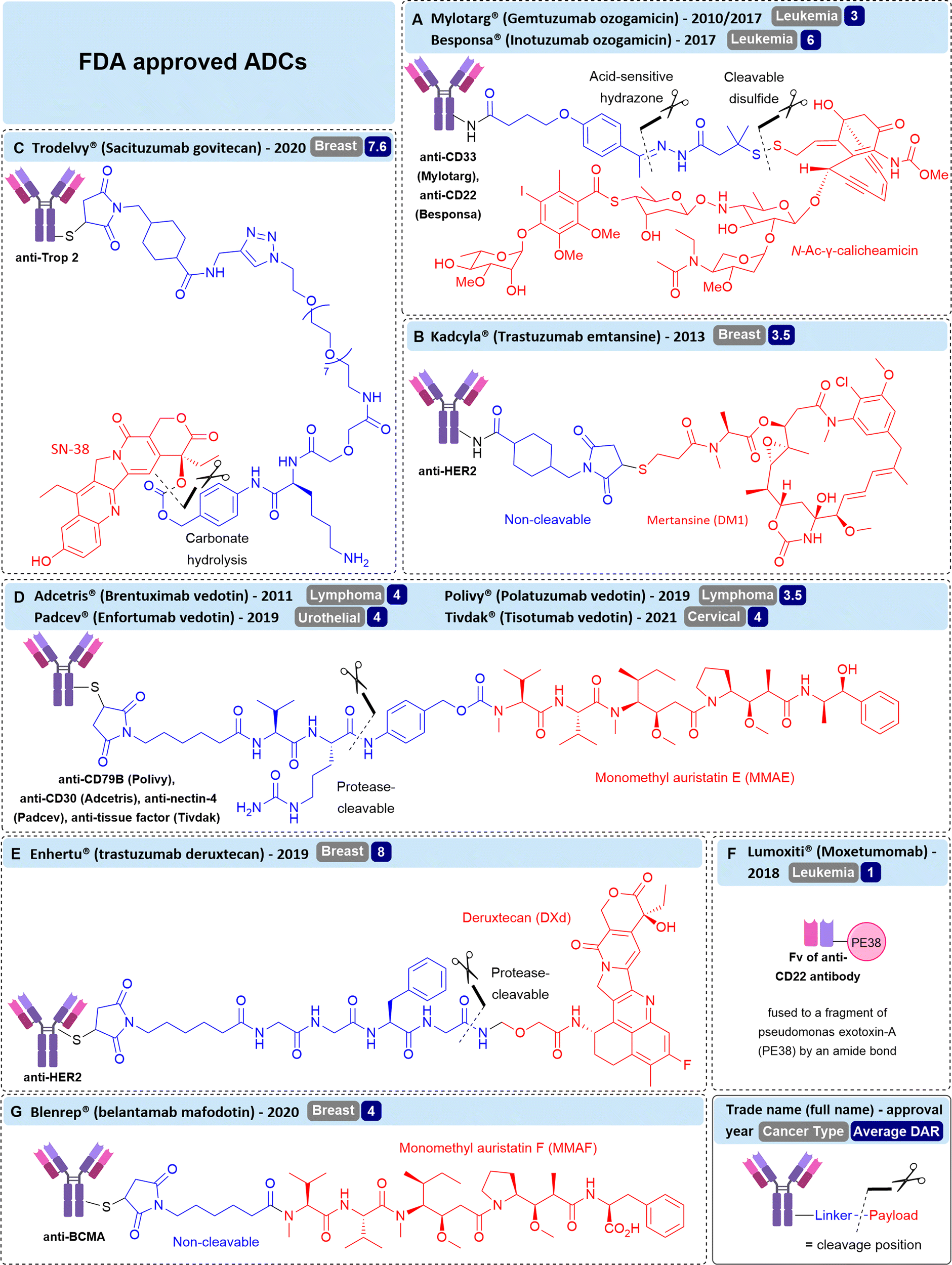 | ||
| Fig. 3 The structure of FDA-approved ADCs including their antigen target, cancer indication, linker structure, payload, and cleavage mechanism. (A) Mylotarg® & Besponsa®, (B) Kadcyla®, (C) Trodelvy® (D) Adcetris®, Polivy®, Padcev® and Tivdak®, (E) Enhertu®, (F) Lumoxiti® and (G) Blenrep®. | ||
The majority (7/12) of approved ADCs target non-solid tumour types, i.e. cancers of the blood including leukemia, lymphoma and myelomas. Although ADCs against solid tumours of the breast, cervix and bladder have successfully gained approval, non-internalising ADCs may be required to expand the range of cancer targets beyond these, especially to treat cancers with dense tumour stroma (the structural component of tumours, such as connective tissue).
 | ||
| Fig. 4 (A) Common cleavable linkers used in ADCs. (B) Common non-cleavable linkers used in ADCs and their released charged payload species after lysosomal antibody degradation which are unable to exert the bystander effect. | ||
In contrast, cleavable linkers possess a chemical trigger that can be cleaved in the specific environment of cancer (typically intracellularly). Often a self-immolative spacer is combined with a cleavable linker that facilitates the release of unmodified payload. Common cleavable linkers include: acid-labile hydrazones, which target the acidic pH of the lysosome; reducible disulfides, targeting the elevated concentrations of glutathione in the cytosol of cancer cells and; enzyme-cleavable linkers, such as Valine-Citrulline (Val-Cit) dipeptides, that can be cleaved by the elevated levels of cathepsin proteases present in cancer (Fig. 4A). Many other cleavable linkers have been described,5 but will not be discussed in detail in this review.
Whilst most cleavable linkers were initially designed to be susceptible to intracellular cleavage, evidence has emerged that some of these chemical or enzymatic cleavage triggers are present extracellularly.6,7 Additionally, dying tumour cells can release high concentrations of intracellular species, such as glutathione or proteases, into the extracellular tumour microenvironment.8 This phenomenon has facilitated the growth of non-internalising ADC research, whereby linkers classically thought to require internalisation have been utilised to give rise to efficacious non-internalising ADCs.
This review will give an overview into the recent developments in non-internalising ADCs, including non-internalising membrane proteins and extracellular protein targets, as well as the linker chemistry that enables extracellular payload release. It should be noted that throughout this review, the term “non-internalising” should not be taken as absolute. “non-internalising” targets are poorly internalising, but it is likely that there is internalisation to a small extent. Thus, the efficacy of ADCs may arise from contribution from both internalised ADC and payload released extracellularly. However, in most of the cases discussed, the non-internalised species are the major contributor to efficacy, since analogues which require internalisation for payload release are markedly less potent.
Targeting membrane proteins
Multiple features of the target antigen are important in determining the success of ADCs. For example, it is desirable that the target antigen is highly expressed on target cancer cells but not on healthy cells, to ensure specificity of the therapeutic and to avoid off-target toxicity. The nature of the antibody–antigen binding interaction dictates whether internalisation of the construct occurs (and how rapidly). For successful internalising ADCs, the antibody must induce rapid receptor internalisation, endosomal trafficking, and lysosomal processing (Fig. 2A). Conversely, for non-internalising ADCs, since internalisation does not need to occur at a considerable rate after antibody binding, these intracellular steps are precluded. As interest is growing in non-internalising ADCs, more cases of poorly internalising antibody–antigen interactions are being discovered.B-cell antigens CD20, CD21 and CD72
As part of an investigation into the mechanism of action of a variety of ADCs, Polson et al. examined the efficacy of non-internalising ADCs against B-cell CD20, CD21 and CD72 antigens.9 In this work, a variety of ADCs were generated with non-cleavable or cleavable linkers incorporating disulfides or Val-Cit dipeptides (Fig. 5). They found that cleavable disulfide ADCs were efficacious even against very poorly internalised antigen targets CD20, CD21 and CD72 in vivo. It was postulated that the disulfide linker can be cleaved extracellularly and therefore act by a non-internalising mechanism. Also in this work, the MMAE payload of an anti-CD21 ADC comprising a Val-Cit linker was replaced with MMAF, an analogue containing a charged carboxylic acid terminus (Fig. 5C) which hinders its ability to diffuse across cell membranes. This change eliminated the activity of the ADC, indicating that diffusion of the released payload across the cell membrane is essential for activity and thus supporting the proposed extracellular mechanism of action. Clearly, this data contradicted the classical belief that disulfide and Val-Cit cleavable linkers require internalisation for payload release and demonstrated that extracellular linker cleavage is possible, leading to efficacious ADCs. | ||
| Fig. 5 (A) The library of ADCs against various B-cell antigens studied by Polson et al.9 (B) The linker structures indicating their conjugation to the antibody and (C) the payload structures. | ||
CD20 was also demonstrated to be an effective target for non-internalising ADCs by DiJoseph et al.10 Calicheamicin was conjugated to anti-CD20 antibody rituximab via an acid-labile or acid stable (amide) linker (Fig. 6). The ADC containing the acid-cleavable 4-(4-acetylphenoxy)butanoic acid (AcBut) linker resulted in potent growth inhibition in mice B-cell lymphoma xenografts. However, the non-cleavable amide linker was markedly less effective with similar performance to unconjugated rituximab, indicating the importance of acid-mediated linker cleavage and implying a lack of significant ADC internalisation of the constructs. This was verified by comparison to analogous ADCs targeting the internalising CD22 antigen. In this case, the efficacy of the acid labile vs non-cleavable ADC was similar. Therefore, the acid-labile ADC is expected to be susceptible to cleavage extracellularly due to the relatively acidic (pH 5.5 – 7.0)11 extracellular environment of tumour cells.
 | ||
| Fig. 6 (A) The structure of the acid-cleavable rituximab ADC, (B) non-cleavable acid stable rituximab ADC and (C) the calicheamicin payload, investigated by DiJoseph et al.10 | ||
Similarly, Mylotarg® (gemtuzumab ozogamicin, Fig. 3A), an FDA-approved ADC targeting internalising CD33 containing an acid-sensitive hydrazone linker, has been investigated for its efficacy against CD33-negative cancer.12 “Passive targeting” of the ADC has been postulated, allowing accumulation of the ADC in tumour tissue and efficacious tumour growth inhibition in various subcutaneous xenografts, despite undetectable amounts of target CD33 antigen on tumour cells.13 The acid-labile linker was crucial for efficacy, as when it was replaced with an acid-stable linker, activity was diminished. Hence, extracellular cleavage in the acidic tumour microenvironment is a likely mechanism of drug release from this approved ADC, previously thought to require internalisation for efficacy. Instead, the efficacy is attributed to the enhanced permeability and retention effect (EPR), whereby the ADC accumulates at the site of action due to the enhanced vascular permeability of tumours. In combination with the extracellularly cleavable acid-sensitive linker, payload can be released at the site-of-action without active targeting of the cancer cells by the antibody.
ADCs incorporating the FDA-approved avelumab antibody in both IgG and alternative antibody formats have previously been demonstrated to internalise.20 A study of a radiolabelled antibody against human PD-L1 by Boerman et al. demonstrated that it was only slowly internalised, with 75% still membrane-bound after 24 hours of incubation with PD-L1 positive MDA-MB-231 cells.21 Additionally, Xiao et al. found that an avelumab-Val-Cit-MMAE ADC was not effective in vitro against PD-L1 positive cell lines, giving IC50's similar to unconjugated avelumab. This is rationalised by the lack of internalisation of the ADC, which may be necessary for lysosomal dipeptide cleavage. Flow cytometry analysis revealed that <20% of avelumab internalised in the MC-38, SK-MES and MDA-MB-231 cell lines, but 40% of alternative antibody atezolizumab was internalised within 2 h in MDA-MB-231 cells. Evidently, it is not easy to predict whether an antibody-conjugate against PD-L1 will internalise, so validation of the final ADC is needed. However, given the limited internalisation of avelumab-conjugates, avelumab could be suitable for development of anti-PD-L1 non-internalising ADCs.
Although in previous studies cleavable Val-Cit linkers were observed to be cleaved extracellularly, in this study internalisation was required, presumably because the levels of extracellular linker cleavage were insufficient. The extracellular levels of proteases or other linker triggers vary between cell lines, tumour types and indeed between pre-clinical mouse models and humans. Therefore, the in vitro and in vivo application of extracellularly cleavable linkers may require case-by-case validation of the levels of available endogenous triggers.
Goldenberg et al. examined the efficacy of anti-CEACAM5 labetuzumab ADCs in mouse models of human colonic carcinoma, human pancreatic tumours, and systemic lymphoma xenografts.22 All ADCs contained linkers with acid-labile carbonate or ester moieties, with some substrates possessing an additional protease-cleavable phenylalanine-lysine (Phe-Lys) residue (Fig. 7). The acid-labile linkers possessed intermediate stability, with half-lives of 10-65 hours in human serum.23 Gratifyingly, although CEACAM5 is poorly internalised,24 ADCs induced 100% survival in the target CEACAM5-positive lymphoma tumour mode, with the best in vivo performance from ADCs bearing the dual cleavable linker CL2 (Fig. 7).22
 | ||
| Fig. 7 The structure of anti-CEACAM5 non-internalising ADCs with (A) cleavable ester linkage or (B) dual cleavable linker comprising a cleavable carbonate with additional protease-cleavable peptide. | ||
The authors propose a non-internalising mechanism of action whereby SN-38 is slowly released from the ADC extracellularly, allowing for high local accumulation of free drug.24 The relatively high lability of the carbonate/ester linkage is deemed appropriate for use with SN-38 due to its moderate cytotoxicity compared to auristatins or maytansinoid payloads, allowing facile drug release without significant off-target toxicity. Indeed, FDA approved ADC Trodelvy® contains SN-38 conjugated via the CL2A linker (Fig. 3C).25 Although it targets highly internalising antigen Trop-2, substitution of the CL2A linker with a highly stable linkage significantly reduced efficacy, suggesting the gradual release of SN-38 from the CL2A linker contributes to efficacy.26
 | ||
| Fig. 8 The mechanism of action of NKA-inhibiting non-internalising ADC. | ||
Targeting the tumour microenvironment
Alternative strategies to target tumours without exploiting membrane antigens have now emerged. In fact, reliance on targeting membrane antigens has several drawbacks since few cancers homogeneously overexpress antigens, and resistance can be acquired through changes in antigen expression.3,29 Instead, other approaches seek to eradicate malignant cells indirectly by targeting components of the extracellular tumour microenvironment, such as secreted or extracellular proteins, or to target the abundant connective tissues and blood vessels of the tumour stroma. As with any targeted therapy, the target must be preferentially displayed in cancer but not in healthy tissue.Targeting proteins in the extracellular matrix
 | ||
| Fig. 9 The mechanism and structure of non-internalising anti-Gal-3-BP ADC. (A) Initial linker cleavage from dying cancer cells results in cancer cell killing and thus increased levels of reductants such as glutathione (GSH) to be released in the tumour microenvironment. (B) This amplifies linker cleavage, which then amplifies cancer cell killing by the released payload species. | ||
Chudasama et al. confirmed that LRG1 remains extracellular following its secretion, allowing the development of non-internalising ADCs comprising magacizumab conjugated to MMAE via a protease-cleavable Val-Cit linkers (Fig. 10).34In vitro, their ADCs were initially ineffective against human-LRG1-positive B16F0 cells due to the limited concentration of extracellular cathepsins in cell culture. Although ineffective, this confirmed the non-internalising nature of the LRG1 ADCs. They postulated that co-treatment with a small dose of cisplatin (5 μM) would enable sufficient release of cathepsins from dying cells to activate their ADCs. This combination approach afforded an IC50 of 15 pM for the ADC, indicating that drug release from their ADCs was improved with the increased cell death from co-administering an additional chemotherapeutic to kick-start the chain reaction. Furthermore, they demonstrated significant inhibition of tumour growth in vivo in a mouse melanoma model of their ADC alone (i.e. without pre-treatment with cathepsins or co-administration of cisplatin). However, ADC treatment was not curative. This may be explained by the known susceptibility of the Val-Cit linker to carboxylesterase 1c (Ces1C) present in mouse plasma.35–37 Premature circulatory hydrolysis of the linker by Ces1C likely impedes full delivery of the cytotoxin at the site-of-action, resulting in inaccurate and potentially lower efficacy than would be expected in human patients.
 | ||
| Fig. 10 (A) The bioconjugation to generate anti-LRG1 DAR 4 ADCs, (B) the structure of the linker-payload. | ||
Therefore, it is still unclear whether Val-Cit linkers are solely activated at the tumour site by target cathepsins or if their ability to release payload extracellularly is due to circulatory instability in mouse models. This is a key shortfall of the in vivo evaluation of Val-Cit ADCs in mice thus far. To truly disambiguate if these linkers are suitable for non-internalising ADCs in humans, studies with Ces1c knockout mice should be conducted, or linkers should be employed which are hydrolytically stable in mouse plasma. Nevertheless, LRG1 appears to be a promising target for the future development of non-internalising ADCs.
 | ||
| Fig. 11 (A) The mechanism of action of non-internalising anti-MMP9 ADC, (B) the structure of linker-payload and the bioconjugation method to generate DAR 8 MMP9 ADC. | ||
Stroma or tumour vasculature targeting
Many solid tumours exhibit dense intercellular stromata: a network of connective tissues and blood vessels which impede the penetration of large macromolecules deep into the tumour (Fig. 12). This is often a barrier to the successful treatment of solid tumours by ADCs. An emerging strategy alternative to targeting tumoral cells that lie behind the stromal barrier, is to target the stroma itself. ADC accumulation in the stroma could enable extracellular payload release in the tumour microenvironment, which can then diffuse into nearby tumour cells. Since the diffusion of small molecules is greater than mAbs, penetration of the active drug into the solid tumour is expected to be improved. This is termed “Cancer Stromal Targeting Therapy” (CAST). Additionally, the treatment of several solid tumours (colorectal, lung, pancreatic) by classic ADCs is limited due to their heterogenous target antigen expression.40 Therefore, an alternative targeting approach is desirable. Components of the modified tumour extracellular matrix include collagens, fibronectin, tenascin-C and fibrin, which are attractive targets for ADC delivery given their abundance, stability and selective presence in the tumour stroma or vasculature.41 | ||
| Fig. 12 (A) Classical internalising ADCs fail to treat solid tumours with dense extracellular stroma. (B) The approach of Cancer Stromal Targeting Therapy (CAST), whereby a non-internalising ADC targeting a protein in the stroma can release payload extracellularly. | ||
 | ||
| Fig. 13 (A) Bioconjugation of anti-collagen 4 antibody to generate a DAR 7-8 ADC, (B) the linker-payload structure. | ||
 | ||
| Fig. 14 (A) Structure of ADC and hinge region of anti-Fibrin ADC, (B) structure of branched linker that enables high DAR of 24. | ||
Matsumura and colleagues once again demonstrated fibrin targeting by using the fibrin-specific mAb combined with MMAE via a plasmin-cleavable tripeptide Val-Leu-Lys linker.43 Plasmin is a proteolytic enzyme produced in its active form only in the presence of fibrin, therefore potentially facilitating additional selectivity for tumoral payload release. In contrast to the ester linkages previously employed, this tripeptide was stable in human plasma over 7 days but efficiently released MMAE in the presence of plasmin. In vivo efficacy was examined in a mouse xenograft pancreatic tumour model. With an ADC dose of 20 mg kg−1, significant tumour growth suppression was observed compared to a non-targeted control ADC or MMAE alone. Although the treatment was not curative, the authors have demonstrated that cleavable tripeptide linkers targeting plasmin are suitable for incorporation into non-internalising ADCs.
The L19 antibody has been established as a high-affinity binder of the EDB domain of fibronectin and non-internalising ADCs have been generated by Zardi et al.45 Unlike classical ADCs in which a drug is chemically conjugated via a linker to the antibody, the authors fused the genes of the single-chain variable fragment (scFv, Fig. 1) L19 antibody and interleukin 2 (IL2) to generate a fusion protein. This fused antibody-cytokine does not require cleavage for IL2 to initiate cell-mediated immune responses and induce tumour regression. Accordingly, the fusion protein was efficacious in mice teratocarcinoma models, overcoming the rapid clearance and limiting cytotoxicity of IL2 treatment by tumour-vasculature specific delivery.
For targeting the EDA domain of fibronectin, Neri et al. utilised a linkerless disulfide conjugation strategy to conjugate thiol-containing dolastatins to the F8 antibody specific for the EDA domain of fibronectin (Fig. 15).8 The linkerless disulfide technology allows simple thiol-drug conjugation via formation of a mixed disulfide with antibody cysteines, which can then be cleaved in the presence of extracellular reductants released from dying tumour cells, such as glutathione. Following cleavage, only free drug and the parent antibody is released, therefore it is deemed “traceless”. The F8 antibody was used in a small-immune protein format (SIP, Fig. 1D) over an IgG format due to its superior tumour-targeting properties. The conjugate was moderately stable in mouse plasma, however, the conjugate rapidly reacted with glutathione to release free payload under conditions similar to the extracellular tumour environment. Potent tumour regression and prolonged survival was observed in mice with tumour subcutaneous grafts with high dosing (43 mg kg−1), although this was well tolerated. With lower dosing (8 mg kg−1) only 50% tumour growth suppression was observed, but neither treatment was curative. Improved therapeutic efficacy was later achieved when the F8 antibody was combined with the more potent maytansinoid payload DM1.46 Several cures and long-lasting remissions were obtained with 3 doses at 7 mg kg−1. Thus, the authors have demonstrated the successful delivery of potent payloads to the tumour vasculature by utilising linkerless disulfides which preclude the need for complex linker synthesis, generate homogenous conjugates and result in the release of only two species: the free drug and the parent antibody. This linkerless technology may be superior to more complex linker constructs which could impart undesired immunogenicity or other side effects.
 | ||
| Fig. 15 (A) The “linkerless” bioconjugation method to generate anti-fibronectin non-internalising ADCs with antibodies in small-immune protein format (B) the structure of the payloads. | ||
Neri and colleagues have also explored combination therapy of IL2 and cytotoxic cemadotin thiol (Cem), each conjugated to the SIP F8 against EDA of fibronectin (F8-IL2 and F8-Cem).47 Combination therapy exhibited curative anticancer properties in a mouse model of leukemia. In another work, they then explored the conjugation of IL2 and Cem to the same antibody, generating multi-payload ADCs for tumour vascular delivery. This time, the F8 antibody was used in diabody format, genetically fused to IL2 and connected to Cem or DM1 via a linkerless disulfide. The resulting multi-payload ADCs retained the properties of all components: tumour homing, cytotoxic activity (after linker cleavage) and cytokine activity. The ADCs preferentially accumulated in the tumour and the F8-IL2-SS-DM1 ADC had a potent antitumour effect in vivo.
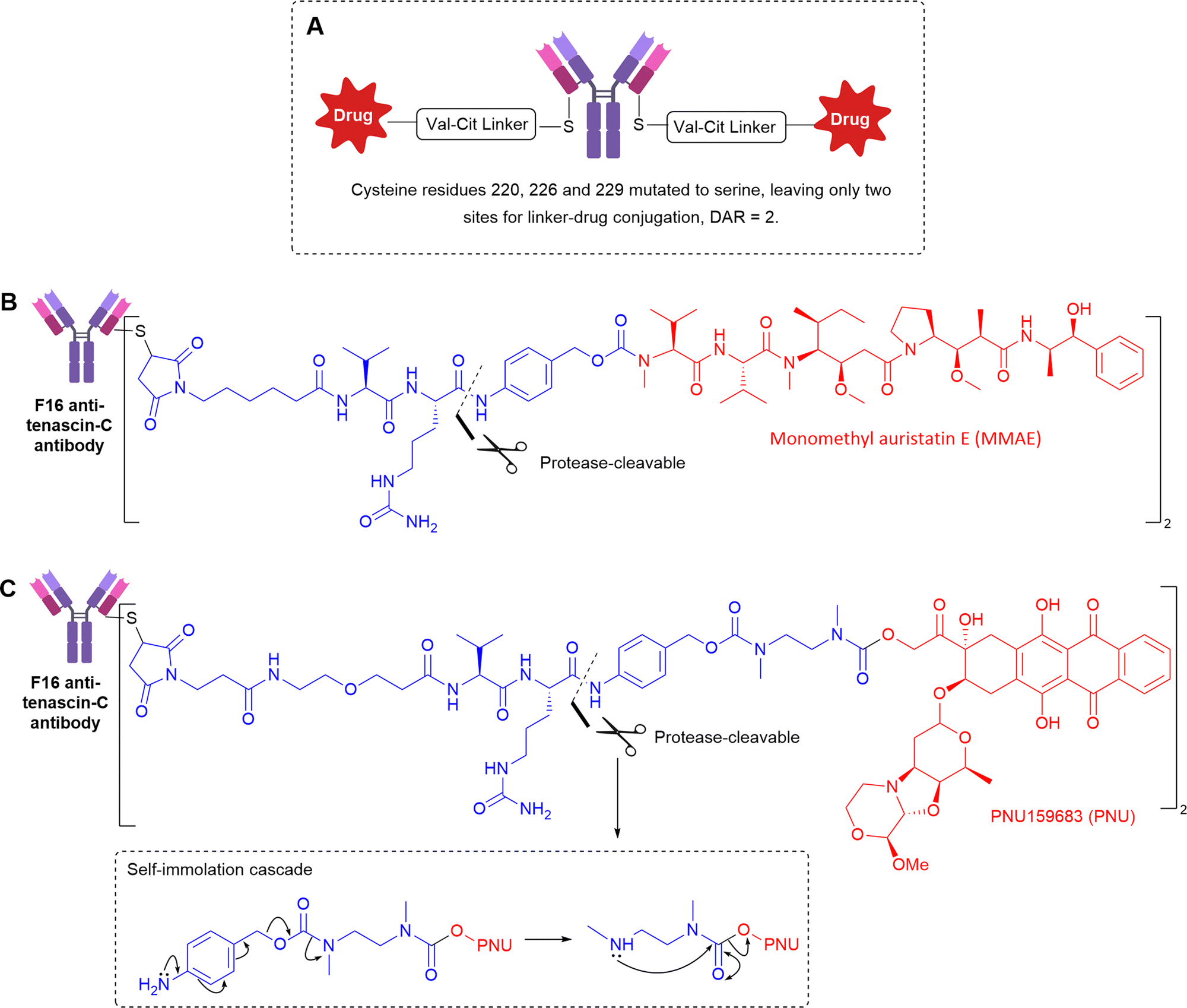 | ||
| Fig. 16 (A) The general structure of anti-Tenascin C non-internalising ADCs (B) the structure of anti-tenascin C targeting ADC with a protease-cleavable linker to release MMAE (C) the structure of anti-tenascin C targeting ADC conjugating PNU alcohol payload by an extended self-immolative spacer. | ||
Utilising a slightly different Val-Cit linker, with an extended self-immolative spacer to release alcohol-containing PNU159682 (Fig. 16C), the Val-Cit linker was stable in mouse serum over 5 days in vitro, but there is no comment on its stability in vivo.52 The authors validated the extracellular mechanism of action by exploring analogues bearing MMAE or MMAF, the latter of which contains a charged carboxylic acid, significantly hindering the ability of the released payload to cross cell membranes. Accordingly, the F16-MMAF conjugate did not display detectable therapeutic activity in vivo, whereas the MMAE analogue had potent cytotoxic effect. Therefore, diffusion of the drug through cell membranes is essential, indicative of extracellular payload release.
To gain increased understanding of the utility of dipeptides for extracellular linker cleavage, Neri et al. explored the use of Val-Cit, Val-Arg, Val-Lys and Val-Ala dipeptidic linkers in tenascin-C targeting non-internalising ADCs.53 The ADCs were well tolerated up to 50 mg kg−1, and the best tumour growth inhibition in vivo was achieved by the Val-Ala ADC. Linker stability was investigated after intravenous injection into mice, which revealed that Val-Lys and Val-Arg dipeptides (containing basic side chains) were cleaved between the two amino acid residues instead of the predicted cleavage site that would enable self-immolation to release free drug. Therefore, these linkers were deemed unsuitable for further development. Despite the efficacy of the Val-Cit and Val-Ala containing ADCs, the source of enzymatic cleavage was still not distinguished between Ces1c-mediated hydrolysis in plasma, or cleavage by target cancer-associated cathepsins.
Extracellular release via exogenous triggers
Whilst most ADCs rely on endogenous triggers for payload release such as tumour-associated enzymes or distinct tumoral pH, an alternative approach has been developed whereby a small molecule trigger is introduced exogenously and separately from the ADC. After the cleavable, non-internalising ADC is administered, sufficient time is allowed for ADC binding to the target antigen and sufficient time for clearance of any unbound, untargeted ADC from the blood to negate off-target effects. Subsequently, the exogenous release trigger is administered, only activating the ADC at the site-of-action (Fig. 17). Unlike traditional ADCs, this approach does not rely on cleavage by natural biological activation mechanisms, such as presence or specific concentrations of certain enzymes for payload release, which may vary widely between cancers and between patients. Thus, the bioorthogonal activation may allow more predictable drug-release rates, with spatial and temporal control.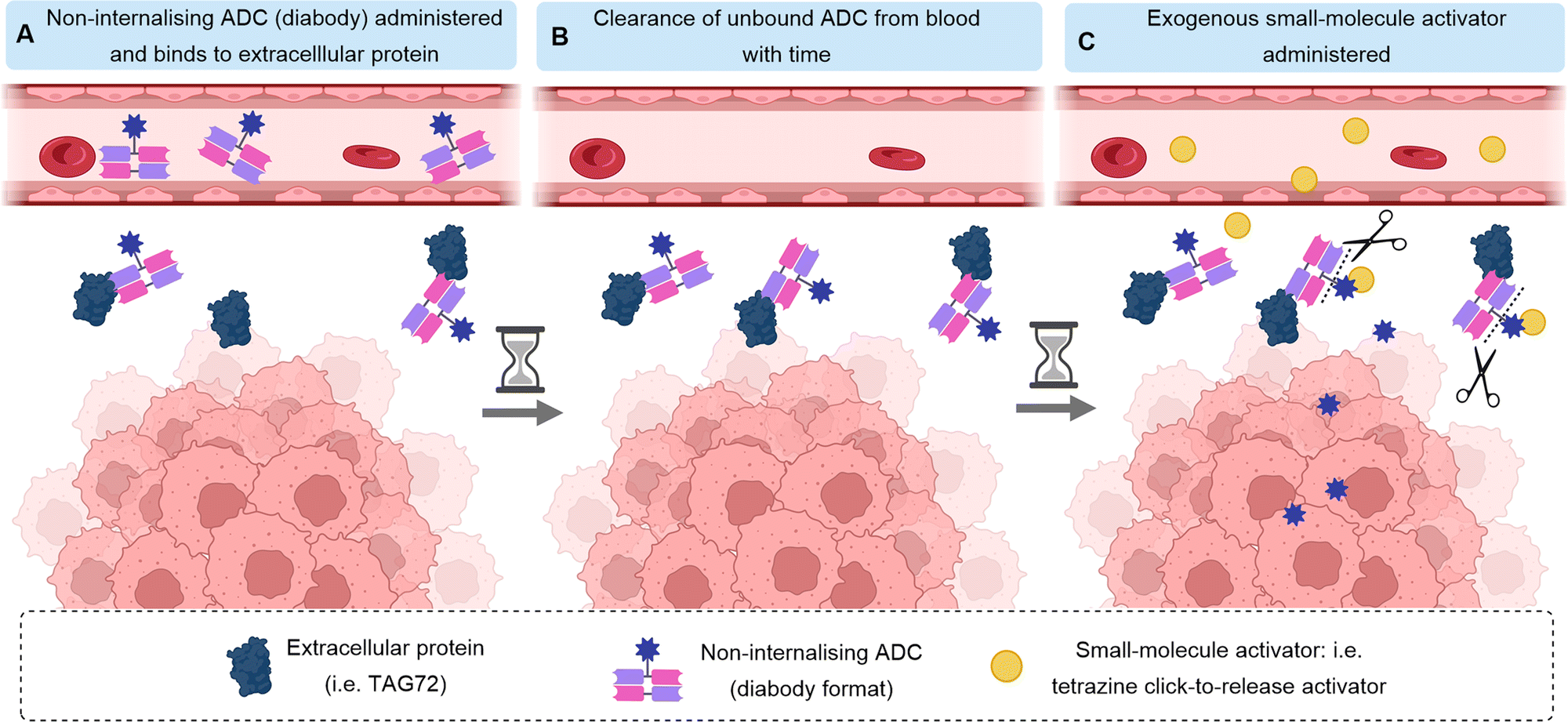 | ||
| Fig. 17 Mechanism of action of exogenously activated ADCs. Shown here is an antibody in diabody format targeting an extracellular protein. After clearance of the ADC from the bloodstream, the small-molecule exogenous activator is administered to cause linker cleavage and payload release only at the site-of-action. | ||
Click-to-release
Tumour-associated glycoprotein 72 (TAG72) is overexpressed on the surface of cancer cells in a wide range of solid tumours. Its suitability as a non-internalising ADC target has been demonstrated by Rossin et al. who utilised the bioorthogonal inverse-electron demand Diels Alder (IEDDA) reaction for generation of a non-internalising anti-TAG-72 ADC.54,55 Incorporation of a trans-cyclooctene linker into the ADC enabled facile “click-to-release” chemistry with exogenously administered small-molecule tetrazine probes. A pyridazine elimination reaction is triggered, allowing cleavage of allylic carbamates from trans-cyclooctenes, releasing free amine-containing cytotoxins (Fig. 18). Thus, a non-internalising ADC is generated which can release the payload extracellularly after linker cleavage by an exogenous trigger. | ||
| Fig. 18 (A) The generic structure and mechanism of “click-to-release” from anti-TAG72 non-internalising ADC by click reaction of the trans-cyclooctene with a tetrazine exogenous activator (B) the structure of the linker (C) the structure of the tetrazine exogenous activator. M = Lu/117Lu, 111In. | ||
As previously mentioned, before the exogenous probe is introduced, clearance of unbound ADC in the blood is required to avoid off-target activation and payload release which would lead to systemic toxicity. Therefore, the authors used ADCs in diabody format due to the faster clearance and deeper tumour penetration. Retaining the high tumour localisation, the diabody-conjugate with MMAE allowed administration of the tetrazine click activator 2 days later, giving efficient extracellular activation and MMAE release. High tumour selectivity was afforded by the low retention of the diabody-conjugate in healthy non-target tissues.
In vivo, the TAG-72 non-internalising click-to-release diabody ADC was compared to analogous TAG-72 diabody-ADC featuring a Val-Cit cleavable linker. Improved tumour-growth suppression was observed in vivo for the click-to-release ADC, achieving EC50s of 185 and 35 pM (in colorectal and ovarian cancer xenografts). In contrast, the Val-Cit ADC was not effective due to limited extracellular release of MMAE. The click-to-release strategy was well tolerated, and no toxicity was attributed to use of the ADC, tetrazine activator or the combination.
Although this approach requires clinical development of two separate components (the ADC and the activator), it is expected that the approach could be applied to broad-spectrum cancer targets (such as components of the stroma present in many solid tumours) with use of the same activator. In summary, the authors demonstrate the suitability of the IEDDA pyridazine elimination to allow temporally controlled and traceless ADC linker cleavage, activating payload release only at the site-of-action without the reliance on endogenous cancer biology.
Exogenous palladium
Bernardes et al. developed an elegant bifunctional thioether-containing propargyl carbamate linker which allows palladium-mediated cleavage to release amine-cytotoxins (Fig. 19).56 A HER2-nanobody ADC was constructed via cysteine bioconjugation with a thioether propargyl carbamate linker connected to doxorubicin (Dox, Fig. 19). In vitro, in HER2-positive cancer cells, the Pd-activatable ADC was as toxic as free Dox when administered with Pd(COD)Cl2, highlighting the success of their activatable approach. However, since their antigen target is internalising, even without exogenous Pd-activator, the same concentration of ADC was highly cytotoxic, due to lysosomal processing of the antibody-linker after internalisation, releasing free drug or toxic-drug derivatives. Thus, a non-internalising ADC format would be interesting for application of this technology, whereby cytotoxicity should exclusively be afforded by exogenous, extracellular activation, allowing better selectivity of payload release and potentially improved bystander effect. Additionally, less toxic Pd-complexes should be considered, since the Pd(COD)Cl2 activator is unlikely to be well tolerated in vivo.5 | ||
| Fig. 19 The structure of the bifunctional thioether propargyl carbamate linker used to release doxorubicin after exogenous administration of Pd-activator. Dox = doxorubicin. | ||
Exogenous platinum
Oliveira et al. have described the generation of a non-internalising ADC based on the F16 antibody against stromal tenascin-C that utilises bioorthogonal metal-mediated decaging for payload release.57 F16 was conjugated to MMAE via a carbonyl acrylic bioconjugation handle, containing a reactive alkyne for metal-catalysed decaging with platinum complexes K2PtCl4 or anti-cancer agent Cisplatin (CisPt, Fig. 20). | ||
| Fig. 20 The structure of non-internalising anti-tenascin C ADC activated exogenously by platinum-mediated decaging. | ||
In vitro, platinum-decaging of F16-MMAE ADC with non-toxic concentrations of K2PtCl4 afforded concentration-dependent cell killing. Although they did not explore the efficacy of ADCs in mammals, they did show that a simple alkynyl-prodrug of 5-fluorouracil was able to induce a significant cytostatic and cytotoxic effect in zebrafish, when combined with CisPt treatment compared to a control non-cleavable fluorouracil analogue, and without the presence of exogenous platinum.
This an impressive addition to the toolbox of bioorthogonal release strategies enabling decaging in aqueous environments with high reaction rates and yields. However, the conversion of activated products was hampered by the presence of nucleophiles such as glutathione (known to be elevated in cancer), giving markedly slower reaction rates. With CisPt, nucleophiles were found to deactivate the metal, resulting in only modest yields. Hence, further development is required to enable activators to be compatible with glutathione and other nucleophiles in vivo.
Although proof-of-concept studies of ADCs with exogenous payload release have been successful, drawbacks of the strategy remain. Notably, the administration of a completely un-vectorised exogenous trigger inherently results in uncontrolled distribution which may lead to off-target toxicity. Furthermore, higher concentrations of the trigger are required since minimal amounts localize to the tumour site, which may also contribute to toxicity. The strategy would also inevitably be more therapeutically complex, requiring multiple dosing and evaluation of the pharmacodynamics/kinetics of two species.
Conclusions
Within the last decade of ADC research, increasing attention has turned towards the development of non-internalising ADCs with extracellular payload release mechanisms, with the intention of improving on some of the drawbacks associated with classical internalising ADCs (a summary of the strategies described in this review can be found in Table 1). For example, their reliance on high, homogenous antigen expression excludes a number of cancer types from target selection and can become problematic when cancers develop resistance related to the internalisation process. Non-internalising ADCs therefore represent a promising opportunity to target a wider range of cancers that lack suitable antigen expression for treatment by internalising ADCs.| a Wavy lines indicate portions of the linker structure not represented in the figure. Bioconjugation position is a representation only. |
|---|

|

|
Despite the traditional belief that internalisation is crucial for the efficacy of ADCs bearing cleavable linkers such as protease-cleavable dipeptides or reducible disulfides, increasing evidence has emerged to the contrary. Instead, it is becoming recognised that dying cancer cells are able to release the typically intracellular linker triggers into the extracellular tumour microenvironment. Thus, these linkers are suitable for incorporation into non-internalising ADCs. However, these preliminary findings in tumour mouse models remain unvalidated in primates, whose concentration and distribution of cleavage-triggers differ. Importantly, the instability of protease-cleavable dipeptides in mouse plasma due to the Ces1C hydrolase complicates evaluation of non-internalising ADCs.
Alternative linkers examined for non-internalising ADCs have moved away from the notion that the linkers must be extremely stable in circulation until internalisation and breakdown within the cancer cell. Instead, linkers incorporating esters or carbonate moieties with moderate stability in circulation have been used in combination with antibodies targeting components of the tumour stroma, allowing sustained drug release local to the tumour. Such carbonate linkers have already been validated in the clinic (i.e., Trodelvy®), but may only be suitable for use with moderately cytotoxic payloads such as SN-38. For improved potency, more cytotoxic payloads could be considered, but off-target toxicity due to the labile linkage should be closely examined.
In addition to the linking chemistry, increasing numbers of poorly internalising cell-surface antigen targets have been identified. For example, non-internalising ADCs targeting CD20,9,10 CD21,9 CD72,9 TAG72,54,55 CEACAM5,22 and NKA27 have been described. Although this approach still requires the expression of a specific cell-surface antigen, removed reliance on internalisation and extracellular payload release may afford improved bystander killing and deeper tumour penetration. Efforts have also focused on targeting proteins other than those expressed on the cancer cell surface, for example, secreted proteins Gal3BP,30 LRG1,34 and MMP9.39
The field has also moved towards targeting the unique physiology of cancer: tumours possess abundant stroma and unique vasculature containing collagen,40 fibrin,43 fibronectin,8,45,46 and tenascin-C51–53,57 – all of which have recently been explored for targeting by non-internalising ADCs. Promising preliminary results have been obtained with stromal-targeting ADCs, this approach may offer a much-needed alternative to internalising ADCs in the future for the treatment of stroma-rich solid tumours.
Finally, a fledgling area of research is described towards the activation of non-internalising ADCs by exogenous small-molecule triggers, such as metals (Pd, Pt) or click-chemistry partners. The use of exogenous triggers avoids the potential variance of cancer biology between patients and cancer types but requires more development for in vivo application. In particular, there is concern over the lack of selectivity of the exogenous trigger, which results in high dosing and potential off-target toxicity.
Future work in the field is likely to see more combinations of non-internalising/extracellular targets with different linker technologies. New linker technologies should explore the balance between linker stability in circulation vs. activation extracellularly to reach a compromise between high stability of the linker in circulation, with high reactivity to enable facile and quick release in the tumour microenvironment. Finally, the discovery of new antibody–antigen binding interactions that do not induce internalisation may enable the generation of novel non-internalising ADCs for different antigen targets.
Overall, non-internalising ADCs are an exciting, emerging area of ADC research likely to assist in overcoming some of the barriers to the clinical success of internalising ADCs and expected to enable the targeting of a wider range of cancer types in the future.
Author contributions
N. A. wrote the original draft. J. D. B. and D. R. S contributed to manuscript revisions, editing, and supervising the review. Figures were made using Biorender.com.Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Q. Dean, S. Luo, J. D. Twomey and B. Zhang, MABS, 2021, 13, 1–23 CrossRef PubMed.
- M. Barok, H. Joensuu and J. Isola, Breast Cancer Res., 2014, 6, 130–146 Search PubMed.
- F. Loganzo, X. Tan, M. Sung, G. Jin, J. S. Myers, E. Melamud, F. Wang, V. Diesl, M. T. Follettie, S. Musto, M. H. Lam, W. Hu, M. B. Charati, K. Khandke, K. S. K. Kim, M. Cinque, J. Lucas, E. Graziani, A. Maderna, C. J. O’Donnell, K. T. Arndt and H. P. Gerber, Mol. Cancer Ther., 2015, 14, 952–963 CrossRef CAS PubMed.
- N. Joubert, C. Denevault-Sabourin, F. Bryden and M. C. Viaud-Massuard, Eur. J. Med. Chem., 2017, 142, 393–415 CrossRef CAS PubMed.
- J. D. Bargh, A. Isidro-Llobet, J. S. Parker and D. R. Spring, Chem. Soc. Rev., 2019, 48, 4361–4374 RSC.
- A. Warnecke, I. Fichtner, G. Saß and F. Kratz, Arch. Pharm., 2007, 340, 389–395 CrossRef CAS PubMed.
- J. M. Rothberg, K. M. Bailey, J. W. Wojtkowiak, Y. Ben-Nun, M. Bogyo, E. Weber, K. Moin, G. Blum, R. R. Mattingly, R. J. Gillies and B. F. Sloane, Neoplasia, 2013, 15, 1125–1137 CrossRef PubMed.
- G. J. L. Bernardes, G. Casi, S. Trüssel, I. Hartmann, K. Schwager, J. Scheuermann and D. Neri, Angew. Chem., Int. Ed., 2012, 51, 941–944 CrossRef CAS PubMed.
- A. G. Polson, J. Calemine-Fenaux, P. Chan, W. Chang, E. Christensen, S. Clark, F. J. De Sauvage, D. Eaton, K. Elkins, J. Michael Elliott, G. Frantz, R. N. Fuji, A. Gray, K. Harden, G. S. Ingle, N. M. Kljavin, H. Koeppen, C. Nelson, S. Prabhu, H. Raab, S. Ross, J. P. Stephan, S. J. Scales, S. D. Spencer, R. Vandlen, B. Wranik, S. F. Yu, B. Zheng and A. Ebens, Cancer Res., 2009, 69, 2358–2364 CrossRef CAS PubMed.
- J. F. DiJoseph, M. M. Dougher, D. C. Armellino, L. Kalyandrug, A. Kunz, E. R. Boghaert, P. R. Hamann and N. K. Damle, Cancer Immunol. Immunother., 2007, 56, 1107–1117 CrossRef CAS PubMed.
- C. R. Justus, L. Dong and L. V. Yang, Front. Physiol., 2013, 4, 1–9 Search PubMed.
- E. R. Boghaert, K. Khandke, L. Sridharan, D. Armellino, M. Dougher, J. F. DiJoseph, A. Kunz, P. R. Hamann, A. Sridharan, S. Jones, C. Discafani and N. K. Damle, Int. J. Oncol., 2006, 28, 675–684 CAS.
- Y. Takakura, T. Fujita, M. Hashida and H. Sezaki, Pharm. Res., 1990, 7, 339–346 CrossRef CAS PubMed.
- T. Okazaki and T. Honjo, Int. Immunol., 2007, 19, 813–824 CrossRef CAS PubMed.
- E. S. Kim, Drugs, 2017, 77, 929–937 CrossRef PubMed.
- A. Markham, Drugs, 2016, 76, 1227–1232 CrossRef CAS PubMed.
- Y. Y. Syed, Drugs, 2017, 77, 1369–1376 CrossRef CAS PubMed.
- D. Xiao, L. Luo, J. Li, Z. Wang, L. Liu, F. Xie, J. Feng and X. Zhou, Bioorg. Chem., 2021, 116, 105366 CrossRef CAS PubMed.
- C. W. Li, S. O. Lim, E. M. Chung, Y. S. Kim, A. H. Park, J. Yao, J. H. Cha, W. Xia, L. C. Chan, T. Kim, S. S. Chang, H. H. Lee, C. K. Chou, Y. L. Liu, H. C. Yeh, E. P. Perillo, A. K. Dunn, C. W. Kuo, K. H. Khoo, J. L. Hsu, Y. Wu, J. M. Hsu, H. Yamaguchi, T. H. Huang, A. A. Sahin, G. N. Hortobagyi, S. S. Yoo and M. C. Hung, Cancer Cell, 2018, 33, 187–201.e10 CrossRef CAS PubMed.
- L. He, L. Wang, Z. Wang, T. Li, H. Chen, Y. Zhang, Z. Hu, D. S. Dimitrov, J. Du and X. Liao, J. Med. Chem., 2021, 64, 15716–15726 CrossRef CAS PubMed.
- S. Heskamp, W. Hobo, J. D. M. Molkenboer-Kuenen, D. Olive, W. J. G. Oyen, H. Dolstra and O. C. Boerman, Cancer Res., 2015, 75, 2928–2936 CrossRef CAS PubMed.
- S. V. Govindan, T. M. Cardillo, S. Moon, H. J. Hansen and D. M. Goldenberg, Clin. Cancer Res., 2009, 15, 6052–6061 CrossRef CAS PubMed.
- S. J. Moon, S. V. Govindan, T. M. Cardillo, A. Christopher, D. Souza, H. J. Hansen, D. M. Goldenberg, C. A. D’Souza, H. J. Hansen and D. M. Goldenberg, J. Med. Chem., 2008, 51, 6916–6926 CrossRef CAS PubMed.
- R. M. Sharkey, H. Karacay, S. V. Govindan and D. M. Goldenberg, Mol. Cancer Ther., 2011, 10, 1072–1081 CrossRef CAS PubMed.
- Y. Y. Syed, Drugs, 2020, 80, 1019–1025 CrossRef CAS PubMed.
- D. M. Goldenberg, T. M. Cardillo, S. V. Govindan, E. A. Rossi and R. M. Sharkey, Oncotarget, 2020, 11, 942 CrossRef PubMed.
- D. J. Marshall, S. S. Harried, J. L. Murphy, C. A. Hall, M. S. Shekhani, C. Pain, C. A. Lyons, A. Chillemi, F. Malavasi, H. L. Pearce, J. S. Thorson and J. R. Prudent, Mol. Ther., 2016, 24, 1760–1770 CrossRef CAS PubMed.
- Y. Ino, M. Gotoh, M. Sakamoto, K. Tsukagoshi and S. Hirohashi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 365–370 CrossRef CAS PubMed.
- G. Li, J. Guo, B. Q. Shen, D. B. Yadav, M. X. Sliwkowski, L. M. Crocker, J. A. Lacap and G. D. L. Phillips, Mol. Cancer Ther., 2018, 17, 1441–1453 CrossRef CAS PubMed.
- F. Giansanti, E. Capone, S. Ponziani, E. Piccolo, R. Gentile, A. Lamolinara, A. Di Campli, M. Sallese, V. Iacobelli, A. Cimini, V. De Laurenzi, R. Lattanzio, M. Piantelli, R. Ippoliti, G. Sala and S. Iacobelli, J. Controlled Release, 2019, 294, 176–184 CrossRef CAS PubMed.
- K. Furukawa, K. Kawamoto, H. Eguchi, M. Tanemura, T. Tanida, Y. Tomimaru, H. Akita, N. Hama, H. Wada, S. Kobayashi, Y. Nonaka, S. Takamatsu, S. Shinzaki, T. Kumada, S. Satomura, T. Ito, S. Serada, T. Naka, M. Mori, Y. Doki, E. Miyoshi and H. Nagano, Pancreas, 2015, 44, 93–98 CrossRef CAS PubMed.
- S. Surinova, M. Choi, S. Tao, P. J. Schüffler, C. Chang, T. Clough, K. Vysloužil, M. Khoylou, J. Srovnal, Y. Liu, M. Matondo, R. Hüttenhain, H. Weisser, J. M. Buhmann, M. Hajdúch, H. Brenner, O. Vitek and R. Aebersold, EMBO Mol. Med., 2015, 7, 1166–1178 CrossRef CAS PubMed.
- J. F. Fahrmann, L. E. Bantis, M. Capello, G. Scelo, J. B. Dennison, N. Patel, E. Murage, J. Vykoukal, D. L. Kundnani, L. Foretova, E. Fabianova, I. Holcatova, V. Janout, Z. Feng, M. Yip-Schneider, J. Zhang, R. Brand, A. Taguchi, A. Maitra, P. Brennan, C. Max Schmidt and S. Hanash, J. Natl. Cancer Inst., 2019, 111, 372–379 CrossRef PubMed.
- F. Javaid, C. Pilotti, C. Camilli, D. Kallenberg, C. Bahou, J. Blackburn, J. R. Baker, J. Greenwood, S. E. Moss and V. Chudasama, RSC Chem. Biol., 2021, 2, 1206–1220 RSC.
- P. Strop, S. Liu, M. Dorywalska, K. Delaria, R. G. Dushin, T. Tran, W. Ho, S. Farias, M. G. Casas, Y. Abdiche, D. Zhou, R. Chandrasekaran, C. Samain, C. Loo, A. Rossi, M. Rickert, S. Krimm, T. Wong, S. M. Chin, J. Yu, J. Dilley, J. Chaparro-riggers, G. F. Filzen, C. J. O. Donnell, F. Wang, J. S. Myers, J. Pons, D. L. Shelton and A. Rajpal, Chem. Biol., 2013, 20, 161–167 CrossRef CAS PubMed.
- M. Dorywalska, P. Strop, J. A. Melton-Witt, A. Hasa-Moreno, S. E. Farias, M. Galindo Casas, K. Delaria, V. Lui, K. Poulsen, C. Loo, S. Krimm, G. Bolton, L. Moine, R. Dushin, T.-T. Tran, S.-H. Liu, M. Rickert, D. Foletti, D. L. Shelton, J. Pons and A. Rajpal, Bioconjugate Chem., 2015, 26, 650–659 CrossRef CAS PubMed.
- M. Dorywalska, R. Dushin, L. Moine, S. E. Farias, D. Zhou, T. Navaratnam, V. Lui, A. Hasa-Moreno, M. G. Casas, T. T. Tran, K. Delaria, S. H. Liu, D. Foletti, C. J. O’Donnell, J. Pons, D. L. Shelton, A. Rajpal and P. Strop, Mol. Cancer Ther., 2016, 15, 958–970 CrossRef CAS PubMed.
- C. Joseph, M. Alsaleem, N. Orah, P. L. Narasimha, I. M. Miligy, S. Kurozumi, I. O. Ellis, N. P. Mongan, A. R. Green and E. A. Rakha, Breast Cancer Res. Treat., 2020, 182, 267–282 CrossRef CAS PubMed.
- E. A. Love, A. Sattikar, H. Cook, K. Gillen, J. M. Large, S. Patel, D. Matthews and A. Merritt, ChemBioChem, 2019, 20, 754–758 CrossRef CAS PubMed.
- M. Yasunaga, S. Manabe, D. Tarin and Y. Matsumura, Bioconjugate Chem., 2011, 22, 1776–1783 CrossRef CAS PubMed.
- D. Neri and R. Bicknell, Nat. Rev. Cancer, 2005, 5, 436–446 CrossRef CAS PubMed.
- M. Yasunaga, S. Manabe and Y. Matsumura, Cancer Sci., 2011, 102, 1396–1402 CrossRef CAS PubMed.
- H. Fuchigami, S. Manabe, M. Yasunaga and Y. Matsumura, Sci. Rep., 2018, 8, 1–9 CAS.
- U. Niesner, C. Halin, L. Lozzi, M. Günthert, P. Neri, H. Wunderli-Allenspach, L. Zardi and D. Neri, Bioconjugate Chem., 2002, 13, 729–736 CrossRef CAS PubMed.
- B. Carnemolla, L. Borsi, E. Balza, P. Castellani, R. Meazza, A. Berndt, S. Ferrini, H. Kosmehl, D. Neri and L. Zardi, Blood, 2002, 99, 1659–1665 CrossRef PubMed.
- E. Perrino, M. Steiner, N. Krall, G. J. L. Bernardes, F. Pretto, G. Casi and D. Neri, Cancer Res., 2014, 74, 2569–2578 CrossRef CAS PubMed.
- T. List, G. Casi and D. Neri, Mol. Cancer Ther., 2014, 13, 2641–2652 CrossRef CAS PubMed.
- L. Borsi, B. Carnemolla, G. Nicolò, B. Spina, G. Tanara and L. Zardi, Int. J. Cancer, 1992, 52, 688–692 CrossRef CAS PubMed.
- M. Dueck, S. Riedl, U. Hinz, A. Tandara, P. Möller, C. Herfarth and A. Faissner, Int. J. Cancer, 1999, 82, 477–483 CrossRef CAS PubMed.
- M. Adams, J. L. Jones, R. A. Walker, J. H. Pringle and S. C. Bell, Cancer Res., 2002, 62, 3289–3297 CAS.
- R. Gébleux, M. Stringhini, R. Casanova, A. Soltermann and D. Neri, Int. J. Cancer, 2017, 140, 1670–1679 CrossRef PubMed.
- A. Dal Corso, R. Gébleux, P. Murer, A. Soltermann and D. Neri, J. Controlled Release, 2017, 264, 211–218 CrossRef CAS PubMed.
- A. Dal Corso, S. Cazzamalli, R. Gébleux, M. Mattarella and D. Neri, Bioconjugate Chem., 2017, 28, 1826–1833 CrossRef CAS PubMed.
- R. Rossin, R. M. Versteegen, J. Wu, A. Khasanov, H. J. Wessels, E. J. Steenbergen, W. Ten Hoeve, H. M. Janssen, A. H. A. M. Van Onzen, P. J. Hudson and M. S. Robillard, Nat. Commun., 2018, 9, 1–11 CrossRef CAS PubMed.
- R. Rossin, P. R. Verkerk, S. M. Van Den Bosch, R. C. M. Vulders, I. Verel, J. Lub and M. S. Robillard, Angew. Chem., Int. Ed., 2010, 49, 3375–3378 CrossRef CAS PubMed.
- B. J. Stenton, B. L. Oliveira, M. J. Matos, L. Sinatra and G. J. L. Bernardes, Chem. Sci., 2018, 9, 4185–4189 RSC.
- B. L. Oliveira, B. J. Stenton, V. B. Unnikrishnan, C. R. De Almeida, J. Conde, M. Negrão, F. S. S. Schneider, C. Cordeiro, M. G. Ferreira, G. F. Caramori, J. B. Domingos, R. Fior and G. J. L. Bernardes, J. Am. Chem. Soc., 2020, 142, 10869–10880 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |