
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlycosidase activated prodrugs for targeted cancer therapy
Harlei
Martin
 a,
Laura Ramírez
Lázaro
ab,
Thorfinnur
Gunnlaugsson
*ab and
Eoin M.
Scanlan
*ab
a,
Laura Ramírez
Lázaro
ab,
Thorfinnur
Gunnlaugsson
*ab and
Eoin M.
Scanlan
*ab
aSchool of Chemistry and Trinity Bioscience Institute, The University of Dublin, Trinity College Dublin, Dublin 2, Ireland. E-mail: gunnlaut@tcd.ie; scanlae@tcd.ie
bSFI Synthesis and Solid State Pharmaceutical Centre (SSPC), Ireland
First published on 9th November 2022
Abstract
In this review glycosidase activated prodrugs that target cancer cells are discussed. Glycosylated prodrugs undergo enzymatic bioconversion, cleaving the prodrug to release the anticancer drug at the desired site of action, hence minimising the toxic side effects associated with many current anticancer drugs. In addition, the presence of the carbohydrate moiety increases the aqueous solubility of the drugs, allowing for a more effective treatment. In the past decade, significant advancements have been made in this field that have led to the development of many novel carbohydrate-based prodrugs – ranging from simple glycoconjugates to complex self-assemblies and materials, which are discussed in detail herein.
 Harlei Martin | Harlei Martin completed her PhD in 2019 in Maynooth University, Ireland, in the group of Assoc. Prof. Trinidad Velasco-Torrijos. Following her PhD, she worked as an assistant lecturer in the Chemistry Department of Maynooth University. She then worked as a postdoctoral researcher with Dr Célia Bonnet in the Centre for Molecular Biophysics, CNRS Orléans campus, France. Currently, Harlei is an IRC postdoc in Trinity College Dublin in the groups of Prof. Eoin Scanlan and Prof. Thorfinnur Gunnlaugsson. Her work is focused on the development of glycosylated prodrugs for targeted cancer therapy. |
 Laura Ramírez Lázaro | Laura Ramírez Lázaro obtained her BSc degree at the University of Alcalá de Henares, Madrid. She completed the last year of her undergraduate degree at Trinity College Dublin where she worked on glycosylated naphthalimide probes for imaging tumor cells utilizing the ADEPT mechanism. She was granted the Trinity College Dublin Postgraduate Research Studentship and a Studentship from the SFI funded Synthesis and Solid State Pharmaceutical Centre (SSPC), and is currently in the third year of her PhD studies. Her project is focused on the development of sugars/amino-acids conjugated naphthalimide Tröger's base scaffolds as probes for biochemical imaging. |
 Thorfinnur Gunnlaugsson | Thorfinnur ‘Thorri’ Gunnlaugsson MRIA is Professor of Chemistry at the School of Chemistry, Trinity College Dublin. His current research focuses on developing various aspects of supramolecular, material and medicinal chemistries. His work has been recognised with several awards, including a Membership to The Royal Irish Academy in 2011, the 2021 Molecular Sensors and Molecular Logic gates (MSMLG) Czarnik Award (at the 7th MSMLG meeting in Dublin July 2022), the 2014 The Institute of Chemistry of Ireland (ICI) Annual Award for Chemistry, and the 2006 Bob Hay Awarded, of the Royal Society of Chemistry, Macrocycle and Supramolecular Chemistry Interest Group. |
 Eoin M. Scanlan | Eoin Scanlan completed his undergraduate degree at NUI Galway and his PhD at the University of St. Andrews. Following postdoctoral work at the University of Bern, Switzerland and at the University of Oxford, UK he joined the School of Chemistry in Trinity College Dublin in 2008 where he is Associate Professor of Organic and Medicinal Chemistry and a PI in the Trinity Biomedical Sciences Institute. He leads an international research team in Trinity College with a focus on new synthetic methods and the discovery of novel therapeutics, diagnostics and biomaterials. He is co-founder and CSO of Glycome Biopharma, a biotech start-up company based in Dublin. |
1. Introduction
According to the World Health Organization (WHO), cancer was the second leading cause of death in 2020 worldwide with 19.3 million cases and 10 million deaths.1 The National Institute for Health (NIH) has anticipated that this number will increase up to 29.5 million and 16.4 million cases and deaths, respectively, by 2040.2 Many current therapeutic strategies, including the drugs used in chemotherapy, usually target cells that are proliferating. Therefore, they are limited in their specificity, targeting not only tumour cells but healthy cells as well. Furthermore, most of the administered drugs remain circulating in the bloodstream, increasing potential side effect on healthy tissue.3 In addition to those drawbacks, current cancer treatments have high systemic toxicity which makes them only applicable in low doses. Hence, there is an urgent need to identify specific cancer-targeting therapies.Prodrug therapy is one approach to overcome specificity issues involved in chemotherapy and anticancer treatments. Adrien Albert first coined the term ‘prodrug’ or ‘proagent’,4 also referred to as ‘latentiated drugs’, ‘bioreversible derivatives’, and ‘congeners’.5,6 This approach has improved cancer drug therapy since the early 1970s.7 Usually a prodrug consists of the drug and a chemical moiety that are covalently linked to form the inactive substrate. Upon activation with specific biological media, such as stomach acid as in the case of Aspirin, or in a more targeted way, by an enzyme that carries out a specific biochemical transformation, the active drug is released in vivo to exert its therapeutic effect.
Carbohydrates are the most abundant group of macromolecules found in the body, and play pivotal roles in many cellular interactions such as signalling or cell surface receptors.8 Due to the rapid proliferation of cancer cells, there exists a high-energy demand. Glucose transporters (GLUTs), which are found to be overexpressed in cancer cells, solve the problem by increasing the uptake of glucose at a higher rate than normal cells, a phenomenon known as the “Warburg Effect”.9 This effect garnered attention by the science community for the design and development of sugar-based targeted drug delivery.10 It has also been extensively reported that various glycosidase enzymes are overexpressed in different cancer types (see Table 1). For example, β-glucosidase is upregulated in a number of cancers including breast,11 gastric12 and liver.13 This overexpression may be exploited by using glycosidase activated prodrugs for targeting many different cancers.
| Glycosidase | Cancer type |
|---|---|
| β-Glucosidase | Breast,11 gastric,12 liver13 |
| Glucosidase II | Lung,14 bladder,15 gastric,16 melanoma17 |
| N-Acetyl-β-D-glucosaminidase | Ovarian,18 liver,19 leukemia,19 thyroid,20 breast21 |
| α-Glucosidase | Liver,19 leukemia19 |
| β-Galactosidase | Liver,19 ovarian,22 prostate,23 colon,24 breast,24 gliomas25 |
| β-Glucuronidase | Lung,26 breast,27 ovarian,28 pancreatic,29 colorectal30 |
| α-Mannosidase | Breast,31 cervical,32 clear cell renal carcinoma33 |
| β-Mannosidase | Liver,19 leukemia19 |
| α-Fucosidase | Ovarian,18 gliomas,25 colon,34 pancreatic34 |
The vast majority of carbohydrate-based prodrugs are designed to improve pharmacokinetic properties. They exhibit high solubility in water, low toxicity, and high biocompatibility. It has been shown that several cytotoxic agents e.g. glufosfamide, chlorambucil, docetaxel, 3-paclitaxel, etc., (Fig. 1) have been glycosylated and found to be less toxic to noncancer cells than the parental aglycons.35 Tumour-associated carbohydrate antigens (TACAs) are specific targets and therefore considered good biomarkers for cancer detection as well. They are essential for carbohydrate-based cancer vaccines to improve immunological response.36,37 Also, the lysosomal enzyme β-galactosidase is overexpressed in some cancers such as lung and ovarian cancers,22,38,39 allowing for the application of galactosidase responsive prodrugs suitable for the treatment of solid tumours. In prodrug monotherapy (PMT), a nontoxic prodrug is administered and cleaved to release the cytotoxic drug upon enzymatic activation, where such enzyme is already overexpressed at the site of action.26 Since in prodrug monotherapy the enzyme is not administrated, the selectivity must be achieved through key differences in tumour environments compared to healthy tissue such as the lower pH of cancer tissue, hypoxia or specific enzymes overexpressed in the tumour environment.10,40 β-Glucuronidase and β-galactosidase, for instance, are found in high concentration in solid tumours, such as lung, breast or ovarian cancer, which makes it the perfect target for the development of drug carriers.26,41 Recent studies have also suggested that mannosidase enzymes are overexpressed in various breast tumors,31 cervical cancers32 and clear cell renal carcinoma (ccRCC).33 Since many glycosidase enzymes are upregulated in various cancers, they provide the perfect target to use in glycosidase activated prodrugs for cancer. Extensive research has been reported in the area of sensing glycosidase enzymes intracellularly,42 using, for example, fluorescent probes,43 near-IR probes,44 and AIE-based glycoclusters.45 This allows the identification of glycosidase enzymes that are overexpressed in cancer tissue and hence, allow for the development of prodrugs that would utilise these enzymes for targeted cancer therapy.
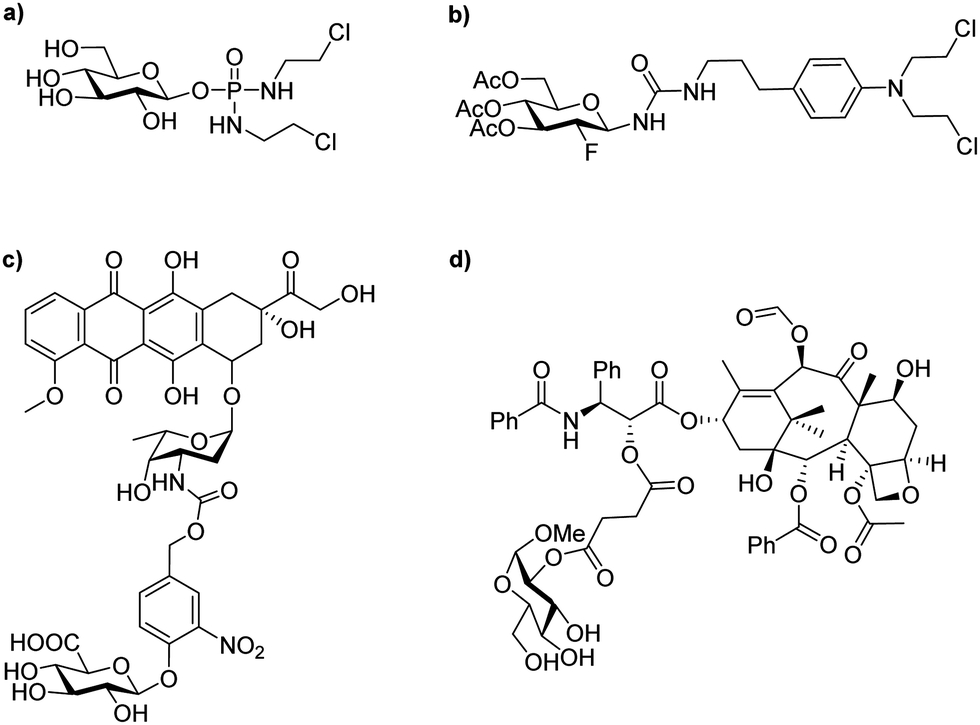 | ||
| Fig. 1 Examples of typical glycosylated anticancer drugs; (a) glufosfamide (or D-glucose isophosphoramide mustard); (b) peracetylated 2-fluorodeoxyglucose conjugated chlorambucil; (c) glucuronic acid conjugated doxorubicin; (d) glucose conjugated paclitaxel. | ||
In contrast to PMT, directed enzyme prodrug therapies such as ADEPT (antibody-directed enzyme prodrug therapy), GDEPT (Gene-DEPT), MDEPT (Magnetically-DEPT) and VDEPT (Virus-DEPT), all require the administration of an enzyme to the site of action. For example, in ADEPT an enzyme-antibody (Ab) conjugate is administrated to the patient. The antibody then binds to the antigen expressed on the surface of the tumour cell and accumulates in the desired site of action. The second step consists of the injection of a non-toxic prodrug, which will be converted into the active drug once it reaches the tumour cell by a catalyst reaction with the enzyme previously linked to the cell. Thus, the concentration of drug within tumours is higher than that which can be achieved through classical drug therapies.46
In this review, some of the most promising recent carbohydrate-based prodrug approaches investigated, such as PMT or DEPT, including ADEPT, GDEPT and VDEPT, will be discussed alongside examples that have been reported in the literature over the past decade. There are many examples of Pt-based cancer prodrugs in the literature, and as these have already been discussed thoroughly in several reviews,47–49 the main focus herein will be on featuring “fully organic” prodrugs, that are activated by glycosidase enzymes to exert their anticancer properties. The scope of the review also includes recent developments in supramolecular assemblies and materials as prodrugs.
2. Glycosidase activated prodrugs
2.1 Simple glycoconjugates
Most glycosidase activated prodrugs reported to date consist of a known anticancer drug directly linked via a glycosidic bond to a sugar unit, or in most cases a known anticancer drug joined to a sugar unit via a linker (Scheme 1a). The linkers are usually self-immolative, meaning they cleave the covalent bond with the active drug after a stimulus.50,51 In this case, a glycosidase enzyme will hydrolyse the glycosidic bond, causing the self-immolative linker to collapse, releasing the anticancer drug. Commonly used sugar moieties used include glucose, glucuronic acid and galactose, but other sugars such as rhamnose and xylose have also been utilised. The most popular linkers are based on a phenolic scaffold that upon glycosidic bond hydrolysis, eliminate from the carbamate-substituted benzyl system with formation of a reactive methylene quinone moiety along with an unstable carbamic acid which quickly decomposes to give carbon dioxide and the corresponding amine, as shown in the example in Scheme 1b. This system was first used by Monnert, Koch and Gesson who prepared prodrugs of the anticancer drug daunorubicin incorporating an α-galactoside trigger (see prodrug 1 in Scheme 1b).52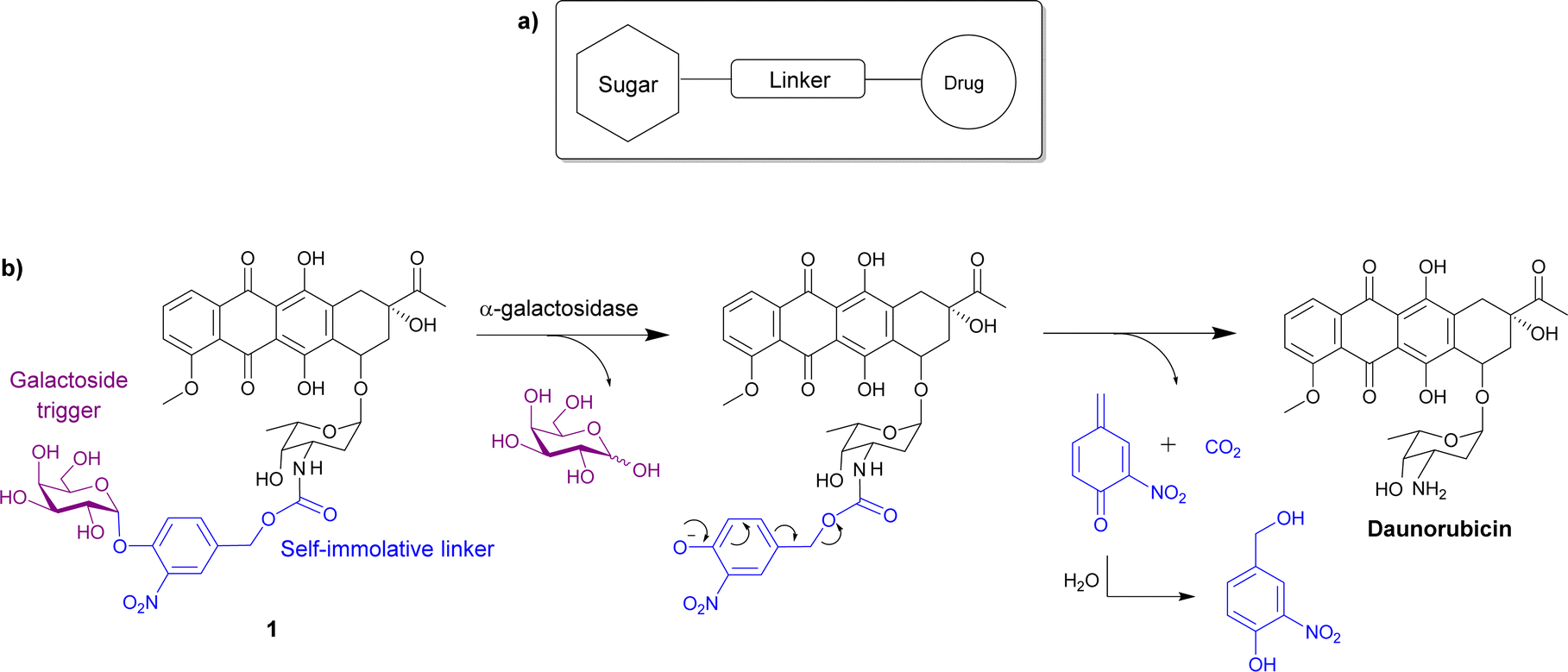 | ||
| Scheme 1 (a) Schematic showing the general structure of glycosidase activated prodrugs; (b) the structure of one of the first daunorubicin prodrugs 1 incorporating an α-galactoside trigger and a self-immolative phenolic linker, and the reaction with the galactosidase that releases the parent drug. | ||
Since this first report in 1992, many examples of glycosylated prodrugs have been reported and the topic has been thoroughly reviewed.53–55 Hence, this review will mainly focus on some of the more recent developments in this field, which have taken place over the past decade or so. With this in mind, the first section of this overview will focus on the development of simple glycoconjugates consisting of a sugar and an anticancer drug, which have been ‘bioconjugated’ with and without the use of covalent linkers between the two components. In the second section, advanced glycoconjugates are discussed, which are composed of a sugar, an anticancer drug, a self-immolative linker and an additional component. Beyond the traditional glycosylated prodrug systems, the third section of this review will cover non-traditional supramolecular and macromolecular systems, such as nanoparticles and gels wherein glycosidase activity is required as a triggering step for the drug release.
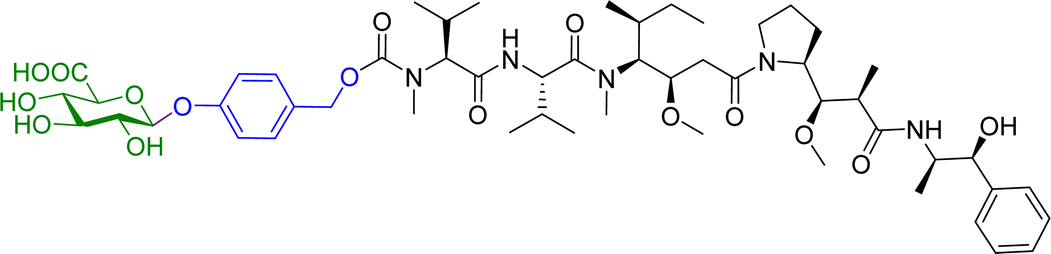
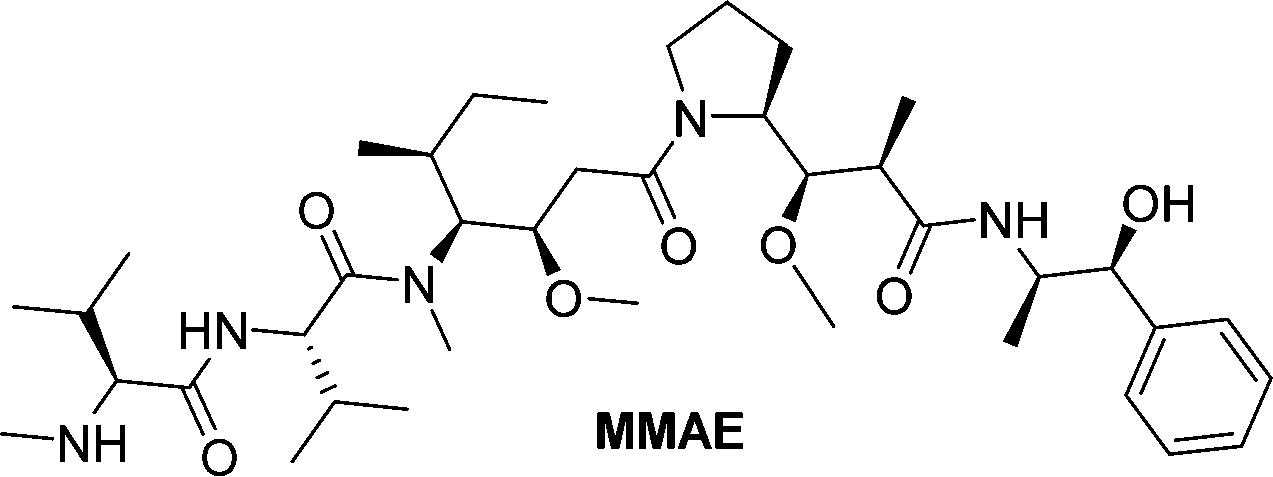
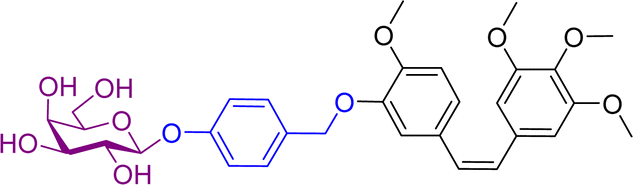
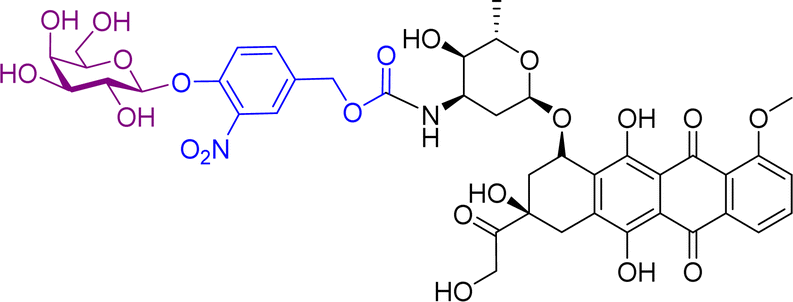
Monomethylauristan E (MMAE) is a potent inhibitor of tubulin polymerization. It is extremely cytotoxic with subnanomolar activity against numerous cancer cell lines. A glucuronide prodrug of MMAE 3 (Table 2) was developed containing a self-immolative linker, which was found to be significantly less toxic than the parent drug. β-Glucuronidase efficiently releases MMAE with IC50 values of 0.16 to 0.52 nM in the presence of various cancer cells – A549 (human lung carcinoma), KB (human squamous carcinoma) and MDA-MB-231 (human breast adenocarcinoma). The antiproliferative activity of the prodrug was also evaluated on primary cultures of patients possessing non-small cell lung cancer. The findings showed that the prodrug did not affect the cell viability when incubated alone at 0.5 or 1 nM, however, addition of the enzyme increased the cytotoxicity, killing about 76% of cells at 1 nM concentration. In vivo experiments using C57BL/6 mice with a subcutaneous murine Lewis Lung Carcinoma (LLC) showed that the prodrug caused significant antitumour activity. The MMAE-prodrug 3 demonstrated comparable efficacy to its doxorubicin analogue when administered at a 237-fold lower dose.57
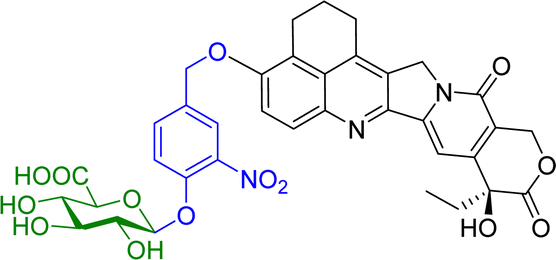
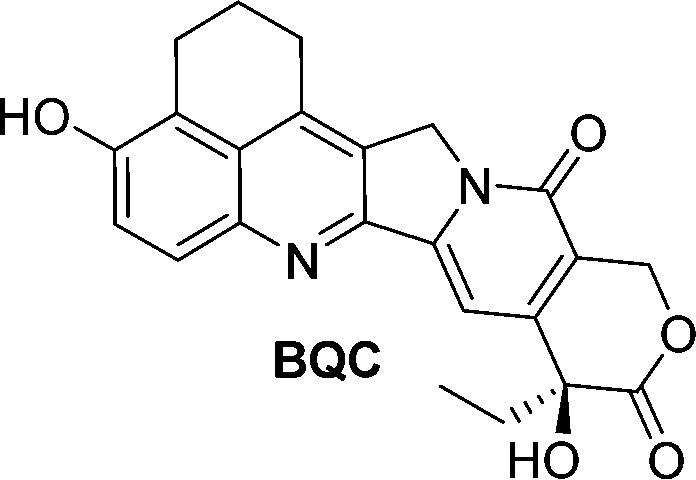
Camptothecin is a topoisomerase inhibitor leading to S-phase cell death. However, there are major limitations with its use as an anticancer agent, including the toxicity, non-selectivity, water insolubility and its inactivation by human serum albumin (HSA). A camptothecin derivative, 5,6-dihydro-4H-benzo[de]quinoline-camptothecin (BQC) was found to have a greater cytotoxicity against cancer cells than other clinically used camptothecin derivatives. A glucuronide prodrug of BQC 4 (Table 2) containing a self-immolative linker had 4000 times greater water solubility and 20–40 times lower cytotoxicity then BQC. Furthermore, the BQC-prodrug was efficiently hydrolysed by β-glucuronidase, leading to release of cytotoxic BQC (IC50 = 13 nM), even in the presence of HSA. Treatment of mice bearing human colon cancer xenografts with the BQC-prodrug 4 produced significant antitumour activity. Xenografts with naturally or artificially elevated levels of β-glucuronidase both showed significant reduction in the mean tumour size from day 2 or day 4 after initiation therapy, respectively.58
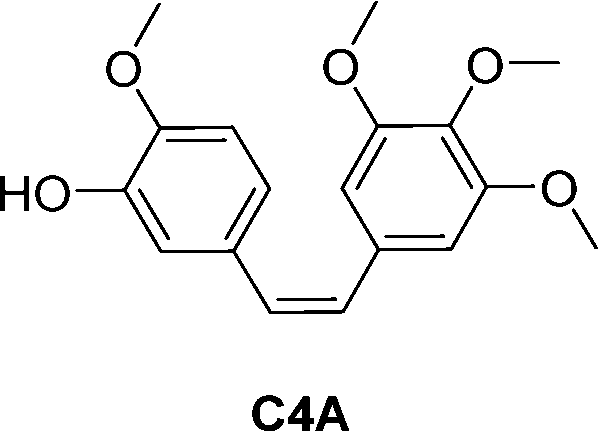
Combretastatin A4 (CA4) is a potent tubulin polymerization inhibitor. Microtubule disrupting agents are used for ovarian cancer chemotherapy. Since β-galactosidase activity is enhanced in ovarian cancer, prodrugs of C4A containing a galactose moiety were developed. In one of the prodrugs the galactose moiety was linked directly to C4A (structure not shown), while in second prodrug there is a self-immolative benzyl linker between the sugar and drug unit, prodrug 5 (Table 2). Enzymatic investigations confirmed that both prodrugs released the parent drug, however prodrug 5 did so at a quicker rate. In cytotoxicity assays using two ovarian cell lines, prodrug 5 exhibited similar toxicity to the parent drug, while the other prodrug was significantly less cytotoxic. This may be explained by the rapid transformation of prodrug 5 into C4A. This was confirmed by inhibiting intracellular β-galactosidase activity, the cytotoxicity levels of both prodrugs decreased substantially. Using fluorescent dyes and fluorescence confocal microscopy, it was confirmed that the cytotoxicity of these prodrugs to ovarian cancer cells was the results of disruption of the microtubule architecture and subsequent apoptosis.59
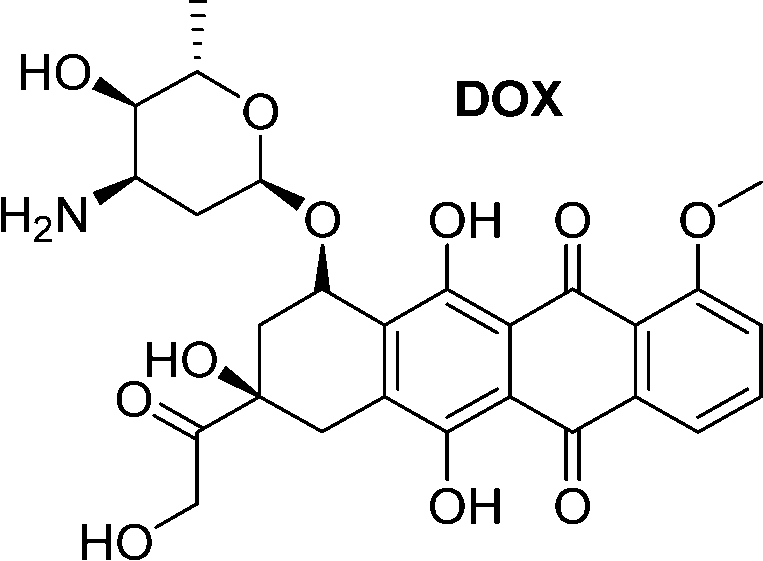
A small-molecule theranostic prodrug 6 (Table 2) of doxorubicin (DOX) was reported. This prodrug contained a galactose moiety, which was utilised to target asialoglycoprotein receptors on colon cancer cells and to enable enzyme-responsive activation strategies. Following activation by β-galactosidase enzymes in the colon, the parent drug is released leading to a ‘turn on’ of fluorescence, which allows the monitoring of both localization and the site of action of the drug. The selectivity was confirmed using HT-29, colon adenocarcinoma cell line, and HepG2 cells, a well-known cancer cell line displaying ASGP receptors. These cells showed fluorescence while the negative control (HeLa cells) exhibited little fluorescence. Cytotoxicity assays using HT-29 and HeLa cells showed that Gal-DOX 6 exhibited a 3-fold more potent therapeutic effect in the HT-29 cells than the HeLa cells, demonstrating that not only was cellular uptake improved, but also the enhancement of the anticancer effect. Using a fluorescence imaging system, the tumour-targeting ability of Gal-DOX 6 was confirmed in HT-29 tumour-bearing mice (Fig. 2a and b), along with the therapeutic efficacy (Fig. 2c and d).60
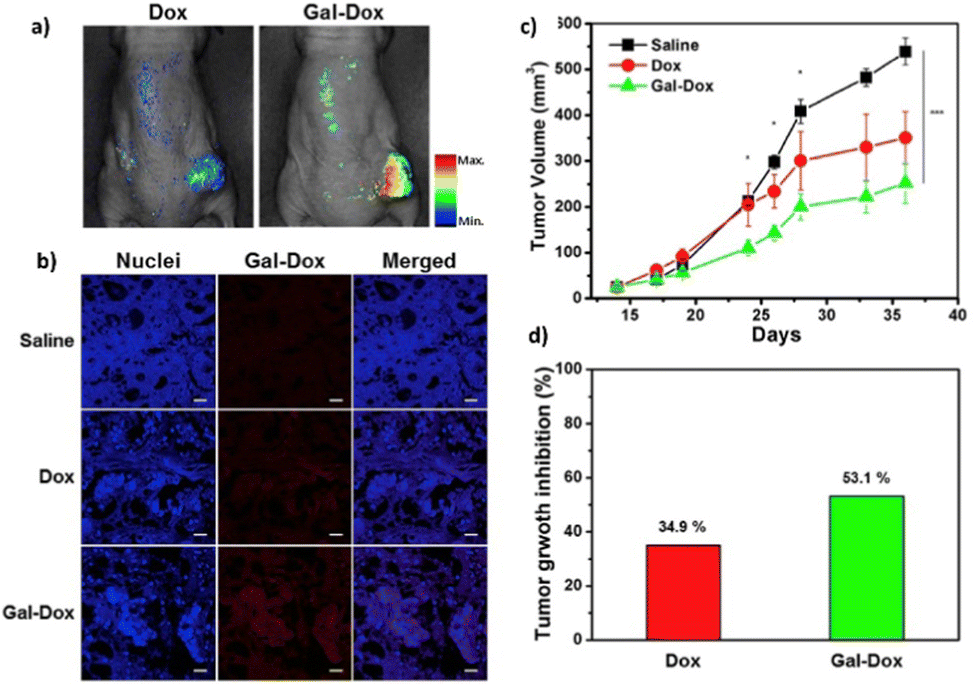 | ||
| Fig. 2 The antitumour activity of prodrug 6 (Gal-DOX). (a) Fluorescence imaging of Gal-DOX 6 treated mice with HT-29 cancer xenografts; (b) fluorescence images of paraffin-embedded tumour tissue; (c) tumour growth inhibition in mice with HT-29 cancer xenografts, injected with saline, DOX, or Gal-DOX 6 at 3 mg kg−1 four times every two days; (d) tumour growth inhibition after treatment with DOX and Gal-DOX 6 after 36 days. Reprinted from ref. 60 with permission from Elsevier. | ||
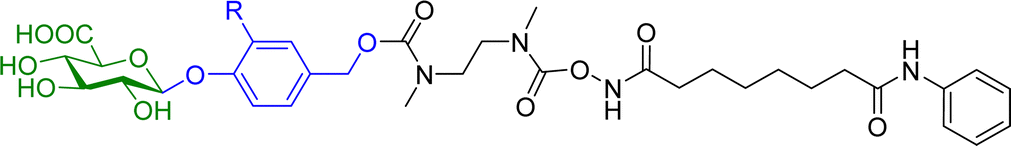
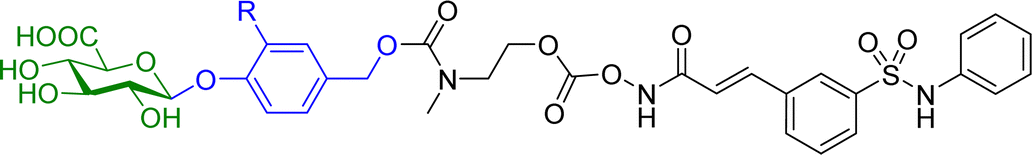
Hydroxamic acid-based histone deacetylase inhibitors such as Vorinostat and Belinostat have been reported to exhibit antitumour effects, but there have been limitations to its use as an anticancer agent due to its poor aqueous solubility. To help overcome this problem, β-glucuronic acid-based prodrugs of Vorinostat 7a and b and Belinostat 8a and b (Table 2) were developed, which were 400- to 750-fold more water soluble than the parent drugs. Cytotoxicity assays were then conducted on HT-29 and Hut-78 tumour cell lines. The IC50 values of the Vorinostat derivatives 7a and b were >10 μM in the absence of β-glucuronidase, however in the presence of the enzyme the inhibitory effects of the prodrugs were comparable to the parent drug, indicating the successful release of the drug upon enzyme-mediated hydrolysis of the glycosidic bond. However, for the Belinostat derivatives 8a and b, no change was observed in the IC50 values, both in the absence or presence of β-glucuronidase. Furthermore, the derivative with an amine functional group on the linker 7b had relatively higher IC50 values than those with a nitro functional group 7a.61
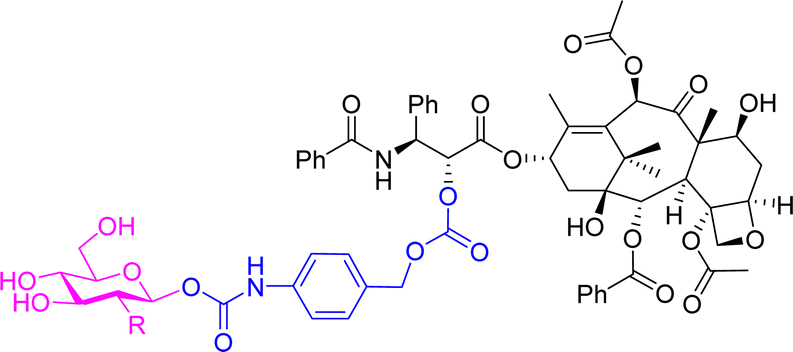
A 2-fluorodeoxyglucose conjugate of the anticancer drug paclitaxel 9a (Table 2) was synthesised and compared to the parent drug and the glucose conjugate (9b with no fluorine) in terms of solubility, stability, cytotoxicity, and selectivity towards cancer cells. A glucose conjugate was selected since most cancers overexpress glucose transporters. The incorporation of fluorine into drugs allows for adjustments of the electronic, lipophilic and steric properties. In previous studies it was found that the 2-deoxyfluoro glucosides had the slowest rate of hydrolysis when compared to other deoxy-glucosides or fluorinated glucosides. It was found that the aqueous solubility of the glucose-prodrug 9b and the 2-deoxyfluoro glucoside 9a was drastically improved compared to the parent drug. When analysing the chemical stability of the prodrugs in the presence of β-D-glucosidase, the 2-deoxyfluoro conjugate 9a exhibited sustained release with less than 50% hydrolysis detected after 12 hours. The non-fluorinated glucose conjugate 9b, however, was unstable and resulted in spontaneous release of the free paclitaxel. In cytotoxicity assays, the fluorinated prodrug 9a showed increased cytotoxicity compared to paclitaxel on carcinoma cells of HepG2 and NCI-H460. Both prodrugs showed excellent nanomolar toxicity to breast cancer cells MCF7. Meanwhile, paclitaxel and the glucose analogue 9b showed similar cytotoxicity to the normal cell line HUVEC, while the fluorinated analogue showed no obvious cytotoxicity up to 10 μM. This demonstrated that the prodrug could reduce the cytotoxicity of paclitaxel while improving selectivity towards cancer cells over normal cells.62
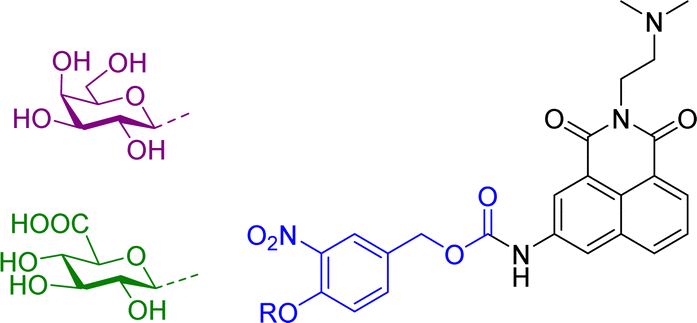
More recently, we have reported theranostic prodrugs of the toxic anticancer drug Amonafide 10a and b (Table 2). Amonafide exhibited excellent activity in phase II breast cancer clinical trials, however failed the phase III trials due to acute side effects and bone marrow toxicity. Spectroscopic characterization and time-dependent enzymatic release studies showed that the enzyme mediated activation of 10a and b occurs rapidly under physiological conditions. Cellular uptake and cell viability of these glyconaphthalimide compounds showed that upon enzymatic treatment, Amonafide was released and taken up into cells more rapidly than the glycosylated form. The IC50 values were consistent across three cancer cell lines (HeLa, HCT-116 and HepG2), with no cytotoxicity observed in the presence of 10a or 10b after 24 hours and low cytotoxicity values observed after incubation with the enzyme. Importantly, release of the therapeutic component was also triggered by the activity of endogenous enzymes that selectively hydrolyse the glycosidic bond, a key finding which was verified using confocal microscopy, whereby emission of the naphthalimide unit was monitored in real-time. This process facilitates selective delivery of the Amonafide to the desired site of action and allows for real-time bioimaging in cancer cell lines; this has also been verified by recording the emission spectra within the cells after delivery, which was red shifted due to enhanced internal charge transfer mechanism in the delivered compounds vs. the prodrug.63 This development was based on some prior work from our laboratory, where in a related study we employed the same principle for the delivery of naphthalimide imaging probes into cells. This system, which we described as a pro-probe (e.g. vs. prodrug) became activated only when a glycosidase activation of the glycosylated naphthalimide occurred, which released the fluorescent imaging agent into cancer cells in vitro, as was observed using real-time imaging, as only minimal cellular uptake was observed in the absence of the enzyme.64
2.1.1.1 Enzyme directed immunostimulants. The glycosidase mediated release strategy has also been widely utilised in the development of enzyme directed immunostimulants. Immunotherapeutics have an ability to instruct the immune system to develop an immune response to tumours. Imidazoquinoline immunostimulants elicit an antitumour immune response when applied to solid tumours. However, they cause severe inflammatory toxicity after systemic administration. To overcome this challenge, a directed enzyme prodrug therapy (DEPT) approach may be utilised using an enzyme-directed imidazoquinoline pro-immunostimulant. Using this methodology, the enzyme-enriched cancer cells generate the immunostimulant and diffusion of this immunostimulant within the cancer microenvironment enables activation of bystander immune cells.
Imidazoquinoline immunostimulants are agonists for Toll-like receptors 7 and 8 present on innate immune cells, and hence activation of these receptors within the tumour microenvironment results in a robust innate and adaptive antitumour immune response. The imidazoquinoline imiquimod (Fig. 3) was appended with a galactose moiety via a self-immolative linker (prodrug 11a), which yields imiquimod following treatment with exogenous β-galactosidase or β-galactosidase-enriched B16 melanoma cells. The enzyme-directed liberation of the immunostimulant resulted in activation of RAW-Blue macrophage and JAWSII monocyte immune cell lines.65
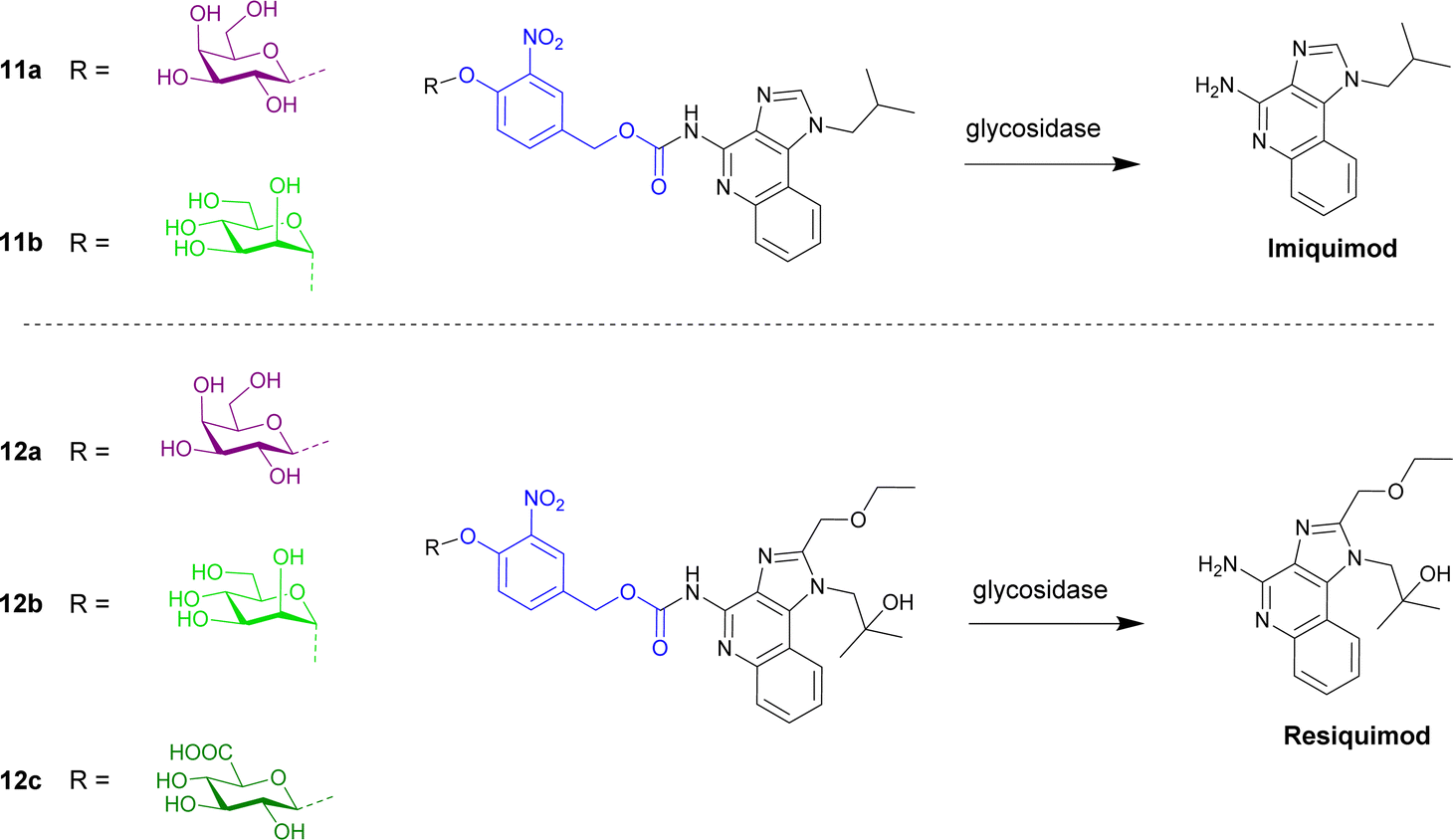 | ||
| Fig. 3 Structure of enzyme-directed immunostimulants (11a and b and 12a–c) explored for cancer therapy. | ||
Further work by the Neilsen and Mancini groups reported the first imidazoquinoline-mannoside pro-immunostimulant 11b that was activated by α-mannosidase.66 AT3B-1 prostate cancer cell lines were used since α-mannosidase is reportedly upregulated in multidrug resistant prostate cancers. AT3B-1 cells convert the pro-immunostimulant to imiquimod, leading to efflux of the drug into the extracellular space. Bystander immune cells are subsequently activated.
The same group then synthesised an enzyme-directed immunostimulant prodrug based on another imidazoquinoline, resiquimod (Fig. 3), covalently modified with β-glucuronic acid, β-galactose and α-mannose.67 The metabolism of the three prodrugs 12a–c were examined across three cancer cell lines (B16 melanoma, TC2 prostate, and 4T1 breast cancer). It was found that B16 cells elicited the highest immunogenicity, however the immunogenicity was comparable for a given cell type and independent of the glycosidase substrate in the prodrug or differences in functional glycosidase activity between cell lines. It was also found that the immunogenicity was governed by the efflux potential of cancer cells; this efflux occurring through P-glycoprotein-dependent and P-glycoprotein-independent transport mechanisms.
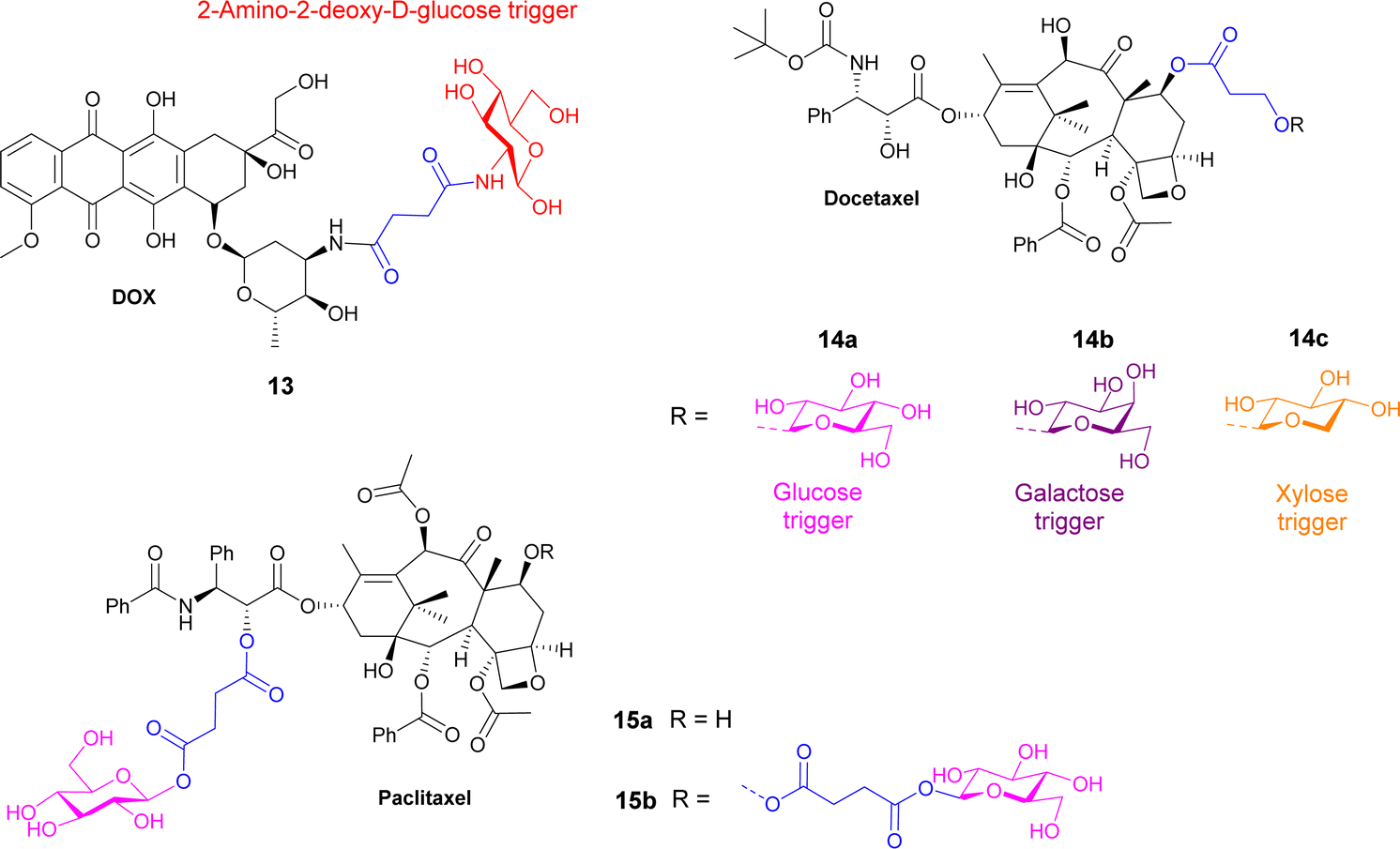 | ||
| Fig. 4 Structure of prodrugs 13–15 containing hydroxypropionic acid and succinate linkers. | ||
Docetaxel, an anticancer drug showing cytotoxicity against leukaemia and other cancers such as ovarian, breast and lung, has low water solubility and high toxicity to normal cells. Highly water-soluble sugar conjugates of docetaxel 14a–c (Fig. 4) were synthesised using a chem-enzymatic procedure, using the relevant enzymes to stereoselectively install the carbohydrate unit onto the hydroxypropionic acid linker, which was subsequently coupled to the docetaxel. Conjugates of glucose, galactose and xylose were synthesised with the glucose conjugate 14a being 52-fold more water soluble than the parent drug. 7-Propionyldocetaxel 3′′-O-β-D-glucopyranoside 14a and 7-propionyldocetaxel 3′′-O-β-D-xylopyranoside 14c were effectively hydrolysed by glycosidase enzymes releasing docetaxel and showed in vitro cytotoxicity against human cancer cells lines. However, the galactose derivative 14b was not successfully hydrolysed by endogenous enzymes in the KB human cancer cells and exerted low cytotoxicity.69
Paclitaxel is a chemotherapy drug used to treat different cancers including breast, ovarian and non-small cell lung cancer. However, due to its low aqueous solubility, the toxic surfactant Cremophor EL (polyethoxylated castor oil) must be used as a formulation vehicle in clinical practices. In this study, two prodrugs of paclitaxel were reported; a single glucose-conjugated paclitaxel 15a and a double glucose-conjugated paclitaxel 15b (Fig. 4), where the glucose moieties were attached via succinate linkers.70 Both prodrugs showed improved aqueous solubility and drug-like lipophilicity over the parent drug. Of the two, the single glucose conjugate 15a had a more efficient release in serum, where a high percentage of the parent drug was detected following hydrolysis with β-glucuronidase. This conjugate also exhibited good solubility in a low toxic formulation with no ethanol and lower percentage of Cremophor EL. Both prodrugs maintained effective cytotoxicity against breast cancer cells and apoptosis inducing effect, with the single glucose conjugate 15a, indeed performing better in both assays.
Gene-directed enzyme prodrug therapy (GDEPT) has also been used to improve cancer drug localization. A recombinant adeno-associated virus (rAAV) was utilised to deliver β-galactosidase; a week later the treatment group received adenocarcinoma cells in the same location of the initial injection (Fig. 5b). Then 1 hour later an inactive prodrug of geldanamycin 16 (Fig. 5a) was administered. Prodrug 16 consists of geldanamycin, a potent antitumour antibiotic from Streptomyces hygroscopicus, with a galactose moiety connected via an O-ethylene linker. It was found that tumour growth was significantly reduced in animals whose muscles were genetically modified to express the enzyme β-galactosidase compared to the control, indicating that the presence of the enzyme releases the parent drug efficiently. Serum assays indicated that there was minimal damage to non-target organs, demonstrating that using this GDEPT technique allows for improved localized activation of chemotherapeutic agents.71
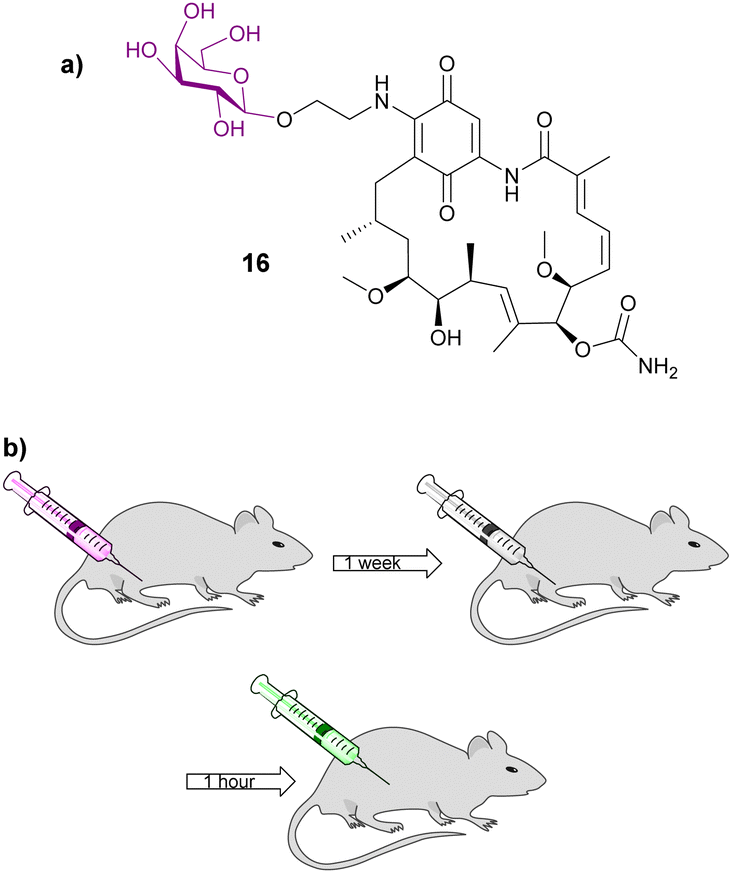 | ||
| Fig. 5 (a) Structure of galactosylated prodrug of geldanamycin 16; (b) diagram depicting the GDEPT procedure used in this study. Diagram adapted from ref. 71. | ||
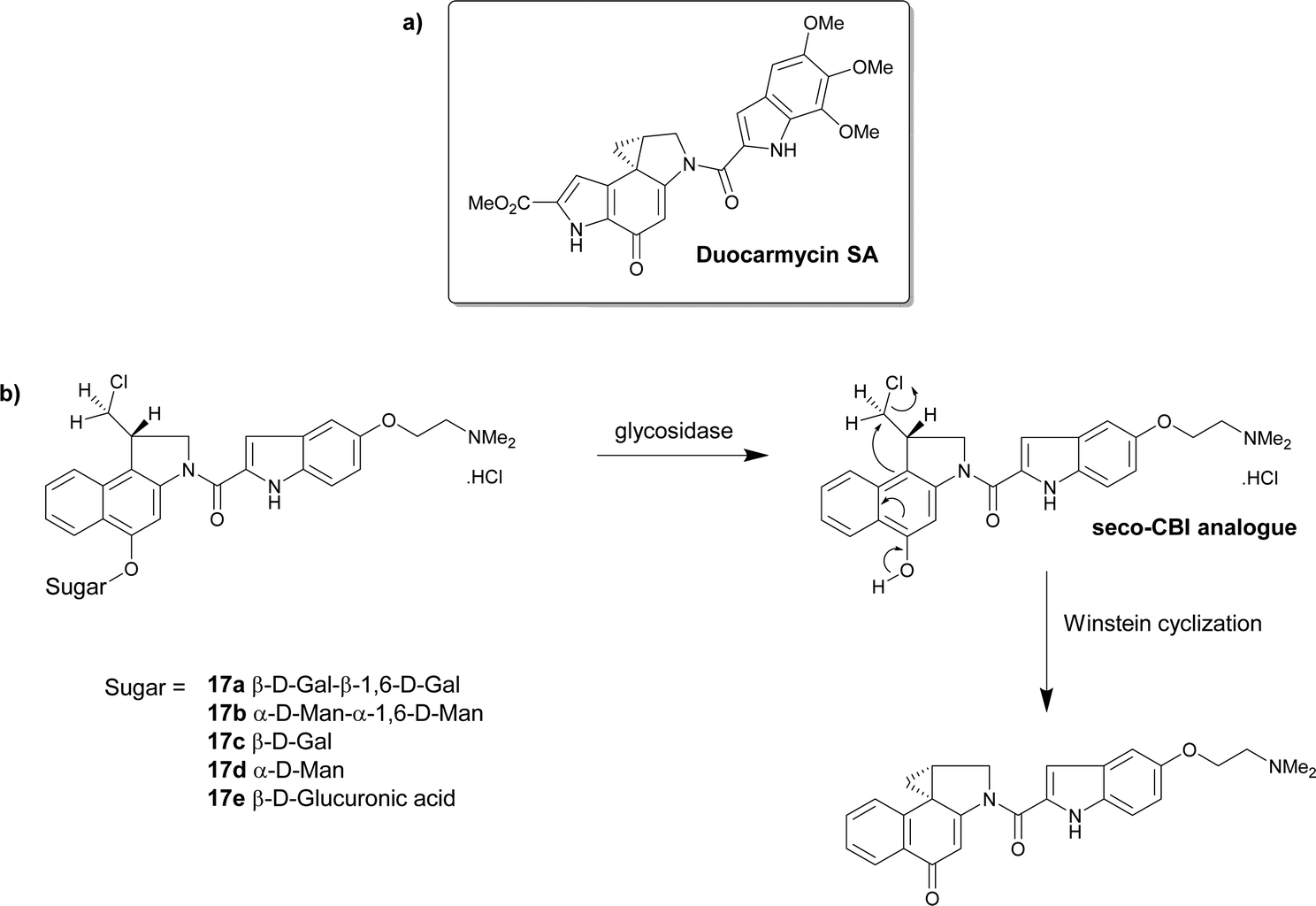 | ||
| Scheme 2 (a) The structure of duocarmycin SA; (b) the structure of the prodrugs 17a–e that under hydrolysis give the seco-CBI analogues that finally produce the cytotoxic drug. | ||
The group further explored the glucuronic acid derivative 17e as an enzyme prodrug mono-therapeutic. The activation of the prodrug by intracellular and extracellular β-glucuronidase was investigated. The cytotoxicity of the prodrug 17e was measured against human cell lines that exhibited varying levels of endogenous β-glucuronidase, including β-glucuronidase-deficient fibroblasts and β-glucuronidase knockdown cancer lines displaying low levels of the enzyme. The glucuronide prodrug 17e was 1000–5000 times less cytotoxic than the parent drug irrespective of the β-glucuronidase levels. Cancer cells expressing membrane-tethered β-glucuronidase were more sensitive to the prodrug indicating that extracellular β-glucuronidase activity was more important than intracellular levels. This glucuronide prodrug 17e (2.5 mg kg−1) displayed less systemic toxicity and greater antitumour activity than clinically approved anticancer drug carboplatin (50 mg kg−1).76
Clioquinol (Scheme 3), a well-known 8-hydroxyquinoline (OHQ), is widely used as an antibiotic. It was also reported as a modulator of metal homeostasis in neurodegenerative disorders such as Alzheimer's disease. This compound also displays anticancer effect both in in vitro and in vivo preclinical models. It and other 8-hydroxyquinolines must interact with Cu2+ ions to exert anticancer activity. Cu2+ ions are co-factors required for tumour growth and angiogenesis, with high levels of the cation found in many cancer types. To exploit the glucose avidity, the elevated glycolysis of tumour cells, and the over-expression of glucose transporter, new clioquinol glucoconjugates were synthesised (GluOHQA 18a and GluCQ 18b). These prodrugs improved the aqueous solubility and since they do not complex Cu2+ ions in their glycosylated form, side effects of systemic chelation are prevented. Following hydrolysis with β-glucosidase, the active aglycone is released. However, after studying the antiproliferative activity of the parent compounds and the glycosylated derivatives, the glycoconjugates are less active than their parent compounds. It was hypothesised that enzyme mediated release of the parent drugs remains incomplete.77
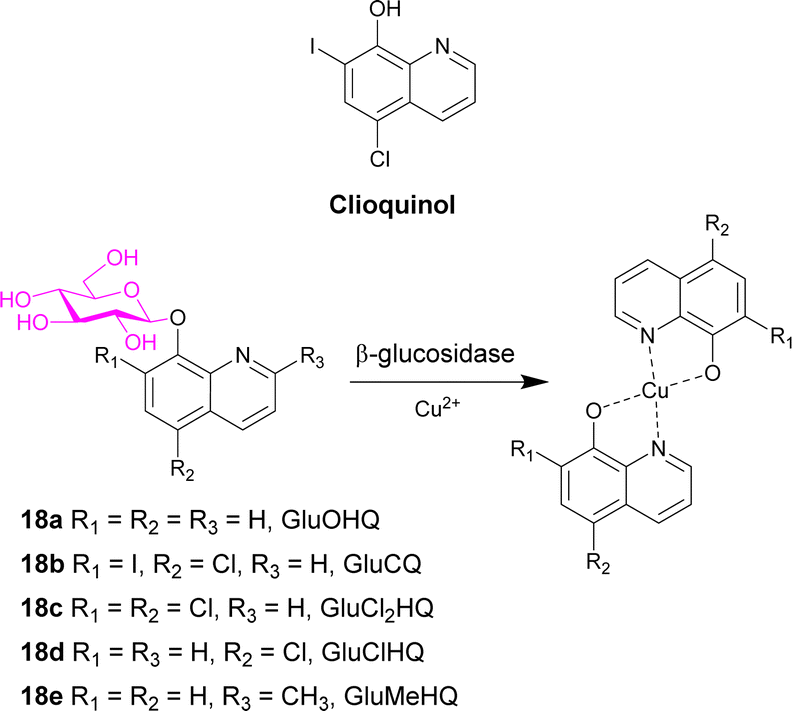 | ||
| Scheme 3 Structure of clioquinol and the prodrugs 18a–e designed in this study. Also shows the complex formed in the presence of copper ions. | ||
The same group then synthesised three new glycoconjugates of OHQs 18c–e, which demonstrated that the glycosylated compounds had similar activity to the parent compounds, with the exception of the methyl derivative (GluMeHQ 18e). Computational docking studies of these compounds with human cytosolic β-glucosidase indicate that GluMeHQ 18e had the best affinity for the binding site, while GluCl2HQ 18c showed the lowest affinity. It is suggested that the compounds that are tightly bound to the enzyme are more slowly hydrolysed, and therefore release of the active compound occurs at a slower rate, reducing the cytotoxicity. The presence of Cu2+ ions increases their antiproliferative activity by an order of magnitude and the pharmacological profile of the glycoconjugates are dependent on the intracellular β-glycosidase activity.78
In another study, galactose was conjugated to the fluorescent dye MPA to evaluate the tumour affinities and hence the cancer targeting ability of galactose. An in vitro cell study showed that the galactose-MPA conjugate had higher affinity to tumour cells (HepG2, MCF-7 and A549) than normal liver cells. They then conjugated galacturonic acid onto the anticancer drug doxorubicin (DOX) for targeted tumour therapy. This prodrug conjugate 19 (Fig. 6) had better therapeutic results than DOX suggesting that galactosylated prodrugs may have a potential use in tumour targeted therapy.79
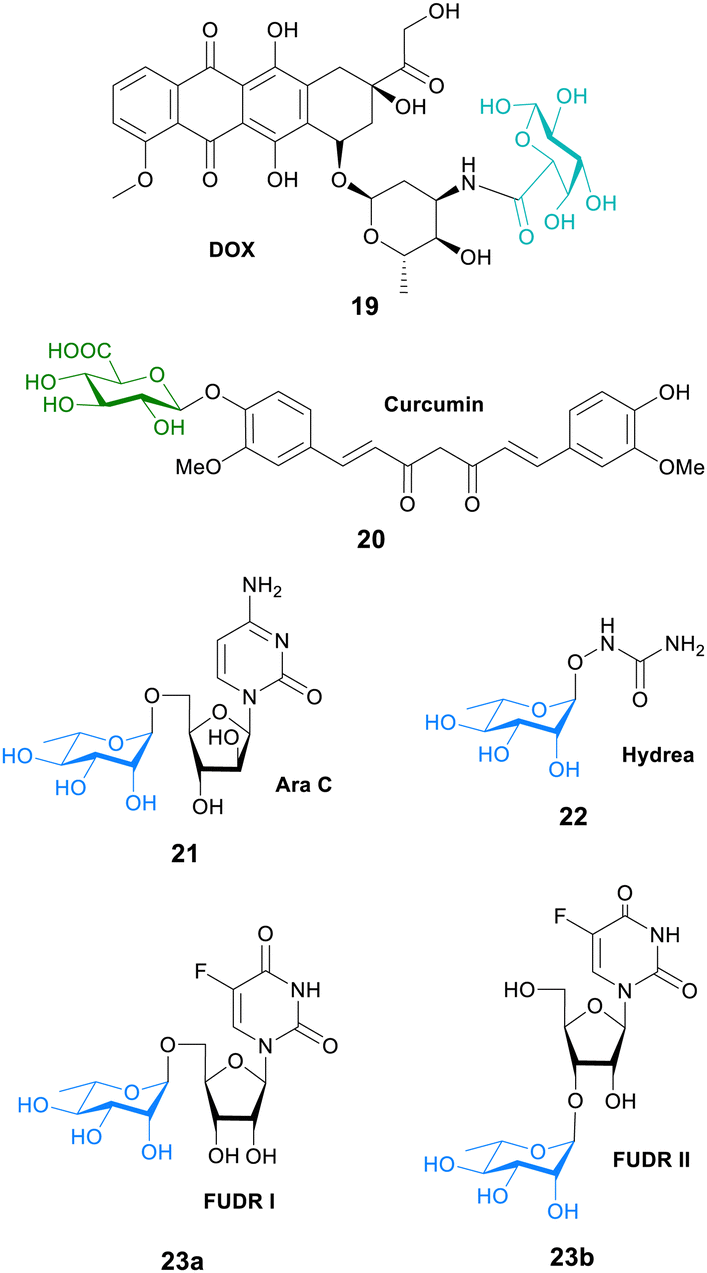 | ||
| Fig. 6 Structure of prodrugs 19–23, where the carbohydrate unit is bonded directly to the anticancer agent. | ||
Curcumin is a natural polyphenol found in turmeric that was been linked to a lower rate of cancer. However, there is low bioavailability of curcumin due to elimination through faeces and metabolism into inactive glucuronides. Although, the mode of action is not known, it is proposed that curcumin glucuronide 20 (Fig. 6) is an inflammation-responsive prodrug that is hydrolysed back to curcumin at the site of action. It is also known that levels of β-glucuronidase are elevated in some cancers such as breast cancer. In mouse tumour models, high levels of curcumin were generated at the tumour after both oral and intravenous administration. Long-term daily dosing of curcumin resulted in the accumulation of curcumin in tumour tissue and led to the inhibition of tumour growth where high levels of β-glucuronidase were expressed.80
The anticancer properties of curcumin were further explored in another study. Colorectal cancer is typically treated with the Pt-complex oxaliplatin. However, it is known that KRAS mutation, common in colorectal cancer patients, hampers the effect of oxaliplatin. NF-κB activation is an important step in oxaliplatin resistance. This NF-κB pathway may be inhibited with curcumin. In this study a water-soluble injectable glucuronide-prodrug of curcumin 20 (Fig. 6) was prepared, and its efficacy, safety and pharmacokinetics in a mouse xenograft model was investigated. KRAS/TP53 mutations increased the IC50 values of oxaliplatin in HCT116 cells in vitro. The glucuronide-curcumin prodrug 20 exhibited better anticancer effects compared to oxaliplatin in an oxaliplatin-resistant xenograft model, with a combination therapy exhibiting additive effects in these models without increasing the toxicity. Importantly, pharmacokinetic analysis showed high levels of free curcumin in tumour tissue after 48 hours, but was not detected in other organs such as the heart, liver and spleen.81
In a novel approach to glycosyl prodrug synthesis, an α-L-rhamnosidase from Alternaria sp. L1 (RhaL1) was employed to catalyse rhamnosylation through reverse hydrolysis reaction of known anticancer drugs, namely 2′-deoxy-5-fluorouridine (FUDR), cytosine arabinoside (Ara C), and hydroxyurea (Hyrea). The rhamnosylated drugs 21–23 (Fig. 6) exhibited little cytotoxicity, with the toxicity restored only when incubated with exogenous α-L-rhamnosidase. This proves their potential use in enzyme-activated prodrug systems. Also in the study, the cancer-targeting ability of the rhamnose moiety was evaluated by conjugating the monosaccharide to the fluorescent dye rhodamine B. The fluorescent probe displayed higher cell affinity and cellular internalization in oral cancer cell KB and breast cancer cell MDA-MB-231 than that of normal epithelial cells MCF 10A. This suggests that the rhamnose moiety could play a key role in targeting the active therapeutic to cancer cells.82
2.1.3.1 Targeting senescent cells. Senescent cells accumulate during ageing and are associated with cancer, fibrosis and many age-related pathologies. They display elevated activity of the lysosomal β-galactosidase. Different galactose-modified duocarmycin prodrugs preferentially kill senescent cells, in a lysosomal β-galactosidase-dependent manner. The effects of a galactosylated seco-duocarmycin analogue dimer 24 (Fig. 7) previously described by Tietze83 was analysed on the survival of IMR90 ER:RAS cells (a model of oncogene-induced senescence). This prodrug can selectively eliminate a wide range of senescent cells, including bystander senescent cells. In an adamantinomatous craniopharyngioma (ACP, a pituitary paediatric tumour) mouse model, treatment with prodrug 24 selectively reduced the number of β-catenin-positive preneoplastic senescent cells.84
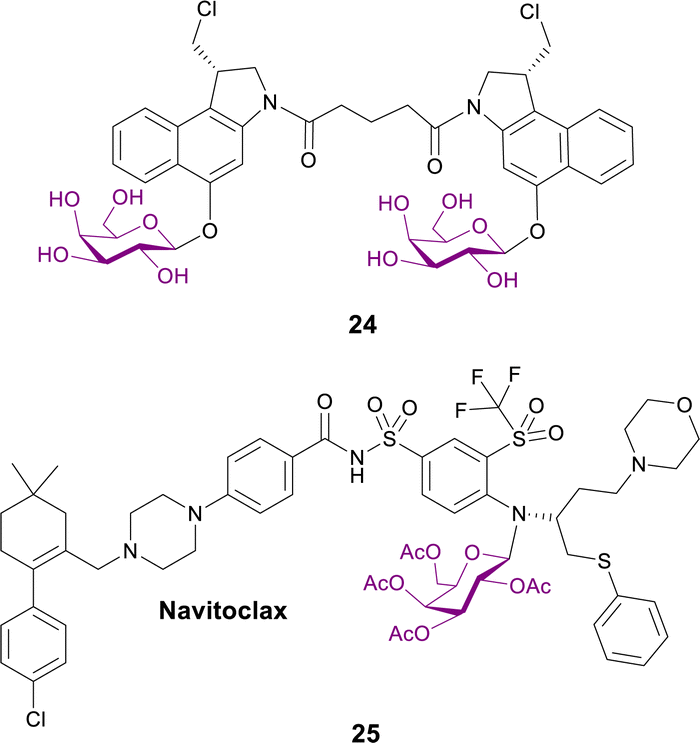 | ||
| Fig. 7 Structure of prodrugs 24 and 25 that target senescent cells. | ||
Senescent cells were targeted in another report, where the BCL-2 family inhibitor Navitoclax was conjugated with galactose to exploit the lysosomal β-galactosidase activity of senescent cells. Prodrug 25 (Fig. 7) selectively induces apoptosis of senolytic cells. Interestingly, this prodrug enhances the cytotoxicity of standard cisplatin-induced senescent lung cancer A5A9 cells. Concomitant treatment with cisplatin and prodrug 25in vivo resulted in the elimination of senescent lung cancer cells and the reduction of tumour growth. Furthermore, galactose conjugation reduces Navitoclax-induced platelet apoptosis in both human and mouse blood samples and reduces thrombocytopenia in mouse lung cancer models.85
In summary, numerous anticancer drugs have been conjugated with a carbohydrate moiety, with and without a linker. The resulting ‘simple’ glycosylated prodrugs exhibit an improvement of their aqueous solubility and their selectively towards cancer cells with a reduction in the toxic side effects.
2.2 Advanced glycoconjugates
In this section, advanced glycosylated prodrugs are discussed. These prodrugs are composed of a glycosidic trigger, a self-immolative linker, an anticancer drug, and unlike that discussed in the previous section, they all contain an additional component. These additional components include extra drug molecules, hydrophilic chains, targeting ligands and mechanically interlocked systems. The aim of these constructs is to increase the potency, solubility and/or selectively of the anticancer drugs.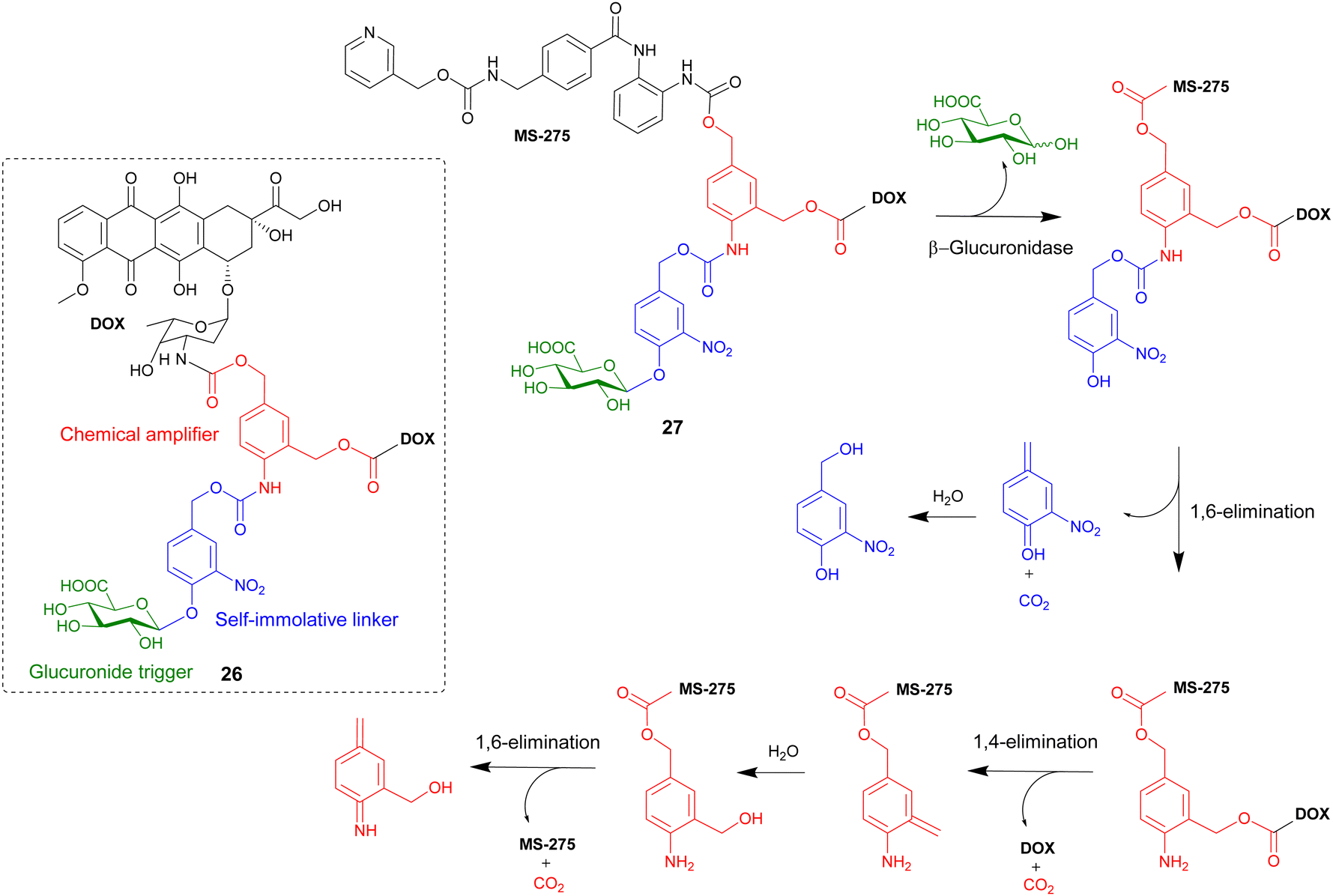 | ||
| Scheme 4 The β-glucuronidase-mediated drug release mechanism of heterodimeric prodrug 27. Inset shows the structure of homodimeric prodrug 26. | ||
A similar heterodimeric system was also reported (Scheme 4), where two different anticancer drugs were released upon enzyme activation. The previously used doxorubicin and a well-known histone deacetylase inhibitor MS-275 were incorporated into prodrug 27. Furthermore, in this study the role of the chemical amplifier was investigated. As well as being responsible for the signal amplification, the amplifier is converted into a highly toxic species that, in combination with the two other drugs, leads to potent cancer tritherapy. In antiproliferative assays against H290 lung mesothelioma cells, confocal microscopy confirmed the β-glucuronidase-mediated release of doxorubicin. Western blot analysis confirmed that enzymatic activation of prodrug 27 also restored the ability of MS-275 to inhibit histone deacetylase. However, since the activated heterodimer 27 was five-fold more toxic than the combination of the two anticancer drugs, it was confirmed that the release of the chemical amplifier as an azaquinone methide unit contributed significantly in the cytotoxicity of prodrug 27.87
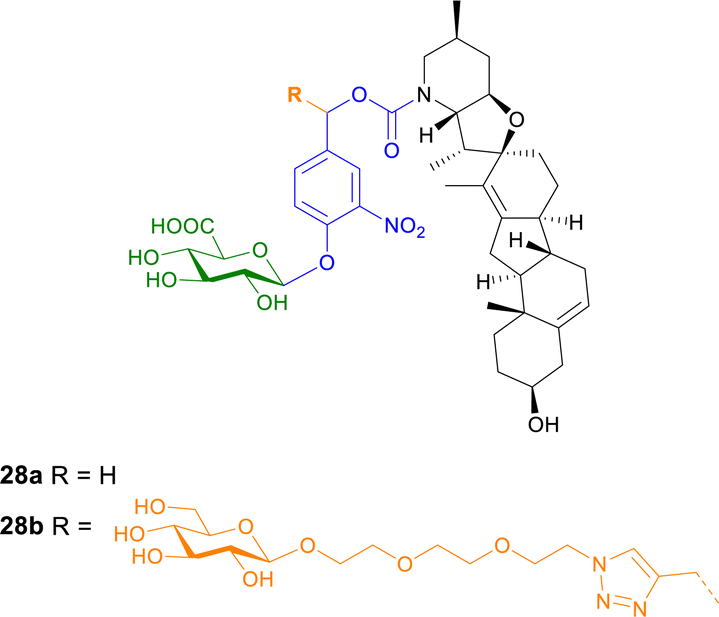 | ||
| Fig. 8 Structure of prodrugs 28a and b of cyclopamine. Prodrug 28b contains an additional hydrophilic linker to enhance water solubility. | ||
The cytotoxic properties of prodrug 28b were also analysed in vitro, ex vivo and in vivo in C6 rat glioblastoma multiforme (GBM) cells, an aggressive primary brain tumour where Hh pathway activation is also observed. The β-glucuronidase activated prodrug was toxic and down regulated the expression of a Hh target gene in cancer cells but not in normal rat astrocytes. When the prodrug was administered to rats bearing fluorescent C6-derived GBM, the tumour density was reduced more efficiently than using cyclopamine alone.90
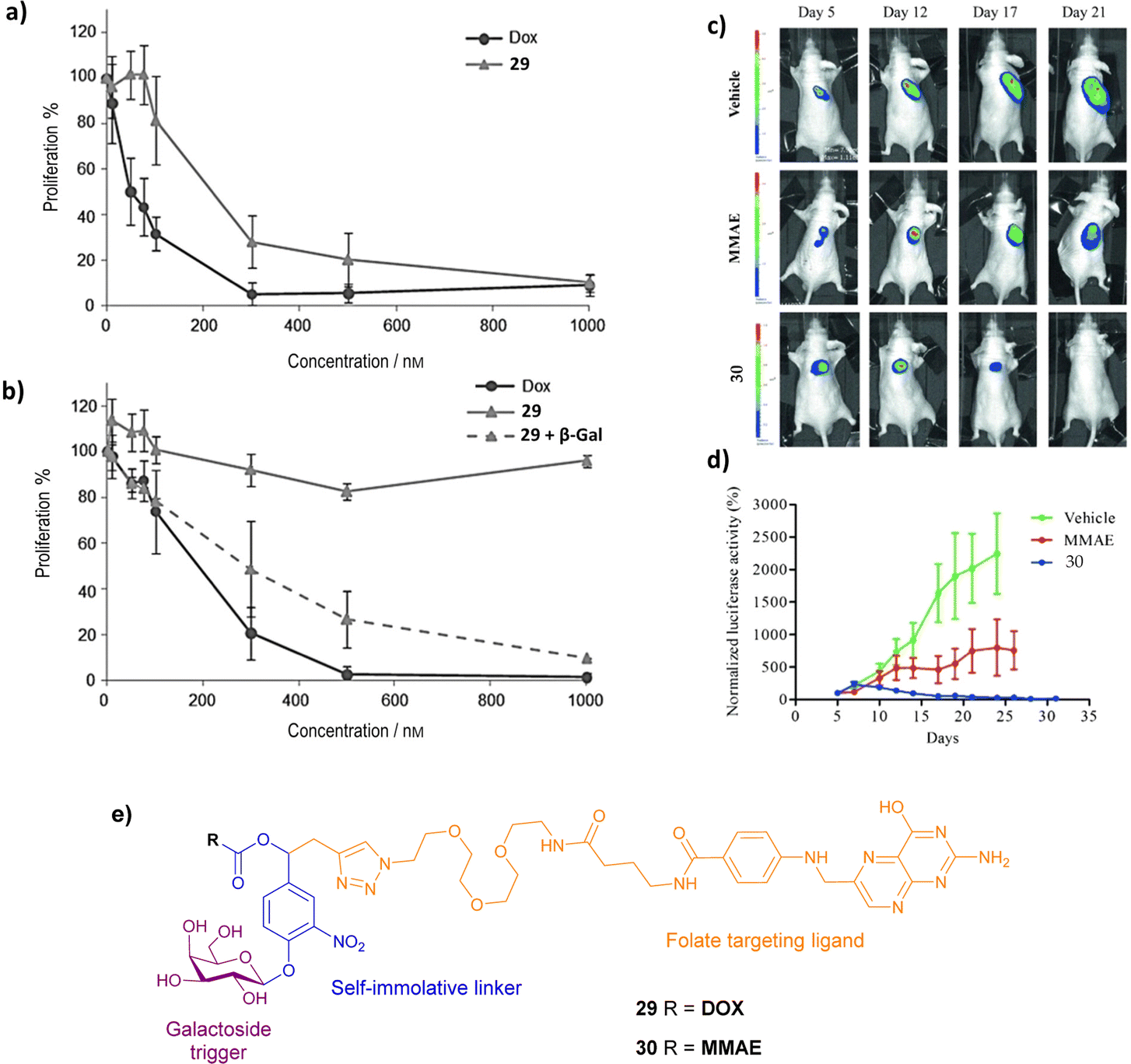 | ||
| Fig. 9 Proliferative activity of (a) HeLa and (b) A549 cells treated with DOX, prodrug 29 and prodrug 29 + β-galactosidase. (Reprinted from ref. 91 with permission from John Wiley and Sons); (c) bioluminescence imaging of KB xenografts following treatment with vehicle (5% DMSO in PBS buffer), MMAE and prodrug 30; (d) tumour growth inhibition in the presence of MMAE and prodrug 30. (Reprinted from ref. 92 with permission from John Wiley and Sons); (e) structure of prodrugs 29 and 30. | ||
To advance this strategy, the concept of the dual drug release was combined with the folate-targeting system, creating novel prodrugs (structure not shown) that target the folate receptors as before, and release two anticancer drugs after one enzyme activation step i.e. two MMAE molecules,93 or two DOX molecules.94 As expected, the dimer prodrugs had superior cytotoxicity towards cancers cells than their monomeric counterparts, demonstrating the increased release of MMAE or DOX within the tumour cells.
Gennari and co-workers have explored the conjugation of different anticancer drugs to a peptidomimetic compound cyclo(DKP-RDG), a low nanomolar ligand of integrin αvβ3, a transmembrane glycoprotein overexpressed in a variety of cancers (e.g. melanoma, glioblastoma, lung, breast, ovary, colon). Two integrin-targeting prodrugs of MMAE containing a glucuronide trigger and a self-immolative linker were reported. The integrin-targeting ligand are connected to the self-immolative linker via two different spacers: (1) a glutaric acid derivative in prodrug 31a; (2) a triazole and PEG4 spacer in prodrug 31b (Fig. 10). The cytotoxicity of prodrugs 31a and b were examined by incubating the RDG-MMAE conjugates with αvβ3-expressing cancer cells, in the absence and presence of β-glucuronidase. The anticancer activity of prodrug 31b increased dramatically upon enzyme activation, with IC50 values similar to MMAE in the low nanomolar range. On the other hand, the enhanced anticancer activity of prodrug 31a was not observed, regardless of β-glucuronidase addition. In this study, the use of a long, hydrophilic PEG4 spacer, connecting the integrin ligand to the linker moiety, was essential for the overall efficiency of the prodrug, which they suggest is most likely due to reduced steric hinderance around the cleavable glycosidic bond.95
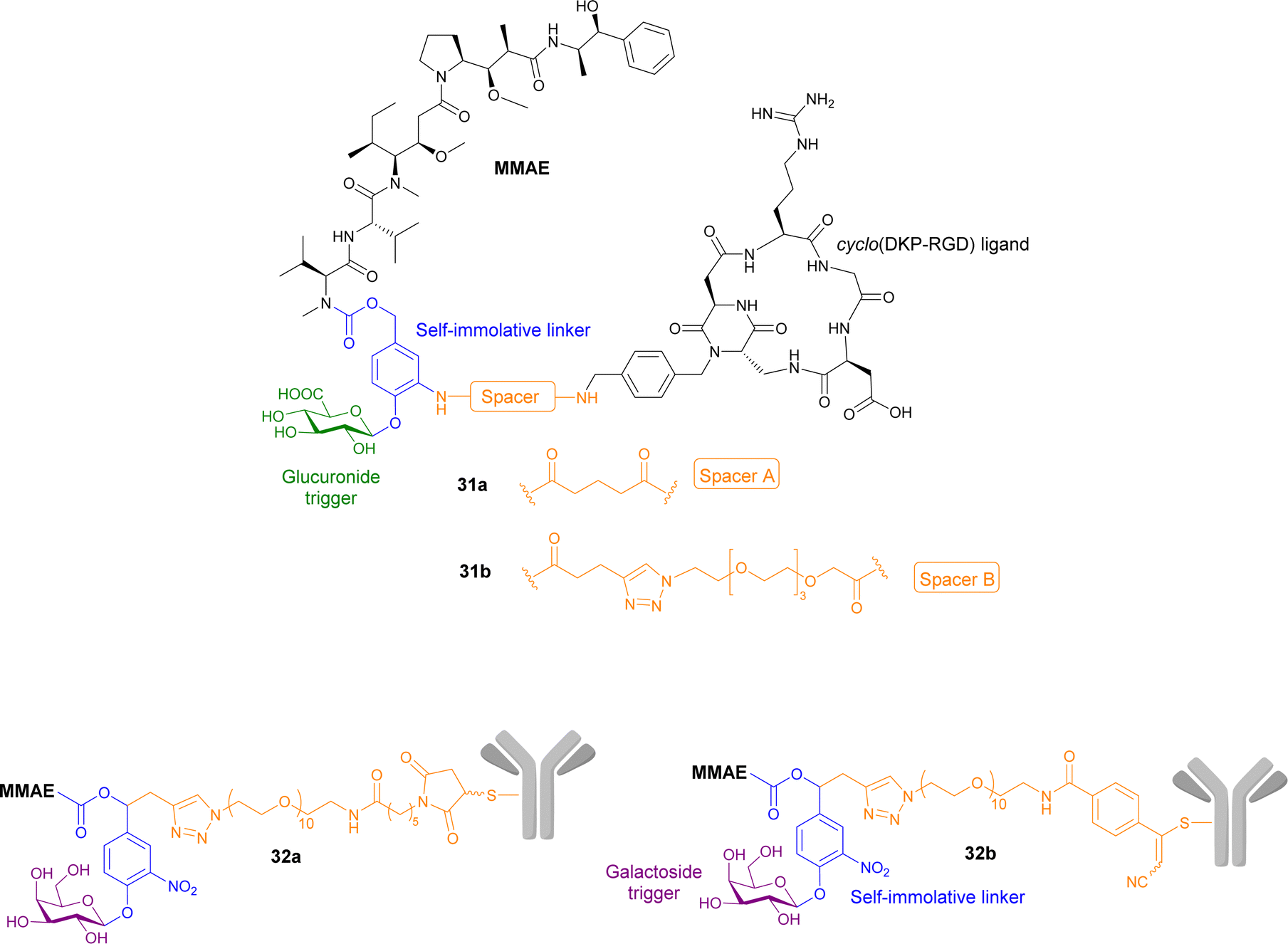 | ||
| Fig. 10 Structures of prodrugs 31a and b, and 32a and b containing a targeting peptidomimetics and antibodies with two different spacers. | ||
Antibodies have also been utilised in targeted cancer therapies. Trastuzumab, a monoclonal antibody that recognises growth factor receptors that are overexpressed in some breast cancers, was functionalised using its cysteine residues onto prodrugs of MMAE containing a galactoside trigger and a self-immolative linker. Two derivatives were reported containing different spacers between the antibody and the self-immolative linker, both containing a decathylene glycol side chain terminated by (1) a maleimidocaproyl functional group (prodrug 32a, Fig. 9); and (2) an arylpropiolonitrile group (prodrug 32b). These antibody-prodrug conjugates exhibited selective cytotoxicity against HER2+ cancer cell lines with IC50 values in the picomolar range. Furthermore, prodrugs 32a and b exhibited a significant antitumour effect in vivo after administration of a single 1 mg kg−1 dose, both displaying 57–58% reduction of tumour volume 31 days post-injection, indicating that the spacer group used was not significant.96
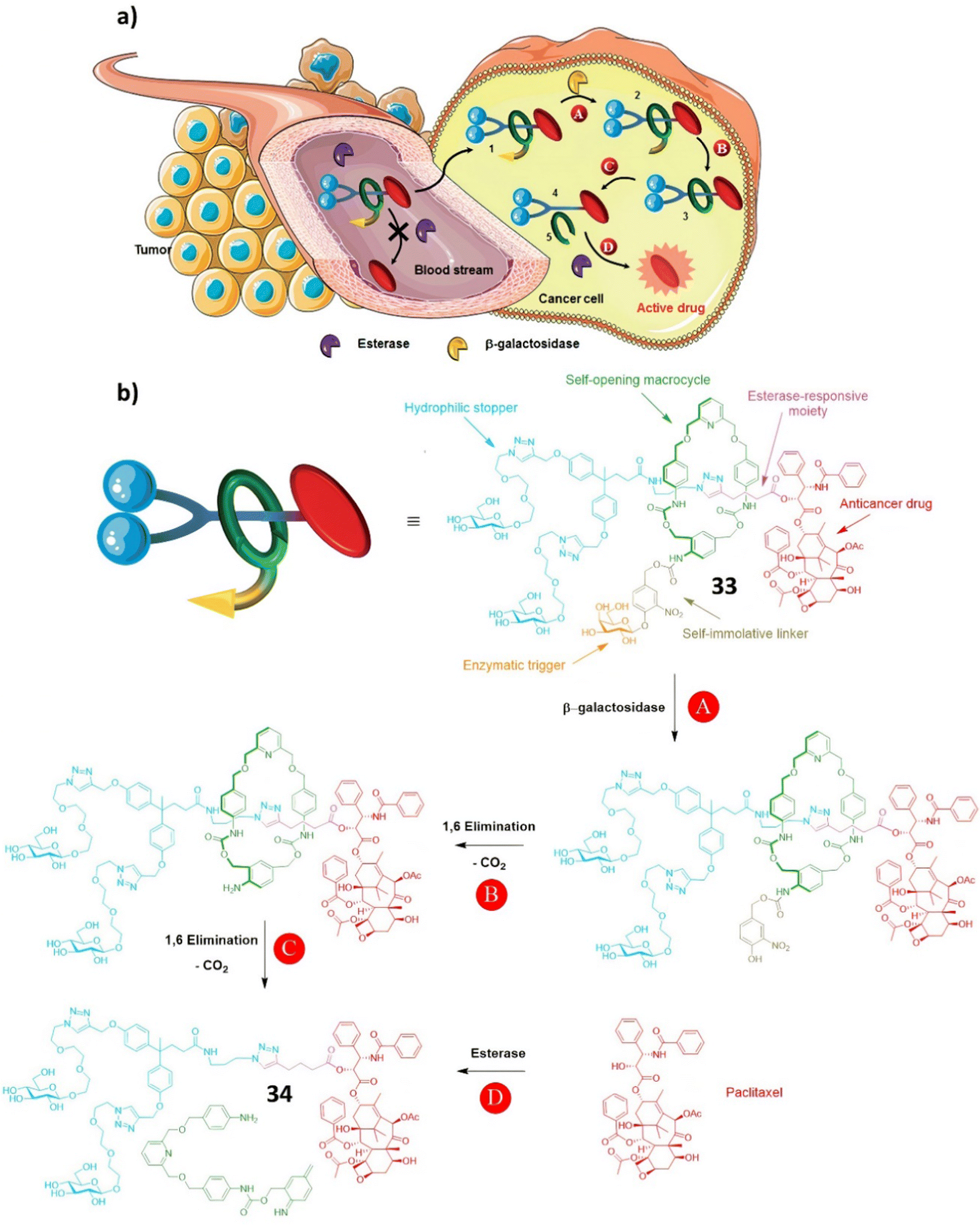 | ||
| Scheme 5 (a) The principle of drug delivery for the interlocked prodrug 33; (b) the structure of prodrug 33 and the paclitaxel release mechanism. (Reproduced from ref. 97 with permission from the Royal Society of Chemistry.) | ||
2.3 Macromolecular and multimolecular systems
In this section, multimolecular systems incorporating glycosidase activated prodrugs are discussed. The main rationale for the development of these multi molecular systems is the enhanced permeability and retention (EPR) effect. EPR is a property of solid tumours, whereby macromolecular drugs and nanoparticles accumulate preferentially in tumour tissues than in normal tissue due to increased vascular permeability.98,99 To take advantage of the EPR effect, research has focused on developing larger supramolecular and macromolecular systems incorporating the prodrug concept. These include, but are not limited to, polymers, micelles, nanoparticles and protein conjugates.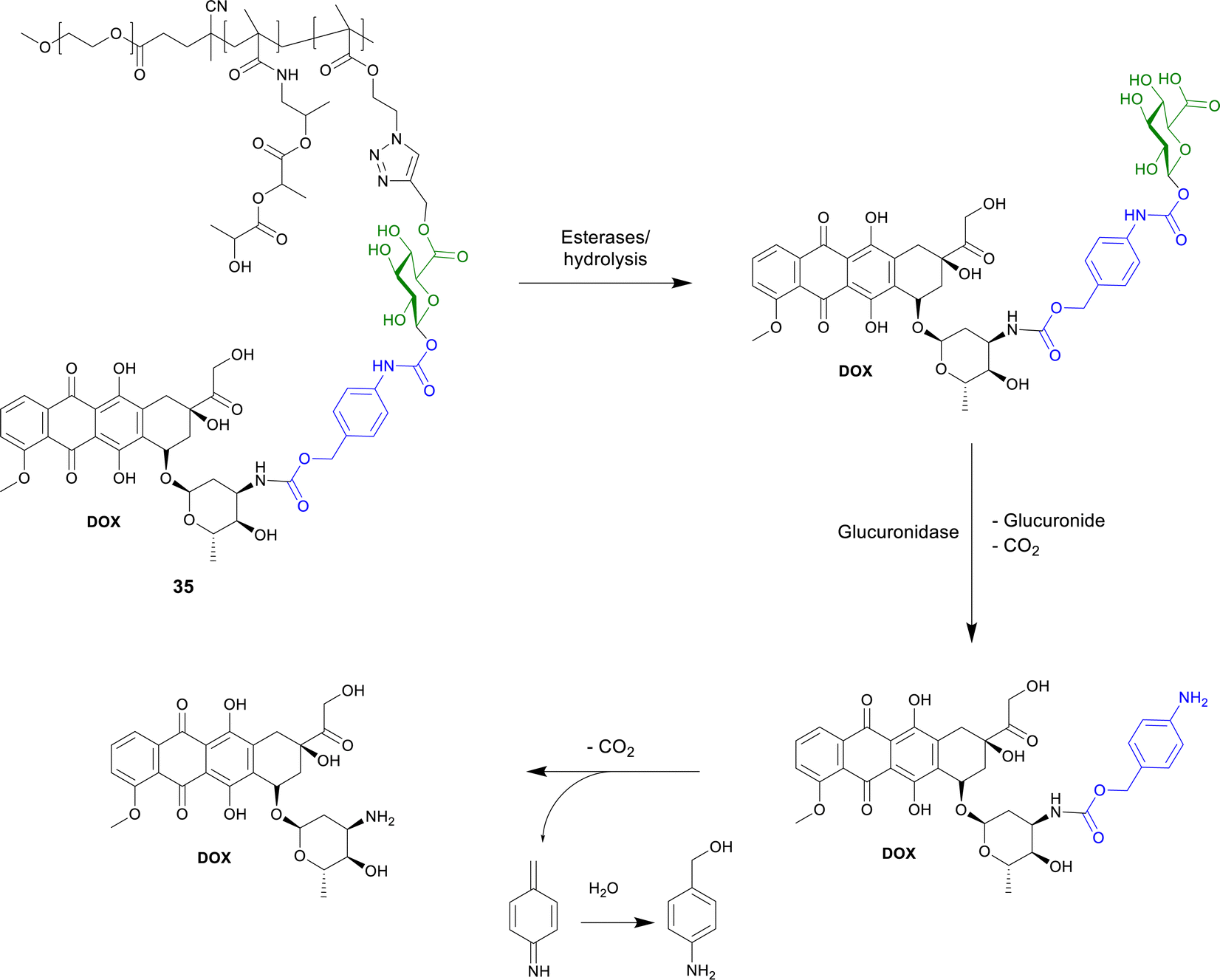 | ||
| Scheme 6 Structure of the polymer-bound DOX-prop-GA3 35 and the conversion by chemical hydrolysis and/or by esterases and subsequent release of DOX by β-glucuronidase. | ||
5-Fluorourical is a clinical anticancer drug that suffers from poor bioavailability. Many chemically modified derivatives of this drug have been reported, however there are still difficulties in targeting particular cancers. Negi and co-workers have reported a xylan-5-fluorourical-1-acetic acid conjugate 36 (Fig. 11) as a colon specific prodrug.101 Xylan was used since it remains intact in the upper gastrointestinal tract and it degraded by xylanases and β-xylosidase enzymes present in the colon. In vitro drug release profiles in the presence of rat's gastrointestinal contents, showed that small amounts of the drug were released in the gastric and small intestine contents, while 53–61% were released in the cecum and colonic contents. The ester linkage of the conjugate is resistant to ester hydrolysing enzymes in the upper GI tract due to the steric hindrance of the polymeric xylan chain. In a human colorectal cancer cell line (HTC-15 and HT-29), the conjugates were more cytotoxic than the parent drug.
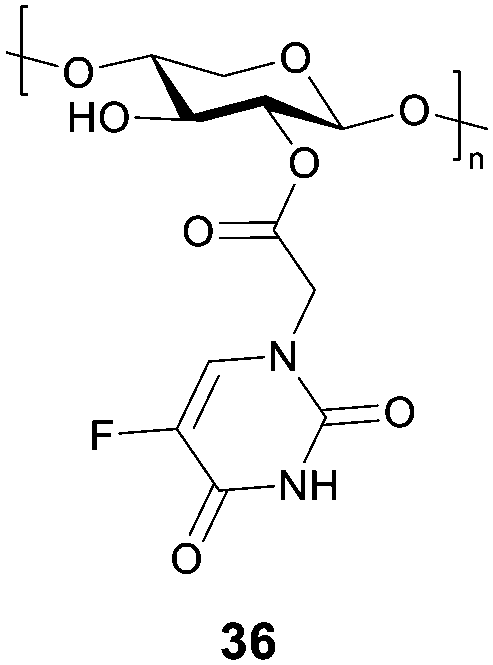 | ||
| Fig. 11 Structure of the xylan-5-fluorouracil-1-acetic acid conjugate 36. | ||
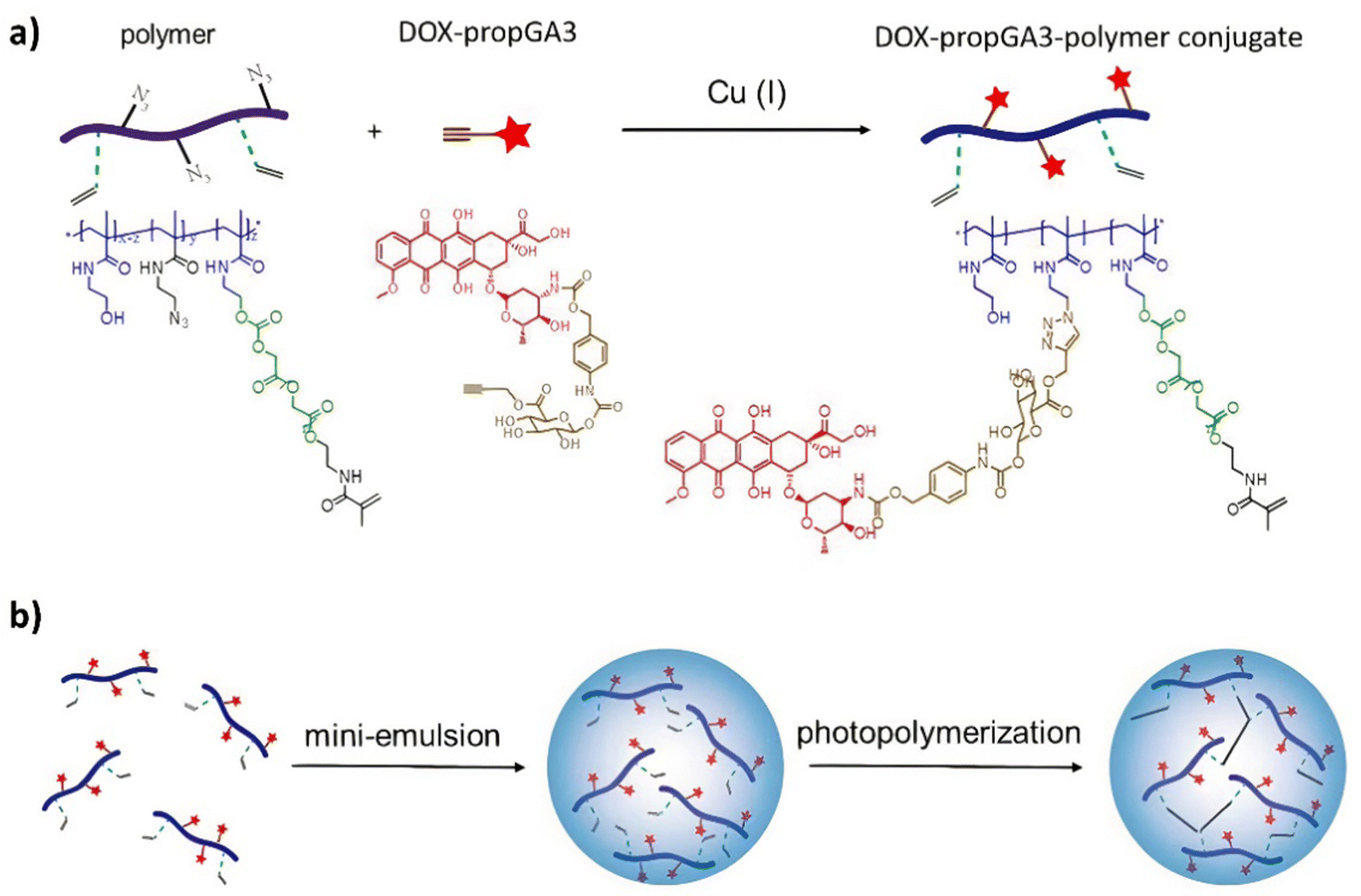 | ||
| Scheme 7 (a) The synthesis of the doxorubicin–glucuronide prodrug (DOX-propGA3)-polymer using click chemistry; (b) shows the preparation of the nanogels using inverse mini-emulsion photopolymerization. Reprinted from ref. 102 with permission from MDPI. | ||
The protein-conjugate approach was next utilised to administer another anticancer drug, monomethylauristatin E (MMAE), a highly toxic anticancer therapeutic that can only be administered when conjugated onto a monoclonal antibody (compound 38, Fig. 12). Using the same design as the previous example, this compound covalently binds to cysteinyl residues on serum albumin through Michael addition, facilitating the accumulation of the prodrug in tumours where extracellular β-glucuronidase triggers the release of the drug. Once again, it was confirmed that the parent drug was released in the presence of the glycosidase, becoming cytotoxic only after the drug was released. This prodrug had remarkable therapeutic efficacy on orthotopic triple-negative mammary and pancreatic tumours in mice, where there was a large reduction or disappearance of tumours, e.g. 50% and 33% of mice with respective tumours, were cured. Importantly, treatment with this drug delivery system did not induce any side effects.104
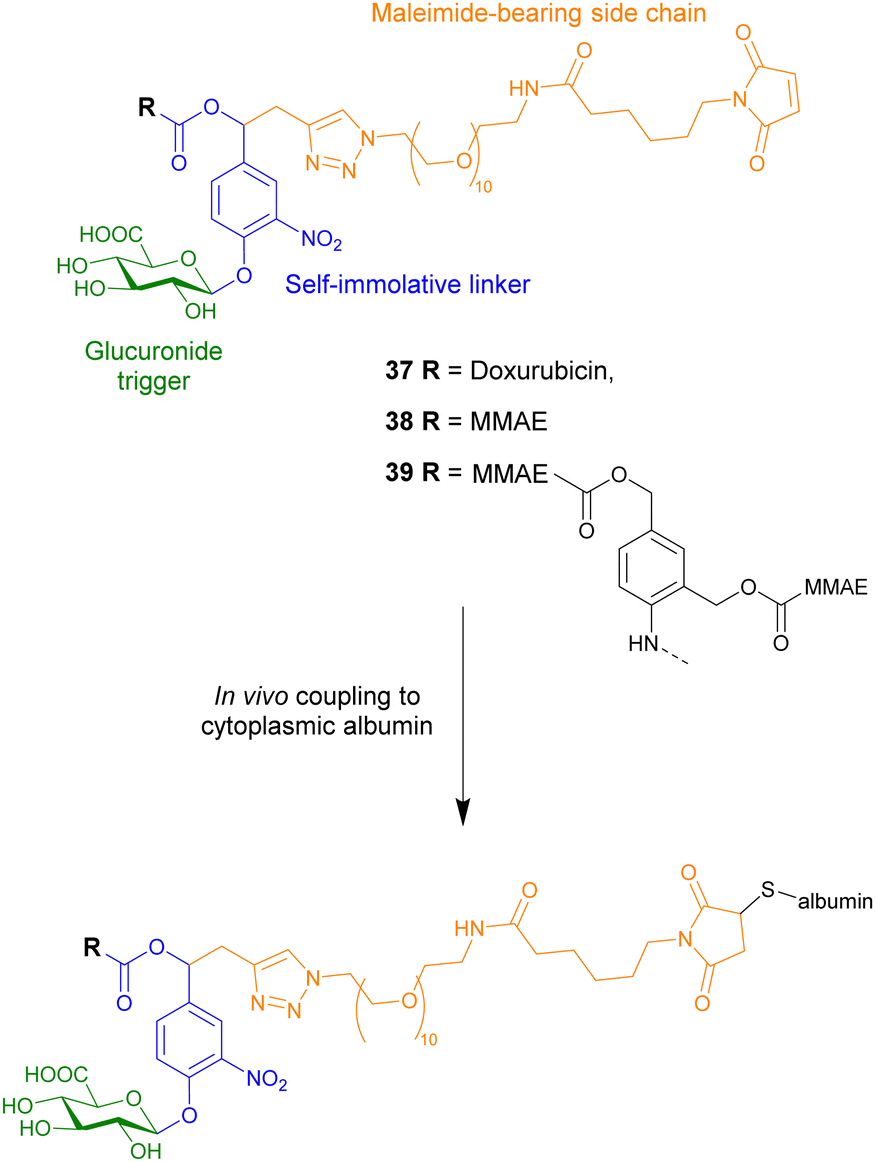 | ||
| Fig. 12 Structure of three β-glucuronidase-responsive albumin-binding prodrugs 37–39, showing how they couple to albumin. | ||
To increase the efficacy of the targeting strategy, a dimeric β-glucuronidase-responsive albumin-binding prodrug of MMAE, compound 39, was developed. Compared to the previously mentioned monomeric MMAE conjugate, the dimeric prodrug contained a chemical amplifier which upon enzymatic activation releases two molecules of the anticancer drug.105 This study represented the first in vivo studies of drug delivery systems containing these molecular constructs. The antitumour efficacy of the dimeric prodrug was tested in Balb/c athymic mice bearing subcutaneous LS174T colorectal adenocarcinoma xenografts, a model known to exhibit reduced levels of β-glucuronidase. Although the tumour volumes decreased, the dimeric prodrug did not exhibit improved efficacy compared to the monomeric prodrug 38.
To increase the potency of the monomeric conjugate 38, a triple-loaded albumin conjugate (compound 40, Fig. 13) was investigated.106 In line with previous studies, following intravenous administration, the maleimide unit of the prodrug reacted with the cysteine-34 of the plasmic albumin. The albumin conjugate then accumulated in malignant tissue where β-glucuronidase triggered the release of the drug. In this case, the enzymatic processes release three MMAE molecules into the extracellular space. The drugs then penetrate into surrounding cancer cells where they exerted their cytotoxicity. In the absence of β-glucuronidase, prodrug 40 was non-cytotoxic. However, in the presence of β-glucuronidase, prodrug 40 was more cytotoxic than the previously reported MMAE conjugate 38. This drug delivery system was then administered to mice bearing intra-pancreatic MIA PaCa-2 tumours at 2.16, 4.32 or 6.48 mg kg−1 (corresponding to 0.35, 0.7 or 1.1 μmol kg−1) per injection. The tumour volumes were recorded as a function of time by 3D echography. It was found that this compound had antitumour activity at all tested doses, without weight loss or side effects. A large reduction in tumour size was observed in animals treated at the two higher doses, with 1/8 and 3/8 mice, respectively, experiencing complete remission. When compared to the previous MMAE conjugate, the trimer 40 resulted in an 18-fold better reduction in tumour size compared to the monomer 38.106
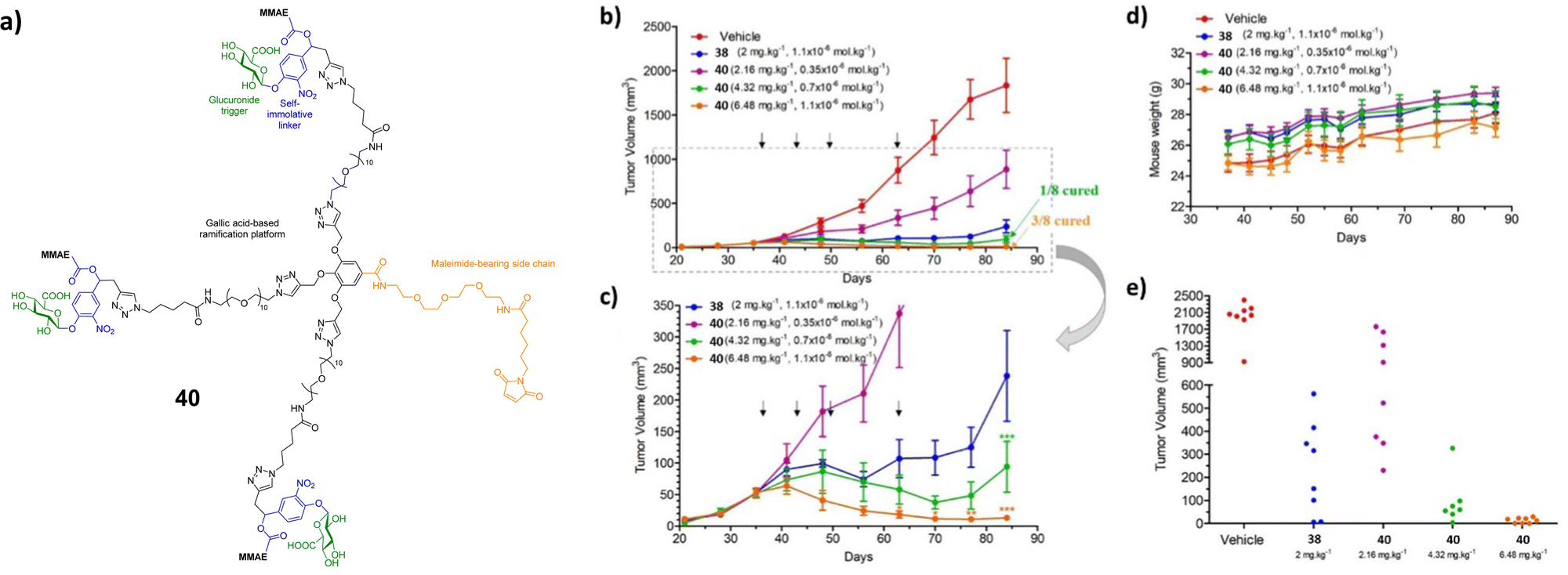 | ||
| Fig. 13 (a) Structure of the triple-loaded albumin target 40; (b) antitumour activity of the β-glucuronidase-responsive albumin-binding prodrug 40 on MIA PaCa-2 orthotopic models. MIA PaCa-2 tumour growth inhibition under therapy with vehicle, 38 (2.16, 4.32 and 6.48 mg kg−1) and 40 (2 mg kg−1). (c) MIA PaCa-2 tumour growth inhibition under therapy with 38 and 40. (d) Mean body weights of each group of mice bearing MIA PaCa-2 xenografts. (e) Tumour volumes at day 84 of mice bearing MIA PaCa-2 xenografts treated with vehicle, 38 and 40. Reprinted from ref. 106 with permission from Elsevier. | ||
Zelikin and co-workers also explored enzyme prodrug therapies by exploiting the enzyme-rich microenvironment of the tumour.107 They set out to maximize the prodrug delivery to the tumour by engineering molecular, macromolecular, and supramolecular prodrugs of the anticancer drug monomethyl auristatin E (MMAE). All prodrugs incorporated the o-substituted p-hydroxybenzyl alcohol scaffold, since it contains three suitable reactive sites at which to append the effector molecule (MMAE), the trigger for drug release (glucuronic acid) and a protraction arm. The protraction arm varied from PEG groups, such as 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE, compound 41, Fig. 14), 4-(p-iodophenyl)butyric acid (AlbuTag, compound 43), and the trityl group (Tr, compound 42). All of the reported prodrugs masked the toxicity of the parent drug, which was released upon enzymatic bioconversion. However, only one prodrug exhibited significant anticancer effects in vivo. The Tr-PEG glucuronide prodrug 42 displayed statistically significant suppression of tumour growth, while a close analogue without an albumin-binding group did not. Conversely, prodrugs containing the other albumin-binding groups (DSPE and AlbuTag, compounds 41 and 43) were not active; indicating the albumin affinity alone is not sufficient for success. Pharmacokinetic studies revealed that tumour accumulation was the only parameter that showed correlation with the in vivo anticancer effects.
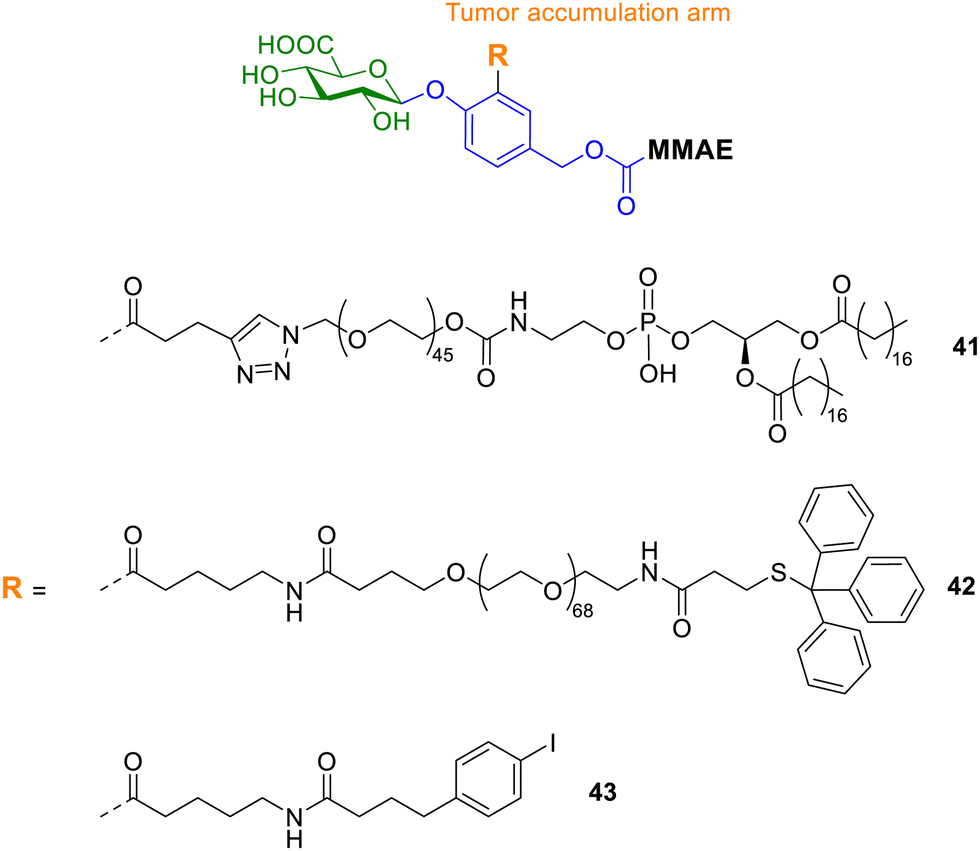 | ||
| Fig. 14 Structure of supramolecular prodrugs 30–32 containing albumin-binding moieties. | ||
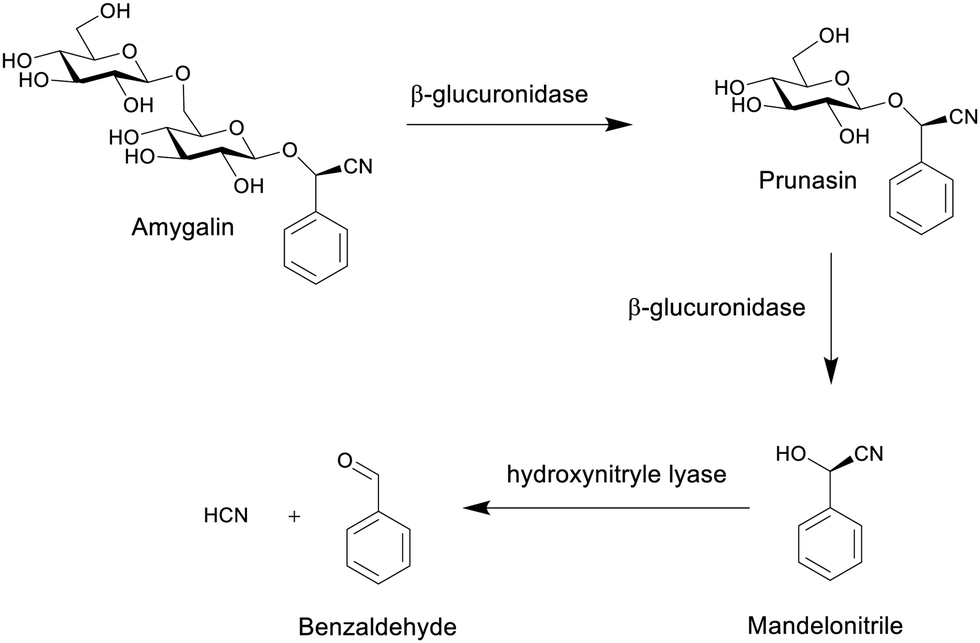 | ||
| Scheme 8 The formation of hydrogen cyanide from the hydrolysis of amygdalin.80 | ||
To improve the activation of prodrugs in vivo, Guo and co-workers developed an innovative strategy for synthesising ketal glycoside prodrugs that have dual-activation properties. Glucosyl acetone-based ketal-linked glycoside prodrugs of etoposide (ETP) 44a–d (Fig. 15a), a topoisomerase II inhibitor, underwent self-assembly into glucose-decorated nanoparticles. These could then be internalized into tumour cells containing enzyme-rich acidic organelles, where they could subsequently be activated through hydrolysis of the glycosidic linkage by glycosidase activity or through hydrolysis of the ketal linkage by acid. This triggers the self-immolative hydrolysis of the other linkage, concomitantly releasing the drug, with the formation of both glucose and acetone as by-products (Fig. 15b). Four prodrugs were reported; the two α-linked isomers 44a and b, and two β-linked isomers 44c and d to the 2′′ and 3′′-hydroxyl groups of the ETP molecule. An A549 xenograft tumour model was used to assess the intratumoural accumulation and antitumour activity of the prodrugs. It was found that the pH and β-glucosidase sensitivity depended on the anomeric nature of the prodrugs, whereby the α-linked prodrug NPs only underwent acid-activated hydrolysis, whereas the β-linked prodrugs underwent hydrolysis by both acid and β-glycosidase. In vivo biodistribution studies showed that prodrug 44c (3′′-positional β-anomeric prodrug) NPs exhibited greater tumour accumulation than free ETP. Also, the NPs were successfully hydrolysed in tumour tissue and exhibited better efficacy in reducing tumour size than free ETP, which was confirmed using hematoxylin and eosin (H&E) and Ki67 immunohistochemical staining (Fig. 15c).110
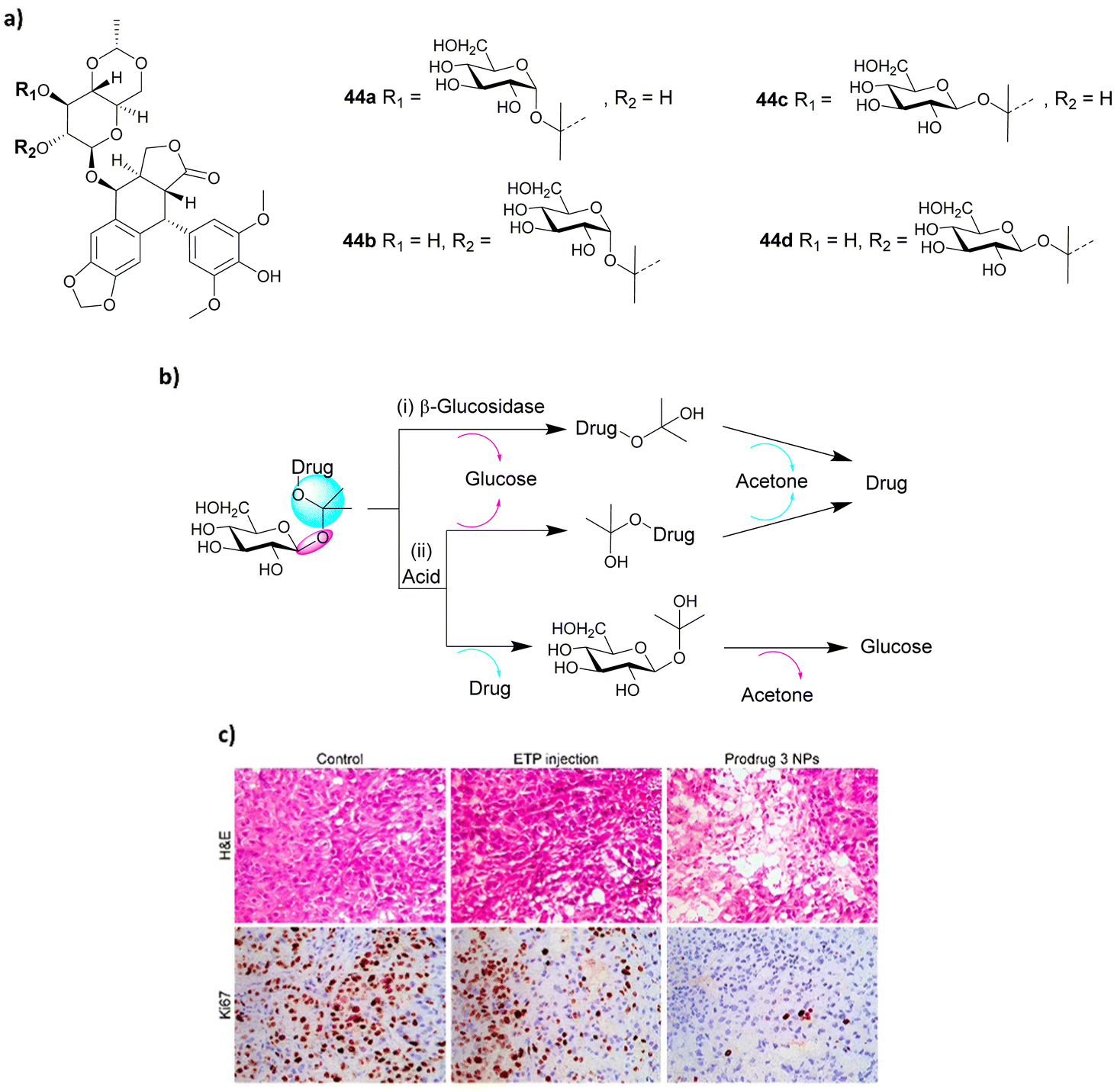 | ||
| Fig. 15 (a) Structure of four prodrugs 44a–d; (b) dual glycosidase- and acid-catalyzed self-immolative hydrolysis of ketal glycoside prodrugs; hydrolysis of the glycosidic or ketal linkage triggers hydrolysis of the other linkage, resulting in spontaneous self-immolation; (c) staining of cancer cells. Reprinted from ref. 110 with permission from the American Chemical Society. | ||
As outlined earlier in Section 2.1.2, gene-directed enzyme prodrug therapy (GDEPT) has been utilised in successful cancer treatment to reduce harmful side effects and to improve pharmacokinetics. Using this approach, cancer cells are transfected with a plasmid containing the bacterial gene lacZ, which promotes the expression of β-galactosidase (Fig. 16). Dendrimer-like mesoporous silica nanoparticles (DMSNs) were loaded with a prodrug consisting of doxorubicin conjugated to a galactose residue through a self-immolative linker (structure similar to prodrug 6). The DMSNs were then capped with a disulfide-containing polyethyleneglycol gatekeeper. It was confirmed that the prodrug remained in the nanoparticles until after the addition of glutathione (GSH), which cleaved the disulfide bonds and released the prodrug, which could then be activated by β-galactosidase. The combined treatment of β-galactosidase transfected cells and the DMSNs-prodrugs resulted in a more effective therapy than that observed for the prodrug alone.111
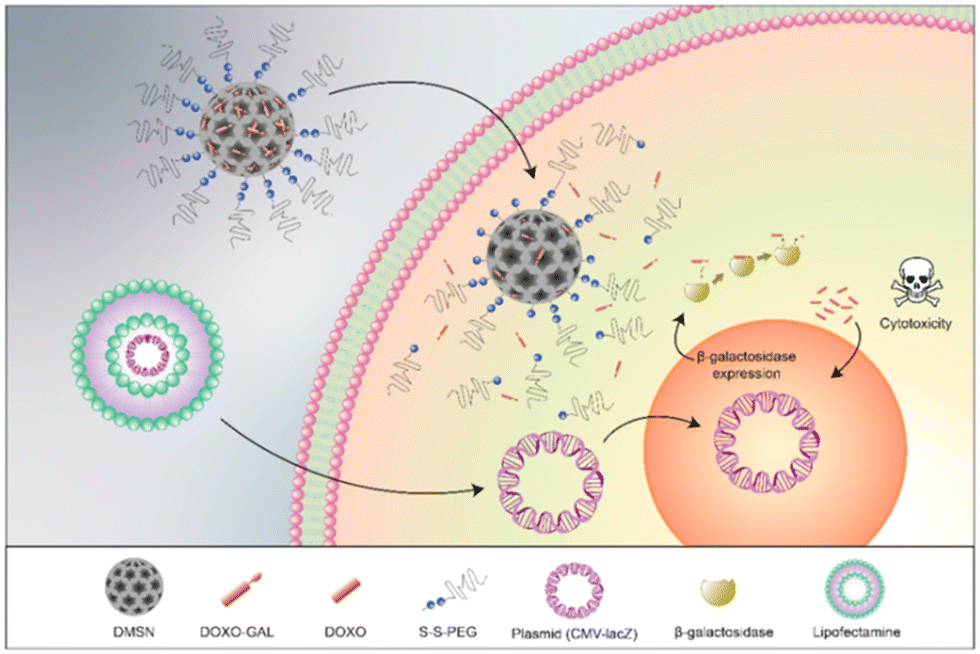 | ||
| Fig. 16 Mechanism of action of the GDEPT system. Reprinted from ref. 111 with permission from MDPI. | ||
3. Conclusion
The glycosidase activated prodrug approach has been utilised in many ways in recent years, incorporating various sugars, linkers and known anticancer drugs. The most common sugars used in this strategy are glucuronic acid and galactose residues, as it is known that their corresponding glycosidases are highly expressed in the tumour microenvironment, allowing the release of the anticancer drugs are the desired site of action. Additional functionality has been incorporated into the simple glycoconjugates, including hydrophilic linkers and targeting ligands to aid in their aqueous solubility and their selectively for cancer cells. More sophisticated approaches are now being developed that incorporate the prodrugs into supramolecular and macromolecular systems such as gels and nanoparticles. Also, to exploit the EPR effect, albumin-binding prodrugs have been developed that are retained in solid tumours, exerting their anti-cancerous effects in the desired site of action and reducing cytotoxic side effects. Herein, we have featured some select examples of such designs which demonstrate their importance and potential applications. The future direction of this area will undoubtedly continue to develop novel prodrugs incorporating more diverse anticancer agents. Utilising the ADEPT concept, more complex glycosides will also be incorporated into the prodrug design. New strategies will certainly be developed to target cancers more effectively thereby eliminating dangerous side effects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Irish Research Council (IRC) (Postdoctoral Fellowship to H. M. GOIPD/2021/186) and Science Foundation Ireland (SFI) (for SFI Centre Award for SSPC Research Centres Phase 2 to E. M. S. and T. G. 12/RC/2275_P2) for financial support and TCD for Provost Award (L. R. L).Notes and references
- World Health Organisation, Cancer, https://www.who.int/news-room/fact-sheets/detail/cancer, accessed 20/01/2022, 2022 Search PubMed.
- National Cancer Institute at the National Institutes of Health, Cancer Statistics, https://www.cancer.gov/about-cancer/understanding/statistics, accessed 20/01/2022, 2020 Search PubMed.
- S. N. Khleif, O. Rixe and R. Skeel, Skeel's Handbook of Cancer Therapy, Wolters Kluwer Health, 2016 Search PubMed.
- A. Albert, Nature, 1958, 182, 421–423 CrossRef CAS PubMed.
- V. J. Stella, W. N. A. Charman and V. H. Naringrekar, Drugs, 1985, 29, 455–473 CrossRef CAS PubMed.
- A. A. Sinkula and S. H. Yalkowsky, J. Pharm. Sci., 1975, 64, 181–210 CrossRef CAS PubMed.
- H.-K. Han and G. L. Amidon, AAPS PharmSci., 2000, 2, 48–58 CrossRef PubMed.
- B. K. Brandley and R. L. Schnaar, J. Leukocyte Biol., 1986, 40, 97–111 CrossRef CAS PubMed.
- O. Warburg, Science, 1956, 123, 309–314 CrossRef CAS PubMed.
- W. A. Denny, Cancer Invest., 2004, 22, 604–619 CrossRef CAS PubMed.
- X. Zhou, Z. Huang, H. Yang, Y. Jiang, W. Wei, Q. Li, Q. Mo and J. Liu, Biomed. Pharmacother., 2017, 91, 504–509 CrossRef CAS PubMed.
- Z. Li, D. Xu, X. Tong and C. Shan, Clin. Res. Hepatol. Gastroenterol., 2021, 45, 101456 CrossRef CAS PubMed.
- Y. Zhang, K. Zhu, X. Miao, X. Hu and T. Wang, Biomarkers, 2016, 21, 249–256 Search PubMed.
- B. Suradej, S. Pata, W. Kasinrerk and R. Cressey, Oncol. Rep., 2013, 30, 2511–2519 CrossRef CAS PubMed.
- Q. Lin, L. Pei, Z. Zhao, X. Zhuang and H. Qin, BMC Cancer, 2022, 22, 817 CrossRef CAS PubMed.
- Y. Qin, L. Zhao, X. Wang, D. Tong, C. Hoover, F. Wu, Y. Liu, L. Wang, L. Liu, L. Ni, T. Song and C. Huang, Glycobiology, 2017, 27, 306–317 CAS.
- Z. Boussadia, J. Lamberti, F. Mattei, E. Pizzi, R. Puglisi, C. Zanetti, L. Pasquini, F. Fratini, L. Fantozzi, F. Felicetti, K. Fecchi, C. Raggi, M. Sanchez, S. D’Atri, A. Carè, M. Sargiacomo and I. Parolini, J. Exp. Clin. Cancer Res., 2018, 37, 245 CrossRef PubMed.
- S. K. Chatterjee, M. Bhattacharya and J. J. Barlow, Cancer Res., 1979, 39, 1943–1951 CAS.
- R. J. Bernacki, M. J. Niedbala and W. Korytnyk, Cancer Metastasis Rev., 1985, 4, 81–101 CrossRef CAS PubMed.
- A. Krzeslak, L. Pomorski and A. Lipinska, Int. J. Mol. Med., 2010, 25, 643–648 CAS.
- C. Slawson, J. Pidala and R. Potter, Biochim. Biophys. Acta, Mol. Basis Dis., 2001, 1537, 147–157 CrossRef CAS.
- X. Li, Y. Pan, H. Chen, Y. Duan, S. Zhou, W. Wu, S. Wang and B. Liu, Anal. Chem., 2020, 92, 5772–5779 CrossRef CAS PubMed.
- J. Wagner, N. Damaschke, B. Yang, M. Truong, C. Guenther, J. McCormick, W. Huang and D. Jarrard, PLoS One, 2015, 10, e0124366 CrossRef PubMed.
- H. B. Bosmann and T. C. Hall, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 1833–1837 CrossRef CAS PubMed.
- P. Wielgat, U. Walczuk, S. Szajda, M. Bień, L. Zimnoch, Z. Mariak and K. Zwierz, J. Neuro-Oncol., 2006, 80, 243–249 CrossRef CAS PubMed.
- K. Bosslet, R. Straub, M. Blumrich, J. Czech, M. Gerken, B. Sperker, H. K. Kroemer, J. P. Gesson, M. Koch and C. Monneret, Cancer Res., 1998, 58, 1195–1201 CAS.
- N. Albin, L. Massaad, C. Toussaint, M.-C. Mathieu, J. Morizet, O. Parise, A. Gouyette and G. G. Chabot, Cancer Res., 1993, 53, 3541–3546 CAS.
- P. H. J. Houba, E. Boven, I. H. V. D. Meulen-Muileman, R. G. G. Leenders, J. W. Scheeren, H. M. Pinedo and H. J. Haisma, Br. J. Cancer, 2001, 84, 550–557 CrossRef CAS PubMed.
- B. Sperker, U. Werner, T. E. Mürdter, C. Tekkaya, P. Fritz, R. Wacke, U. Adam, M. Gerken, B. Drewelow and H. K. Kroemer, Naunyn-Schmiedeberg's Arch. Pharmacol., 2000, 362, 110–115 CrossRef CAS PubMed.
- S. Feng and J. D. Song, World J. Gastroenterol., 1997, 3, 251–252 CrossRef CAS PubMed.
- K. Fujita, M. Kamiya, T. Yoshioka, A. Ogasawara, R. Hino, R. Kojima, H. Ueo and Y. Urano, ACS Cent. Sci., 2020, 6, 2217–2227 CrossRef CAS PubMed.
- C. R. F. Silveira, M. Cipelli, C. Manzine, S. H. Rabelo-Santos, L. C. Zeferino, G. Rodríguez Rodríguez, J. B. de Assis, S. Hebster, I. Bernadinelli, F. Laginha, E. Boccardo, L. L. Villa, L. Termini and A. P. Lepique, PLoS One, 2019, 14, e0213184 CrossRef PubMed.
- C. Li, L. Guo, F. Chen, W. Yu, T. Rao and Y. Ruan, Oncol. Res. Treat., 2020, 43, 264–275 CrossRef CAS PubMed.
- S. D. Szajda, N. Waszkiewicz, A. Stypułkowska, J. Dadan and K. Zwierz, Folia Histochem. Cytobiol., 2010, 48, 351–357 CrossRef PubMed.
- E. C. Calvaresi and P. J. Hergenrother, Chem. Sci., 2013, 4, 2319–2333 RSC.
- P. Rosenbaum, C. Artaud, S. Bay, C. Ganneau, M. Campone, S. Delaloge, C. Gourmelon, D. Loirat, J. Medioni, F. Pein, M.-P. Sablin, O. Tredan, A. Varga and C. Leclerc, Cancer Immunol. Immunother., 2020, 69, 703–716 CrossRef CAS PubMed.
- S. Nishat and P. R. Andreana, Vaccines, 2016, 4, 19 CrossRef PubMed.
- E.-J. Kim, R. Kumar, A. Sharma, B. Yoon, H. M. Kim, H. Lee, K. S. Hong and J. S. Kim, Biomaterials, 2017, 122, 83–90 CrossRef CAS PubMed.
- T. Wirth, K. Schmuck, L. F. Tietze and S. A. Sieber, Angew. Chem., Int. Ed., 2012, 51, 2874–2877 CrossRef CAS PubMed.
- F. M. de Groot, E. W. Damen and H. W. Scheeren, Curr. Med. Chem., 2001, 8, 1093–1122 CrossRef CAS PubMed.
- B. Y. Lee, J. A. Han, J. S. Im, A. Morrone, K. Johung, E. C. Goodwin, W. J. Kleijer, D. DiMaio and E. S. Hwang, Aging Cell, 2006, 5, 187–195 CrossRef CAS PubMed.
- H.-W. Liu, L. Chen, C. Xu, Z. Li, H. Zhang, X.-B. Zhang and W. Tan, Chem. Soc. Rev., 2018, 47, 7140–7180 RSC.
- G. Jiang, G. Zeng, W. Zhu, Y. Li, X. Dong, G. Zhang, X. Fan, J. Wang, Y. Wu and B. Z. Tang, Chem. Commun., 2017, 53, 4505–4508 RSC.
- T.-C. Cheng, S. R. Roffler, S.-C. Tzou, K.-H. Chuang, Y.-C. Su, C.-H. Chuang, C.-H. Kao, C.-S. Chen, I. H. Harn, K.-Y. Liu, T.-L. Cheng and Y.-L. Leu, J. Am. Chem. Soc., 2012, 134, 3103–3110 CrossRef CAS PubMed.
- L. Dong, M.-Y. Zhang, H.-H. Han, Y. Zang, G.-R. Chen, J. Li, X.-P. He and S. Vidal, Chem. Sci., 2022, 13, 247–256 RSC.
- K. D. Bagshawe, Expert Rev. Anticancer Ther., 2006, 6, 1421–1431 CrossRef CAS PubMed.
- C. Fei Chin, D. Yuan Qiang Wong, R. Jothibasu and W. Han Ang, Curr. Top. Med. Chem., 2011, 11, 2602–2612 CrossRef PubMed.
- D. Gibson, ChemMedChem, 2021, 16, 2188–2191 CrossRef CAS PubMed.
- Z. Wang, Z. Deng and G. Zhu, Dalton Trans., 2019, 48, 2536–2544 RSC.
- P. L. Carl, P. K. Chakravarty and J. A. Katzenellenbogen, J. Med. Chem., 1981, 24, 479–480 CrossRef CAS PubMed.
- R. V. V. S. Edupuganti, D. A. J. Tyndall and B. A. Gamble, Recent Pat. Anti-Cancer Drug Discovery, 2021, 16, 479–497 CrossRef PubMed.
- S. Andrianomenjanahary, X. Dong, J. C. Florent, G. Gaudel, J. P. Gesson, J. C. Jacquesy, M. Koch, S. Michel, M. Mondon, C. Monneret, P. Petit, B. Renoux and F. Tillequin, Bioorg. Med. Chem. Lett., 1992, 2, 1093–1096 CrossRef CAS.
- F. L. Tietze and T. Feuerstein, Curr. Pharm. Des., 2003, 9, 2155–2175 CrossRef PubMed.
- L. F. Tietze and K. Schmuck, Curr. Pharm. Des., 2011, 17, 3527–3547 CrossRef CAS PubMed.
- L. F. Tietze and B. Krewer, Chem. Biol. Drug Des., 2009, 74, 205–211 CrossRef CAS PubMed.
- R. Adidala, H. Devalapally, C. Srivari, K. Devarakonda R and A. Raghuram Rao, Drug Dev. Ind. Pharm., 2012, 38, 1047–1053 CrossRef CAS PubMed.
- T. Legigan, J. Clarhaut, B. Renoux, I. Tranoy-Opalinski, A. Monvoisin, C. Jayle, J. Alsarraf, M. Thomas and S. Papot, Eur. J. Med. Chem., 2013, 67, 75–80 CrossRef CAS PubMed.
- Z. M. Prijovich, P.-A. Burnouf, H.-C. Chou, P.-T. Huang, K.-C. Chen, T.-L. Cheng, Y.-L. Leu and S. R. Roffler, Mol. Pharmaceutics, 2016, 13, 1242–1250 CrossRef CAS PubMed.
- T. Doura, K. Takahashi, Y. Ogra and N. Suzuki, ACS Med. Chem. Lett., 2017, 8, 211–214 CrossRef CAS PubMed.
- A. Sharma, E.-J. Kim, H. Shi, J. Y. Lee, B. G. Chung and J. S. Kim, Biomaterials, 2018, 155, 145–151 CrossRef CAS PubMed.
- J. Li, Y. Zhu, M. Xie, Q. Zhang and W. Du, Chem. Biol. Drug Des., 2019, 94, 1760–1767 CrossRef CAS PubMed.
- X. Meng, X. Lian, X. Li, Q. Ya, T. Li, Y. Zhang, Y. Yang and Y. Zhang, Carbohydr. Res., 2020, 493, 108034 CrossRef CAS PubMed.
- E. Calatrava-Pérez, L. A. Marchetti, G. J. McManus, D. M. Lynch, R. B. P. Elmes, D. C. Williams, T. Gunnlaugsson and E. M. Scanlan, Chem. – Eur. J., 2022, 28, e202103858 CrossRef PubMed.
- E. Calatrava-Pérez, S. A. Bright, S. Achermann, C. Moylan, M. O. Senge, E. B. Veale, D. C. Williams, T. Gunnlaugsson and E. M. Scanlan, Chem. Commun., 2016, 52, 13086–13089 RSC.
- J. D. Hantho, T. A. Strayer, A. E. Nielsen and R. J. Mancini, ChemMedChem, 2016, 11, 2496–2500 CrossRef CAS PubMed.
- A. J. Burt, J. D. Hantho, A. E. Nielsen and R. J. Mancini, Biochemistry, 2018, 57, 2184–2188 CrossRef CAS PubMed.
- A. T. Ryan, A. J. Pulukuri, M. Davaritouchaee, A. Abbasi, A. T. Hendricksen, L. K. Opp, A. J. Burt, A. E. Nielsen and R. J. Mancini, Acta Pharmacol. Sin., 2020, 41, 995–1004 CrossRef CAS PubMed.
- J. Cao, S. Cui, S. Li, C. Du, J. Tian, S. Wan, Z. Qian, Y. Gu, W. R. Chen and G. Wang, Cancer Res., 2013, 73, 1362–1373 CrossRef CAS PubMed.
- K. Shimoda and N. Kubota, Molecules, 2011, 16, 6769–6777 CrossRef CAS PubMed.
- Y. Mao, Y. Zhang, Z. Luo, R. Zhan, H. Xu, W. Chen and H. Huang, Molecules, 2018, 23, 3211 CrossRef PubMed.
- K. H. Carruthers, G. Metzger, M. J. During, A. Muravlev, C. Wang and E. Kocak, Cancer Gene Ther., 2014, 21, 434–440 CrossRef CAS PubMed.
- H. J. Schuster, B. Krewer, J. M. von Hof, K. Schmuck, I. Schuberth, F. Alves and L. F. Tietze, Org. Biomol. Chem., 2010, 8, 1833–1842 RSC.
- L. F. Tietze, H. J. Schuster, B. Krewer and I. Schuberth, J. Med. Chem., 2009, 52, 537–543 CrossRef CAS PubMed.
- L. F. Tietze, H. J. Schuster, K. Schmuck, I. Schuberth and F. Alves, Bioorg. Med. Chem., 2008, 16, 6312–6318 CrossRef CAS PubMed.
- L. F. Tietze, K. Schmuck, H. J. Schuster, M. Müller and I. Schuberth, Chem. – Eur. J., 2011, 17, 1922–1929 CrossRef CAS PubMed.
- K.-C. Chen, K. Schmuck, L. F. Tietze and S. R. Roffler, Mol. Pharmaceutics, 2013, 10, 1773–1782 CrossRef CAS PubMed.
- V. Oliveri, M. L. Giuffrida, G. Vecchio, C. Aiello and M. Viale, Dalton Trans., 2012, 41, 4530–4535 RSC.
- V. Oliveri, M. Viale, G. Caron, C. Aiello, R. Gangemi and G. Vecchio, Dalton Trans., 2013, 42, 2023–2034 RSC.
- Y. Ma, H. Chen, S. Su, T. Wang, C. Zhang, G. Fida, S. Cui, J. Zhao and Y. Gu, J. Cancer, 2015, 6, 658–670 CrossRef PubMed.
- G. Liu, V. Khanna, A. Kirtane, A. Grill and J. Panyam, FASEB J., 2019, 33, 9453–9465 CrossRef CAS PubMed.
- H. Ozawa-Umeta, A. Kishimoto, A. Imaizumi, T. Hashimoto, T. Asakura, H. Kakeya and M. Kanai, Cancer Sci., 2020, 111, 1785–1793 CrossRef CAS PubMed.
- L. Xu, X. Liu, Y. Li, Z. Yin, L. Jin, L. Lu, J. Qu and M. Xiao, Appl. Microbiol. Biotechnol., 2019, 103, 7997–8008 CrossRef CAS PubMed.
- L. F. Tietze, J. M. von Hof, M. Müller, B. Krewer and I. Schuberth, Angew. Chem., Int. Ed., 2010, 49, 7336–7339 CrossRef CAS PubMed.
- A. Guerrero, R. Guiho, N. Herranz, A. Uren, D. J. Withers, J. P. Martínez-Barbera, L. F. Tietze and J. Gil, Aging Cell, 2020, 19, e13133 CrossRef CAS PubMed.
- E. González-Gualda, M. Pàez-Ribes, B. Lozano-Torres, D. Macias, J. R. Wilson Iii, C. González-López, H.-L. Ou, S. Mirón-Barroso, Z. Zhang, A. Lérida-Viso, J. F. Blandez, A. Bernardos, F. Sancenón, M. Rovira, L. Fruk, C. P. Martins, M. Serrano, G. J. Doherty, R. Martínez-Máñez and D. Muñoz-Espín, Aging Cell, 2020, 19, e13142 CrossRef PubMed.
- M. Grinda, J. Clarhaut, B. Renoux, I. Tranoy-Opalinski and S. Papot, MedChemComm, 2012, 3, 68–70 RSC.
- M. Grinda, J. Clarhaut, I. Tranoy-Opalinski, B. Renoux, A. Monvoisin, L. Cronier and S. Papot, ChemMedChem, 2011, 6, 2137–2141 CrossRef CAS PubMed.
- B. Renoux, T. Legigan, S. Bensalma, C. Chadéneau, J.-M. Muller and S. Papot, Org. Biomol. Chem., 2011, 9, 8459–8464 RSC.
- A. Balbous, B. Renoux, U. Cortes, S. Milin, K. Guilloteau, T. Legigan, P. Rivet, O. Boissonnade, S. Martin, C. Tripiana, M. Wager, R. J. Bensadoun, S. Papot and L. Karayan-Tapon, Mol. Cancer Ther., 2014, 13, 2159–2169 CrossRef CAS PubMed.
- S. Bensalma, C. Chadeneau, T. Legigan, B. Renoux, A. Gaillard, M. de Boisvilliers, C. Pinet-Charvet, S. Papot and J. M. Muller, J. Mol. Neurosci., 2015, 55, 51–61 CrossRef CAS PubMed.
- M. Thomas, J. Clarhaut, P.-O. Strale, I. Tranoy-Opalinski, J. Roche and S. Papot, ChemMedChem, 2011, 6, 1006–1010 CrossRef CAS PubMed.
- T. Legigan, J. Clarhaut, I. Tranoy-Opalinski, A. Monvoisin, B. Renoux, M. Thomas, A. Le Pape, S. Lerondel and S. Papot, Angew. Chem., Int. Ed., 2012, 51, 11606–11610 CrossRef CAS PubMed.
- J. Alsarraf, E. Péraudeau, P. Poinot, I. Tranoy-Opalinski, J. Clarhaut, B. Renoux and S. Papot, Chem. Commun., 2015, 51, 15792–15795 RSC.
- M. Grinda, T. Legigan, J. Clarhaut, E. Peraudeau, I. Tranoy-Opalinski, B. Renoux, M. Thomas, F. Guilhot and S. Papot, Org. Biomol. Chem., 2013, 11, 7129–7133 RSC.
- P. López Rivas, C. Müller, C. Breunig, T. Hechler, A. Pahl, D. Arosio, L. Belvisi, L. Pignataro, A. Dal Corso and C. Gennari, Org. Biomol. Chem., 2019, 17, 4705–4710 RSC.
- S. Kolodych, C. Michel, S. Delacroix, O. Koniev, A. Ehkirch, J. Eberova, S. Cianférani, B. Renoux, W. Krezel, P. Poinot, C. D. Muller, S. Papot and A. Wagner, Eur. J. Med. Chem., 2017, 142, 376–382 CrossRef CAS PubMed.
- R. Barat, T. Legigan, I. Tranoy-Opalinski, B. Renoux, E. Péraudeau, J. Clarhaut, P. Poinot, A. E. Fernandes, V. Aucagne, D. A. Leigh and S. Papot, Chem. Sci., 2015, 6, 2608–2613 RSC.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- J. Y. Yhee, S. Son, S. Son, M. K. Joo and I. C. Kwon, in Cancer Targeted Drug Delivery: An Elusive Dream, ed. Y. H. Bae, R. J. Mrsny and K. Park, Springer New York, New York, NY, 2013, pp. 621–632 Search PubMed.
- M. Talelli, K. Morita, C. J. F. Rijcken, R. W. M. Aben, T. Lammers, H. W. Scheeren, C. F. van Nostrum, G. Storm and W. E. Hennink, Bioconjugate Chem., 2011, 22, 2519–2530 CrossRef CAS PubMed.
- Sauraj, S. U. Kumar, P. Gopinath and Y. S. Negi, Carbohydr. Polym., 2017, 157, 1442–1450 CrossRef CAS PubMed.
- H. C. Besse, Y. Chen, H. W. Scheeren, J. M. Metselaar, T. Lammers, C. T. W. Moonen, W. E. Hennink and R. Deckers, Pharmaceutics, 2020, 12, 536 CrossRef CAS PubMed.
- T. Legigan, J. Clarhaut, B. Renoux, I. Tranoy-Opalinski, A. Monvoisin, J.-M. Berjeaud, F. Guilhot and S. Papot, J. Med. Chem., 2012, 55, 4516–4520 CrossRef CAS PubMed.
- B. Renoux, F. Raes, T. Legigan, E. Péraudeau, B. Eddhif, P. Poinot, I. Tranoy-Opalinski, J. Alsarraf, O. Koniev, S. Kolodych, S. Lerondel, A. Le Pape, J. Clarhaut and S. Papot, Chem. Sci., 2017, 8, 3427–3433 RSC.
- B. Renoux, L. Fangous, C. Hötten, E. Péraudeau, B. Eddhif, P. Poinot, J. Clarhaut and S. Papot, MedChemComm, 2018, 9, 2068–2071 RSC.
- R. Châtre, J. Lange, E. Péraudeau, P. Poinot, S. Lerondel, A. Le Pape, J. Clarhaut, B. Renoux and S. Papot, J. Controlled Release, 2020, 327, 19–25 CrossRef PubMed.
- M. T. Jarlstad Olesen, R. Walther, P. P. Poier, F. Dagnæs-Hansen and A. N. Zelikin, Angew. Chem., Int. Ed., 2020, 59, 7390–7396 CrossRef CAS PubMed.
- E. Jaszczak-Wilke, Ż. Polkowska, M. Koprowski, K. Owsianik, A. E. Mitchell and P. Bałczewski, Molecules, 2021, 26, 2253 CrossRef CAS PubMed.
- J. Zhou, J. Hou, J. Rao, C. Zhou, Y. Liu and W. Gao, Int. J. Nanomed., 2020, 15, 4639–4657 CrossRef CAS PubMed.
- N. Yu, T. Liu, X. Zhang, N. Gong, T. Ji, J. Chen, X.-J. Liang, D. S. Kohane and S. Guo, Nano Lett., 2020, 20, 5465–5472 CrossRef CAS PubMed.
- V. Candela-Noguera, G. Vivo-Llorca, B. Díaz de Greñu, M. Alfonso, E. Aznar, M. Orzáez, M. D. Marcos, F. Sancenón and R. Martínez-Máñez, Nanomaterials, 2021, 11, 1298 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |