
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation, characterization, evaluation and mechanistic study of organic polymer nano-photocatalysts for solar fuel production
Mariia V.
Pavliuk
 *,
Sina
Wrede
,
Aijie
Liu
,
Andjela
Brnovic
,
Sicong
Wang
,
Martin
Axelsson
and
Haining
Tian
*
*,
Sina
Wrede
,
Aijie
Liu
,
Andjela
Brnovic
,
Sicong
Wang
,
Martin
Axelsson
and
Haining
Tian
*
Department of Chemistry, Ångström Laboratory, Uppsala University, Box 523, 75120 Uppsala, Sweden. E-mail: mariia.pavliuk@kemi.uu.se; haining.tian@kemi.uu.se
First published on 1st August 2022
Abstract
Production of renewable fuels from solar energy and abundant resourses, such as water and carbon dioxide, via photocatalytic reactions is seen as a promising strategy to adequately address the climate challenge. Photocatalytic systems based on organic polymer nanoparticles (PNPs) are seen as one avenue to transform solar energy into hydrogen and other solar fuels. Semiconducting PNPs are light-harvesting materials with exceptional optical properties, photostability, low cost and low cytotoxity, whose performance surpasses conventional organic dyes and inorganic semiconductors. This review introduces the optimization strategies for the preparation methods of PNP via cocatalyst loading and morphology tuning. We present an analysis on how the preparative methods will impact the physico-chemical properties of these materials, and thus the catalytic activity. A list of experimental techniques is presented for characterization of the physico-chemical properties (optical, morphological, electrochemical and catalytic properties) of PNPs. We provide detailed analysis of PNP photochemistry during photocatalysis with focus on the mechanistic understanding of processes of internal charge generation and transport to the catalyst. This tutorial review provides the reader with the guidelines on current strategies used to optimize PNP performance highlighting the future directions of polymer nano-photocatalysts development.
 Mariia V. Pavliuk | Mariia Pavliuk is a researcher in the Haining Tian group at Uppsala University (UU). She is a distinguished graduate of the Taras Shevchenko National University of Kyiv. She completed her PhD in Physical Chemistry at UU in 2019, before starting her postdoc on hydrogenase-based biohybrid assemblies for proton reduction. Her research interests are in photo-driven chemistry and redox catalysis. Her current work focuses on the synergy between polymeric light harvesters and biocatalysts, and their application in solar fuel production. |
 Sina Wrede | Sina Wrede is currently studying for a doctoral degree in Uppsala University, Sweden, in the group of Haining Tian. She obtained her bachelor's degree in Chemistry from Ludwig-Maximilian Universität (LMU), Germany, in 2017 and her MSc degree in chemistry for renewable energy from Uppsala University in 2019. Her current research focuses mainly on molecular devices, such as dye-sensitized photocathodes for solar energy conversion and storage with an interest on charge transport processes. |
 Aijie Liu | Dr Aijie Liu obtained her PhD in 2017 from the University of Twente, the Netherlands, under the supervision of Prof. Jeroen J. L. M. Cornelissen. Since 2018, she has been working as postdoc in Haining Tian's group. Her research interests are in the fields of (macro)molecules self-assembly and their application in photocatalysis. |
 Andjela Brnovic | Andjela Brnovic is a PhD student in Physical Chemistry at the Department of Chemistry – Ångstrom at Uppsala University. She has completed bachelor's and MSc degree in chemistry from the Faculty of Sciences in Novi Sad, Serbia. Her work focuses on mechanistic study and application of photoreduction processes of organic polymer and molecule nanoparticles. |
 Sicong Wang | Sicong Wang is currently a PhD student in Physical Chemistry at Department of Chemistry-Ångström Laboratory, Uppsala University. His research interest covers polymeric materials and their application in photocatalysis. |
 Martin Axelsson | Martin Axelsson was born in Vattholma, Sweden in 1993. He received his BS in chemistry from Uppsala University in 2017 where he then started his master's level studies before joining Haining Tian's research group for a PhD in physical chemistry. He is currently studying the intrinsic mechanisms of fuel formation reactions with organic catalysts, both in small molecules and in polymers. |
 Haining Tian | Haining Tian is an Associate Professor (Universitetslektor) and Docent at Uppsala University, Sweden, leading a research group of Molecular Devices for Artificial Photosynthesis. He obtained his PhD in Applied Chemistry at Dalian University of Technology (DUT) in 2009 and then moved to Royal Institute of Technology (KTH) as Postdoc and senior researcher until 2014. He has been awarded Göran Gustafsson Prize for young researchers (2016 small and 2020 large), Young Investigator from European Photochemistry Association (2019) and Wallenberg Academy Fellow (2019). His research interests focus on development and investigation of sustainable materials including inorganic materials, molecules and polymers for solar energy conversion and storage. |
Key learning points(1) Preparation principle and methods of obtaining polymer nano-photocatalysts for solar fuel production.(2) Techniques and methods used to characterize polymer nano-photocatalysts. (3) Techniques and methods used to investigate charge generation and charge transport in polymer nano-photocatalysts. (4) Parameters that are key for understanding and evaluating the photocatalytic performance and stability of polymer nano-photocatalysts. |
1. Introduction
With global warming being one of the main challenges of the century for humankind, finding possible solutions to make the transition from fossil fuels is now more imperative than ever. Scientists have been given the task to find new and sustainable forms of energy in order to meet the demands of a rapidly increasing worldwide energy consumption, which is expected to double by 2050.1 At the forefront of this growth lies the rise of developing countries and an increased living standard which makes finding a sustainable solution essential to achieve a socially just and equitable future.2Among all of the renewable energy sources, solar energy is considered as the most attractive because it is by far the largest exploitable source and omnipresent on our planet – it provides the earth with more energy in 1 hour than the annual worldwide energy consumption.3 However, due to its intermittent nature (both temporal and spatial), efficient storage and distribution of this energy must be made feasible, for example in the form of chemical bonds, as a solar fuel. This has motivated the development of a wide array of artificial photosynthetic (ΔG > 0) and photocatalytic (ΔG < 0) systems that upon light excitation, promote a chemical reaction which stores the energy.4 The performance of these systems relies on distinguishable functional parameters – for example, the photocatalytic device depends on surface area under optimal light conditions whereas artificial photosynthesis devices are limited by carrier mobility and mass transport or charge transfer selectivity.4
In its most fundamental way, artificial photosynthesis can be described as two half reactions that occur simultaneously where the generated charge carriers interact with a reactant (R) to produce a reduced and an oxidized product, Pred and Pox.
| R + ne− → Pred | (1) |
| R + nh+ → Pox | (2) |
In order to move the reaction forward, unfavorable reaction pathways must be prevented, such as unwanted charge recombination processes in (eqn (1) and (2)), and the potential direct reaction of Pred with Pox. While both reactions (eqn (1) and (2)) are wanted for processes such as solar water splitting, a common approach is to spatially separate the two half reactions from each other to prevent unwanted side reactions and compartmentalize the two reactions. An alternative is a non-separated system where charge-transfer selectivity is accomplished by chemical modification of the surface.4 A large portion of new materials for solar fuel production intended for the full reaction are first studied for one of the half reactions in the presence of a scavenger that takes care of the unwanted charge. Both hydrogen evolution and CO2 reduction with such a sacrificial agent in combination with a particle suspension or solution are thermodynamically downhill and can thus be considered in photocatalytic systems.4
The field of photocatalysis has long been dominated by inorganic semiconductors with wide band gaps and with limited activity in the ultraviolet (UV) region. Bearing in mind that band gaps of most inorganic semiconductors are difficult to be tuned and that UV radiation makes up only 6.6% of the solar spectrum, organic semiconductors started emerging as photocatalysts for water splitting in 1985 as earth-abundant alternatives that offer superior optical properties reaching 44.7% of the solar spectrum.5 Furthermore, facile preparation of organic photocatalysts under mild conditions and environmental friendliness make them attractive for large-scale deployment.
Although organic semiconductors are advantageous in many aspects, they still exhibit lower solar to energy conversion efficiencies compared to inorganic semiconductors. This can be explained by their key difference in charge photogeneration: whereas photoexcitation of inorganic semiconductors results in free charge carrier generation, organic semiconductors generate electrostatically bound excited electron and hole pairs (called excitons) upon photoexcitation. The explanation for this lies in the difference in environments in which the charges are generated, specifically differences in the dielectric constants. Whereas the value of the dielectric constant is high for inorganic semiconductors, meaning it has a high ability to dissociate charges; this value is low for organic semiconductors, resulting in poor charge separation and thus localized charges. Instead of Wannier-Mott model generally used for inorganic photocatalysts,6 Frenkel model more accurately describes the high exciton binding energies and small exciton Bohr radii for organic photocatalysts.7 Since the crucial step for any redox reaction is the generation of free charge carriers, organic semiconductors first need to dissociate an exciton into free charge carriers in a low dielectric medium, in order to improve their solar energy conversion efficiency.8
Nonetheless, organic semiconductors have found their place as a viable addition to inorganic semiconductors in the field of photocatalysis and have come a long way since the first organic photocatalyst for water splitting in 1985 – poly(p-phenylene) (PPP). PPP was quickly modified in the years after, giving rise to multiple variations of the polymer, however it was not very active under visible light conditions.9 It took until 2009 for a major breakthrough in the field of organic semiconductors when graphitic carbon nitride was discovered as a promising photocatalyst with a band gap of 2.7 eV.10 From then onwards, a multitude of organic semiconductors for photocatalysis have emerged, along with conjugated organic frameworks or microporous polymer network (a more in depth list in ref. 8).
π-Conjugation along the polymer backbone enables polymers to be efficient photocatalysts to carry out the desired redox reactions (Fig. 1 and 2).11 Most of these polymer photocatalysts are composed of alternating donor–acceptor monomers due to the necessary enhanced separation and charge transport across the interfacial area between interacting domains. For water splitting, however, an additional challenge has been the insolubility of most bulk polymers in water that limit the availability of active surface area. Since surface area plays a key role in photocatalysis, it is not surprising that a lot of the development of conjugated frameworks and polymer networks focuses on increasing the active area. One of the approaches to increase solubility and surface area is utilization of the inherent hydrophobicity of conjugated polymers for self-assembly to form water dispersible polymer nanoparticles abbreviated as PNP or CPN.11,12 Polymer dots (Pdots) are a subgroup of polymer nanoparticles that show particle size less than 30 nm13 or 100 nm.14
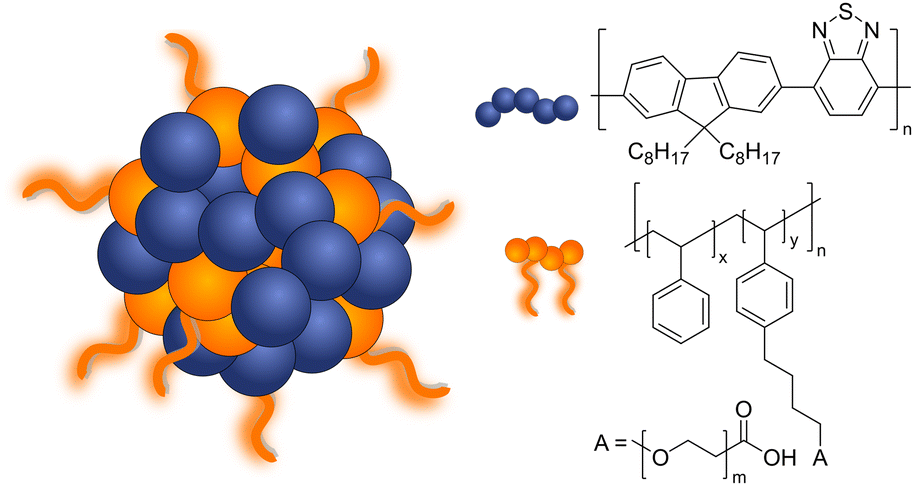 | ||
| Fig. 1 Schematic diagram of Pdots composed of PFBT (F8BT) polymer and PS-PEG-COOH co-polymer used for light-driven hydrogen evolution. | ||
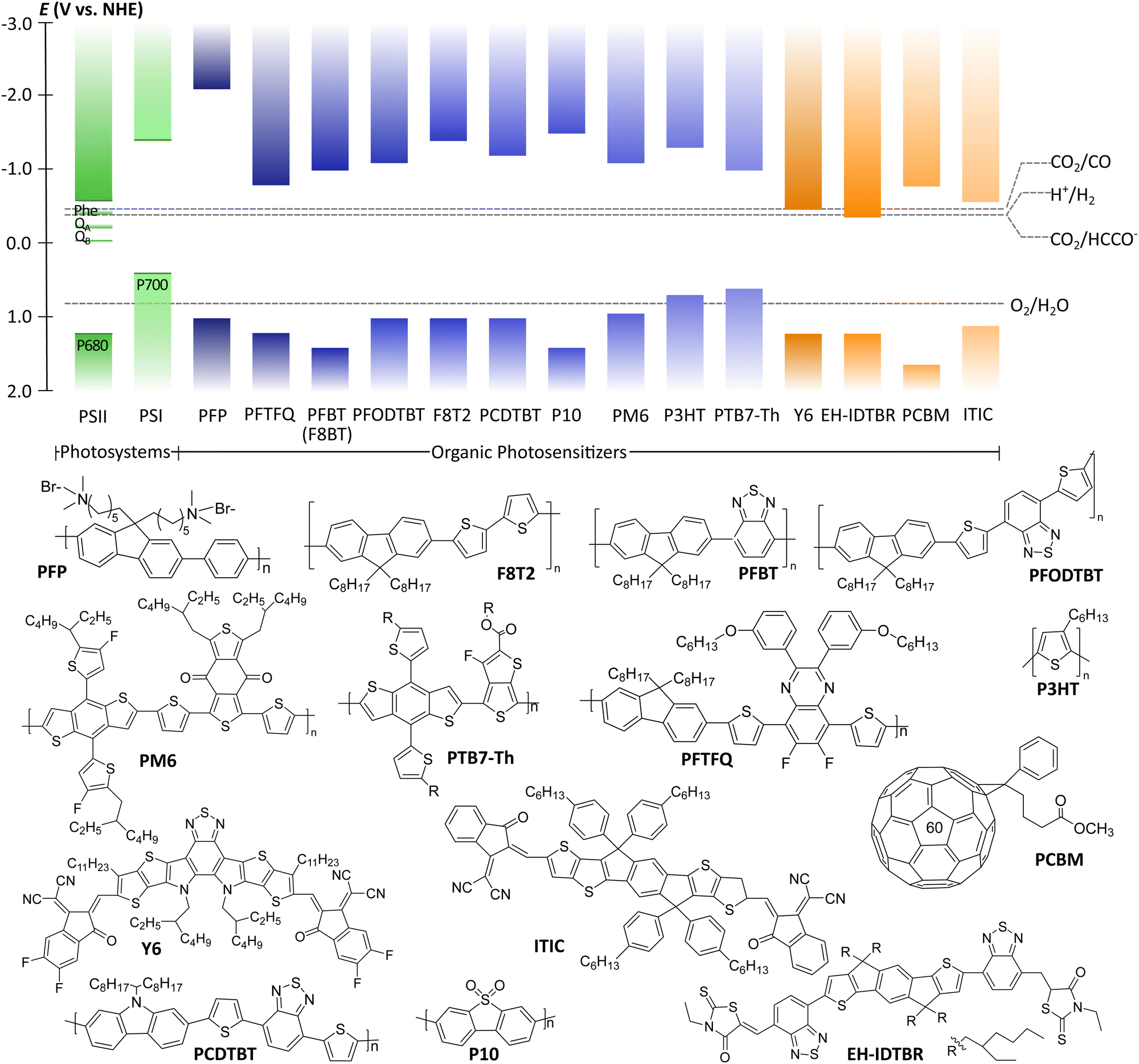 | ||
| Fig. 2 Energy band edges of natural photosystems (Pheo: pheophytin; QA: quinone A; QB: quinone B) and polymeric photocatalysts. Thermodynamic potentials of various reactions are taken at pH 7. The structures of polymeric photocatalysts are presented below the energy diagram. | ||
Pdots have first drawn a lot of attention in biomedical fields such as cell labeling, bioimaging, lymph nodes mapping, cancer phototherapy, drug delivery, and biosensing due to their advantageous fluorescent nature.15 Some of the features Pdots have shown in these fields such as non-toxicity, photoluminescence, and high photostability, are also attractive for photocatalysis. Consequently, in 2016, Pdots were first introduced as photocatalysts for visible-light-driven hydrogen generation from absolute aqueous solutions by Wang et al.11 This work adapted the PFBT Pdots prepared from nano-precipitation method to improve dispersibility of hydrophobic polymeric photocatalysts in solution (Fig. 1) and showed significant enhancement in hydrogen production (ca. 5 orders of magnitude) compared to the pristine PFBT polymer in water. Conversion of hydrophobic polymers into nanosized Pdots contributed to the increased surface area which elevated polymer–water interfacial contact.8 An additional benefit of the much smaller size was the shorter distance that photogenerated charges needed to cover in order to reach the surface. Thus, the recombination rate was suppressed and photocatalytic quantum yield was improved.11 Moreover, Pdots have proton channels in their particles which facilitates proton diffusion and enhances photocatalytic performance.16
In this tutorial review, we aim to provide an overview and a practical guide on the preparation (Section 2) and characterization of photophysical and physico-chemical properties of polymer nanoparticles, Pdots specifically, using various experimental techniques (Section 3). In Section 4 we will unveil current understandings of the mechanism of solar fuel formation using PNP as photocatalysts, highlighting strategies that can be applied to eliminate the problem of initially poor exciton dissociation typical for organic photocatalysts. Section 5 will introduce the reader with ways of accurate reporting of the hydrogen evolution rates and the overall performance of PNP during photocatalysis. Finally, Section 6 will highlight the insights into the future development of the renewable energy field, based on the current understandings of PNP.
2. Preparative methods
Polymer nanoparticles can be prepared from pre-synthesized conjugated polymers (postpolymerization method) or by direct polymerization in heterophase systems.17,18 Both of these methods have their weak and strong sides. The first method, postpolymerization, has received wider application as it allows utilization of commercially available polymers. Nanoprecipitation (or reprecipitation) and miniemulsion approaches are included in postpolymerization methods. Method of direct polymerization in heterophase systems has been suggested to offer more wide-ranging control in terms of nanoparticle size and structure. Moreover, direct polymerization method is not restricted to polymer solubility in organic solvents, but it requires harsh structure design and synthetic work. Table 1 summarizes advantages and limitations of both postpolymerization and direct polymerization methods.| Method | Advantages/drawbacks | Schematic |
|---|---|---|
| Nanoprecipitation (postpolymerisation) |
 Particle sizes are adjustable from 5 to 100 nm Particle sizes are adjustable from 5 to 100 nm |
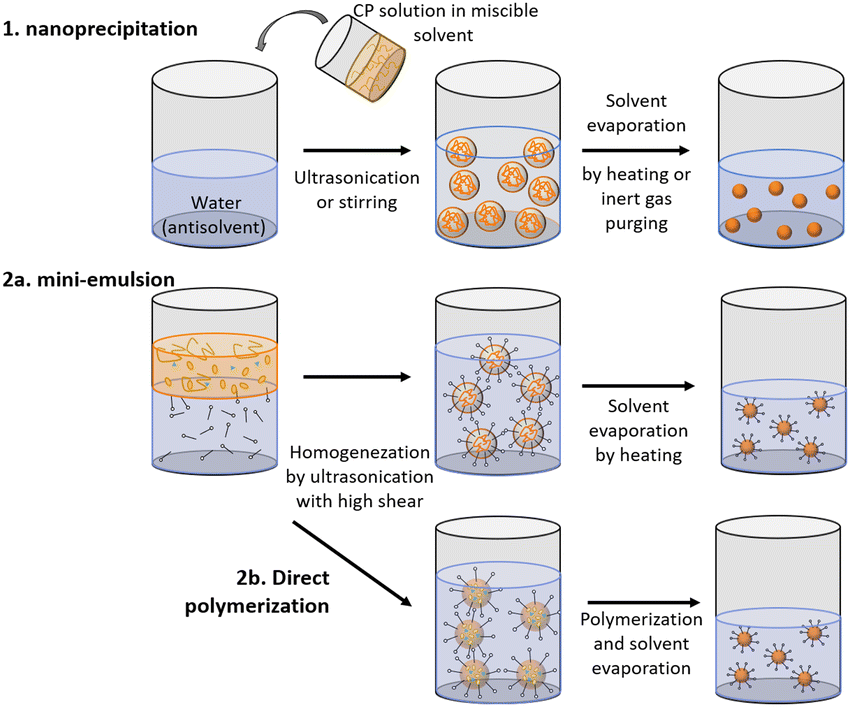
|
 Stable polymer nanoparticle solutions can be obtained only in a limited concentration range, but can be centrifuged to get concentrated solutions Stable polymer nanoparticle solutions can be obtained only in a limited concentration range, but can be centrifuged to get concentrated solutions |
||
 Polymers are required to be soluble in organic solvents which are miscible with water Polymers are required to be soluble in organic solvents which are miscible with water |
||
| Mini-emulsion (postpolymerization) |
 Organic solvents which are not miscible with water can be used Organic solvents which are not miscible with water can be used |
|
 Polymers are still required to be soluble in these organic solvents Polymers are still required to be soluble in these organic solvents |
||
| Direct polymerization |
 Only monomers are required to be soluble in solvents. This method can be used to prepare nanoparticles where the polymers are not soluble in organic solvents. Only monomers are required to be soluble in solvents. This method can be used to prepare nanoparticles where the polymers are not soluble in organic solvents. |
2.1. Postpolymerization methods
Within this strategy PNP are formed upon reprecipitation of the polymer during rapid mixing of organic solution containing polymer with aqueous solution (nanoprecipitation method) or upon solvent removal from emulsified solution droplets inside immiscible media (miniemulsion method). In both approaches, nanoparticles are often stabilized by a surfactant (Table 2).| Method | Surfactant | Sample | Solvent | Stability* (day) | Size (nm) | Ref. |
|---|---|---|---|---|---|---|
| Nanoprecipitation | PS-PEG-COOH | PFBT | THF | >30 | 30–40 | 19 |
| F8T2 | ||||||
| PFODTBT | ||||||
| PS-PEG-COOH | PFTFQ | THF | N/A | 40 | 20 | |
| PS-PEG-COOH | PFBDD | THF | N/A | 47 | 21 | |
| PFTBDD | 36 | |||||
| PFBTA | 35 | |||||
| PFTBTA | 40 | |||||
| PS-PEG-COOH | PFBT | THF | >30 | 30–50 | 11 | |
| F127 | PBQ-QF:o-IDTBR | THF | >90 | 68 | 22 | |
| Tween 80 | P3HT | THF | >60 | 23 | ||
| Mini-emulsion | SDS | PDPP5T-2:PC71BM | CHCl3 | >60 | 36–107 | 24 |
| SDS | PTNT:PC71BM | o-Xylene | N/A | 27 | 25 | |
| SDS | PDPP5T:PC61BM | CHCl3 | >10 | 34–62 | 26 | |
| BDAB | PNDI-TVT | CHCl3 | N/A | 92 | 27 | |
Polymer nanoparticles for light-driven proton reduction was firstly reported as Pdots with poly[(9,9′-dioctylfluorenyl-2,7-diyl)-co-(1,4-benzo-{2,1′,3}thiadiazole)] (PFBT or F8BT) that was stabilized by an amphiphilic copolymer PS-PEG-COOH (Fig. 1 and Table 2).11,19 A reasonable enhancement of both colloidal and photocatalytic stability was further achieved by increasing the amount of the used polymer surfactant.31 Heterojunction Pdots composed of conjugated donor polymer(s) and small molecule acceptors synthesized via nanoprecipitation method have shown dramatically enhanced photocatalytic activities,31,33 as a result of more efficient light harvesting and charge separation (for details see Section 4).31
As a special kind of nanoprecipitation, microfluidic approach helps to control the particle size and dispersity of polymer nanoparticles more effectively by tuning the total flow rate and flow rate ratio of aqueous and organic components. Hereby, homogeneous “seed”36,37 are allowed to grow through the rapid mixing of aqueous and organic phase, which in turn results in particles with smaller size and better dispersity in contrast to particles synthesized by common nanoprecipitation method. Recently, Yu et al. reported a flash nano-precipitation method for monodispersed PNP preparation which allowed to scale up the preparation of PNP for photocatalytic hydrogen production.38
Using mini-emulsion method, Hashim et al. synthesized quantum-dot-sized PNP functionalized by poly(ethylene-glycol) surfactant with mean diameters ranging between 2 and 5 nm.41 Cho et al. systematically investigated the influence of 18 different cationic, anionic and non-ionic surfactants towards their ability to generate small and uniform nanoparticles from polymers regardless of their polarity and molecular structure.27
It was found that the amount of used surfactant needs to be controlled in order to omit significant depletion forces between particles that can cause formation of large ununiform clusters. Mini-emulsion method can be also utilized to prepare multiphase nanoparticles. Following this synthetic approach, Kosco et al. synthesized PNP with photocatalytic activities that could be greatly enhanced by varying surfactants which could interact differently with the polymers that compose PNP, thus affecting the resulting particle morphology (see Section 2.4 for details).42
2.2. Direct polymerization (mini-emulsion polymerization)
The above introduced postpolymerization dispersion methods are relatively simple and straightforward, however they are restricted to usage of polymers soluble in organic solvent which can be miscible with water. Long alkyl chains are required in order to increase the solubility of conjugated polymers in organic solvents, which in turn not only complicates the difficulty in the synthetic procedure but also often influences the electronic properties of conjugated polymers. In contrast to postpolymerization methods, direct polymerization allows the utilization of polymers that are insoluble in any solvent, thus accessing wider types of applicable conjugated polymers. Within this strategy, monodispersed PNP are formed from low-molecular-weight monomers that are polymerized to sub-micrometer oil droplets in a heterophase medium. The morphology of PNP produced by direct polymerization will highly depend on the building blocks of the polymers, allowing for shape tunability, e.g. spherical, ring-shape, rod shape and irregular shape of Pdots.43PNP composed of linear polymers and conjugated microporous polymers (CMPs) that could not be processed by postpolymerization methods were shown to be synthesized via palladium-catalyzed Suzuki–Miyaura and Sonogashira–Hagihara cross-coupling polycondensation reactions in oil-in-water miniemulsions.43,44 Six-fold enhancement of photocatalytic activity was observed for nanoparticles of dibenzo[b,d]thiophene sulfone (P10-e) formed via direct polymerization method in contrast to bulk P10 polymer.45
2.3. Cocatalyst loading
In order to enhance the performance of PNP towards water splitting, cocatalysts can be introduced. The catalytic activity will be boosted due to ability of cocatalysts to: (1) reduce the activation energy or overpotential of the photocatalytic reaction; (2) provide the effective sites for substrate adsorption;42 (3) provide the efficient trap centers for charges from the photoexcited polymer dots, thus facilitating electron–hole separation,32 and (4) prolong the photocatalytic lifetime by preventing polymer photodegradation. In this section, we focus our attention on methods used for cocatalyst loadings, rather than synthetic pathways (e.g. Suzuki–Miyaura coupling, Yamamoto coupling) that result in residual “intrinsic” Pd co-catalyst.Strategies used to functionalize PNP (Table 3) with cocatalysts include (i) direct cocatalyst addition; (ii) covalent linking of the cocatalyst in the backbone of the comonomer with the following polymerization; and (iii) introduction of the metal nanoparticles by the in situ photodeposition of the corresponding precursor directly in the reaction system.
| Method | Principle of adding cocatalyst, advantages and drawbacks | Schematic |
|---|---|---|
| (a) Direct cocatalyst addition | Cocatalysts are introduced to the reaction mixture during the photocatalytic experiment. There is no/limited direct contact between light harvesting nanoparticle and cocatalyst |
|
 Simplicity Simplicity |
||
 Weak communication with the photosensitizer that generally leads to low hydrogen evolution rates Weak communication with the photosensitizer that generally leads to low hydrogen evolution rates |
||
| (b) Covalent linking | Conjugated polymer nanoparticles are presynthesized directly with the cocatalyst as a comonomer |
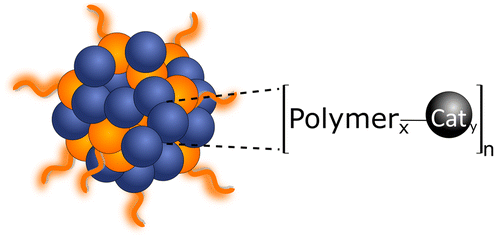
|
 Enhanced interaction between light harvesting centers and photocatalyst, thus showing higher hydrogen evolution rates Enhanced interaction between light harvesting centers and photocatalyst, thus showing higher hydrogen evolution rates |
||
 The complexity of the synthesis, where corresponding cocatalyst precursor needs to be presynthesized in advance The complexity of the synthesis, where corresponding cocatalyst precursor needs to be presynthesized in advance |
||
| (c) In situ photodeposition | Cocatalysts are photodeposited on PNP conjugated polymer nanoparticles. The reduction potential of the metal that is photodeposited needs to be more positive than the reduction potential of the polymer or if oxidation potential is more negative than the oxidation potential of the polymer |
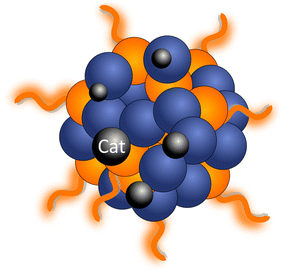
|
 (1) Facilitated electron transfer and more intimate interaction between light harvesting centers and photocatalyst, thus higher hydrogen evolution rates; (2) possibility to deposit both cocatalysts for efficient water oxidation and proton reduction (1) Facilitated electron transfer and more intimate interaction between light harvesting centers and photocatalyst, thus higher hydrogen evolution rates; (2) possibility to deposit both cocatalysts for efficient water oxidation and proton reduction |
(i) Direct cocatalyst addition. Within the first strategy (i) Ru, Pt, and Pd nanoparticles are generally used as cocatalysts due to their lowest overpotential for photocatalytic proton reduction and large work functions for trapping photogenerated charge carriers.46 Addition of excessive quantities of metal nanoparticles is known to decrease the amount of produced H2, due to the increased light scattering that prevents efficient light harvesting by the PNP.47 Molecular cocatalysts based on earth-abundant elements, have recently gained attention. Yong et al. investigated a water-soluble DuBois-type NiP catalyst with conjugated polymers reaching an activity of 429 mmol h−1 gCP−1.20 In most cases, the distances between PNP and cocatalysts were outside the effective electron-transfer radius. This lead to diffusion-controlled case, when cocatalysts needed to migrate in order to accept electrons from the polymer (undirected/randomized electron transfer). In order to minimize the electron diffusion distance, Pavliuk et al. co-adsorbed proton reduction catalyst, namely [FeFe]-hydrogenase enzyme, via suitable surface groups.48
(ii) Covalent linking of the cocatalyst. Using covalent linking method (ii) Tseng et al. prepared a series of Pdots with a presynthesized platinum complex unit as a comonomer. The resulting nanoparticles outperformed Pdots where the Pt-complex was simply blended in.21 Additionally, the same group introduced cycloplatinated nanoparticles that showed a prolonged photocatalytic reaction time with higher hydrogen production.21 Gradual increase of the Pt-complex content up to 15 mol % led to enhancement of hydrogen evolution rates (HERmax) up to 12.7 mmol h−1 g−1. Nevertheless, when the ratio of Pt-complex reached 25 mol%, HER decreased, assigned to saturation effect of the metal cocatalyst.47
(iii) Cocatalyst photodeposition. Cocatalyst photodeposition is seen as a more straightforward method, where facile electron transfer is achieved due to more intimate interaction between the PNP and the anchored cocatalyst.33,42 Hereby, kinetically feasible electron transfer to the cocatalyst occurs, if the surface of PNP has permeable layers where cocatalyst can easily go through and be deposited into. Metallic particles can be introduced as cocatalysts by photodeposition when it is thermodynamically feasible for the metal ion to be reduced/oxidized by the polymer. This is quite advantageous for designing future systems for the overall water splitting where both cocatalysts for efficient water oxidation49 and proton reduction31 will be required. Detailed conditions of the photodeposition (e.g. pH, concentration, excitation wavelength, influence of sacrificial reagents surrounding), and their impact on particle size distribution, generated metal's oxidation state and resulting photocatalytic activity can be found in ref. 50.
Within the photodeposition method (iii), the nature of surfactants and side chains on the PNP surface (their charge, shape, and structure) affect their interaction with the cocatalyst.42 In the assembly of oppositely charged subunits, the distance between polymer and cocatalyst will shorten, thus boosting the interfacial charge transfer. It has been shown that cationic polymer micelles overperform anionic ones, due to a more intimate electrostatic attraction to the cocatalyst's precursor, [PtCl4]2−, in the early stage of Pt nanoparticles formation.51 The structure of the side chains on the surface of PNP also plays a role on the interaction with the cocatalyst. For instance, Hu et al. showed enhanced charge transfer to the cocatalyst from conjugated polymer coated with oligoethyleneglycol (OEG) side chains. OEG side chains robustly interacted with Pt-cocatalyst, additionally providing adsorption sites for H+ loading, thus causing beneficial input for H2 evolution.52 The length and hydrophilicity of the side chains also affect the HERs. PNP with the longer hydrophilic OEG side chain have shown a higher HER (15.9 mmol h−1 g−1), outperforming PNP composed of the polymer without side chains, or the polymer with shorter hydrophilic side chains as well as PNP composed of the polymer with hydrophobic alkyl side chains.52 This work emphasized the key role of the polymer's side chain nature on the photocatalytic activity.
Using the photodeposition method (iii) it was possible to achieve not only efficient hydrogen evolution, but also to make water oxidation to molecular oxygen possible.49 Oxygen evolution rates (OER) obtained by Bai et al. for cobalt-loaded organic polymers were much higher than those observed for related triazine-based frameworks under similar photocatalytic conditions. By exchanging photodeposited cobalt to IrO2, the same research group provided the first example of overall water splitting utilizing organic photocatalyst.53
To summarize, higher reaction rates (for HER and OER) are generally observed for PNP with cocatalysts. However, the interaction mechanism between the interface of the polymer and cocatalyst is not yet fully understood (e.g. surface recombination, charge trapping at the interface), as well as the influence of reaction media (e.g. pH, temperature and type of used solvent). A table comparing the performance/photocatalytic conditions of PNP without and with cocatalysts loaded via methods (i)–(iii) is presented above (Table 4).
| Photocatalyst | Cocatalyst | Synthesis/cat. loading | Light source | Conditions | Activity (μmol h−1 g−1) | AQY (%) | Stability (h) | Morphology |
|---|---|---|---|---|---|---|---|---|
| Hydrogen Evolution (HER) | ||||||||
| PFP20 | Without | —/— | Xe (100 mW cm−2) 320–2500 nm | EDTA (0.1 M), pH 6 | 20 | 0.64@380 nm | 4 | — |
| PFTFQ21 | Without | N/— | LED (20 W), λ > 420 nm | Diethylamine | 1.3 | 0.04@515 nm | 6 | Spherical |
| PFBT11 | Without | N/— | LED (17 W), λ > 420 nm | Ascorbic acid (0.2 M), pH 4 | 8300 | 0.5@445 nm | 1 | — |
| PFODTBT19 | Without | N/— | LED (17 W), λ > 420 nm | Ascorbic acid (0.2 M), pH 4 | 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
0.6@550 nm | 4 | — |
| PFBT/PFODTBT/ITIC31 | Without | N/— | LED (17W), λ > 420 nm | Ascorbic acid (0.2 M), pH 4 | 1.29 | N/A | >25 | Quazi-spherical |
| 50 mW cm−2 | ||||||||
| PFP20 | DuBois-type NiP catalyst | (i) | Xe 320–2500 nm | EDTA (0.1 M), pH 6 | 429000 |
0.64 | 4 | — |
| PPV | 8000 | |||||||
| PT1 | 15000 |
|||||||
| PT2 | 10000 |
|||||||
| PT3 | 9000 | |||||||
| F8T2/ABA48 | [FeFe]-H2ase enzyme | N/(i) | LED (17 W) λ > 420 nm | 20% TEOA, pH 7 | 88460 |
1.1@405 nm | 150 | Hollow/spherical |
| PFTFQ-PtPy521 | Pt (5 mol%) | N/(ii) | LED (20 W) λ > 420 nm | Diethylamine | 4100 ± 100 | 0.02@515 nm | 12 | — |
| PFTFQ-PtPy15 | Pt (15 mol%) | 12700 ± 600 |
0.4@515 nm | |||||
| PFTFQ-PtPy25 | Pt (25 mol%) | 7700 ± 200 | 0.09@515 nm | |||||
| PFTFQ-PtIq5 | Pt (5 mol%) | 4200 ± 100 | 0.05@515 nm | |||||
| PFTFQ-PtIq5 | Pt (15 mol%) | 11100 ± 300 |
0.42@515 nm | |||||
| PFTFQ-PtIq5 | Pt (25 mol%) | 1600 ± 200 | 0.03@515 nm | |||||
| PFN-Br51 | Pt (1.5wt%) | (ii) | Xe (300 W), λ > 300 nm | TEOA | 4280 | N/A | 20 | — |
| PFNDPP-Br | Pt (3wt%) | Xe (300 W), λ > 300 nm | Ascorbic acid (0.2 M), pH 4 | 4600 | 0.12@550 nm | |||
| Pt (5wt%) | 2700 | 0.40@600 nm | ||||||
| Pt (3 wt%) | 11160 |
0.44@650 nm | ||||||
| PFBDD-PtPy54 | Pt (4.9 wt%) | N/(ii) | LED (20 W) λ > 420 nm | Triethylamine | 960 ± 90 | 0.27@420 nm | 9 | — |
| PFTBDD-PtPy | Pt (0.08 wt%) | 2600 ± 350 | ||||||
| PFBTA-PtPy | Pt (0.11 wt%) | 840 ± 50 | ||||||
| PFTBTA-PtPy | Pt (2.3 wt%) | 7340 ± 800 | ||||||
| PBDTBT-7EO52 | Pt (3 wt%) | (ii) | Xe (300 W), λ > 300 nm | Ascorbic acid (0.2 M), pH 4 | 15900 | 0.30@600 nm | N/A | — |
| PF8DTBT55 | Pt (3 wt%) | N/(iii) | Xe (300 W) λ (N/A) | Ascorbic acid (0.2 M), pH 4 | 3790 | N/A | 5 | — |
| PCDTBT/PC60BM33 | Pt (3 wt%) | N/(iii) | Xe (300 W) λ > 420 nm | Ascorbic acid (0.2 M) | 179000 |
3.72@420 nm | 18 | Mixed blend |
| Rh (3 wt%) | Ascorbic acid (0.04 M) | ∼4000 | 3.42@490 nm | |||||
| Au (3 wt%) | ∼12000 |
3.16@515 nm | ||||||
| Cu (3 wt%) | ∼1000 | 3.0@595 nm | ||||||
| PTB7-Th/EH-IDTBR42 | Pt (10 wt %) | M/(iii) | Xe (300 W) 350–800 nm | Ascorbic acid (0.2 M) | 64426 ± 7022 |
5.6@660 nm | 16 | Mixed blend/spherical |
| 6.2@700 nm | ||||||||
| PFBT/PFODTBT/ITIC31 | Pt (6 wt%) | N/(iii) | LED (17 W) λ > 420 nm | Ascorbic acid (0.2 M), pH 4 | 60800 |
6.5@550 nm | 120 | Quazi-spherical |
| 7.1@600 nm | ||||||||
| 6.1@650 nm | ||||||||
| Oxygen evolution (OER) | ||||||||
| P1049 | Co (1 wt %)s | (iii) | Xe (300 W), both full arc and λ > 420 nm | AgNO3(0.01M) La2O3 (200 mg) | 16.6 | N/A | 6 | — |
| P24 | 1.9 | |||||||
| P26 | 0.2 | |||||||
| P28 | 4.9 | |||||||
| P29 | 0.4 | |||||||
| P30 | 0.9 | |||||||
| P31 | 1.2 | |||||||
| P35 | 1.0 | |||||||
2.4. Morphology tuning.
Morphology of PNP plays a crucial role on the photophysical properties of these materials and thus influences the activity towards solar fuel production. In this section we first introduce methods used to tune the morphology of conjugated polymer nanoparticles from solid particles (discussed in Section 2.1) to mixed blend, core–shell or hollow nanoparticles (Fig. 3). In the second part we will provide examples on how morphology tuning is used to improve photocatalytic activity of polymeric light harvesters.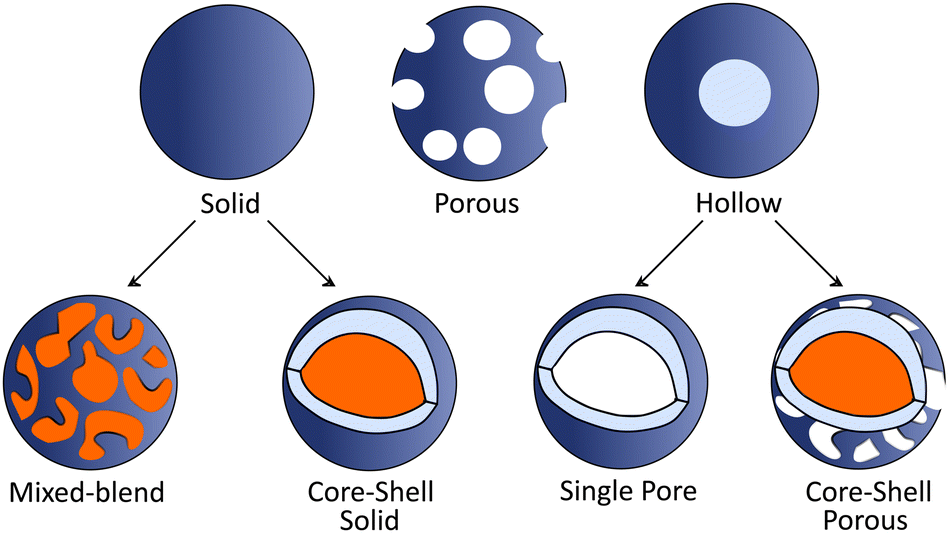 | ||
| Fig. 3 Schematic representation of various PNP morphologies. For clarification, different polymers are represented in blue and orange colors. | ||
Core–shell PNP based on polythiophene can be prepared via a two-step postfunctionalization approach. At first, P3HT nanoparticles are synthesized via nanoprecipitation method. Then, oxygenation of the exterior surface of P3HT nanoparticles with different equivalents of HOF·CH3CN results in the formation of PNP with a core–shell morphology.57 Sochor et al. investigated in detail the influence of the loading of three different but structurally similar ABA triblock copolymers on the morphology of resulting nanoparticles. This approach enabled the formation of core–shell nanoparticles with varying thickness of the shells surrounding the core.58 Furthermore, Richards et al. studied the impact of different dispersion media and synthetic conditions on the internal morphology.59 Core–shell particles based on polymers that can be interchangeably located either in the core or shell were synthesized, and their intrinsic charge generation abilities were compared.
Hollow polymer particles are spherical particles with a single pore,60 while particles with many pores are called porous polymers.61 Furthermore, hollow particles can be subdivided into two groups (Fig. 3): single pore particles and core–shell particles that have porous shell. Hollow structures can be manufactured by tuning the polymer configuration61 with utilization of copolymers or monomers (e.g. methyl methacrylate, butyl acrylate, butyl methacrylate, and vinyl acetate)62 that will internally promote formation of hollow architectures. Using methacrylate-based copolymer as a building block, Pavliuk et al. synthesized hollow Pdots with donut-like morphology following a self-assembly approach.48 Pdots with the porous polymer shell that allowed fast proton diffusion were synthesized by introducing ultrasonication in pair with argon stripping during mixing of a solvent and an anti-solvent.16
Internal morphology of PNP should be considered in future work as it also plays an important role in photocatalysis. Flexible control over nanoparticles morphology opens the opportunity for a rational design of particles with specific photophysical properties suitable for solar fuel production. Among desired properties that can be easily tuned by the morphology and are of importance for solar fuel production are (a) efficient light harvesting and charge separation; (b) surface area optimization; (c) surface functionality and side chain tuning, and (d) surface permeability to reactants, e.g. protons.
(a) Efficient light harvesting and charge separation. Structures that enable light scattering or reflection induced re-absorption inside the particle show positive impact on photocatalytic performance.45 Liu et al. designed hollow polymer vesicles, which enhanced light scattering within the nanostructure and enhanced charge separation within thin polymer membrane due to an increased interface area. This had a positive impact on the photocatalytic activity compared to the activity of analogous solid particles.16 Moreover, a precise control of the nanomorphology of particles by varying the stabilizing surfactant designed by Kosco et al. revealed that more efficient charge separation, and thus enhanced photocatalytic activity can be achieved.42 A surfactant with strong interaction to small molecule acceptor enables the formation of an intermixed donor/acceptor blend morphology with more efficient charge extraction compared to acceptor core-donor shell structures.
(b) Surface area. Particles in the nanometer range (<100 nm) with higher surface area will provide more efficient charge separation at the interface, thus eliminating the limits settled by small diffusion length of exciton in organic polymers.30,65 Five orders of magnitude enhancement towards hydrogen evolution was observed for PFBT polymer, when PFBT nanoparticles (30–50 nm), instead of PFBT polymer suspension were utilized.11
(c) Surface functionality and side chain tuning. Local environment of the polymer's active center highly affects the photocatalytic performance.66,67 High surface hydrophilicity (or water wettability) has a positive impact on proton reduction.68 There are several strategies to enhance the water permittivity, such as the introduction of water-soluble side chains,51,52,68 and use of amphiphilic surfactants.11,32,45,69,70 For instance, the introduction of more polar groups (e.g. dibenzo[b,d]thiophene sulfone or oligo(ethyleneglycol)) with finely tuned side chains showed positive impact on HERs.52,68 Moreover, surface charge and morphology play a crucial role on the interaction between conjugated polymer nanoparticles and cocatalyst as discussed earlier in Section 2.3. Suitable surface groups and donut-like morphology of Pdots facilitated encapsulation of a hydrogenase enzyme as a proton reduction catalyst. Resulting intimate interaction within the biohybrid assembly showed a positive impact on overall photocatalytic performance.48
(d) Surface permeability. Permeability of the nanoparticles surface towards reactants, e.g. proton diffusion, can also affect the photocatalytic activity. Hollow polymer vesicles16 showed advantageous permeability of surface to protons that resulted in 50 times higher hydrogen-generation rate as compared to the solid ones.
3. Characterization of polymer nanoparticles
A wide array of methods can be utilized to characterize polymer nanoparticle systems. The following section aims to provide the reader with the understanding and practical knowledge of how these steady-state methods can be used and what insights can be gained for polymer nanoparticles (Table 5).| Method | Application (advantages and challenges) | Schematic |
|---|---|---|
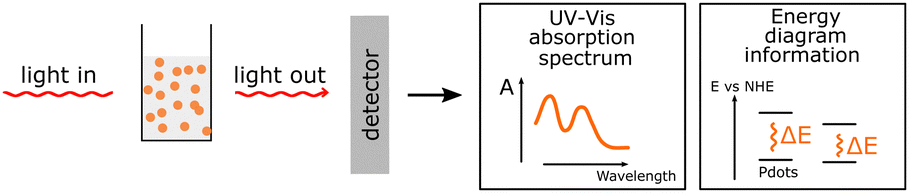
| Steady-state UV-Vis |
 UV-Vis absorption is a non-destructive method that provides information about the possible optical energy transitions in the material. It gives valuable insight necessary for energy diagrams and can be used for various applications, such as the quantification of the polymer blend inside the PNP. UV-Vis absorption is a non-destructive method that provides information about the possible optical energy transitions in the material. It gives valuable insight necessary for energy diagrams and can be used for various applications, such as the quantification of the polymer blend inside the PNP. |
|
| Steady-state PL |
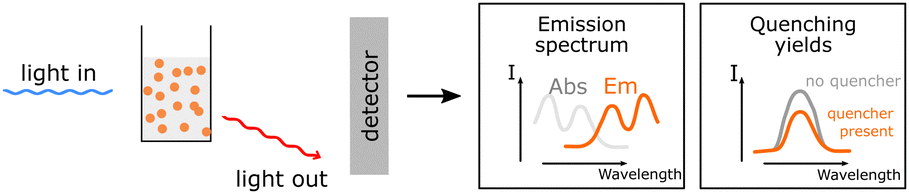
 Information about the emission of excited species. Information about the emission of excited species. |
|
 Together with UV-Vis this method is utilized to obtain the zero-zero transition energy and provide information about quenching possibilities, giving insight into possible energy transfer pathways. Together with UV-Vis this method is utilized to obtain the zero-zero transition energy and provide information about quenching possibilities, giving insight into possible energy transfer pathways. |
||
 Possible sample damage and photobleaching. Possible sample damage and photobleaching. |
||
| NMR |
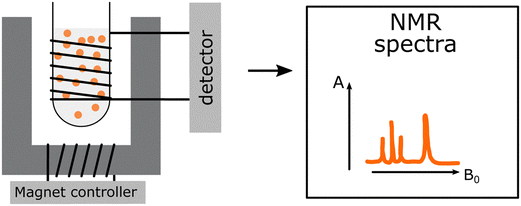
 Identification and structural assignment of monomers and polymers used for PNP preparation. Identification and structural assignment of monomers and polymers used for PNP preparation. |
|
 Characterization of functional side chains. Characterization of functional side chains. |
||
 Significant changes of PNP morphology and dispersion stability due to utilization of deuterated solvents. Significant changes of PNP morphology and dispersion stability due to utilization of deuterated solvents. |
||
| FTIR/Raman |
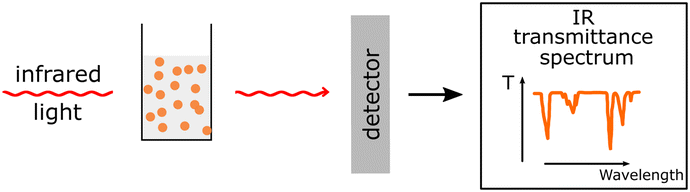
 Analysis of PNP functional groups. Analysis of PNP functional groups. |
|
 Poor spectra resolution due to strong absorption from H2O (in case of FTIR). Poor spectra resolution due to strong absorption from H2O (in case of FTIR). |
||
| ATR-FTIR71 |
 Sample damage due to heating and light source. Sample damage due to heating and light source. |
|
 Analysis of PNP functional groups. Analysis of PNP functional groups. |
||
 Well resolved spectra of aqueous PNP dispersions. Well resolved spectra of aqueous PNP dispersions. |
||
| DLS |
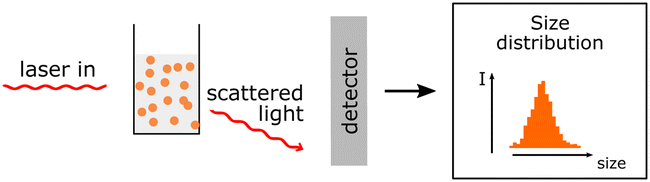
 Hydrodynamic size and polydispersity can be determined. Zeta potential measurements allow determination of the surface charge. Hydrodynamic size and polydispersity can be determined. Zeta potential measurements allow determination of the surface charge. |
|
 Actual size of PNP cannot be determined only the hydrodynamic sizes. Actual size of PNP cannot be determined only the hydrodynamic sizes. |
||
| SEM/TEM |
 Actual sizes and PNP size distribution can be determined. Actual sizes and PNP size distribution can be determined. |
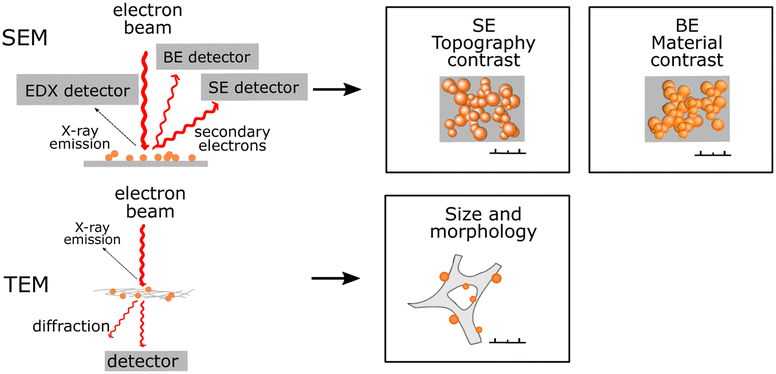
|
 Information about inner (TEM) or surface (SEM) morphology. Information about inner (TEM) or surface (SEM) morphology. |
||
 Sample beam damage. Sample beam damage. |
||
 Significant changes in PNP structure as a result of sample drying. Significant changes in PNP structure as a result of sample drying. |
||
| STEM |
 Actual sizes and morphology of PNP can be determined. Actual sizes and morphology of PNP can be determined. |
|
 Gentle conditions and slightly lower sample beam damage. Gentle conditions and slightly lower sample beam damage. |
||
 Significant changes in PNP structure as a result of sample drying. Significant changes in PNP structure as a result of sample drying. |
||
| Cryo-TEM |
 Cryo-TEM images particles in their native state as aqueous dispersions. There are no changes in PNP structure. Cryo-TEM images particles in their native state as aqueous dispersions. There are no changes in PNP structure. |
|
| E-Chem (cyclic voltammetry spectro-EChem) |
 Electrochemical methods provide information on the energy levels of the components, on the kinetics of the involved electrochemical steps and on the spectral visualization of the oxidized or reduced polymer species. Electrochemical methods provide information on the energy levels of the components, on the kinetics of the involved electrochemical steps and on the spectral visualization of the oxidized or reduced polymer species. |
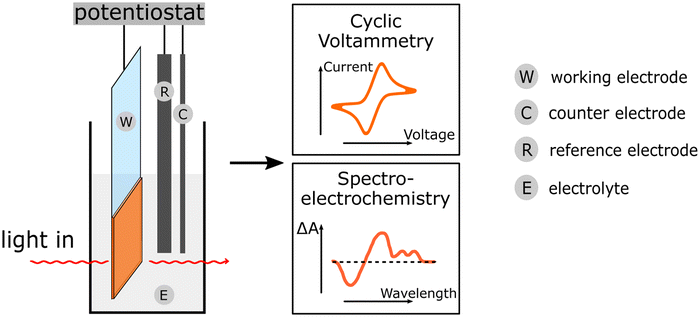
|
 Can be used for mechanistic investigations of polymer in solution. Can be used for mechanistic investigations of polymer in solution. |
||
 Narrow potential window of water that limits determination of the energy levels of the PNP components. Narrow potential window of water that limits determination of the energy levels of the PNP components. |
||
 Deposition of PNPs on electrode surface influences the E-Chem signal. Deposition of PNPs on electrode surface influences the E-Chem signal. |
||
| SANS |
 Small angle neutron scattering provides information on the internal distribution of different polymers, e.g. donor and acceptor polymer, within dispersed nanoparticles in large volume. Small angle neutron scattering provides information on the internal distribution of different polymers, e.g. donor and acceptor polymer, within dispersed nanoparticles in large volume. |
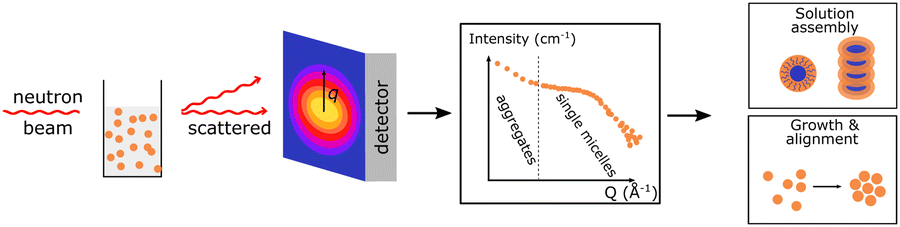
|
 It can also show the growth and assembly of polymers. It can also show the growth and assembly of polymers. |
||
 In pair with structural modelling nanoparticles size, shape and phase separation length scale can be quantified. In pair with structural modelling nanoparticles size, shape and phase separation length scale can be quantified. |
||
| ICP |
 Content of trace amounts of elements such as residual Pd from synthesis can be quantified. Content of trace amounts of elements such as residual Pd from synthesis can be quantified. |
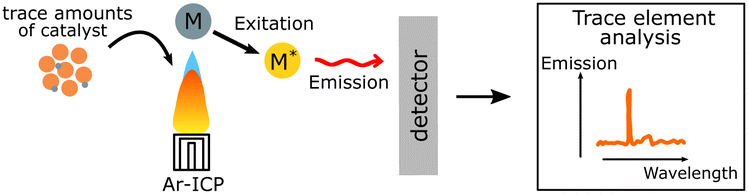
|
 Light elements cannot be analysed. Light elements cannot be analysed. |
||
| PXRD |
 Crystallinity of PNP can be probed with powder X-ray diffraction. Crystallinity of PNP can be probed with powder X-ray diffraction. |
|
 Amorphous samples cannot be analysed. Amorphous samples cannot be analysed. |
||
3.1. Methods to study optical properties
One of the first steps when it comes to identifying optical properties of PNP suitable for solar fuel formation is the analysis of steady-state ultraviolet-visible absorption (UV-Vis) and fluorescence spectra of these materials. Hereby, the optical energy gap (Eg), also recognized as zero–zero transition energy (E0–0) for molecule can be determined from the intersection wavelength (λint) of normalized absorption and emission spectra (eqn (3)).19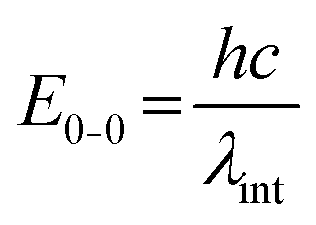 | (3) |
Electronic transitions between two energy levels, e.g. from bonding (σ, π) or non-bonding to anti-bonding (σ* or π* energy levels), with the energy difference of 200–800 nm will be visible in UV-Vis spectroscopy. For solar fuel application it is beneficial to have PNP that absorb light covering the entire UV-Vis range or even extending to the near-IR region (Fig. 4a). In contrast to inorganic materials, light harvesting properties of PNP can be easily tuned by rational polymer backbone engineering though structural and electronic optimization strategies. Absorption spectra of PNP are usually red shifted in respect to spectra of corresponding polymers in organic solvent as a result of increased inter-/intrachain interactions upon PNP formation.73 Polymer chain packing and chain interaction influence the intraparticle energy migration efficiency and overall photophysical properties of PNP.74
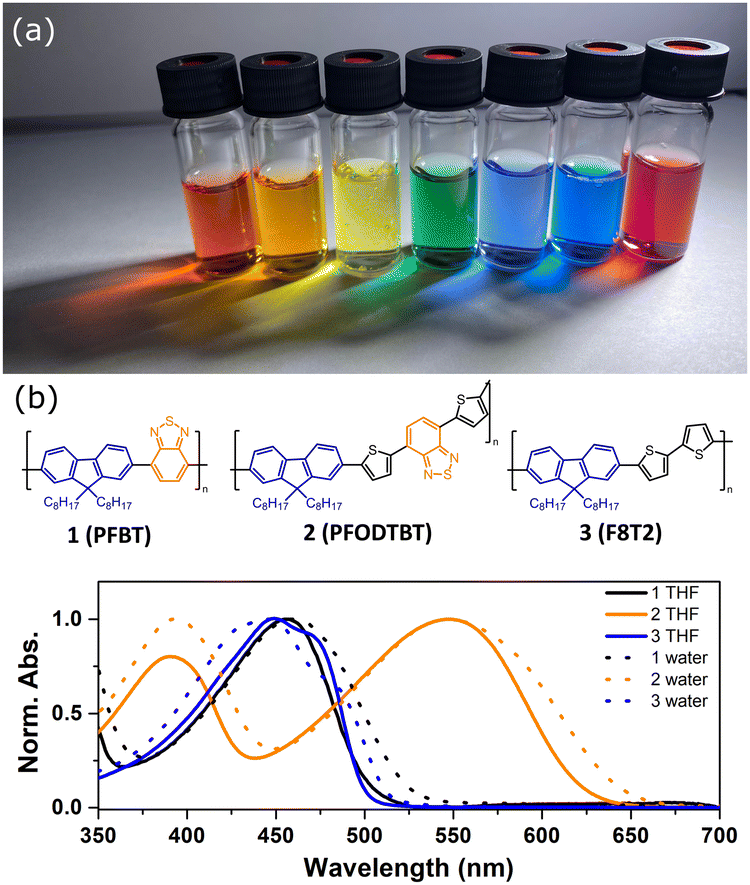 | ||
| Fig. 4 (a) Photograph of Pdots under LED light. (b) Absorption spectra of Pdots in water (dashed line) and corresponding polymers in THF (solid line). Molecular structures of utilized polymers are presented. Reproduced from ref. 19 with permission from Tian et al. and with permission from Royal Society of Chemistry, copyright 2017. | ||
Usually the final concentration of polymer inside the PNP does not match the starting concentration during PNP preparation. This occurs because samples have different degrees of precipitation during PNP formation and PNP are normally purified in the end by filtration to remove large particle aggregates. UV-Vis spectroscopy is a very helpful tool that can be used to determine the final concentration of polymers inside the PNP. At first, the PNP solutions need to be dried by rotatory evaporation or freeze-dried, and then re-dissolved in certain amount of a suitable organic solvent. As the extinction coefficient (ε) is the same for the polymer itself and polymer in PNP, concentration of polymer in PNP (c2) can be quantified by eqn (4) using Lambert–Beer law (taking into account that the path length (L) is also the same).75 Hereby, A1 and A2 are absorbances of the standard sample (polymer in THF) and the tested sample (PNP), and c1 and c2 is the concentration of the standard sample and the tested sample (in μg mL−1).
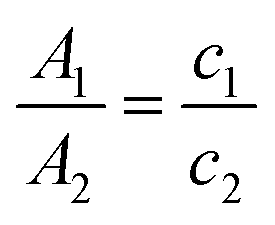 | (4) |
As mentioned in introduction, prior utilization of polymer nanoparticles in photocatalysis, PNP and Pdots specifically have been widely applied in biological fluorescence imaging due to high fluorescence brightness per volume ratio.73 The absorption of light by PNP is generally followed by the decay processes that result in emission at longer wavelengths.76,77 For PNP composed of a given conjugated polymer, nanoparticles will exhibit different emission colors.31 Similarly to absorption spectra, the emission spectra of PNP are additionally red-shifted in respect to the emission spectra of composite polymers in organic solvent due to enhanced inter- and intrachain interactions.
Combination of steady state UV-Vis absorption and photoluminescence methods can be used to investigate preliminary charge and energy transfer steps in different types of heterojunction PNPs.78 Based on energy level alignment of donor and acceptor polymers that are blended inside one PNP, the following types of heterojunction PNP are differentiated (Fig. 5): PNP with a straddling gap (type I), PNP with a staggered gap (type II, that has similar energy level alignment as the Z-Scheme), and PNP with a broken gap (type III). In some cases, it is preferable to perform complete water splitting on systems, where two PNPs are mixed together, PNP A and PNP B (Fig. 5b), for reduction and oxidation reactions respectively. In this case charge transfer would occur via the Z-Scheme with and without the Redox shuttle. In a Z-Scheme without the redox mediator the direct electron transfer between two PNPs remains challenging because of large distances between interacting PNPs.31 Upon excitation at the maximum absorption wavelength of one of the polymers in the PNP composition, fluorescence can be probed. Furthermore, by plotting the emission intensity determined at various concentration of either donor or acceptor polymer versus the emission intensity of individual PNP composed of one of the polymers, the quenching yield can be determined. High quenching yields are indicative for efficient charge separation or energy transfer within the blended polymers. It is suggested that smaller particles will have more efficient energy transfer to various fluorescence quenching sites. Recently Liu et al. observed Förster resonance energy transfer (FRET) for blended Pdots composed of different polymers with overlapped emission and absorption spectra.31 Energy transfer was further supported by the overlapping excitation spectra with the absorption spectra of binary Pdots. If polymers were not blended within one Pdot, energy transfer did not occur as the distance between interacting polymers was out of the Förster range. However, detailed mechanism of possible charge and/or energy transfer need to be confirmed with a detailed analysis of combined transient spectroscopy methods and spectroelectrochemistry (see Section 4 for details).
 | ||
| Fig. 5 (a) Schematic representation of different PNP types based on energy level alignment of donor and acceptor that are blended inside one PNP (here conjugated polymer semiconductors or molecular donors/acceptors units can be used for PNP construction). Red arrows highlight the possible charge transfer pathways. (b) Z-scheme heterojunction mechanism for full reactions based on different PNP particles with/without redox mediator. | ||
3.2. PNP structural changes
Fourier-transform infrared spectroscopy (FTIR), Raman and nuclear magnetic resonance spectroscopy (NMR) can be applied to characterize the polymer nanoparticles, specifically to analyze the PNPs functional groups and involved structural changes. Currently PNP functionalization studies by means of NMR, and FTIR are largely represented in the field of drug delivery, while there are only a few reports with application of these techniques in PNP photocatalysis.20,48,791H NMR and 13C NMR are typically used for identification and structural assignment of monomers and polymers that are used for polymer nanoparticles preparation.20,79 Successful synthesis of PNP was confirmed by the presence of characteristic peaks from monomers in 1H NMR and 13C NMR spectra, by the broadened aromatic hydrogen peaks and by the disappearance of the characteristic peaks from coupling units in the monomers.79,80 Using 1H NMR Wang et al. characterized the conjugated polymers with different length side chains that were later used for PNP synthesis via nanoprecipitation approach.81 Pavliuk et al. utilized 1H NMR to characterize the functional side chains of tertiary amine-terminated groups that were grafted to facilitate the electrostatic interaction with the catalyst during photocatalysis. It was shown that both ethyl and methyl protons signals from (–O–(CH2)2–N(CH3)2) groups were successfully grafted to the polymers side chains.48
FTIR is a qualitative and quantitative technique that allows characterization of major polymer-based compounds that absorb IR light. Among advantages of FTIR, is the distribution of a large number of characteristic absorption bands with a limited overlap over a wide spectral range. While in most cases FTIR spectra are recorded for solid samples (as KBr discs), it is preferable to characterize polymer nanoparticles in their native state as aqueous dispersions. Recently, Pavliuk et al. reported well resolved attenuated total reflection ATR-FTIR spectra of aqueous Pdots solutions.48 To achieve fine spectral resolution of Pdots in their unperturbed state a custom-made gas titration cell was utilized.71 In this case, Pdots solutions were at first concentrated to remove ∼60% of water under the stream of dry N2 gas, and then ATR-FTIR spectra were recorded for the rehydrated samples by purging “wet” aerosol that formed hydroscopic Pdots films.
Combination of NMR with FTIR is generally applied to more explicitly characterize the functionalization of polymer nanoparticles. Lin et al. have shown that amphiphilic PNP contained both hydroxyl and carboxyl functional groups by means of FTIR, while 1H NMR provided evidence of PNP functionalization with cyclohexyl group and phenyl ring moieties.82
Stimulated Raman scattering (SRS) is a complementary to FTIR non-linear vibrational technique that offers up to 108 excitation efficiency enhancement. In contrast to surface enhanced Raman scattering, SRS does not rely on metallic nanostructures, and thus is more applicable for PNP characterization. Incorporation of vibrational labels results in formation of Raman-active polymer nanoparticles that can be studied by SRS.19,83
3.3. Methods to study morphology
The morphology of PNP has a strong impact on their photocatalytic performance (as mentioned in Section 2.4.2): for a polymeric photocatalyst, particles with higher surface area are expected to result in higher HERs. Hereby methods that allow detailed characterization of size, surface charge and shape of Pdots, with a focus on both advantages and limitations of selected techniques are introduced.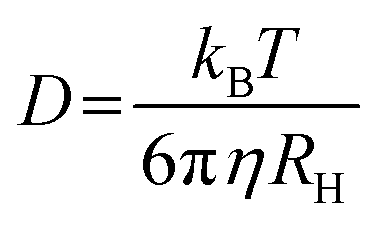 | (5) |
The size of PNP is determined in the presence of solvent molecules such as water that interacts with PNP through a variety of non-covalent interactions. As a result, DLS provides information on hydrodynamic radii which includes nanoparticles with solvent molecules attached or adsorbed on the surface, rather than the actual size of PNP.82 As size-dependent hydrogen evolution has been proven to be an important parameter that influences the photocatalytic activity, knowing the size of nanoparticles is particularly useful information when it comes to comparing different PNP systems.16,32
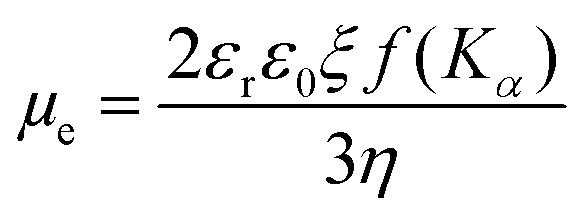 | (6) |
Zeta potential values can be correlated to the stability of PNP dispersions. Zeta potentials above +30 mV and below −30 mV indicate that particles strongly repel each other and, accordingly, that PNP dispersion is stable.
Hu et al. used zeta potential measurements in order to investigate the interactions between conjugated polyelectrolytes and Pt co-catalysts.51 It was shown that net electric charge of Pt co-catalysts is negative and therefore the interaction between conjugated polyelectrolytes with net positive charge will be stronger. Furthermore, Pavliuk et al. used zeta potential studies to show that efficient interaction is achieved when the surface of the Pdots is positively charged while the surface of cocatalyst (hydrogenase enzyme) is negatively charged.48
Energy dispersive X-ray analysis of PNP can be useful in identifying certain polymer subunits,25 or detect element traces that have a unique element signature. In combination with SEM, EDX mapping can additionally be used to locate the distribution of certain elements in the SEM image. SEM poses challenges due to sample drying prior the measurements, thus PNP samples may have a significantly different morphology from that observed with SEM.
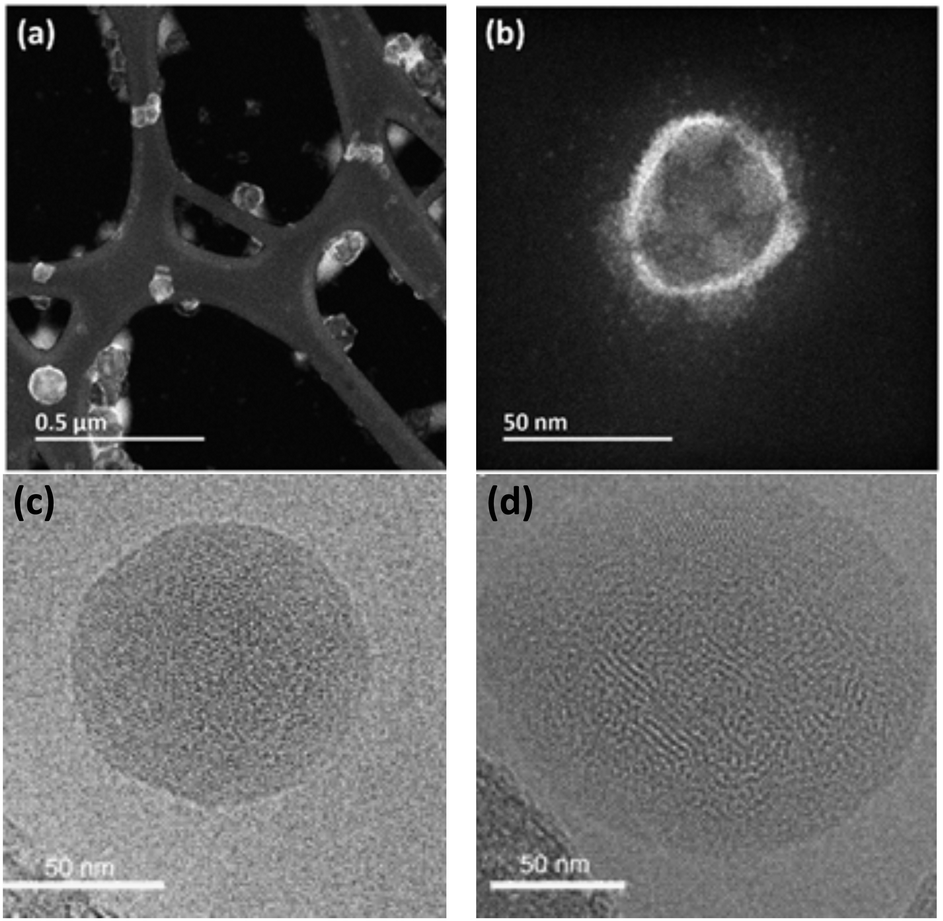 | ||
| Fig. 6 HAADF-STEM image with low magnification (a) and high magnification (b) that highlights the hollow morphology of PFODTBT Pdots. Reproduced from ref. 16 with permission from Tian et al. and with permission from Royal Society of Chemistry, copyright 2019. Bright-field cryo-TEM images of nanoparticles composed of PTB7-Th and EH-IDTBR polymers with the ratio of 30:70 that have been synthesized using SDS (c, crystalline core, amorphous shell morphology) or TEBS (d, mixed blend morphology with distributed blend of crystalline and amorphous domains) surfactants. Reproduced from ref. 42 with permission from Nature Research, copyright 2020. | ||
3.3.6.1 Scanning transmission X-ray microscopy (STXM). Among the techniques that can study the distribution of polymers inside the nanoparticle volume is scanning transmission X-ray microscopy. This becomes feasible when polymers blended within a single PNP exhibit a difference in the absorption cross section of soft X-rays. Using scanning transmission X-ray microscopy, Holmes et. al. identified a core–shell morphology of single nanoparticles with internal fullerene-rich regions and external P3HT shell.90 In spite of its ability to characterize a defined number of single nanoparticles, STXM cannot be applied for the analysis of multiple dispersed particles.
3.3.6.1. Small angle neutron scattering (SNAS). can be used to determine the internal distribution of different polymers, e.g. donor and acceptor polymers, within dispersed nanoparticles in a large probed volume. These findings can be further utilized to gain insight into the internal structure of the PNP and draw parallels with intrinsic ability to separate charges.59 If polymers blended inside one particle cannot be distinguished due to insufficient contrast, partial or complete deuteration of one component can be applied. This would preserve the chemical identity of either component, while additionally allowing to distinguish the internal structure. Moreover, the fundamental scattering body for SANS is the nucleus that enables to differentiate between the scattering length density of polymers and most common solvents, in contrast to small angle X-ray scattering (SAXS). Together with structural modeling, nanoparticle size, shape, composition and phase-separation length scale can be quantified. In this case measured scattering intensity (I(Q), eqn (7)) is defined as a function of form and structure factors (F(Q) and S(Q) respectively) as well as size distribution function (Δ(Q), for details on model fitting we encourage the reader to check ref. 58.
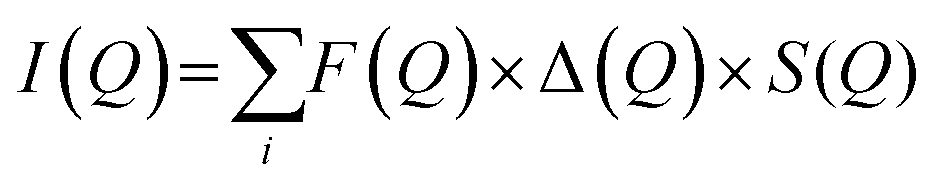 | (7) |
Richards et al. used contrast variation SANS to identify the internal structure of various composite nanoparticles formed by manipulation of the processing conditions.59 Particles with core–shell, eccentric morphology and particles with uniformly blended polymers were identified. Detailed analysis of SANS data together with simulations allowed to corelate structure of particles with intrinsic performance characteristics (e.g. charge extraction and recombination rates). Zheng et al. studied the formation of various supramolecular structures of D–A polymer in solution trying to understand the impact of microstructure on charge-transport properties. Hereby, SANS was used to discern between different 1D structures (rod, wire and fiber) and 2D lamellar structures formed upon dissolving the D–A polymer in good and bad solvents respectively. It was shown that particles with better interaggregate connection, such as 1D structures, had higher carrier mobility, in contrast to 2D structures with hindered carrier migration.91 Sochor et al. used SANS to distinguish between particles with different number of shells around the core, as well between formation of larger structures (aggregates) or single particles.58 Among examples of SANS application for solar fuel research is the recent work from Kosco et al.42 Hereby, SANS was used to verify the formation of mixed blend nanoparticles (with TEBS surfactant) and core–shell nanoparticles (with SDS surfactant) as suggested earlier by cryo-TEM studies.42
3.4. Other methods
In a standard electrochemical experiment, a three-electrode cell is composed of reference electrode, counter electrode and working electrode. In practice, polymers or PNP can be measured both in solution or alternatively, attached to a surface, such as FTO glass. During cyclic voltammetry, the potential is swept with a constant rate and the resulting current at the working electrode is measured. The observed current arises due to electron transfer reactions at the electrode which allows for the determination of the reduction (E(P−/P)) and oxidation (E(P+/P)) potentials of PNP or polymers. In most cases, the reduction and oxidation potentials are directly taken as LUMO and HOMO or conduction band (CB) and valence badn (VB) of the polymers, respectively.78,92
Another way is to use these potentials to calculate the energy levels of the corresponding excited states e.g., E(P−/P*) and E(P*/P+) which can be used to evaluate different photochemical reactions such as oxidation and reduction.31 The energy level of the excited species (eqn (8) and (9)) can be calculated by subtracting or adding the energy of the 0–0 transition energy (E0–0) between the lowest vibrational levels in the ground and excited states to/from the reduction or oxidation potential of the polymer in the ground state respectively.20,48
| E(P−/P*) = E(P−/P) + E0–0 | (8) |
| E(P*/P+) = E(P/P+) − E0–0 | (9) |
These data are particularly useful once there are different possible charge transfer pathways happening in a photocatalytic system. For example, if the photocatalyst first gives electron away from the excited state (oxidative quenching), the potentials of E(P−/P*) and E(P−/P) can be used to evaluate the charge transfer feasibility. In another way round, E(P*/P+) and E(P/P+) are more useful.
Often the reduction and oxidation peaks of the polymers or PNP are not reversible and also the potentials are different in various test conditions (such as solid or dispersed samples) which create messy data reported. How and where (peak, half-wave or foot-wave) to choose the potential value from CV measurement are not consistent in publications. Most important is to keep a consistent way in each study to evaluate potentials, particularly for comparison of potentials of different polymers in the study.
In pair with cyclic voltammetry, chronoamperometry methods are normally utilized, namely controlled-potential (bulk) electrolysis. During bulk electrolysis, the current is measured over time while a constant potential is applied, allowing for a stepwise oxidation/reduction between the oxidation states. Both cyclic voltammetry and chronoamperometry can be easily combined with different spectroscopic methods, e.g. ultraviolet-visible spectroscopy or infrared spectroscopy, which gives rise to the field of spectroelectrochemistry. This technique allows to obtain spectra for the generated oxidized or reduced species, providing necessary information for transient spectroscopy methods.
An example of complementary studies between spectroelectrochemistry and transient absorption data is shown in the work by Liu et al. on ternary Pdots.31 In order to understand how the charge transfer occurs in the ternary system, spectroelectrochemistry of acceptor polymer (ITIC) was carried out to characterize the absorption of reduced ITIC molecule (ITIC˙−) which shows a characteristic absorption between 440–510 nm. The data from transient absorption spectrum (TAS) showed negative absorption at 400 nm and 525 nm which can be assigned to the ground state bleach of oxidized donor polymer (PFODTBT+˙) and the positive absorption between 440–510 nm which matches well with the absorption spectrum of ITIC˙−. Combining spectroelectrochemical and TAS data (Fig. 9b), suggested that the formation of ITIC˙− was caused by a hole injection from excited ITIC to excited PFODTBT.
Inductively coupled plasma-mass spectroscopy (ICP-MS) is a widely used technique that allows quantification of trace amounts of metal elements lower to 1 ppm. In this technique, the tested liquid sample is ionized into atomic ions by inductively coupled plasma which are further detected by mass spectroscopy, where the signal intensity is proportional to the concentration of the studied element.
For instance, with the help of ICP, Kosco et al. showed that residual Pd even at very low concentrations (Pd < 40 ppm) had significant impact on photocatalytic hydrogen evolution. Nearly linear dependence of the hydrogen evolution rate of F8BT nanoparticles versus Pd concentration was observed up to 100 ppm of Pd, when system reached the saturation point.32 In their followed-up work Sachs et al. investigated the effect of Pd concentration on exciton quenching and relaxation pathways to the ground state in different polymer nanoparticles, which helped to further understand the role of the residual metal in photocatalytic process.30
| nλ = 2dsinθ | (10) |
4. Mechanistic study of polymer nanoparticles during photocatalysis
PNP have their own toolbox that includes several key methods used (1) to understand the internal photophysical processes and their kinetics within the PNP, (2) to prove or disprove a suggested mechanism or theoretical model that describes the involved charge transfer processes, and (3) to identify routes for future PNP design and research direction. In addition to steady-state spectroscopic methods, time-dependent studies such as transient absorption and photoluminescence spectroscopies are among the techniques that are generally used for mechanistic studies (Table 6). In this section, some examples of the applications using these techniques are introduced to understand the photochemistry of PNP during photocatalysis.| Technique | Application | Schematic |
|---|---|---|
| Time-resolved absorption spectroscopy (TAS) | (1) Examination of polymer's excited-state dynamics |
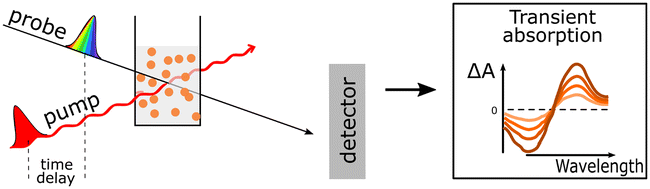
|
| (2) Monitoring the formation and identification of new reaction intermediates (e.g. oxidized/reduced polymer, oxidized/reduced catalysts, charge-separated states) in real time from fs to s | ||
| (3) Determination of the exciton diffusion length via singlet exciton–singlet exciton annihilation experiments by analysing TAS data obtained at different excitation fluences. | ||
| (4) Probing the impact of particle morphology on charge generation dynamics | ||
| (5) Probing charge accumulation under operando catalytic conditions with the help of photoinduced absorption spectroscopy | ||
| Time-resolved photoluminescence techniques | (1) Determination of the radiative electron lifetimes in the excited state |
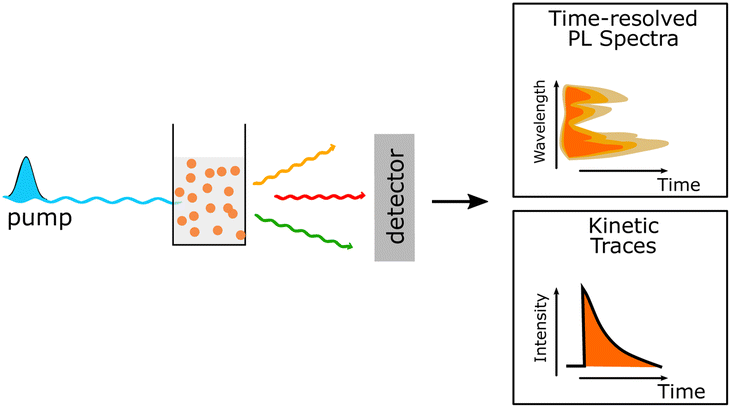
|
| (2) understanding the energy and electron transfer processes between the donor and acceptor polymer units | ||
Photocatalysis using PNP for solar fuel generation can be subdivided into the following steps presented in Fig. 7. In a simplified case (Fig. 7a), light harvesting by PNP induces exciton formation and dissociation to free charges (Step 1). In reality the mechanism is much more complicated, exhibiting multiple additional pathways (Fig. 7b). Starting from poor exciton dissociation,8,95 typically high reorganization energies upon relaxation of the excited state, significantly limit the ultrafast electron transfer from the photoexcited species. This suggests that a system for solar fuel production must be able to efficiently overcome both high exciton binding energies from hundreds meV to ∼1 eV96 (caused by low dielectric constants that prevent charge screening, and strong interaction of the exciton with the molecular backbone);7 as well as to overcome high reorganization energies present in polymeric materials with overall minimal losses of the free energy (Section 4.1). Migration of the charges to the catalytic active sites (Step 2) strongly depends on the polymer microstructure, solvent polarity, as well as permeability of PNP surface to reactants through the specific channels in the PNP structure during photocatalysis, and is discussed in Section 4.2. Moreover, prior to redox reactions that form valuable solar fuels (Section 4.3), charges that are supposed to reach the catalytic active sites might undergo recombination on both the surface of PNP and in bulk that is seen as disadvantageous processes that has to be bypassed (Section 4.4).
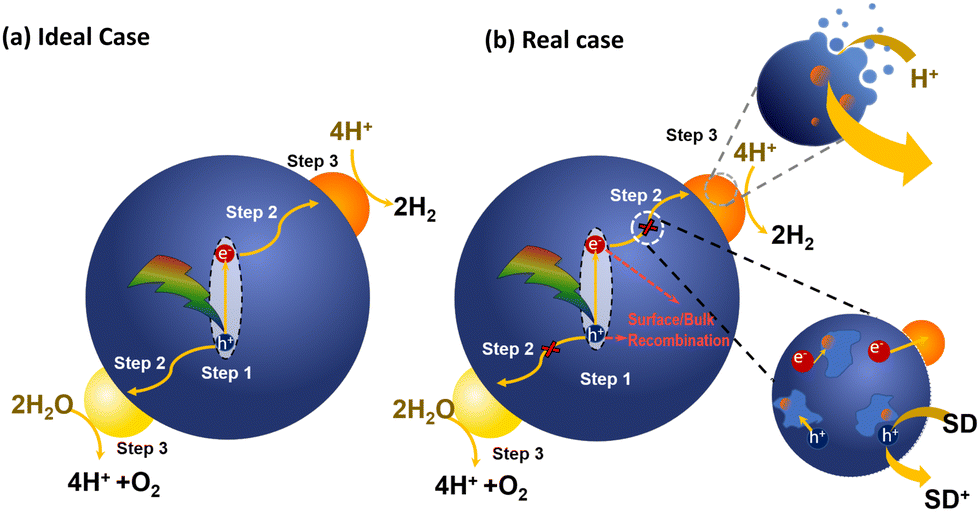 | ||
| Fig. 7 Schematic representation of photocatalytic steps that lead to light-driven water splitting using PNP in ideal case (a) and real case (b). | ||
4.1 Light harvesting and exciton dissociation (step 1)
Broad light harvesting within the entire visible range is one of the steps towards efficient solar fuel formation. However, in order to get insights into the kinetics of the involved processes, monochromatic laser pulse excitation is usually being applied in transient absorption/photoluminescence spectroscopy.A typical transient absorption spectrum of polymeric photocatalyst after excitation is composed of negative absorption features (e.g. ground-state bleach, stimulated emission) and positive absorption features (e.g. excited-state absorption and absorption of newly generated oxidized/reduced species). Using transient absorption spectroscopy Sachs et al.66 investigated a series of conjugated polymers with fluorene backbone and assigned the positive absorption signal from 800 nm and towards the near-infrared to the formation of singlet excitons (Fig. 8),97 while characteristic stimulated emission from polymer's excitons appeared as a negative absorption feature around 450–700 nm. Excited state absorption signals from singlet excitons were found to decay dramatically fast on the picosecond timescale for conjugated polymers,66 thus resulting in very poor charge generation. This highlighted the weak side of organic photocatalysts, namely poor exciton dissociation.
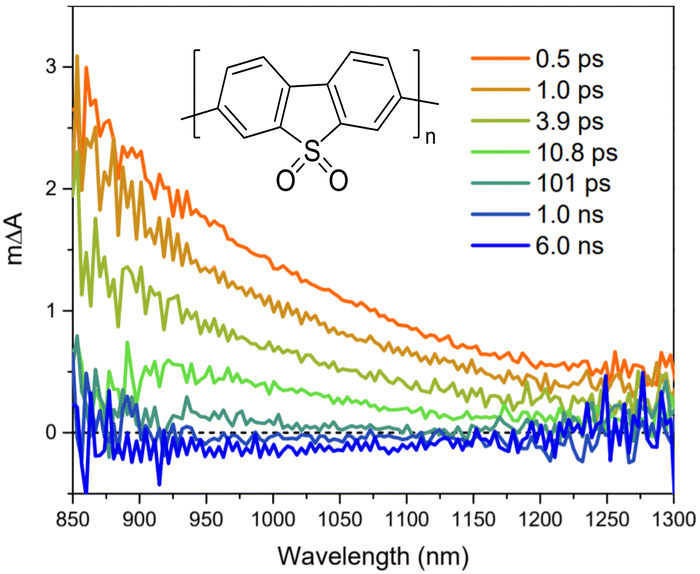 | ||
| Fig. 8 TA spectrum of P10 suspension in a solvent mixture consisting of equal volumes of H2O, MeOH, and TEA (λexc. = 355 nm, a fluence 0.08 mJ cm−2. Reproduced from ref. 66 with permission from Nature Research, copyright 2018. | ||
In this section we provide the most common strategies used to overcome the problem of initially poor charge separation for polymers. The desired excitons with longer lifetimes are bound stronger and may require larger energy alignment offset to break them apart.96 Translational motions of excitons cannot be influenced by the electric fields as excitons are electrically neutral. However, charge generation that has to be formed by dissociation of the diffused excitons at the polymer–water interface can be facilitated to some extent by the use of media with relatively high permittivity, making high dispersibility of polymers in water a necessity.98,99
(a) Size tuning. A first strategy towards efficient exciton dissociation includes size tuning of polymer nanoparticles. In order to initiate redox reactions, excitons need to dissociate prior to relaxation to the ground state within the exciton diffusion length (LD).30,100 Exciton diffusion length settles limits for the movement of free charge carriers towards the surface of the PNP or the catalytic active site. On average LD in organic semiconductors is limited to 5–20 nm,30,65 suggesting that in case of smaller size PNP, the diffusion length will not be an obstacle towards efficient exciton dissociation and charge separation at the interface.
(b) Introduction of heterojunction. One of the most effective strategies to enhance exciton dissociation finds its roots in solar cells with donor–acceptor (D–A) heterojunctions,101 where ultrafast charge separation was obtained by directing the electron via an energetically downhill path. It was suggested that intermolecular electric fields that arise between donor and acceptor polymers with different electrostatic potentials stand behind subpicosecond long-range charge separation.56,102,103 In pair with ultrafast charge separation, the addition of an acceptor unit may result in generation of long-lived species in contrast to neat nanoparticles, solely based on a donor polymer.56 Energy level alignment between donor and acceptor polymers plays a crucial role on the feasibility of charge separation (Fig. 5) and overall energy conversion efficiency. Heterojunction PNP42,104,105 and Pdots31 have been recently introduced in photocatalysis, showing enhanced performance compared to single polymer-based PNP. Ternary Pdots using this D–A heterojunction model have shown light harvesting within the entire visible range via consecutive steps of energy and electron transfer events (Fig. 9).31 It was demonstrated that energy transfer between polymers occurred within 400–600 fs, and could only be observed if polymers were blended inside the same Pdot, highlighting the key role of donor–acceptor distance for efficient energy transfer.106 Recently, Kosco et al. reported the accumulation of photogenerated long-lived charges that survived on a millisecond to second timescale in PM6:PCBM and PM6:Y6 nanoparticles even in the absence of electron or hole scavengers due to beneficial heterojunction architecture.105 Efficient charge separation was achieved either by electron transfer from donor to acceptor or by hole transfer from acceptor to donor polymers, or the combination of the above steps that took place on a subpicosecond timescale within 200 fs. It was suggested that charge transfer with subpicosecond risetime occurred as a result of extensive exciton delocalization without the necessity for its diffusion within the polymer network.107
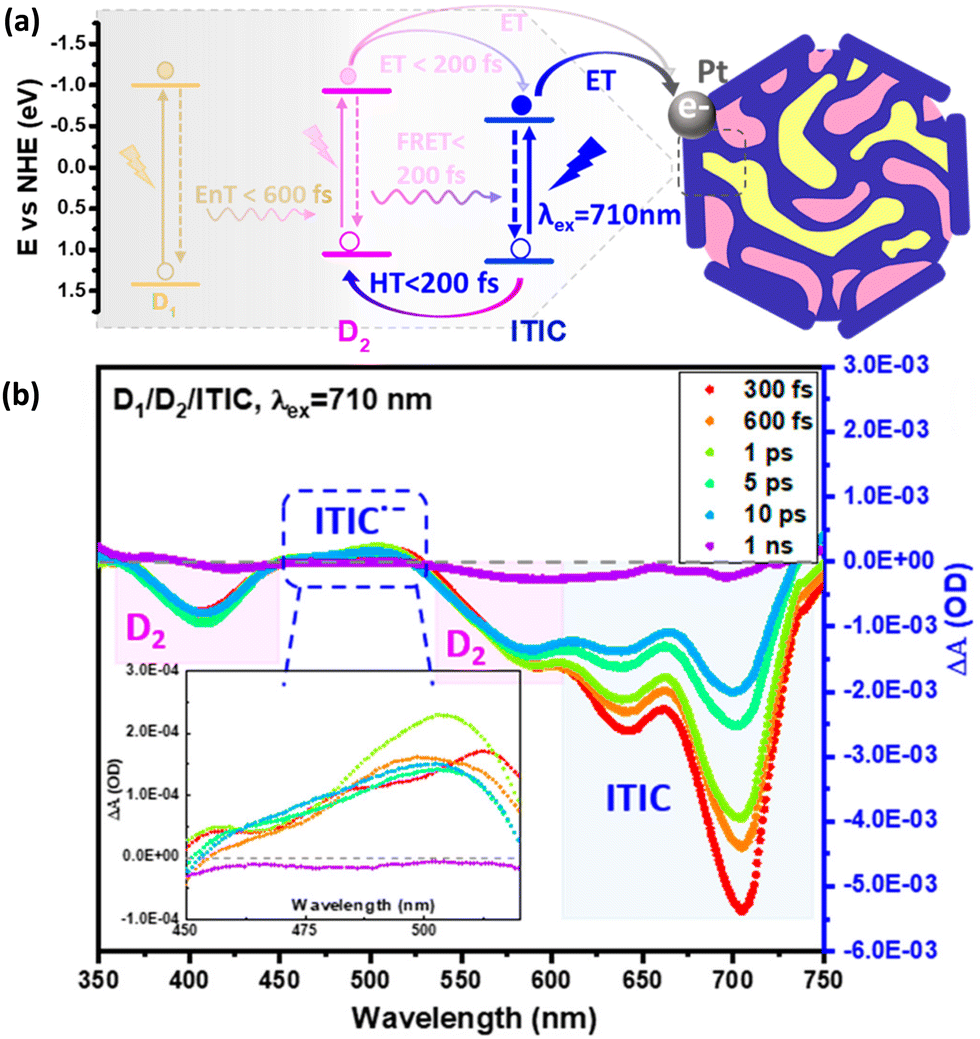 | ||
| Fig. 9 (a) Energy diagram summarizing the photophysical pathways involved in D1/D2/ITIC ternary Pdots, where D1 is PFBT polymer and D2 is PFODTBT polymer respectively. (b) Transient absorption spectrum of ternany Pdots (λexc. = 710 nm). Formation of reduced ITIC upon hole transfer from D2 polymer is presented in inset figure. Reproduced from ref. 31 with permission from American Chemical Society, copyright 2021. | ||
(c) Introduction of the polar glycol side chains. A third strategy towards overcoming the high exciton binding energy includes introduction of the polar glycol side chains in conjugated polymers used for PNP preparation. Ultrafast exciton separation in pair with increased yield of long-lived photogenerated charges was observed by Kosco et al. for glycolated PNP even without D–A heterojunction.108 At the same time implementation of the heterojunction could further promote exciton dissociation for glycolated PNP. This work showed that hydrophilic glycol side chains enhance εr of the PNP environment, promoting water molecules to penetrate inside the PNP and reducing coulombic attraction between photogenerated electrons and holes.108
4.2. Charge-carrier transport (step 2)
Charge carrier transport that takes place inside the volume of a single PNP must reach the interface and be transported out of the particle. Once excitons have dissociated to free charge carriers, electrons and holes can participate in either oxidative or reductive quenching in a SD/P/SA system, where SD is a sacrificial electron donor (e.g. TEOA, TEA, EDTA), P is light harvesting polymer, and SA is a sacrificial electron acceptor. On the one hand, the photoexcited polymer (P*) may act at first as a reductant to the electron accepting unit, while later being oxidized accept electron from SD within the oxidative quenching cycle. On the other hand, a photoexcited polymer may act as an oxidant, at first accepting an electron from SD, and then donating an electron from the generated reduced species to SA in case of reductive quenching cycle. The prevailing mechanism will be the one with the highest quenching rate as determined for example by Stern–Volmer quenching experiments. Potentials alignment of the separate D/P/A units, driving force, or simply the availability of either electron donor or electron acceptor in the closer proximity to photoexcited species will direct the reaction in a specific way.109Using microsecond to second TAS Kosco et al.105 reported that addition of sacrificial electron acceptors, e.g. Pt, typically results in largely enhanced amplitude and longer-lived charges due to suppressed bimolecular recombination kinetics. At the same time addition of sacrificial electron donors, e.g. ascorbic acid, results in accelerated decay of long-lived charges due to hole transfer to ascorbic acid. Determination of the exact charge transfer mechanism still remains challenging due to polymer chain heterogeneity and low concentration of mobile charges. It is suggested that in organic photocatalysts charge transport occurs mainly via hopping.65 Zhou et al.110 observed faster charge carriers mobilities for Pdots upon changing the polyfluorene unit (F8DTBT Pdots) to polyindofluorene unit (PIFDTBT).
The impact of polymer microstructure, degree of its solvation and the solvent polarity on the involved charge transfer and thermodynamic driving forces towards proton reduction have been investigated in detail by Cooper et al.66 In the presence of triethanolamine (TEA) an additional positive feature appeared at 630 nm in TAS spectra of polymers with sulfone groups in their backbone, in contrast to polymer with hydrophobic backbone. This band was assigned to the formation of excitons with delocalized over the polymer network electrons and holes, namely polarons (Fig. 10a).111,112 Spectroscopic signature from polarons generated via hole scavenging by TEA was observed from femtosecond to second time window. Productive generation of polarons via hole scavenging was found to stand behind higher photocatalytic proton reduction yields of polymers with sulfone backbone. Localization of more polar solvents molecules around the polymer chain resulted in the generation of a higher population of long-lived electron polarons, thus favoring the electron transfer from the sacrificial electron donor, to the sulfone-containing polymer. This was achieved by controlling the polaron formation with the solvent media and further directing of the hopping electrons through the polymer network. Both excited state ionization potentials and excited state electron affinity were shown to be affected by the solvent, resulting in overall ∼0.5 eV shift in the driving force towards proton reduction. Thermodynamically downhill charge transfer to TEA was observed for polymer with sulfone groups, in contrast to thermodynamically uphill charge transfer for polymer with a hydrophobic backbone (Fig. 10b).
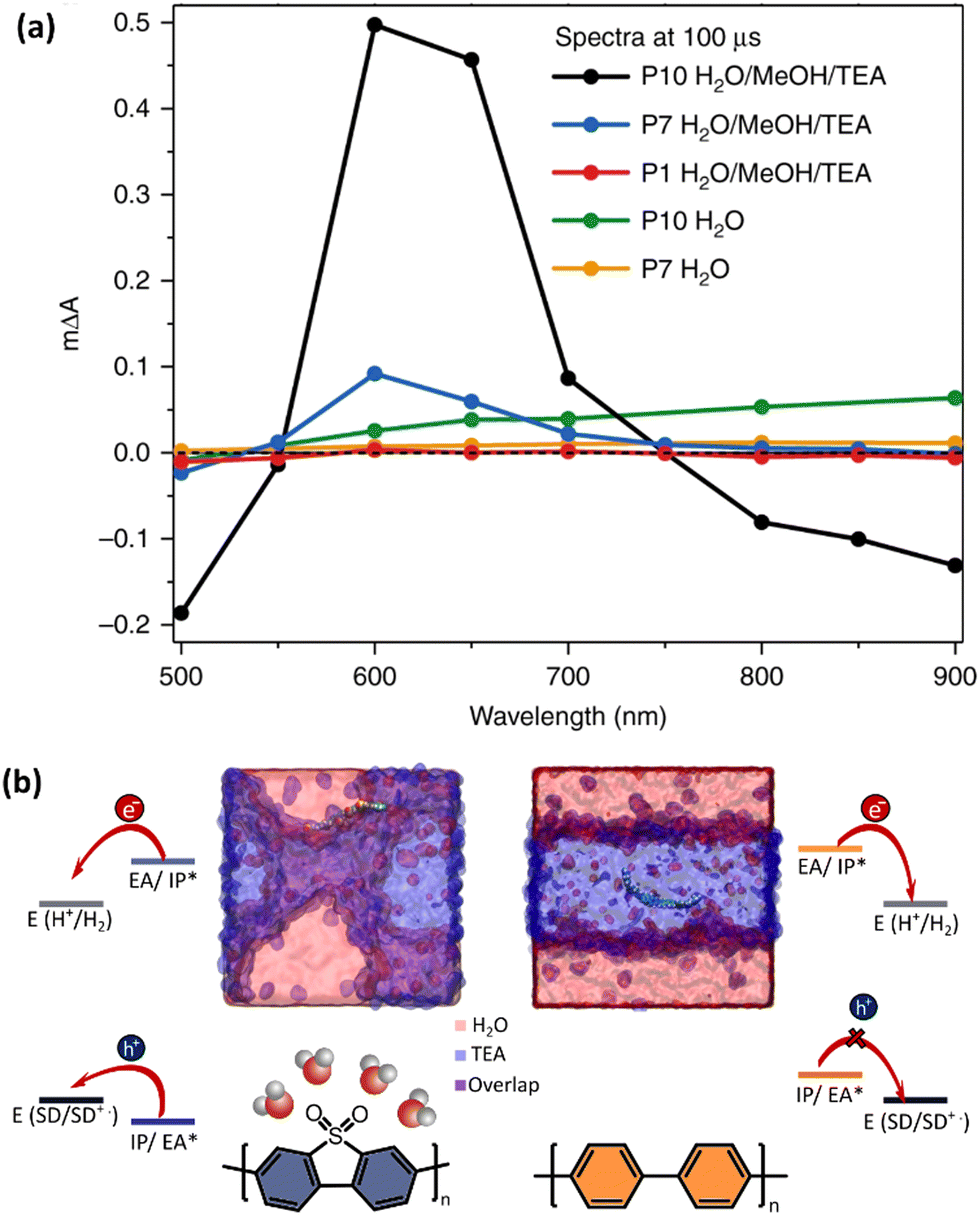 | ||
| Fig. 10 (a) TA spectra probed 100 μs after excitation (λexc. = 355 nm). (b) Snapshots of atomistic molecular-dynamics simulations for polymers with sulfone (P10, blue-shaded) and hydrophobic (P1, orange-shaded) backbone. The sulfone groups of P10 polymer resides at the interface of water and triethylamine (TEA), whereas non-polar P1 hides in the TEA phase. This difference in solvation affects the driving force for hole transfer to TEA, that is thermodynamically downhill for sulfone-containing P10 polymer, and thermodynamically uphill for P1 without sulfone groups. Ionization potentials (IP), electron affinity (EA) for ground-state oligomers of P1and P10 together with the exciton electron affinity (EA*) and ionization potential (IP*), and sacrificial electron donor (SD) in comparison with the potentials for proton reduction (H2/H+). Reproduced from ref. 66 with permission from. Nature Research, copyright 2018. | ||
Electrons are not the only species to be transported through the PNP – the reactants such as protons need to be also available within the close proximity to the catalytic active site. Permeability of protons through the specific channels within the PNP structure have been shown in ref. 16. As suggested by Sachs et al.,66 polymers with functional groups that attract water, e.g. sulfonated polymers, will provide faster rates of both charge and proton transfer. Moreover, filling of the pores of PNP with water with high dielectric constant will have beneficial impact on the overall dielectric permittivity of the PNP, in spite of low dielectric constants observed for bare organic polymers. The detailed mechanism of the electron and proton transfer have not yet been explored in-depth (e.g. concerted electron proton transfer, electron transfer followed by proton transfer or proton transfer followed by electron transfer mechanisms).113 Further studies may unveil the exact role of solvent reorganization on understanding the reaction cycle mechanism.
4.3. Electron transport to the cocatalyst (step 3)
Cocatalysts play a crucial role in charge separation, by utilizing long-lived accumulated electrons from the polymer (e.g. acceptor polymer within D–A blend structures), thus suppressing the unwanted recombination. Durrant et al. used transient spectroscopy methods to demonstrate how differences in timescales of electron transfer from polymer (F8BT and P10) to the catalyst translate into hydrogen evolution activity (Fig. 11a).30 Both femtosecond TA spectrum (fs-TAS) and time-correlated single photon counting (TCSPC) measurements were used to monitor quenching of the polymer excitons lifetime upon stepwise addition of the Pd cocatalyst. In total 54% of photogenerated F8BT excitons were quenched by the cocatalyst (Fig. 11b), whereas 10% of those excitons were quenched even before 100 fs, and the next 23% were quenched faster than 200 ps. With the help of spectroelectrochemistry, spectra for the reduced catalyst species were identified and used for further monitoring of long-lived electron localization on either polymer or cocatalyst itself. Two possible mechanisms were proposed to stand behind efficient and fast exciton quenching for F8BT, namely energy and charge transfer to Pd sites. In contrast to relatively fast (fs–ps) electron transfer to the catalyst, quenching via a sacrificial electron donor occurred for F8BT on late ps–ns timescale. The opposite situation with fast reductive quenching by sacrificial electron donor and slow electron transfer to the catalytic Pd sites was observed for the P10 polymer. Higher overall photocatalytic activity of P10 compared to F8BT was explained by the ability of P10 polymer to generate higher population of long-lived electron polarons (Fig. 11c). Their work highlights that fast reductive quenching, fast charge transfer to the cocatalyst, as well as the ability to accumulate long-lived charges are necessary for the further development of polymers with cocatalyst. Elsayed et al. have shown that electron transfer to a Pt cocatalyst can be enhanced by changing from amphiphilic polymer (surfactant) typically used for Pdots preparation to a low-molecular weight conjugated polyelectrolyte.79 Introduced polyelectrolyte could act as energy donor via Förster resonance energy transfer (FRET) mechanism, additionally promoting wettability of Pdots.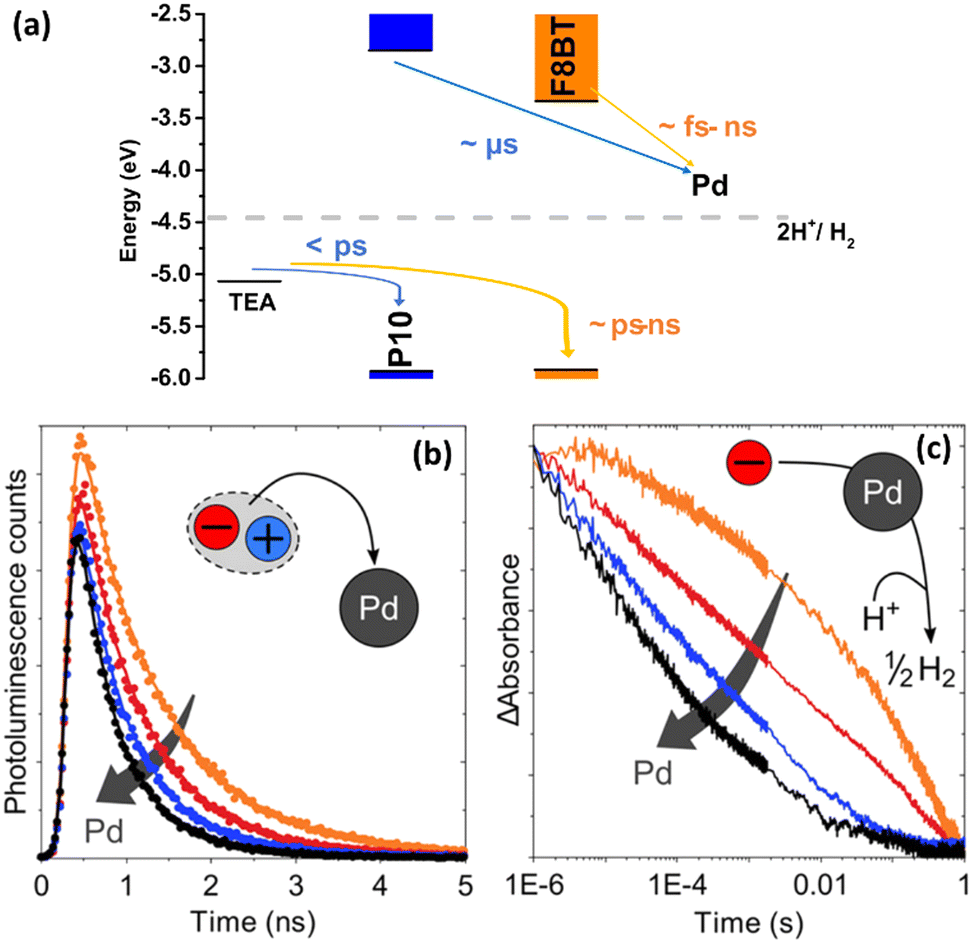 | ||
| Fig. 11 (a) Energy diagram that demonstrates different time scales of the involved processes for F8BT, P3HT and P10. For F8BT, in contrast to P10, electron polarons encounter Pd clusters much more quickly, which implies that electrons already reside on Pd sites before the typical time scale of hydrogen evolution. In spite of generated an order of magnitude higher number of long-lived electrons, electron polarons in P10 take a long time to transfer to catalytic Pd sites, and their transfer to Pd clusters can therefore be considered the kinetic bottleneck of hydrogen evolution. At the same time reductive quenching in F8BT occurs only from late ps in contrast to fast quenching in P10 (b) Photoluminescence decays of F8BT nanoparticle suspensions with different Pd concentrations (λexc. = 467 nm, probed at 545 nm). (c) TA decay kinetics of long-lived electron polarons in P10 synthesized via a Ni0 mediated Yamamoto coupling (orange solid line) and P10 with different content of Pd (λexc. = 355 nm, 630 nm probe). Reproduced from ref. 30 with permission from American Chemical Society, copyright 2020. | ||
Ultrafast electron transfer from the ternary Pdots (PFBT-PFODTBT-ITIC) to the Pt cocatalyst was observed by Liu et al.31 The Pt cocatalyst was found to play a role of an additional electron acceptor that stimulated spatial charge separation and prevented recombination between oxidized donor polymer (PFODTBT˙+) and ITIC˙−. In contrast to efficient charge transfer from ITIC˙−, no direct charge transfer from both donor polymers (PFBT and PFODTBT) in the ternary system to Pt was observed, suggesting that Pt nanoparticles were deposited closer to the ITIC surface.
The possibility of a catalytic mechanism for hydrogen evolution only on the molecular backbone of the polymer itself, without any addition of a co-catalyst has been proposed first by Pati et al.29 Recently, Axelsson et al. showed that a small molecule based on benzothiadiazole, a common acceptor unit in the polymers used in PNP (PFBT, PFODTBT, etc.) can work as an electrocatalyst for hydrogen evolution.114 The mechanism was mainly studied using electrochemical methods, where cyclic voltammetry was used to identify specific reductions and protonation events and bulk electrolysis could confirm catalytic hydrogen evolution. The catalytic intermediates were characterised using spectro-electrochemical methods to capture spectra of the intermediates in both UV-vis and IR. The spectra were then compared to DFT calculations of the possible intermediates. The importance of this type of electrocatalytic mechanism of benzothiadiazole unit, particularly the formation of organic hydride, in photocatalytic reactivity of PNP is yet to be determined if it occurs at all.
4.4. Surface and bulk recombination
Recombination processes that occur both at the interface of PNP (for example with cocatalyst) and in the bulk of PNP are seen as disadvantageous processes that limit the efficiency and performance of PNP. Such back-electron transfer reactions usually occur on a microsecond to millisecond time scale and follow the multiexponential decay time law. The excitation of polymers most often generates local charge separated states well described in the form of excitons, thus particle size is expected to have negligible impact on recombination rates. Since geminate recombination is always assumed to be fast and local, it is the dissociation of the exciton that is the key process for charge generation in the PNP, as described in chapter 4.1 of the review. However, particle morphology can potentially have a large impact on non-geminate recombination rates, both in single polymer systems as well as in heterojunction and co-catalyst systems. Richards et al. observed limited charge transport of photogenerated electrons from the nanoparticles with core–shell morphology due to higher internal recombination events.59 At the same time, efficient transport of electrons and holes was observed, when donor and acceptor polymer were uniformly blended.59 It was suggested that in well blended nanoparticles there is a large amount of p–n junction interface for excitons to dissociate, which results in high rates of charge transport out of the particle, suppressing non-geminate recombination.105 This effect can be amplified by adding a co-catalyst to the PNP which will use the long-lived charges and thus further suppressing non-geminate recombination.Another way to enhance charge transport and simultaneously bypass the recombination inside PNP is by introducing (semi)crystalline polymers inside the mixed blends. Implementation of phase separation promotes fast charge generation without unnecessary losses due to diffusion of the excitons to the interface,56 thus suppressing unwanted geminate recombination.115,116 Enhanced hole mobility and overall charge carrier generation was observed for P3HT:PCBM system with low degree of intra- and interchain disorder.56 Recently, Kosco et al. have shown that both geminate and nongeminate recombination can be efficiently slowed down from typically observed ps–ns timescales up to μs timescale by introduction of glycol side to the polymer backbone used for PNP preparation.108
5. Evaluation of PNP for solar fuel production
Here we introduce parameters used to evaluate the photocatalytic performance. Traditional parameters include reaction rates (e.g., hydrogen evolution rate), external quantum yields (EQYs), internal quantum yields (IQY), solar to hydrogen conversion efficiency (STH), and characterization of the overall photocatalytic stability.5.1. Comparison of hydrogen evolution rates
There are several considerations for accurate evaluation of heterogeneous photocatalytic systems.117–119 How much product is generated during a certain time is a commonly used parameter to evaluate the performance of the polymeric photocatalyst. For example, molp h−1 means how many moles of product is produced during one hour. As this parameter is significantly influenced by the catalyst concentration and the volume of solution, as well as the light intensity, dividing this parameter with catalyst amount or solution volume can render two alternative parameters. For example, molp gcat−1 h−1 means how many moles of product is produced by one-gram of catalyst during one hour, and molp mL−1 h−1 means how many moles of product is produced by one milliliter of solution during one hour.31 The former parameter is similar to turnover frequency commonly used for catalysis. One should be aware that doubling concentration of the catalyst would not double this parameter. The initial intention of comparing the hydrogen evolution rates in terms of molp gcat−1 h−1 was trying to optimize the polymer structure on the molecular level in order to obtain the most photoactive polymeric photocatalyst. Therefore, one may find out that many groups have reported initially the mass normalized hydrogen evolution rates.11,19,33 In practice, photocatalytic hydrogen evolution rate can be influenced by many parameters, such as reaction condition, photocatalyst origin (presence or absence of cocatalyst), reactor type (shape, light pathlength, etc.), and reaction medium.117 For example, residual Pd remains often within polymer and originates from the Pd complex catalyst that is used for the synthesis of the polymer. By using PFBT nanoparticles as a model system, Kosco et al. systematically investigated that residual Pd affects the hydrogen evolution.32 Considering that the amount of residual Pd usually varies, it was suggested that reporting the amount of Pd is important in order to elucidate its impact on the overall performance of the polymeric nanoparticles. Presenting the hydrogen evolution rates as molp mL−1 h−1 describes the overall production capacity of the system. To conclude, for evaluation of the hydrogen evolution rates for the PNP photocatalytic system it is more informative to report the hydrogen evolution rates in terms of both molp gcat−1 h−1 and molp mL−1 h−1.5.2. External quantum yield, internal quantum yield and IQY, and solar-to-hydrogen conversion efficiency
To compare the performance of PNP to other materials, it is highly recommended to report established parameters, namely the external quantum yield (EQY), and the solar to hydrogen conversion efficiency (STH). While STH has become the most reported parameter to characterize photoelectrochemical (PEC) water splitting,120 so far EQY or apparent quantum yield (AQY) have been more widely reported for the evaluation of the polymer nanoparticles for the solar fuel production. At the same time, reporting the internal quantum yield (IQY) is underrepresented, most likely because its estimation involves inaccuracy of direct determination of the absorbed photons due to the inevitable scattering events.121 EQY and STH can be determined as following (eqn (11) and (12)):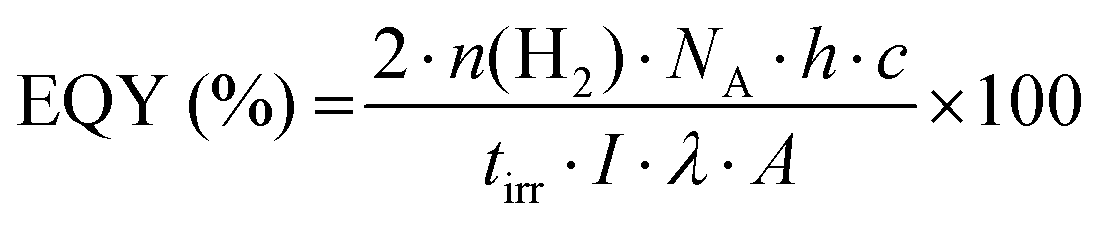 | (11) |
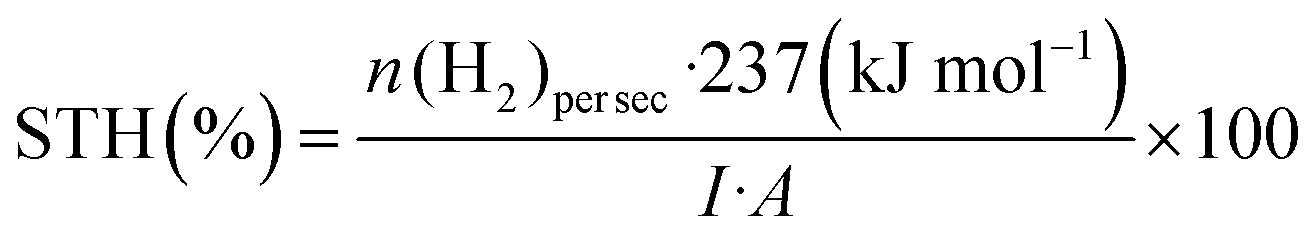 | (12) |
Determination of EQY and STH needs to be performed with care using certified equipment. For EQY quantification, samples need to be excited with nearly monochromatic light source. To have comparable values in publications, it is better to determine STH under a light source that has an intensity of 100 mW cm−2 with AM1.5G solar spectrum or similar.
In pair with reported EQY and STH values, it is good to provide the following information as much as possible in order to evaluate the polymer nanoparticle photocatalysts properly:
– Incident light spectrum (lamp type);
– Incident photon flux;
– Light response study (in order to distinguish whether it is true photoinduced catalysis reaction), particularly for reporting a new system;
– Concentration dependent study (in order to report the optimal hydrogen evolution rate);
– Reaction conditions: catalyst concentration, co-catalyst concentration, solvent, sacrificial reagents and their concentrations, pH, volume (as well as volume of headspace), reactor's shape and volume.
By presenting detailed information discussed above, we will be able to have more accurate and meaningful comparison of average hydrogen evolution rate, EQY and/or STH as reference to other researchers.
5.3. Photocatalytic stability
Photocatalytic stability is one of the important parameters to evaluate the semiconducting polymeric particles in photocatalysis. To test the stability of the photocatalytic system, a long-term photocatalytic experiment is normally performed. Plotting product amount as a function of time can give the direct information on stability of the photocatalytic system (Fig. 12). Once the system reaches a plateau or the product amount is significantly decreased, there are two actions to reevaluate the stability of the system: addition of fresh components (Fig. 12, light-blue dashed curve) and removal of the product (Fig. 12, grey dashed line) to reinitiate the photocatalysis (Fig. 12, orange curve). Addition of one of fresh components such as PNP or co-catalyst to the reaction mixture, is a way to check which component is essentially unstable and if solar fuel formation can be reinitiated.48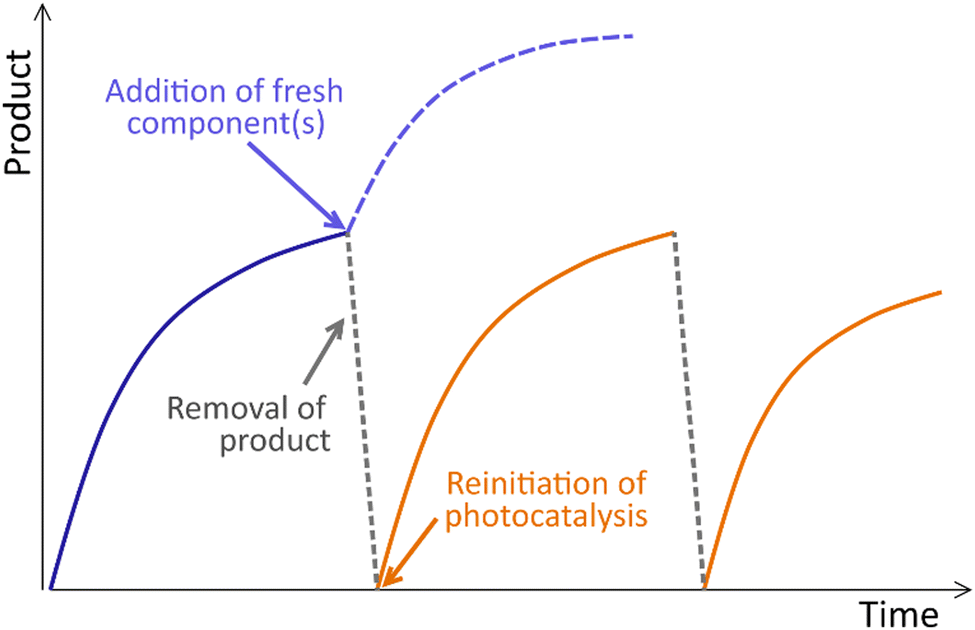 | ||
| Fig. 12 The strategies to evaluate the PNP photocatalytic stability. | ||
Gradually increased concentration of product in the solution can also slack the photocatalytic process. Therefore, removing product and checking if the photocatalytic process can be reinitiated is an effective method to verify this. Liu et al. purged the Pdots photocatalytic system to remove produced hydrogen and found the photocatalytic reaction can be reinitiated,16 indicating that the produced hydrogen could block the proton channels in Pdots by the patio-pressure created by generated gas and therefore slow down the reaction. For a half-reaction stability test, a sacrificial electron or hole scavenger is usually added in excess amount to avoid that the consumption of sacrificial agent affects the stability of the system. However, for a very efficient and stable system running for long time, re-addition of the sacrificial agent is also a way to check if the performance of the system can be resumed. Moreover, the sacrificial agent could generate some undesired side-products which can absorb light or deactivate the photocatalysts, eventually affecting the performance of the system.31
The intrinsic photocatalytic stability of the polymeric particle photocatalysts can be affected by (a) particle stability, (b) material stability and (c) co-catalyst stability (if applicable). (a) Particle stability refers to the change of particle size during the photocatalytic reaction. Aggregation is the most common issue that deactivates the photocatalytic ability of the polymer particles.11 In some cases, aggregation can be visually seen, otherwise DLS, SEM or TEM can be used to further determine the degree of aggregation after the reaction. (b) Material stability refers to the stability of the material itself, for example, if the polymer is degraded during the photocatalytic reaction due to unstable excited state, reduced state or oxidized state. UV-vis, NMR, FTIR TAG and DSC are useful methods to determine the degradation degree of the materials. Sometimes, the system exhibits both particle stability and material stability issues. Then it is necessary to recycle the material and determine the final amount of the polymer photocatalyst remaining in the system by comparing it with a standard sample. (c) Co-catalyst stability refers to if the co-catalyst itself is detached or degraded from the polymer particle surface during the photocatalytic reaction. Even, if a system has no stability issues concerning particle size and the material itself, it is very important to check the stability of the co-catalyst. As mentioned before, addition of fresh co-catalyst to check if the photocatalytic reaction can be regenerated is a way to conclude that co-catalysts are stable or not. Meanwhile, as the co-catalyst is usually metal-based, such as metallic particles, metal oxides/sulfides and metal complexes, XPS and SEM/TEM/EDX are also vital techniques to check if the co-catalyst has changed or detached from the surface.31
Furthermore, other factors such as the light intensity, the particle size and its morphology, the concentration of the particles and even the photocatalytic system itself can also influence the stability. As the production of excited photocatalyst ideally is linearly proportional to the light intensity, stronger light will of course generate more activated intermediates and give more products. If the degradation of the activated intermediates dominates the instability of the system, it gives an impression that the system is less stable under a stronger light illumination. As the particle surface or co-catalyst surface on the PNP, where the excitons get dissociated, typically functions as the active site for photocatalytic reactions, much of the degradation happens therefore on the surface instead of the bulk. This surface degradation is caused mainly by insufficient electron or hole utilization and accumulated charges in the materials.
For certain photocatalysts, the particle size and morphology can also become a vital parameter influencing the stability. Let's assume a perfect spherical photocatalyst particle with a diameter D (Fig. 13a): we consider that active sites with a diameter d are uniformly located at the interface between the particle and the solution where the photocatalytic reaction happens. If the stability of a single active site is t, then the observed stability of the particle should be t·(D/2d). If d is much less than D, then the observed catalytic stability is much longer than the real stability of the active site. If the same photocatalyst particle becomes mesoporous (Fig. 13b), then the observed stability of a mesoporous particle becomes t·(D/n2d), where D/n (n > 1). Hence, the mesoporous particle shows a shorter lifetime than that of a solid one. However, the observed stability does not represent the intrinsic stability of the photocatalytic material.
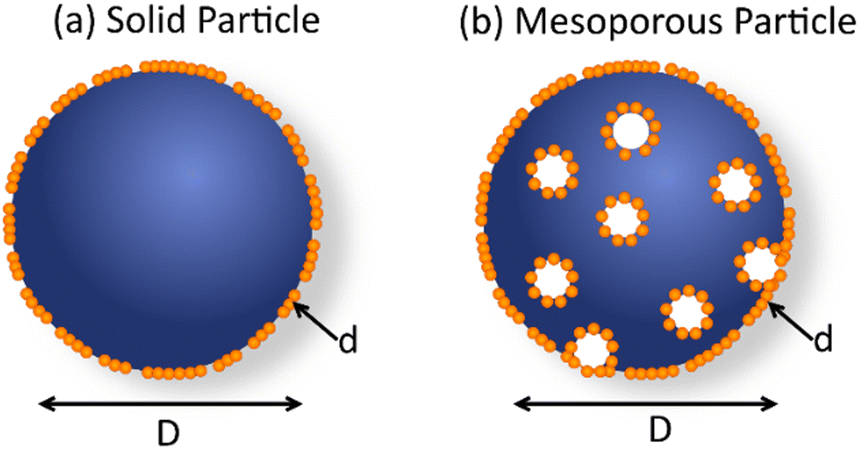 | ||
| Fig. 13 Schematic drawing of solid and mesoporous polymeric photocatalyst particle with dimeter D and active sites (orange dots) with a diameter d. | ||
Moreover, for a system with concentrated samples, the “dark” photocatalysts that cannot absorb light, and therefore don’t participate in photocatalysis, will of course contribute to slow degradation and prolong the observed photocatalytic time. It is also possible for an inappropriate test system to create a lot of “dark” photocatalysts, for example if the light illumination area is much smaller than the container front window.122
All of these parameters make it hard to compare the stability data of different systems reported so far from the literature. However, it is still an unavoidable parameter when a new photocatalytic system is reported and intrinsic stability tests are therefore encouraged. Using 1 sun illumination and the dilute solution system under light illumination of the entire front sample window of the test system can make the stability data more reliable and meaningful.
6. Perspective
During past years, conjugated organic polymers are of high interest for photocatalytic fuel production. Making the conjugated organic polymers into nanoparticles, PNP (e.g. Pdots), is an effective strategy not only to increase the surface area, but also to shorten the diffusion path of light generated exciton as well as to create a proton-favorable environment for the corresponding reactions. PNP creates both challenges and opportunities in the polymeric photocatalyst research field. Excellent dispersibility of PNP in water allows scientists to use various spectroscopic methods to characterize and investigate the PNP photocatalysts from different angles such as proton diffusion, inner structure, catalytic sites, charge transfer and energy transfer within the PNP matrix. The generated knowledge and understanding further guide the design of advanced polymeric and PNP photocatalysts.To enhance the photocatalytic performance of PNP, the design of new polymers with broad absorption which covers the whole UV-vis region up to near IR region is always a viable strategy. Considering the bandgap and energy levels limitation of a single polymer, heterojunction systems have advantages to adopt narrower band gap polymers or small molecular acceptor in photocatalysis. In heterojunction system, ultrafast charge separation and/or energy transfer and slow charge recombination between components can enhance the quantity and lifetime of photo-generated electrons and holes, therefore showing large potential to further boost photocatalytic performance of organic polymeric photocatalysts. Although PNP photocatalysts show several orders of magnitude higher reactivity as compared to bulk polymeric photocatalyst (if one evaluates the performance by use of molp gcat−1 h−1), the production capacity evaluated based on volume molp mL−1 h−1 is still unsatisfactory even with almost 100% light harvesting efficiency, which could be caused by serious light scattering or fast charge recombination. This could be addressed by particle preparation and morphology tuning. Therefore, to achieve high solar fuel generation yields with PNP, it is important to understand the impact of PNP morphology on its photophysical properties and photocatalysis. Morphology and composition tuning to enhance the reaction kinetics of each catalytic step is also a good strategy to consider. Although different morphologies of PNP have been reported, an understanding for how to well design and how to optimize the morphology in a hypothesis-driven way is still lacking in this field.
Ideally, polymeric photocatalytic system should be scaled up and eventually used in industry. Although PNP are reported to be intrinsically very stable dispersions, the observed stability issues of PNP during photocatalysis involving light and heat still needs more attention. The design of new materials with robust building blocks to improve the intrinsic photo- and thermal-stability of the polymers is a recommended direction to go. Choosing optimal surfactants to well stabilize the PNP from aggregation during photocatalysis can also be used to improve the stability of the PNP photocatalysts. To reliably evaluate the stability of the PNP photocatalyst, dark photocatalysts that do not absorb light and participate in the reaction should be avoided as much as possible.
The field of solar fuel production constantly expands and finds new application routes. Recently it was shown that PNP can be also utilized not only for proton reduction, but also for water oxidation to H2O2 and high-value chemicals (e.g. formate).123 Moreover, the report on utilization of polymer nanoparticles in overall water splitting under visible light have appeared recently.53 Even though PNP have been well used for proton reduction, other reactions such as CO2 reduction and N2 fixation are still reserved to be further investigated in the future. Having these reactions being successfully completed with PNP photocatalysts, how to connect the reduction reaction and oxidation reaction to form valuable chemicals by an efficient way will be an important scientific question in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to gratefully thank Wallenberg Academy Fellow program by K&A Wallenberg Foundation (2019.0156), Olle Engkvist Foundation (200-0523), the Swedish Research Council (2017-03757) and Uppsala University for the financial support.References
- IEA, World Energy Outlook 2019, OECD, 2019 Search PubMed.
- N. S. Lewis, MRS Bull., 2007, 32, 808–820 CrossRef.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- F. E. Osterloh, ACS Energy Lett., 2017, 2, 445–453 CrossRef CAS.
- J. Kosco, F. Moruzzi, B. Willner and I. McCulloch, Adv. Energy Mater., 2020, 10, 2001935 CrossRef CAS.
- K. Takanabe, ACS Catal., 2017, 7, 8006–8022 CrossRef CAS.
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Nat. Rev. Mater., 2021, 6, 168–190 CrossRef CAS.
- M. Rahman, H. Tian and T. Edvinsson, Angew. Chem., Int. Ed., 2020, 59, 16278–16293 CrossRef CAS PubMed.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Ser. Chem. Commun., 1985, 474–475 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- L. Wang, R. Fernández-Terán, L. Zhang, D. L. A. A. Fernandes, L. Tian, H. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 12306–12310 CrossRef CAS PubMed.
- S. Kundu and A. Patra, Chem. Rev., 2016, 117, 712–757 CrossRef PubMed.
- J. Yu, Y. Rong, C. T. Kuo, X. H. Zhou and D. T. Chiu, Anal. Chem., 2017, 89 Search PubMed.
- L. Guo, J. Ge and P. Wang, Photochem. Photobiol., 2018, 94, 916–934 CrossRef CAS PubMed.
- E. Solhi and M. Hasanzadeh, TrAC, Trends Anal. Chem., 2019, 118, 840–852 CrossRef CAS.
- A. Liu, C. W. Tai, K. Holá and H. Tian, J. Mater. Chem. A, 2019, 7, 4797–4803 RSC.
- J. Pecher and S. Mecking, Chem. Rev., 2010, 110, 6260–6279 CrossRef CAS PubMed.
- C. M. Aitchison and R. S. Sprick, Nanoscale, 2021, 13, 634–646 RSC.
- P. B. Pati, G. Damas, L. Tian, D. L. A. Fernandes, L. Zhang, I. B. Pehlivan, T. Edvinsson, C. M. Araujo and H. Tian, Energy Environ. Sci., 2017, 10, 1372–1376 RSC.
- W. W. Yong, H. Lu, H. Li, S. Wang and M. T. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 10828–10834 CrossRef CAS PubMed.
- P.-J. J. Tseng, C.-L. L. Chang, Y.-H. H. Chan, L.-Y. Y. Ting, P.-Y. Y. Chen, C.-H. H. Liao, M.-L. L. Tsai and H.-H. H. Chou, ACS Catal., 2018, 8, 7766–7772 CrossRef CAS.
- C. Xie, T. Heumüller, W. Gruber, X. Tang, A. Classen, I. Schuldes, M. Bidwell, A. Späth, R. H. Fink, T. Unruh, I. McCulloch, N. Li and C. J. Brabec, Nat. Commun., 2018, 9, 5355 CrossRef PubMed.
- A. Fraleoni-Morgera, S. Marazzita, D. Frascaro and L. Setti, Synth. Met., 2004, 147, 149–154 CrossRef CAS.
- C. Xie, A. Classen, A. Späth, X. Tang, J. Min, M. Meyer, C. Zhang, N. Li, A. Osvet, R. H. Fink and C. J. Brabec, Adv. Energy Mater., 2018, 8, 1702857 CrossRef.
- X. Pan, A. Sharma, D. Gedefaw, R. Kroon, A. Diaz de Zerio, N. P. Holmes, A. L. D. Kilcoyne, M. G. Barr, A. Fahy, M. Marks, X. Zhou, W. Belcher, P. C. Dastoor and M. R. Andersson, Org. Electron., 2018, 59, 432–440 CrossRef CAS.
- F. J. M. Colberts, M. M. Wienk and R. A. J. Janssen, ACS Appl. Mater. Interfaces, 2017, 9, 13180–13389 CrossRef PubMed.
- J. Cho, S. Yoon, K. Min Sim, Y. Jin Jeong, C. Eon Park, S. K. Kwon, Y. H. Kim and D. S. Chung, Energy Environ. Sci., 2017, 10, 2324–2333 RSC.
- C. Szymanski, C. Wu, J. Hooper, M. A. Salazar, A. Perdomo, A. Dukes and J. McNeill, J. Phys. Chem. B, 2005, 109, 8543–8546 CrossRef CAS PubMed.
- J. Aubry, F. Ganachaud, J.-P. Cohen Addad and B. Cabane, Langmuir, 2009, 25, 1970–1979 CrossRef CAS PubMed.
- M. Sachs, H. Cha, J. Kosco, C. M. Aitchison, L. Francas, S. Corby, C.-L. Chiang, A. A. Wilson, R. Godin, A. Fahey-Williams, A. I. Cooper, R. S. Sprick, I. McCulloch and J. R. Durrant, J. Am. Chem. Soc., 2020, 142, 14574–14587 CrossRef CAS PubMed.
- A. Liu, L. Gedda, M. Axelsson, M. Pavliuk, K. Edwards, L. Hammarström and H. Tian, J. Am. Chem. Soc., 2021, 143, 2875–2885 CrossRef CAS PubMed.
- J. Kosco, M. Sachs, R. Godin, M. Kirkus, L. Francas, M. Bidwell, M. Qureshi, D. Anjum, J. R. Durrant and I. McCulloch, Adv. Energy Mater., 2018, 8, 1802181 CrossRef.
- H. Yang, X. Li, R. S. Sprick and A. I. Cooper, Chem. Commun., 2020, 56, 6790–6793 RSC.
- L. Feng, C. Zhu, H. Yuan, L. Liu, F. Lv and S. Wang, Chem. Soc. Rev., 2013, 42, 6620–6633 RSC.
- G. Feng, J. Liu, R. Liu, D. Mao, N. Tomczak and B. Liu, Adv. Sci., 2017, 4, 1600407 CrossRef PubMed.
- B. K. Johnson and R. K. Prud’homme, Phys. Rev. Lett., 2003, 91, 118302 CrossRef PubMed.
- K. Abstiens and A. M. Goepferich, J. Drug Delivery Sci. Technol., 2019, 49, 433–439 CrossRef CAS.
- M. Yu, W. Zhang, Z. Guo, Y. Wu and W. Zhu, Angew. Chem., Int. Ed., 2021, 60, 15590–15597 CrossRef CAS PubMed.
- K. Landfester, Adv. Mater., 2001, 13, 765–768 CrossRef CAS.
- D. Tuncel and H. V. Demir, Nanoscale, 2010, 2, 484–494 RSC.
- Z. Hashim, P. Howes and M. Green, J. Mater. Chem., 2011, 21, 1797–1803 RSC.
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. Gonzalez Lopez, L. Zou, A. Wadsworth, W. Zhang, L. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Nat. Mater., 2020, 19, 559–565 CrossRef CAS PubMed.
- B. C. Ma, S. Ghasimi, K. Landfester, F. Vilela and K. A. I. Zhang, J. Mater. Chem. A, 2015, 3, 16064–16071 RSC.
- F. J. Schork, G. W. Poehlein, S. Wang, J. Reimers, J. Rodrigues and C. Samer, Colloids Surf., A, 1999, 153, 39–45 CrossRef CAS.
- C. M. Aitchison, R. S. Sprick and A. I. Cooper, J. Mater. Chem. A, 2019, 7, 2490–2496 RSC.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- M. V. Pavliuk, M. Lorenzi, D. R. Morado, L. Gedda, S. Wrede, S. H. Mejias, A. Liu, S. Glover, K. Edwards, G. Berggren and H. Tian, J. Am. Chem. Soc., 2022 DOI:10.1021/jacs.2c03882.
- R. S. Sprick, Z. Chen, A. J. Cowan, Y. Bai, C. M. Aitchison, Y. Fang, M. A. Zwijnenburg, A. I. Cooper and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 18695–18700 CrossRef CAS PubMed.
- K. Wenderich and G. Mul, Chem. Rev., 2016, 116, 14587–14619 CrossRef CAS PubMed.
- Z. Hu, X. Zhang, Q. Yin, X. Liu, X. F. Jiang, Z. Chen, X. Yang, F. Huang and Y. Cao, Nano Energy, 2019, 60, 775–783 CrossRef CAS.
- Z. Hu, Z. Wang, X. Zhang, H. Tang, X. Liu, F. Huang and Y. Cao, iScience, 2019, 13, 33–42 CrossRef CAS PubMed.
- Y. Bai, C. Li, L. Liu, Y. Yamaguchi, B. Mounib, H. Yang, A. Gardner, M. A. Zwijnenburg, N. D. Browning, A. J. Cowan, A. Kudo, A. I. Cooper and R. S. Sprick, Angew. Chem., Int. Ed., 2022, 61, e202201299 CAS.
- C. L. Chang, W. C. Lin, C. Y. Jia, L. Y. Ting, J. Jayakumar, M. H. Elsayed, Y. Q. Yang, Y. H. Chan, W. S. Wang, C. Y. Lu, P. Y. Chen and H. H. Chou, Appl. Catal., B, 2020, 268, 118436 CrossRef CAS.
- X. Zhang, F. Shen, Z. Hu, Y. Wu, H. Tang, J. Jia, X. Wang, F. Huang and Y. Cao, ACS Sustainable Chem. Eng., 2019, 7, 4128–4135 CrossRef CAS.
- K. N. Schwarz, S. B. Farley, T. A. Smith and K. P. Ghiggino, Nanoscale, 2015, 7, 19899–19904 RSC.
- F. Di Maria, A. Zanelli, A. Liscio, A. Kovtun, E. Salatelli, R. Mazzaro, V. Morandi, G. Bergamini, A. Shaffer and S. Rozen, ACS Nano, 2017, 11, 1991–1999 CrossRef CAS PubMed.
- B. Sochor, Ö. Düdükcü, M. M. Lübtow, B. Schummer, S. Jaksch and R. Luxenhofer, Langmuir, 2020, 36, 3494–3503 CrossRef CAS PubMed.
- J. J. Richards, C. L. Whittle, G. Shao and L. D. Pozzo, ACS Nano, 2014, 8, 4313–4324 CrossRef CAS PubMed.
- R. A. Ramli, RSC Adv., 2017, 7, 52632–52650 RSC.
- D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959–4015 CrossRef CAS PubMed.
- C. F. Lee, M. L. Hsu, C. H. Chu and T. Y. Wu, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3441–3451 CrossRef CAS.
- Y. Bai, L. Wilbraham, B. J. Slater, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Am. Chem. Soc., 2019, 141, 9063–9071 CrossRef CAS PubMed.
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li, R. Clowes, N. Rankin, B. Harris, R. S. Sprick and A. I. Cooper, Nature, 2020, 583, 237–241 CrossRef CAS PubMed.
- A. Bruno, L. X. Reynolds, C. Dyer-Smith, J. Nelson and S. A. Haque, J. Phys. Chem. C, 2013, 117, 19832–19838 CrossRef CAS.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef PubMed.
- R. S. Sprick, K. J. Cheetham, Y. Bai, J. Alves Fernandes, M. Barnes, J. W. Bradley and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 7125–7129 RSC.
- D. J. Woods, S. A. J. Hillman, D. Pearce, L. Wilbraham, L. Q. Flagg, W. Duffy, I. McCulloch, J. R. Durrant, A. A. Y. Guilbert, M. A. Zwijnenburg, R. S. Sprick, J. Nelson and A. I. Cooper, Energy Environ. Sci., 2020, 13, 1843–1855 RSC.
- C. Dai and B. Liu, Energy Environ. Sci., 2020, 13, 24–52 RSC.
- L. R. MacFarlane, H. Shaikh, J. D. Garcia-Hernandez, M. Vespa, T. Fukui and I. Manners, Nat. Rev. Mater., 2021, 6, 7–26 CrossRef CAS.
- S. T. Stripp, ACS Catal., 2021, 11, 7845–7862 CrossRef CAS.
- H. Kisch, Angew. Chem., Int. Ed., 2013, 52, 812–847 CrossRef CAS PubMed.
- C. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeill, ACS Nano, 2008, 2, 2415–2423 CrossRef CAS PubMed.
- H. Piwoński, T. Michinobu and S. Habuchi, Nat. Commun., 2017, 8, 15256 CrossRef PubMed.
- K. Dyamenahalli, A. Famili and R. Shandas, Characterization of shape-memory polymers for biomedical applications, Elsevier Ltd, 2015 Search PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, 2006.
- W.-F. Su, Principles of Polymer Design and Synthesis, Lecture Notes in Chemistry 82, 2013, vol. 82 Search PubMed.
- M. Liras, M. Barawi and V. A. De La Peña O’Shea, Chem. Soc. Rev., 2019, 48, 5454–5487 RSC.
- M. Hammad Elsayed, M. Abdellah, Y.-H. Hung, J. Jayakumar, L.-Y. Ting, A. M. Elewa, C.-L. Chang, W.-C. Lin, K.-L. Wang, M. Abdel-Hafiez, H.-W. Hung, M. Horie and H.-H. Chou, ACS Appl. Mater. Interfaces, 2021, 13, 56554–56565 CrossRef PubMed.
- K. Wang, X. Zhang, X. Zhang, X. Fan, Z. Li, Z. Huang, Q. Zhang and Y. Wei, RSC Adv., 2015, 5, 75823–75830 RSC.
- Q. B. Wang, C. Xu, Y. B. Jiang, X. Zhang, J. S. Yao, C. De Qiao, Q. Z. Liu and Y. H. Zhang, AIMS Mater. Sci., 2018, 5, 770–780 Search PubMed.
- F. Lin, H. Liu, Q. Zhou, S. Zhang, Y. Zhou, Y. Feng and J. Li, Int. J. Biol. Macromol., 2021, 183, 2152–2161 CrossRef CAS PubMed.
- F. Hu, S. D. Brucks, T. H. Lambert, L. M. Campos and W. Min, Chem. Commun., 2017, 53, 6187–6190 RSC.
- P. M. Carvalho, M. R. Felício, N. C. Santos, S. Gonçalves and M. M. Domingues, Front. Chem, 2018, 6 Search PubMed.
- J. Stetefeld, S. A. McKenna and T. R. Patel, Biophys. Rev., 2016, 8, 409–427 CrossRef CAS PubMed.
- M. Kaszuba, J. Corbett, F. M. Watson and A. Jones, Philos. Trans. Ser. A, Math. Phys. Eng. Sci., 2010, 368, 4439–4451 CAS.
- W. D. Pyrz and D. J. Buttrey, Langmuir, 2008, 24, 11350–11360 CrossRef CAS PubMed.
- W. Zhou and H. F. Greer, Eur. J. Inorg. Chem., 2016, 941–950 CrossRef CAS.
- J. Kuntsche, J. C. Horst and H. Bunjes, Int. J. Pharm., 2011, 417, 120–137 CrossRef CAS PubMed.
- N. P. Holmes, N. Nicolaidis, K. Feron, M. Barr, K. B. Burke, M. Al-Mudhaffer, P. Sista, A. L. D. Kilcoyne, M. C. Stefan, X. Zhou, P. C. Dastoor and W. J. Belcher, Sol. Energy Mater. Sol. Cells, 2015, 140, 412–421 CrossRef CAS.
- Y. Q. Zheng, Z. F. Yao, T. Lei, J. H. Dou, C. Y. Yang, L. Zou, X. Meng, W. Ma, J. Y. Wang and J. Pei, Adv. Mater., 2017, 29, 1701072 CrossRef PubMed.
- W. J. Albery, P. V. Bartlett, J. Electroanal Chem, S. Hiinig, J. L. Brédas, R. Silbey, D. S. Boudreaux and R. R. Chance, J. Am. Chem. Soc., 1983, 105, 6555–6559 CrossRef.
- J. Kim and T. S. Lee, Small, 2018, 14, 1702758 CrossRef PubMed.
- M. Sim, J. Shin, C. Shim, M. Kim, S. Byeok Jo, J.-H. Kim and K. Cho, J. Phys. Chem. C, 2014, 118, 760–766 CrossRef CAS.
- S. E. Gledhill, B. Scott and B. A. Gregg, J. Mater. Res., 2005, 20, 3167–3179 CrossRef CAS.
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736–6767 CrossRef CAS PubMed.
- S. Xu, V. I. Klimov, B. Kraabel, H. Wang and D. W. McBranch, Phys. Rev. B: Condens. Matter Mater. Phys, 2001, 64, 193201 CrossRef.
- S. Kajimoto, N. Yoshii, J. Hobley, H. Fukumura and S. Okazaki, Chem. Phys. Lett., 2007, 448, 70–74 CrossRef CAS.
- P. Guiglion, C. Butchosa and M. A. Zwijnenburg, Macromol. Chem. Phys., 2016, 217, 344–353 CrossRef CAS.
- D. E. Markov, E. Amsterdam, P. W. M. Blom, A. B. Sieval and J. C. Hummelen, J. Phys. Chem. A, 2005, 109, 5266–5274 CrossRef CAS PubMed.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- S. Gélinas, A. Rao, A. Kumar, S. L. Smith, A. W. Chin, J. Clark, T. S. Van Der Poll, G. C. Bazan and R. H. Friend, Science, 2014, 343, 512–516 CrossRef PubMed.
- H. Yao, Y. Cui, D. Qian, C. S. Ponseca, A. Honarfar, Y. Xu, J. Xin, Z. Chen, L. Hong, B. Gao, R. Yu, Y. Zu, W. Ma, P. Chabera, T. Pullerits, A. Yartsev, F. Gao and J. Hou, J. Am. Chem. Soc., 2019, 141, 7743–7750 CrossRef CAS PubMed.
- H. Yang, X. Li, R. S. Sprick, A. I. Cooper, R. Sebastian Sprick and A. I. Cooper, Chem. Commun., 2020, 56, 6790–6793 RSC.
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, T. Fei, Y. Dong, R. Sougrat, G. T. Harrison, Y. Firdaus, R. Sheelamanthula, B. Purushothaman, F. Moruzzi, W. Xu, L. Zhao, A. Basu, S. De Wolf, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Nat. Energy, 2022, 7, 340–351 CrossRef CAS.
- G. A. Jones and D. S. Bradshaw, Front. Phys., 2019, 7, 100 CrossRef.
- N. Banerji, J. Mater. Chem. C, 2013, 1, 3052–3066 RSC.
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, W. Zhang, M. Moser, R. Sheelamanthula, L. Zhao, B. Willner, T. C. Hidalgo, H. Faber, B. Purushothaman, M. Sachs, H. Cha, R. Sougrat, T. D. Anthopoulos, S. Inal, J. R. Durrant and I. McCulloch, Adv. Mater., 2021, 2105007 Search PubMed.
- W. C. Lin, J. Jayakumar, C. L. Chang, L. Y. Ting, M. H. Elsayed, M. Abdellah, K. Zheng, A. M. Elewa, Y. T. Lin, J. J. Liu, W. S. Wang, C. Y. Lu and H. H. Chou, Appl. Catal., B, 2021, 298, 120577 CrossRef CAS.
- W. Zhou, T. Jia, D. Zhang, Z. Zheng, W. Hong and X. Chen, Appl. Catal., B, 2019, 259, 118067 CrossRef CAS.
- J. G. Müller, U. Lemmer, J. Feldmann and U. Scherf, Phys. Rev. Lett., 2002, 88, 147401 CrossRef PubMed.
- E. Frankevich, H. Ishii, Y. Hamanaka, T. Yokoyama, A. Fuji, S. Li, K. Yoshino, A. Nakamura and K. Seki, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 2505–2515 CrossRef CAS.
- T. Liu, R. Tyburski, S. Wang, R. Fernández-Terán, S. Ott and L. Hammarström, J. Am. Chem. Soc., 2019, 141, 17245–17259 CrossRef CAS PubMed.
- M. Axelsson, C. F. N. Marchiori, P. Huang, C. Moyses Araujo and H. Tian, J. Am. Chem. Soc., 2021, 143, 21229–21233 CrossRef CAS PubMed.
- I. A. Howard, R. Mauer, M. Meister and F. Laquai, J. Am. Chem. Soc., 2010, 132, 14866–14876 CrossRef CAS PubMed.
- P. E. Keivanidis, T. M. Clarke, S. Lilliu, T. Agostinelli, J. Emyr Macdonald, J. R. Durrant, D. D. C. Bradley and J. Nelson, J. Phys. Chem. Lett., 2010, 1, 734–738 CrossRef CAS.
- S. Cao and L. Piao, Angew. Chem., Int. Ed., 2020, 59, 18312–18320 CrossRef CAS PubMed.
- H. Kisch and D. Bahnemann, J. Phys. Chem. Lett., 2015, 6, 1907–1910 CrossRef CAS PubMed.
- M. Qureshi and K. Takanabe, Chem. Mater., 2017, 29, 158–167 CrossRef CAS.
- J. R. Bolton, Sol. Energy, 1996, 57, 37–50 CrossRef CAS.
- S. Pokrant, Nature, 2020, 581, 386–388 CrossRef CAS PubMed.
- G. C. Roda, V. Loddo, L. Palmisano and F. Parrino, Heterogeneous Photocatalysis: Relationships with Heterogeneous Catalysis and Perspectives, 2019 Search PubMed.
- S. Wang, B. Cai and H. Tian, Angew. Chem., Int. Ed., 2022, e202202733 CAS.
| This journal is © The Royal Society of Chemistry 2022 |